 Mena Abdelsayed
Mena Abdelsayed Dana Page1
Dana Page1 Peter C. Ruben
Peter C. Ruben- 1Department of Biomedical Physiology and Kinesiology, Simon Fraser University, Burnaby, BC, Canada
- 2Stanford Cardiovascular Institute, Stanford University, Stanford, CA, United States
- 3Department of Medicine, Stanford University, Stanford, CA, United States
Background: Most therapeutics targeting cardiac voltage-gated sodium channels (Nav1.5) attenuate the sodium current (INa) conducted through the pore of the protein. Whereas these drugs may be beneficial for disease states associated with gain-of-function (GoF) in Nav1.5, few attempts have been made to therapeutically treat loss-of-function (LoF) conditions. The primary impediment to designing efficacious therapies for LoF is a tendency for drugs to occlude the Nav1.5 central pore. We hypothesized that molecular candidates with a high affinity for the fenestrations would potentially reduce pore block.
Methods and Results: Virtual docking was performed on 21 compounds, selected based on their affinity for the fenestrations in Nav1.5, which included a class of sulfonamides and carboxamides we identify as ARumenamide (AR). Six ARs, AR-051, AR-189, AR-674, AR-802, AR-807 and AR-811, were further docked against Nav1.5 built on NavAb and rNav1.5. Based on the virtual docking results, these particular ARs have a high affinity for Domain III-IV and Domain VI-I fenestrations. Upon functional characterization, a trend was observed in the effects of the six ARs on INa. An inverse correlation was established between the aromaticity of the AR’s functional moieties and compound block. Due to its aromaticity, AR-811 blocked INa the least compared with other aromatic ARs, which also decelerated fast inactivation onset. AR-674, with its aliphatic functional group, significantly suppresses INa and enhances use-dependence in Nav1.5. AR-802 and AR-811, in particular, decelerated fast inactivation kinetics in the most common Brugada Syndrome Type 1 and Long-QT Syndrome Type 3 mutant, E1784K, without affecting peak or persistent INa.
Conclusion: Our hypothesis that LoF in Nav1.5 may be therapeutically treated was supported by the discovery of ARs, which appear to preferentially block the fenestrations. ARs with aromatic functional groups as opposed to aliphatic groups efficaciously maintained Nav1.5 availability. We predict that these bulkier side groups may have a higher affinity for the hydrophobic milieu of the fenestrations, remaining there rather than in the central pore of the channel. Future refinements of AR compound structures and additional validation by molecular dynamic simulations and screening against more Brugada variants will further support their potential benefits in treating certain LoF cardiac arrhythmias.
Introduction
The cardiac voltage-gated sodium channel (Nav1.5) is responsible for electrical depolarization in cardiac tissue through the conduction of sodium current (INa). Mutations in the SCN5a gene, encoding Nav1.5, result in loss-of-function (LoF) or gain-of-function (GoF) abnormalities in the protein, leading to an array of cardiac disorders (Dumaine et al., 1996; Antzelevitch et al., 2007; Makita et al., 2008; Gui et al., 2010; Biswas et al., 2019). The common clinical manifestation of LoF mutations is Brugada Syndrome Type 1, whereas Long-QT syndrome Type 3 is caused by GoF (Antzelevitch et al., 2005; Beaufort-Krol et al., 2005; Bankston et al., 2007). Both these arrhythmogenic syndromes increase the likelihood of sudden cardiac arrest in patients (Kaufman, 2009; Skinner et al., 2019). The most common Brugada Syndrome Type 1 and Long-QT Syndrome Type 3 mutant is E1784K (Makita et al., 2008; Peters et al., 2020).
Non-genetic factors common in disease states can also trigger LoF and GoF in Nav1.5 through the activation of multiple cellular signaling cascades that enhance intracellular kinases and cytosolic calcium (Song et al., 2001; Baartscheer et al., 2003; Wagner et al., 2006). Increased phosphorylation in Nav1.5 augments late or persistent INa and enhances entry into fast and slow inactivation (Wagner et al., 2006, 2011; Herren et al., 2013). An elevated sympathetic drive increases the heart rate and promotes rapid onset into fast inactivation in Nav1.5, prematurely terminating peak INa. With time, channels also accumulate into slow inactivation, which lowers the available pool of Nav1.5 required to sustain normal heart rhythmicity (Ashpole et al., 2012; Herren et al., 2013).
To date, the major pharmaceuticals targeting Nav1.5 treat GoF. These agents, like ranolazine, phenytoin, and lidocaine, to name a few, generally block the central pore of Nav1.5 by interacting with residues in the inner vestibule (Balser et al., 1996a, 1996b; An et al., 1996; Antzelevitch, 2004; Sokolov et al., 2013; Abdelsayed et al., 2018). Very few attempts, however, have been made to target LoF. Toxins, such as batrachotoxin, were regarded as potential hits for treating LoF since they bind to NaV and stabilize the channel in the open state (Du et al., 2011; Wang and Wang, 2014; Finol-Urdaneta et al., 2019; MacKenzie et al., 2022). Open-state stabilization, however, may produce other adverse side effects that further exacerbate the pathophysiology (MacKenzie et al., 2022). Inadequate targeting of LoF leaves behind a plethora of untreated NaV-related diseases like Brugada Syndrome Type 1.
Targeting other sites besides the pore regions in Nav1.5 is the most prudent approach in treating LoF to avoid blocking peak INa. Rearrangement in the four fenestrations of NaV has been implicated as an important pathway for lipophilic drug entry into the channel pore (Payandeh et al., 2011, 2012; Kaczmarski and Corry, 2014; Jiang et al., 2020). In 1977, Hille proposed that, unlike charged compounds, lipophilic compounds would enter the channel through these fenestrations (Hille, 1977). His prediction was correct, and structural and electrophysiological studies proved that bulky drugs, especially ones like flecainide elicit their state-dependent effects on Nav via the fenestrations (Kaczmarski and Corry, 2014; Gamal El-Din et al., 2018; Montini et al., 2018).
Comparison between the structural models of the closed and open Nav from Arcobacter butzleri (NavAb) and Magnetococcus marinus (NavMs) confirmed that at rest, drugs, especially lipophilic ones, have a high access into the channel through the dilated fenestrations and await the dilation of the central pore to elicit their pharmacological action (Payandeh et al., 2011; Martin and Corry, 2014; Montini et al., 2018). However, other drug-binding modes, such as during open-state or the inactivated-state, are not explained by this mechanism of action, although they may involve the fenestrations (Martin and Corry, 2014). Open-state drug binding is likely to occur via the open activation gate through the intracellular pathway (Lenaeus et al., 2017). Classic antiarrhythmics, such as benzocaine and lidocaine, have low thermodynamic stability in the fenestrations; thus, they rapidly move to the inner vestibule and bind to their receptor sites (Kaczmarski and Corry, 2014). Benzocaine’s binding site in NavAb is F203 that is a surrogate for a conserved phenylalanine found in Domain IV S6 of eukaryotic Nav (Boiteux et al., 2014).
The recently solved cryo-EM structure of rat Nav1.5 (rNav1.5) at 3.2–3.5 Å resolution was captured in a state in which all four voltage sensors were partially activated; hence, the channel was also partially inactivated. The four fenestrations were identified: Domain II-III fenestration was the largest compared to the others (Jiang et al., 2020). Flecainide associates with residues in the central cavity via Domain II-III fenestration. Other studies suggest that Domain III-IV fenestration, can also provide access for the drug (Nguyen et al., 2019); however, this fenestration was too narrow in this model.
A major caveat of the cyro-EM structure of rNav1.5 is that it does not fully depict fenestration size at rest, nor in activated or inactivated states. Crystal structures obtained of NavAb inactivated states are compatible with slow inactivation in eukaryotic Nav channels, since NavAb lacks the fast inactivation particle (Payandeh et al., 2011, 2012). Upon inactivation, the wild-type NavAb undergoes reshaping of the fenestrations where two opposing fenestrations dilate and the other two constrict (Payandeh et al., 2012).
The mammalian skeletal muscle voltage-gated sodium channel, Nav1.4, homology model built on NavAb, also confirmed the previous finding in that two fenestrations, Domains II-III and IV-I, are narrower than the adjacent two (Domain I-II and III-IV) and their radial size is determined mainly by isoleucine and phenylalanine residues in S5s (Kaczmarski and Corry, 2014). The cryo-EM structure solved for the American cockroach voltage-gated sodium channel (NavPas) contains only one small fenestration formed by the pore-forming segments in Domains III-IV (Shen et al., 2017). Using MD simulations, Tao and Corry (2022) have characterized the fenestrations in the Nav subtypes based on the recent published structures (Tao and Corry, 2022). In contrast to the results reported by Jiang et al. (2020) in NaV1.5, DI-II and DIII-IV fenestrations are dilated, and DII-III and DI-IV fenestrations are constricted (Tao and Corry, 2022).
Our hypothesis is based on the premise that compounds dock in the dilated fenestrations during activation, and the fenestrations are prevented from constricting during inactivation, thereby maintaining a dilated and unblocked central pore, thus maintaining INa conduction. In this study, we used virtual docking and whole-cell patch clamp to assess the efficacy of a new class of compounds, ARumenamide (AR), that dock within the fenestrations, in an effort to target LoF. We screen high profile ARumenamides against E1784K to validate their effect in attenuating LoF.
Materials and methods
Homology modeling
Homology modeling was performed using the Swiss-Model server (swissmodel.expasy.org) (Bordoli et al., 2008). We generated various homology models of Nav1.5 using several templates of NaV crystallized or solved via cryo-EM. Not all were ideal for docking ARumenamide compounds since many of their fenestrations were too constricted or obscure from targeting.
The pre-activated bacterial crystal structure of NavAb (2.7 Å resolution) was used as a template against the Nav1.5 sequence. Modeling was done according to the protocol established by Bordoli et al. (2008). PyMOL-pdb viewer was used for optimization and structure visualization. The cryo-EM structure of rNav1.5 at 3.2–3.5 Å resolution was also used as a template against Nav1.5. Domain II-III fenestration was the largest compared to other fenestrations (Jiang et al., 2020).
