 Shan He1
Shan He1 Xinyue He1
Xinyue He1 Lei Liu2
Lei Liu2 Wenbo Zhang3
Wenbo Zhang3 Lanlan Yu3Zhun Deng3
Lanlan Yu3Zhun Deng3 Zhang Feiyi3,4
Zhang Feiyi3,4 Shanshan Mo3Yue Fan1Xinyue Zhao1Lun Wang1
Shanshan Mo3Yue Fan1Xinyue Zhao1Lun Wang1 Chenxuan Wang3*
Chenxuan Wang3* Shuyang Zhang1*
Shuyang Zhang1*- 1Department of Cardiology, Peking Union Medical College Hospital, Peking Union Medical College and Chinese Academy of Medical Sciences, Beijing, China
- 2Department of Chemistry, University of Wisconsin-Madison, Madison, WI, United States
- 3State Key Laboratory of Medical Molecular Biology, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
- 4Institute for Advanced Materials, Jiangsu University, Zhenjiang, China
Substantial controversies exist in the exploration of the molecular mechanism of heart failure (HF) and pose challenges to the diagnosis of HF and the discovery of specific drugs for the treatment. Recently, cardiac transthyretin (TTR) amyloidosis is becoming recognized as one of major causes of underdiagnosed HF. The investigation and modulation of TTR misfolding and amyloidal aggregation open up a new revenue to reveal the molecular mechanisms of HF and provide new possibilities for the treatment of HF. The aim of this review is to briefly introduce the recent advances in the study of TTR native and misfolding structures, discuss the correlation between the genotype and phenotype of cardiac TTR amyloidosis, and summarize the therapeutic applications of TTR structural stabilizers in the treatment of TTR amyloidosis-associated HF.
Introduction
Heart failure (HF) is a clinical syndrome that originates from the insufficient cardiac output to accommodate the need of peripheral tissues or organs due to the cardiac systolic or diastolic dysfunction (McMurray and Pfeffer, 2005). HF is a global pandemic, affecting at least 40 million people worldwide. Its prevalence increases with aging populations (Triposkiadis et al., 2019). Roughly 2% of adults suffered from this disease and this ratio further increases by 4–8% for the senior population with ages of 65 years or older (GBD 2015 Disease and Injury Incidence and Prevalence Collaborators, 2016). Nowadays, HF is emerging as one of leading causes of hospitalization among elderly people. The treatment of symptomatic HF is a notorious challenge with a 5-years survival rate of 50% (Metra and Teerlink, 2017). HF has imposed a huge economic burden to global healthcare systems. Globally, about $108 billion must be spent annually for the treatment of HF including $65 billion in direct medical costs and $43 billion attributed to indirect costs (GBD 2015 Disease and Injury Incidence and Prevalence Collaborators, 2016). Due to the high prevalence, poor prognosis, and enormous burden on healthcare systems, it raises an urgent requirement for a better understanding of the mechanism of HF. There are many factors that cause HF including cardiac factors (e.g., coronary artery disease, heart attack, hypertension, faulty heart valves, cardiomyopathy, myocarditis, congenital heart defects, and heart arrhythmias), and non-cardiac factors (such as diabetes, emphysema, HIV, hyperthyroidism, hypothyroidism, hemochromatosis, severe forms of anemia, amyloidosis, drug or alcohol misuse, and certain medications). The treatment for HF include pharmacologic treatments (such as the administration of angiotensin-converting enzyme inhibitor (or angiotensin II receptor blockers) and beta blocker), aerobic exercise, and symptom treatment (such as diuretic, sodium restriction, digoxin). The mechanism research providing valuable targets for drug discovery can be translated to improve the treatment of HF. However, many pathogenic mechanisms leading to HF have been proposed, such as neurohormonal activation, inflammation, abnormal immunity activation, amyloidogenesis, increased hemodynamic overload, ischemia-related dysfunction, which cause the comprehension in the clinical treatment of HF. Substantial challenges and debates remain in the exploration of the molecular mechanism of HF.
Currently, extensive attempts have been made toward understanding the mechanisms that cause the development of HF. Distinct hypotheses have been made to explain the occurrence of HF from different perspectives including neurohormonal activation, inflammation, and immunity (Zhang et al., 2017; Lam et al., 2018; Stanciu, 2019). 1) Neurohormonal activation (Lam et al., 2018). This theory hypothesizes that when the cardiac output is greatly reduced at the early stage of HF, the activation of the adrenergic nervous system and the renin-angiotensin-aldosterone system (RAAS) generates compensatory feedbacks (strengthened cardiac contractility, peripheral vasoconstriction, and sodium and fluid retention) to maintain the effective circulating volume. The consequences of neurohormonal activation are two-sided: a sufficient circulation in a short-term period and a cardiac remodeling as a long-term negative outcome. Increased plasma levels of several neurohormonal biomarkers (e.g., plasma renin, aldosterone, norepinephrine, and endothelin 1) promote fibroblast proliferation, extracellular matrix deposition, and increased oxidative stress. Thus, it leads to a series of decompensated cardiac dysfunctions, such as cardiac fibrosis, dilated chamber, weakened contractile function, and decreased stroke volume (Lam et al., 2018). 2) Inflammation (Stanciu, 2019). Pro-inflammatory cytokines, including interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α) are produced by the stimulated cardiomyocytes. Pro-inflammatory cytokines promote the expansion of macrophages in the cardiac tissues and the recruitment of peripheral blood monocytes to heart. As a result, the inflammatory cascade is magnified and triggers cardiac hypertrophy and cardiac fibrosis (Stanciu, 2019). 3) Immunity (Zhang et al., 2017). In the state of heart damage, dendritic cells (DCs) are switched to a mature state with enhanced antigen-presenting capacity and facilitate the release of pro-inflammatory cytokines. Subsequently, mature DCs induce the differentiation of native T cells into CD4+ T cells and CD8+ T cells, leading to a breaking self-tolerance, i.e., the cytotoxicity and adaptive immune response to heart tissue (Zhang et al., 2017).
