 Sergey I. Dikalov
Sergey I. Dikalov Vladimir I. Mayorov
Vladimir I. Mayorov Alexander V. Panov
Alexander V. Panov- 1Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, United States
- 2Division of Basic Medical Sciences, Mercer University School of Medicine, Macon, GA, United States
- 3Institute of Molecular Biology and Biophysics, Russian Academy of Sciences, Novosibirsk, Russia
Mitochondria are the major source of superoxide radicals and superoxide overproduction contributes to cardiovascular diseases and metabolic disorders. Endothelial dysfunction and diminished nitric oxide levels are early steps in the development of these pathological conditions. It is known that physiological production of nitric oxide reduces oxidative stress and inflammation, however, the precise mechanism of “antioxidant” effect of nitric oxide is not clear. In this work we tested the hypothesis that physiological levels of nitric oxide diminish mitochondrial superoxide production without inhibition of mitochondrial respiration. In order to test this hypothesis we analyzed effect of low physiological fluxes of nitric oxide (20 nM/min) on superoxide and hydrogen peroxide production by ESR spin probes and Amplex Red in isolated rat brain mitochondria. Indeed, low levels of nitric oxide substantially attenuated both basal and antimycin A-stimulated production of reactive oxygen species in the presence of succinate or glutamate/malate as mitochondrial substrates. Furthermore, slow releasing NO donor DPTA-NONOate (100 μM) did not change oxygen consumption in State 4 and State 3. However, the NO-donor strongly inhibited oxygen consumption in the presence of uncoupling agent CCCP, which is likely associated with inhibition of the over-reduced complex IV in uncoupled mitochondria. We have examined accumulation of dinitrosyl iron complexes and nitrosothiols in mitochondria treated with fast-releasing NO donor MAHMA NONOate (10 μM) for 30 min until complete release of NO. Following treatment with NO donor, mitochondria were frozen for direct detection of dinitrosyl iron complexes using Electron Spin Resonance (ESR) while accumulation of nitrosothiols was measured by ferrous-N-Methyl-D-glucamine dithiocarbamate complex, Fe(MGD)2, in lysed mitochondria. Treatment of mitochondria with NO-donor gave rise to ESR signal of dinitrosyl iron complexes while ESR spectra of Fe(MGD)2 supplemented mitochondrial lysates showed presence of both dinitrosyl iron complexes and nitrosothiols. We suggest that nitric oxide attenuates production of mitochondrial superoxide by post-translational modifications by nitrosylation of protein cysteine residues and formation of protein dinitrosyl iron complexes with thiol-containing ligands and, therefore, nitric oxide reduction in pathological conditions associated with endothelial dysfunction may increase mitochondrial oxidative stress.
Introduction
Antioxidant properties of endothelial-derived nitric oxide (NO•) are strongly associated with inhibition of platelet activation, suppression of vascular smooth muscle proliferation, inhibition of leukocyte adherence (Somers and Harrison, 1999) and reduction of myocardial injury during ischemia(Wolfrum et al., 2003). Decreased NO• production by endothelial nitric oxide synthase (eNOS) constitutes an early step in the pathogenesis of vascular disease (Harrison and Cai, 2003) and is associated with increased superoxide (O2•) generation (Spiekermann et al., 2003). However, little is known about regulation of O2• production by NO•.
Nitric oxide is synthesized from L-arginine by three major isoforms of NO synthase eNOS, iNOS, nNOS, and, possibly, by mitochondrial isoform (mtNOS), but its properties still remain uncertain. NO• can rapidly react with O2• to produce highly reactive peroxynitrite. However, NO• is highly lipophilic molecule, therefore, due to its high concentration in the hydrophobic environment (lipid membrane and proteins) it can exert its regulatory effects via heme nitrosylation, binding to FeS centers and S-nitrosylation (Kagan et al., 2001; Gow et al., 2002; Pieper et al., 2003). It has been demonstrated that reduced bioavailability of tetrahydrobiopterin, a critical co-factor for eNOS, and L-arginine, eNOS substrate, lead to eNOS “uncoupling” which produces O2• rather than NO• (Xia et al., 1996; Kuzkaya et al., 2003). Impaired flow-dependent endothelium-mediated vasodilatation in cardiovascular patients and metabolic conditions, at least in part, occurs due to accelerated degradation of nitric oxide and diminished nitric oxide production (Landmesser et al., 2002; Walther et al., 2015). Diminished nitric oxide levels increases intracellular Ca2+ due to impaired cellular cGMP pathway and reduced NO-mediated post-translational modifications leading to cellar dysregulations (Adachi et al., 2004; Thomas et al., 2006).
Mitochondrial O2• plays an important role in the ischemia/reperfusion injury, neurodegeneration and aging (Cadenas and Davies, 2000). We have recently demonstrated an important role of mitochondrial O2• in endothelial dysfunction and hypertension (Dikalova et al., 2010; Dikalov et al., 2014). We hypothesize that reduced nitric oxide levels may contribute to elevation in mitochondrial O2•. Indeed, endothelial dysfunction is accompanied by increased intracellular Ca2+ and deletion of Ca2+ sensitive regulatory subunit of the mitochondrial permeability transition pore Cyclophilin D reduces mitochondrial O2•, improves endothelial function and attenuates hypertension (Kroller-Schon et al., 2014; Itani et al., 2016). Complex I S-nitrosylation is cardio-protective (Burwell et al., 2006; Nadtochiy et al., 2007) and mitochondrial nitroso-proteomes reveal that endogenous NO is associated with SNO-proteins in energy and redox regulation, transport, iron homeostasis, translation, mitochondrial morphology, and apoptosis (Satohisa et al., 2014).
