 Hooriyah S. Rizavi1,2
Hooriyah S. Rizavi1,2 Kayla A. Chase3
Kayla A. Chase3 Chunyu Liu2,4
Chunyu Liu2,4 Hannah Gavin1
Hannah Gavin1 Cherise Rosen1Cuihua Xia5
Cherise Rosen1Cuihua Xia5 Alessandro Guidotti1
Alessandro Guidotti1 Rajiv P. Sharma1,2*
Rajiv P. Sharma1,2*- 1Department of Psychiatry, The Psychiatric Institute, University of Illinois at Chicago, Chicago, IL, United States
- 2Jesse Brown Veterans Affairs Medical Center, Chicago, IL, United States
- 3Department of Biochemistry and Molecular Biology, University of Illinois at Chicago, Chicago, IL, United States
- 4Department of Psychiatry, SUNY Upstate Medical University, Syracuse, NY, United States
- 5School of Life Sciences, Central South University, Changsha, China
The existence of repressive and durable chromatin assemblies along gene promoters or networks, especially in the brain, is of theoretical and therapeutic relevance in a subset of individuals diagnosed with schizophrenia who experience a chronic, persistent, and treatment-resistant trajectory. We used chromatin immunoprecipitation followed by deep sequencing (ChIP-Seq) to generate an epigenomic map that includes differential sites occupied by di-methylated lysine 9 of histone 3 (H3K9me2), a repressive modification that is yet unexplored in human postmortem brain tissue. We have discovered over 150 significantly differential promoter sites in the postmortem prefrontal cortex tissue of individuals diagnosed with schizophrenia (n = 15) when compared to controls (n = 15). Potentially dysregulated gene categories include postsynaptic proteins, processing enzymes (for proproteins, lipids, and oxidative stress), cadherin family genes, the complement system, and peptide hormones. Ten genes with significantly increased or decreased H3K9me2 promoter occupation were selected through statistical analysis, function, or previous GWAS association, and Quantitative RT-PCR (qRT-PCR) was performed on an extended sample of postmortem brain tissue, adding an additional 17 controls, 7 individuals with schizophrenia, and 19 individuals with bipolar samples (n = 32 control, 22 schizophrenia, 19 bipolar). This approach revealed that mRNA expression levels correlated with chromatin modification levels in eight of 10 selected genes, and mRNA expression in the total sample could be predicted by the occupancy of H3K9me2. Utilization of this method and replication in a larger sample open a pathway to durable and restrictive epigenomic assemblies whose accumulation across the lifespan of individuals diagnosed with schizophrenia may explain treatment resistance, and advance therapeutic options.
Introduction
The molecular basis of chronic, persistent, and treatment resistance in a subset of individuals diagnosed with schizophrenia (SCZ) can be informed by the study of durable tertiary cellular structures. Heterochromatin is one such structure and is seeded, assembled, and distributed along the chromosome by the post-translational modification of multiprotein components, as well as the underlying DNA strand. Because SCZ is characterized by long-term dysfunction in the frontal cortex, it follows that altered gene expression may be coordinated with the local epigenetic architecture of particular promoters and other regulatory sequences. Conceptually, studying these types of durable epigenetic modifications makes possible a dissection of a spatial/temporal stream of pathological events (immune, metabolic, psychological, stress, pharmacological) deposited in this molecular repository.
Heterochromatin that is characterized by the methylation of lysine 9 along the H3 histone tail (H3K9me2) is a chromatin assembly that is particularly linked to transcriptional repression. This type of chromatin assembly is often called “facultative heterochromatin” but is best described as permissively restrictive. H3K9me2 heterochromatin is located in gene-rich stretches of the genome and can facilitate nuclear localization and compartmentalization (1). Functionally, this modification can be distinguished from H3K9me1 or H3K9me3 and is targeted by histone methyltransferases (HMTs) such as G9a, GLP, and SETDB1 (2). H3K9me2 serves as a “ligand” for ambient “receptor” proteins of the heterochromatin family, particularly heterochromatin protein 1γ (HP1γ), which can be dislodged from this attachment by phosphorylation of the adjacent serine (H3S10-phos). The entire assembly anchored at H3K9me2/3 can include an array of epigenetic repressors such as Histone deacetylases (HDACs), DNA methyltransferases (DNMTs), and anchor platform adaptor proteins such as REST/CoREST as well as several identified lncRNAs that can cobble together these multiple components. Recent findings have identified up to 172 proteins embedded in this H3K9me2/3 seeded assembly (3, 4). This multiprotein complex is typically coordinated with local CpG methylation, another potentially lifelong covalent modification (5), providing a durable anchor for these repressive complexes (6, 7). H3K9me2 heterochromatin is dynamic and can assemble in response to oxidative stress (neurons) or prolonged kinase signaling (e.g., endotoxin tolerance in macrophages) (8–10). Once nucleated, H3K9me2/3 heterochromatin can spread up to distances of 15 kb along the DNA strand (11) and can sequester large regions of the epigenome to the nuclear periphery rendering these regions resistant to gene activation (1, 12, 13).
The ENCODE project has H3K9me2 data on 14 immortalized cell lines and three in vitro differentiated cell types. The three differentiated cell types include one bipolar neuron, one neural cell, and one hepatocyte. The Roadmap Epigenomics and PsychENCODE projects have not yet generated data on this histone modification. In other words, H3K9me2 has not been studied in a human population sample, particularly in the brain. Given the deficiency of scientific literature in this area, there is urgency to develop a reference map of H3K9me2 in human brains and explore H3K9me2 in individuals who experience chronic, persistent, and treatment resistant psychosis and in non-clinical controls.
Our laboratory has previously reported elevated global levels of H3K9me2 protein in the parietal cortex from postmortem brain tissue samples of individuals diagnosed with SCZ (14). Additionally, we identified elevated mRNA levels of the enzymes responsible for catalysis of H3K9me2 (GLP, G9a, and SETDB1) in postmortem brain tissue, as well as peripheral blood mononuclear cells (PBMCs) from individuals with SCZ (14, 15). Finally, we reported that this assembly is reversible by small molecule inhibition of its methyltransferases (16, 17) as well as kinase inhibitors modifying histone phosphorylation on site specific promoters (17). Because global protein levels do not identify the regulation of individual genes, we advanced these findings with a promoter-targeted approach. Here, we pioneered the examination of genome-wide occupancy of H3K9me2 in the postmortem frontal cortex tissue of individuals with schizophrenia and attempted to understand whether causation can be implied through the increase or decrease of gene expression in networks crucial to natural development and function.
Chromatin immunoprecipitation followed by deep sequencing (ChIP-Seq) for H3K9me2 in the postmortem brain tissue of individuals with schizophrenia has not previously been performed and can provide an illustrative example of the benefits of epigenome mapping. For logistical reasons, we have restricted the ChIP-Seq analysis to promoter regions in 30 prefrontal cortex samples [15 non-psychiatric controls (NPC) and 15 SCZ]. We selected H3K9me2 since it is a strongly repressive type of heterochromatin associated with euchromatin sectors of the genome, and conceptually could be reversed to allow facile expression of genes in the underlying and coordinate regions. In order to determine if the identified expression changes secondary to heterochromatin were specific to individuals with SCZ we directly measured mRNA levels using qRT-PCR in a larger and inclusive cohort of brain samples (NPC, n = 32, SCZ, n = 22, BPD, n = 19) that included individuals diagnosed with bipolar disorder (BPD). Finally, we performed a correlational analysis between mRNA expression and H3K9me2 promoter occupation in the brain.
Materials and methods
Postmortem brain tissue samples
Human postmortem brain tissue samples [Prefrontal cortex, Brodmann area 9 (PFC)] were obtained from the Harvard Brain Tissue Resource Center that were donated by individuals diagnosed with schizophrenia, bipolar, and controls. ChIP-Seq for the repressive modification H3K9me2 was performed on 30 postmortem PFC from individuals with SCZ (n = 15) and NPC (n = 15). ChIP-Seq mRNA validation using qRT-PCR in a larger cohort of brain samples from the same collection included three diagnostic groups NPC (n = 32), individuals with SCZ (n = 22), and individuals with BPD (n = 19). This expanded set included the 15 NPC and 15 individuals with SCZ samples that were initially analyzed with ChIP-seq. A summary of demographic parameters is given in Table 1.
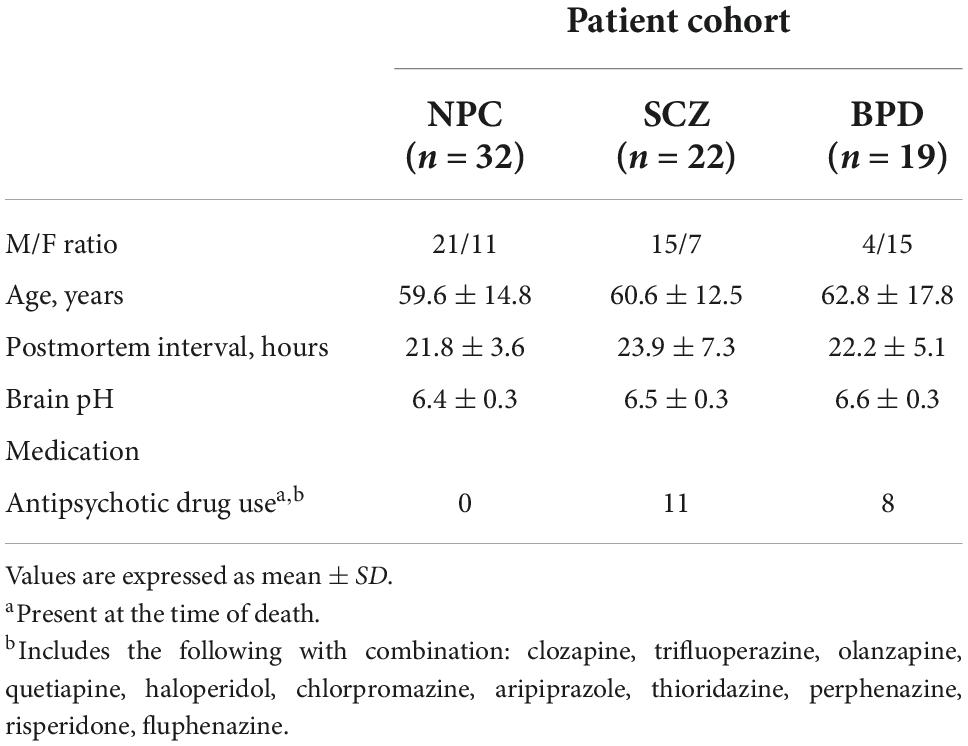
Table 1. Subject demographics and clinical characteristics.