The mechanisms discussed above have been applied to guide the therapeutic pharmacy design. However, the discovered drugs are unsuccessful in the treatment of the HF patients with characteristic amyloid deposits in myocardium. A recent investigation of cardiac amyloidosis prevalence revealed that the incidence rate of cardiac amyloidosis among the hospitalized patients in the United States increased substantially since 2000 from 18 per 100,000 person-years to 55 per 100,000 person-years in 2012 (Gilstrap et al., 2019). The mainly amyloidal components that infiltrate in the heart causing cardiac amyloidosis are immunoglobulin light chain amyloid fibril protein (AL) and transthyretin amyloidosis (ATTR), and thus cardiac amyloidosis can be classified into AL-cardiac amyloidosis (AL-CA) and TTR-cardiac amyloidosis (ATTR-CA) (Benson et al., 2018). Emerging results suggest that AL-CA is responsible for 70% of patients with cardiac amyloidosis (Oerlemans et al., 2019). The population prevalence of ATTR-CA is less certain, may be responsible for 30% of HF patients with preserved ejection fraction aged 75 or older (Maurer et al., 2017; Oerlemans et al., 2019). TTR is mainly synthesized in the liver and the brain choroid plexus, circulating in peripheral plasma and cerebrospinal fluid (Fleming et al., 2009). The main physiological activity exerted by TTR is thought to be a transporter of thyroxine and retinol. TTR may also participate in the maintenance of normal cognitive processes and nerve regeneration during ageing (Fleming et al., 2009). As shown in Figure 1A, pathological TTR amyloidosis is a multiple-step protein assembly process involving: 1) amino acid mutations lead to a dissociation of native TTR tetramers into abnormally TTR monomers; 2) misfolded TTR monomers associate into oligomers and protofibrils; 3) TTR oligomers and protofibrils elongate to form mature amyloidal fibrils (Dobson, 2003). The exposure of TTR amyloidal intermediates, i.e., oligomers and protofibrils, to myocardial cells induces caspase-3 activation, triggering the onset of programmed cell death (Sousa et al., 2001). It leads to the degradation of myocardial cells, cardiac atrophy, and progressive infiltration, in which the receptors of advanced glycosylation end products (RAGE) also participate the organ injury generated by ATTR. Consequently, the deposition of ATTR results in cardiac conduction abnormalities and diastolic dysfunction (Marcoux et al., 2015; Michels da Silva et al., 2019). The prevalence of TTR amyloid deposit in heart is estimated to be 13% in the HF patients aged 60 or older with preserved ejection fraction. This proportion further increases to 32% for the patients aged 75 and older (González-López et al., 2015). The average age of onset for wild-type ATTR is 75 years and for hereditary ATTR is 45 years (Nakagawa et al., 2016; He et al., 2019). Cardiac ATTR is frequently accompanied by debilitating neurological with or without cardiac complications (Traynor et al., 2019). For the HF patients aged 60 or older with ATTR, they may also suffer from hypertension, coronary heart disease, and chronic obstructive pulmonary disease, etc. The amyloidosis of TTR is regarded as an underdiagnosed cause of HF and provides a new avenue to understand the mechanism of HF development.
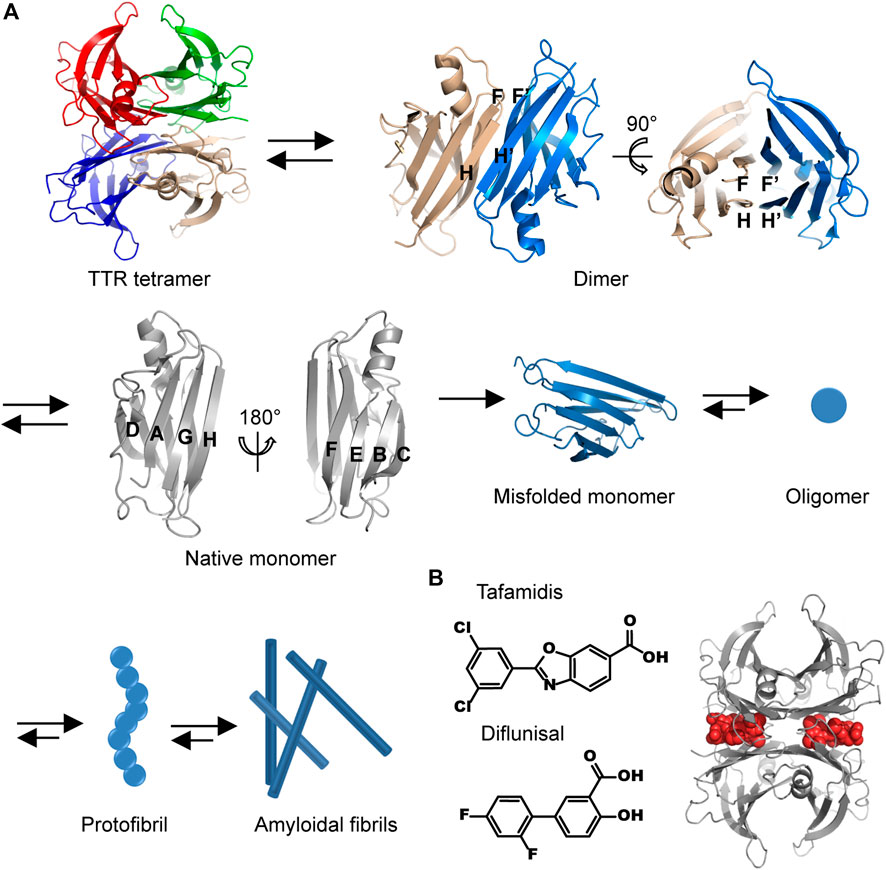
FIGURE 1. The molecular pathway of TTR amyloidal aggregation. (A) The structural illustration of the dissociation of TTR tetramer into monomer and the aggregation of TTR misfolded monomer. Different protein chains are indicated by different colors. The structures of TTR tetramer, dimer, and monomer are drawn by using the crystallographic data of PDB ID 1F41. (B) The chemical structures of tafamidis and diflunisal (left) and the representative model of structural stabilizers occupying the thyroxine binding site (right, the structure is drawn by using the crystallographic data of PDB ID 2ROX). Gray, a TTR tetramer; red, two binding structural stabilizers.
In this review, we summarize the recent structural and medical advances of cardiac TTR amyloidosis studies. We illuminate the potential relationship between the genotype and phenotype of ATTR, discuss the impact of amino acid mutation on TTR activities, and outline the current therapeutic approaches against ATTR in clinical practice.
The Structural Conversion of TTR Amyloidogenesis
Elucidating the TTR conformational change from native state to misfolded state is crucial for uncovering the molecular mechanism of TTR amyloid fibrosis. Substantial technical efforts have been applied toward studying the structural conversion of TTR using a series of experimental approaches, such as X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and cryo-electron microscopy (Cryo-EM), despite the heterogeneity and polymorphism in TTR folding and assembly manner, which create a challenge for structural characterization.
X-ray crystal diffraction experiments revealed that the native human TTR is folded into a globular structure consisting of eight antiparallel strands (denoted as from A to H) and one short α-helix (Wojtczak et al., 2010; Palaninathan, 2012) (Figure 1A). The eight ß-strands are associated into two groups of twisted ß-sheets. Specifically, A, D, G, and H strands are tethered by interstrand hydrogen bonds into an inner ß-sheet, whereas B, C, E, and F strands constitute an outer ß-sheet. The inner and outer ß-sheets are packed with each other in a layer-by-layer manner. The H and F strands are located at the edges of ß-sheets, providing active sites for interacting with the H′ and F′ strands from the adjacent TTR monomer, respectively. Thus, two TTR monomers are held together to form a dimer via interstrand hydrogen bonds. The dimerization of TTR renders two inner ß-sheets to form an extended ß-sheet pleat containing of A, D, G, H, and A′, D′, G′, H’ strands. Two inner extended ß-sheet pleats further wrap around each other, generating a dimer-dimer interface. Consequently, native human TTR is organized as a tetrameric cluster possessing an inner core (A, D, G, and H strands) and presenting an outer surface (B, C, E, and F strands).
