 Daram Choi1,2
Daram Choi1,2 Thomas C. Harrington3
Thomas C. Harrington3 David C. Shaw1
David C. Shaw1 Jane E. Stewart4
Jane E. Stewart4 Ned B. Klopfenstein5
Ned B. Klopfenstein5 Duncan R. Kroese2
Duncan R. Kroese2 Mee-Sook Kim2*
Mee-Sook Kim2*- 1Department of Forest Engineering, Resources and Management, Oregon State University, Corvallis, OR, United States
- 2Pacific Northwest Research Station, USDA Forest Service, Corvallis, OR, United States
- 3Department of Plant Pathology, Entomology and Microbiology, Iowa State University, Ames, IA, United States
- 4Department of Agricultural Biology, Colorado State University, Fort Collins, CO, United States
- 5Rocky Mountain Research Station, USDA Forest Service, Moscow, ID, United States
Leptographium wageneri is a native fungal pathogen in western North America that causes black stain root disease (BSRD) of conifers. Three host-specialized varieties of this pathogen were previously described: L. wageneri var. wageneri on pinyon pines (Pinus monophylla and P. edulis); L. wageneri var. ponderosum, primarily on hard pines (e.g., P. ponderosa, P. jeffreyi); and L. wageneri var. pseudotsugae on Douglas-fir (Pseudotsuga menziesii). Morphological, physiological, and ecological differences among the three pathogen varieties have been previously determined; however, DNA-based characterization and analyses are needed to determine the genetic relationships among these varieties. The objective of this study was to use DNA sequences of 10 gene regions to assess phylogenetic relationships among L. wageneri isolates collected from different hosts. The multigene phylogenetic analyses, based on maximum likelihood and Bayesian inference, strongly supported species-level separation of the three L. wageneri varieties. These results, in conjunction with previously established phenotypic differences, support the elevation of L. wageneri var. ponderosum and L. wageneri var. pseudotsugae to the species level as L. ponderosum comb. nov. and L. pseudotsugae comb. nov., respectively, while maintaining L. wageneri var. wageneri as Leptographium wageneri. Characterization of the three Leptographium species, each with distinct host ranges, provides a baseline to further understand the ecological interactions and evolutionary relationships of these forest pathogens, which informs management of black stain root disease.
Introduction
Black stain root disease (BSRD) is a vascular wilt disease of conifers that is damaging to forests in western North America, especially in the western United States (Washington, Oregon, and California), and southwestern Canada (British Columbia) (Lockman and Kearns, 2016). BSRD was first found producing a dark sapwood stain in hard pines [Jeffrey pine (Pinus jeffreyi) and ponderosa pine (P. ponderosa)] and single-leaf pinyon (P. monophylla) in 1938 and 1941, respectively, in California (Wagener and Mielke, 1961). Although BSRD was not well recognized in the Pacific Northwest of United States before 1969, it has emerged as one of the five most-damaging root diseases in western forests of the United States (Hadfield et al., 1986; Cobb, 1988). The disease is caused by a native, insect-vectored ascomycete fungal pathogen, originally named Verticladiella wageneri W.B Kendr. (Kendrick, 1962) and later transferred to Leptographium wageneri (W.B Kendr.) M.J. Wingf. (Wingfield, 1985).
Leptographium wageneri can be introduced to hosts in two ways: 1) via root contacts or grafts between healthy and infected trees, or 2) via insect vectors. Wounds may be required for non-root graft infection because the pathogen’s hyphae cannot penetrate bark or break down cellulose (Hessburg, 1984), though small roots may be infected directly (Cobb, 1988). Once the BSRD fungal pathogen enters the host, it colonizes tracheid cells in sapwood xylem, and dark-brown to purple-black staining starts to appear when the xylem is colonized by hyphae (Figure 1) (Wagener and Mielke, 1961; Hessburg et al., 1995). Other than the characteristic staining, infected hosts often show general wilt disease symptoms, such as tufted needles, needle loss, chlorosis, and/or reduced growth (Supplementary Figure 1), typically followed by tree mortality. Basal resin flow may also occur, and a stress cone crop may be produced in Douglas-fir (Pseudotsuga menziesii). Because the crown symptoms are similar to other root diseases, the best way to confirm BSRD in the field is by verifying the presence of the dark stains in sapwood of roots and/or at the base of the stem (Figure 1) (Hadfield et al., 1986).

Figure 1 Black streaking of sapwood, which is associated with black stain root disease (caused by Leptographium wageneri varieties s.l.), appears in (A) root collars above ground and (B) underground roots from a single-leaf pinyon (Pinus monophylla). Staining appears above ground level in (C) ponderosa pine (P. ponderosa) and (D) Douglas-fir (Pseudotsuga menziesii).
The sexual stage of the BSRD pathogen was described as Ceratocystis wageneri Goheen & F.W. Cobb [syn. Ophiostoma wageneri (Goheen & F.W. Cobb) T.C. Harr. & F.W. Cobb; syn, Grosmannia wageneri (Goheen & F.W. Cobb) Zipfel, Z.W. de Beer & M.J. Wingf.] (Goheen and Cobb, 1978; Harrington and Cobb, 1987; Zipfel et al., 2005). Perithecia and ascospores were found in an insect gallery in a root of a ponderosa pine in a diseased stand in California (Goheen and Cobb, 1978), but no culture of the teleomorph is available, and subsequent searches in this same location failed to find perithecia of L. wageneri (Harrington, 1988). No further reports of a sexual stage have been made. Further, the sexual stage of other heterothallic Leptographium species are readily produced in culture if opposite mating types are paired (Duong et al., 2016), but pairing of isolates from ponderosa pine and other hosts failed to produce perithecia (Harrington and Cobb, 1986).
In culture, the BSRD pathogens produce stalked conidiophores with sticky drops of conidia, which appear suitable for insect dispersal, and such conidia are presumably formed in galleries produced by the insect vectors. When young, adult insects emerge from infected roots, their exoskeleton may be contaminated by conidia. These conidia can be subsequently introduced to roots of healthy trees when these adult insect vectors bore into roots for egg laying or maturation feeding (Hessburg et al., 1995; Ferguson, 2009). Root-feeding insects, such as bark beetles (Hylastes nigrinus and H. macer) and weevils (Pissodes fasciatus and Stereminius carinatus) (Coleoptera: Curculionidae), are the primary vectors involved in spread of the BSRD pathogen (Hessburg et al., 1995; Ferguson, 2009). Among these potential vectors, the conifer seedling weevil, S. carinatus, is considered an important agent for within stand spread because it is flightless (Hadfield et al., 1986; Ferguson, 2009).
Leptographium wageneri was known to have three varieties: L. wageneri var. ponderosum, primarily on ponderosa, Jeffrey, and lodgepole (P. contorta) pines; L. wageneri var. pseudotsugae, primarily on Douglas-fir; and the originally described species (L. wageneri var. wageneri), a pathogen on single-leaf pinyon and pinyon pines (P. edulis) (Harrington and Cobb, 1986). These three varieties can be distinguished by their distinct range of host preferences (Harrington and Cobb, 1984), minor morphological and physiological differences (Harrington and Cobb, 1987), and isozyme variation (Zambino and Harrington, 1989; Zambino and Harrington, 1992).
