 Marijn Lijffijt1,2
Marijn Lijffijt1,2 Charles E. Green3,4
Charles E. Green3,4 Nicholas Balderston5
Nicholas Balderston5 Tabish Iqbal1,2
Tabish Iqbal1,2 Megan Atkinson6,7
Megan Atkinson6,7 Brittany Vo-Le1,2
Brittany Vo-Le1,2 Bylinda Vo-Le1,2
Bylinda Vo-Le1,2 Brittany O’Brien1,2
Brittany O’Brien1,2 Christian Grillon4Alan C. Swann2,8Sanjay J. Mathew2,8*
Christian Grillon4Alan C. Swann2,8Sanjay J. Mathew2,8*- 1Research Service Line, Michael E. DeBakey VA Medical Center, Houston, TX, United States
- 2Menninger Department of Psychiatry and Behavioral Sciences, Baylor College of Medicine, Houston, TX, United States
- 3Department of Psychiatry and Behavioral Sciences, UTHealth McGovern Medical School, University of Texas Health Science Center at Houston, Houston, TX, United States
- 4Department of Pediatrics - Center for Evidence Based Medicine, UTHealth McGovern Medical School, University of Texas Health Science Center at Houston, Houston, TX, United States
- 5Section on Neurobiology of Fear and Anxiety, National Institute of Mental Health, National Institutes of Health, Bethesda, MD, United States
- 6Department of Anesthesiology, Michael E. DeBakey VA Medical Center, Houston, TX, United States
- 7Department of Anesthesiology, Baylor College of Medicine, Houston, TX, United States
- 8Mental Health Care Line, Michael E. DeBakey VA Medical Center, Houston, TX, United States
Background: Individuals with post-traumatic stress disorder (PTSD) have a heightened sensitivity to subsequent stressors, addictive drugs, and symptom recurrence, a form of behavioral sensitization. N-methyl-D-aspartate receptors (NMDARs) are involved in the establishment and activation of sensitized behavior.
Objective: We describe a protocol of a randomized placebo-controlled Phase 1b proof-of-mechanism trial to examine target engagement, safety, tolerability, and possible efficacy of the NMDAR antagonist lanicemine in individuals with symptoms of PTSD (Clinician Administered PTSD Scale [CAPS-5] score ≥ 25) and evidence of behavioral sensitization measured as enhanced anxiety-potentiated startle (APS; T-score ≥ 2.8).
Methods: Subjects (n = 24; age range 21–65) receive three 60-min intravenous infusions of placebo or 100 mg lanicemine over 5 non-consecutive days. Primary endpoint is change in APS from pre-treatment baseline to after the third infusion. NMDAR engagement is probed with resting state EEG gamma band power, 40 Hz auditory steady state response, the mismatch negativity amplitude, and P50 sensory gating. Change in CAPS-5 scores is an exploratory clinical endpoint. Bayesian statistical methods will evaluate endpoints to determine suitability of this agent for further study.
Conclusion: In contrast to traditional early-phase trials that use symptom severity to track treatment efficacy, this study tracks engagement of the study drug on expression of behavioral sensitization, a functional mechanism likely to cut across disorders. This experimental therapeutics design is consistent with recent NIMH-industry collaborative studies, and could serve as a template for testing novel pharmacological agents in psychiatry.
Clinical Trial Registration: www.ClinicalTrials.gov, identifier NCT03166501.
Introduction
With few exceptions, the explosion in basic neuroscience knowledge over the past two decades has not led to the development of mechanistically novel treatments for serious psychiatric disorders. It has been argued that the traditional approach to psychiatric clinical trials, which solely rely on symptom rating scales as endpoints, has stymied progress. To address this problem, the United States National Institute of Mental Health (NIMH) has adopted an experimental therapeutics approach to early-phase clinical trials, which require proof-of-mechanism (POM) studies to determine whether an experimental intervention is viable for testing in larger randomized controlled trials (www.nimh.nih.gov/about/strategic-planning-reports/strategic-research-priorities/index.shtml). The experimental therapeutics approach mandates (i) a functional mechanism that is likely to be associated with both clinical phenotype and therapeutic response, (ii) an intervention that is likely to act on a target that engages the functional mechanism [target specificity], (iii) a biomarker to show that the intervention engages the hypothesized target [target engagement], and (iv) a biomarker to show that the intervention engages the hypothesized functional mechanism [POM] (1, 2). Selection of the functional mechanism can be guided by Research Domain Criteria (RDoC). RDoC regards psychiatric disorders not as DSM-defined clinical phenotypes but as disease-overlapping functional domains associated with neuroscience-inspired neural circuitries that are at the crossroads of genotype and clinical phenotype (3, 4).
In this paper we describe the rationale and protocol for an NIMH-funded POM study to determine whether the N-methyl-D-aspartate receptor (NMDAR) antagonist lanicemine (BHV-5500) engages the NMDAR to block expression of behavioral sensitization underlying symptoms of post-traumatic stress disorder (PTSD).
Post-Traumatic Stress Disorder
PTSD is a trauma-induced condition marked by intrusive thoughts about and re-experiencing of trauma (life-like flashbacks, nightmares), avoidance of trauma-associated stimuli and thoughts, negative cognitions and mood (anhedonia, hopelessness), and hyperarousal (anger outbursts, hypervigilance, exaggerated startle) (5). An estimated 6% of the U.S. population has a lifetime history of PTSD (6, 7), with a higher prevalence among first responders (8), military veterans (9–12), and women (6, 7, 12). An additional 6% of U.S. adults could have a lifetime history of partial PTSD defined as fewer, but not less frequent, intense or severe, PTSD symptoms (13). Mean age of PTSD onset is 24 (7), although about half of the individuals with lifetime PTSD meet criteria before age 18 (6). While there are several evidence-based psychotherapies for PTSD, there remain only two U.S. Food and Drug Administration (FDA)-approved drugs for PTSD, the selective serotonin reuptake inhibitors paroxetine and sertraline. Both of these approved medications have only low to moderate efficacy for improving PTSD symptoms (14). This highlights the importance of finding novel targets for intervention.
Functional Mechanism
Behavioral sensitization refers to a process whereby trauma-associated stress (but also repeated use of substances of abuse, mood or anxiety episodes, and suicide attempts) sensitize behavioral, motivational and stress systems, thereby increasing the behavioral and physiological reactivity to subsequent stressors or other sensitizing agents even after a prolonged absence of those agents (15–18). Consistent with findings in animals, research in humans showed at least three different aspects of behavioral sensitization: induction, the development of behavioral sensitization to a sensitizing agent, including uncontrollable stressors (19), substances of abuse (18, 20, 21), and, in PTSD, repeated illness episodes (22); expression, exaggerated behavioral or physiological responses to a sensitizing agent even after prolonged absence of that agent (18, 21, 23); and cross-sensitization, the process by which sensitization to one agent results in sensitization to other agents (e.g., facilitation of behavioral sensitization to psychostimulants after exposure to uncontrollable stress) (24). Animal research showed that all three aspects of behavioral sensitization require activation of NMDARs albeit via different neural pathways. Repeated cocaine administration increased NMDAR sensitivity only in rats that developed a sensitized motor response (25). Blockade of NMDARs by non-competitive NMDAR antagonist ketamine or MK-801 prevented induction of behavioral sensitization to ethanol (26, 27), apomorphine (28), stimulants (29–36), stress (36), and nicotine (37, 38), blocked expression of behavioral sensitization to alcohol (39), stimulants (29–32, 35, 40), stress (41), and nicotine (38), and blocked cross-sensitization between ethanol and stimulants (40, 42) and between stress and stimulants (36, 43, 44). Administration of MK-801 in the nucleus accumbens prevented induction but not expression of behavioral sensitization to stimulant administration (45), consistent with involvement of different neural pathways in induction and expression of behavioral sensitization.
Our premise for this study is that PTSD symptoms are associated with behavioral sensitization (46–48). We further propose that this type of sensitization could be blocked rapidly with an NMDAR antagonist (36, 43, 44) to relieve PTSD symptoms.
Primary Outcome Measure: Biomarker of Functional Mechanism
Behavioral sensitization in PTSD is associated with hyperarousal of the extended amygdala—the basolateral amygdala (BLA), central amygdala, medial amygdala, bed nucleus of the stria terminalis (BNST), shell of the nucleus accumbens, and their interconnectivity (48, 49). In animals, uncontrollable stressors, including trauma, enhanced sensitivity of the extended amygdala to future, milder, stressors (19, 50). In humans, enhanced reactivity of the amygdala could predispose to development of PTSD symptoms (51, 52) and is associated with less resilience to stressors of everyday life (53). Sensitization of the extended amygdala cuts across all RDoC functional domains (49) and has been associated with most PTSD symptom clusters (48).