Docking
Virtual docking was preferentially performed against the Nav1.5-NavAb model, which has two accessible fenestrations (Payandeh et al., 2012). The Nav1.5 homology model built on NavAb (Nav1.5-NavAb) was docked against the ZINC free database using DOCK Blaster server (blaster.docking.org) (Morris et al., 2009). The highest 20 hits (Table 1), selected based on their binding affinity (kcal/mol) to Nav1.5-NavAb, were then virtually screened against Nav1.5-NavAb using AutoDock4 (Morris et al., 2009). PyMOL-pdb viewer was used for optimization and visualization of the auto-docking results.

TABLE 1. Chemical name and formula
Given the unique features of the Nav1.5-NavAb compared to other homology models, we selected six ARs for functional characterization using patch clamp electrophysiology. We based this decision on each AR compound’s affinity and probability of adhering to the fenestrations (Figure 1). We subsequently docked the same compounds against the rNav1.5 channel (Jiang et al., 2020) as a means of determining the potential binding state of the compounds during fast inactivation. In either channels (Nav1.5-NavAb or rNav1.5), compounds were screened against individual fenestrations: Domain I-II, Domain II-III, Domain III-IV, and Domain IV-I. Finally, the compounds were screened against the complete channel, with the four domains intact.
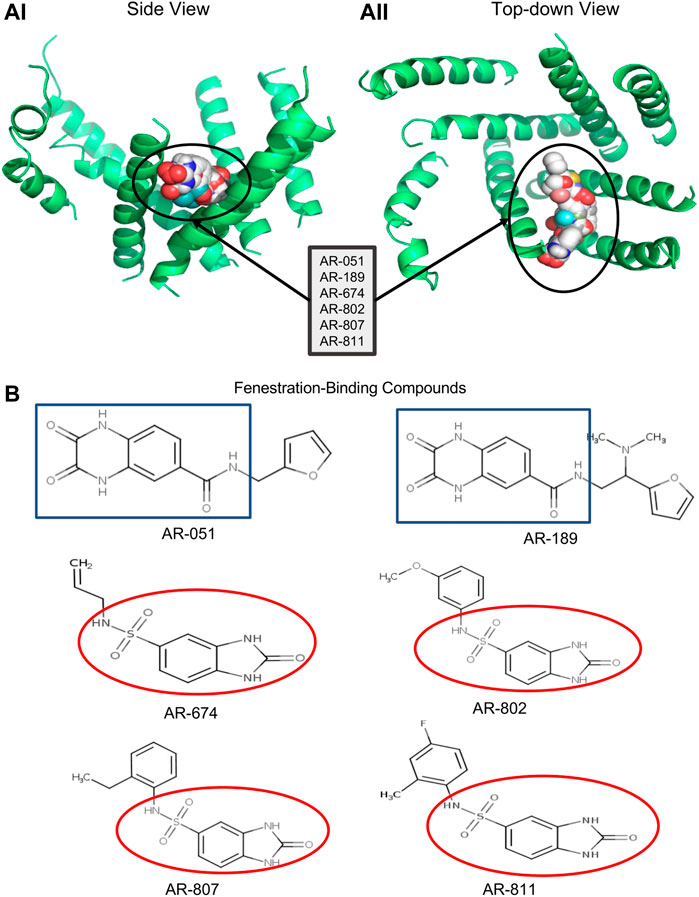
FIGURE 1. Docking ARumenamide. (AI, AII) display the virtual docking results for ARumenamide compounds screened against Nav1.5 -NavAb from the side and top-down views, respectively. The highest binding affinity mode is shown for the six ARumenamide compounds: AR-051; AR-189; AR-674; AR-802; AR-807; and AR-811. All these compounds have a favorable affinity to Domain III-IV fenestration (circled). The compounds are shown as spherical representations. (B) shows the chemical structure of the docked ARumenamide compounds. The common backbone is either squared (blue) if it contains a carboxamide or circled if it contains sulfonamide (red). The functional groups (non-squared and non-circled) are the key moieties and determinants of the ARumenamide’s efficacy.
Compounds
The six chosen ARumenamide compounds (Figure 1) can be generally categorized into carboxamides and sulfonamides: AR-051, AR-189, AR-674, AR-802, AR-807 and AR-811. ARumenamide compounds were purchased from MolPort SIA (Riga, Latvia) in powder form. They were dissolved in 100% DMSO to create a stock solution of 50 mM and diluted with external solution to the desired final concentration.
AR-051 (MolPort-002-662-051/ZINC38767171/STK615542)
AR-189 (MolPort-004-279-189/ZINC40014265/Z26997243)
AR-674 (MolPort-001-911-674/ZINC39699427/STK742976)
AR-802 (MolPort-010-669-802/ZINC64470729/G801-0062)
AR-807 (MolPort-010-669-807/ZINC64470737/G801-0091)
AR-811 (MolPort-010-669-811/ZINC64470745/G801-0104)
Vehicle (0.05% DMSO) controls were often performed to enable correction for any compound-independent decrease of currents. Since NaV1.5 is TTX-resistant, we used lidocaine to confirm the ARumenamide effects observed in E1784K.
Cell culture
HEK293 cells were grown at pH 7.4 in a DMEM (1x) nutrient medium (Life Technologies, NY, United States), supplemented with 10% FBS and maintained in a humidified environment at 37°C with 5% CO2. The α-subunits (WT or E1784K) were transiently co-transfected with the β1 subunit and green fluorescent protein, eGFP (1.50 μg: 0.75 μg: 1.50 μg, respectively). The cDNA mixture was then allowed to incubate with the HEK293 cells before plating on coverslips.
Electrophysiology
Whole-cell patch clamp recordings were performed in extracellular solution containing (mM): 140 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, and 10 HEPES (pH 7.4 adjusted with CsOH). Solutions were titrated with CsOH to pH 7.4. Pipettes were fabricated with a P-1000 puller using borosilicate glass (Sutter Instruments, CA, United States), dipped in dental wax to reduce capacitance, then thermally polished to a resistance of 1.0–1.5 MΩ. Low resistance electrodes were used to minimize series resistance between pipette and intracellular solution resulting in typical access resistances of 3.5 MΩ or less, thereby minimizing voltage measurement error. Pipettes were filled with intracellular solution (in mM): 130 CsF, 10 NaCl, 10 HEPES, and 10 EGTA titrated to pH 7.4.
All recordings were made using an EPC-9 patch-clamp amplifier (HEKA Elektronik, Lambrecht, Germany) digitized at 20 kHz using an ITC-16 interface (HEKA Elektronik, Lambrecht, Germany). Data were acquired and low-pass-filtered (5 kHz) using PatchMaster/FitMaster software (HEKA Elektronik, Lambrecht, Germany) running on an Apple iMac (Apple Computer, Cupertino, CA). Leak subtraction was performed online using a P/4 procedure. Bath solution temperature was controlled using a Peltier device driven by a TC-10 Temperature Controller (Dagan, Minneapolis, MN), and maintained at 37°C. When screening the effects of AR-802 and AR-811 on E1784K, bath solution temperature was maintained at 22°C since it was difficult to record at higher temperatures due to the inherent instability of cells.
The six ARumenamide compounds chosen for biophysical characterization were screened against wild type (WT) and E1784K Nav1.5 by bath perfusion following an initial series of voltage clamp protocols (below).
Voltage clamp protocols
Conductance was measured by a protocol that depolarized the membrane from -100 mV to +80 mV in increments of +5 mV for 19 ms. Prior to the test pulse, channels were allowed to recover from fast inactivation at −130 mV for 197 ms. Channel conductance was calculated from peak INa
where GNa is sodium channel conductance, INa is peak sodium current in response to the command potential V, and ERev is the reversal potential. Calculated values for conductance were fitted with the Boltzmann function:
where G/Gmax is the normalized conductance amplitude, VM is the command potential, z is the apparent valency, e0 is the elementary charge, V1/2 is the midpoint voltage, k is the Boltzmann constant, and T is temperature in °K.
Fast Inactivation Onset: At voltages greater than −50 mV, the fast inactivation τ values were calculated from the mono-exponential fit to the decay of sodium current.
where I is current amplitude, Iss is the plateau amplitude, α is the amplitude at time 0 for time constant τ, and t is time.
Persistent INa current was measured by holding the potential at -110 mV, −90 mV, or −70 mV for 1 s followed by a depolarization to 0 mV for 200 ms and repolarization to −110 mV for 50 ms. The fast inactivation τ values were also calculated from the mono-exponential fit to the decay of sodium current measured in this protocol.
Use-Dependence was used to indirectly measure slow inactivation (SI), which is a physiologically relevant protocol. The protocol includes a series of 110 ms depolarizing pulses to 0 mV followed by a –90 mV recovery pulse for 55 ms at a frequency 6 Hz. With repetitive depolarizations, Nav1.5 channels accumulate into slow inactivation. Normalized current amplitude as a function of time was fit with a double exponential.
where I is current amplitude, Iss is the plateau amplitude, α1 and α2 are the amplitudes at time 0 for time constants τ1 and τ2, and t is time.
Statistical analysis
Statistical analysis was performed using a one-factor completely randomized design ANOVA followed by a post hoc Tukey test. Our statistical model was a full factorial in which all the factors were allowed to interact together. Data are shown as mean ± SEM. Statistical significance was considered at p < 0.05.
Intellectual property
ARumenamides described here are under IP protection (Publication Number WO/2020/161606; International Application No. PCT/IB 2020/050853).
Results
Docking in the fenestrations
Six ARumenamide hits, AR-051, AR-189, AR-674, AR-802, AR-807, and AR-811, are shown in Figure 1 docked, in their highest affinity, to the Domain III-IV fenestration in Nav1.5-NavAb. In addition, there were states in which the compounds interacted with lower affinities with other channel regions, including other fenestrations, the central pore, the pore-forming and voltage-sensing domains. The existence of additional interaction sites is important when interpreting our electrophysiological findings. The ARumenamides displayed the same binding affinity (in kcal/mol) whether they were docked against the individual fenestrations or the entire channel.
None of the six ARumenamide hits screened against rNav1.5 interacted with Domain I–II and Domain II–III fenestrations (Table 2). The compounds, however, had a high affinity for Domain III–IV and Domain IV-I fenestrations. When docked only against Domain III–IV fenestration, AR-674, AR-807, and AR-811 displayed the highest binding affinities compared to the other compounds. All compounds displayed high affinity to Domain IV-I fenestration in rNav1.5 (Table 2). When all four domains were reannealed together to form an intact channel, virtual docking confirmed that most compounds preferentially interacted with Domain IV–I fenestration compared to Domain III–IV fenestration (Table 2).