Isozyme variation and random amplified polymorphic DNA (RAPD) have previously distinguished the three varieties of L. wageneri (Otrosina and Cobb, 1987; Zambino and Harrington, 1989; Zambino and Harrington, 1992; Witthuhn et al., 1997), and population genomics have been used to distinguish differences between two varieties, L. wageneri var. wageneri and L. wageneri var. pseudotsugae (Bennett et al., 2021). Previous studies have suggested that very limited genetic variation is found within L. wageneri. Few genetic loci have been sequenced for isolates of L. wageneri, and limited numbers of sequences are publicly available (National Center for Biotechnology Information; NCBI, GenBank, https://www.ncbi.nlm.nih.gov/genbank/), including some sequences for rDNA [e.g., internal transcribed spacer (ITS) regions, 28S (large subunit, LSU), and 18S (small subunit, SSU)], actin (ACT), β-tubulin (TUB), calmodulin (CAL), elongation factor-1alpha (TEF-1α) gene, mating type genes (MAT1-2-1 and MAT1-1-3), and RNA polymerase II (RPB2) (Jacobs et al., 2001; Hausner et al., 2005; Zipfel et al., 2005; Duong et al., 2016; Marincowitz et al., 2017; Bennett et al., 2021; de Beer et al., 2022). Some of these deposited sequences are from isolates of unknown geographic and host origin, which could provide information to help identify the L. wageneri variety. In every locus except TUB, only a few isolates for each L. wageneri variety are available. Other phylogenetic studies of Leptographium and related species (e.g., Grosmannia) have used DNA sequences from a limited number of L. wageneri isolates representing several loci, such as the ITS region 2 (ITS2) and LSU rDNA, ACT, TUB, CAL, and TEF-1α (Duong et al., 2012; Linnakoski et al., 2012; Yin et al., 2015). Because the ITS region is difficult to amplify and sequence for L. wageneri s.l. and related fungi, the ITS region has not been widely used as a barcoding region for distinguishing among some Ophiostomatales (Witthuhn et al., 1997; Harrington et al., 2010).
We hypothesized that the three varieties of L. wageneri are phylogenetically distinct and genetic differences are sufficient to warrant their recognition at the species level. Thorough phylogenetic analyses of L. wageneri requires ample isolates and accurate sequences from a sufficient number of diverse loci. The overall objective of this study was to use DNA sequences derived from multiple loci to genetically characterize the three L. wageneri varieties collected from western North America and determine their phylogenetic relationships.
Materials and methods
Study sites and sample collection
During 2019-2021, a total of 24 BSRD samples were collected from various National Forests where BSRD was previously reported in Idaho, Oregon, and California (Supplementary Table 1). Samples were collected from three single-leaf pinyon pines, six ponderosa pines, two Jeffrey pines, and nine Douglas-fir trees. The number of samples collected from each host species varied depending on site accessibility, disease incidence, and previous records. Trees with BSRD crown symptoms were examined for black or dark-brown streaks in the sapwood near the roots and/or basal stems. For each tree, three to five wood samples (ca. 2 x 7 x 1 cm) were collected directly from the stained tissue and stored in a sealed plastic bag assigned to each tree with paper towels dampened in sterile water. Samples were kept cool by storage on ice or refrigeration before fungal isolation was performed. Precise locations were collected using a GPSMAP 64s GPS receiver (Garmin, Olathe, KS, USA). Diameter at breast height (DBH), health status (dead/live/declining and crown status), and other noticeable field conditions (e.g., elevation, vegetation, stand conditions) were also recorded. Two-thirds of the collected samples were sent to USDA Forest Service – Pacific Northwest Research Station, Forest Heath Laboratory (Corvallis, OR) for isolation within 48 hours of collection, and one-third of samples were isolated on-site.
Sample isolation
For surface disinfestation, the wood samples were cut into smaller pieces containing mostly stained areas (Supplementary Figure 2). The wood pieces were submerged and soaked in 95% ethanol for 30 seconds, and transferred to 10% commercial bleach (0.05% sodium hypochlorite) for a 3-minute soak. The samples were then rinsed four times in a separate beaker with sterile, deionized H2O. Damp surfaces were dried with sterile tissues, and the samples were cut into pieces (ca. 10 x 2.5 x 2.5 mm) for culturing. Approximately ten pieces were embedded in a Petri dish containing a selective medium with malt extract agar (MEA) amended with cycloheximide and streptomycin sulfate (CSMA) (one-fourth strength MEA media: 0.75% malt, 0.75% dextrose, 0.25% peptone, and 1.5% agar with 200 ppm cycloheximide and 200 ppm streptomycin sulfate) (Harrington, 1992). Three to five Petri dishes were established for samples from each tree. During incubation, samples were stored at 15°C, checked daily, and discarded if contamination occurred. After 2-3 weeks, when L. wageneri conidiophores were observed on the wood piece, drops of conidia were transferred from the conidiophores onto MEA medium (3% malt, 3% dextrose, 1% peptone, 1.5% agar). After 1-2 weeks of incubation, when fungal hyphae started to grow from the transferred conidia, hyphal tips were transferred onto fresh MEA medium with the use of a flame-sterilized dissecting pin. In addition, 15 isolates were studied from collections made in the 1980s (Harrington and Cobb, 1986; Harrington and Cobb, 1987). These previously collected isolates were used in descriptions of the varieties, including ex-types (CAP-19 and CAD-18), and they are now archived at the USDA Forest Service – Pacific Northwest Research Station, Forest Heath Laboratory (Corvallis, OR) (Supplementary Table 1).
DNA extraction and PCR
For DNA extractions, a total of 39 L. wageneri hyphal-tipped isolates were grown on 0.2-μm-pore, nylon-membrane filters (Millipore Corporation, Burlington, MA, USA) overlaid on MEA medium and grown at 15°C for 2-3 weeks. Mycelia, ca. 50-100 mg, were scraped directly from the filters and homogenized in extraction buffer (BashingBead™ Buffer, Zymo Research, Irvine, CA, USA) using FastPrep-24™ 5G (MP Biomedicals, Santa Ana, CA, USA). DNA extraction was conducted according to the ZR Fungal/Bacterial DNA MiniPrep Kit protocols supplied by Zymo Research (Irvine, CA, USA). DNA concentration was measured using a NanoDrop™ 2000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Species identification of L. wageneri s.l. isolates was confirmed by comparisons with TUB sequences in GenBank using BLAST (Basic Local Alignment Search Tool) (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
Polymerase Chain Reaction (PCR) was used to amplify the following ten loci for sequencing: 28S LSU, ACT* (primers Lepact-F & Lepact-R) and ACT** (primers ACT512F & ACT783R), TUB, CAL, TEF-1α, a mating-type gene (MAT1-1-3), RPB2, glyceraldehyde-3-phosphate dehydrogenase (GPD), and chitin synthase (CHS). Two regions of ACT were amplified using two different primer sets, and they are differentiated as ACT* and ACT** (Supplementary Table 2).
The PCR mixture contained 4 µL template DNA (ca. 25ng/µL), 5 µL 10X PCR reaction buffer, 1 µL dNTP, 5 µL (5 nM) of each primer, and 0.5 µL (2.5 units) Green Taq DNA polymerase (GenScript Biotech, Piscataway, NJ, USA) in a total volume of 50 µL. The PCR reaction was conducted using a C1000 Touch ™ Thermal Cycler (Bio-Rad, Hercules, CA, USA), and the thermocycling setting for each primer pair followed the published references with minor modifications (Supplementary Table 3). The peak sizes of the PCR amplicons were determined using an automated electrophoresis analyzer, QIAxcel® Advanced (QIAGEN, Hilden, Germany). Amplified PCR products were then purified with ExoSAP-IT™ Express (Thermo Fisher Scientific, Waltham, MA, USA), following the manufacturer-suggested protocol. Purified PCR products were sent to Psomagen, Inc. (Rockville, MD, USA) for Sanger sequencing.
Phylogenetic analyses
The resulting sequences of each L. wageneri s.l. isolate consisted of forward and reverse ABI sequencer data files (.ab1). Geneious Prime version 2022.2.2 (https://www.geneious.com/) was used to pair and edit the sequences following IUPAC (International Union of Pure and Applied Chemistry) codes. The 39 paired sequences for each of the 10 loci were generated as consensus sequences, and they were aligned using Clustal Omega 1.2.2. (Sievers et al., 2011) with default parameters and trimmed to the same length for comparison.