The effect of NO• on mitochondria is still controversial. Some studies show inhibition of mitochondrial respiration by nitric oxide (Galkin and Moncada, 2007; Moncada, 2015). This effect however was observed at high nitric oxide or SNO donor levels. We suggested that normal low physiological levels of nitric oxide should not cause mitochondrial dysfunction and may potentially reduce production mitochondrial O2• via post-translational modifications of mitochondrial proteins. We have previously measured physiological NO• production in aorta as 20 nM/min (Dikalov and Fink, 2005). In this work we have investigated the effect of low physiological fluxes of NO• on O2• and H2O2 production, respiration and accumulation of mitochondrial dinitrosyl iron complexes and nitrosothiols.
Materials and Methods
Animal Use and Ethics Statement
All animal experiments were performed in accordance with NIH Guide for the Care and Use of Laboratory Animals and approval by the Institutional Animal Care and Use Committee at Emory University and Vanderbilt University Medical Center. All surgery was performed under anesthesia and all efforts were made to minimize animal suffering. Two- to three-month-old male Sprague-Dawley rats and male C57Bl/6J mice were used for isolation of the kidney and brain mitochondria.
Isolation of Brain and Kidney Mitochondria
Both brain and kidney mitochondria were isolated in medium that contained (in mM) 225 mannitol, 75 sucrose, 20 MOPS (pH 7.2), 1 EGTA, and 0.1% BSA. Kidney mitochondria (LM) were isolated by conventional differential centrifugation with a final spin at 8,600 g. Brain mitochondria were isolated from the pooled forebrains of three rats. We used the modified method of Sims (53) to isolate and purify brain mitochondria (BM) in a Percoll gradient. The modifications were as follows: brain tissue was homogenized with 15 strokes of a loose pestle in a Dounce homogenizer, and 5-ml volumes per tube of 15, 23, and 40% (vol/vol) of Percoll solutions were used to purify the brain mitochondria. After the final sedimentation of mitochondria at 8,600 g, the mitochondria were suspended in 250 mM sucrose and 10 mM MOPS (pH 7.2) (Panov et al., 2007).
Measurements of H2O2 Release by Mitochondria
H2O2 was determined using Amplex red (Molecular Probes) method. In the presence of horseradish peroxidase, the following reaction occurs: Amplex red + H2O2 → resorufin + O2. Resorufin is a stable and highly fluorescent compound with a wavelength spectra excitation/emission of 570/585 nm. The fluorescence of resorufin was determined in 1-ml incubations in a medium (medium A) containing (in mM) 125 KCl, 10 MOPS, pH 7.2, 2 MgCl2, 2 KH2PO4, 10 NaCl, 1 EGTA, 0.7 CaCl2, and 0.2 mg/ml mitochondrial protein, 5 μM Amplex red, and 3 units of horseradish peroxidase, as previously described (Panov et al., 2007). We measured H2O2 production in the presence of glutamate 20 mM + malate 2 mM, or succinate 5 mM. Fluorimetric measurements were made using a fluorometer from C&L (Middletown, PA).
Measurements of O2• Release by Mitochondria
The cyclic hydroxyl-amine PPH (Enzo Life Sciences, Inc., Farmingdale, NY) was used for measurements of O2• release by mitochondria (Panov et al., 2005). PPH reacts with O2• producing stable PP-nitroxide detected with ESR spectroscopy (Dikalov et al., 2011). Briefly, 10 mM PPH was dissolved in deoxygenated media with 50 μm deferoxamine. Mitochondria preparations and PPH stock solutions were kept on ice (50 μg of protein mixed with 1 mm PPH and mitochondrial substrates in 100 μl of Medium. Detection of radical was confirmed by inhibition of the ESR signal with 50 units/ml of SOD. Accumulation of PP-nitroxide was measured using a Bruker EMX ESR spectrometer. Superoxide production was detected by following the low-field peak of the nitroxide ESR spectra using time scans with the following ESR settings: microwave frequency 9.78 GHz, modulation amplitude 2 G, microwave power 10 dB, conversion time 1.3 s, and time constant 5.2 s.
Statistics
Data are presented as a mean ± S.E. for four or five separate measurements of a parameter. For comparison of two groups, a two-tailed t-test was employed using Excel software. Statistical significance was assumed when p < 0.05.
Results
Effect of Low Nitric Oxide on O2• Release by Rat Brain Mitochondria
In order to study the effects of NO on O2• production by mitochondria we used the slow releasing NO-donor DPTA-NONOate (half-life time is 5 h) (Keefer et al., 1996). We have determined that 100 μM DPTA-NONOate generates NO at 20 nM/min which corresponds to normal endothelial NO production (Dikalov and Fink, 2005). Our experiments with rat brain mitochondria demonstrated strong inhibition of O2• production by 20 μM NONOate (Figure 1A). Inhibition was concentration dependent and reached maximum at 100 μM NONOate (Figure 1A). It was found that NONOate only partially inhibited mitochondrial O2• production stimulated by Antimycin A (Figure 1B). Of note, the amount of inhibited O2• in the presence of Antimycin A was equivalent to the amount of O2• blocked in the absence of Antimycin A (Figures 1A,B). It is important to note that NO can potentially react with O2• producing highly reactive peroxynitrite. Meanwhile, PPH spin probe detects both O2• and peroxynitrite, therefore, potential peroxynitrite formation should not diminish the ESR signal (Dikalov et al., 1998). Furthermore, NO-mediated inhibition of O2• production was not affected by complex III inhibitor Antimycin A despite 3-fold increase of O2• production in the presence of Antimycin A suggesting site specific regulatory effect of NO. These data suggest that NO inhibits superoxide production by complex I but does not affect superoxide release by complex III stimulated by addition of Antimycin A.