In vitro experiments revealed that acidic pH conditions and the mutations identified from patients induce the dissociation of TTR tetramer into monomers and trigger the misfolding of monomer into amyloidal aggregates (Vieira and Saraiva, 2014). The destabilization of TTR tetrameric structure can be tracked by NMR. The acidification of solution triggers the flexible loop region between A and B to interact with A strand, leading to conformational changes occurring in the inner core domain (Lim et al., 2016). The impact of single amino acid mutation on the stability of native TTR tetramer is attributed to a propagation of structural perturbation from the mutation site to the entire inner/outer ß-sheet domains (Leach et al., 2018). The transmission electron micrographs showed that the TTR amyloid aggregates are rigid and unbranched fibrils with several micrometers in length and 70 to 130 Å in diameter (Serpell et al., 1995). A TTR fibril composes of two to three protofilaments. Each protofilaments are 25–35 Å in diameter and interwind with each other (Serpell, 1996). Synchrotron X-ray fiber diffraction pattern of TTR aggregate showed an intense 4.7 to 4.8 Å reflection on the meridian (Serpell, 1996; Sunde et al., 1997). It demonstrates that the periodicity of TTR protein arrangement along the fibril axis is 4.7–4.8 Å. This result suggests that the misfolded TTR proteins adopt a typical cross-β conformation and are stacked perpendicular to the elongation of protofilament (Serpell, 1996; Sunde et al., 1997). Multiple complementary methods have been used to reveal the atomic level structural model for the misfolded TTR aggregate, including scanning tunneling microscopy (STM) and X-ray crystallography for the key segments of TTR, and Cryo-EM for the full length of TTR. By using STM, the assembly structures of TTR 105–115 segment were observed by Yu et al. at a single-molecular level (Yu et al., 2018). In the STM images, TTR 105–115 segments present as linear lamella structures. Individual peptides are parallel to each other and packed into a protofilament-like structure. The distance between two adjacent peptide axes is 4.0 ± 0.2 Å, which is consistent with the structural features of cross-β conformation. Saelices et al. analyzed the crystal structures of 13 hexapeptide segments derived from the key amyloidogenic domains of TTR and found distinct assembly behaviors encoded by different TTR regions (Saelices et al., 2018). The segments from the C-terminal region of TTR (corresponding to the residues from 50 to 127) were observed to form in-register ß-sheets and pair as steric zippers. For instance, the crystalline structure of TTR 80–85 segment (80KALGIS85) shows that the TTR 80–85 ß-strands are assembled into anti-parallel ß-sheets. The side chains protruding from the surface of ß-sheet drive two ß-sheets to stack in layer-by-layer and form a tightly interdigitating steric zipper interface that is free from water. In contrast, the segments from the N-terminus (corresponding to the residues from 1 to 50) form out-of-register ß-sheets. For example, the anti-parallel ß-sheet formed by TTR 28–33 segment (28VAVHVF33) exhibits a wet and loosely stacking interface to interact with another TTR 28–33 ß-sheet, whereas the side chains from the same ß-sheet are self-complementing. To understand the folding structure of a full length TTR protein, Schmidt et al. investigated the molecular arrangement in a TTR fibril extracted from the tissue of a patient with hereditary Val30Met ATTR by Cryo-EM (Matthias Schmidt et al., 2019). The Cryo-EM experiment revealed that the misfolded TTR Val30Met protein consists of three ß-arch regions (TTR 11–35, 70–111, and 106–123) and 13 ß-strands (i.e., TTR 12–16, 19–21, 25–34, 60–62, 64–66, 70–73, 75–77, 79–80, 91–99, 103–104, 107–108, 114–115, and 118–122). Consistent with the X-ray crystallography results from TTR segments, the C-terminal motif of full length TTR Val30Met Cryo-EM structure is folded into multiple in-register ß-sheets. Whereas the N-terminal motif of TTR Cryo-EM structure presents one out-of-register ß-hairpin region involving the residues from 11 to 35 and two unstructured regions including the regions 1–10 and 36–50. Different from the key segment crystal structures, the full length TTR adopts a parallel ß-sheet conformation in the Cryo-EM structure.
TTR Genotype and Cardiac Phenotype
ATTR is relatively common in the men older than 60 years and often regarded as senile amyloidosis (Grogan et al., 2016). Any step during the conversion from the soluble native state to amyloidal fibrils may significantly alter the progression of fibril formation, such as acidification, proteolysis, and the co-assembly with other non-fibrous substances. For example, the co-existence of glycosaminoglycan and serum amyloid P component obviously accelerates the misfolding and association of TTR (Kisilevsky, 2000; Zhang and Li, 2010). Point mutation in the TTR gene is another key factor that causes the occurrence of TTR-associated HF. Hereditary transthyretin amyloid cardiomyopathy (ATTR-CM) is an autosomal dominant single-gene genetic disease (Damrauer et al., 2019). Up to now, more than 120 types of point mutations have been identified (Ruberg et al., 2019). The common symptoms of hereditary ATTR (hATTR) typically include progressive infiltration of amyloidal deposit, early right-sided heart failure, and late systolic ejection fraction reduction. Patients could also have syndromes of peripheral neuropathy, autonomic neuropathy, cardiomyopathy, eye disease, and spinal amyloidosis (Koga et al., 2003; Rapezzi et al., 2010; Maurer et al., 2017). Changes in the heart function caused by TTR amyloidal deposition include cardiac systolic or diastolic dysfunction. Patients usually suffer from orthostatic hypotension and cannot tolerate RAAS inhibitor treatment. Some severe cases could result in HF and suffer from syncope caused by arrhythmia or heart block, and angina or infarction caused by amyloid deposits in the coronary arteries (Finsterer et al., 2018). In addition to heart diseases, other illnesses generated by hereditary TTR include polyneuropathy, sensorimotor polyneuropathy, glaucoma, intravitreal deposition, nephrotic syndrome, and renal failure (Michels da Silva et al., 2019). General diagnosis of hereditary TTR requires extracardiac tissue biopsy (lower fat, small salivary glands, rectal mucosa or kidney, and nerve) (Ando et al., 2013; Jamet et al., 2015). The TTR amyloidal deposit appears as an amorphous transparent substance in the optical microscopic image and can be labeled by Congo red or thioflavin T immunohistology (Schönland et al., 2012). The diagnosis of ATTR-CM requires an intracardiac tissue biopsy to manifest the presence of TTR amyloid deposits in the myocardium (Ruberg et al., 2012). In the clinical examination, a low electrocardiography (ECG) voltage in which the amplitudes of all the QRS complexes in the limb are smaller than 0.5 mV in the limb is frequently observed (Damy et al., 2016). Some patients may also exhibit one or more of the following diagnostic characteristics: a speckled appearance of the myocardium on echocardiography with end-diastolic thickness of the ventricular septum >1.2 cm, excessive uptake of technetium-99 m (Tc-99m) scintillation agent by the heart with ATTR amyloidosis, delayed enhancement of subendothelial gadolinium on cardiac magnetic resonance imaging (MRI), multiple occurrences of osteolytic lesions of myeloma, and cystic bone lesions of dialysis-related amyloidosis (Sousa et al., 2001; Jurcut et al., 2010). It is necessary to point out that imaging techniques only provide supporting evidences, and the gold standard for the diagnosis of ATTR is still lacking. The current diagnosis in clinical practice for ATTR-CM includes: 1) a noninvasive nuclear scintigraphy by using planar and SPECT imaging; 2) an invasive endomyocardial biopsy, i.e., the histology with Congo red staining with apple-green birefringence; and 3) a genetic testing to determine if the disease is due to a hereditary mutation in the TTR gene (Gillmore et al., 2016; Maurer et al., 2018; Dorbala et al., 2019).