Chromatin immunoprecipitation with sequencing
ChIP was performed according to a previously published procedure (16). Briefly, an aliquot of human brain tissue was used for each experiment and homogenized in 500 μl of RPMI1640 media (GIBCO #11875-093). Proteins were cross-linked to DNA by adding 48.25 μl of methanol-free formaldehyde (Thermo #28908) and incubated at 37°C for 5 min. The sample was then quenched with 70.5 μl of 1M glycine, spun down, the supernatant removed, and washed with PBS in the presence of 1:100 protease inhibitor (Calbiochem #539134). The samples were spun-down again, the supernatant removed, and resuspended in SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.1), again in the presence of 1:100 protease inhibitor. Samples were then sonicated for 20 min at 10%df in a Covaris m220 sonicator. Further processing was performed using a ChIP assay kit (#17-295; Millipore, Upstate), and DNA was precipitated by standard ethanol precipitation. Lysates were immunoprecipitated (IP) with H3K9me2 monoclonal antibody (Abcam, ab1220) that was validated by both peptide ELISA and Western blot and is specific to only H3K9me2, not H3K9, H3K9me1, H3K9me3, H3K27me2/3, or H3K4me1/2/3, as verified in previous ChIP studies (17–19). Samples were then examined for quality prior to sequencing through qPCR for previously established negative (GAPDH) and positive (ZNF333 and UGT1A10) control genes for the H3K9me2 modification (20) (Supplementary Figure 1A).
Quality control
Sample quality was examined in the following ways. First, a small quantity of the sonicated chromatin was run on an agarose gel to ensure fragment size was within 500–1,000 bp. Secondly, after IP and DNA extraction, DNA concentrations of the input and IP were measured via Qubit fluorometric assay. Finally, pulldown efficiency was determined by qPCR of the IP product. We verified GAPDH as a transcriptionally active promoter (not containing H3K9me2) and two genes ZNF333 and UGT1A10 as transcriptionally inactive promoters and thus would contain high H3K9me2 occupancy (Supplementary Figure 1) (20). There was no qPCR amplification of GAPDH promoter, while the ZNF333 and UGT1A10 show amplification and thus successful pulldown by the antibody. Samples that exhibited GAPDH promoter pulldown were discarded because of cross-reactivity or contamination and were not used for library preparation or sequencing (for example, sample #1113 in Supplementary Figure 1). These samples were reprocessed using fresh brain tissue for successful pulldown if necessary.
Library construction and sequencing
After completion of the ChIP-Seq protocol, frozen immunoprecipitates were sent to the University of Illinois at Urbana Champaign for library construction and sequencing on an Illumina HiSeq 2500, followed by a data quality control pipeline. Samples were run at four samples per lane, returning 130–180 million reads, putting the average sequencing depth for each sample at approximately 40 million single-end reads (21, 22). We utilized single-end reads due to our focus on promoter regions, as the information on duplications, inversions, splice variants, or SNPs provided by double-end reads was not a priority. The quality of the runs was determined by FASTQC. An example of an acceptable FASTQC output from our data is provided in Supplementary Figure 1B.
Alignment of sequenced reads
Reads were trimmed at both ends using Trimmomatic (23). Genome alignment was performed using Bowtie2, which was selected for its utility in aligning short DNA sequence reads to long mammalian genomes (24, 25). These alignments were visualized using the UCSC Genome Browser; examples for two selected genes are shown in Supplementary Figure 2. Multiple aligned sequences and duplicate reads were discarded before normalization. We limited our region of interest to a 10 kb window centered at the transcription start site (TSS)—a defined promoter region of fixed but approximate length since the upstream boundaries of most promoter regions are not well defined.
GWAS enrichment analysis
Starting with the Working Group of Psychiatric Genomics Consortium 1 (PGC) SCZ GWAS summary statistics, we searched the significant differentially modified promoters from our data set against the 25,069 genes. Next we performed the hypergeometric distribution test using the software R 4.1.0 to see which promoters were enriched in the PGC3 SCZ GWAS significant associated genes. The PGC3 SCZ GWAS summary statistics (PGC3_SCZ_wave3.primary.autosome.public.v3.vcf.tsv.gz) was downloaded from PGC website,1 and the SNPs were annotated to genes using the software ANNOVAR (26) based on hg19_avsnp142 database (Supplementary Table 2).
Identification and analysis of H3K9me2 sites in cases and controls
DiffReps were used to detect differential H3K9me2 modification sites, again limiting our search to the promoter regions of annotated genes (27). According to the evaluation of tools for ChIP-seq data analysis, this is the best choice for data analysis with biological replicates without predefined regions of interest (28). DiffReps analyzes an entire ChIP-Seq dataset using a fix-sized sliding genomic window, where only the reads falling inside the window are counted. The sliding window analyzes a fixed length as it is moved along the genome in fixed steps (e.g., window size 1 Kb moving along at 100 bp per step). The sliding window strategy is advantageous for a more “smeared” modification such as H3K9me2, since it is independent of any “peak” calling program. DiffReps output data includes not only nearby gene annotation but also other gene features (e.g., gene body, centromere, promoter, gene desert) allowing us to focus on gene promoters. DiffReps calculates case-control group difference directly, allowing us to identify gene-promoters based on fold change as well as both raw and Padj values (FDR) to determine a significant difference between cases and controls.
Quantitative RT-PCR
Expanding the sample of postmortem brain tissue to include the same original ChIP-seq samples and an additional 17 NPC, 7 individuals with SCZ, and 19 individuals with BPD, total RNA was isolated using TRIzol reagent (Life Technologies) and treated with DNase (Ambion/Life Technologies #AM1906) after extraction. Total RNA was used to prepare cDNA via the Applied Biosystems High Capacity cDNA Reverse Transcription Kit (#4368814). For detection and measurement of expression, Fermentas Maxima SYBR Green/ROX qPCR Master Mix (#K0222) was used. PCR mixtures were run on a Thermo Scientific PikoReal real-time PCR System using manufacturer cycling conditions. Cycle threshold (CT) value was used for relative quantification, and all values were normalized to GAPDH, and run in duplicate. Ten genes were selected for qPCR assessment, taking into account combinatorial best fold change and adjusted p-values from our ChIP-seq data Table. Primer sequences are provided in Supplementary Table 3.
Results
Differential heterochromatin occupancy of 159 promoters
DiffReps output data contained 159 differentially modified promoters based on a raw p-value of p < 0.01, once all other gene features (intergenic areas, pericentromere, etc.) were cleared from the output (Supplementary Table 1). Of the 159 promoters, 82 have increased H3K9me2 occupancy, while 77 have decreased occupancy in individuals with SCZ when compared to NPC. Five genes (AKT3, C4B, FURIN, GPX6, and NRGN) were common to the PGC2 SCZ GWAS data set available to us during the project, and three of these genes (AKT3, FURIN, GPX6) remained common to the most recent PGC3 SCZ GWAS data set. Three genes, FURIN, GPX6, and NRGN have increased H3K9me2 promoter occupancy and two genes, AKT3 and C4B show decreased promoter occupancy. Five additional genes (GPHN, OXT, CDHR2, CDH20, LIPJ) were selected, two (GPHN, OXT) based on previous associations in the SCZ literature (29, 30) and three (CDHR2, CDH20, LIPJ) are novel with no consistent link to individuals with SCZ. C4B was included with an additional focus of its identification with the PGC2 GWAS analysis (2014) and its central role in immune function; an isotypic allele C4A was noted to be significantly higher in the brain of individuals with SCZ (31). The statistical analysis of diffReps on the ten regions chosen to be analyzed using qPCR is reported in Table 2. The full list of 159 promoter regions with differentially modified H3K9me2 occupancy is provided in Supplementary Table 1.