NMDARs antagonists may improve PTSD symptoms by affecting behavioral sensitization. In preclinical models, NMDAR antagonists blocked induction (36) and expression (41) of behavioral sensitization by stress, and blocked cross-sensitization between stress and stimulants (36, 43, 44). In humans, intraoperative administration of NMDAR antagonist ketamine may reduce PTSD risk (54, 55), and a single infusion of 0.5 mg/kg ketamine compared to midazolam in patients with PTSD resulted in rapid (within 24 h) and sustained (at least 7 days) improvement in PTSD symptoms (56, 57). Finally, the low-affinity NMDAR antagonist memantine improved hyperarousal and depressive symptoms in individuals with PTSD (58).
In this study we operationalize expression of behavioral sensitization as an aversion-potentiated startle amplitude expressed as T-score obtained on the No-threat, Predictable-threat, Unpredictable-threat (NPU) test (59–61). Sudden intense stimuli elicit an eye blink startle reflex that can be potentiated in negative emotional states (62, 63). Phasic negative emotional states potentiate startle via activation of the central amygdala in response to an explicit cue that signals threat of an uncontrollable aversive stimulus, assessing fear [fear-potentiated startle (FPS)]. Tonic negative emotional states potentiate startle primarily via activation of the BNST in response to a context, not a specific cue, that signals threat of an uncontrollable aversive stimulus, assessing anxiety [anxiety-potentiated startle (APS)] (62, 63). PTSD is associated with an enhanced APS but not FPS (64, 65), which could be related to increased activation of BNST excitatory glutamatergic neurons relative to BNST inhibitory GABA neurons (66) or with decreased regulation of the prefrontal cortex over the amygdala (67) associated with diminished prefrontal glutamate concentration (68).
We propose a POM study using lanicemine 100 mg in patients with symptoms of PTSD with evidence of behavioral sensitization operationalized as an enhanced APS. Using air puffs to the forehead as startle probes and loud acoustic sounds as aversive stimuli, the NPU-threat test discriminated between patients with PTSD (n = 16; APS T-score mean ± SD = 6.5 ± 1.4) and healthy controls (n = 34; APS T-score = 0.6 ± 1.1) (65). For this trial, we select participants with at least moderate symptoms of PTSD operationalized as a CAPS-5 score ≥25, and a state of behavioral sensitization operationalized as an APS T-score at least two standard deviations above the mean among healthy volunteers (APS T-score ≥2.8). We anticipate approximately 60% of PTSD patients to meet APS entry criteria (Grillon, unpublished data). The primary endpoint of this study is change in APS T-score from pre-treatment baseline to that following the last of three lanicemine infusions. Paradigm specifics are described in Supplemental Materials.
Experimental Drug
Lanicemine is an intravenously administered NMDAR antagonist that crosses the blood-brain barrier, binding within channel pores (Ki 0.5–3 µM) of NR2A and NR2B NMDAR complexes (IC50 4–40 µM) to block the flow of charged cations. In contrast to other NMDAR antagonists such as ketamine and MK-801, lanicemine has a fast off-rate and is low-trapping, properties associated with favorable safety and tolerability profiles (69). In addition, ketamine’s actions include effects on systems beyond NMDAR channel blockade, including at opiate, sigma, and muscarinic receptors (70), whereas lanicemine has negligible off-target pharmacological effects and provides a more selective NMDAR probe. Lanicemine has been examined in preclinical and early-phase clinical studies in patients with stroke, sleep apnea, and treatment-resistant depression (TRD) and is considered safe in humans. A recently completed phase 2b trial found that lanicemine 100 mg dose was effective in individuals with TRD who had the most severe depression or suicidal ideation at the start of the study (71), potentially suggestive of effects on behavioral sensitization.
Prior studies exposed healthy individuals and patients with treatment resistant depression to single or repeated infusions of lanicemine 50 mg or 100 mg (69, 71, 72). At least one adverse event (AE) was reported by 77.1% of subject in the lanicemine arms, compared to 70% of subjects in the placebo arm. Although most AEs were of mild or moderate intensity, a greater proportion of subjects discontinued the trials due to an AE for lanicemine 100 mg (9.0%) than lanicemine 50 mg (2.0%) or placebo (4.0%). Dizziness was the most common AE and appeared to be dose-related. Other reported side effects included: changes in balance, feeling drunk, blurred vision, headache, sleepiness, weakness, impaired concentration, abnormal sensations (tingling of hands, feet, feelings of crawling ants within body), nausea, and vomiting. Lanicemine has been associated also with dose-dependent transient mild elevations in blood pressure with no evidence of sustained changes in blood pressure or pulse rate. There were no clinically meaningful differences between lanicemine and placebo groups for mean changes in clinical chemistry, hematology, or urinalysis. Treatment with lanicemine was not associated with any decline in psychomotor function, attention, working memory, learning, or general cognitive function. An important difference between lanicemine and ketamine concerns dissociative side effects. Although 8% of patients with treatment resistant depression in the lanicemine 100 mg arm spontaneously reported dissociative symptoms compared to 4% in both the lanicemine 50 mg and placebo arms, only 1.1% of patients in both lanicemine groups had a Clinician-Administered Dissociative States Scale (CADSS) score classified as high (11–25) at any time point. In contrast, 50% of patients with treatment resistant depression showed high dissociation with a 40 minute subanesthetic infusion of ketamine (73).
Secondary Outcome Measures
Biomarkers of NMDAR Target Engagement
NMDAR target engagement is examined with neurophysiological measures that are translatable between animal and human studies.
Subanesthetic doses of NMDAR antagonist ketamine increased resting-state gamma band (30–100 Hz) power in animals (74–76) and humans as a function of time since start of ketamine infusion, rapidly normalizing after end of infusion (69, 77, 78). This response has been associated with activation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPAR) (79) or activation of fast-spiking gamma-aminobutyric acid (GABA)-ergic parvalbumin interneurons (80) following a ketamine-driven increase in prefrontal glutamate (81). Lanicemine 75 mg and 150 mg increased midline electrodes gamma band activity in a dose-response fashion in healthy volunteers (69). This increase was unrelated to increased motor activity, separating it from ketamine-increased gamma band power that did correlate with enhanced motor activity (69). Ketamine-increased gamma band power was unrelated to treatment response in patients with TRD (78). We hypothesize that resting state gamma band power will increase with lanicemine 100 mg compared to placebo.
The 40 Hz auditory steady state response (40 Hz ASSR) measures stimulus-evoked gamma band (40 Hz) power and phase synchronization. In awake rats, a single intravenous bolus of 1, 3, 10, and 30 mg/kg ketamine increased intracranial frontocentral 40 Hz power, but the time was delayed or duration prolonged linearly with increase in dose (82). Phase synchrony decreased at the lowest dose and increased at the highest dose, with changes correlating negatively with ketamine NMDAR occupancy (82). A single dose of MK-801 increased intracranial 40 Hz phase synchrony but did not affect power (83). In healthy humans and humans with schizophrenia, memantine 20 mg increased frontal 40 Hz power and phase locking; memantine 10 mg had no effects (84). We expect lanicemine 100 mg to increase 40 Hz ASSR power and phase synchrony.
The auditory Mismatch Negativity (MMN) is evoked between 100 and 250 ms after an unexpected auditory event, and is measured as the difference between (frequent) expected and (infrequent) unexpected stimuli in a passive oddball task (85). MMN amplitude could be increased in PTSD (64, 86, 87), although this has not been found uniformly (88). MMN in healthy volunteers was increased under threat of an aversive stimulus (89) suggesting it could be sensitive to behavioral sensitization. Ketamine suppressed MMN amplitude (90–92), perhaps via blockage of NR2B receptor units (93), without affecting temporally overlapping ERP components (92). This effect is sustained for at least 30 min after end of infusion (92). Memantine could increase MMN amplitude (94). MMN amplitude is included as a measure of target engagement as well as a measure associated with functional mechanism. We expect suppression of MMN amplitude with lanicemine compared to placebo, potentially associated with change in APS.