Recorded TTR mutations associated with cardiac phenotypes includes Val122Ile, Thr60Ala, Leu111Met, Ile68Leu, Val30Met, Val30Met, Phe33Leu, Asp38Val, Asp38Val, Gly47Glu, Gly53Glu, Glu54Lys, Leu55Pro, Glu61Lys, Tyr69His, Ser77Tyr, His88Arg, and Tyr114Cys (Maurer et al., 2016; Damy et al., 2019). Point mutations mostly locate at the terminal regions of the TTR protein, resulting in an abnormal folding structure and diminished tetramer stability. TTR mutation sites are diverse and could lead to a variety of clinical symptoms. Familial amyloid polyneuropathy (FAP), familial amyloid cardiomyopathy (FAC), and familial leptomeningeal amyloidosis are the three main phenotypes of inherited ATTR amyloidosis. The main phenotype of heart disease in hATTR patients is aggressive cardiomyopathy (Sun et al., 2018). Val122Ile, Val30Met, Thr60Ala, Leu111Met, Ile68Leu showed obvious cardiac phenotypes in the large sample cohort (Reilly et al., 1995; de Carvalho et al., 2000; Sattianayagam et al., 2012; Quarta et al., 2015). The clinical manifestations are more frequent in middle-age and older men with severely damaged cardiac structure. Examined by echocardiography, invasive cardiomyopathy where strong echoes occur in the left ventricular myocardium is accompanied by pericardial effusion and atrial enlargement (González-López et al., 2017). Val122Ile is the frequently occurred mutation that causes FAC (Quarta et al., 2015). Patients with congestive heart failure, conduction block, and intractable arrhythmia need the implantation of pacemakers with/without cardioverter defibrillators (Maurer et al., 2016). Val30Met mutation usually causes FAP. Cardiac manifestations are mainly manifested in different types of heart block, and most patients require pacemaker implantation (Damy et al., 2019). Patients with Thr60Ala mutation can develop late-onset restrictive cardiomyopathy as well as sensorimotor and autonomic polyneuropathy (Zanazzi et al., 2019). Patients with Leu111Met mutation have a higher propensity to develop carpal tunnel syndrome as the initial manifestation and a liver transplantation is necessary for the treatment (Liepnieks and Benson, 2007). In a family study of patients with Ile68Leu mutation, the presence of amyloid aggregates was observed in the spinal canal of patients (Salvi et al., 2003). Despite the heterogeneity of mutation sites, the clinical characteristics and natural history of cardiac phenotype also generate difficulty for the diagnosis and treatment of ATTR. A long-term follow-up study of the Asian population (23 patients) revealed that the overall survival rates at 12, 24, 36, 48, and 60 months after diagnosis were 77.8, 55.6, 38.9, 27.8, and 11.1%, respectively (He et al., 2019). The low prognosis of ATTR is correlated with two facts. First, the onset symptoms were not obvious in the heart. Within the 23 patients, only five patients were observed to possess a low left ventricle ejection fraction (ejection fraction <50%). Second, the clinical characteristics of ATTR were heterogeneous, e.g., 78.3% of patients had abnormal electrocardiography and 56.5% of patients displayed pseudoinfarct pattern. This result highlights the complexity in the ATTR phenotypes and the difficulty to avoid misdiagnosis in practice (He et al., 2019).
Motivated by the different clinical phenotypes of the above-mentioned TTR mutants, a series of studies were approached to investigate the pathogenic mechanisms of TTR mutants. For example, Wright et al. used 19F-NMR technology to monitor in real time and found that amyloidal aggregation was caused by V122I mutation (Benjamin et al., 2018). The dissociation of TTR tetramer into monomer is revealed to be the rate-limiting step. Jiang et al. demonstrated that the mutation of V122I makes the Gibbs free energy of tetramer 3 times higher than that of the wild-type, and thus the energetic barrier of tetramer dissociation is significantly reduced by the mutation of V122I (Jiang et al., 2001). The dissociation of TTR tetramers is accelerated by the V122I mutant in favor of kinetics and thermodynamics. It is worth to note that, the observation with TTR V122I mutant that the single site mutation at the key site of TTR promotes the dissociation of TTR tetramer is probably a general mechanism existing in the misfolding process of other types of TTR pathogenic mutations. As a summary, the cardiac phenotype-related pathogenic site accelerates the rate-limiting step of dissociation of tetramers into monomers, resulting in massive formation of TTR amyloidal aggregates and the accumulation of the clinical phenotype of HF.
The Clinical Practice of TTR Structural Stabilizer
Current therapeutic approaches toward the treatment of ATTR-CM focus on drug administration and surgical operation. Because 95% of plasma TTR is synthesized from liver, liver transplantation was firstly proved as the only potentially curative treatment for TTR amyloidosis in 1990s (Adams et al., 2019). The transplanted healthy liver produced normal and stable form of tetramers, which lowers the concentration of amyloidogenic monomers in blood and halts disease progress (Muchtar et al., 2017). However, the transplantation faces several challenges including the limited supply of livers and the long-term usage of immune suppressive therapy (Heymann and Tacke, 2016). Therefore, the majority of patients with ATTR need more affordable and accessible options. The structural stabilizer preventing the dissociation of TTR tetramer is a promising candidate. Several stabilizers have been approved by the Food and Drug Administration (FDA) for the treatment of ATTR-CM worldwide. Herein, we describe the pharmacological mechanism of stabilizers and summarize its clinical data.