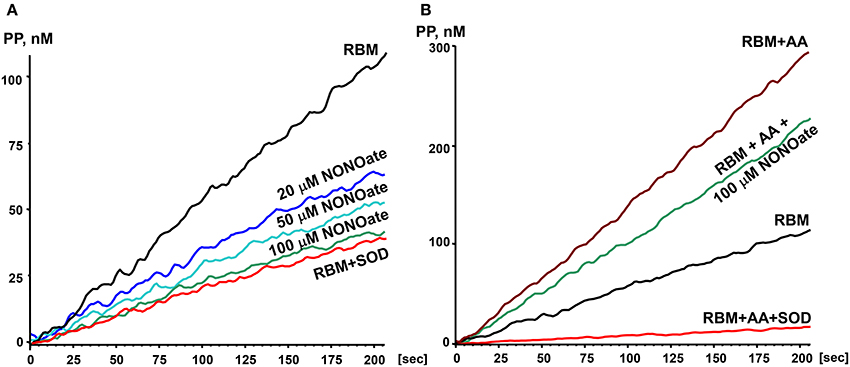
Figure 1. Effect of nitric oxide on O2• release by rat brain mitochondria. Rat brain mitochondria (RBM) were placed in respiration media with Glutamate/Malate as a substrate and acutely treated with slow releasing NO-donor DPTA Nonoate (A). To test the potential effect of NO on complex III-mediated O2• production mitochondria were supplemented with complex III blocker Antimycin A (1 μM) (B). Release of mitochondrial O2• was measured by spin probe PPH (1 mM) and accumulation of PP-nitroxide followed by ESR spectrometer as described in Material and Methods. Addition of Cu,Zn-superoxide dismutase (SOD, 10 Units/ml) confirms specific detection of extramitochondrial O2•. Figure shows typical ESR data of four independent experiments.
Effect of Nitric Oxide on O2• Release by Rat Brain Mitochondria
It is known that several sites on complex I, II, and III may be involved in mitochondrial O2• production. Most of them, however, release O2• in the matrix, where O2• is quickly dismutate by Mn-SOD to H2O2. Therefore, the analysis of H2O2 reflects total mitochondrial O2• production, while extarmitochondrial O2• detection may not take into account O2• produced in the matrix (Figure 2). In order to test the effect of NO• on total mitochondrial O2• we measured H2O2 production by brain mitochondria in the absence and presence of NO-donor (Figure 2). It was found that NO• partially inhibited H2O2 production both with succinate and glutamate/malate as mitochondrial substrates. It is interesting that inhibition of H2O2 production with succinate was similar to the effect of rotenone, which blocks O2• production on complex I due to reverse electron flow. Inhibition of H2O2 production with glutamate/malate resembled the one observed with O2• measurements by ESR (Figure 1). The amount of inhibited O2• in the presence of Antimycin A was similar to the amount blocked in the absence of Antimycin A (Figure 2, Glutamate+Malate). One of the main sites of O2• production with glutamate/malate is associated with complex I, which can generate O2• both to the matrix and intermembrane space. It is possible that NO may inhibit O2• production at the site facing the intermembrane space, but does not affect O2• production in the matrix.
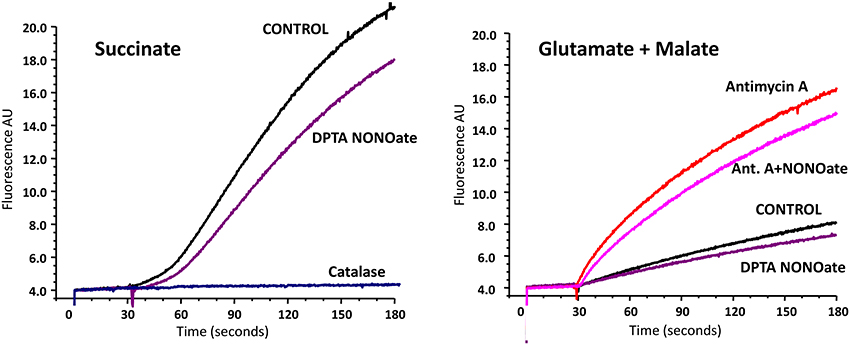
Figure 2. Effect of nitric oxide on H2O2 release by rat brain mitochondria measured with Amplex Red. Rat brain mitochondria (RBM) were supplemented with slow releasing NO-donor DPTA NONOate (100 μM). To test the potential effect of NO on complex III-mediated H2O2 production mitochondria were treated with complex III blocker Antimycin A (1 μM). Release of mitochondrial H2O2 was measured by Amplex Red assay as described in Material and Methods. Addition of catalase (20 μg/ml) confirms specific detection of mitochondrial H2O2. Figure shows typical data of four independent experiments.
Effect of Slow Releasing NO-Donor on Mitochondrial Respiration
On the next step we tested if slow release DPTA-NONOate did inhibit mitochondrial respiration. Rat brain and kidney mitochondria were isolated as we have described previously (Panov et al., 2004). We have compared respiration of mitochondria with glutamate-malate in the absence or presence of DPTA-NONOate (Figure 3). It was found that 100 μM DPTA-NONOate did not significantly change oxygen consumption in State 4 and State 3. However, the NO-donor strongly inhibited oxygen consumption in the presence of uncoupling agent CCCP, which is likely associated with inhibition of the reduced complex IV. This experiment argues that NO-donor DPTA-NONOate did not affect mitochondrial respiration in State 4, and inhibition of the superoxide release was due to specific effect on superoxide production rather than inhibition of mitochondrial respiration. NO, though, rapidly inhibited the uncoupled mitochondrial respiration, when complex IV was more reduced than in State 4.