P50 auditory sensory gating measures P50 amplitude suppression following presentation of the same auditory stimulus in short temporal proximity, reflecting pre-attentional information filtering (95–98). P50 gating is impaired in PTSD compared to trauma controls (99), and after acute stress in healthy individuals (100, 101). Lanicemine may normalize P50 gating if lanicemine attenuates stress reactivity. However, ketamine 0.3 mg/kg (bolus) in healthy volunteers impaired P50 gating through ketamine-induced increases in gamma band activity for the second stimulus of the repetition (102) which was also found in rats (76). Thus, lanicemine may normalize or worsen P50 gating. We study effects of lanicemine on P50 gating to examine this apparent discrepancy and in order to characterize its effects on aspects of information filtering.
Further specifics of the paradigms can be found in Supplemental Materials.
Symptom Severity Rating Scales
The Clinician Administered PTSD Scale (CAPS-5) (103) is a structured interview to assess intensity and frequency of DSM-5 PTSD symptoms. The CAPS-5 is administered at screening to determine subject eligibility (104), before the first infusion, and 3 days after the last infusion (day 8) to preliminarily examine treatment efficacy (105). Further specifics of the CAPS-5 and of other clinical measures can be found in the accompanying Supplemental Materials.
Sample Size
The primary statistical contrast is lanicemine compared to placebo on APS from baseline to the end of the third infusion. Assuming an observed posterior probability of at least 0.75 of Cohen’s d < -0.4 in the active condition relative to placebo and a correlation r = 0.5 due to repeated measures, K = 500 Monte Carlo simulations indicated that a sample size of 20 allows an 82% chance of detecting superiority of lanicemine. Allowing for attrition, we will recruit 24 subjects. To date, we randomized 23 of the 24 subjects.
Study Design
This study is funded by the NIMH under an R61/R33 Phased Innovation Award (5R61MH110540). Biohaven Pharmaceutical Holding Company LTD provided material support. The study drug is administered as an intravenous solution under FDA IND number 134304; the trial is registered at ClinicalTrials.gov under NCT03166501. All study-related procedures and materials have been approved by the Baylor College of Medicine Institutional Review Board, and the MEDVAMC Office of Research and Development. An NIMH-appointed data and safety monitoring board (DSMB) provides oversight.
This study uses a randomized, double-blind, parallel-arm, placebo-controlled fixed dose design to test lanicemine (100 mg) compared to saline placebo in up to 24 male and female outpatients between the ages of 21 and 65 who have significant PTSD symptoms (CAPS-5 score of at least 25, and a Clinical Global Impression of Severity [CGI-S] score of at least 4), and physiological manifestations of behavioral sensitization (APS T-score of at least 2.8). Inclusion and exclusion criteria are displayed in Supplemental Material.
To qualify for randomization, entry criteria must be met at screening and the morning of the first lanicemine infusion. Subjects are randomized in a 1:1 ratio to receive three 60-min intravenous infusions of lanicemine or placebo on non-consecutive days over a 5-day period. Table 1 provides the timeline of all study procedures. Table 2 provides the timeline of study procedures at the first infusion (day 1) and third infusion (day 5).
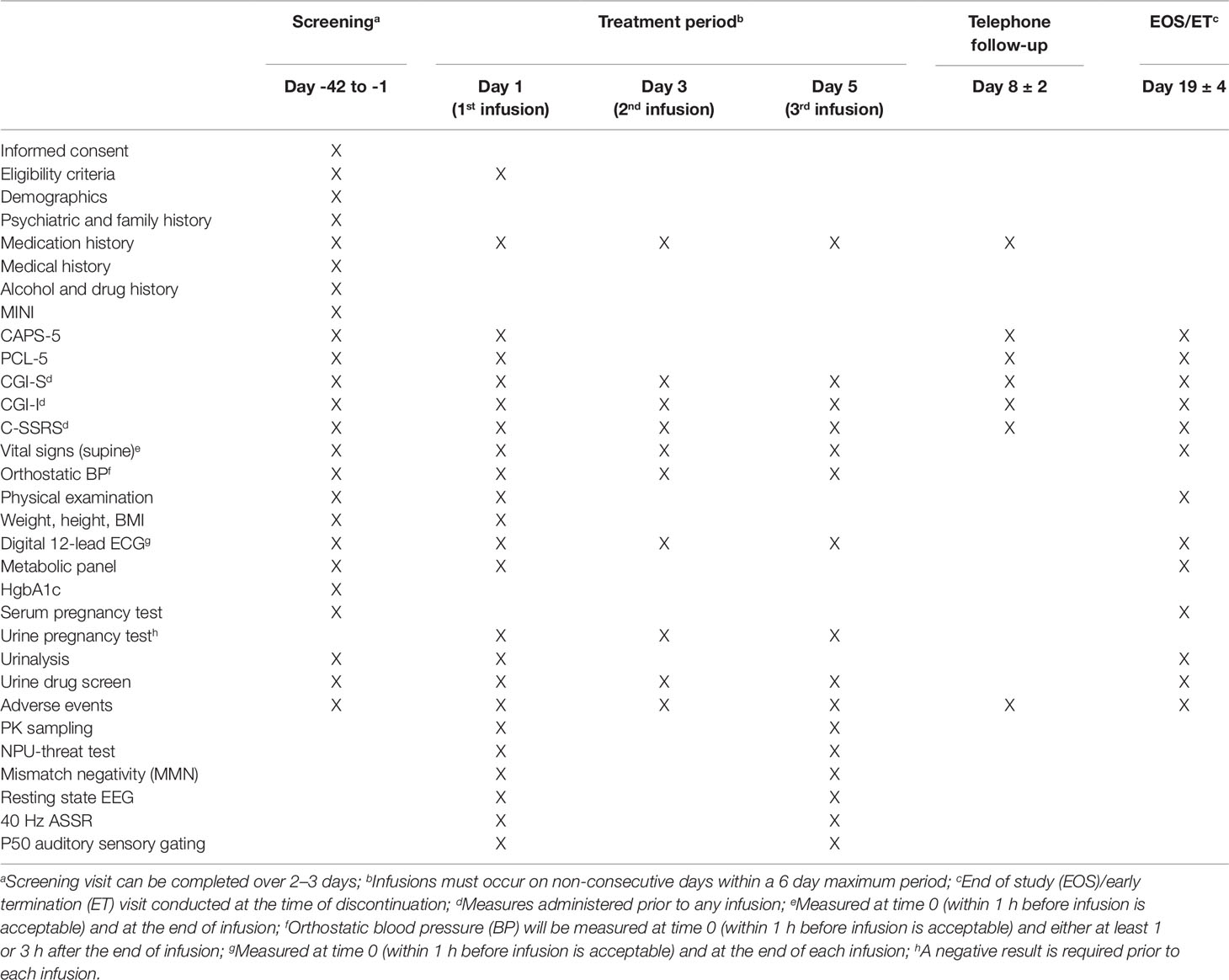
Table 1 Schedule of events.
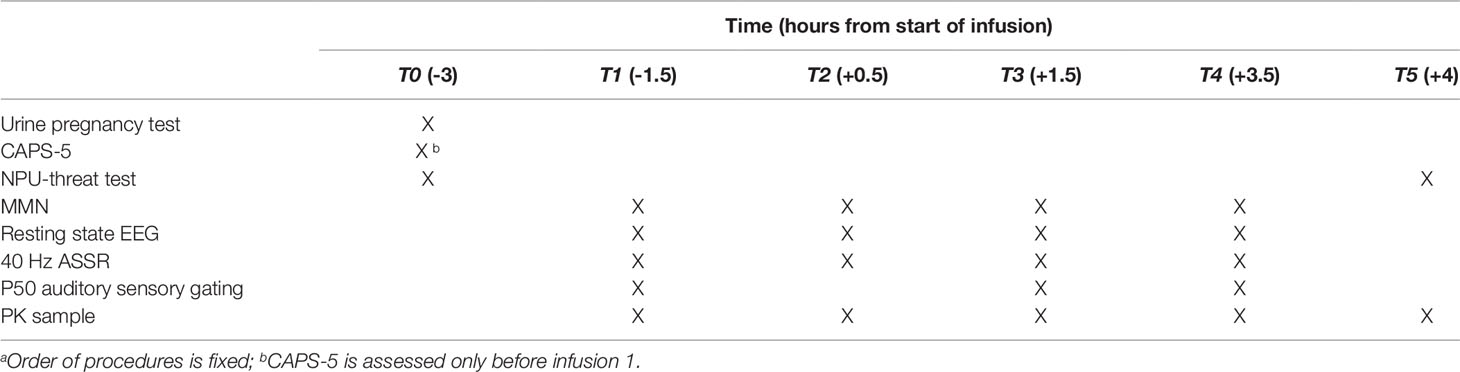
Table 2 Study procedures during infusion 1 (day 1) and infusion 3 (day 5) a.