Vigorous screening experiments and structure-based drug design studies have been carried out to identify TTR tetramers stabilizers at molecule level. More than 1,000 small aromatic molecules dynamically stabilizing TTR tetramers have been designed and synthesized (Gavrin et al., 2012). Most of these compounds were designed as a mimic of thyroxine, a natural hormone that specifically binds to the cavity of tetrameric TTR. These species share a similar chemical skeleton: two aromatic rings conjugated by a linker. The introduction of a polar group, halogen, or alkyl group into the aromatic ring facilitates the formation of electrostatic interactions and hydrophobic interactions between the thyroxine mimics and the binding pocket in the TTR protein (Gavrin et al., 2012). As manifested in the co-crystal structure of TTR binding with thyroxine mimic, the synthetic mimic occupies the thyroxine binding site and does not disturb TTR’s native structure (Gavrin et al., 2012). At low pH conditions, where TTR tetramers are prone to aggregate, the presence of thyroxine mimic in TTR solution greatly improve the stability of TTR against denaturation (Wiseman et al., 2005). The plausible mechanism underlying the stabilizing impact by thyroxine mimic is attributed to the elevated energy barrier of TTR tetramer dissociation leading to less amyloidal fibril formation and thus reducing the rate of amyloidogenesis progression (Johnson et al., 2012). As for increasing the bioavailability of compound, it is important to avoid the non-specific binding of thyroxine mimic to the abundant proteins in serum and increase the selectivity of the compounds toward thyroxine TTR binding site (Connelly et al., 2010).
Two TTR kinetic stabilizers, tafamidis and diflunisal, have been selected and used in clinical trials (chemical structures are shown in Figure 1B). Tafamidis, a non-steroidal anti-inflammatory drug (non-NSAID) benzoxazole derivative, is the first stabilizer approved to halt the progression of cardiac impairment in TTR familial amyloid cardiomyopathy (Maurer et al., 2018). The tafamidis phase III transthyretin amyloidosis cardiomyopathy (ATTR-ACT) trial is a multicenter blinded study in 441 patients with ATTR-CM who received placebo 20 mg, tafamidis 20 mg, or tafamidis 80 mg daily with a randomization ratio of 2:2:1 (Table 1). This investigation exhibits a striking cardiovascular outcome in the patients with oral intake of tafamidis at 80 mg/day (Maurer et al., 2018). Over a period of 30 months, all-cause mortality decreased from 42.9% to 29.5%, and cardiovascular-related hospitalizations reduced from 0.70 to 0.48 annually. Both of mortality and morbidity endpoints all achieved statistical significance. This remarkable result proves that 80 mg dose of tafamidis not only relieves symptoms of HF but also improves a long-term prognosis of ATTR-CM. The general molecular mechanism of structural stabilizer can be applied to interpret the pharmacological activity that tafamidis selectively occupies the thyroxine binding site, preventing the structural conversion from native tetramer to denatured monomers (Figure 1B). Consequently, the possibility of TTR misfolding and amyloidal aggregation is substantially reduced (Bulawa et al., 2012). Tafamidis circulates in serum or plasma mainly in the form of binding with plasma proteins (99%) and undergoes the metabolic pathway through glucuronidation (Bulawa et al., 2012). Up to date, Tafamidis has been approved as the first-line exclusive drug in the treatment of TTR amyloidosis in US, Europe, and Japan.
Diflunisal is a NSAID that was previously administered in the clinical practice as an antipyretic, analgesic, and anti-inflammatory agent (Enthoven et al., 2016). Recent investigations revealed that diflunisal can be applied to facilitate nervous and cardiac repair and regeneration (Miller et al., 2004; Sekijima et al., 2006). In a clinical trial including 120 patients with TTR cardiac amyloidosis demonstrates that an increased survival is associated with diflunisal treatment (Rosenblum et al., 2017) (Table 1). In an international randomized double-blind trial, a total of 130 patients received diflunisal or placebo twice daily for 2 years (diflunisal 250 mg, n = 64; placebo group, n = 66) (Berk et al., 2013) (Table 1). Compared to the placebo group, diflunisal significantly slowed down the progression of nervous amyloidal disease and enhanced the cardiac functions. Nowadays, diflunisal has been approved by FDA for the clinical application, but some side effects have also been reported. For example, diflunisal can lead to a decline (6%) in estimated glomerular filtration rate (Ikram et al., 2018). A chronical administration of diflunisal can slow down the renal blood flow and thus becomes inadvisable to some patients with renal insufficiency. Therefore, Diflunisal is a second line drug for ATTR-CM. In the future, randomized trials with large samples are needed to collect more evidence for evaluate the safety of diflunisal in the treatment of ATTR-CM.
Conclusion
The investigations of TTR misfolding and amyloidal aggregation at molecular level have greatly improved the understanding the mechanism of HF and promoted the treatment of HF. The destabilization and dissociation of TTR tetramers into abnormally TTR monomers lead to the misfolded TTR monomers, which abnormally assemble into oligomers, protofibrils, and mature amyloidal fibrils. Structural stabilizers such as tafamidis and diflunisal can block the dissociation of TTR tetramer and facilitate the treatment of patients with TTR amyloidosis. With the gradual deepening of the research on the structure of TTR protein, there will be a profound understanding of the mechanism of TTR-associated HF. We note that, there still remain challenges in the research that need to be urgently studied, including: 1) the discovery of compounds to reverse the association state of TTR from amyloidal state to native folding state; 2) the misfolding structures of TTR amyloidal oligomers and the structure-based drug design to specifically target the amyloidal intermediates of TTR; 3) the therapeutic approaches to promote cardiac repair and preserve cardiac function against the dysfunction caused by TTR amyloidosis. The efforts made in these aspects will benefit the medical treatment of TTR-related patients in the future.

TABLE 1. Clinical trials of structural stabilizers in patients with ATTR-CM.
Author Contributions
SH, XH, LL, WZ, LY, ZD, FZ, YF, SM, XZ, LW, CW, and SZ. discussed and wrote the manuscript.