The sequences of the loci were analyzed in four parts: Analysis 1) individual loci; Analysis 2) concatenated sequences of two (CAL + ACT*) and three (CAL + ACT* + TEF-1α) loci; Analysis 3) concatenated sequences of 10 loci; and Analysis 4) concatenated sequences of two loci (CAL + ACT*) analyzed with multiple outgroups (Supplementary Table 4). Leptographium douglasii was used as an outgroup for single outgroup analyses (Analysis 1, 2, and 3). The sequences of ACT**, GPD, and CHS of L. douglasii were not available in GenBank, therefore, these sequences were generated using the same primer sets and protocols as for L. wageneri s.l. For Analysis 2, the two- and three-loci combinations were selected because each of the loci have relatively large numbers of parsimony-informative sites. Parsimony-informative sites were identified using IQ-TREE (Nguyen et al., 2014). For Analysis 4, six outgroup taxa (L. douglasii, L. rhodanense, L. gracile, L. castellanum, G. alacris, and G. serpens) were selected based on the phylogenetic tree from de Beer et al. (2022) and availability of sequences in GenBank.
A partition homogeneity test was performed using PAUP* version 4.0a169 (Swofford and Sullivan, 2003) to determine whether the sequence data for the 10 loci were congruent for concatenation. A p-value over 0.05 indicates that partitions can be combined, and the sequence data of the 10 loci resulted in a p-value = 0.08. The best-fit substitution model for each locus was selected by Bayesian Information Criterion (BIC) calculated in ModelFinder in IQ-TREE (Nguyen et al., 2014; Kalyaanamoorthy et al., 2017) (Supplementary Table 5). Phylogenetic analyses were implemented using maximum likelihood and Bayesian inference. Maximum likelihood trees were constructed using IQ-TREE (Nguyen et al., 2014; Hoang et al., 2018) with 1,000 bootstrap pseudo-replications. Maximum likelihood trees were imported and visualized using Geneious Prime, with bootstrap support values (BS) over 50%. MrBayes (Huelsenbeck and Ronquist, 2001) plugin in Geneious Prime was used for the Bayesian inference. Because Tamura–Nei (TN) models could not be implemented using MrBayes in Geneious Prime, the next best models were selected. Bayesian trees were generated using 1,100,000 generations with the four heated-chains setting in Markov Chain Monte Carlo (MCMC). The first 100,000 chains were discarded as burn-in, posterior probability (PP) for each dataset was calculated, and a consensus tree was produced with PP over 0.80 illustrated on the branches.
Results
Sequences from 39 L. wageneri s.l. isolates (eight, 14, and 17 for L. wageneri var. wageneri, L. wageneri var. ponderosum, and L. wageneri var. pseudotsugae, respectively) were obtained for the 10 loci (Table 1).
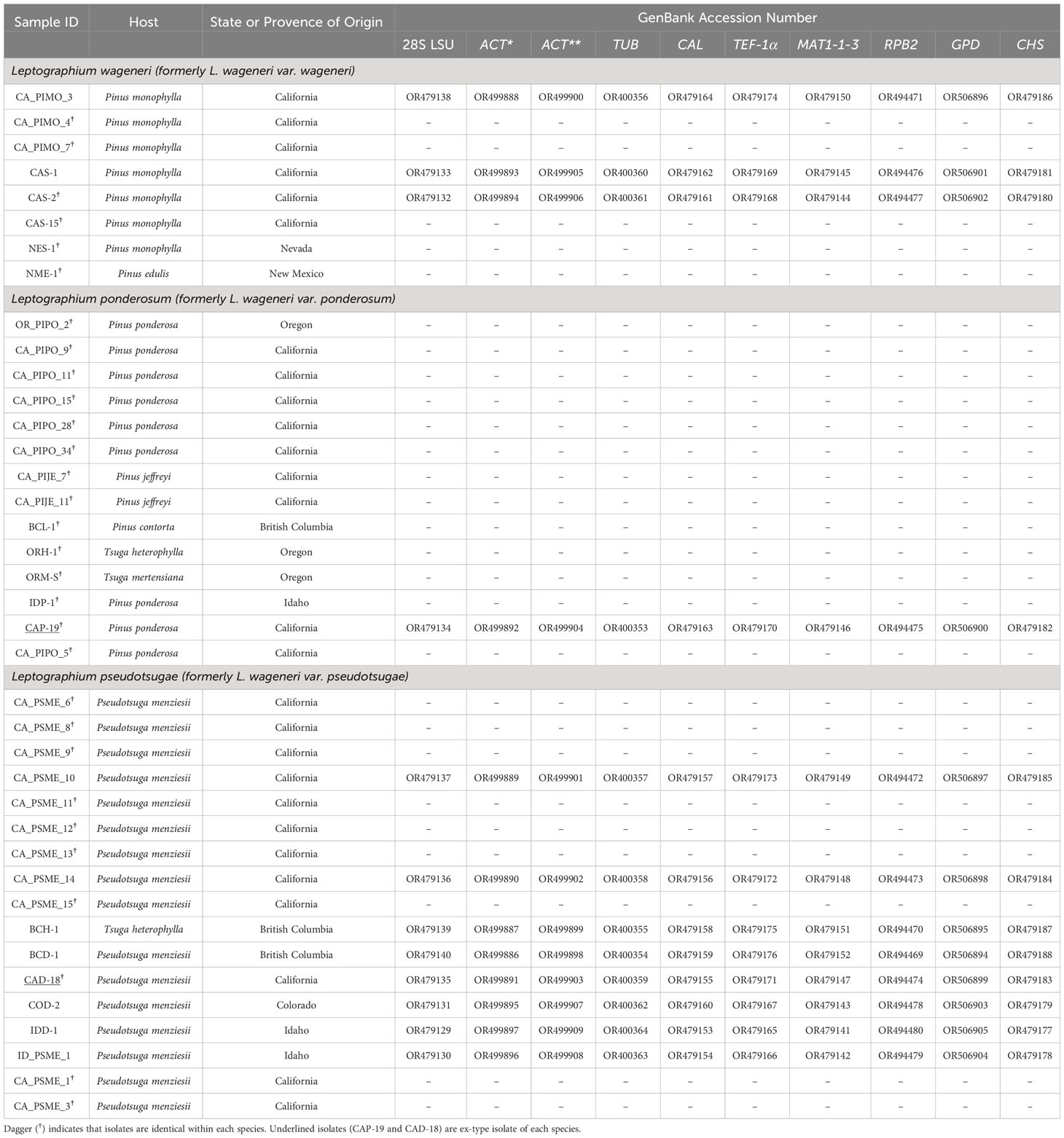
Table 1 GenBank accession numbers and host information for 8 Leptographium wageneri (formerly L. wageneri var. wageneri), 14 L. ponderosum (formerly L. wageneri var. ponderosum), and 17 L. pseudotsugae (formerly L. wageneri var. pseudotsugae) isolates for 10 loci [28S large subunit rDNA (LSU), actin (ACT*; Lepact-F & Lepact-R), actin (ACT**; ACT512F & ACT783R), β-tubulin (TUB), calmodulin (CAL), elongation factor-1alpha (TEF-1α) gene, a mating-type gene (MAT 1-1-3), RNA polymerase II (RPB2), glyceraldehyde-3-phosphate dehydrogenase (GPD), and chitin synthase (CHS)].
Individual loci
No parsimony-informative sites were found among the three L. wageneri varieties at the LSU, TUB, and RPB2 loci. The sequences of LSU and RPB2 were identical among the 39 isolates, and only a single isolate of L. wageneri var. ponderosum showed a single base substitution for TUB.
One variety, L. wageneri var. ponderosum, was separated from the other two varieties based on single-base substitutions in the sequences for ACT** (Supplementary Figure 3), MAT1-1-3 (Supplementary Figure 4), GPD (Supplementary Figure 5), and CHS (Supplementary Figure 6) (with 95.49%, 99.33%, 99.63%, and 98.53% constant sites, respectively). In the ACT** tree, L. wageneri var. pseudotsugae and var. wageneri were separated from var. ponderosum (0.99 PP and 67 BS) with a few exceptions. In the MAT1-1-3 tree, separation of L. wageneri var. wageneri isolates was well-supported (0.98 PP and 63 BS); however, isolate COD-2 (L. wageneri var. pseudotsugae) was included in the var. wageneri clade. The GPD tree showed a well-supported (0.99 PP and 61 BS) separation of L. wageneri var. ponderosum. In the CHS tree, L. wageneri var. pseudotsugae isolates, except COD-2, had identical sequences and were separated from the other varieties (0.99 PP and 65 BS); the CHS sequence of isolate COD-2 (L. wageneri var. pseudotsugae) grouped with the other two varieties at some loci.