Data Analysis Plan
Primary Endpoint
We expect that relative to placebo, three infusions of lanicemine will normalize the APS response after the last infusion. The primary analysis for this endpoint is regression of the APS measured on the fifth day of treatment (third infusion) onto treatment group after controlling for APS measured at baseline of the first day of treatment (first infusion).
Secondary Endpoints
We expect that relative to placebo, lanicemine will demonstrate effects on neurophysiology measured on the fifth day of treatment, and on the CAPS-5 measured 3 days after the last infusion (day 8). The primary analyses for these endpoints are the regression of neurophysiology measures, measured after infusion 3, and CAPS-5, measured on day 8, onto treatment group after covariation for the respective baseline values. Secondary analyses will use multilevel models to evaluate changes as a function of stratification variables, treatment, time and the interaction of treatment and time. In addition, Bayesian Structural Equation Modeling will be used to test the hypothesis that treatment effects on day 5 APS or neurophysiology mediate treatment effects on day 8 CAPS-5.
Analysis Sets
Analyses will be performed on a:
Modified intent-to-treat (mITT) analysis set: this analysis set will include all randomized patients who took study medication, and who have a baseline APS and at least 1 post-baseline behavioral rating. The mITT analysis set will be used for exploratory efficacy analyses.
Per-protocol (PP) analysis set: This analysis set will include only those mITT patients without significant protocol deviation, and who received the treatment to which they were randomized. The analysis of the primary efficacy variable will be repeated on this set, and additional analyses of efficacy variables may be performed using this set to assess the robustness of the treatment effects.
Safety analysis set: This analysis set will include all randomized patients who were given at least one dose of study medication, on whom any post-dose data are available, and who are classified according to the treatment actually received (i.e., erroneously treated patients will be accounted for in their actual treatment group). This set will be used to assess safety and tolerability variables.
Specific Analytic Strategies
Efficacy and safety variables will be summarized using descriptive statistics and graphs. Continuous variables will be summarized by descriptive statistics. Categorical variables will be summarized in frequency tables. Pharmacokinetic variables will be summarized by the geometric mean and coefficient of variation. Lanicemine will be compared with placebo. Where appropriate, we will report model-based point estimates together with their 95% credible intervals.
Preliminary data analyses examining group differences for demographic and baseline variables will use cross-tabulation, ANOVA’s, and examination of correlations between baseline variables and specified outcomes. For the purposes of evaluating the comparability of groups, posterior probabilities of ≥95% will constitute evidence for statistically reliable differences. Baseline or demographic variables on which group differences are detected, and which are correlated with outcomes meet the definition of confounders (106, 107), and will result in two sets of analyses: one in which the relevant variable is included as a covariate and one in which it is not. This will permit determination of the degree to which any covariate might confound conclusions regarding treatment. The data analytic strategy will use generalized linear modeling (GLM) and multilevel models for both continuous and discrete outcomes. Cross-sectional continuous, count, dichotomous and time to event data will be evaluated using GLM and proportional hazards regression, respectively. All analyses will be conducted on a mITT analysis set.
Bayesian approaches will implement joint modeling of observed outcomes and the missing data which is robust to ignorable missingness (108). Sensitivity analyses will evaluate robustness of analytic conclusions to missing data. Non-ignorable missing data patterns will be addressed through pattern-mixture modeling methods (109). Evaluation of posterior distributions will permit statements regarding the probability that effects of varying magnitudes exist, given the data. Specification of diffuse, neutral priors will reflect the initial uncertainty regarding effect sizes. For all generalized linear mixed models, priors for regression coefficients will be specified as ∼Normal (µ = 0, σ2 = 1 x 106); level one error variances will be specified as ∼Folded t-distribution (ν = 3, µ = 0, σ = 1000). Prior distributions for level-two variances in multilevel models will follow ∼Folded t-distribution (ν = 3, µ = 0, σ = 1000). Priors for the comparison of proportions will be specified as ∼Beta (α = 0.5, β = 0.5). Sensitivity analysis using optimistic and pessimistic, skeptical priors will evaluate prior assumptions (110). Decisions regarding the degree to which treatment confers benefit, and whether further confirmatory trials are warranted will be based on the posterior distribution of effect sizes. If the observed posterior probability of a Cohen’s d < -0.4 is at least 0.75, this will be sufficient evidence to proceed to a larger POC clinical trial.
Assessing the convergence of Bayesian analyses on the posterior distributions via Monte-Carlo Markov chain (MCMC) will use graphical (Trace Plot, Autocorrelation Plot) and quantitative (Gelman-Rubin Diagnostics and Effective Sample Size) evidence. Mediational modeling will permit estimates of the indirect effects of treatment on primary and secondary endpoints using the product coefficient method (111). Bayesian Structural Equation Modelling (BSEM) prior specification will adapt recommendations from Muthén and Asparouhov (112). A Bayesian estimate for the indirect effect employs the posterior distribution of the parameter (i.e. the product coefficient): a density denoting the probability that the different values of the parameter obtain given the observed data. This posterior distribution may be further partitioned to evaluate the probability that the true parameter falls within a specific range of values. This will facilitate decision-making regarding the relative merits of continued investigation of the compound. Use of the MCMC approach in Bayesian analyses has demonstrated superiority for small sample performance compared to maximum likelihood-based approaches in continuous, normally distributed data (111). These properties are likely due to the MCMC approach’s lack of reliance on large sample size assumptions (113). Sensitivity analysis using optimistic and pessimistic, skeptical priors evaluates prior assumptions (110).
Conclusion
This POM clinical trial examines a novel target—trauma-induced behavioral sensitization—hypothesized to be associated with PTSD symptoms. The expression of behavioral sensitization is measured as an exaggerated APS reflecting enhanced reactivity of the BNST, a component of the extended amygdala, to uncontrollable stressors. Behavioral sensitization in general, and BNST reactivity specifically, may be under the control of NMDARs (66). Lanicemine potentially addresses the interaction between behavioral sensitization and acute behavioral and cognitive disturbances that characterize PTSD through its interaction with NMDAR that we will test using neurophysiological measures sensitive to NMDAR agents. If lanicemine engages the functional target and is safe and efficacious in this difficult-to-treat population, there is genuine translational potential for this compound or similar agents in treatment of PTSD and of other disorders potentially involving behavioral sensitization, such as bipolar disorder or substance use disorders. This study does have limitations that limit the generalizability of outcomes. The outcomes are limited to individuals with high APS. In addition, we exclude individuals with disorders that frequently co-exist with PTSD, and with medications that are frequently used for PTSD. Finally, studies with lanicemine and placebo in individuals with treatment resistant depression have revealed a more pronounced placebo response for individuals with fewer symptoms of depression, absent or milder suicidal ideation, and no treatment with antipsychotic medication (71). Our decision to enroll individuals with symptoms of PTSD instead of individuals who meet full PTSD criteria may benefit the placebo arm over the lanicemine arm. However, the findings regarding placebo apply to effects on symptoms of depression and may not generalize to symptoms of PTSD, and findings may not generalize to biomarkers of functional mechanisms or to biomarkers of target engagement.
This study is funded under an R61/R33 Phased Innovation Award (R61 MH10540-01). This award consists of an R61 POM study followed by an R33 phase to test clinical efficacy of a study drug. The R33 study would only commence if the POM study shows evidence of engagement by lanicemine of the functional mechanism (change in APS) and the biological target (change in neurophysiological measures). The go/no-go framework for the R61 study is operationalized such that if there is significant symptom improvement without a clear effect of functional mechanism and evidence of target engagement, the drug would not be tested further in an R33. A potential risk of this approach is that the measures of functional mechanism and of target engagement selected may not be appropriate even if the drug does engage the proposed mechanisms. This approach is in marked contrast to usual industry-supported early phase trials in which early signals of clinical efficacy drive the decision to move forward with larger Phase 2 studies, irrespective of information gleaned from biomarkers.
In conclusion, this study tracks engagement of the study drug on a functional mechanism likely to cut across disorders, which is consistent with recent NIMH-industry collaborative studies (114) and “Fast-Fail” trials (2, 115), and could serve as a template for testing pharmacological agents in psychiatry.
Ethics Statement
The study drug is administered as an intravenous solution under FDA IND number 134304; the trial is registered at ClinicalTrials.gov under NCT03166501. All study-related procedures and materials have been approved by the Baylor College of Medicine Institutional Review Board, and the MEDVAMC Office of Research and Development. An NIMH-appointed data and safety monitoring board (DSMB) provides oversight.