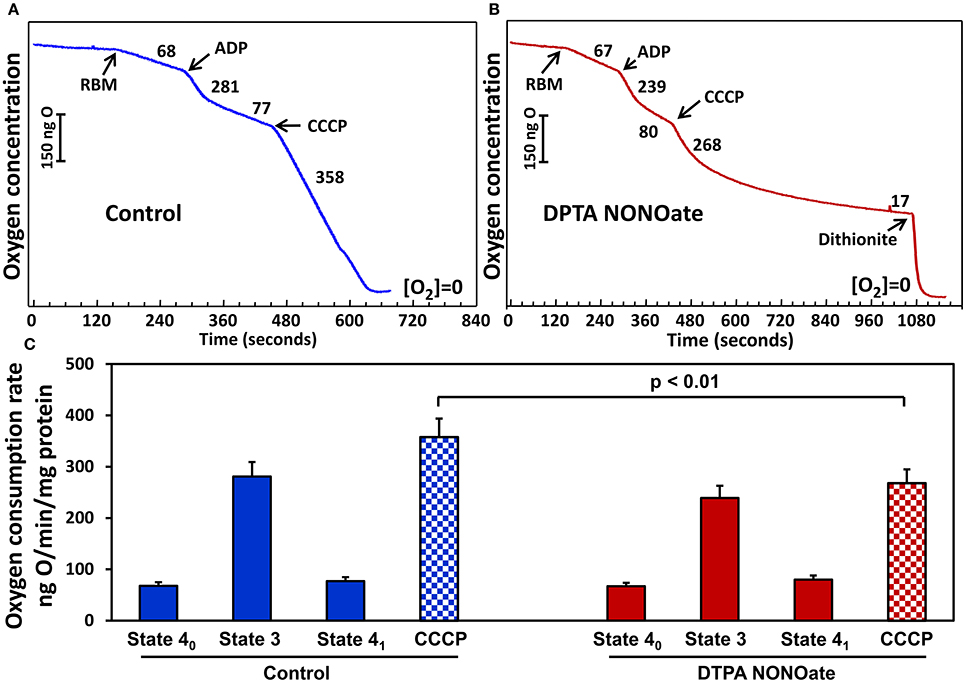
Figure 3. Effect of nitric oxide on respiration with glutamate/malate by rat brain mitochondria (RBM) (A). Mitochondria (0.5 mg/ml) were supplemented with slow releasing NO-donor DPTA NONOate (100 μM) (B). (C) Oxygen consumption rate is expressed as ng O2/min/mg protein. Clark oxygen electrode was calibrated with air saturated water at 25°C. Instrumental zero oxygen level ([O2] = 0) was confirmed at the end of every experiment by addition of sodium dithionite as shown in (B). State 40, State 3 and State 41 were defined as previously described in Panov (2014). Results represent mean ± SEM (n = 4). *P < 0.01 vs. NONOate/CCCP.
Accumulation of Dinitrosyl Iron Complexes and Nitrosothiols in NO-Donor Treated Mitochondria
In additional experiments we have studied NO-mediated post-translational modifications in mitochondria. For this aim we have treated isolated mouse kidney mitochondria with fast releasing NO donor MAHMA-NONOate (half-life time is 3 min) (Keefer et al., 1996). Mitochondria were incubated with 10 μM MAHMA-NONOate for 30 min until complete release of NO which was confirmed in the separate experiments in respiration media without mitochondria using NO spin trap Fe(MGD)2 (Komarov et al., 2000). Following incubation with MAHMA-NONOate or vehicle mitochondria (10 mg protein/ml) were placed in insulin syringes and snap frozen in the liquid nitrogen for ESR analysis. Untreated mitochondria show characteristic heme spectrum (Figure 4A), however, NO-donor treated mitochondria showed a robust accumulation of dinitrosyl iron complexes with thiol-containing ligands (Figure 4B) detected by specific ESR spectrum of dinitrosyl iron complexes as has been previously reported by Dr. Vanin (Vanin, 2016). In order to test the accumulation of nitrosothiols we have lysed mitochondria by freeze-thaw cycles and added 0.1 mM Fe(MGD)2 which rapidly converts SNO to NO-Fe(MGD)2 complex detectable by ESR (Komarov et al., 2000; Vanin et al., 2004). Indeed, supplementation of Fe(MGD)2 to NO-treated mitochondria substantially changed ESR spectra by adding the specific triplet ESR signal of NO-Fe(MGD)2 complex (Figure 4C). Additional analysis of ESR spectra showed the presence of both dinitrosyl iron complexes and nitrosothiols in NO-donor treated mitochondria (Figures 4B,D). To quantify the accumulation of dinitrosyl iron complexes and nitrosothiols in NO-donor treated mitochondria we have used reference calibration sample containing 2 μM GSNO plus 0.1 mM Fe(MGD)2 (Figure 4E). Analysis of double integral intensities of ESR spectra showed that the accumulation of dinitrosyl iron complexes and nitrosothiols in MAHMA-NONOate treated mitochondria is close to 2 μM.
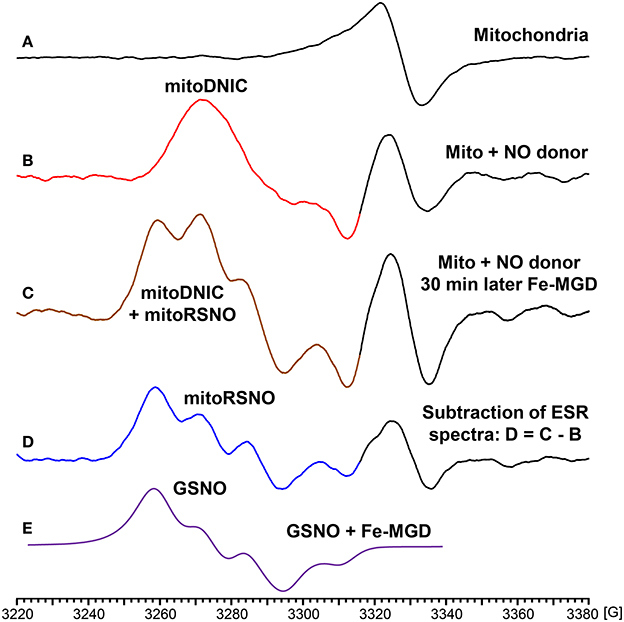
Figure 4. Formation of dinitrosyl iron complexes and nitrosothiols in nitric oxide-treated mitochondria. Mitochondria were isolated from mouse kidney (10 mg/ml) and incubated in respiration media at 25°C for 30 with vehicle or NO-donor MAHMA Nonoate (10 μM). Then mitochondria were snap-frozen in liquid nitrogen for Electron Spin Resonance (ESR) studies. (A) Untreated mitochondria; (B) NO-donor treated mitochondria; (C) Lysed NO-donor treated mitochondria with 0.1 mM Fe(MGD)2 complex; (D) Subtraction of dinitrosyl iron complex ESR spectrum (B) from spectrum (C) revealed the presence of mitochondrial nitrosothiols (mitoRSNO); (E) Reference ESR spectrum of 2 μM GSNO plus 0.1 mM Fe(MGD)2. The average integral amount of mitochondrial dinitrosyl iron complexes (mitoDNIC) and nitrosothiols ESR signal is 2 μM (standard error <15%). Figure shows typical ESR spectra of four independent experiments.