Sequences of CAL (Supplementary Figure 7), ACT* (Figure 2), and TEF-1α (Supplementary Figure 8) contained nine, four, and two parsimony-informative sites, with 98.41%, 99.15%, and 99.12% constant sites, respectively. In the CAL-based tree, the clade of L. wageneri var. ponderosum and var. wageneri was well-supported (1 PP and 97 BS). In the ACT*-based tree, the clades of L. wageneri var. ponderosum and var. wageneri were well-supported (1 PP and 86 BS; 1 PP and 71 BS, respectively); however, isolate IDD-1 (L. wageneri var. pseudotsugae) was an exception and grouped with the var. wageneri clade. Out of the 10 individual loci, single locus-based separation of the three varieties was observed only for ACT*. The TEF-1α-based tree separated L. wageneri var. wageneri (0.99 PP and 72 BS) from the other varieties.
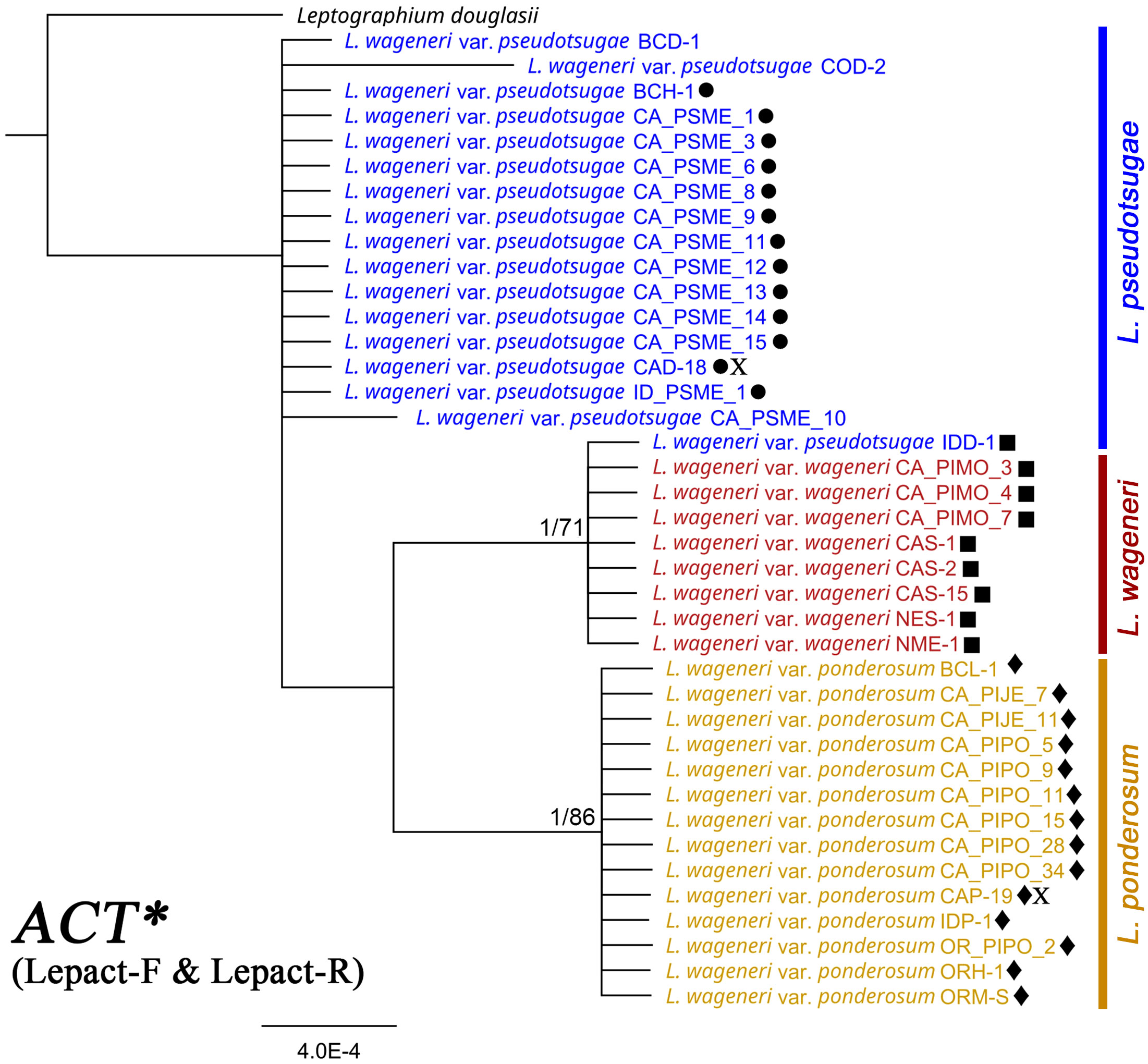
Figure 2 Phylogeny of actin (ACT*) with Leptographium douglasii (GenBank accession #KY424502) used as the outgroup. Leptographium wageneri (formerly L. wageneri var. wageneri), L. ponderosum (formerly L. wageneri var. ponderosum), and L. pseudotsugae (formerly L. wageneri var. pseudotsugae) are color-coded in red, yellow, and blue, respectively. The round, square, and diamond shapes inserted to the right of the isolate name indicate identical sequences. Numbers at each node indicate supports posterior probabilities (PP) greater than 0.80 and bootstrap support (BS) values greater than 50% (PP/BS). ACT* indicates actin dataset using Lepact-F & Lepact-R primers. Letter X indicates ex-type cultures of each variety.
Concatenation of two and three loci
Analyses using the combined data of the most polymorphic loci clearly separated the L. wageneri varieties. The three varieties were well separated in the tree generated with the two loci, CAL and ACT* (Supplementary Figure 9). The clade containing the L. wageneri var. ponderosum isolates was well-supported (0.97 PP and 88 BS), and a L. wageneri var. pseudotsugae clade was also supported (0.93 PP and 56 BS). The three varieties were also clearly separated based on concatenated sequences of CAL, ACT*, and TEF-1α (Supplementary Figure 10). In the phylogenetic tree based on the concatenated sequences of these three loci, each variety was well-supported: L. wageneri var. wageneri at 1 PP and 80 BS, var. ponderosum at 0.96 PP and 83 BS, and var. pseudotsugae at 0.96 PP and 63 BS.
Concatenated of 10 loci
In the phylogenetic tree based on concatenated sequences of the 10 loci (LSU, TUB, RPB2, MAT1-1-3, CAL, ACT*, TEF-1α, ACT**, GPD, and CHS), each of the three varieties was well-supported as a separate clade: L. wageneri var. wageneri (1 PP and 91 BS), L. wageneri var. ponderosum (1 PP and 95 BS), and L. wageneri var. pseudotsugae (0.92 PP and 57 BS) (Figure 3).