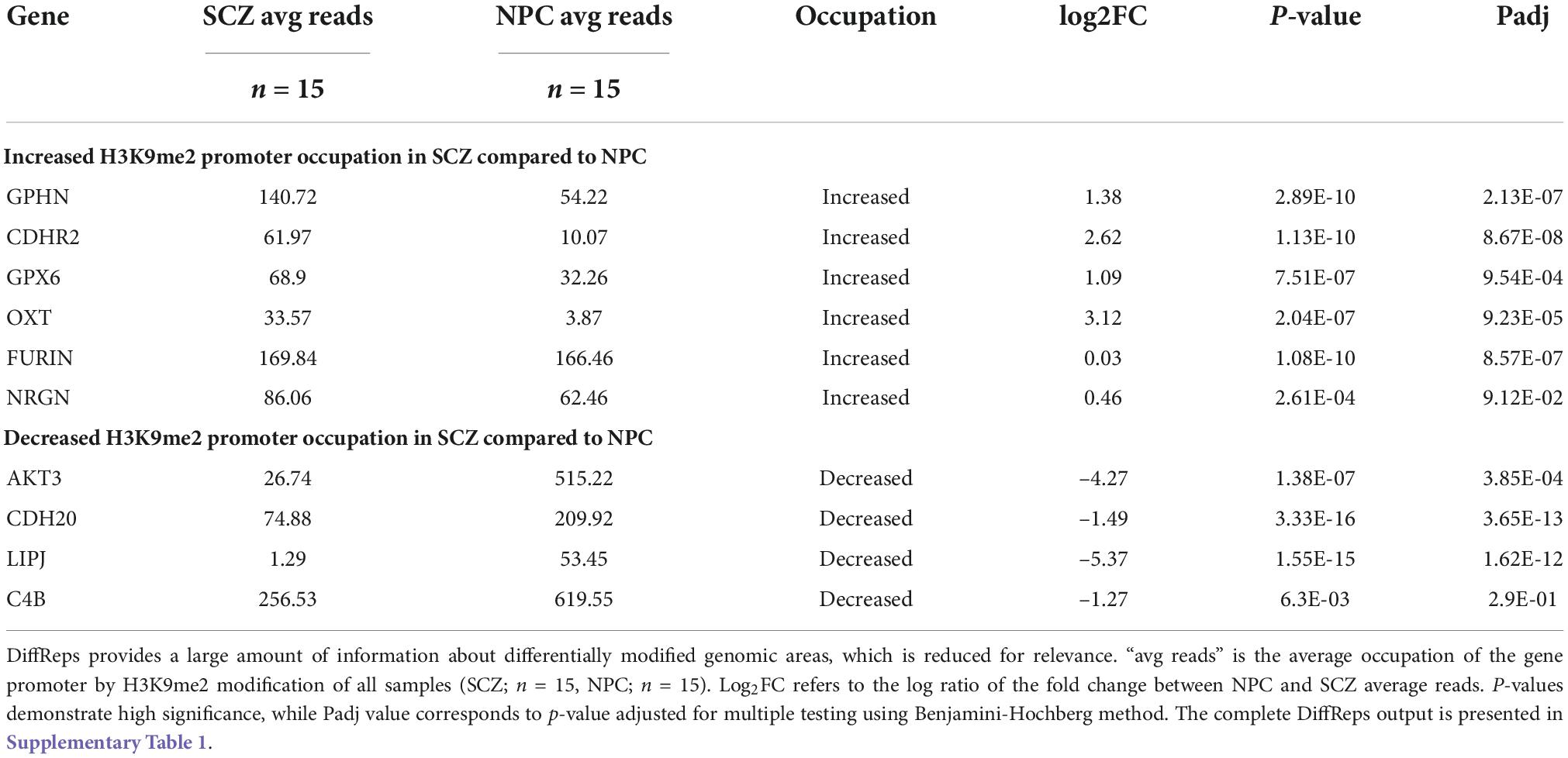
Table 2. DiffReps output.
mRNA expression is coordinated with H3K9me2 occupancy
Ten genes were selected for qPCR analysis using a larger cohort. Five genes (GPHN, CDHR2, FURIN, GPX6, and OXT) of the six genes which showed higher H3K9me2 promoter occupancy also resulted in significantly decreased mRNA expression in individuals with SCZ compared to NPC (Figure 1). Three of the four genes with decreased H3K9me2 promoter occupation in individuals with SCZ (AKT3, C4B, and CDH20) showed significantly increased mRNA expression in individuals with SCZ compared to NPC. When we include the individuals with BPD samples, they exhibit a similar level of mRNA expression to NPC for all 10 genes examined, indicating the effect is specific to individuals with SCZ (Figure 1). Next we performed a correlation analysis between individual promoter H3K9me2 ChIP-Seq reads and mRNA expression of the occupied gene (Figure 2). For all subjects, seven of the 10 genes analyzed were found to have highly significant negative correlations, as shown in Figure 2, supportive of the repressive nature of the H3K9me2 assembly. Of the genes that did not reach significance in their correlations (FURIN, NRGN, and LIPJ), only FURIN achieved significance between individuals with SCZ compared to NPC in the mRNA expression data. We suspect this to be the case due to the closeness in average promoter reads (169.84 vs. 166.46) between the NPC and individuals with SCZ groups. NRGN and LIPJ are perhaps the most curious of all genes analyzed, as the diffReps analysis provided what appeared to be significant enrichment of H3K9me2 on promoters between the NPC and individuals with SCZ groups. Still, neither gene reached significance in either the mRNA expression data or correlational analysis.
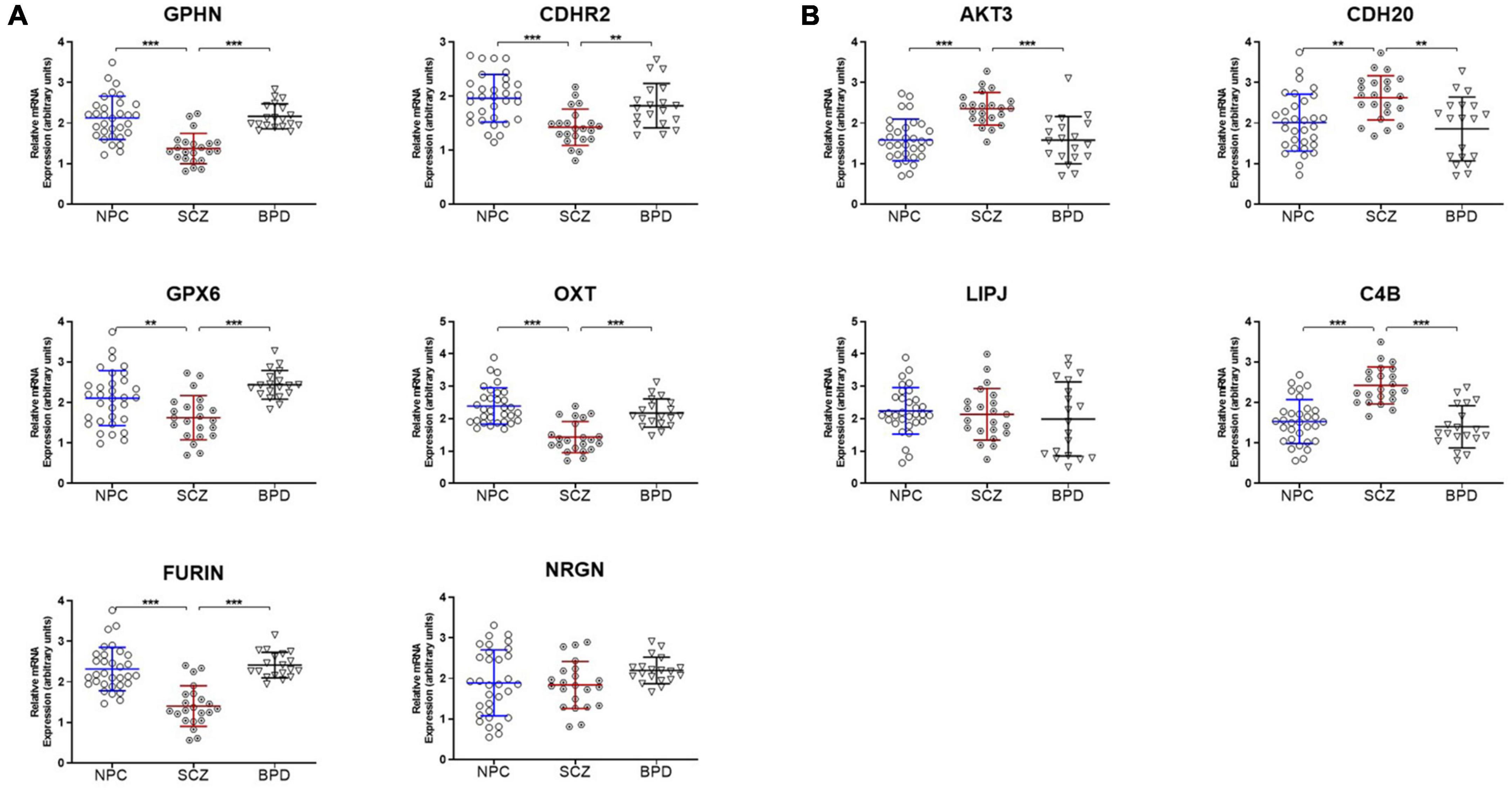
Figure 1. mRNA expression is coordinated with promoter H3K9me2 occupation. (A) Genes with increased H3K9me2 occupation on their promoters in SCZ samples demonstrate decreased mRNA expression. GPHN, CDHR2, FURIN, GPX6, OXT, and NRGN all demonstrated significantly increased promoter occupancy of H3K9me2. After the ChIP-seq discovery phase in the initial 15 NPC and 15 SCZ brain samples, an additional 17 NPC, 7 individuals with SCZ, and 19 individuals with BPD samples were included and processed for mRNA expression of these genes, all but NRGN showed decreased expression in SCZ when compared to NPC or individuals with BPD samples. **p < 0.01, ***p < 0.005. A table of statistical analysis is provided in Supplementary Table 1. (B) Genes with decreased H3K9me2 occupation on their promoters in SCZ samples demonstrate increased mRNA expression. AKT3, C4B, CDH20, and LIPJ were four genes found to have decreased H3K9me2 promoter occupancy in the SCZ brain samples. When processed for mRNA expression, AKT3, C4B, and CDH20 showed increased mRNA expression, but LIPJ did not.