Discussion
This study provides the first evidence that physiological levels of nitric oxide reduce mitochondrial superoxide production without significant inhibition of mitochondrial respiration. We have employed two independent assays for measurements of mitochondrial reactive oxygen species. Analysis of superoxide release was done by mitochondria-impermeable spin probe PPH while release of H2O2 was measured by Amplex Red assay (Panov et al., 2005). It was found that low physiological levels of nitric oxide reduced both O2• and H2O2 production. Our data show that treatment of mitochondria with NO-donor lead to significant accumulation of dinitrosyl iron complexes and nitrosothiols suggesting the role of NO-mediated post-translational modifications in down-regulation of mitochondrial O2• production.
Physiological production of nitric oxide is mainly mediated by endothelial nitric oxide synthase (eNOS) in the vasculature and kidney while neural nitric oxide synthase (nNOS) is important in the brain, heart and skeletal muscle (Silberman et al., 2010; Gonzalez et al., 2015). It is interesting that these tissues are highly metabolically active and it appears to develop adaptation to avoid the interference of low fluxes of nitric oxide with mitochondrial respiration. Furthermore, nitric oxide in these tissues modulates mitogenesis and reduces intracellular Ca2+ which attenuates mPTP opening and improves mitochondrial function (Hassid et al., 1994; Khan and Hare, 2003; Lira et al., 2010). Our data show that nitric oxide gas in mitochondria is converted into nitrosyl, NO+, which does not affect respiration and mediates post-translational modifications of cysteine residues into dinitrosyl iron complexes and nitrosothiols. Our ESR study of nitrosylation of mitochondrial targets is consistent with previous ESR analysis of cellular dinitrosyl iron complexes known as DNIC (Kleschyov et al., 2007). There are low molecular DNIC complexes such as low molecular weight DNIC–cysteine and protein DNIC. Our preliminary data showed accumulation of DNIC and nitrosothiols both in the mitochondrial membrane fraction and mitochondrial matrix (supernatant) suggesting nitrosylation of low molecular and protein targets. It has been previously shown that S-nitrosylation of NADPH oxidase reduces O2• production (Selemidis et al., 2007) and we suggest that similar post-translational modifications may contribute to down-regulation of mitochondrial O2•. The precise molecular mechanisms of “anti-oxidant” effect of NO in mitochondria are not clear and it may include not only inhibition of O2• production but also upregulation of mitochondrial antioxidants such as superoxide dismutase and redox buffering of peroxides.
The discrepancy that exists in the literature about contribution of respiratory chain complexes I and III into mitochondrial ROS production to a large extent can be explained by the fact that mitochondria from different organs have different rates of State 4 and State 3 respiration and different regulatory mechanisms, as well as, different antioxidant activities (Herrero and Barja, 1997). Cardiac and skeletal muscles have antioxidant concentrations more than one order of magnitude lower than those of other highly aerobic tissues like liver or kidney (Herrero and Barja, 1997). Another important issue in studies of ROS generation is the location of the site of O2• generation. The sites that are located close to the inner surface of the mitochondrial membrane will more likely release O2• into the matrix, whereas the sites located close to the intermembrane space, will more likely release O2• into the cytosolic side. Therefore, extramitochondrial probes register only O2• that was formed close to the intermembrane space. Meanwhile, H2O2 is a neutral molecule and will easily leave mitochondria regardless of mitochondrial energization. Indeed, we have found that in brain mitochondria measurements of total ROS generation can be best followed by H2O2 production. Thus, in order to study NO-mediated modulation of mitochondrial reactive oxygen species we have investigated both O2• and H2O2 production by mitochondria. Our data show that the amount of inhibited O2• in the absence of Antimycin A was similar to the amount blocked in the presence of Antimycin A which is consistent with modulation of mitochondrial complex I by nitrosylation leading to reduced O2• generation at this site.
The previous report show that high levels of DNIC (100 μM) induces apoptosis (Kleschyov et al., 2006). Indeed, inflammation is accompanied with robust activation of inducible nitric oxide synthase (iNOS) leading to NO overproduction, nitrosative stress and metabolic dysfunction (Kaneki et al., 2007). It is important to note that accumulation of mitochondrial DNIC and nitrosothiols in our experiments with physiological fluxes of NO• did not exceed 2 μM and DNIC at concentrations <10 μM did not induce apoptosis (Kleschyov et al., 2006). One should take into account substantial differences between NO• and NO+ (DNIC and nitrosothiols) in reactivity and life-time. NO• is much more reactive and has a shorter life-time compared to NO+. Therefore, micromolar concentrations of bolus NO• have very distinct effect compared to NO+. It has been shown that NO• (bolus 1 μM) significantly increases production of O2• and H2O2 by mitochondria (Riobo et al., 2001). It has been found that NO• causes nitrosylation of complex IV, modifications of iron-sulfur clusters of complex I, S-nitrosation of cysteine residues, glutathionylation of cysteine residues, N-nitrosation of secondary amines, nitration of tyrosine residues (Brown and Borutaite, 2001, 2004). This nitrosative stress is mediated by NO•-derived reactive nitrogen species such as, • NO2, N2O3, and ONOO−. It has been suggested that ONOO−-mediated complex I nitration mimics rotenone action (Riobo et al., 2001). However, the exact mechanism of the NO•-mediated increase in O2• production by mitochondria is not clear. Our own preliminary data have shown that the inhibitory effect of NO• on complex IV strongly depends on the metabolic state of mitochondria: NO• inhibits respiration when mitochondria have low membrane potential and complex IV is present mainly in reduced form. It is important to note that high doses of NO• cause nitrosative stress, while smaller amounts of NO• may have an antioxidant action. Indeed, our data showed that low physiological fluxes of NO• (20 nM/min) reduced O2• release by mitochondria respiring in the resting metabolic state (State 4), and significantly inhibited H2O2 production. This effect was likely associated with NO•, but not with ONOO−. Thus, the effect of NO• on mitochondria may depend not only on the amount of NO• but also on the functional state and antioxidant status of mitochondria. Of note, the effect of NO• on mitochondria can depend on the type of cells and species because of organ-specific and species-specific variability of mitochondrial function (Panov et al., 2007). However, factors, which determine the balance of antioxidant/pro-oxidant action of NO•, are not well-defined.