Funding
This work was funded by CAMS Innovation Fund for Medical Sciences (2018-I2M-3–006, 2016-I2M-1–002), the National Natural Science Foundation of China (31901007), China Postdoctoral Science Foundation (2020T130006ZX), State Key Laboratory Special Fund 2,060,204, the National Key Research and Development Program of China (2016YFC0901500), and Center for Rare Diseases Research, Chinese Academy of Medical Sciences, Beijing, China (2016ZX310174–4).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adams, D., Koike, H., Slama, M., and Coelho, T. (2019). Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat. Rev. Neurol. 15 (7), 387–404. doi:10.1038/s41582-019-0210-4
Ando, Y., Coelho, T., Berk, J. L., Cruz, M. W., Ericzon, B. G., Ikeda, S., et al. (2013). Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J. Rare Dis. 8, 31. doi:10.1186/1750-1172-8-31
Benson, M. D., Buxbaum, J. N., Eisenberg, D. S., Merlini, G., Saraiva, M. J. M., Sekijima, Y., et al. (2018). Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid 25 (4), 215–219. doi:10.1080/13506129.2018.1549825
Berk, J. L., Suhr, O. B., Obici, L., Sekijima, Y., Zeldenrust, S. R., Yamashita, T., et al. (2013). Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. J. Am. Med. Assoc. 310 (24), 2658–2667. doi:10.1001/jama.2013.283815
Bulawa, C. E., Connelly, S., Devit, M., Wang, L., Weigel, C., Fleming, J. A., et al. (2012). Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc. Natl. Acad. Sci. U.S.A. 109 (24), 9629–9634. doi:10.1073/pnas.1121005109
Castano, A., Helmke, S., Alvarez, J., Delisle, S., and Maurer, M. S. (2012). Diflunisal for ATTR cardiac amyloidosis. Congest Heart Fail. 18 (6), 315–319. doi:10.1111/j.1751-7133.2012.00303.x
Connelly, S., Choi, S., Johnson, S. M., Kelly, J. W., and Wilson, I. A. (2010). Structure-based design of kinetic stabilizers that ameliorate the transthyretin amyloidoses. Curr. Opin. Struct. Biol. 20 (1), 54–62. doi:10.1016/j.sbi.2009.12.009
Damrauer, S. M., Chaudhary, K., Cho, J. H., Liang, L. W., Argulian, E., Chan, L., et al. (2019). Association of the V122I hereditary transthyretin amyloidosis genetic variant with heart failure among individuals of african or hispanic/latino ancestry. J. Am. Med. Assoc. 322 (22), 2191–2202. doi:10.1001/jama.2019.17935
Damy, T., Maurer, M. S., Rapezzi, C., Planté-Bordeneuve, V., Karayal, O. N., Mundayat, R., et al. (2016). Clinical, ECG and echocardiographic clues to the diagnosis of TTR-related cardiomyopathy. Open Heart 3 (1), e000289. doi:10.1136/openhrt-2015-000289
Damy, T., Kristen, A. V., Suhr, O. B., Maurer, M. S., Planté-Bordeneuve, V., Yu, C. R., et al. (2019). Transthyretin cardiac amyloidosis in continental western europe: an insight through the transthyretin amyloidosis outcomes survey (THAOS). Eur. Heart J. doi:10.1093/eurheartj/ehz173
de Carvalho, M., Moreira, P., Evangelista, T., Ducla-Soares, J. L., Bento, M., Fernandes, R., et al. (2000). New transthyretin mutation V28M in a Portuguese kindred with amyloid polyneuropathy. Muscle Nerve 23 (7), 1016–1021. doi:10.1002/1097-4598(200007)23:7<1016::aid-mus3>3.0.co;2-w
Dobson, C. M. (2003). Protein folding and misfolding. Nature 426 (6968), 884–890. doi:10.1038/nature02261
Dorbala, S., Ando, Y., Bokhari, S., Dispenzieri, A., Falk, R. H., Ferrari, V. A., et al. (2019). ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: Part 1 of 2-evidence base and standardized methods of imaging. J. Nucl. Cardiol. 26 (6), 2065–2123. doi:10.1007/s12350-019-01760-6
Enthoven, W. T., Roelofs, P. D., Deyo, R. A., van Tulder, M. W., and Koes, B. W. (2016). Non-steroidal anti-inflammatory drugs for chronic low back pain. Cochrane Database Syst. Rev. 2 (2), CD012087. doi:10.1002/14651858.cd012087
Finsterer, J., Stöllberger, C., Rauschka, H., and Gatterer, E. (2018). Coronary ectasia in amyloid cardiomyopathy and neuropathy due to the transthyretin mutation c.323a>g. Heart Lung 47 (2), 127–129. doi:10.1016/j.hrtlng.2017.11.007
Fleming, C. E., Nunes, A. F., and Sousa, M. M. (2009). Transthyretin: more than meets the eye. Prog. Neurobiol. 89 (3), 266–276. doi:10.1016/j.pneurobio.2009.07.007
Gavrin, L. K., Denny, R. A., and Saiah, E. (2012). Small molecules that target protein misfolding. J. Med. Chem. 55 (24), 10823–10843. doi:10.1021/jm301182j
GBD 2015 Disease and Injury Incidence and Prevalence Collaborators (2016). Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: a systematic analysis for the global burden of disease study 2015. Lancet 388 (10053), 1545–1602. doi:10.1016/S0140-6736(16)31678-6
Gillmore, J. D., Maurer, M. S., Falk, R. H., Merlini, G., Damy, T., Dispenzieri, A., et al. (2016). Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 133 (24), 2404–2412. doi:10.1161/CIRCULATIONAHA.116.021612
Gilstrap, L. G., Dominici, F., Wang, Y., El-Sady, M. S., Singh, A., Di Carli, M. F., et al. (2019). Epidemiology of cardiac amyloidosis-associated heart failure hospitalizations among fee-for-service medicare beneficiaries in the United States. Circ Heart Fail 12, e005407. doi:10.1161/CIRCHEARTFAILURE.118.005407
González-López, E., Gallego-Delgado, M., Guzzo-Merello, G., de Haro-del Moral, F. J., Cobo-Marcos, M., Robles, C., et al. (2015). Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur. Heart J. 36 (38), 2585–2594. doi:10.1093/eurheartj/ehv338
González-López, E., López-Sainz, Á., and Garcia-Pavia, P. (2017). Diagnosis and treatment of transthyretin cardiac amyloidosis. Progress and hope. Rev. Esp. Cardiol. 70 (11), 991–1004. doi:10.1016/j.rec.2017.05.036
Grogan, M., Scott, C. G., Kyle, R. A., Zeldenrust, S. R., Gertz, M. A., Lin, G., et al. (2016). Natural history of wild-type Transthyretin Cardiac amyloidosis and Risk stratification using a Novel Staging system. J. Am. Coll. Cardiol. 68 (10), 1014–1020. doi:10.1016/j.jacc.2016.06.033
He, S., Tian, Z., Guan, H., Li, J., Fang, Q., and Zhang, S. (2019). Clinical characteristics and prognosis of Chinese patients with hereditary transthyretin amyloid cardiomyopathy. Orphanet J. Rare Dis. 14 (1), 251. doi:10.1186/s13023-019-1235-x
Heymann, F., and Tacke, F. (2016). Immunology in the liver--from homeostasis to disease. Nat. Rev. Gastroenterol. Hepatol. 13 (2), 88–110. doi:10.1038/nrgastro.2015.200
Ikram, A., Donnelly, J. P., Sperry, B. W., Samaras, C., Valent, J., and Hanna, M. (2018). Diflunisal tolerability in transthyretin cardiac amyloidosis: a single center's experience. Amyloid 25 (3), 197–202. doi:10.1080/13506129.2018.1519507
Jamet, M. P., Gnemmi, V., Hachulla, É., Dhaenens, C. M., Bouchindhomme, B., Delattre, C., et al. (2015). Distinctive patterns of transthyretin amyloid in salivary tissue: a clinicopathologic study of 92 patients with amyloid-containing minor salivary gland biopsies. Am. J. Surg. Pathol. 39 (8), 1035–1044. doi:10.1097/pas.0000000000000430
Jiang, X., Buxbaum, J. N., and Kelly, J. W. (2001). The V122I cardiomyopathy variant of transthyretin increases the velocity of rate-limiting tetramer dissociation, resulting in accelerated amyloidosis. Proc. Natl. Acad. Sci. U.S.A. 98 (26), 14943–14948. doi:10.1073/pnas.261419998