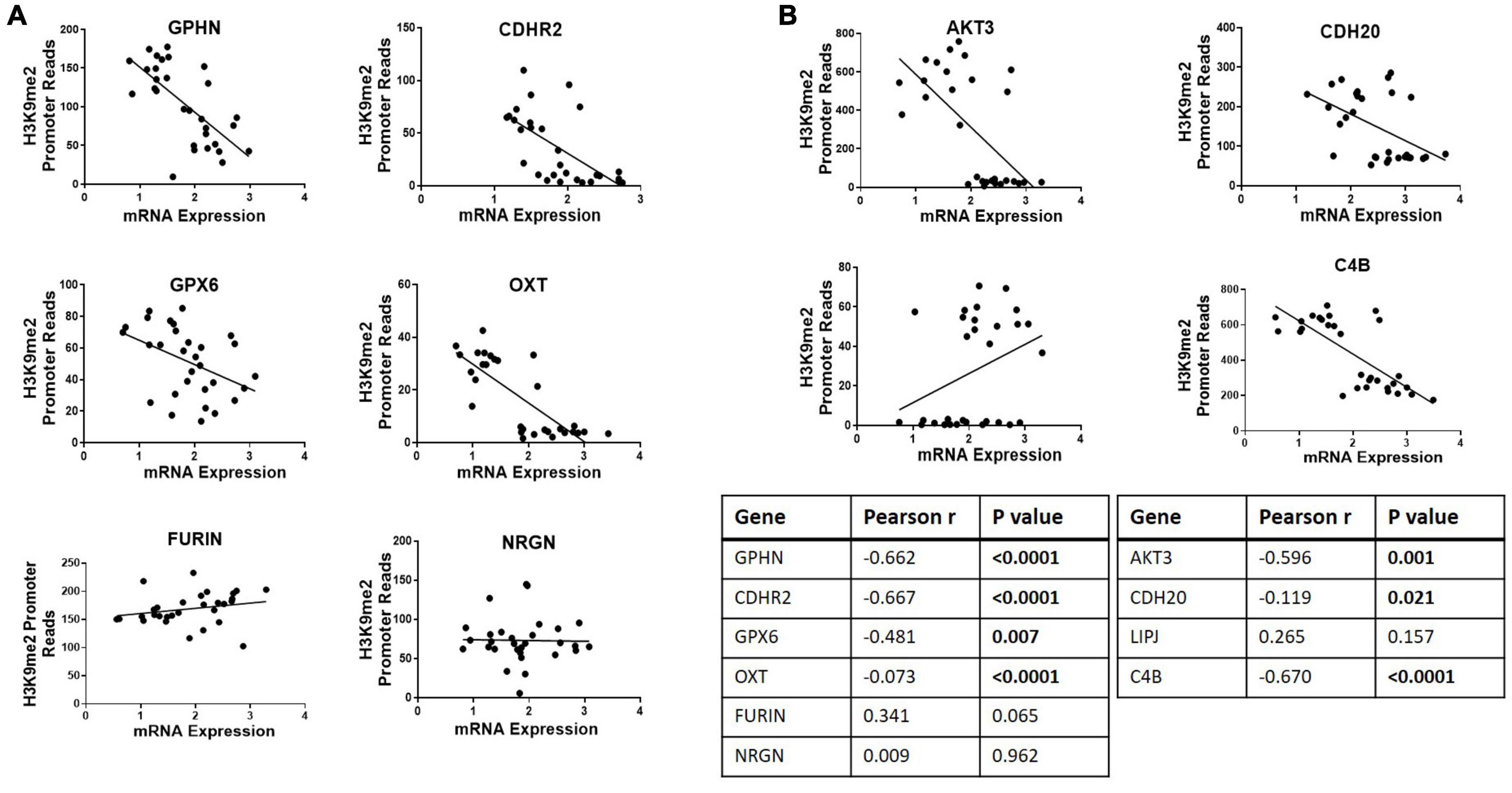
Figure 2. Correlational analysis of H3K9me2 promoter occupation and mRNA expression. Scatterplots showing correlations between H3K9me2 promoter DiffReps reads and mRNA levels of the corresponding gene. (A) Promoter loci where H3K9me2 occupancy is significantly increased in SCZ compared to NPC. Promoters GPHN, CDHR2, GPX6, and OXT show H3K9me2 diffReps reads have significant negative correlation with their corresponding mRNA levels. (B) Promoter loci where H3K9me2 occupancy is significantly decreased in SCZ compared to NPC. Promoters AKT3, CDH20, and C4B show H3K9me2 diffReps reads have significant negative correlation with their corresponding mRNA levels. Pearson’s r and P-values for each loci are shown in the insert table.
Enrichment of GWAS signals from the PGC3 schizophrenia analysis
To match our list of heterochromatinized gene promoters to the most recent PGC3 SCZ GWAS findings we filtered at the FDR < 0.05 level. We matched 143 promoters that are differentially occupied with H3K9me2 to the significantly associated gene identities form the PGC3 SCZ study (32). We identified 19 promoters (5 with increased heterochromatin and 14 with reduced heterochromatin) which also had significant (P < 5e-08) SCZ GWAS signals (Supplementary Table 2). This PGC3 enrichment coordinated with promoter heterochromatin levels was significant (hypergeometric distribution test p = 0.022).
Potential confounding variables
SPSS was used to analyze the possible effects of several confounding factors on the diffReps and qPCR data. There was no significant difference in either diffReps or qPCR data when we compared the individuals with SCZ group with and without antipsychotics, the individuals with BPD group with and without antipsychotics, individuals with SCZ and individuals with BPD grouped together and compared with and without antipsychotics. We found no significant effect of demographic variables; age, sex, brain pH, PMI among the three groups (NPC, SCZ, and BPD), indicating that these variables will not affect our conclusions.
Epigenetic enzymes/proteins underwriting heterochromatin assembly
Because the target molecule in this study was the assembly of heterochromatin, we also measured mRNA levels of several canonical enzymes critical in producing heterochromatin. These include three HMTs responsible for the H3K9 methylation; GLP and G9A primary for H3K9me2 and SETDB1 for H3K9me3. In addition, we also measured the REST protein (RE1-Silencing Transcription factor), an adaptor/platform protein coordinating heterochromatin assembly alongside other repressor proteins (33, 34). Binding of REST protein to its cognate motif results in an accumulation of H3K9me2/3 heterochromatin (34). The results are presented in Figure 3, indicating an increase in the three measured methyltransferases (GLP, G9A, and SETDB1) and a decrease in the REST protein. Together these results could suggest a chromatin environment conducive to heterochromatin formation in the brain of individuals diagnosed with SCZ.
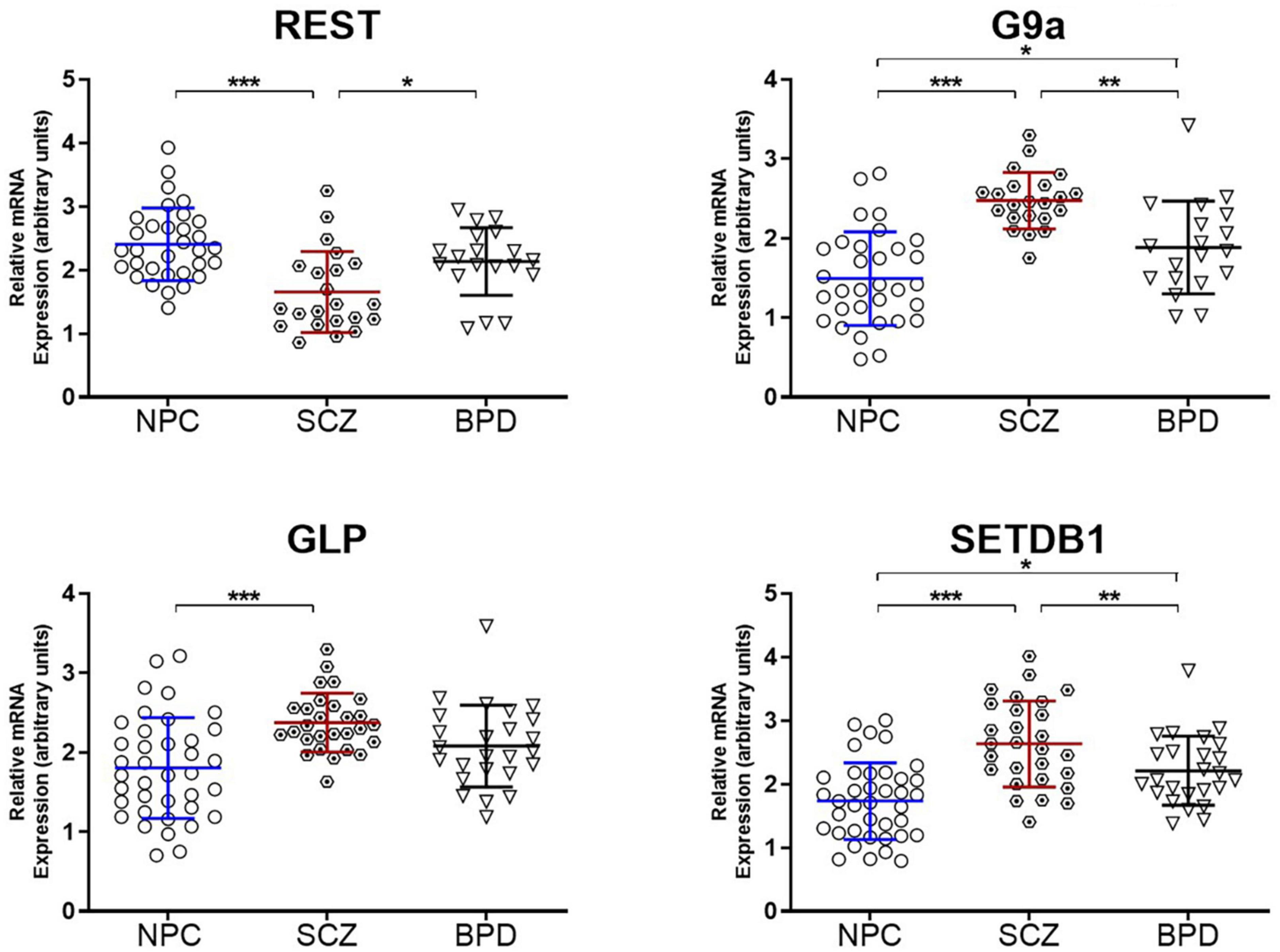
Figure 3. Epigenetic enzymes underwriting heterochromatin formation. Relative mRNA expression levels in the PFC of Repressor element-1 silencing transcription factor (REST) and H3K9 histone methyltransferases (G9a, GLP, and SETDB1). Data is shown as fold change (FC) ± S.E.M. (error bars) after normalization to GAPDH and case compared to NPC. NPC (n = 32), individuals with SCZ (n = 22), individuals with BPD (n = 19). *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Discussion
We have demonstrated that genome-wide sequencing of H3K9me2-bound regions of DNA can provide valuable insight into differentially modified regions and inform correlated mRNA expression levels in the postmortem frontal cortex tissue of individuals with schizophrenia and control subjects. As this was the first sequencing study of its nature for this modification, some gene selections were based on previous disease associations, but also some novel exploratory genes were selected based on their statistical significance and fold change. Using the stringApp in Cytoscape we retrieved functional enrichment analysis results for the 159 H3K9me2 differentially occupied promoter sites listed in Supplementary Table 1 (Figure 4). To visualize and identify groups of proteins that exhibit similar changes in H3K9me2 occupancy we used the K-means clustering method in STRING. This resulted in a network with three clusters, an average local clustering coefficient of 0.36, and an average node degree of 1.04. Identifying enriched genes involved mainly in neurotransmitter/receptor neuronal function, hormone/hormone receptor function, and the immune system. Additionally, we see a high degree of connectivity of AKT3-MAPK3-MAPK10 protein network. All analyses were performed using Cytoscape version 3.9.1, stringApp version 1.7.0, and clusterMaker2 version 2.2. Connections with the Kinase signaling network noted is exciting given the role of MAPK pathways in modifying heterochromatin (35). The nuclear kinase mitogen- and stress-activated protein kinase 1 regulates hippocampal chromatin remodeling in memory formation. Furthermore, our lab has previously demonstrated the role of kinase signaling in “disassembling” heterochromatin using the antipsychotic risperidone (36).