It is interesting that targeting mitochondrial O2• improves endothelial function and reduces hypertension (Dikalova et al., 2010, 2017). It is conceivable that decreased NO level in cardiovascular conditions (Landmesser and Harrison, 2001) may contribute to mitochondrial dysfunction and strategies to improve NO-production by eNOS such as tetrahydrobiopterin supplementation can also improve mitochondrial function. On the other hand, a number of pathological conditions dealing with hepatotoxicity and neurodegeneration are associated with NO• overproduction by iNOS as well as with the increased oxidative stress (Venkatraman et al., 2004) and, therefore, specific inhibition of iNOS can be beneficial (Ljubisavljevic and Stojanovic, 2015). It is possible that effect of nitric oxide on mitochondrial functions has a bell shaped curve and, therefore, require an optimal NO level to balance the oxidative stress and metabolic activity.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was funded by National Institute of Health (R01HL124116) and Mercer University School of Medicine.
References
Adachi, T., Weisbrod, R. M., Pimentel, D. R., Ying, J., Sharov, V. S., Schoneich, C., et al. (2004). S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat. Med. 10, 1200–1207. doi: 10.1038/nm1119
Brown, G. C., and Borutaite, V. (2001). Nitric oxide, mitochondria, and cell death. IUBMB Life 52, 189–195. doi: 10.1080/15216540152845993
Brown, G. C., and Borutaite, V. (2004). Inhibition of mitochondrial respiratory complex I by nitric oxide, peroxynitrite and S-nitrosothiols. Biochim. Biophys. Acta 1658, 44–49. doi: 10.1016/j.bbabio.2004.03.016
Burwell, L. S., Nadtochiy, S. M., Tompkins, A. J., Young, S., and Brookes, P. S. (2006). Direct evidence for S-nitrosation of mitochondrial complex I. Biochem. J. 394(Pt 3), 627–634. doi: 10.1042/BJ20051435
Cadenas, E., and Davies, K. J. (2000). Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 29, 222–230. doi: 10.1016/S0891-5849(00)00317-8
Dikalov, S., and Fink, B. (2005). ESR techniques for the detection of nitric oxide in vivo and in tissues. Meth. Enzymol. 396, 597–610. doi: 10.1016/S0076-6879(05)96052-7
Dikalov, S., Grigor'ev, I. A., Voinov, M., and Bassenge, E. (1998). Detection of superoxide radicals and peroxynitrite by 1-hydroxy-4-phosphonooxy-2,2,6,6-tetramethylpiperidine: quantification of extracellular superoxide radicals formation. Biochem. Biophys. Res. Commun. 248, 211–215. doi: 10.1006/bbrc.1998.8936
Dikalov, S. I., Kirilyuk, I. A., Voinov, M., and Grigor'ev, I. A. (2011). EPR detection of cellular and mitochondrial superoxide using cyclic hydroxylamines. Free Radic. Res. 45, 417–430. doi: 10.3109/10715762.2010.540242
Dikalov, S. I., Nazarewicz, R. R., Bikineyeva, A., Hilenski, L., Lassegue, B., Griendling, K., et al. (2014). Nox2-induced production of mitochondrial superoxide in angiotensin II - mediated endothelial oxidative stress and hypertension. Antioxid. Redox Signal. 20, 281–294. doi: 10.1089/ars.2012.4918
Dikalova, A. E., Bikineyeva, A. T., Budzyn, K., Nazarewicz, R. R., McCann, L., Lewis, W., et al. (2010). Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 107, 106–116. doi: 10.1161/CIRCRESAHA.109.214601
Dikalova, A. E., Itani, H. A., Nazarewicz, R. R., McMaster, W. G., Fessel, J. P., Flynn, C. R., et al. (2017). Sirt3 impairment and SOD2 hyperacetylation in vascular oxidative stress and hypertension. Circ. Res. 121, 664–774. doi: 10.1161/CIRCRESAHA.117.310933
Galkin, A., and Moncada, S. (2007). S-nitrosation of mitochondrial complex I depends on its structural conformation. J. Biol. Chem. 282, 37448–37453. doi: 10.1074/jbc.M707543200
Gonzalez, J. P., Crassous, P. A., Schneider, J. S., Beuve, A., and Fraidenraich, D. (2015). Neuronal nitric oxide synthase localizes to utrophin expressing intercalated discs and stabilizes their structural integrity. Neuromuscul. Disord. 25, 964–976. doi: 10.1016/j.nmd.2015.09.011
Gow, A. J., Chen, Q., Hess, D. T., Day, B. J., Ischiropoulos, H., and Stamler, J. S. (2002). Basal and stimulated protein S-nitrosylation in multiple cell types and tissues. J. Biol. Chem. 277, 9637–9640. doi: 10.1074/jbc.C100746200
Harrison, D. G., and Cai, H. (2003). Endothelial control of vasomotion and nitric oxide production. Cardiol. Clin. 21, 289–302. doi: 10.1016/S0733-8651(03)00073-0
Hassid, A., Arabshahi, H., Bourcier, T., Dhaunsi, G. S., and Matthews, C. (1994). Nitric oxide selectively amplifies FGF-2-induced mitogenesis in primary rat aortic smooth muscle cells. Am. J. Physiol. 267(3 Pt 2), H1040–H1048.