Johnson, S. M., Connelly, S., Fearns, C., Powers, E. T., and Kelly, J. W. (2012). The transthyretin amyloidoses: from delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency-approved drug. J. Mol. Biol. 421 (2-3), 185–203. doi:10.1016/j.jmb.2011.12.060
Jurcut, R., Giusca, S., La Gerche, A., Vasile, S., Ginghina, C., and Voigt, J. U. (2010). The echocardiographic assessment of the right ventricle: what to do in 2010?. Eur. J. Echocardiogr. 11 (2), 81–96. doi:10.1093/ejechocard/jep234
Kisilevsky, R. (2000). The relation of proteoglycans, serum amyloid P and apo E to amyloidosis current status, 2000. Amyloid 7 (1), 23–25. doi:10.3109/13506120009146820
Koga, T., Ando, E., Hirata, A., Fukushima, M., Kimura, A., Ando, Y., et al. (2003). Vitreous opacities and outcome of vitreous surgery in patients with familial amyloidosis polyneuropathy. Am. J. Ophthalmol. 135 (2), 188–193. doi:10.1016/s0002-9394(02)01838-x
Lam, C. S. P., Voors, A. A., de Boer, R. A., Solomon, S. D., and van Veldhuisen, D. J. (2018). Heart failure with preserved ejection fraction: from mechanisms to therapies. Eur. Heart J. 39 (30), 2780–2792. doi:10.1093/eurheartj/ehy301
Leach, B. I., Zhang, X., Kelly, J. W., Dyson, H. J., and Wright, P. E. (2018). NMR measurements reveal the structural basis of transthyretin destabilization by pathogenic mutations. Biochemistry 57 (30), 4421–4430. doi:10.1021/acs.biochem.8b00642
Liepnieks, J. J., and Benson, M. D. (2007). Progression of cardiac amyloid deposition in hereditary transthyretin amyloidosis patients after liver transplantation. Amyloid 14 (4), 277–282. doi:10.1080/13506120701614032
Lim, K. H., Dasari, A. K., Hung, I., Gan, Z., Kelly, J. W., and Wemmer, D. E. (2016). Structural changes associated with transthyretin misfolding and amyloid formation revealed by solution and solid-state NMR. Biochemistry 55 (13), 1941–1944. doi:10.1021/acs.biochem.6b00164
Marcoux, J., Mangione, P. P., Porcari, R., Degiacomi, M. T., Verona, G., Taylor, G. W., et al. (2015). A novel mechano-enzymatic cleavage mechanism underlies transthyretin amyloidogenesis. EMBO Mol. Med. 7 (10), 1337–1349. doi:10.15252/emmm.201505357
Maurer, M. S., Elliott, P., Comenzo, R., Semigran, M., and Rapezzi, C. (2017). Addressing common questions encountered in the diagnosis and management of cardiac amyloidosis. Circulation 135 (14), 1357–1377. doi:10.1161/circulationaha.116.024438
Maurer, M. S., Hanna, M., Grogan, M., Dispenzieri, A., Witteles, R., Drachman, B., et al. (2016). Genotype and phenotype of transthyretin cardiac amyloidosis: thaos (transthyretin amyloid outcome survey). J. Am. Coll. Cardiol. 68 (2), 161–172. doi:10.1016/j.jacc.2016.03.596
Maurer, M. S., Schwartz, J. H., Gundapaneni, B., Elliott, P. M., Merlini, G., Waddington-Cruz, M., et al. (2018). Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N. Engl. J. Med. 379 (11), 1007–1016. doi:10.1056/NEJMoa1805689
McMurray, J. J., and Pfeffer, M. A. (2005). Revised American College of cardiology/American heart association guidelines for the management of heart failure. Prev. Cardiol. 8 (9474), 254–256. doi:10.1016/s0140-6736(05)66621-4
Metra, M., and Teerlink, J. R. (2017). Heart failure. Lancet 390 (10106), 1981–1995. doi:10.1016/S0140-6736(17)31071-1
Michels da Silva, D., Langer, H., and Graf, T. (2019). Inflammatory and molecular pathways in heart failure-ischemia, HFpEF and transthyretin cardiac amyloidosis. Int. J. Mol. Sci. 20 (9). doi:10.3390/ijms20092322
Miller, S. R., Sekijima, Y., and Kelly, J. W. (2004). Native state stabilization by nsaids inhibits transthyretin amyloidogenesis from the most common familial disease variants. Lab. Invest. 84 (5), 545–552. doi:10.1038/labinvest.3700059
Muchtar, E., Grogan, M., Dasari, S., Kurtin, P. J., and Gertz, M. A. (2017). Acquired transthyretin amyloidosis after domino liver transplant: phenotypic correlation, implication of liver retransplantation. J. Neurol. Sci. 379, 192–197. doi:10.1016/j.jns.2017.06.013
Nakagawa, M., Sekijima, Y., Yazaki, M., Tojo, K., Yoshinaga, T., Doden, T., et al. (2016). Carpal tunnel syndrome: a common initial symptom of systemic wild-type ATTR (ATTRwt) amyloidosis. Amyloid 23 (1), 58–63. doi:10.3109/13506129.2015.1135792
Oerlemans, M. I. F. J., Rutten, K. H. G., Minnema, M. C., Raymakers, R. A. P., Asselbergs, F. W., and de Jonge, N. (2019). Cardiac amyloidosis: the need for early diagnosis. Neth. Heart J. 27, 525–536. doi:10.1007/s12471-019-1299-1
Palaninathan, S. K. (2012). Nearly 200 x-ray crystal structures of transthyretin: what do they tell us about this protein and the design of drugs for TTR amyloidoses?. Curr. Med. Chem. 19 (15), 2324–2342. doi:10.2174/092986712800269335
Quarta, C. C., Buxbaum, J. N., Shah, A. M., Falk, R. H., Claggett, B., Kitzman, D. W., et al. (2015). The amyloidogenic V122I transthyretin variant in elderly black americans. N. Engl. J. Med. 372 (1), 21–29. doi:10.1056/NEJMoa1404852
Rapezzi, C., Quarta, C. C., Riva, L., Longhi, S., Gallelli, I., Lorenzini, M., et al. (2010). Transthyretin-related amyloidoses and the heart: a clinical overview. Nat. Rev. Cardiol. 7 (7), 398–408. doi:10.1038/nrcardio.2010.67
Reilly, M. M., Adams, D., Booth, D. R., Davis, M. B., Said, G., Laubriat-Bianchin, M., et al. (1995). Transthyretin gene analysis in european patients with suspected familial amyloid polyneuropathy. Brain 118 (Pt 4), 849–856. doi:10.1093/brain/118.4.849
Rosenblum, H., Castano, A., Alvarez, J., Helmke, S., and Maurer, M. S. (2017). Improved survival in patients with transthyretin cardiac amyloidosis treated with TTR stabilizers. J. Card. Fail. 23 (8), S20. doi:10.1016/j.cardfail.2017.07.039
Ruberg, F. L., Grogan, M., Hanna, M., Kelly, J. W., and Maurer, M. S. (2019). Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. J. Am. Coll. Cardiol. 73 (22), 2872–2891. doi:10.1016/j.jacc.2019.04.003
Ruberg, F. L., Maurer, M. S., Judge, D. P., Zeldenrust, S., Skinner, M., Kim, A. Y., et al. (2012). Prospective evaluation of the morbidity and mortality of wild-type and V122I mutant transthyretin amyloid cardiomyopathy: the transthyretin amyloidosis cardiac study (TRACS). Am. Heart J. 164 (2), 222–228. e1. doi:10.1016/j.ahj.2012.04.015
Saelices, L., Sievers, S. A., Sawaya, M. R., and Eisenberg, D. S. (2018). Crystal structures of amyloidogenic segments of human transthyretin. Protein Sci. 27 (7), 1295–1303. doi:10.1002/pro.3420
Salvi, F., Scaglione, C., Michelucci, R., Linke, R. P., Obici, L., Ravani, A., et al. (2003). Atypical familial motor neuropathy in patients with mutant TTR Ile68Leu. Amyloid 10 (3), 185–189. doi:10.3109/13506120308998999
Sattianayagam, P. T., Hahn, A. F., Whelan, C. J., Gibbs, S. D., Pinney, J. H., Stangou, A. J., et al. (2012). Cardiac phenotype and clinical outcome of familial amyloid polyneuropathy associated with transthyretin alanine 60 variant. Eur. Heart J. 33 (9), 1120–1127. doi:10.1093/eurheartj/ehr383
Schönland, S. O., Hegenbart, U., Bochtler, T., Mangatter, A., Hansberg, M., Ho, A. D., et al. (2012). Immunohistochemistry in the classification of systemic forms of amyloidosis: a systematic investigation of 117 patients. Blood 119 (2), 488–493. doi:10.1182/blood-2011-06-358507
Sekijima, Y., Dendle, M. A., and Kelly, J. W. (2006). Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid 13 (4), 236–249. doi:10.1080/13506120600960882.