Author Contributions
SJM, ACS, CEG, and ML conceived and developed the project. ML, NB, TI, MA, BrV-L, By V-L, BOB, CG, ACS, and SJM contributed to implementing the project. ML, CEG, and SJM wrote the draft version and the final version of this manuscript. NB, TI, MA, BrV-L, ByV-L, BOB, CG, and ACS contributed to the draft version and the final version of this manuscript.
Funding
This work was supported by the National Institute of Mental Health through grant 5R61MH110540. The views expressed in this publication do not necessarily reflect the official policies of the U.S. Department of Veterans Affairs.
Conflict of Interest
SM is PI on a Biohaven-sponsored clinical trial.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declare that this study received material support from Biohaven Pharmaceuticals. Biohaven Pharmaceuticals was not involved in the study design, data collection, data analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Acknowledgments
The authors would like to thank Robert Berman, MD, and Lia Donahue, MA, of Biohaven for providing material and logistical support. Dr. Mathew is supported through the use of facilities and resources at the Michael E. DeBakey VA Medical Center.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpsyt.2019.00846/full#supplementary-material
References
1. Szabo ST, Kinon BJ, Brannan SK, Krystal AK, van Gerven JMA, Mahableshwarkar A, et al. Lessons learned and potentials for improvement in CNS drug development: ISCTM section on designing the right series of experiments. Innov Clin Neurosci (2015) 12:11S–25S.
2. Krystal AD, Pizzagalli DA, Mathew SJ, Sanacora G, Keefe R, Song A, et al. The first implementation of the NIMH FAST-FAIL approach to psychiatric drug development. Nat Rev Drug Discovery (2018) 18:82–4. doi: 10.1038/nrd.2018.222
3. Insel T, Cuthbert B, Garvey M, Heinssen R, Pine DS, Quinn K, et al. Research domain criteria (RDoC): toward a new classification framework for research on mental disorders. Am J Psychiatry (2010) 167:748–51. doi: 10.1176/appi.ajp.2010.09091379
4. Insel TR. The NIMH Research Domain Criteria (RDoC) project: precision medicine for psychiatry. Am J Psychiatry (2014) 171:395–7. doi: 10.1176/appi.ajp.2014.14020138
5. Friedman MJ. Finalizing PTSD in DSM-5: getting here from there and where to go next. J Trauma Stress (2013) 26:548–56. doi: 10.1002/jts.21840
6. Kessler RC, Petukhova M, Sampson NA, Zaslavsky AM, Wittchen H-U. Twelve-month and lifetime prevalence and lifetime morbid risk of anxiety and mood disorders in the United States. Int J Methods Psychiatr Res (2012) 21:169–84. doi: 10.1002/mpr.1359
7. Goldstein RB, Smith SM, Chou SP, Saha TD, Jung J, Zhang H, et al. The epidemiology of DSM-5 posttraumatic stress disorder in the United States: results from the National Epidemiologic Survey on Alcohol and Related Conditions-III. Soc Psychiatry Psychiatr Epidemiol (2016) 51:1137–48. doi: 10.1007/s00127-016-1208-5
8. Berger W, Coutinho ESF, Figueira I, Marques-Portella C, Luz MP, Neylan TC, et al. Rescuers at risk: a systematic review and meta-regression analysis of the worldwide current prevalence and correlates of PTSD in rescue workers. Soc Psychiatry Psychiatr Epidemiol (2012) 47:1001–11. doi: 10.1007/s00127-011-0408-2
9. Dursa EK, Reinhard MJ, Barth SK, Schneiderman AI. Prevalence of a positive screen for PTSD among OEF/OIF and OEF/OIF-era veterans in a large population-based cohort. J Trauma Stress (2014) 27:542–9. doi: 10.1002/jts.21956
10. Magruder K, Serpi T, Kimerling R, Kilbourne AM, Collins JF, Cypel Y, et al. Prevalence of posttraumatic stress disorder in Vietnam-era women veterans: the health of Vietnam-era women’s study (HealthVIEWS). JAMA Psychiatry (2015) 72:1127–34. doi: 10.1001/jamapsychiatry.2015.1786
11. Goldberg J, Magruder KM, Forsberg CW, Friedman MJ, Litz BT, Vaccarino V, et al. Prevalence of post-traumatic stress disorder in aging Vietnam-era veterans: veterans administration cooperative study 569: course and consequences of post-traumatic stress disorder in Vietnam-era veteran twins. Am J Geriatr Psychiatry (2016) 24:181–91. doi: 10.1016/j.jagp.2015.05.004
12. Lehavot K, Katon JG, Chen JA, Fortney JC, Simpson TL. Post-traumatic stress disorder by gender and veteran status. Am J Prev Med (2018) 54:e1–9. doi: 10.1016/j.amepre.2017.09.008
13. Pietrzak RH, Goldstein RB, Southwick SM, Grant BF. Prevalence and Axis I comorbidity of full and partial posttraumatic stress disorder in the United States: results from Wave 2 of the National Epidemiologic Survey on Alcohol and Related Conditions. J Anxiety Disord (2011) 25:456–65. doi: 10.1016/j.janxdis.2010.11.010
14. Forman-Hoffman V, Cook Middleton J, Feltner C, Gaynes BN, Palmieri Weber R, Bann C, et al. Psychological and Pharmacological Treatments for Adults With Posttraumatic Stress Disorder: A Systematic Review Update. Rockville, MD: Agency for Healthcare Research and Quality (AHRQ) (2018). doi: 10.23970/AHRQEPCCER207
15. Kalivas PW, Stewart J. Dopamine transmission in the initiation and expression of drug- and stress-induced sensitization of motor activity. Brain Res Rev (1991) 16:223–44. doi: 10.1016/0165-0173(91)90007-U
16. Lijffijt M, O’Brien B, Salas R, Mathew SJ, Swann AC. Interactions of immediate and long-term action regulation in the course and complications of bipolar disorder. Philos Trans R Soc B: Biol Sci (2018) 374:20180132. doi: 10.1098/rstb.2018.0132
17. McLaughlin KA, Koenen KC, Bromet EJ, Karam EG, Liu H, Petukhova M, et al. Childhood adversities and post-traumatic stress disorder: evidence for stress sensitisation in the World Mental Health Surveys. Br J Psychiatry (2017) 211:280–8. 17m11634. doi: 10.1192/bjp.bp.116.197640
18. Swann AC. Beyond dugs: addictions in the context of recurrent/progressive psychiatric illness. In: Neurobiology of Addiction. (New York: Oxford University Press).
19. Lijffijt M. Stress and Addiction. In: Neurobiology of Addiction. (New York: Oxford University Press). p. 153–75.
20. Swann AC, Lijffijt M, Lane SD, Steinberg JL, Moeller FG. Interacting mechanisms of impulsivity in bipolar disorder and antisocial personality disorder. J Psychiatr Res (2011) 45:1477–82. doi: 10.1016/j.jpsychires.2011.06.009
21. Lijffijt M, Hu K, Swann AC. Stress modulates illness-course of substance use disorders: a translational review. Front In Psychiatry (2014) 5:83. doi: 10.3389/fpsyt.2014.00083
22. Maeng LY, Milad MR. Post-Traumatic stress disorder: the relationship between the fear response and chronic stress. Chronic Stress (2017) 1:247054701771329. doi: 10.1177/2470547017713297
23. Swann AC. The strong relationship between bipolar and substance-use disorder. Ann N Y Acad Sci (2010) 1187:276–93. doi: 10.1111/j.1749-6632.2009.05146.x
24. Booij L, Welfeld K, Leyton M, Dagher A, Boileau I, Sibon I, et al. Dopamine cross-sensitization between psychostimulant drugs and stress in healthy male volunteers. Transl Psychiatry (2016) 6:e740. doi: 10.1038/tp.2016.6
25. Churchill L, Swanson CJ, Urbina M, Kalivas PW. Repeated cocaine alters glutamate receptor subunit levels in the nucleus accumbens and ventral tegmental area of rats that develop behavioral sensitization. J Neurochem (1999) 72:2397–403. doi: 10.1046/j.1471-4159.1999.0722397.x
26. Broadbent J, Weitemier AZ. Dizocilpine (MK-801) prevents the development of sensitization to ethanol in DBA/2J mice. Alcohol Alcohol (1999) 34:283–8. doi: 10.1093/alcalc/34.3.283
27. Camarini R, Frussa-Filho R, Monteiro MG, Calil HM. MK-801 blocks the development of behavioral sensitization to the ethanol. Alcohol Clin Exp Res (2000) 24:285–90. doi: 10.1111/j.1530-0277.2000.tb04609.x
28. Druhan JP, Jakob A, Stewart J. The development of behavioral sensitization to apomorphine is blocked by MK-801. Eur J Pharmacol (1993) 243:73–7. doi: 10.1016/0014-2999(93)90169-i
29. Kalivas PW. Interactions between dopamine and excitatory amino acids in behavioral sensitization to psychostimulants. Drug Alcohol Depend (1995) 37:95–100. doi: 10.1016/0376-8716(94)01063-q
30. Kalivas PW, Alesdatter JE. Involvement of N-methyl-D-aspartate receptor stimulation in the ventral tegmental area and amygdala in behavioral sensitization to cocaine. J Pharmacol Exp Ther (1993) 267:486–95.