Herrero, A., and Barja, G. (1997). ADP-regulation of mitochondrial free radical production is different with complex I- or complex II-linked substrates: implications for the exercise paradox and brain hypermetabolism. J. Bioenerg. Biomembr. 29, 241–249. doi: 10.1023/A:1022458010266
Itani, H. A., Dikalova, A. E., McMaster, W. G., Nazarewicz, R. R., Bikineyeva, A. T., Harrison, D. G., et al. (2016). Mitochondrial cyclophilin D in vascular oxidative stress and hypertension. Hypertension 67, 1218–1227. doi: 10.1161/HYPERTENSIONAHA.115.07085
Kagan, V. E., Kozlov, A. V., Tyurina, Y. Y., Shvedova, A. A., and Yalowich, J. C. (2001). Antioxidant mechanisms of nitric oxide against iron-catalyzed oxidative stress in cells. Antioxid. Redox Signal. 3, 189–202. doi: 10.1089/152308601300185160
Kaneki, M., Shimizu, N., Yamada, D., and Chang, K. (2007). Nitrosative stress and pathogenesis of insulin resistance. Antioxid. Redox Signal. 9, 319–329. doi: 10.1089/ars.2006.1464
Keefer, L. K., Nims, R. W., Davies, K. M., and Wink, D. A. (1996). “NONOates” (1-substituted diazen-1-ium-1,2-diolates) as nitric oxide donors: convenient nitric oxide dosage forms. Meth. Enzymol. 268, 281–293. doi: 10.1016/S0076-6879(96)68030-6
Khan, S. A., and Hare, J. M. (2003). The role of nitric oxide in the physiological regulation of Ca2+ cycling. Curr. Opin. Drug Discov. Devel. 6, 658–666.
Kleschyov, A. L., Strand, S., Schmitt, S., Gottfried, D., Skatchkov, M., Sjakste, N., et al. (2006). Dinitrosyl-iron triggers apoptosis in Jurkat cells despite overexpression of Bcl-2. Free Radic. Biol. Med. 40, 1340–1348. doi: 10.1016/j.freeradbiomed.2005.12.001
Kleschyov, A. L., Wenzel, P., and Munzel, T. (2007). Electron paramagnetic resonance (EPR) spin trapping of biological nitric oxide. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 851, 12–20. doi: 10.1016/j.jchromb.2006.10.006
Komarov, A. M., Wink, D. A., Feelisch, M., and Schmidt, H. H. (2000). Electron-paramagnetic resonance spectroscopy using N-methyl-D-glucamine dithiocarbamate iron cannot discriminate between nitric oxide and nitroxyl: implications for the detection of reaction products for nitric oxide synthase. Free Radic. Biol. Med. 28, 739–742. doi: 10.1016/S0891-5849(00)00156-8
Kroller-Schon, S., Steven, S., Kossmann, S., Scholz, A., Daub, S., Oelze, M., et al. (2014). Molecular mechanisms of the crosstalk between mitochondria and NADPH oxidase through reactive oxygen species-studies in white blood cells and in animal models. Antioxid. Redox Signal. 20, 247–266. doi: 10.1089/ars.2012.4953
Kuzkaya, N., Weissmann, N., Harrison, D. G., and Dikalov, S. (2003). Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: implications for uncoupling endothelial nitric-oxide synthase. J. Biol. Chem. 278, 22546–22554. doi: 10.1074/jbc.M302227200
Landmesser, U., and Harrison, D. G. (2001). Oxidative stress and vascular damage in hypertension. Coron. Artery Dis. 12, 455–461. doi: 10.1097/00019501-200109000-00004
Landmesser, U., Spiekermann, S., Dikalov, S., Tatge, H., Wilke, R., Kohler, C., et al. (2002). Vascular oxidative stress and endothelial dysfunction in patients with chronic heart failure: role of xanthine-oxidase and extracellular superoxide dismutase. Circulation 106, 3073–3078. doi: 10.1161/01.CIR.0000041431.57222.AF
Lira, V. A., Brown, D. L., Lira, A. K., Kavazis, A. N., Soltow, Q. A., Zeanah, E. H., et al. (2010). Nitric oxide and AMPK cooperatively regulate PGC-1 in skeletal muscle cells. J. Physiol. 588(Pt 18), 3551–3566. doi: 10.1113/jphysiol.2010.194035
Ljubisavljevic, S., and Stojanovic, I. (2015). Neuroinflammation and demyelination from the point of nitrosative stress as a new target for neuroprotection. Rev. Neurosci. 26, 49–73. doi: 10.1515/revneuro-2014-0060
Moncada, P. S. (2015). Nitric oxide and oxygen: actions and interactions in health and disease. Redox Biol. 5, 421. doi: 10.1016/j.redox.2015.09.034
Nadtochiy, S. M., Burwell, L. S., and Brookes, P. S. (2007). Cardioprotection and mitochondrial S-nitrosation: effects of S-nitroso-2-mercaptopropionyl glycine (SNO-MPG) in cardiac ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 42, 812–825. doi: 10.1016/j.yjmcc.2007.01.010
Panov, A. (2014). Practical Mitochondriology. Pitfalls and Problems in Studies of Mitochondria. CreateSpace Independent Publishing Platform.