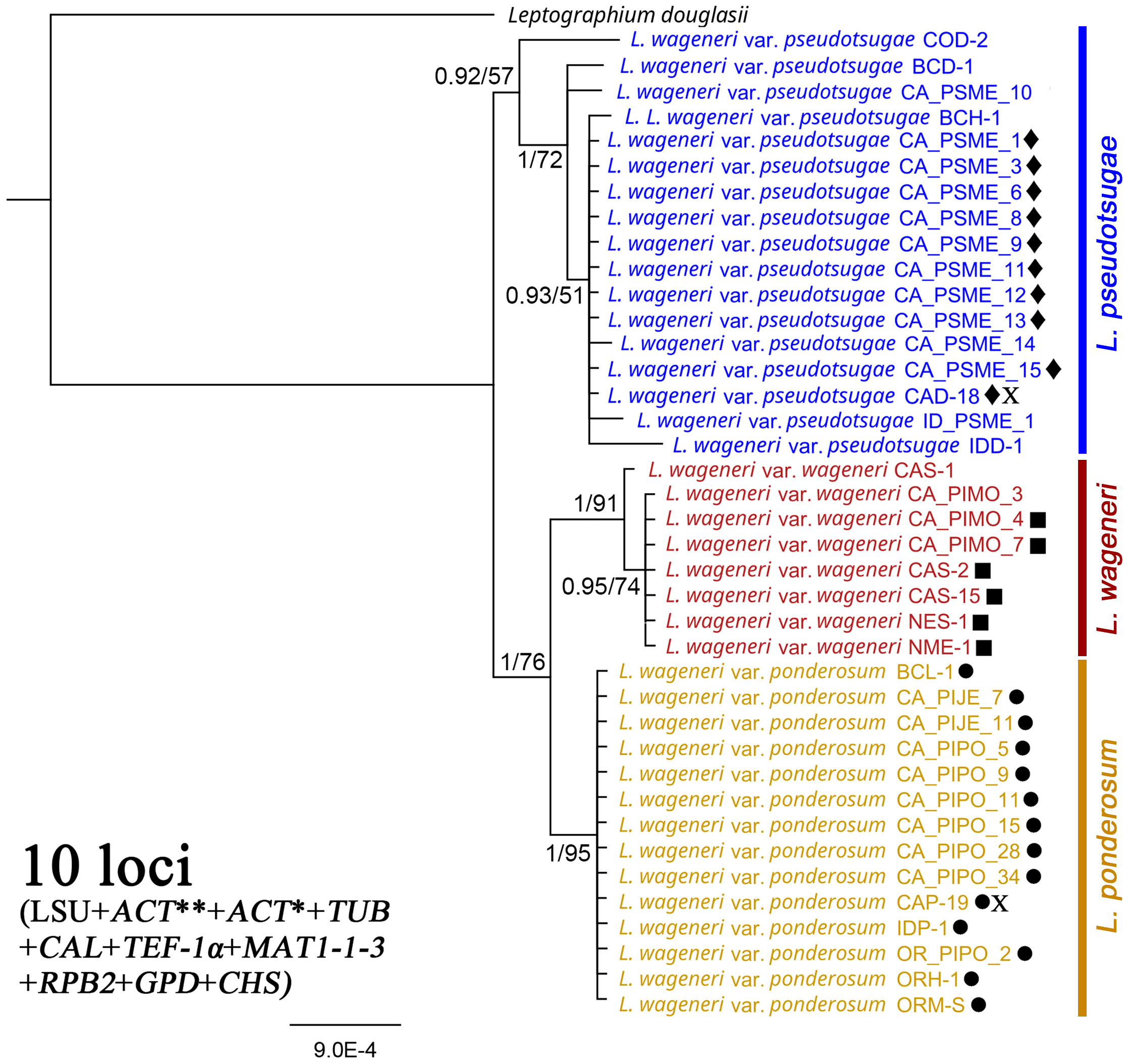
Figure 3 Phylogeny of the combined 28S large subunit (LSU) rDNA, actins (ACT** and ACT*), β-tubulin (TUB), calmodulin (CAL), translation elongation factor 1-alpha (TEF-1α), mating-type gene (MAT1-1-3), RNA polymerase II subunit (RPB2), glyceraldehyde-3-phosphate dehydrogenase (GPD), and chitin synthase (CHS) loci with Leptographium douglasii used as an outgroup. Leptographium wageneri (formerly L. wageneri var. wageneri), L. ponderosum (formerly L. wageneri var. ponderosum), and L. pseudotsugae (formerly L. wageneri var. pseudotsugae) are color-coded in red, yellow, and blue, respectively. The round, square, and diamond shapes inserted to the right of the isolate name indicate identical sequences. Numbers at each node indicate posterior probabilities (PP) greater than 0.80 and bootstrap support (BS) values greater than 50% (PP/BS). ACT* and ACT** indicate actin dataset using Lepact-F & Lepact-R and ACT512F & ACT783R primers, respectively. Letter X indicates ex-type cultures of each variety.
Concatenation of two loci with multiple outgroup taxa
For two loci, sequences were publicly available for six related Leptographium and Grosmannia species. A phylogenetic tree was generated based on concatenated sequences of CAL and ACT* of the L. wageneri s.l. isolates with those of L. douglasii, L. rhodanense, L. gracile, L. castellanum, G. alacris, and G. serpens (Figure 4). In this tree, each L. wageneri variety was well-supported: L. wageneri var. wageneri with 0.84 PP and 73 BS, L. wageneri var. ponderosum with 1 PP and 92 BS, and L. wageneri var. pseudotsugae with 0.96 PP and 61 BS.
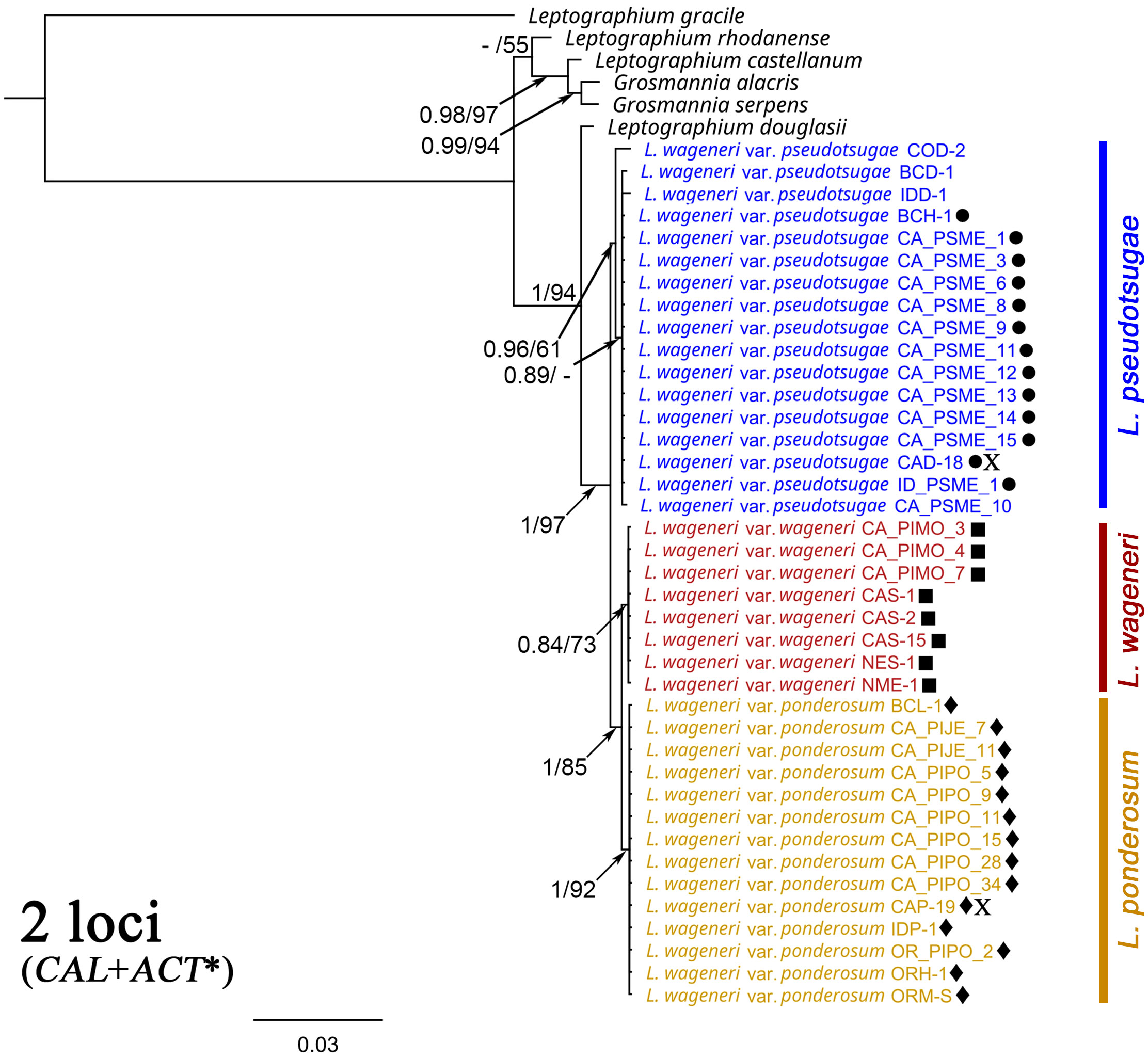
Figure 4 Phylogeny trees of the combined calmodulin (CAL) and actin (ACT*) loci with Leptographium douglasii, L. rhodanense, L. gracile, L. castellanum, Grosmannia alacris, and G. serpens [GenBank accession numbers (CAL/ACT*), KY424522/KY424502, KY424527/KY424506, MG205782/KM491324, JN135299/JN135324, JN135296/JN135318, and JN135300/JN135325, respectively] used as outgroup taxa. Leptographium wageneri (formerly L. wageneri var. wageneri), L. ponderosum (formerly L. wageneri var. ponderosum), and L. pseudotsugae (formerly L. wageneri var. pseudotsugae) are color-coded in red, yellow, and blue, respectively. The round, square, and diamond shapes inserted to the right of the isolate name indicate identical sequences. Numbers at each node indicate posterior probabilities (PP) greater than 0.80 and bootstrap support (BS) values greater than 50% (PP/BS). ACT* indicates actin dataset using Lepact-F & Lepact-R primers. Letter X indicates ex-type cultures of each variety.