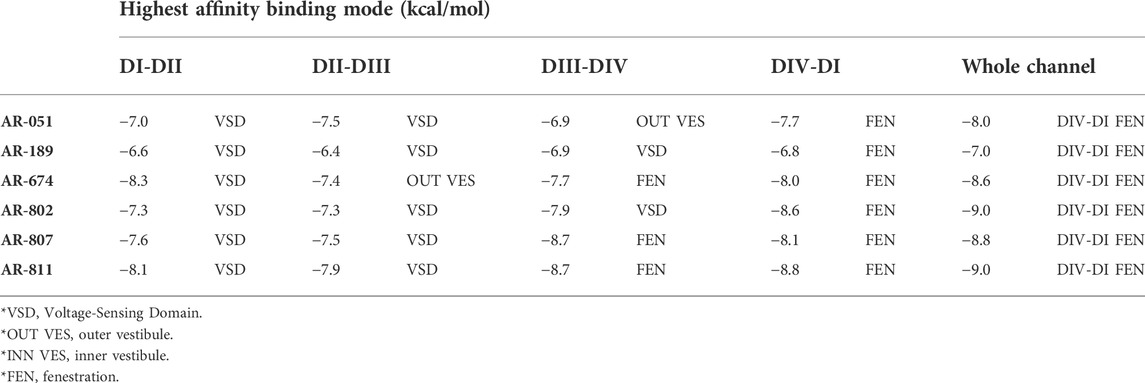
TABLE 2. Binding sites and affinities for compounds docked against rNav1.5
Screening the six ARumenamide compounds on wild-type NaV1.5
All six ARumenamide compounds assessed virtually were functionally screened against wild type Nav1.5 using whole-cell patch clamp. The compounds displayed different affinities to block peak INa at −20 mV (Figure 2AI-FI and Table 3). AR-674 blocked peak INa at −20 mV by 34% (p < 0.05) at 10 µM and progressively more with higher compound concentrations. AR-051, AR-189, AR-802, and AR-807 all significantly blocked peak INa at −20 mV at concentrations of 50 µM and higher. AR-811 was the only compound that did not block peak INa within the concentrations tested (1–100 µM) (Figure 3 and Table 3). A dose-response curve is shown for peak INa block at −20 mV in Figure 3 along with the Hill function parameters in Table 4. The trends observed in these parameters confirm that AR-674 is the most potent in blocking peak INa compared to the rest of the compounds.
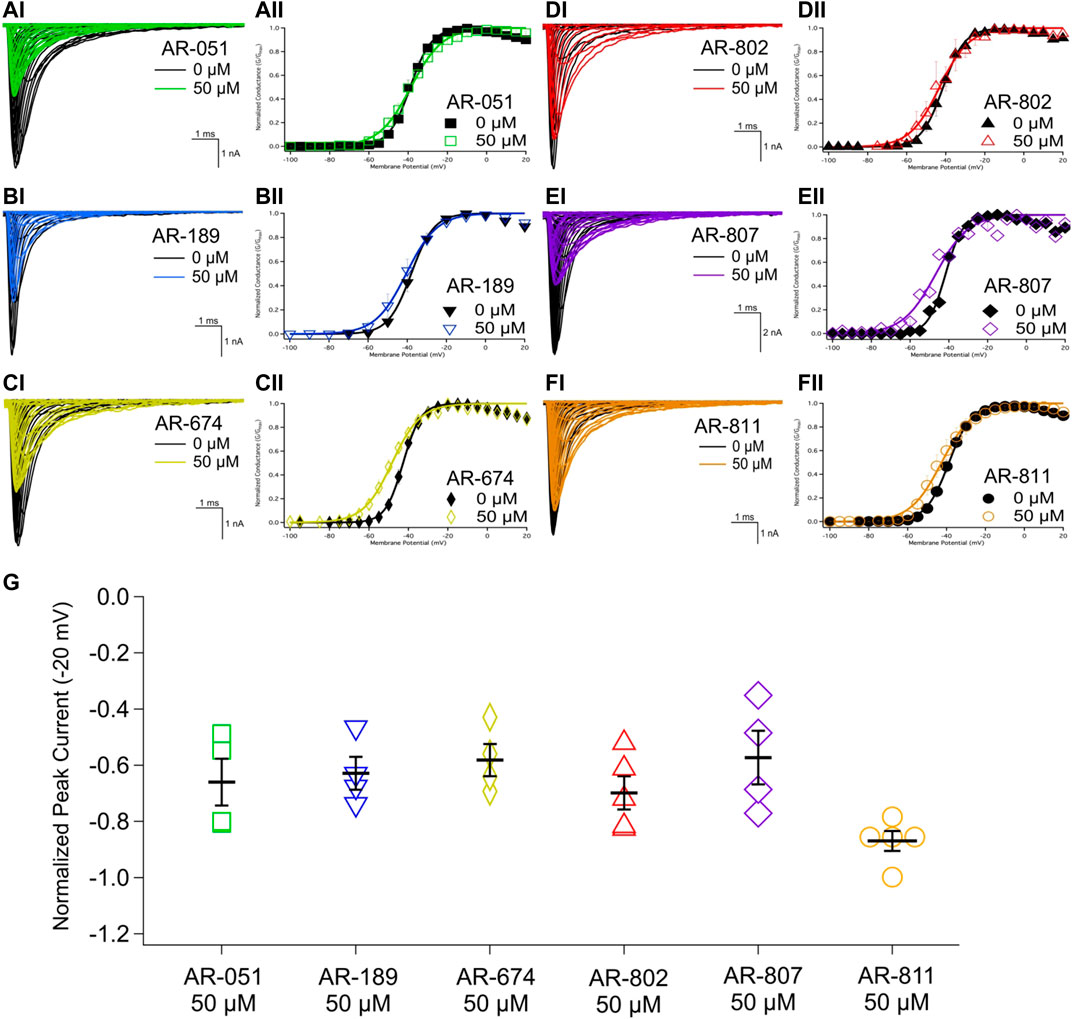
FIGURE 2. Peak INA and conductance. (AI-FI) show representative traces of peak INa in vehicle (black) and compound treatment at 50 µM (varied colors). (AII-FII) shows the voltage-dependence of conductance fit with a Boltzmann curve for vehicle (black) and compound treatment at 50 µM (varied colors). (G) shows a box-plot for compound-induced shifts in normalized peak INa (normalized to control) at −20 mV. Statistical significance is reported in Table 3.
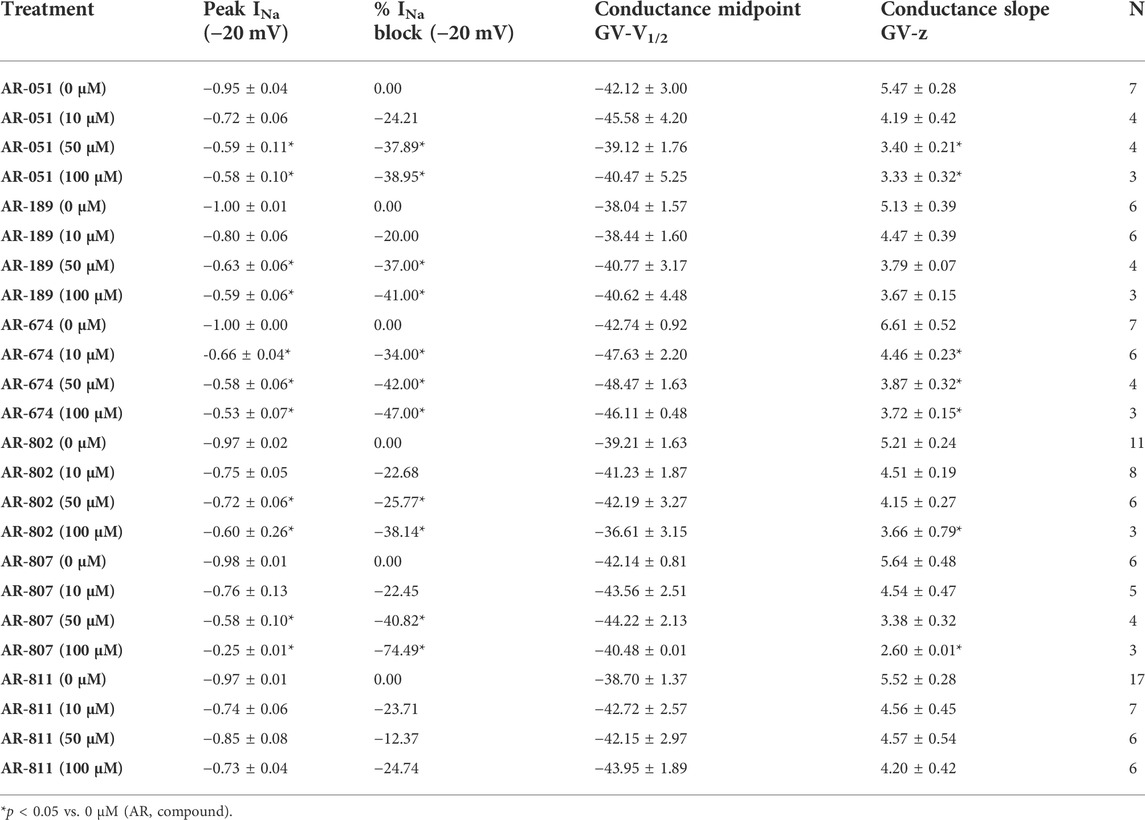
TABLE 3. Peak INA and Conductance.