Serpell, B. L. (1996). Synchrotron X-ray studies suggest that the core of the transthyretin amyloid fibril is a continuous β-sheet helix. Structure 4 (8), 989–998. doi:10.1016/S0969-2126(96)00104-9
Serpell, L. C., Sunde, M., Fraser, P. E., Luther, P. K., Morris, E. P., Sangren, O., et al. (1995). Examination of the structure of the transthyretin amyloid fibril by image reconstruction from electron micrographs. J. Mol. Biol. 254 (2), 113–118. doi:10.1006/jmbi.1995.0604
Sousa, M. M., Cardoso, I., Fernandes, R., Guimarães, A., and Saraiva, M. J. (2001). Deposition of transthyretin in early stages of familial amyloidosis polyneuropathy: evidence for toxicity of nonfibrillar aggregates. Am. J. Pathol. 159 (6), 1993–2000. doi:10.1016/s0002-9440(10)63050-7
Stanciu, A. E. (2019). Cytokines in heart failure. Adv. Clin. Chem. 93, 63–113. doi:10.1016/bs.acc.2019.07.002
Sun, X., Dyson, H. J., and Wright, P. E. (2018). Kinetic analysis of the multistep aggregation pathway of human transthyretin. Proc. Natl. Acad. Sci. U. S. A. 115 (27), E6201–e6208. doi:10.1073/pnas.1807024115
Sunde, M., Serpell, L. C., Bartlam, M., Fraser, P. E., Pepys, M. B., and Blake, C. C. (1997). Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J. Mol. Biol. 273 (3), 729–739. doi:10.1006/jmbi.1997.1348
Traynor, B. P., Shamsi, A., and Voon, V. (2019). Multi-modality imaging in transthyretin amyloid cardiomyopathy. World J. Cardiol. 11 (11), 266–276. doi:10.4330/wjc.v11.i11.266
Triposkiadis, F., Xanthopoulos, A., and Butler, J. (2019). Cardiovascular aging and heart failure: JACC review topic of the week. J. Am. Coll. Cardiol. 74 (6), 804–813. doi:10.1016/j.jacc.2019.06.053
Vieira, M., and Saraiva, M. J. (2014). Transthyretin: a multifaceted protein. Biomol. Concepts 5 (1), 45–54. doi:10.1515/bmc-2013-0038
Wiseman, R. L., Johnson, S. M., Kelker, M. S., Foss, T., Wilson, I. A., and Kelly, J. W. (2005). Kinetic stabilization of an oligomeric protein by a single ligand binding event. J. Am. Chem. Soc. 127 (15), 5540–5551. doi:10.1021/ja042929f
Wojtczak, A., Cody, V., Luft, J. R., and Pangborn, W. (2010). Structure of rat transthyretin (rTTR) complex with thyroxine at 2.5 A resolution: first non-biased insight into thyroxine binding reveals different hormone orientation in two binding sites. Acta Crystallogr D Biol Crystallogr 57 (8), 1061–1070. doi:10.1107/s0907444901007235
Yu, L. L., Zheng, Y. F., Xu, J., Qu, F. Y., Lin, Y. C., Zou, Y. M., et al. (2018). Site-specific determination of TTR-related functional peptides by using scanning tunneling microscopy. Nano Res 11 (1), 577–585. doi:10.1007/s12274-017-1825-7
Zanazzi, G., Arshad, M., Maurer, M. S., Brannagan, T. H., and Tanji, K. (2019). Demyelinating neuropathy in a patient treated with revusiran for transthyretin (Thr60Ala) amyloidosis. J. Clin. Neuromuscul. Dis. 20 (3), 120–128. doi:10.1097/cnd.0000000000000242
Zhang, X., and Li, J. P. (2010). Heparin sulfate proteoglycans in amyloidosis. Prog Mol Biol Transl Sci 93, 309–334. doi:10.1016/S1877-1173(10)93013-5
Keywords: Transthyretin, amyloidal aggregation, protein structural stabilizers, cardiac amyloidosis, protein folding
Citation: He S, He X, Liu L, Zhang W, Yu L, Deng Z, Feiyi Z, Mo S, Fan Y, Zhao X, Wang L, Wang C and Zhang S (2021) The Structural Understanding of Transthyretin Misfolding and the Inspired Drug Approaches for the Treatment of Heart Failure Associated With Transthyretin Amyloidosis. Front. Pharmacol. 12:628184. doi: 10.3389/fphar.2021.628184
Received: 11 November 2020; Accepted: 13 January 2021;
Published: 18 February 2021.
Edited by:
Lei Xi, Virginia Commonwealth University, United StatesReviewed by:
Tianqing Peng, Western University, CanadaDongze Qin, Albert Einstein College of Medicine, United States
Copyright © 2021 He, He, Liu, Zhang, Yu, Deng, Feiyi, Mo, Fan, Zhao, Wang, Wang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chenxuan Wang, wangcx@ibms.pumc.edu.cn; Shuyang Zhang, shuyangzhang103@nrdrs.org