Taxonomy
Clear phylogenetic distinction as well as several minor morphological and physiological characters (Harrington and Cobb, 1986; Harrington and Cobb, 1987) (Table 2) provide strong justification for separating these three host-specialized, Leptographium pathogens as species. The low level of genetic diversity in the L. wageneri complex suggests that the three varieties represent three predominantly asexual lineages. The connection between Leptographium wageneri var. ponderosum and perithecia of Ceratocystis wageneri Goheen & F.W. Cobb [= Ophiostoma wageneri (Goheen & F.W. Cobb) T.C. Harr., = Grosmannia wageneri (Goheen & F.W. Cobb) Zipfel, Z.W. de Beer & M.J. Wingf., (MB#500831)] was not firmly established and was likely in error (Harrington, 1988). If not the teleomorph of L. wageneri, the ascospore shape and lack of a thick outer wall would place the purported teleomorph in Grosmannia (de Beer et al., 2022). Discounting the connection between G. wageneri and L. wageneri, the following three Leptographium species are proposed as the causes of black strain root disease on pinyons, hard pines, and Douglas-fir, respectively.
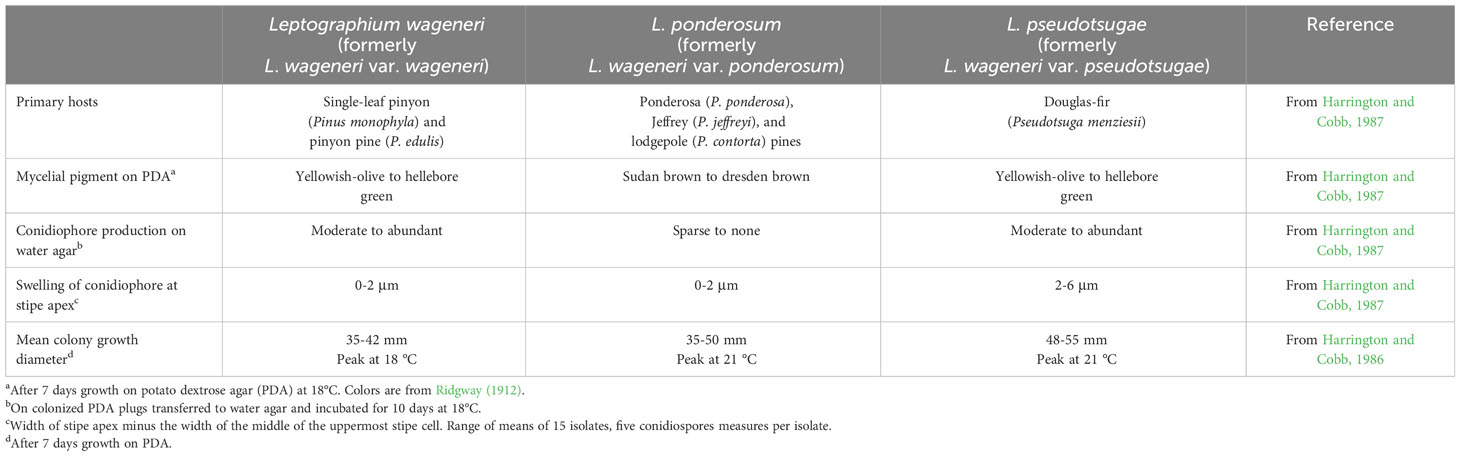
Table 2 The key ecological (host specificity), morphological, and physiological differences of three species: Leptographium wageneri (formerly L. wageneri var. wageneri), L. ponderosum (formerly L. wageneri var. ponderosum), and L. pseudotsugae (formerly L. wageneri var. pseudotsugae).
Leptographium wageneri (W.B. Kendr.) M.J. Wingf., Trans. Brit. Mycol. Soc. 85 (1): 92 (1985) [MB#105458].
Leptographium wageneri var. wageneri (?) [MB#418941].
Leptographium ponderosum (T.C. Harr. & F.W. Cobb) D. Choi, M.-S. Kim & T.C. Harr., comb. nov. [MB#849475].
Verticicladiella wageneri var. ponderosa T.C. Harr. & F.W. Cobb, Mycologia 78 (4): 566 (1986) [MB#117426].
Leptographium wageneri var. ponderosum (T.C. Harr. & F.W. Cobb) T.C. Harr. & F.W. Cobb, Mycotaxon 30: 505 (1987) [MB#133361].
Holotype: California, Siskyou County, McCloud Flat, from Pinus ponderosa, collected by T. C. Harrington, July 1980, UCB 1475041, ex-type isolate ATCC 58575 (= CAP-19).
Leptographium pseudotsugae (T.C. Harr. & F.W. Cobb) D. Choi, M.-S. Kim & T.C. Harr., comb. nov. [MB#849460].
Leptographium wageneri var. pseudotsugae T.C. Harr. & F.W. Cobb, Mycotaxon 30: 505 (1987) [MB#133362].
Holotype: California, El Dorado County, Union Valley Res., from Pseudotsuga menziesii, collected by T. C. Harrington, July 1980, UCB 1475052, ex-type isolate ATCC 64196 (= CAD-18).
Discussion
Because morphological features were once the predominate method available for classifying fungi (Hawksworth et al., 1996), early classification of the three L. wageneri varieties was based on their phenotypic characteristics, such as host-specialization, pigmentation of mycelia and conidiophore features (Harrington and Cobb, 1986; Harrington and Cobb, 1987) (Table 2). Subsequently, isozyme variation and RAPD markers supported distinction of the three varieties (Otrosina and Cobb, 1987; Zambino and Harrington, 1989; Zambino and Harrington, 1992; Witthuhn et al., 1997). Phylogenetic analyses based on rDNA (ITS2 and LSU) sequences did not provide clear separation of the three L. wageneri varieties (Jacobs et al., 2001). The present study is the first to demonstrate robust separation of the three L. wageneri varieties using multiple, non-rDNA loci. Although Bennett et al. (2021) demonstrated strong separation between two varieties (L. wageneri var. wageneri and L. wageneri var. pseudotsugae) based on over 100,000 binary SNPs, their analyses included isolates from limited geographic areas (e.g., L. wageneri var. pseudotsugae isolates from Oregon and L. wageneri var. wageneri isolates from California). And, sequences of the three most parsimony informative loci (e.g., CAL, ACT*, and TEF-1α) from L. wageneri var. pseudotsugae isolates were identical to those within the genomic sequences of L. wageneri var. pseudotsugae isolates from Oregon used in the study of Bennett et al. (2021). The clear phylogenetic separation warrants species designation.