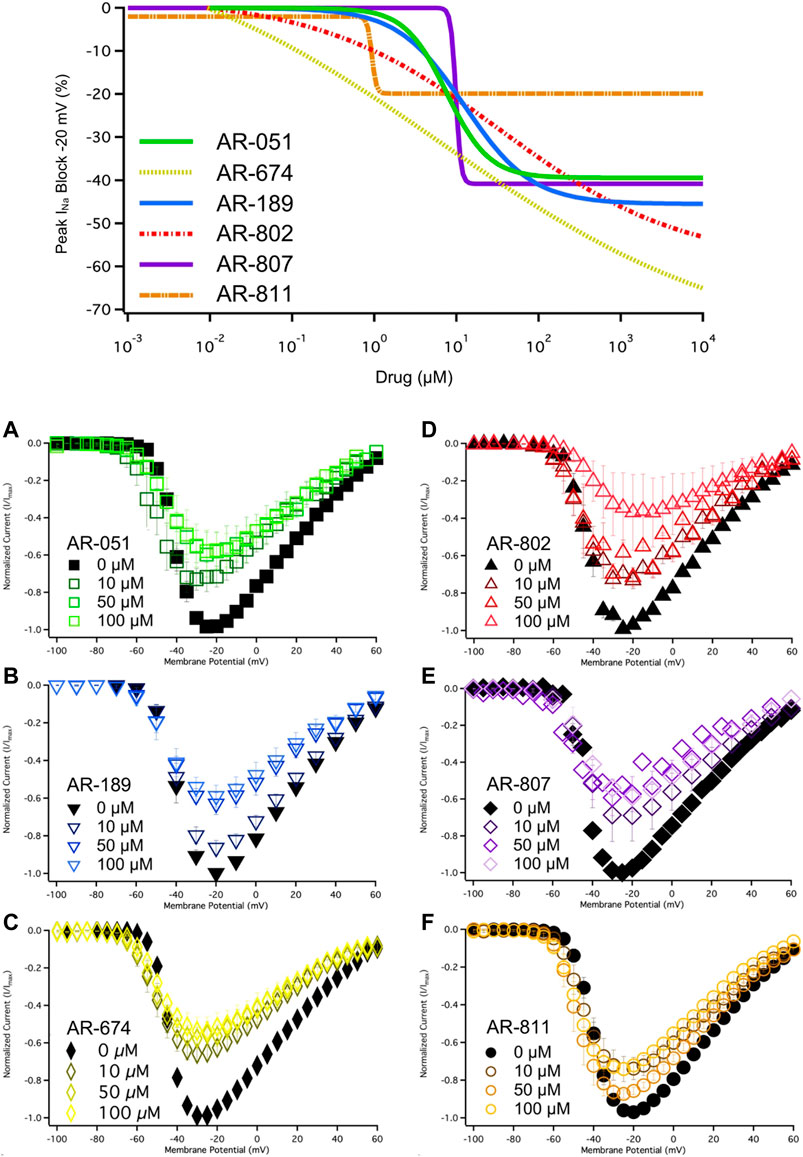
FIGURE 3. Dose-Response and current-voltage relationships. A dose-response curve was fitted to the compound concentrations for peak INa block at –20 mV, generating hill curves shown at the top of the figure. Parameters from the Hill function curve are reported in Table 4. (A-F) show the current-voltage relationship for vehicle (black) and compound treatment between 0–100 μMs (varied colors). Statistical significance is reported in Table 3.
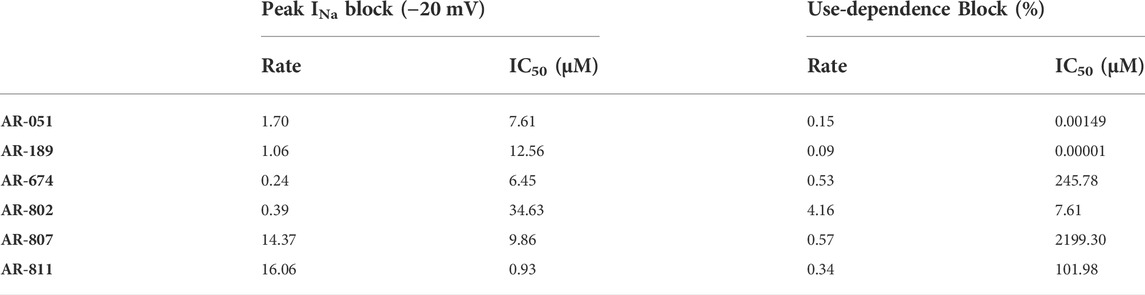
TABLE 4. Hill function parameters for peak INA and use-dependence block.
The voltage-dependence of conductance was assessed using a series of depolarizing test pulses. None of the ARumenamide compounds affected the midpoint potential value of conductance (Figure 2AII-FII and Table 3). AR-674 was highly potent (at 10 µM) in decreasing voltage-sensitivity of conductance as measured from the conductance slope (Table 3). Other compounds like AR-051 significantly decreased (p < 0.05) the conductance slope at 50 µM and higher. While AR-802 and AR-807 significantly decreased (p < 0.05) the conductance slope at 100 µM (Table 3).
Open-state fast inactivation was measured at voltages greater than -50 mV (Figure 4). The compounds did not induce significant shifts in fast inactivation kinetics (p > 0.05) at −50 mV. However, AR-802 and AR-807 slowed the onset of fast inactivation (higher τ - Table 5) at 50 µM and higher. AR-807 was more potent than AR-802 in slowing fast inactivation kinetics at −30 mV, −10 mV, and +10 mV as low as 10 µM (Table 5). AR-051 was less effective in slowing fast inactivation kinetics and had only a subtle effect on τ at −30 mV at 100 µM. Similarly, AR-811 was only effective at 100 µM in decelerating fast inactivation at −30 mV, −10 mV, and +10 mV (Table 5).
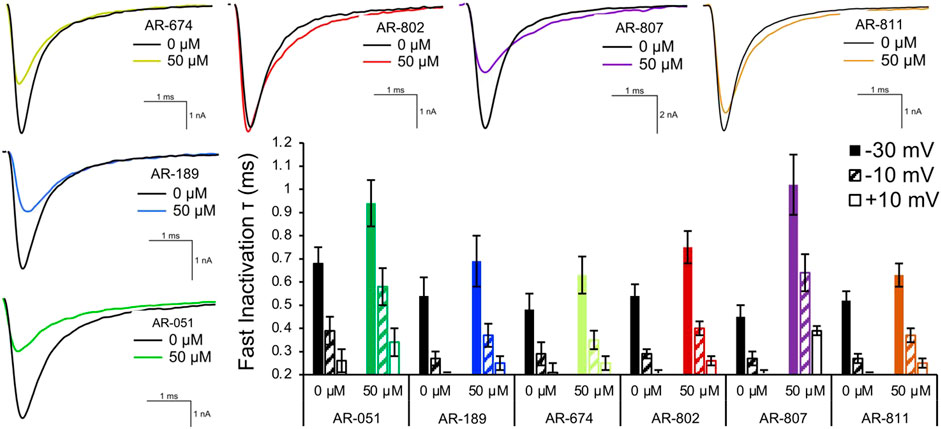
FIGURE 4. Fast inactivation onset. Representative traces are shown for vehicle (black) and compound treatment (varied colors) at –20 mV. The bar graph displays the effects of the compound (50 μM) on the fast inactivation time constant (τ in milliseconds) at –30 mV, –10 mV, and +10 mV. Statistical significance is reported in Table 5.
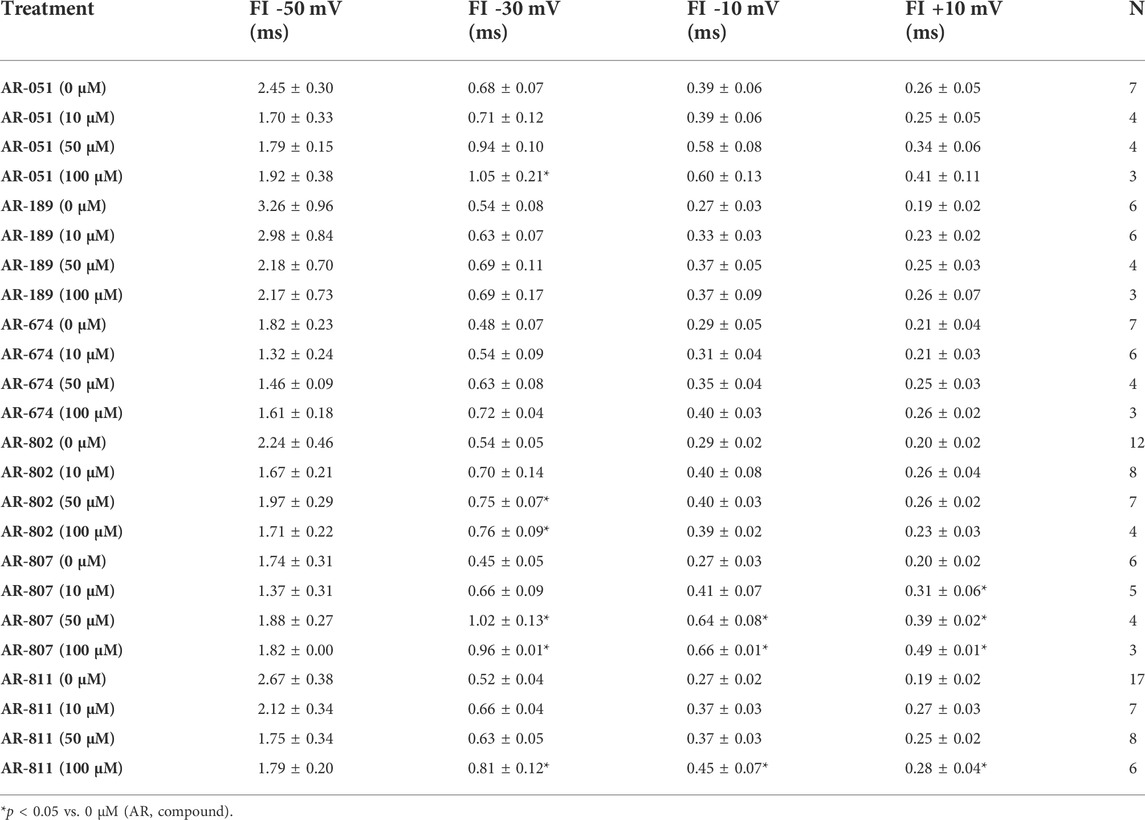
TABLE 5. Fast inactivation onset.
The six ARumenamide compounds displayed similar trends in their effect on use-dependent decrease of current amplitude as they did with peak INa block (Figure 5). AR-674 significantly enhanced (p < 0.05) use-dependence in Nav1.5 at 10 µM and higher concentrations (y0 - Table 6). AR-189 also enhanced use-dependence but only at 50 µM and higher (Table 6). The rest of the compounds, namely AR-051, AR-802, AR-807, and AR-811 did not induce a significant on use-dependence. No compound-induced shifts were observed in t1 and t2, the parameters of the double-exponential fit to use-dependence (Table 6). The dose-response curve in Figure 5 shows the effect of AR-674 to profoundly increase Nav1.5 use-dependence (IC50 values and rates of Hill curves - Table 4).