Panov, A., Dikalov, S., Shalbuyeva, N., Hemendinger, R., Greenamyre, J. T., and Rosenfeld, J. (2007). Species- and tissue-specific relationships between mitochondrial permeability transition and generation of ROS in brain and liver mitochondria of rats and mice. Am. J. Physiol. Cell Physiol. 292, C708–C718. doi: 10.1152/ajpcell.00202.2006
Panov, A., Dikalov, S., Shalbuyeva, N., Taylor, G., Sherer, T., and Greenamyre, J. T. (2005). Rotenone model of Parkinson disease: multiple brain mitochondria dysfunctions after short term systemic rotenone intoxication. J. Biol. Chem. 280, 42026–42035. doi: 10.1074/jbc.M508628200
Panov, A. V., Andreeva, L., and Greenamyre, J. T. (2004). Quantitative evaluation of the effects of mitochondrial permeability transition pore modifiers on accumulation of calcium phosphate: comparison of rat liver and brain mitochondria. Arch. Biochem. Biophys. 424, 44–52. doi: 10.1016/j.abb.2004.01.013
Pieper, G. M., Halligan, N. L., Hilton, G., Konorev, E. A., Felix, C. C., Roza, A. M., et al. (2003). Non-heme iron protein: a potential target of nitric oxide in acute cardiac allograft rejection. Proc. Natl. Acad. Sci. U.S.A. 100, 3125–3130. doi: 10.1073/pnas.0636938100
Riobo, N. A., Clementi, E., Melani, M., Boveris, A., Cadenas, E., Moncada, S., et al. (2001). Nitric oxide inhibits mitochondrial NADH:ubiquinone reductase activity through peroxynitrite formation. Biochem. J. 359(Pt 1), 139–145. doi: 10.1042/bj3590139
Satohisa, S., Zhang, H. H., Feng, L., Yang, Y. Y., Huang, L., and Chen, D. B. (2014). Endogenous NO upon estradiol-17beta stimulation and NO donor differentially regulate mitochondrial S-nitrosylation in endothelial cells. Endocrinology 155, 3005–3016. doi: 10.1210/en.2013-2174
Selemidis, S., Dusting, G. J., Peshavariya, H., Kemp-Harper, B. K., and Drummond, G. R. (2007). Nitric oxide suppresses NADPH oxidase-dependent superoxide production by S-nitrosylation in human endothelial cells. Cardiovasc. Res. 75, 349–358. doi: 10.1016/j.cardiores.2007.03.030
Silberman, G. A., Fan, T. H., Liu, H., Jiao, Z., Xiao, H. D., Lovelock, J. D., et al. (2010). Uncoupled cardiac nitric oxide synthase mediates diastolic dysfunction. Circulation 121, 519–528. doi: 10.1161/CIRCULATIONAHA.109.883777
Somers, M. J., and Harrison, D. G. (1999). Reactive oxygen species and the control of vasomotor tone. Curr. Hypertens. Rep. 1, 102–108. doi: 10.1007/s11906-999-0080-z
Spiekermann, S., Landmesser, U., Dikalov, S., Bredt, M., Gamez, G., Tatge, H., et al. (2003). Electron spin resonance characterization of vascular xanthine and NAD(P)H oxidase activity in patients with coronary artery disease: relation to endothelium-dependent vasodilation. Circulation 107, 1383–1389. doi: 10.1161/01.CIR.0000056762.69302.46
Thomas, D. D., Ridnour, L. A., Espey, M. G., Donzelli, S., Ambs, S., Hussain, S. P., et al. (2006). Superoxide fluxes limit nitric oxide-induced signaling. J. Biol. Chem. 281, 25984–25993. doi: 10.1074/jbc.M602242200
Vanin, A. F. (2016). Dinitrosyl iron complexes with thiol-containing ligands as a “working form” of endogenous nitric oxide. Nitric Oxide 54, 15–29. doi: 10.1016/j.niox.2016.01.006
Vanin, A. F., Papina, A. A., Serezhenkov, V. A., and Koppenol, W. H. (2004). The mechanisms of S-nitrosothiol decomposition catalyzed by iron. Nitric Oxide 10, 60–73. doi: 10.1016/j.niox.2004.02.005
Venkatraman, A., Landar, A., Davis, A. J., Ulasova, E., Page, G., Murphy, M. P., et al. (2004). Oxidative modification of hepatic mitochondria protein thiols: effect of chronic alcohol consumption. Am. J. Physiol. Gastrointest. Liver Physiol. 286, G521–G527. doi: 10.1152/ajpgi.00399.2003
Walther, G., Obert, P., Dutheil, F., Chapier, R., Lesourd, B., Naughton, G., et al. (2015). Metabolic syndrome individuals with and without type 2 diabetes mellitus present generalized vascular dysfunction: cross-sectional study. Arterioscler. Thromb. Vasc. Biol. 35, 1022–1029. doi: 10.1161/ATVBAHA.114.304591
Wolfrum, S., Grimm, M., Heidbreder, M., Dendorfer, A., Katus, H. A., Liao, J. K., et al. (2003). Acute reduction of myocardial infarct size by a hydroxymethyl glutaryl coenzyme A reductase inhibitor is mediated by endothelial nitric oxide synthase. J. Cardiovasc. Pharmacol. 41, 474–480. doi: 10.1097/00005344-200303000-00017
Keywords: mitochondria, superoxide, nitric oxide, dinitrosyl iron complexes, nitrosothiols, electron spin resonance
Citation: Dikalov SI, Mayorov VI and Panov AV (2017) Physiological Levels of Nitric Oxide Diminish Mitochondrial Superoxide. Potential Role of Mitochondrial Dinitrosyl Iron Complexes and Nitrosothiols. Front. Physiol. 8:907. doi: 10.3389/fphys.2017.00907
Received: 07 September 2017; Accepted: 25 October 2017;
Published: 07 November 2017.
Edited by:
Andrey V. Kozlov, Institute for Experimental and Clinical Traumatology (LBG), AustriaReviewed by:
Andreas Daiber, Johannes Gutenberg-Universität Mainz, GermanyDavid F. Stowe, Medical College of Wisconsin, United States
Copyright © 2017 Dikalov, Mayorov and Panov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sergey I. Dikalov, sergey.dikalov@vanderbilt.edu