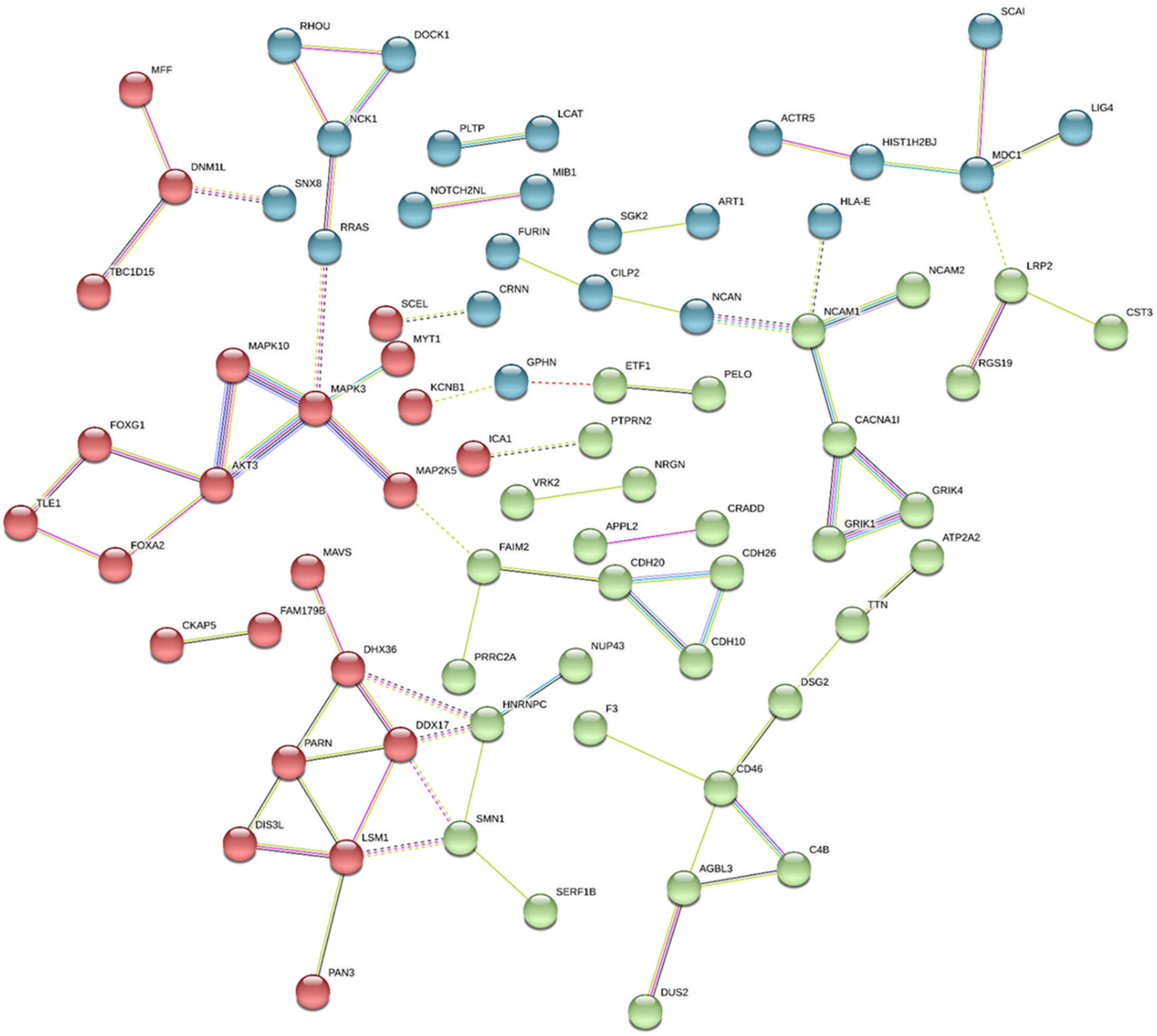
Figure 4. Pathway and Gene Ontology analysis. Using the Cytoscape STRING app, a network was retrieved for the 159 promoters with differentially modified H3K9me2 occupancy. Genes with significantly increased or decreased H3K9me2 levels were selected by the diffReps program using a p-value > 0.05 and were entered in the “Network and Enrichment Analysis” within the STRING app. With a confidence cutoff of 0.4, kmeans clustering was used to generate 3 clusters (red, blue, and green). Resulting associations within and between clusters is depicted with solid and dotted lines, respectively. Different color lines indicates different type of interactions. (Cyan-from curated databases; Magenta-experimentally determined; Blue-gene co-occurrence; green-from text mining; Black-coexpression; lilac-protein homology; Red gene fusions).
H3K9me2 is known to occupy gene-rich territories and is known to be modifiable using pharmacology directed to one of its primary methylating enzymes G9a. Heterochromatin has durability and is likely responsible for the reduced expression of non-neuronal genes as in the heterochromatin assemblies coordinated with the REST/Co-REST adaptor proteins (37).
Heterochromatin regulation of synaptic genes
Gephyrin (GPHN) is a scaffold protein that docks GABAa receptors to the postsynaptic membrane (38). Abnormal GPHN clustering during neurodevelopment is associated with the pathogenesis of neurological, neurodevelopmental, and psychiatric disorders (39), with exonic deletions implicated in the risk for schizophrenia, autism, and epilepsy development (40, 41). GPHN SNPs have also been associated with schizophrenia (41). Furthermore, a recent study showed that enrichment of copy-number variations in GABAergic genes from individuals with schizophrenia that were identified duplications and deletions in the region of GPHN (42). Deficient GPHN function, as indicated by decreased expression in our samples, impairs inhibitory GABAergic synaptic activity, which results in hyper-excitability of neural cells, as seen in epilepsy (43).
Cadherin family members CDHR2 and CDH20 are intercellular adhesion molecules associated with psychiatric disorders (44–46). Variable expression of these proteins may alter synaptic connectivity and information processing in the developing brain. Epilepsy, autism, bipolar, and schizophrenia are associated with cadherin dysfunction, mostly in genome-wide association studies (44, 46). Our expression analysis revealed increased H3K9me2 and decreased expression of CDHR2; decreased H3K9me2, and increased expression of CDH20, suggesting that symptoms typically reported by individuals with schizophrenia may be caused by dysregulation spread throughout the cadherin system.
Neurogranin (NRGN) is a neuron-specific calmodulin binding protein, expressed in postsynaptic dendritic spines modulating the activity of downstream calmodulin-Ca2+-dependent enzymes that function in the neuroplasticity mechanisms of learning and memory (47). Altered mRNA expression is demonstrated in postmortem brain tissue of individuals with schizophrenia (48–50). Altered CSF levels of NRGN are associated with the impairments in cognition seen in both Alzheimer’s and Parkinson’s disease (51).
Heterochromatin regulation of catalytic activity
FURIN is necessary for axonal growth and development of dendritic arbors, a process linked to neurological diseases of the CNS (52). Knockdown of FURIN in human neural progenitor cells results in abnormal neuronal migration (53). In a study examining RNA-seq from over 500 dorsolateral prefrontal cortices, FURIN emerged as one of five single-gene loci implicated in schizophrenia. Decreased expression of this gene due to heterochromatinization could result in disruption of many key systems, potentially contributing to comorbidities associated with schizophrenia.
Glutathione Peroxidase 6 (GPX6) is part of a family of enzymes that protect cells from oxidative stress, resulting from an imbalance between reactive oxygen species (ROS) production and deficiency of antioxidants to process the ROS, leading to deleterious peroxidations of lipids, proteins, and DNAs. The brain is especially vulnerable to oxidative damage due to its high utilization of oxygen, high content of oxidizable polyunsaturated fatty acids, and the presence of redox-active metals (54). Oxidative stress is hypothesized to contribute to symptomology reported by individuals with schizophrenia, specifically along the Tryptophan/Kynureine/Quinolinic acid pathway, which can be induced by cytokines such as IFNγ, long noted to be abnormally regulated in schizophrenia (55). Developmental dysregulation of glutathione synthesis combined with environmental risk factors that generate oxidative stress may result in deficits in neural connectivity and synchronization observed in individuals with schizophrenia (56–58).