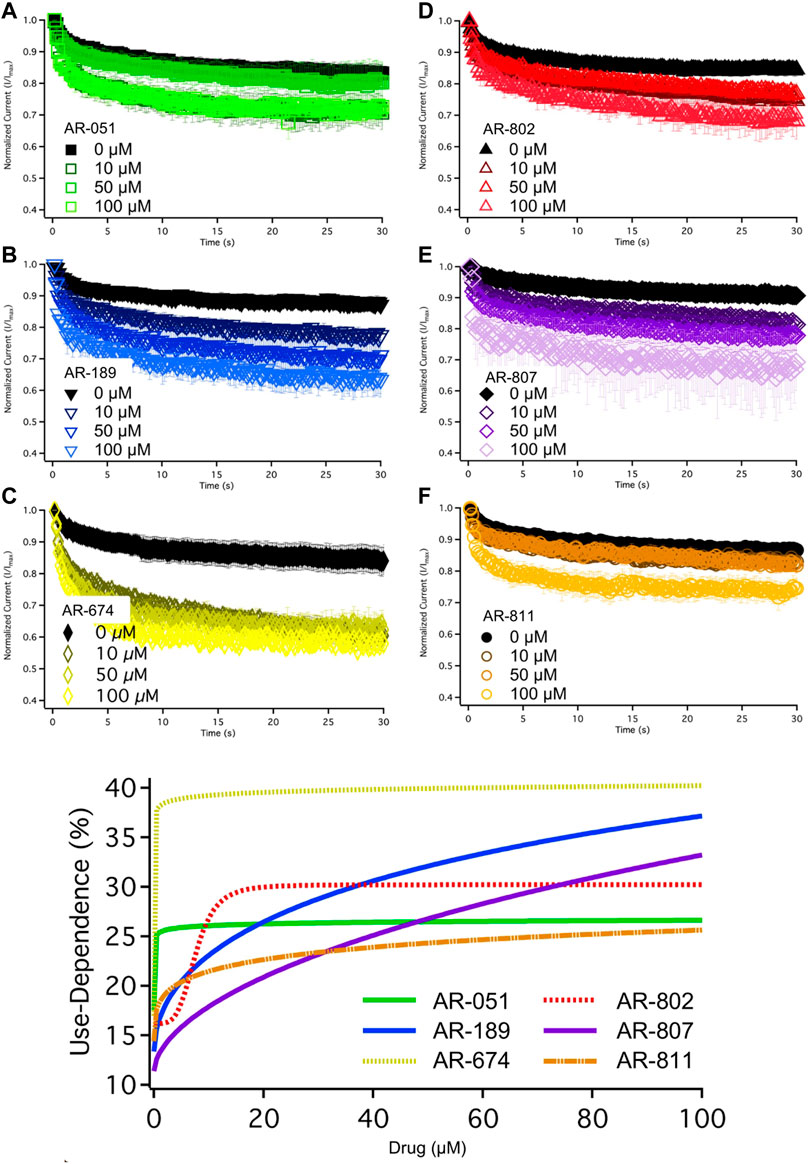
FIGURE 5. Use-Dependence. Nav1.5 use-dependence is shown for vehicle (black) and compound treatment (varied colors). A dose-response curve was fitted to the compound concentrations for use-dependence (%), generating hill curves shown at the bottom of the figure. Statistical significance is reported in Table 6.
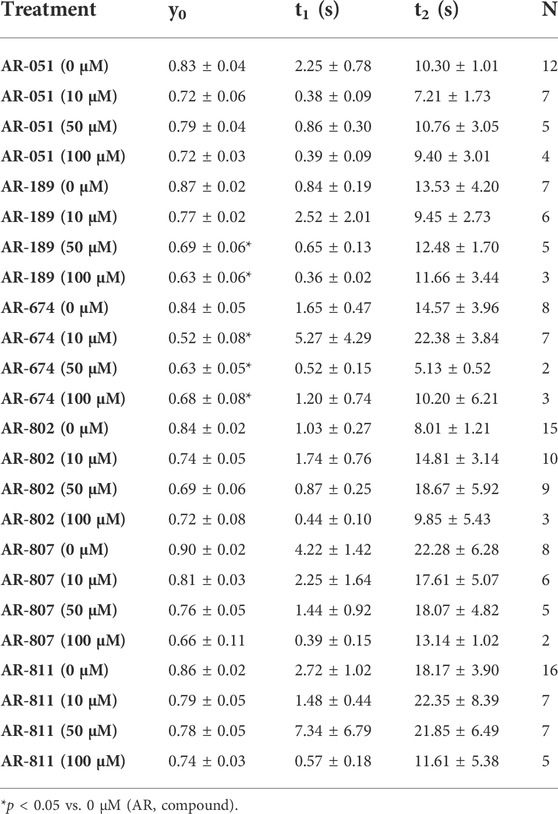
TABLE 6. Use-dependence.
Screening AR-802 and AR-811 on E1784K
To confirm their effects in potentiating INa, we screened representative ARumenamide compounds, namely AR-802 and AR-811, on the Brugada Syndrome Type 1 and Long-QT syndrome Type 3 mutant, E1784K. Both compounds had no significant effect on peak INa measured at −20 mV compared to the vehicle control (Figure 6AI-BIII and Table 7). Neither compound affected the midpoint potential value or the slope of conductance (Table 7). Fast inactivation onset kinetics were assessed at voltages greater than −50 mV recorded with the current-voltage protocol. AR-802 and AR-811 had no significant shifts on fast inactivation onset kinetics compared to vehicle (Table 8). However, AR-802 and AR-811 significantly decelerated fast inactivation onset kinetics compared to vehicle in peak INa measured at 0 mV after E1784K channels were pre-conditioned to −110 mV, −90 mV, and 70 mV for 1 s (Figure 6CI-CII). AR-811 seems to have a larger effect in decelerating fast inactivation kinetics compared to AR-802. AR-802 and AR-811 had no significant effect on peak and persistent INa (Figure 6BI-BIII insets).
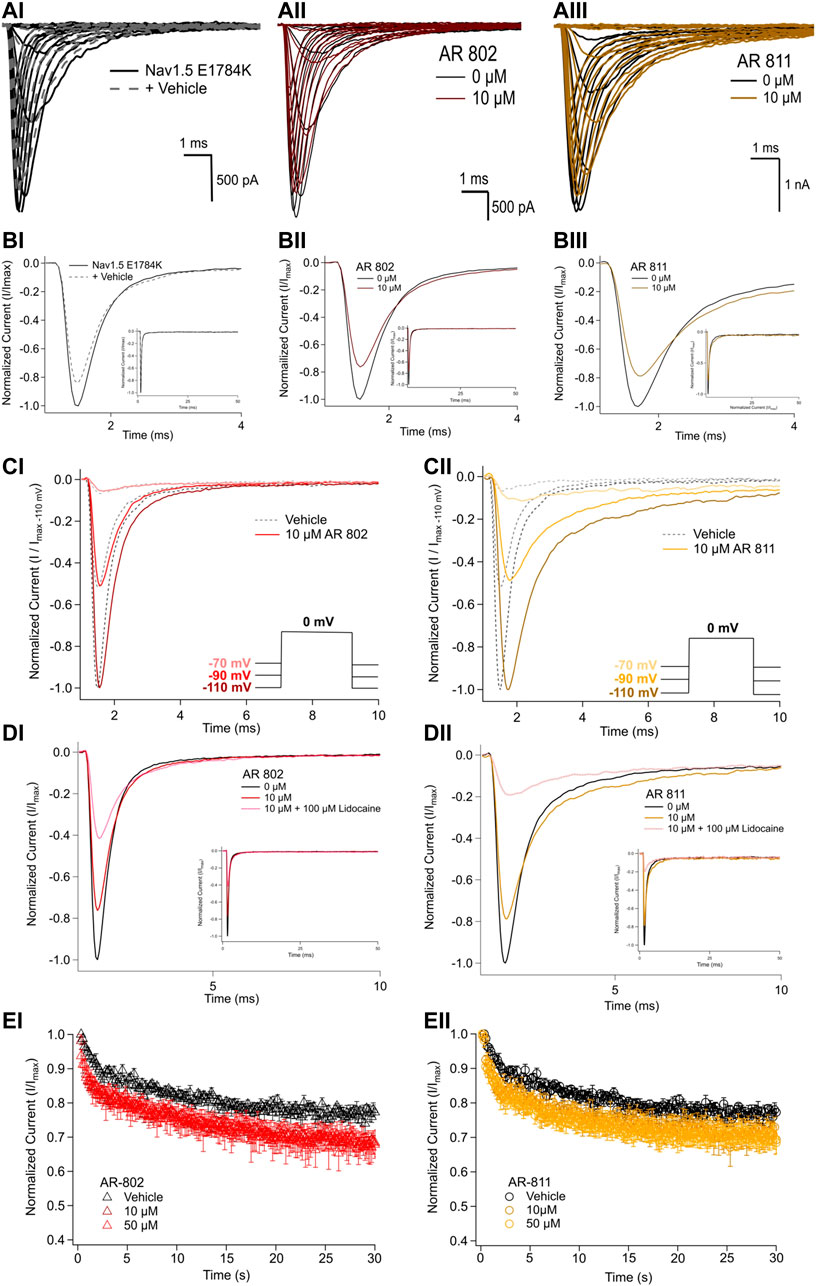
FIGURE 6. Effects of AR-802 and AR-811 on E1784K. (AI-III) show representative traces for peak INa in vehicle (black) and compound treatment (varied colors, 10 µM) in E1784K. (BI-III) show representative traces emphasizing the deceleration in fast inactivation onset kinetics caused by AR-802 and AR-811 compared to vehicle. AR-802 and AR-811 had no significant effect on peak and persistent INa as shown in the insets. (CI-CII) show the deceleration effect of AR-802 and AR-811 on fast inactivation onset when peak INa is measured after pre-conditioning channels at −110 mV, −90 mV, and −70 mV for 1 s as described in the Methods. (DI-DII) show the effects of both compounds with the application of 100 µM lidocaine. (EI-EII) show the effects of AR-802 and AR-811 on use-dependence.