Phylogenetic separation of L. wageneri, L. ponderosum and L. pseudotsugae was better resolved when multi-locus analyses were used rather than a single locus. In general, these three Leptographium species showed very little genetic variation, and most of the loci showed only minor sequence differences. When analyzing with a single locus, these three species tended to be separated differently depending on the locus being analyzed. For example, for MAT1-1-3 and TEF-1α, L. wageneri was distinct, while for ACT** (ACT512F & ACT783R) and GPD, L. ponderosum was distinct, and for CHS and CAL, L. pseudotsugae was distinct. The very limited genetic variation within each of these three Leptographium species, pathogens generally believed to be native to western North America, strongly suggests that they are predominantly asexual.
In most cases, ACT* (Lepact-F & Lepact-R) could be used to identify the three Leptographium species. ACT*-based separation of three species was generally well-supported, with the exception of one isolate, IDD-1 (L. pseudotsugae), which was placed in the L. wageneri clade. The TUB locus has been used for identification at the species level; however, TUB-based identification of L. ponderosum is distinguished by only a single-base substitution.
Because the majority of parsimony-informative sites were found in the CAL, ACT* (Lepact-F & Lepact-R), and TEF-1α loci, combinations among these loci were found to be most useful in separating the Leptographium species. Of these three loci, CAL had the most parsimony-informative sites. However, with the CAL-based tree, only L. pseudotsugae was separated. A phylogeny based on CAL with ACT* (Lepact-F & Lepact-R), which had the next most parsimony-informative sites, resulted in divergence and separation of three species. In the phylogenetic analysis of three combined-loci, CAL, ACT* (Lepact-F & Lepact-R), and TEF-1α, the separation of all three Leptographium species was clear and strongly supported. These results suggest that cumulative phylogenetic information provides more precise divergence with stronger support values for examining evolutionary relationships within and among the species (Taylor et al., 2000).
Distinguishing different species of closely related fungi is a complex process that considers various factors including morphology, biological reproduction, biochemical properties, ecological behaviors, genetic differences, and other factors (Mayr, 1940; Hennig, 1965; Hawksworth et al., 1996; Harrington and Rizzo, 1999; Taylor et al., 2000). In the case of L. wageneri, the three host-specialized pathogens (Harrington and Cobb, 1984) were previously described as varieties instead of species, largely because phylogenetic data were lacking to distinguish these taxa. However, differences among the three L. wageneri varieties were previously noted in terms of minor morphological characteristics, isozyme variations, RAPD profiles, and ecological habitat (host species) (Harrington and Cobb, 1986; Harrington and Cobb, 1987; Otrosina and Cobb, 1987; Zambino and Harrington, 1989; Zambino and Harrington, 1992; Witthuhn et al., 1997). When recognizing new species within a genus, multiple genes have been used in phylogenetic analyses to allow for concordance (Taylor et al., 2000), and several new Leptographium species have been described using multi-locus datasets (Kim et al., 2004; Lee et al., 2005; Jacobs et al., 2006). In this study, the three varieties of L. wageneri were clearly separated phylogenetically on the basis of concatenated sequences of 10 loci and on the two-loci dataset with multiple outgroup taxa. Thus, solid genetic evidence is provided that the three L. wageneri varieties should be elevated to species status. Recognition of the L. wageneri varieties as three separate Leptographium species will help to understand ecological interactions and evolutionary history of these forest pathogens, which can contribute to improved management of the black stain root disease pathosystem.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
DC: Data curation, Formal analysis, Methodology, Writing – original draft, Software, Visualization. TH: Data curation, Methodology, Investigation, Validation, Writing – review & editing. DS: Investigation, Writing – review & editing, Resources, Supervision. JS: Conceptualization, Data curation, Formal analysis, Funding acquisition, Methodology, Validation, Writing – review & editing. DK: Writing – review & editing, Data curation, Formal analysis, Methodology, Software. NK: Writing – review & editing, Conceptualization, Funding acquisition, Investigation, Resources. M-SK: Conceptualization, Funding acquisition, Investigation, Resources, Writing – review & editing, Data curation, Formal analysis, Methodology, Project administration, Supervision, Validation.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The project has been supported by USDA Forest Service (FS), State, Private, and Tribal Forestry, Forest Health Protection – Special Technology and Development Program (STDP R5-2020-3) and FS Research & Development Bipartisan Infrastructure Law Ecosystem Restoration Research Project - ERR09.
Acknowledgments
We thank USDA FS FHP Region 5 Forest Pathologists (Martin MacKenzie, Bill Woodruff, Ashley Hawkins, Phil Cannon), CAL Fire Forest Health Specialists (Chris Lee, Kim Corella), Sukhyun Joo, and Magdalena Kacprzyk who contributed to the sample collection. We also thank Sean Toporek and Jessa Ata for their help with the initial genetic analyses, and Patrick Bennett for his consultations.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
This study was partially supported by the employees of the USDA Forest Service, R&D, Pacific Northwest Research Station and Rocky Mountain Research Station. The findings and conclusions in this publication are those of the authors and should not be construed to represent any official USDA or U.S. Government determination or policy.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1286157/full#supplementary-material
References
Bennett, P. I., Tabima, J. F., Leon, A. L., Browning, J., Wingfield, M. J., Leboldus, J. M. (2021). Spatial genetic structure of the insect-vectored conifer pathogen Leptographium wageneri suggests long distance gene flow among Douglas-fir plantations in western Oregon. Front. Forests Global Changes 4, 695981. doi: 10.3389/ffgc.2021.695981
Cobb, F. W. Jr. (1988). “Leptographium wageneri cause of black-stain root disease: a review of its discovery, occurrence and biology with emphasis on pinyon and ponderosa piney,” in Leptographium Root Diseases on Conifers. Eds. Harrinton, T. C., Cobb Jr, F. W. (St. Paul, MN: APS Press), 41-62.
de Beer, Z., Procter, M., Wingfield, M., Marincowitz, S., Duong, T. (2022). Generic boundaries in the Ophiostomatales reconsidered and revised. Stud. Mycology. 101, 57-120. doi: 10.3114/sim.2022.101.02
Duong, T. A., de Beer, Z. W., Wingfield, B. D., Wingfield, M. J. (2012). Phylogeny and taxonomy of species in the Grosmannia serpens complex. Mycologia 104, 715–732. doi: 10.3852/11-109
Duong, T. A., de Beer, Z. W., Wingfield, B. D., Wingfield, M. J. (2016). Mating type markers reveal high levels of heterothallism in Leptographium sensu lato. Fungal Biol. 120, 538–546. doi: 10.1016/j.funbio.2016.01.001
Ferguson, B. A. (2009). Black stain root disease ecology and management. Chapter 11.7 Forest insect and disease management guide for the northern and central Rocky Mountains (Missoula, MT: USDA Forest Service, Northern Region, State and Private Forestry).
Goheen, D. J., Cobb, F. (1978). Occurrence of Verticicladiella wagenerii and its perfect state, Ceratocystis wageneri sp. nov., in insect galleries. Phytopathology 68, 1192–1195. doi: 10.1094/Phyto-68-1192
Hadfield, J. S., Goheen, D. J., Filip, G. M., Schmitt, C. L., Harvey, R. D. (1986). Root diseases in Oregon and Washington conifers (Portland, OR; United States Department of Agriculture, Forest Service, Pacific Northwest Region).
Harrington, T. C. (1988). “Leptographium species, their distributions, hosts and insect vectors,” in Leptographium Root Diseases on Conifers. Eds. Harrinton, T. C., Cobb, F. W., Jr (St. Paul, MN: APS Press), 1-39.
Harrington, T. C. (1992). “Leptographium,” in Methods for Research on Soilborne Phytopathogenic Fungi. Eds. Singleton, L. L., Mihail, J. D., Rush, C. M. (St. Paul, MN: APS Press).