31. Wolf ME. The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Prog Neurobiol (1998) 54:679–720. doi: 10.1016/s0301-0082(97)00090-7
32. Wolf ME, White FJ, Hu XT. MK-801 prevents alterations in the mesoaccumbens dopamine system associated with behavioral sensitization to amphetamine. J Neurosci (1994) 14:1735–45. doi: 10.1523/JNEUROSCI.14-03-01735.1994
33. Cador M, Bjijou Y, Cailhol S, Stinus L. D-amphetamine-induced behavioral sensitization: implication of a glutamatergic medial prefrontal cortex-ventral tegmental area innervation. Neuroscience (1999) 94:705–21. doi: 10.1016/s0306-4522(99)00361-9
34. Gaytan O, Sripada S, Swann A, Dafny N. Blockade of sensitization to methylphenidate by MK-801: partial dissociation from motor effects. Neuropharmacology (2001) 40:298–309. doi: 10.1016/s0028-3908(00)00122-2
35. Schenk S, Valadez A, McNamara C, House DT, Higley D, Bankson MG, et al. Development and expression of sensitization to cocaine’s reinforcing properties: role of NMDA receptors. Psychopharmacol (Berl) (1993) 111:332–8. doi: 10.1007/bf02244949
36. Yap JJ, Covington HE, Gale MC, Datta R, Miczek KA. Behavioral sensitization due to social defeat stress in mice: antagonism at mGluR5 and NMDA receptors. Psychopharmacol (Berl) (2005) 179:230–9. doi: 10.1007/s00213-004-2023-3
37. Hong SK, Jung IS, Bang SA, Kim SE. Effect of nitric oxide synthase inhibitor and NMDA receptor antagonist on the development of nicotine sensitization of nucleus accumbens dopamine release: an in vivo microdialysis study. Neurosci Lett (2006) 409:220–3. doi: 10.1016/j.neulet.2006.09.052
38. Shim I, Kim H-T, Kim Y-H, Chun B-G, Hahm D-H, Lee EH, et al. Role of nitric oxide synthase inhibitors and NMDA receptor antagonist in nicotine-induced behavioral sensitization in the rat. Eur J Pharmacol (2002) 443:119–24. doi: 10.1016/s0014-2999(02)01582-0
39. Broadbent J, Kampmueller KM, Koonse SA. Expression of behavioral sensitization to ethanol by DBA/2J mice: the role of NMDA and non-NMDA glutamate receptors. Psychopharmacol (Berl) (2003) 167:225–34. doi: 10.1007/s00213-003-1404-3
40. Chen JC, Liang KW, Huang YK, Liang CS, Chiang YC. Significance of glutamate and dopamine neurons in the ventral pallidum in the expression of behavioral sensitization to amphetamine. Life Sci (2001) 68:973–83. doi: 10.1016/s0024-3205(00)00995-4
41. Garcia LSB, Comim CM, Valvassori SS, Réus GZ, Stertz L, Kapczinski F, et al. Ketamine treatment reverses behavioral and physiological alterations induced by chronic mild stress in rats. Prog Neuropsychopharmacol Biol Psychiatry (2009) 33:450–5. doi: 10.1016/j.pnpbp.2009.01.004
42. Itzhak Y, Martin JL. Effects of cocaine, nicotine, dizocipline and alcohol on mice locomotor activity: cocaine-alcohol cross-sensitization involves upregulation of striatal dopamine transporter binding sites. Brain Res (1999) 818:204–11. doi: 10.1016/s0006-8993(98)01260-8
43. Covington HE, Tropea TF, Rajadhyaksha AM, Kosofsky BE, Miczek KA. NMDA receptors in the rat VTA: a critical site for social stress to intensify cocaine taking. Psychopharmacol (Berl) (2008) 197:203–16. doi: 10.1007/s00213-007-1024-4
44. Pacchioni AM, Gioino G, Assis A, Cancela LM. A single exposure to restraint stress induces behavioral and neurochemical sensitization to stimulating effects of amphetamine: involvement of NMDA receptors. Ann N Y Acad Sci (2002) 965:233–46. doi: 10.1111/j.1749-6632.2002.tb04165.x
45. Cornish JL, Duffy P, Kalivas PW. A role for nucleus accumbens glutamate transmission in the relapse to cocaine-seeking behavior. Neuroscience (1999) 93:1359–67. doi: 10.1016/s0306-4522(99)00214-6
46. Cloitre M, Stolbach BC, Herman JL, van der Kolk B, Pynoos R, Wang J, et al. A developmental approach to complex PTSD: childhood and adult cumulative trauma as predictors of symptom complexity. J Trauma Stress (2009) 22:399–408. doi: 10.1002/jts.20444
47. Davis M, Walker DL, Miles L, Grillon C. Phasic vs sustained fear in rats and humans: role of the extended amygdala in fear vs anxiety. Neuropsychopharmacology (2010) 35:105–35. doi: 10.1038/npp.2009.109
48. Weston CSE. Posttraumatic stress disorder: a theoretical model of the hyperarousal subtype. Front Psychiatry (2014) 5:37. doi: 10.3389/fpsyt.2014.00037
49. Schmidt U, Vermetten E. Integrating NIMH Research Domain Criteria (RDoC) into PTSD Research. Curr Top Behav Neurosci (2018) 38:69–91. doi: 10.1007/7854_2017_1
50. Perusini JN, Meyer EM, Long VA, Rau V, Nocera N, Avershal J, et al. Induction and expression of fear sensitization caused by acute traumatic stress. Neuropsychopharmacology (2016) 41:45–57. doi: 10.1038/npp.2015.224
51. Admon R, Lubin G, Rosenblatt JD, Stern O, Kahn I, Assaf M, et al. Imbalanced neural responsivity to risk and reward indicates stress vulnerability in humans. Cereb Cortex (2013) 23:28–35. doi: 10.1093/cercor/bhr369
52. McLaughlin KA, Busso DS, Duys A, Green JG, Alves S, Way M, et al. Amygdala response to negative stimuli predicts PTSD symptom onset following a terrorist attack. Depress Anxiety (2014) 31:834–42. doi: 10.1002/da.22284
53. Swartz JR, Knodt AR, Radtke SR, Hariri AR. A neural biomarker of psychological vulnerability to future life stress. Neuron (2015) 85:505–11. doi: 10.1016/j.neuron.2014.12.055
54. McGhee LL, Maani CV, Garza TH, Gaylord KM, Black IH. The correlation between ketamine and posttraumatic stress disorder in burned service members. J Trauma (2008) 64:S195–198. doi: 10.1097/TA.0b013e318160ba1d
55. McGhee LL, Maani CV, Garza TH, Slater TM, Petz LN, Fowler M. The intraoperative administration of ketamine to burned U.S. service members does not increase the incidence of post-traumatic stress disorder. Mil Med (2014) 179:41–6. doi: 10.7205/MILMED-D-13-00481
56. Albott CS, Lim KO, Forbes MK, Erbes C, Tye SJ, Grabowski JG, et al. Efficacy, safety, and durability of repeated ketamine infusions for comorbid posttraumatic stress disorder and treatment-resistant depression. J Clin Psychiatry (2018) 79: 17m11634. doi: 10.4088/JCP.17m11634
57. Feder A, Parides MK, Murrough JW, Perez AM, Morgan JE, Saxena S, et al. Efficacy of intravenous ketamine for treatment of chronic posttraumatic stress disorder: a randomized clinical trial. JAMA Psychiatry (2014) 71:681–8. doi: 10.1001/jamapsychiatry.2014.62
58. Battista MA, Hierholzer R, Khouzam HR, Barlow A, O’Toole S. Pilot trial of memantine in the treatment of posttraumatic stress disorder. Psychiatry (2007) 70:167–74. doi: 10.1521/psyc.2007.70.2.167
59. Grillon C, Heller R, Hirschhorn E, Kling MA, Pine DS, Schulkin J, et al. Acute hydrocortisone treatment increases anxiety but not fear in healthy volunteers: a fear-potentiated startle study. Biol Psychiatry (2011) 69:549–55. doi: 10.1016/j.biopsych.2010.12.013
60. Schmitz A, Grillon C. Assessing fear and anxiety in humans using the threat of predictable and unpredictable aversive events (the NPU-threat test). Nat Protoc (2015) 7:527–32. doi: 10.1038/nprot.2012.001