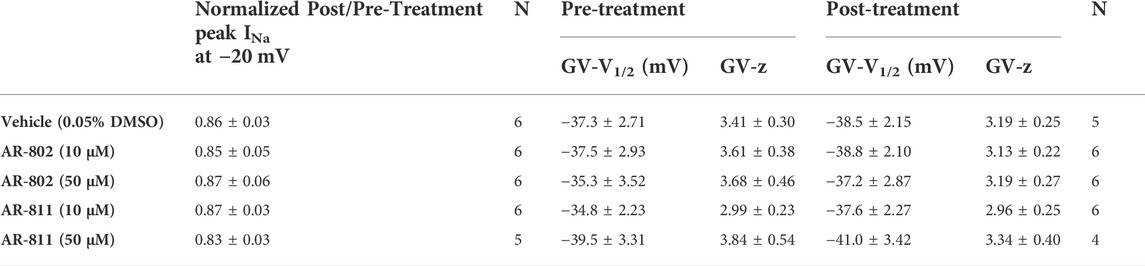
TABLE 7. E1784K peak INA and conductance.
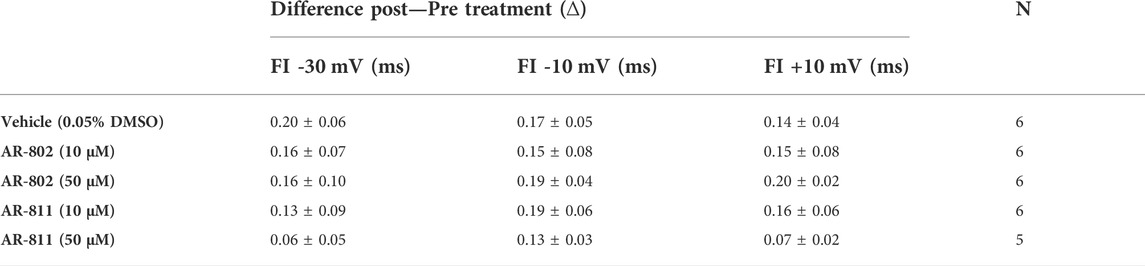
TABLE 8. E1784K FI ONSET from I-V relationship.
The difference between the post and pre-treatment fast inactivation time constant was significantly larger in AR-802 and AR-811 compared to vehicle when measured from a pre-conditioning pulse of −110 mV (Table 9 and Figure 6CI-CII). To confirm this result, we co-administered lidocaine (100 µM) with AR-802 and AR-811. Lidocaine blocked peak INa by ∼60%; however, the AR effects on fast inactivation kinetics persisted even in the presence of lidocaine (Figure 6DI-DII). Although not shown, washout of lidocaine reverts the blockade in peak INa. Both AR-802 and AR-811 had no effect on use-dependence in E1784K.
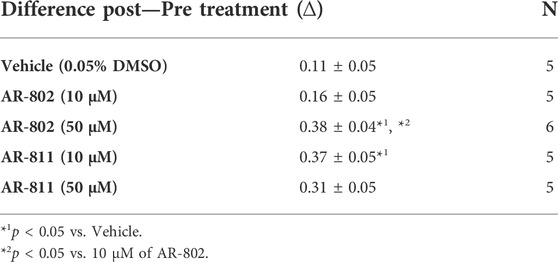
TABLE 9. E1784K FI onset from persistent INA (−110 mV).
Discussion
Here we introduce a novel class of compounds with the potential to correct LoF in Nav1.5 and arrhythmias that result from LoF. Most available antiarrhythmics that target Nav1.5 block the central pore (peak) INa to attenuate channel activity and thereby suppress GoF arrhythmias (Antzelevitch et al., 2007; Holmes et al., 2009; Antzelevitch, 2013; Potet et al., 2015). Most GoF conditions in Nav1.5 result in Long-QT syndromes that often degenerate into more lethal forms of arrhythmias, including torsade-de-pointes or ventricular tachycardia and ventricular fibrillation (Gimrikh et al., 1985; Antzelevitch et al., 1996). The underlying pathophysiology is an instability in channel inactivation; therefore, Nav1.5 becomes active during the refractory period of the cardiac action potential and may trigger early or delayed depolarizations (Ten Tusscher et al., 2004; Flaim et al., 2006; Burashnikov et al., 2008). Suppression of INa is a potent approach for treating these forms of arrhythmias.
Other cardiac conditions, including progressive cardiac conduction disorder (PCCD), sick sinus syndrome, progressive familial heart block, atrial fibrillation, sudden infant death syndrome, and dilated cardiomyopathy, also degenerate into lethal arrhythmias (Savio-Galimberti et al., 2017; Baruteau et al., 2018; Villarreal-Molina et al., 2021). However, they may arise from LoF in Nav1.5 (Amin et al., 2009; 2010b; 2010a). The pathophysiology involves a variety of structural rearrangements in the voltage sensors, pore-forming or voltage-sensing domains, central pore, or the fenestrations (O’Reilly et al., 2012; Payandeh et al., 2012). The effects of LoF on the cardiac action potential are not traditionally treated with Nav1.5 activators but with transient outward potassium current suppressors like quinidine to counteract the repolarizing reserve in light of the suppressed depolarizing current (Brugada et al., 2000; Antzelevitch and Fish, 2006; Wu et al., 2008; Brodie et al., 2018). Toxins like batrachotoxin do have the potential to become Nav1.5 potentiators (Du et al., 2011; Finol-Urdaneta et al., 2019; MacKenzie et al., 2022). However, their harmful effects outweigh their potential benefits.
Although our approach in treating LoF in Nav1.5 is novel, it builds on Hille’s theory (1977) that lipophilic drugs access their binding sites via an alternative route, slowly slithering through the fenestrations into the central pore (Hille, 1977). Structural studies have proven the existence of these fenestrations that Hille correctly predicted (Payandeh et al., 2011, 2012; Kaczmarski and Corry, 2014). The ARumenamide compounds we screened were selected from a cohort of other hits based on their affinity for the fenestrations. Although docking ARumenamides against the Nav1.5 model based on the NavAb template is temperamental, this approach allowed us to shortlist compounds for electrophysiological testing. We tested our hypothesis that ARumenamides block the fenestrations and restrict the motion of Nav1.5 from naturally progressing into inactivation, thereby maintaining INa conduction. Although we are the first to treat LoF by targeting the fenestrations, previous attempts have been made to attenuate hyperexcitability in Nav through “non-blocking” modulation as with riluzole, which indirectly blocks peak INa in Nav1.4 by binding to one of the fenestrations (Földi et al., 2021). Our results suggest that AR-802 and AR-811 may be efficacious in attenuating LoF in E1784K. Future structural modelling using molecular dynamic simulations may help confirm our predictions of how ARs interact with Nav1.5.
Interpretation of results
Our results suggest AR-674 differs from the other AR candidates in its effects on Nav1.5. Although virtual docking suggested its high binding affinity for the fenestrations, AR-674 blocks INa and suppresses Nav1.5 availability with time via use-dependence. Independent of any particular theory, the biophysical shifts differentiating AR-674 from the other compounds, or at least its counter sulfonamides that share the same backbone structure, seems to be that its functional group is aliphatic and not aromatic. We predict that an inverse relationship exists between AR’s aromaticity and INa block. The aliphaticity of AR-674 appears to lower the affinity to bind within the fenestrations. Lower affinity within the fenestrations would be predicted to allow greater access of AR-674 into the central pore, which may help explain the 34% reduction in peak INa at -20 mV and the significant use-dependence at concentrations as low as 10 µM. AR-674 may also interact with the voltage sensors during activation, suppressing their movement and accounting for the decreased slope in conductance.
Aromatic functional groups in ARs seem to increase binding affinity within Nav1.5 fenestrations. AR-802, AR-807, and AR-811 have aromatic benzene in their side chains compared to the aliphatic side chain of AR-674. Compound binding within the fenestrations may impede changes in fenestration size during fast inactivation. These three compounds slow the onset of fast inactivation at different concentrations. The fluorine atom attached to the benzene ring of AR-811 seems to mediate the potentiator effects in Nav1.5; however, this conclusion is based only on comparing AR-811 effects with other relatable sulfonamides. The presence of the fluorine atom may account for the high resistance to peak INa block and the unaltered use-dependence. These results suggest that AR-811 mediates its apparent “potentiating” effects (compared to AR-674) at elevated depolarization rates through an open/inactivated compound-block mechanism. The observed deceleration in fast inactivation kinetics caused by AR-802 and AR-811 is seen only after E1784K channels are pre-conditioned at −110 mV, −90 mV, and −70 mV for 1 s. With prolonged depolarizing pre-conditioning pulses, it seems reasonable to hypothesize that AR-811 mediates its “potentiating” effects by targeting fast and potentially slow inactivation. The benefit of attenuating LoF in E1784K via the aromatic compounds, AR-802 and AR-811, in particular, is that their speculated adherence to the fenestrations reduces their likelihood of blocking peak or persistent INa, a result that we clearly observed.
Compared to the carboxamides, the sulfonamides, AR-051 and AR-189, also inform us about the structural features required to generate Nav1.5 potentiators. Although not statistically significant, AR-189 seems to be slightly better in not suppressing INa as much at 10 µM compared to AR-051. The branched alkyl side chain adjacent to the furan may increase the hydrophobicity of AR-189, increasing its binding affinity within the fenestrations.
Study limitations
Virtual docking and the Nav1.5 homology models have limitations. Not all the fenestrations were targetable, especially in rNav1.5. Crystallography and recent cryo-EM structures do not fully depict the fenestrations in their various voltage-dependent conformations (Payandeh et al., 2011, 2012; Shen et al., 2017; Yan et al., 2017; Jiang et al., 2020). Thus, our homology models and the auto-docking experiments were limited by the constraints of these structures. Due to the existent varieties in structural data, it is difficult to determine the exact state dependence of fenestration size. We cannot completely conclude whether the AR compounds have preference for a particular fenestration based on the virtual docking alone; however, our modeling clearly showed an affinity of AR compounds screened to the Domain III-IV fenestration in Nav1.5-NavAb and Domain VI-I fenestration in rNav1.5. Molecular dynamics simulation could be used to verify AR interaction with the fenestrations in Nav1.5. Additional screening of AR compounds against other Brugada variants of Nav1.5 will help confirm our initial experiments that suggest ARs may have therapeutic potential to treat LoF.