Though Lipase family member J (LIPJ) is not well studied, the general lipase family increases after antipsychotic treatment (59). Therefore, our non-significant mRNA expression results may be a result of the variable antipsychotic dosing present in our sample. We have also shown that antipsychotic treatment in vitro can decrease heterochromatin levels and increase promoter phosphorylation and mRNA expression of genes that are tolerized (heterochromatinized) via an endotoxin tolerance paradigm, while other promoters do not respond in this way (36, 60). This targeted response to antipsychotic treatment could be an explanation for the LIPJ and NRGN genes that did not show coordination with their H3K9me2 promoter occupation levels.
The AKT Serine/Threonine Kinase 3 (AKT3) genomic locus is a replicated GWAS signal in individuals with schizophrenia [Schizophrenia Working Group (61)]. AKT signaling plays a critical role in cell growth, proliferation, survival, differentiation, and metabolism. AKT3 is highly expressed in the brain, with expression peaking during development, indicating a crucial role in cognitive function (62). AKT3 genetic abnormalities result in intellectual disabilities (63–67), and deficits in temporal order discrimination and spatial memory are seen in AKT3 knockout mice (68). Protein levels of AKT3 family member AKT1 are significantly reduced in lymphocytes and the brain of individuals diagnosed with schizophrenia (69). However, our findings seem to conflict, as AKT3 expression is increased in postmortem prefrontal cortices in individuals diagnosed with schizophrenia (Figure 1).
Oxytocin is a peptide hormone acting on both central and peripheral targets to decrease cortisol release in response to social stresses (70). In individuals with schizophrenia, plasma oxytocin levels are elevated in response to trust-related interactions, an effect not seen in normal controls (71). Low oxytocin levels also correlate with negative symptoms in individuals with schizophrenia (72). Our data found significant H3K9me2 enrichment on the OXT promoter, correlating with significantly decreased oxytocin mRNA levels in postmortem brain tissue of individuals with schizophrenia. Due to the function of the hormone, it is hypothetically valid to suggest that heterochromatin promoter occupancy and decreased oxytocin mRNA expression could limit the plastic response to social stress or interactions seen in individuals with schizophrenia.
The C4 locus on chromosome 6 has a strong association with postmortem brain tissue in individuals with schizophrenia (31, 73) and expresses two isotype molecules, C4A and C4B (74). As both complements serve a crucial role in synaptic pruning (75), it follows that dysregulation of this process can result in aberrant connections that can underlie neurological disorders. Most recently, our lab showed that C4A mRNA expression is positively associated with psychotic symptomology, including the severity of delusions (76). Our data support a linkage between aberrations in postmortem brain tissue in individuals with schizophrenia, as we found that at the C4B promoter, H3K9me2 was significantly decreased, resulting in increased mRNA levels.
Epigenetic enzymes/proteins underwriting heterochromatin assembly
We have previously demonstrated elevated levels of both mRNA and protein in two separate tissues obtained from individuals with schizophrenia; parietal cortical samples from the Stanley Foundation Neuropathology Consortium and lymphocyte samples from the University of Illinois at Chicago (UIC) (14). In both tissues types, we measured mRNA expression of HMTs, GLP, G9a, and SETDB1 via real-time RT-PCR and H3K9me2 levels via western blot. We have replicated this in our current study using frontal cortex from the Harvard Brain Tissue Resource Center. Elevation of the mRNA of these HMTs suggest a cellular/neuronal environment conducive to heterochromatin formation. We additionally measured mRNA levels of REST protein, which is a platform adaptor protein that coordinates the multicomponent assembly of Heterochromatin proteins, and note that expression levels are differentially reduced in individuals with schizophrenia.
Limitations
There are several sources of variation that could misalign our results when comparing DNA promoter occupancy (estimation of DNA sequences from the smaller ChIP-Seq subset) to its downstream gene expression (mRNA measured by qRT-PCR and presented in Figure 1). These could include variations in the expanded cohort utilized purely for qRT-PCR, unidentified sources of gene regulation that could impact gene mRNA expression levels, and additional regulatory factors acting downstream of the promoter. For example, the NRGN locus shows significantly increased H3K9me2 occupancy (refer to p-value in Table 2), while the LIPJ locus has significantly decreased H3K9me2 occupancy (refer to p-value in Table 2). While conceptually, we would see the coordination of promoter occupancy and mRNA expression, these two measurements (ChIP-seq vs. qRT-PCR) are uncoupled for these two loci.
We did not perform ChIP-seq on subjects with BPD, and therefore, we can only state that heterochromatin itself is differentially located, in SCZ, on the promoter sites provided in Supplementary Table 1 and note that the mRNA expression of these promoters in BPD is not different from the mRNA expression in NPC, allowing us to conclude that the selected promoters are “heterochromatinized” in SCZ and not BPD samples.
There are universal confounds in all translational or clinical tissue-oriented studies including the effect of pharmacology/psychotropics, obesity, drugs of abuse, trauma, or environmental factors related to psychosocial disparities and these have not been resolved. Limitations of ChIP-Seq directed to a specific histone modification are impacted by the heterogeneous population of neuronal/non-neuronal cells. This is approached with techniques such as laser capture dissection of unique neuronal populations or FACS sorting according to neuronal markers (77), but this was not feasible given the tissue requirements of ChIP-Seq.
Conclusion and summary
We have demonstrated the experimental identification of specific gene promoters differentially occupied by H3K9me2 heterochromatin in the PFC of postmortem brain tissue in individuals diagnosed with schizophrenia. We have also demonstrated a functional effect of promoter occupation by this modification on mRNA gene expression in a coordinated manner. We demonstrate differential modification levels on the promoters of several genes that have previously been linked to individuals with schizophrenia, in addition to genes that are novel to the disorder. Some of the genes featured here offer coherent functional explanations to established hypotheses (i.e., Gephyrin expression and the hypothesis of excitatory and inhibitory synapse imbalance as it relates to individuals with schizophrenia), while others will require further exploration to determine their role in the development or maintenance of disease.
Our ability to demonstrate differences in heterochromatin along specific gene promoters in the brains of individuals diagnosed with schizophrenia opens up new hypotheses and therapeutic targets. We have earlier noted that heterochromatin assemblies are durable and can persist for the life span especially in neurons (78). Theoretically, they possibly serve as repositories for the accumulation of psychological, metabolic or pharmacological inputs over extended periods (79). In treatment resistant individuals, the blockade of gene promoters by durable and stable repressive heterochromatin could explain why targeting of membrane monoamine synaptic receptors is not adequate in disassembling these deep genomic structures and the inability to activate the underlying gene networks (79). Our findings suggest that alternate/parallel signaling pathways capable of targeting genomic heterochromatin may provide a supplemental advantage.
Considering this approach, we have elsewhere demonstrated that signaling along the kinase pathways in cell cultures can target heterochromatin assemblies by regulating histone phosphorylation (36). Because heterochromatin is a stable modification and in equilibrium with its regulatory factors, its measurement along specific gene networks in the neuron would allow a reverse engineering approach to study “what stress” deposits heterochromatin on “which network.” This approach has been utilized using defined immunological stimuli to assemble heterochromatin on pre-defined immune gene promoters (15, 36, 80–82).
To our knowledge, this is the first study of H3K9me2 based heterochromatin promoter assembly in the postmortem brain tissue in individuals diagnosed with schizophrenia and is directly relevant to gene expression. These modifications may be particularly relevant in individuals who are resistant to available psychotropics, suggesting gene networks that are not receptive to usual monoamine receptor signaling. Future studies aiming to reveal new potential targets for treatment advancement will attempt to map the distribution of other restrictive chromatin assemblies such as those originating with H3K27me3, as well as mapping regions of the genome that are primed for transcription such as those occupied by H3K4me1/2/3.
Data availability statement
The original contributions presented in the study are publicly available. The data presented in the study are deposited in the GEO repository, accession number GSE215991. This data can be found here: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE215991.
Ethics statement
The studies involving human brain samples were reviewed and approved by the University of Illinois Institutional Review Board. Brain samples utilized for this study were obtained from the Harvard Brain Tissue Resource Center.
Author contributions
RS was PI for this grant, coordinated the interpretation, data analysis, manuscript preparation and submission. CL and CX helped with data analysis and wrote part of the methods for the manuscript. HR, KC, CR, and HG provided critical scientific input on data analysis and manuscript preparation. AG provided aliquots of brain samples from the Harvard Brain Tissue Resource Center. All authors provided critical edits to this manuscript.
Funding
This work was supported in part by NIH RO1 MH094358 (RS).