61. Grillon C, Chavis C, Covington MF, Pine DS. Two-week treatment with the selective serotonin reuptake inhibitor citalopram reduces contextual anxiety but not cued fear in healthy volunteers: a fear-potentiated startle study. Neuropsychopharmacology (2009) 34:964–71. doi: 10.1038/npp.2008.141
62. Grillon C, Baas J. A review of the modulation of the startle reflex by affective states and its application in psychiatry. Clin Neurophysiol (2003) 114:1557–79. doi: 10.1016/s1388-2457(03)00202-5
63. Vaidyanathan U, Patrick CJ, Cuthbert BN. Linking dimensional models of internalizing psychopathology to neurobiological systems: affect-modulated startle as an indicator of fear and distress disorders and affiliated traits. Psychol Bull (2009) 135:909–42. doi: 10.1037/a0017222
64. Grillon C, Morgan CA. Fear-potentiated startle conditioning to explicit and contextual cues in Gulf War veterans with posttraumatic stress disorder. J Abnorm Psychol (1999) 108:134–42. doi: 10.1037//0021-843x.108.1.134
65. Grillon C, Pine DS, Lissek S, Rabin S, Bonne O, Vythilingam M. Increased anxiety during anticipation of unpredictable aversive stimuli in posttraumatic stress disorder but not in generalized anxiety disorder. Biol Psychiatry (2009) 66:47–53. doi: 10.1016/j.biopsych.2008.12.028
66. Jennings JH, Sparta DR, Stamatakis AM, Ung RL, Pleil KE, Kash TL, et al. Distinct extended amygdala circuits for divergent motivational states. Nature (2013) 496:224–8. doi: 10.1038/nature12041
67. Hayes JP, Hayes SM, Mikedis AM. Quantitative meta-analysis of neural activity in posttraumatic stress disorder. Biol Mood Anxiety Disord (2012) 2:9. doi: 10.1186/2045-5380-2-9
68. Ousdal OT, Milde AM, Craven AR, Ersland L, Endestad T, Melinder A, et al. Prefrontal glutamate levels predict altered amygdala-prefrontal connectivity in traumatized youths. Psychol Med (2018), 49:1822–30. doi: 10.1017/S0033291718002519
69. Sanacora G, Smith MA, Pathak S, Su H-L, Boeijinga PH, McCarthy DJ, et al. Lanicemine: a low-trapping NMDA channel blocker produces sustained antidepressant efficacy with minimal psychotomimetic adverse effects. Mol Psychiatry (2014) 19:978–85. doi: 10.1038/mp.2013.130
70. Sanacora G, Schatzberg AF. Ketamine: promising path or false prophecy in the development of novel therapeutics for mood disorders?. Neuropsychopharmacology (2015) 40:1307. doi: 10.1038/npp.2014.338
71. Sanacora G, Johnson MR, Khan A, Atkinson SD, Riesenberg RR, Schronen JP, et al. Adjunctive lanicemine (AZD6765) in patients with major depressive disorder and history of inadequate response to antidepressants: a randomized, placebo-controlled study. Neuropsychopharmacology (2017) 42:844–53. doi: 10.1038/npp.2016.224
72. Agbo F, Bui KH, Zhou D. Population pharmacokinetic analysis of lanicemine (AZD6765), an NMDA channel blocker, in healthy subjects and patients with major depressive disorder. J Clin Pharm Ther (2017) 42:539–46. doi: 10.1111/jcpt.12541
73. Murrough JW, Iosifescu DV, Chang LC, Al Jurdi RK, Green CE, Perez AM, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry (2013) 170:1134–42. doi: 10.1176/appi.ajp.2013.13030392
74. Hiyoshi T, Kambe D, Karasawa J, Chaki S. Differential effects of NMDA receptor antagonists at lower and higher doses on basal gamma band oscillation power in rat cortical electroencephalograms. Neuropharmacology (2014) 85:384–96. doi: 10.1016/j.neuropharm.2014.05.037
75. Kocsis B. Differential role of NR2A and NR2B subunits in N-methyl-D-aspartate receptor antagonist-induced aberrant cortical gamma oscillations. Biol Psychiatry (2012) 71:987–95. doi: 10.1016/j.biopsych.2011.10.002
76. Nagy D, Stoiljkovic M, Menniti FS, Hajós M. Differential effects of an NR2B NAM and ketamine on synaptic potentiation and gamma synchrony: relevance to rapid-onset antidepressant efficacy. Neuropsychopharmacology (2016) 41:1486–94. doi: 10.1038/npp.2015.298
77. Rivolta D, Heidegger T, Scheller B, Sauer A, Schaum M, Birkner K, et al. Ketamine dysregulates the amplitude and connectivity of high-frequency oscillations in cortical-subcortical networks in humans: evidence from resting-state magnetoencephalography-recordings. Schizophr Bull (2015) 41:1105–14. doi: 10.1093/schbul/sbv051
78. Nugent AC, Ballard ED, Gould TD, Park LT, Moaddel R, Brutsche NE, et al. Ketamine has distinct electrophysiological and behavioral effects in depressed and healthy subjects. Mol Psychiatry (2018). 24:1040–52 doi: 10.1038/s41380-018-0028-2
79. Routley BC, Singh KD, Hamandi K, Muthukumaraswamy SD. The effects of AMPA receptor blockade on resting magnetoencephalography recordings. J Psychopharmacol (Oxford) (2017) 31:1527–36. doi: 10.1177/0269881117736915
80. Carlén M, Meletis K, Siegle JH, Cardin JA, Futai K, Vierling-Claassen D, et al. A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. Mol Psychiatry (2012) 17:537–48. doi: 10.1038/mp.2011.31
81. Abdallah CG, De Feyter HM, Averill LA, Jiang L, Averill CL, Chowdhury GMI, et al. The effects of ketamine on prefrontal glutamate neurotransmission in healthy and depressed subjects. Neuropsychopharmacology (2018) 43:2154–60. doi: 10.1038/s41386-018-0136-3
82. Sivarao DV, Chen P, Senapati A, Yang Y, Fernandes A, Benitex Y, et al. 40 Hz auditory steady-state response is a pharmacodynamic biomarker for cortical NMDA receptors. Neuropsychopharmacology (2016) 41:2232–40. doi: 10.1038/npp.2016.17
83. Sullivan EM, Timi P, Hong LE, O’Donnell P. Effects of NMDA and GABA-A receptor antagonism on auditory steady-state synchronization in awake behaving rats. Int J Neuropsychopharmacol (2015) 18:pyu118. doi: 10.1093/ijnp/pyu118
84. Light GA, Zhang W, Joshi YB, Bhakta S, Talledo JA, Swerdlow NR. Single-dose memantine improves cortical oscillatory response dynamics in patients with schizophrenia. Neuropsychopharmacology (2017) 42:2633–9. doi: 10.1038/npp.2017.81
85. Näätänen R, Kujala T, Winkler I. Auditory processing that leads to conscious perception: a unique window to central auditory processing opened by the mismatch negativity and related responses. Psychophysiology (2011) 48:4–22. doi: 10.1111/j.1469-8986.2010.01114.x
86. Ge Y, Wu J, Sun X, Zhang K. Enhanced mismatch negativity in adolescents with posttraumatic stress disorder (PTSD). Int J Psychophysiol (2011) 79:231–5. doi: 10.1016/j.ijpsycho.2010.10.012
87. Zukerman G, Fostick L, Ben-Itzchak E. Early automatic hyperarousal in response to neutral novel auditory stimuli among trauma-exposed individuals with and without PTSD: an ERP study. Psychophysiology (2018) 55:e13217. doi: 10.1111/psyp.13217
88. Menning H, Renz A, Seifert J, Maercker A. Reduced mismatch negativity in posttraumatic stress disorder: a compensatory mechanism for chronic hyperarousal?. Int J Psychophysiol (2008) 68:27–34. doi: 10.1016/j.ijpsycho.2007.12.003