Treating LoF in Nav1.5 is difficult, given that most compounds will occlude the pore at a certain concentration. Despite the recent advances in designing pharmaceuticals that preferentially inhibit persistent INa and attenuate GoF, drugs like GS-967, eleclazine, ranolazine, and mexiletine analogs block peak INa at varying concentrations (Potet et al., 2020; Johnson et al., 2021). To overcome this challenge, we plan to optimize structures like AR-811 that block peak INa less and decelerate fast inactivation onset. Enhancing the aromaticity of the ARumenamides’ functional groups is key to enhancing their anti-LoF effects.
Significance
Our results suggest that ARumenamides may have therapeutic potential to treat LoF channelopathies like Brugada Syndrome by lodging within the fenestrations. Although other binding sites are possible, our docking results suggest that these compounds preferentially bind to the fenestrations. ARs with aromatic functional groups, as opposed to aliphatic ones, maintained Nav1.5 availability since the bulkier aromatic side groups have a higher affinity for binding within the hydrophobic milieu of the fenestrations. These compounds seem to remain within the fenestrations rather than slithering into the central pore to occlude INa, as opposed to most therapeutic agents targeting Nav1.5. AR-802 and AR-811 (two representative compounds that preferentially delay fast inactivation) attenuate LoF in the most common Brugada Syndrome Type 1 and Long-QT Syndrome Type 3 mutant, E1784K, by decelerating fast inactivation onset kinetics. Neither AR-802 nor AR-811 block peak INa or enhance persistent INa in E1784K, two highly sensitive parameters that must be controlled to avoid exacerbation of phenotype. Successfully optimizing AR compounds and testing them in LoF models related to cardiac disorders may ultimately lead to lowering the economic burden and health costs associated with untreated arrhythmias.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
MA discovered, designed, and developed the ARumenamides. He also collected, assembled, analyzed, and interpreted the data, designed the experiments, and drafted the manuscript. DP collected, assembled, and analyzed the data. PR critically revised the manuscript for important intellectual content. All authors approved the final version of the manuscript and qualify for authorship.
Funding
This study was supported by the Alexander Graham Bell Canada Graduate Scholarships-Doctoral Program from the Natural Science and Engineering Research Council of Canada to MA.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abdelsayed, M., Ruprai, M., and Ruben, P. C. (2018). The efficacy of Ranolazine on E1784K is altered by temperature and calcium. Sci. Rep. 8, 3643. doi:10.1038/s41598-018-22033-1
Amin, A. S., de Groot, E. A. A., Ruijter, J. M., Wilde, A. A. M., and Tan, H. L. (2009). Exercise-induced ECG changes in Brugada syndrome. Circ. Arrhythm. Electrophysiol. 2, 531–539. doi:10.1161/CIRCEP.109.862441
Amin, A. S., Klemens, C. A., Meregalli, P. G., Asghari-Roodsari, A., de Bakker, J. M. T., January, C. T., et al. (2010a). Fever-triggered ventricular arrhythmias in Brugada syndrome and type 2 long-QT syndrome. Neth. Heart J. 18, 165–169. doi:10.1007/BF03091755
Amin, A. S., Tan, H. L., and Wilde, A. A. M. (2010b). Cardiac ion channels in health and disease. Heart rhythm. 7, 117–126. doi:10.1016/j.hrthm.2009.08.005
An, R. H., Bangalore, R., Rosero, S. Z., and Kass, R. S. (1996). Lidocaine block of LQT-3 mutant human Na+ channels. Circ. Res. 79, 103–108. doi:10.1161/01.res.79.1.103
Antzelevitch, C., Belardinelli, L., Zygmunt, A. C., Burashnikov, A., Di Diego, J. M., Fish, J. M., et al. (2004). Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation 110, 904–910. doi:10.1161/01.CIR.0000139333.83620.5D
Antzelevitch, C., Brugada, P., Brugada, J., and Brugada, R. (2005). Brugada syndrome: From cell to bedside. Curr. Probl. Cardiol. 30, 9–54. doi:10.1016/j.cpcardiol.2004.04.005
Antzelevitch, C., and Fish, J. M. (2006). Therapy for the Brugada syndrome. Handb. Exp. Pharmacol. 171, 305–330. doi:10.1007/3-540-29715-4_12
Antzelevitch, C. (2013). J wave syndromes: Molecular and cellular mechanisms. J. Electrocardiol. 46, 510–518. doi:10.1016/j.jelectrocard.2013.08.006
Antzelevitch, C., Pollevick, G. D., Cordeiro, J. M., Casis, O., Sanguinetti, M. C., Aizawa, Y., et al. (2007). Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 115, 442–449. doi:10.1161/CIRCULATIONAHA.106.668392
Antzelevitch, C., Sun, Z.-Q., Zhang, Z.-Q., and Yan, G.-X. (1996). Cellular and ionic mechanisms underlying erythromycin-induced long QT intervals and torsade de pointes. J. Am. Coll. Cardiol. 28, 1836–1848. doi:10.1016/S0735-1097(96)00377-4
Ashpole, N. M., Herren, A. W., Ginsburg, K. S., Brogan, J. D., Johnson, D. E., Cummins, T. R., et al. (2012). Ca 2+/Calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel Na V 1.5 gating by multiple phosphorylation sites. J. Biol. Chem. 287, 19856–19869. doi:10.1074/jbc.M111.322537
Baartscheer, A., Schumacher, C. A., Belterman, C. N. W., Coronel, R., and Fiolet, J. W. T. (2003). SR calcium handling and calcium after-transients in a rabbit model of heart failure. Cardiovasc. Res. 58, 99–108. doi:10.1016/s0008-6363(02)00854-4
Balser, J. R., Nuss, H. B., Orias, D. W., Johns, D. C., Marban, E., Tomaselli, G. F., et al. (1996a). Local anesthetics as effectors of allosteric gating. Lidocaine effects on inactivation-deficient rat skeletal muscle Na channels. J. Clin. Invest. 98, 2874–2886. doi:10.1172/JCI119116
Balser, J. R., Nuss, H. B., Romashko, D. N., Marban, E., and Tomaselli, G. F. (1996b). Functional consequences of lidocaine binding to slow-inactivated sodium channels. J. Gen. Physiol. 107, 643–658. doi:10.1085/jgp.107.5.643
Bankston, J. R., Sampson, K. J., Kateriya, S., Glaaser, I. W., Malito, D. L., Chung, W. K., et al. (2007). A novel LQT-3 mutation disrupts an inactivation gate complex with distinct rate-dependent phenotypic consequences. Channels (Austin) 1, 273–280. doi:10.4161/chan.4956
Baruteau, A.-E., Kyndt, F., Behr, E. R., Vink, A. S., Lachaud, M., Joong, A., et al. (2018). SCN5A mutations in 442 neonates and children: Genotype-phenotype correlation and identification of higher-risk subgroups. Eur. Heart J. 39, 2879–2887. doi:10.1093/eurheartj/ehy412
Beaufort-Krol, G. C. M., van den Berg, M. P., Wilde, A. A. M., van Tintelen, J. P., Viersma, J. W., Bezzina, C. R., et al. (2005). Developmental aspects of long QT syndrome type 3 and Brugada syndrome on the basis of a single SCN5A mutation in childhood. J. Am. Coll. Cardiol. 46, 331–337. doi:10.1016/j.jacc.2005.03.066
Biswas, A., Raza, A., Das, S., Kapoor, M., Jayarajan, R., Verma, A., et al. (2019). Loss of function mutation in the P2X7, a ligand-gated ion channel gene associated with hypertrophic cardiomyopathy. Purinergic Signal. 15, 205–210. doi:10.1007/s11302-019-09660-7
Boiteux, C., Vorobyov, I., French, R. J., French, C., Yarov-Yarovoy, V., and Allen, T. W. (2014). Local anesthetic and antiepileptic drug access and binding to a bacterial voltage-gated sodium channel. Proc. Natl. Acad. Sci. U. S. A. 111, 13057–13062. doi:10.1073/pnas.1408710111
Bordoli, L., Kiefer, F., Arnold, K., Benkert, P., Battey, J., and Schwede, T. (2008). Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 4, 1–13. doi:10.1038/nprot.2008.197
Brodie, O. T., Michowitz, Y., and Belhassen, B. (2018). Pharmacological therapy in Brugada syndrome. Arrhythmia Electrophysiol. Rev. 7, 135. doi:10.15420/aer.2018.21.2
Brugada, R., Brugada, J., Antzelevitch, C., Kirsch, G. E., Potenza, D., Towbin, J. A., et al. (2000). Sodium channel blockers identify risk for sudden death in patients with ST-segment elevation and right bundle branch block but structurally normal hearts. Circulation 101, 510–515. doi:10.1161/01.cir.101.5.510
Burashnikov, A., Shimizu, W., and Antzelevitch, C. (2008). Fever accentuates transmural dispersion of repolarization and facilitates development of early afterdepolarizations and torsade de pointes under long-QT Conditions. Circ. Arrhythm. Electrophysiol. 1, 202–208. doi:10.1161/CIRCEP.107.691931
Du, Y., Garden, D. P., Wang, L., Zhorov, B. S., and Dong, K. (2011). Identification of new batrachotoxin-sensing residues in segment IIIS6 of the sodium channel. J. Biol. Chem. 286, 13151–13160. doi:10.1074/jbc.M110.208496
Dumaine, R., Wang, Q., Keating, M. T., Hartmann, H. A., Schwartz, P. J., Brown, A. M., et al. (1996). Multiple mechanisms of Na+ channel-linked long-QT syndrome. Circ. Res. 78, 916–924. doi:10.1161/01.res.78.5.916
Finol-Urdaneta, R. K., McArthur, J. R., Goldschen-Ohm, M. P., Gaudet, R., Tikhonov, D. B., Zhorov, B. S., et al. (2019). Batrachotoxin acts as a stent to hold open homotetrameric prokaryotic voltage-gated sodium channels. J. Gen. Physiol. 151, 186–199. doi:10.1085/jgp.201812278
Flaim, S. N., Giles, W. R., and McCulloch, A. D. (2006). Contributions of sustained INa and IKv43 to transmural heterogeneity of early repolarization and arrhythmogenesis in canine left ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 291, H2617–H2629. doi:10.1152/ajpheart.00350.2006
Földi, M. C., Pesti, K., Zboray, K., Toth, A. V., Hegedűs, T., Málnási-Csizmadia, A., et al. (2021). The mechanism of non‐blocking inhibition of sodium channels revealed by conformation‐selective photolabeling. Br. J. Pharmacol. 178, 1200–1217. doi:10.1111/bph.15365
Gamal El-Din, T. M., Lenaeus, M. J., Zheng, N., and Catterall, W. A. (2018). Fenestrations control resting-state block of a voltage-gated sodium channel. Proc. Natl. Acad. Sci. U. S. A. 115, 13111–13116. doi:10.1073/pnas.1814928115
Gimrikh, E. O., Popov, S. V., and Pekarskiĭ, V. V. (1985). Electrocardiostimulation in the treatment of recurrent ventricular fibrillation in a patient with long QT syndrome on the ECG. Ter. Arkh. 57, 45–46.