Acknowledgments
We are respectfully grateful to the individuals with schizophrenia, bipolar, and others who donated their postmortem brain to the Harvard Brain Tissue Resource Center and other repositories and to those who participated in the research conducted at UIC, all of which made this work possible. We are also grateful to Benjamin Feiner BS/BA1 for his excellent laboratory work on ChIP preparations.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpsyt.2022.1006109/full#supplementary-material
Footnotes
References
1. Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. (2008) 453:948–51. doi: 10.1038/nature06947
2. Jih G, Iglesias N, Currie MA, Bhanu NV, Paulo JA, Gygi SP, et al. Unique roles for histone H3K9me states in RNAi and heritable silencing of transcription. Nature. (2017) 547:463–7. doi: 10.1038/nature23267
3. Rastogi A, Maheswari U, Dorrell RG, Vieira FRJ, Maumus F, Kustka A, et al. Integrative analysis of large scale transcriptome data draws a comprehensive landscape of Phaeodactylum tricornutum genome and evolutionary origin of diatoms. Sci Rep. (2018) 8:1–14. doi: 10.1038/s41598-018-23106-x
4. McCarthy RL, Kaeding KE, Keller SH, Zhong Y, Xu L, Hsieh A, et al. Diverse heterochromatin-associated proteins repress distinct classes of genes and repetitive elements. Nat Cell Biol. (2021) 23:905–14. doi: 10.1038/s41556-021-00725-7
5. Gavin DP, Sharma RP. Histone modifications, DNA methylation, and schizophrenia. Neurosci Biobehav Rev. (2010) 34:882–8. doi: 10.1016/j.neubiorev.2009.10.010
6. Du J, Johnson LM, Jacobsen SE, Patel DJ. DNA methylation pathways and their crosstalk with histone methylation. Nat Rev Mol Cell Biol. (2015) 16:519–32. doi: 10.1038/nrm4043
7. Rose NR, Klose RJ. Understanding the relationship between DNA methylation and histone lysine methylation. Biochim Biophys Acta Gene Regul Mech. (2014) 1839:1362–72. doi: 10.1016/j.bbagrm.2014.02.007
8. Chervona Y, Costa M. The control of histone methylation and gene expression by oxidative stress, hypoxia, and metals. Free Radic Biol Med. (2012) 53:1041–7. doi: 10.1016/j.freeradbiomed.2012.07.020
9. El Gazzar M, Yoza BK, Chen X, Hu J, Hawkins GA, McCall CE. G9a and HP1 couple histone and DNA methylation to TNFα transcription silencing during endotoxin tolerance. J Biol Chem. (2008) 283:32198–208. doi: 10.1074/jbc.M803446200
10. Kreuz S, Fischle W. Oxidative stress signaling to chromatin in health and disease. Epigenomics. (2016) 8:843–62. doi: 10.2217/epi-2016-0002
11. Kungulovski G, Nunna S, Thomas M, Zanger UM, Reinhardt R, Jeltsch A. Targeted epigenome editing of an endogenous locus with chromatin modifiers is not stably maintained. Epigenet Chromatin. (2015) 8:1–11. doi: 10.1186/s13072-015-0002-z
12. Wen B, Wu H, Shinkai Y, Irizarry RA, Feinberg AP. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat Genet. (2009) 41:246–50. doi: 10.1038/ng.297
13. Lienert F, Mohn F, Tiwari VK, Baubec T, Roloff TC, Gaidatzis D, et al. Genomic prevalence of heterochromatic H3K9me2 and transcription do not discriminate pluripotent from terminally differentiated cells. PLoS Genet. (2011) 7:e1002090. doi: 10.1371/journal.pgen.1002090
14. Chase KA, Gavin DP, Guidotti A, Sharma RP. Histone methylation at H3K9: evidence for a restrictive epigenome in schizophrenia. Schizophr Res. (2013) 149:15–20. doi: 10.1016/j.schres.2013.06.021
15. Sharma RP, Feiner B, Chase KA. Histone H3 phosphorylation is upregulated in PBMCs of schizophrenia patients in comparison to healthy controls. Schizophr Res. (2015) 169:498. doi: 10.1016/j.schres.2015.09.030
16. Zhang T, Cooper S, Brockdorff N. The interplay of histone modifications–writers that read. EMBO Rep. (2015) 16:1467–81. doi: 10.15252/embr.201540945
17. Egelhofer TA, Minoda A, Klugman S, Lee K, Kolasinska-Zwierz P, Alekseyenko AA, et al. An assessment of histone-modification antibody quality. Nat Struct Mol Biol. (2011) 18:91–3. doi: 10.1038/nsmb.1972
18. Hansen AM, Ge Y, Schuster MB, Pundhir S, Jakobsen JS, Kalvisa A, et al. H3K9 dimethylation safeguards cancer cells against activation of the interferon pathway. Sci Adv. (2022) 8:eabf8627.
19. Salzberg AC, Harris-Becker A, Popova EY, Keasey N, Loughran TP, Claxton DF, et al. Genome-wide mapping of histone H3K9me2 in acute myeloid leukemia reveals large chromosomal domains associated with massive gene silencing and sites of genome instability. PLoS One. (2017) 12:e0173723. doi: 10.1371/journal.pone.0173723
20. Frost B, Hemberg M, Lewis J, Feany MB. Tau promotes neurodegeneration through global chromatin relaxation. Nat Neurosci. (2014) 17:357–66. doi: 10.1038/nn.3639
21. Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. (2012) 22:1813–31. doi: 10.1101/gr.136184.111
22. Sims D, Sudbery I, Ilott NE, Heger A, Ponting CP. Sequencing depth and coverage: key considerations in genomic analyses. Nat Rev Genet. (2014) 15:121–32. doi: 10.1038/nrg3642
23. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. (2014) 30:2114–20. doi: 10.1093/bioinformatics/btu170
24. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. (2012) 9:357–9. doi: 10.1038/nmeth.1923
25. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. (2009) 10:1–10. doi: 10.1186/gb-2009-10-3-r25
26. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variations from next-generation sequencing data. Nucleic Acids Res. (2010) 38:e164. doi: 10.1093/nar/gkq603
27. Shen L, Shao N-Y, Liu X, Maze I, Feng J, Nestler EJ. DiffReps: detecting differential chromatin modification sites from ChIP-seq data with biological replicates. PLoS One. (2013) 8:e65598. doi: 10.1371/journal.pone.0065598
28. Steinhauser S, Kurzawa N, Eils R, Herrmann C. A comprehensive comparison of tools for differential ChIP-seq analysis. Brief Bioinform. (2016) 17:953–66. doi: 10.1093/bib/bbv110
29. Goh KK, Chen C-H, Lane H-Y. Oxytocin in schizophrenia: pathophysiology and implications for future treatment. Int J Mol Sci. (2021) 22:2146. doi: 10.3390/ijms22042146
30. Ho NF, Holt DJ, Cheung M, Iglesias JE, Goh A, Wang M, et al. Progressive decline in hippocampal CA1 volume in individuals at ultra-high-risk for psychosis who do not remit: findings from the longitudinal youth at risk study. Neuropsychopharmacology. (2017) 42:1361–70. doi: 10.1038/npp.2017.5
31. Sekar A, Bialas AR, De Rivera H, Davis A, Hammond TR, Kamitaki N, et al. Schizophrenia risk from complex variation of complement component 4. Nature. (2016) 530:177–83. doi: 10.1038/nature16549
32. Trubetskoy V, Pardinas AF, Qi T, Panagiotaropoulou G, Awasthi S, Bigdeli TB, et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature. (2022) 604:502–508.
33. Ooi L, Wood IC. Chromatin crosstalk in development and disease: lessons from REST. Nat Rev Genet. (2007) 8:544–54. doi: 10.1038/nrg2100
34. Zheng D, Zhao K, Mehler MF. Profiling RE1/REST-mediated histone modifications in the human genome. Genome Biol. (2009) 10:1–20. doi: 10.1186/gb-2009-10-1-r9
35. Chwang WB, Arthur JS, Schumacher A, Sweatt JD. The nuclear kinase mitogen-and stress-activated protein kinase 1 regulates hippocampal chromatin remodeling in memory formation. J Neurosci. (2007) 27:12732–42. doi: 10.1523/JNEUROSCI.2522-07.2007
36. Feiner B, Chase K, Melbourne J, Rosen C, Sharma R. Risperidone effects on heterochromatin: the role of kinase signaling. Clin Exp Immunol. (2019) 196:67–75. doi: 10.1111/cei.13250
37. Abrajano JJ, Qureshi IA, Gokhan S, Zheng D, Bergman A, Mehler MF. REST and CoREST modulate neuronal subtype specification, maturation and maintenance. PLoS One. (2009) 4:e7936. doi: 10.1371/journal.pone.0007936
38. Lüscher B, Keller CA. Regulation of GABAA receptor trafficking, channel activity, and functional plasticity of inhibitory synapses. Pharmacol Ther. (2004) 102:195–221. doi: 10.1016/j.pharmthera.2004.04.003
39. Tyagarajan SK, Fritschy J-M. Gephyrin: a master regulator of neuronal function? Nat Rev Neurosci. (2014) 15:141–56. doi: 10.1038/nrn3670
40. Lionel AC, Vaags AK, Sato D, Gazzellone MJ, Mitchell EB, Chen HY, et al. Rare exonic deletions implicate the synaptic organizer Gephyrin (GPHN) in risk for autism, schizophrenia and seizures. Hum Mol Genet. (2013) 22:2055–66. doi: 10.1093/hmg/ddt056
41. Balan S, Yamada K, Iwayama Y, Hashimoto T, Toyota T, Shimamoto C, et al. Comprehensive association analysis of 27 genes from the GABAergic system in Japanese individuals affected with schizophrenia. Schizophr Res. (2017) 185:33–40. doi: 10.1016/j.schres.2017.01.003
42. Pocklington AJ, Rees E, Walters JT, Han J, Kavanagh DH, Chambert KD, et al. Novel findings from CNVs implicate inhibitory and excitatory signaling complexes in schizophrenia. Neuron. (2015) 86:1203–14. doi: 10.1016/j.neuron.2015.04.022
43. Dejanovic B, Djémié T, Grünewald N, Suls A, Kress V, Hetsch F, et al. Simultaneous impairment of neuronal and metabolic function of mutated gephyrin in a patient with epileptic encephalopathy. EMBO Mol Med. (2015) 7:1580–94. doi: 10.15252/emmm.201505323
44. Redies C, Hertel N, Hübner CA. Cadherins and neuropsychiatric disorders. Brain Res. (2012) 1470:130–44. doi: 10.1016/j.brainres.2012.06.020
45. Rivero O, Selten MM, Sich S, Popp S, Bacmeister L, Amendola E, et al. Cadherin-13, a risk gene for ADHD and comorbid disorders, impacts GABAergic function in hippocampus and cognition. Transl Psychiatry. (2015) 5:e655. doi: 10.1038/tp.2015.152
46. Singh SM, Castellani C, O’Reilly R. Autism meets schizophrenia via cadherin pathway. Schizophr Res. (2009) 116:293–4. doi: 10.1016/j.schres.2009.09.031
47. Rossetti T, Banerjee S, Kim C, Leubner M, Lamar C, Gupta P, et al. Memory erasure experiments indicate a critical role of CaMKII in memory storage. Neuron. (2017) 96:207–16.e2. doi: 10.1016/j.neuron.2017.09.010
48. Broadbelt K, Ramprasaud A, Jones LB. Evidence of altered neurogranin immunoreactivity in areas 9 and 32 of schizophrenic prefrontal cortex. Schizophr Res. (2006) 87:6–14. doi: 10.1016/j.schres.2006.04.028
49. Ohi K, Hashimoto R, Yasuda Y, Nemoto K, Ohnishi T, Fukumoto M, et al. Impact of the genome wide supported NRGN gene on anterior cingulate morphology in schizophrenia. PLoS One. (2012) 7:e29780. doi: 10.1371/journal.pone.0029780
50. Umeda-Yano S, Hashimoto R, Yamamori H, Weickert CS, Yasuda Y, Ohi K, et al. Expression analysis of the genes identified in GWAS of the postmortem brain tissues from patients with schizophrenia. Neurosci Lett. (2014) 568:12–6. doi: 10.1016/j.neulet.2014.03.031
51. Casaletto KB, Elahi FM, Bettcher BM, Neuhaus J, Bendlin BB, Asthana S, et al. Neurogranin, a synaptic protein, is associated with memory independent of Alzheimer biomarkers. Neurology. (2017) 89:1782–8. doi: 10.1212/WNL.0000000000004569
52. Salzberg Y, Ramirez-Suarez NJ, Bülow HE. The proprotein convertase KPC-1/furin controls branching and self-avoidance of sensory dendrites in Caenorhabditis elegans. PLoS Genet. (2014) 10:e1004657. doi: 10.1371/journal.pgen.1004657
53. Fromer M, Roussos P, Sieberts SK, Johnson JS, Kavanagh DH, Perumal TM, et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat Neurosci. (2016) 19:1442–53.
54. Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. (2007) 39:44–84. doi: 10.1016/j.biocel.2006.07.001
55. Lovelace MD, Varney B, Sundaram G, Franco NF, Ng ML, Pai S, et al. Current evidence for a role of the kynurenine pathway of tryptophan metabolism in multiple sclerosis. Front Immunol. (2016) 7:246. doi: 10.3389/fimmu.2016.00246
56. Barron H, Hafizi S, Andreazza AC, Mizrahi R. Neuroinflammation and oxidative stress in psychosis and psychosis risk. Int J Mol Sci. (2017) 18:651. doi: 10.3390/ijms18030651
57. Calabrese V, Giordano J, Crupi R, Di Paola R, Ruggieri M, Bianchini R, et al. Hormesis, cellular stress response and neuroinflammation in schizophrenia: early onset versus late onset state. J Neurosci Res. (2017) 95:1182–93. doi: 10.1002/jnr.23967
58. Yao JK, Reddy RD, Van Kammen DP. Oxidative damage and schizophrenia. CNS Drugs. (2001) 15:287–310. doi: 10.2165/00023210-200115040-00004
59. Ustohal L, Mayerova M, Valkova B, Sedlakova H, Kasparek T. Asymptomatic elevation of amylase and lipase after olanzapine treatment. J Clin Psychopharmacol. (2016) 36:181–3. doi: 10.1097/JCP.0000000000000460
60. Chase KA, Feiner B, Ramaker MJ, Hu E, Rosen C, Sharma RP. Examining the effects of the histone methyltransferase inhibitor BIX-01294 on histone modifications and gene expression in both a clinical population and mouse models. PLos One. (2019) 14:e0216463. doi: 10.1371/journal.pone.0216463
61. Pantelis C, Papadimitriou GN, Papiol S, Parkhomenko E, Pato MT, Paunio T, et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature (2014) 511:421–427.
62. Jansen LA, Mirzaa GM, Ishak GE, O’Roak BJ, Hiatt JB, Roden WH, et al. PI3K/AKT pathway mutations cause a spectrum of brain malformations from megalencephaly to focal cortical dysplasia. Brain. (2015) 138:1613–28. doi: 10.1093/brain/awv045
63. Ballif BC, Rosenfeld JA, Traylor R, Theisen A, Bader PI, Ladda RL, et al. High-resolution array CGH defines critical regions and candidate genes for microcephaly, abnormalities of the corpus callosum, and seizure phenotypes in patients with microdeletions of 1q43q44. Hum Genet. (2012) 131:145–56. doi: 10.1007/s00439-011-1073-y
64. Boland E, Clayton-Smith J, Woo VG, McKee S, Manson FD, Medne L, et al. Mapping of deletion and translocation breakpoints in 1q44 implicates the serine/threonine kinase AKT3 in postnatal microcephaly and agenesis of the corpus callosum. Am J Hum Genet. (2007) 81:292–303. doi: 10.1086/519999
65. Lee JH, Huynh M, Silhavy JL, Kim S, Dixon-Salazar T, Heiberg A, et al. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet. (2012) 44:941–5. doi: 10.1038/ng.2329
66. Poduri A, Evrony GD, Cai X, Elhosary PC, Beroukhim R, Lehtinen MK, et al. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron. (2012) 74:41–8. doi: 10.1016/j.neuron.2012.03.010
67. Rivière J-B, Mirzaa GM, O’Roak BJ, Beddaoui M, Alcantara D, Conway RL, et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet. (2012) 44:934–40.
68. Howell KR, Floyd K, Law AJ. PKBγ/AKT3 loss-of-function causes learning and memory deficits and deregulation of AKT/mTORC2 signaling: relevance for schizophrenia. PLoS One. (2017) 12:e0175993. doi: 10.1371/journal.pone.0175993
69. Thiselton DL, Vladimirov VI, Kuo P-H, McClay J, Wormley B, Fanous A, et al. AKT1 is associated with schizophrenia across multiple symptom dimensions in the Irish study of high density schizophrenia families. Biol Psychiatry. (2008) 63:449–57. doi: 10.1016/j.biopsych.2007.06.005
70. Heinrichs M, Baumgartner T, Kirschbaum C, Ehlert U. Social support and oxytocin interact to suppress cortisol and subjective responses to psychosocial stress. Biol Psychiatry. (2003) 54:1389–98. doi: 10.1016/S0006-3223(03)00465-7
71. Keri S, Kiss I, Kelemen O. Sharing secrets: oxytocin and trust in schizophrenia. Soc Neurosci. (2009) 4:287–93. doi: 10.1080/17470910802319710
72. Rich ME, Caldwell HK. A role for oxytocin in the etiology and treatment of schizophrenia. Front Endocrinol. (2015) 6:90. doi: 10.3389/fendo.2015.00090
73. Ripke S, Neale BM, Corvin A, Walters JT, Farh K-H, Holmans PA, et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature. (2014) 511:421. doi: 10.1038/nature13595
74. Belt KT, Carroll MC, Porter RR. The structural basis of the multiple forms of human complement component C4. Cell. (1984) 36:907–14. doi: 10.1016/0092-8674(84)90040-0
75. Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci. (2012) 35:369–89. doi: 10.1146/annurev-neuro-061010-113810
76. Melbourne JK, Rosen C, Feiner B, Sharma RP. C4A mRNA expression in PBMCs predicts the presence and severity of delusions in schizophrenia and bipolar disorder with psychosis. Schizophr Res. (2018) 197:321–7. doi: 10.1016/j.schres.2018.01.018
77. Kundakovic M, Jiang Y, Kavanagh DH, Dincer A, Brown L, Pothula V, et al. Practical guidelines for high-resolution epigenomic profiling of nucleosomal histones in postmortem human brain tissue. Biol Psychiatry. (2017) 81:162–70. doi: 10.1016/j.biopsych.2016.03.1048
78. Grayson DR, Kundakovic M, Sharma RP. Is there a future for histone deacetylase inhibitors in the pharmacotherapy of psychiatric disorders? Mol Pharmacol. (2010) 77:126–35. doi: 10.1124/mol.109.061333
79. Sharma RP, Gavin DP, Chase KA. Heterochromatin as an incubator for pathology and treatment non-response: implication for neuropsychiatric illness. Pharmacogenomics J. (2012) 12:361–7. doi: 10.1038/tpj.2011.64
80. Liu TF, Yoza BK, El Gazzar M, Vachharajani VT, McCall CE. NAD+-dependent SIRT1 deacetylase participates in epigenetic reprogramming during endotoxin tolerance. J Biol Chem. (2011) 286:9856–64. doi: 10.1074/jbc.M110.196790
81. Yoza BK, Hu JY-Q, Cousart SL, Forrest LM, McCall CE. Induction of RelB participates in endotoxin tolerance. J Immunol. (2006) 177:4080–5. doi: 10.4049/jimmunol.177.6.4080
Keywords: schizophrenia, histone methylation, heterochromatin, postmortem brain, ChIP-Seq, gene expression
Citation: Rizavi HS, Chase KA, Liu C, Gavin H, Rosen C, Xia C, Guidotti A and Sharma RP (2022) Differential H3K9me2 heterochromatin levels and concordant mRNA expression in postmortem brain tissue of individuals with schizophrenia, bipolar, and controls. Front. Psychiatry 13:1006109. doi: 10.3389/fpsyt.2022.1006109
Received: 28 July 2022; Accepted: 15 September 2022;
Published: 26 October 2022.
Edited by:
Anilkumar Pillai, University of Texas Health Science Center at Houston, United StatesReviewed by:
Chuanjun Zhuo, Tianjin Anding Hospital, ChinaCarlos M. Opazo, University of Melbourne, Australia
Copyright © 2022 Rizavi, Chase, Liu, Gavin, Rosen, Xia, Guidotti and Sharma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rajiv P. Sharma, rsharma@uic.edu