89. Cornwell BR, Baas JMP, Johnson L, Holroyd T, Carver FW, Lissek S, et al. Neural responses to auditory stimulus deviance under threat of electric shock revealed by spatially-filtered magnetoencephalography. Neuroimage (2007) 37:282–9. doi: 10.1016/j.neuroimage.2007.04.055
90. Heekeren K, Daumann J, Neukirch A, Stock C, Kawohl W, Norra C, et al. Mismatch negativity generation in the human 5HT2A agonist and NMDA antagonist model of psychosis. Psychopharmacol (Berl) (2008) 199:77–88. doi: 10.1007/s00213-008-1129-4
91. Kreitschmann-Andermahr I, Rosburg T, Demme U, Gaser E, Nowak H, Sauer H. Effect of ketamine on the neuromagnetic mismatch field in healthy humans. Brain Res Cognit Brain Res (2001) 12:109–16. doi: 10.1016/s0926-6410(01)00043-x
92. Umbricht D, Schmid L, Koller R, Vollenweider FX, Hell D, Javitt DC. Ketamine-induced deficits in auditory and visual context-dependent processing in healthy volunteers: implications for models of cognitive deficits in schizophrenia. Arch Gen Psychiatry (2000) 57:1139–47. doi: 10.1001/archpsyc.57.12.1139
93. Sivarao DV, Chen P, Yang Y, Li Y-W, Pieschl R, Ahlijanian MK. NR2B antagonist CP-101,606 abolishes pitch-mediated deviance detection in awake rats. Front Psychiatry (2014) 5:96. doi: 10.3389/fpsyt.2014.00096
94. Swerdlow NR, Bhakta S, Chou H-H, Talledo JA, Balvaneda B, Light GA. memantine effects on sensorimotor gating and mismatch negativity in patients with chronic psychosis. Neuropsychopharmacology (2016) 41:419–30. doi: 10.1038/npp.2015.162
95. Lijffijt M, Moeller FG, Boutros NN, Burroughs S, Lane SD, Steinberg JL, et al. The role of age, gender, education, and intelligence in P50, N100, and P200 auditory sensory gating. J Psychophysiol (2009) 23:52–62. doi: 10.1027/0269-8803.23.2.52
96. Lijffijt M, Lane SD, Meier SL, Boutros NN, Burroughs S, Steinberg JL, et al. P50, N100, and P200 sensory gating: relationships with behavioral inhibition, attention, and working memory. Psychophysiology (2009) 46:1059–68. doi: 10.1111/j.1469-8986.2009.00845.x
97. Lijffijt M, Cox B, Acas MD, Lane SD, Moeller FG, Swann AC. Differential relationships of impulsivity or antisocial symptoms on P50, N100, or P200 auditory sensory gating in controls and antisocial personality disorder. J Psychiatr Res (2012) 46:743–50. doi: 10.1016/j.jpsychires.2012.03.001
98. Lijffijt M, Moeller FG, Boutros NN, Steinberg JL, Meier SL, Lane SD, et al. Diminished P50, N100 and P200 auditory sensory gating in bipolar I disorder. Psychiatry Res (2009) 167:191–201. doi: 10.1016/j.psychres.2008.04.001
99. Karl A, Malta LS, Maercker A. Meta-analytic review of event-related potential studies in post-traumatic stress disorder. Biol Psychol (2006) 71:123–47. doi: 10.1016/j.biopsycho.2005.03.004
100. Ermutlu MN, Karamürsel S, Ugur EH, Senturk L, Gokhan N. Effects of cold stress on early and late stimulus gating. Psychiatry Res (2005) 136:201–9. doi: 10.1016/j.psychres.2003.03.002
101. Griskova-Bulanova I, Paskevic J, Dapsys K, Maciulis V, Ruksenas O, Arnfred SM. The level of arousal modulates P50 peak amplitude. Neurosci Lett (2011) 499:204–7. doi: 10.1016/j.neulet.2011.05.062
102. Hong LE, Summerfelt A, Buchanan RW, O’Donnell P, Thaker GK, Weiler MA, et al. Gamma and delta neural oscillations and association with clinical symptoms under subanesthetic ketamine. Neuropsychopharmacology (2010) 35:632–40. doi: 10.1038/npp.2009.168
103. Weathers FW, Bovin MJ, Lee DJ, Sloan DM, Schnurr PP, Kaloupek DG, et al. The Clinician-Administered PTSD Scale for DSM-5 (CAPS-5): development and initial psychometric evaluation in military veterans. Psychol Assess (2018) 30:383–95. doi: 10.1037/pas0000486
104. Weathers FW, Blake DD, Schnurr PP, Kaloupek DG, Marx BP, Keane TM. The Clinician-Administered PTSD Scale for DSM-5 (CAPS-5) – Past Month / Worst Month. (2015). Available at: www.ptsd.va.gov.
105. Weathers FW, Blake DD, Schnurr PP, Kaloupek DG, Marx BP, Keane TM. The Clinician-Administered PTSD Scale for DSM-5 (CAPS-5) – Past Week. (2015). Available at: www.ptsd.va.gov.
106. Pocock SJ, Assmann SE, Enos LE, Kasten LE. Subgroup analysis, covariate adjustment and baseline comparisons in clinical trial reporting: current practice and problems. Stat Med (2002) 21:2917–30. doi: 10.1002/sim.1296
107. Best N, Thomas A. Bayesian graphic models and software for GLMs. In: Generalized linear models: A Bayesian perspective. Marcel Dekker, Inc.. p. 387–402.
108. Fitzmaurice GM, Laird NM. Generalized linear mixture models for handling nonignorable dropouts in longitudinal studies. Biostatistics (2000) 1:141–56. doi: 10.1093/biostatistics/1.2.141
109. Spiegelhalter DJ, Abrams KR, Myles JP. Bayesian approaches to clinical trials and health-care evaluation. West Sussex, England: John Wiley & Sons, Ltd. (2004).
110. Lee S-Y, Song X-Y. Evaluation of the Bayesian and maximum likelihood approaches in analyzing structural equation models with small sample sizes. Multivariate Behav Res (2004) 39:653–86. doi: 10.1207/s15327906mbr3904_4
111. Muthén B, Asparouhov T. Bayesian structural equation modeling: a more flexible representation of substantive theory. Psychol Methods (2012) 17:313–35. doi: 10.1037/a0026802
112. Dunson DB, Palomo J, Bollen K. Bayesian structural equation modeling. Stat Appl Math Sci Inst (2005). http://www.samsi.info/sites/default/files/tr2005-05.pdf.
113. Grabb MC, Cross AJ, Potter WZ, McCracken JT. Derisking psychiatric drug development: The NIMH’s Fast Fail program, a novel precompetitive model. J Clin Psychopharmacol (2016) 36:419–21. doi: 10.1097/JCP.0000000000000536
114. Javitt DC, Carter CS, Krystal JH, Kantrowitz JT, Girgis RR, Kegeles LS, et al. Utility of imaging-based biomarkers for glutamate-targeted drug development in psychotic disorders: a randomized clinical trial. JAMA Psychiatry (2018) 75:11–9. doi: 10.1001/jamapsychiatry.2017.3572
Keywords: behavioral sensitization, NMDA receptor, hyperarousal, neurophysiology, post-traumatic stress disorder, anxiety potentiated startle
Citation: Lijffijt M, Green CE, Balderston N, Iqbal T, Atkinson M, Vo-Le B, Vo-Le B, O’Brien B, Grillon C, Swann AC and Mathew SJ (2019) A Proof-of-Mechanism Study to Test Effects of the NMDA Receptor Antagonist Lanicemine on Behavioral Sensitization in Individuals With Symptoms of PTSD. Front. Psychiatry 10:846. doi: 10.3389/fpsyt.2019.00846
Received: 02 August 2019; Accepted: 24 October 2019;
Published: 13 December 2019.
Edited by:
James Murrough, Icahn School of Medicine at Mount Sinai, United StatesReviewed by:
Manish Kumar Jha, UT Southwestern Medical Center, United StatesRodrigo Machado-Vieira, National Institute of Mental Health (NIMH), United States
Copyright © 2019 Lijffijt, Green, Balderston, Iqbal, Atkinson, Vo-Le, Vo-Le, O’Brien, Grillon, Swann and Mathew. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sanjay J. Mathew, sjmathew@bcm.edu