Harrington, T. C., Aghayeva, D. N., Fraedrich, S. (2010). New combinations in Raffaelea, Ambrosiella, and Hyalorhinocladiella, and four new species from the redbay ambrosia beetle, Xyleborus glabratus. Mycotaxon 111, 337–361. doi: 10.5248/111.337
Harrington, T. C., Cobb, F. W., Jr. (1984). Host specialization of three morphological variants of Verticicladiella wageneri. Phytopathology 74, 286–290. doi: 10.1094/Phyto-74-286
Harrington, T. C., Cobb, F. W., Jr. (1986). Varieties of Verticicladiella wageneri. Mycologia 78, 562–567. doi: 10.1080/00275514.1986.12025290
Harrington, T. C., Cobb, F. W., Jr. (1987). Leptographium wageneri var. pseudotsugae, var. nov., cause of black stain root disease on Douglas-fir. Mycotaxon 30, 501–507.
Harrington, T. C., Rizzo, D. M. (1999). “Defining species in the fungi”, in Structure and Dynamics of Fungal Populations. Ed. Worrall, J. J. (Dordrecht: Springer Netherlands), 43–71.
Hausner, G., Iranpour, M., Kim, J. -J., Breuil, C., Davis, C. N, Gibb, E. A., et al. (2005). Fungi vectored by the introduced bark beetle Tomicus piniperda in Ontario, Canada, and comments on the taxonomy of Leptographium lundbergii, Leptographium terebrantis, Leptographium truncatum, and Leptographium wingfieldii. Botany 83, 1222–1237. doi: 10.1139/b05-095
Hawksworth, D. L., Kirk, P. M., Sutton, B. C., Pegler, D. N. (1996). Ainsworth & Bisby’s Dictionary of the Fungi, 8th ed (Wallingford, UK: CABI).
Hennig, W. (1965). Phylogenetic systematics. Annu. Rev. entomology 10, 97–116. doi: 10.1146/annurev.en.10.010165.000525
Hessburg, P. F. (1984). Pathogenesis and intertree transmission of Verticicladiella wageneri in Douglas-fir (Pseudotsuga menziesii). Ph.D (Corvallis, OR: Oregon State University).
Hessburg, P. F., Goheen, D. J., Bega, R. V. (1995). Black stain root disease of conifers. Forest insect & disease leaflet 145. (Washington, D.C.: U.S. Department of Agriculture, Forest Service).
Hoang, D. T., Chernomor, O., Von Haeseler, A., Minh, B. Q., Vinh, L. S. (2018). UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522. doi: 10.1093/molbev/msx281
Huelsenbeck, J. P., Ronquist, F. (2001). MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755. doi: 10.1093/bioinformatics/17.8.754
Jacobs, K., Eckhardt, L. G., Wingfield, M. J. (2006). Leptographium profanum sp. nov., a new species from hardwood roots in North America. Botany 84, 759–766. doi: 10.1139/b06-030
Jacobs, K., Wingfield, M. J., Wingfield, B. D. (2001). Phylogenetic relationships in Leptographium based on morphological and molecular characters. Can. J. Bot. 79, 719–732. doi: 10.1139/b01-041
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K., von Haeseler, A., Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Kendrick, W. B. (1962). The leptographium complex Verticicladiella hughes. Can. J. Bot. 40, 772–797. doi: 10.1139/b62-072
Kim, J.-J., Lim, Y. W., Wingfield, M. J., Breuil, C., Kim, G.-H. (2004). Leptographium bistatum sp. nov., a new species with a Sporothrix synanamorph from Pinus radiata in Korea. Mycological Res. 108, 699–706. doi: 10.1017/S0953756204000036
Lee, S., Kim, J.-J., Breuil, C. (2005). Leptographium longiclavatum sp. nov., a new species associated with the mountain pine beetle, Dendroctonus ponderosae. Mycological Res. 109, 1162–1170. doi: 10.1017/S0953756205003588
Linnakoski, R., de Beer, Z. W., Duong, T. A., Niemelä, P., Pappinen, A., Wingfield, M. J. (2012). Grosmannia and Leptographium spp. associated with conifer-infesting bark beetles in Finland and Russia, including Leptographium taigense sp. nov. Antonie van Leeuwenhoek 102, 375–399. doi: 10.1007/s10482-012-9747-6
Lockman, I. B., Kearns, H. S. (2016). Black stain and related root diseases Leptographium Species. Forest root diseases across the United States. Gen. Tech. Rep. RMRS-GTR-342 (Ogden, UT: U.S. Department of Agriculture, Forest Service, Rocky Mountain Research Station).
Marincowitz, S., Duong, T. A., Heiniger, U., Wingfield, B. D., Wingfield, M. J., de Beer, Z. W. (2017). A new Leptographium species from the roots of declining Pinus sylvestris in Switzerland. For. Pathol. 47, e12346. doi: 10.1111/efp.12346
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A., Minh, B. Q. (2014). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Otrosina, W. J., Cobb, F. W. (1987). Analysis of allozymes of three distinct variants of Verticicladiella wageneri isolated from conifers in western North America. Phytopathology 77, 1360–1363. doi: 10.1094/Phyto-77-1360
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. doi: 10.1038/msb.2011.75
Swofford, D. L., Sullivan, J. (2003). “Phylogeny inference based on parsimony and other methods using Paup*,” in The Phylogenetic Handbook, A Practical Approach to DNA and Protein Phylogeny. Eds. Salemmi, M., Vandamme, A. M. (New York, NY: Cambridge University Press), 267–312.
Taylor, J. W., Jacobson, D. J., Kroken, S., Kasuga, T., Geiser, D. M., Hibbett, D. S., et al. (2000). Phylogenetic species recognition and species concepts in fungi. Fungal Genet. Biol. 31, 21–32. doi: 10.1006/fgbi.2000.1228
Wagener, W. W., Mielke, J. L. (1961). A staining-fungus root disease of pondersoa, Jeffrey, and pinyon pines. Plant Dis. Rep. 45, 831–835.
Wingfield, M. J. (1985). Reclassification of Verticicladiella based on conidial development. Trans. Br. Mycol. Soc. 85, 81–93. doi: 10.1016/S0007-1536(85)80157-1
Witthuhn, R. C., Wingfield, B. D., Wingfield, M. J., Harrington, T. C. (1997). Comparison of three varieties of Leptographium wageneri using Random Amplified Polymorphic DNA. South Afr. J. Bot. 63, 198–200. doi: 10.1016/S0254-6299(15)30744-4
Yin, M., Duong, T. A., Wingfield, M. J., Zhou, X., de Beer, Z. W. (2015). Taxonomy and phylogeny of the Leptographium procerum complex, including Leptographium sinense sp. nov. and Leptographium longiconidiophorum sp. nov. Antonie van Leeuwenhoek 107, 547–563. doi: 10.1007/s10482-014-0351-9
Zambino, P. J., Harrington, T. C. (1989). Isozyme variation within and among host-specialized varieties of Leptographium wageneri. Mycologia 81, 122–133. doi: 10.1080/00275514.1989.12025632
Zambino, P. J., Harrington, T. C. (1992). Correspondence of isozyme characterization with morphology in the asexual genus Leptographium and taxonomic implications. Mycologia 84, 12–25. doi: 10.1080/00275514.1992.12026100
Keywords: Leptographium, phylogeny, genetic characterization, forest pathogen, taxonomy
Citation: Choi D, Harrington TC, Shaw DC, Stewart JE, Klopfenstein NB, Kroese DR and Kim M-S (2023) Phylogenetic analyses allow species-level recognition of Leptographium wageneri varieties that cause black stain root disease of conifers in western North America. Front. Plant Sci. 14:1286157. doi: 10.3389/fpls.2023.1286157
Received: 31 August 2023; Accepted: 07 November 2023;
Published: 22 December 2023.
Edited by:
Ahmad Fakhoury, Southern Illinois University Carbondale, United StatesReviewed by:
Hui Chen, South China Agricultural University, ChinaSusanna Kerio, Connecticut Agricultural Experiment Station, United States
Copyright © 2023 Choi, Harrington, Shaw, Stewart, Klopfenstein, Kroese and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mee-Sook Kim, meesook.kim@usda.gov