Gui, J., Wang, T., Jones, R. P. O., Trump, D., Zimmer, T., and Lei, M. (2010). Multiple loss-of-function mechanisms contribute to SCN5A-related familial sick sinus syndrome. PLoS ONE 5, e10985. doi:10.1371/journal.pone.0010985
Herren, A. W., Bers, D. M., and Grandi, E. (2013). Post-translational modifications of the cardiac Na channel: Contribution of CaMKII-dependent phosphorylation to acquired arrhythmias. Am. J. Physiol. Heart Circ. Physiol. 305, H431–H445. doi:10.1152/ajpheart.00306.2013
Hille, B. (1977). Local anesthetics: Hydrophilic and hydrophobic pathways for the drug-receptor reaction. J. Gen. Physiol. 69, 497–515. doi:10.1085/jgp.69.4.497
Holmes, M. V., Shah, T., Vickery, C., Smeeth, L., Hingorani, A. D., and Casas, J. P. (2009). Fulfilling the promise of personalized medicine? Systematic review and field synopsis of pharmacogenetic studies. PLoS ONE 4, e7960. doi:10.1371/journal.pone.0007960
Jiang, D., Shi, H., Tonggu, L., Gamal El-Din, T. M., Lenaeus, M. J., Zhao, Y., et al. (2020). Structure of the cardiac sodium channel. Cell 180, 122–134. doi:10.1016/j.cell.2019.11.041
Johnson, M., Gomez-Galeno, J., Ryan, D., Okolotowicz, K., McKeithan, W. L., Sampson, K. J., et al. (2021). Human iPSC-derived cardiomyocytes and pyridyl-phenyl mexiletine analogs. Bioorg. Med. Chem. Lett. 46, 128162. doi:10.1016/j.bmcl.2021.128162
Kaczmarski, J. A., and Corry, B. (2014). Investigating the size and dynamics of voltage-gated sodium channel fenestrations: A molecular dynamics study. Channels 8, 264–277. doi:10.4161/chan.28136
Kaufman, E. S. (2009). Mechanisms and clinical management of inherited channelopathies: Long QT syndrome, Brugada syndrome, catecholaminergic polymorphic ventricular tachycardia, and short QT syndrome. Heart rhythm. 6, S51–S55. doi:10.1016/j.hrthm.2009.02.009
Lenaeus, M. J., Gamal El-Din, T. M., Ing, C., Ramanadane, K., Pomès, R., Zheng, N., et al. (2017). Structures of closed and open states of a voltage-gated sodium channel. Proc. Natl. Acad. Sci. U. S. A. 114, E3051–E3060. doi:10.1073/pnas.1700761114
MacKenzie, T. M. G., Abderemane-Ali, F., Garrison, C. E., Minor, D. L., and Bois, J. D. (2022). Differential effects of modified batrachotoxins on voltage-gated sodium channel fast and slow inactivation. Cell Chem. Biol. 29, 615–624. doi:10.1016/j.chembiol.2021.12.003
Makita, N., Behr, E., Shimizu, W., Horie, M., Sunami, A., Crotti, L., et al. (2008). The E1784K mutation in SCN5A is associated with mixed clinical phenotype of type 3 long QT syndrome. J. Clin. Invest. 118, 2219–2229. doi:10.1172/JCI34057
Martin, L. J., and Corry, B. (2014). Locating the route of entry and binding sites of benzocaine and phenytoin in a bacterial voltage gated sodium channel. PLoS Comput. Biol. 10, e1003688. doi:10.1371/journal.pcbi.1003688
Montini, G., Booker, J., Sula, A., and Wallace, B. A. (2018). Comparisons of voltage-gated sodium channel structures with open and closed gates and implications for state-dependent drug design. Biochem. Soc. Trans. 46, 1567–1575. doi:10.1042/BST20180295
Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., et al. (2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791. doi:10.1002/jcc.21256
Nguyen, P. T., DeMarco, K. R., Vorobyov, I., Clancy, C. E., and Yarov-Yarovoy, V. (2019). Structural basis for antiarrhythmic drug interactions with the human cardiac sodium channel. Proc. Natl. Acad. Sci. U. S. A. 116, 2945–2954. doi:10.1073/pnas.1817446116
O’Reilly, A. O., Eberhardt, E., Weidner, C., Alzheimer, C., Wallace, B. A., and Lampert, A. (2012). Bisphenol A binds to the local anesthetic receptor site to block the human cardiac sodium channel. PLoS ONE 7, e41667. doi:10.1371/journal.pone.0041667
Payandeh, J., Gamal El-Din, T. M., Scheuer, T., Zheng, N., and Catterall, W. A. (2012). Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 486, 135–139. doi:10.1038/nature11077
Payandeh, J., Scheuer, T., Zheng, N., and Catterall, W. A. (2011). The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358. doi:10.1038/nature10238
Peters, C. H., Watkins, A. R., Poirier, O. L., and Ruben, P. C. (2020). E1784K, the most common Brugada syndrome and long-QT syndrome type 3 mutant, disrupts sodium channel inactivation through two separate mechanisms. J. Gen. Physiol. 152, e202012595. doi:10.1085/jgp.202012595
Potet, F., Beckermann, T. M., Kunic, J. D., and George, A. L. (2015). Intracellular calcium attenuates late current conducted by mutant human cardiac sodium channels. Circ. Arrhythm. Electrophysiol. 8, 933–941. doi:10.1161/CIRCEP.115.002760
Potet, F., Egecioglu, D. E., Burridge, P. W., and George, A. L. (2020). GS-967 and eleclazine block sodium channels in human induced pluripotent stem cell–derived cardiomyocytes. Mol. Pharmacol. 98, 540–547. doi:10.1124/molpharm.120.000048
Savio-Galimberti, E., Argenziano, M., and Antzelevitch, C. (2017). “Cardiac arrhythmias related to sodium channel dysfunction,” in Voltage-gated sodium channels: Structure, Function and channelopathies handbook of experimental Pharmacology. Editor M. Chahine (Cham: Springer International Publishing), 331–354. doi:10.1007/164_2017_43
Shen, H., Zhou, Q., Pan, X., Li, Z., Wu, J., and Yan, N. (2017). Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science 355, eaal4326. doi:10.1126/science.aal4326
Skinner, J. R., Winbo, A., Abrams, D., Vohra, J., and Wilde, A. A. (2019). Channelopathies that lead to sudden cardiac death: Clinical and genetic aspects. Heart Lung Circ. 28, 22–30. doi:10.1016/j.hlc.2018.09.007
Sokolov, S., Peters, C. H., Rajamani, S., and Ruben, P. C. (2013). Proton-dependent inhibition of the cardiac sodium channel Nav1.5 by ranolazine. Front. Pharmacol. 4, 78. doi:10.3389/fphar.2013.00078
Song, L. S., Wang, S. Q., Xiao, R. P., Spurgeon, H., Lakatta, E. G., and Cheng, H. (2001). beta-Adrenergic stimulation synchronizes intracellular Ca(2+) release during excitation-contraction coupling in cardiac myocytes. Circ. Res. 88, 794–801. doi:10.1161/hh0801.090461
Tao, E., and Corry, B. (2022). Characterizing fenestration size in sodium channel subtypes and their accessibility to inhibitors. Biophys. J. 121, 193–206. doi:10.1016/j.bpj.2021.12.025
Ten Tusscher, K. H. W. J., Noble, D., Noble, P. J., and Panfilov, A. V. (2004). A model for human ventricular tissue. Am. J. Physiol. Heart Circ. Physiol. 286, H1573–H1589. doi:10.1152/ajpheart.00794.2003
Villarreal-Molina, T., García-Ordóñez, G. P., Reyes-Quintero, Á. E., Domínguez-Pérez, M., Jacobo-Albavera, L., Nava, S., et al. (2021). Clinical spectrum of SCN5A channelopathy in children with primary electrical disease and structurally normal hearts. Genes 13, 16. doi:10.3390/genes13010016
Wagner, S., Dybkova, N., Rasenack, E. C. L., Jacobshagen, C., Fabritz, L., Kirchhof, P., et al. (2006). Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J. Clin. Invest. 116, 3127–3138. doi:10.1172/JCI26620
Wagner, S., Ruff, H. M., Weber, S. L., Bellmann, S., Sowa, T., Schulte, T., et al. (2011). Reactive oxygen species-activated Ca/calmodulin kinase IIδ is required for late I(Na) augmentation leading to cellular Na and Ca overload. Circ. Res. 108, 555–565. doi:10.1161/CIRCRESAHA.110.221911
Wang, G. K., and Wang, S.-Y. (2014). Block of human cardiac sodium channels by lacosamide: Evidence for slow drug binding along the activation pathway. Mol. Pharmacol. 85, 692–702. doi:10.1124/mol.113.091173
Wu, L., Guo, D., Li, H., Hackett, J., Yan, G.-X., Jiao, Z., et al. (2008). Role of late sodium current in modulating the proarrhythmic and antiarrhythmic effects of quinidine. Heart rhythm. 5, 1726–1734. doi:10.1016/j.hrthm.2008.09.008
Keywords: ARumenamide, loss-of-function, Brugada syndrome, Nav1.5, fenestrations, aromaticity, aliphaticity
Citation: Abdelsayed M, Page D and Ruben PC (2022) ARumenamides: A novel class of potential antiarrhythmic compounds. Front. Pharmacol. 13:976903. doi: 10.3389/fphar.2022.976903
Received: 24 June 2022; Accepted: 02 September 2022;
Published: 28 September 2022.
Edited by:
Jean-François Desaphy, University of Bari Aldo Moro, ItalyReviewed by:
Elisabetta Cerbai, University of Florence, ItalyBen Corry, Australian National University, Australia
Copyright © 2022 Abdelsayed, Page and Ruben. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mena Abdelsayed, menaabdelsayed1993@gmail.com; Peter C. Ruben, pruben@sfu.ca