 Lidong Zhao1Jinqiang Zhuang1
Lidong Zhao1Jinqiang Zhuang1 Yihui Wang1Dandan Zhou1Dandan Zhao1Shun Zhu1Jinjun Pu2Hongyu Zhang3Ming Yin4Wenjuan Zhao4
Yihui Wang1Dandan Zhou1Dandan Zhao1Shun Zhu1Jinjun Pu2Hongyu Zhang3Ming Yin4Wenjuan Zhao4 Zejian Wang4*
Zejian Wang4* Jiang Hong1*
Jiang Hong1*- 1Department of Internal and Emergency Medicine, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine (Originally Named “Shanghai First People’ s Hospital”), Shanghai, China
- 2Department of Emergency Medicine, Putuo Hospital Affiliated to Shanghai University of Traditional Chinese Medicine, Shanghai, China
- 3Department of Biomedicine, KG Jebsen Centre for Research on Neuropsychiatric Disorders, University of Bergen, Bergen, Norway
- 4School of Pharmacy, Shanghai Jiao Tong University, Shanghai, China
Background: The cardioprotective effect of propofol on ischemia-reperfusion injury (I/R injury) is partly due to suppressing apoptosis. Mitochondrial dynamics are also involved in apoptosis. Mitochondrial fusion and fission lead to mitochondrial morphological changes. However, whether suppressing apoptosis effect of propofol against ischemia-reperfusion injury in the heart is via regulating mitochondrial morphology remains unclear.
Methods: H9c2 cells underwent oxygen glucose deprivation (OGD) followed by reperfusion to simulate cardiomyocytes ischemia/reperfusion injury. Cell viability, apoptosis ratio and intracellular reactive oxygen species (ROS) were assessed, respectively. Mitochondrial membrane dynamin family proteins, extracellular signal regulated kinase 1 and 2 (ERK1/2), phosphorylated extracellular signal regulated kinase 1 and 2 (p-ERK1/2) and proteins related to intrinsic apoptosis pathways were detected by western blotting. The mitochondrial morphology and the distribution of dynamin-related protein 1 (Drp1) were observed by using laser confocal microscopy.
Results: Propofol enhanced the survival of H9c2 cells, decreased ROS levels and inhibited apoptosis during oxygen glucose deprivation/reperfusion (OGD/R) injury. Mitochondrial fission in H9c2 cells was inhibited by propofol during OGD injury. Propofol alleviated high levels of mitochondrial fusion and fission during OGD/R in H9c2 cells, by regulating mitochondrial membrane remodeling dynamin family proteins. Propofol inhibited Drp1 colocalization with mitochondria in H9c2 cells during OGD/R injury. Moreover, Drp1 phosphorylation was inhibited by propofol through decreasing ERK activation during OGD/R injury. We found that propofol ameliorated H9c2 cells apoptosis during OGD/R via inhibiting mitochondrial cytochrome c release and caspase-9, caspase-6, caspase-7 and caspase-3 activation.
Conclusion: Propofol suppresses H9c2 cells apoptosis during OGD/R injury via inhibiting intrinsic apoptosis pathway, which may be partly due to reducing high levels of mitochondrial fusion and fission induced by OGD/R injury.
Introduction
The intravenous anesthetic propofol (2, 6-diisopropylphenol) is widely used in anesthesia and sedation due to the rapid induction and recovery from anesthesia (Wahr et al., 2000). Many lines of evidence indicate that propofol exhibits cardioprotective effects (Kokita and Hara, 1996; Xia et al., 2003). As for the mechanism, similar to vitamin E, propofol contains a phenolic hydroxyl group, which has been associated with its antioxidant effect (Vasileiou et al., 2009). Researches indicate that propofol can scavenge the free radical, both in vitro and in vivo (Li et al., 2012), thus ameliorating ischemic myocardial contractile dysfunction and arrhythmias (Hanouz et al., 2003), narrowing infarct size, and reducing tissue lesions (Ko et al., 1997). Moreover, propofol has been shown to attenuate ischemia-reperfusion injury (I/R injury) by suppressing apoptosis and preserving mitochondrial function (Jin et al., 2009), but the exact mechanism remains unclear.
Mitochondria are the most important sources of energy in the heart, providing over 90% adenosine triphosphate (ATP) to the heart through oxidative phosphorylation (Schaper et al., 1985). In addition, mitochondria also play a key role in regulating apoptosis and cell growth, and in generating reactive oxygen species (ROS). Additionally, mitochondrial morphology is now recognized as an important factor closely associated with the energetic state of mitochondria (Galloway et al., 2012b). Mitochondrial morphology varies among different cell types. Mitochondria are in the process of continuous fission and fusion mediated by membrane remodeling dynamin family proteins (Ishihara et al., 2009). When oxidative stress occurs during acute I/R injury, mitochondrial fission can be caused in HL-1 cardiac cells (Ong et al., 2010). Dynamin family proteins involve mitofusin 1 (Mfn1), mitofusin 2 (Mfn2) and optic atrophy 1 (Opa1) protein that mediate mitochondrial fusion, whereas dynamin-related protein (Drp1) and fission 1 (Fis1) protein regulate mitochondrial fission.
Cardiomyocyte apoptosis plays an essential role in acute myocardial ischemia-reperfusion injury (I/R injury) (Haunstetter and Izumo, 1998). Apoptosis can be regulated through both intrinsic and extrinsic pathways (Zhang et al., 2002). Mitochondrial-shaping proteins are involved in intrinsic apoptosis pathway (Ong et al., 2017). They play important roles in the mitochondrial outer membrane permeabilization (MOMP) and the release of apoptotic factors, for example, cytochrome c release (Montessuit et al., 2010). However, whether suppressing apoptosis effect of propofol against ischemia-reperfusion injury (I/R injury) in the heart is via an intrinsic mitochondrial mechanism remains unclear. Based on previous studies, we hypothesize that propofol may reduce cardiomyocyte apoptosis induced by acute ischemia-reperfusion injury (I/R injury), via an intrinsic mitochondrial mechanism, by regulating mitochondrial fusion and fission. In this study, we used the H9c2 cell line subjected to oxygen glucose deprivation (OGD) followed by reperfusion (OGD/R) as an in vitro model of cardiomyocytes ischemia and investigated the underlying mechanism of propofol against cells apoptosis.
Materials and Methods
Cell Culture and Reagents
The H9c2 cells, a cardiomyocyte cell line, were purchased from the Shanghai Institute for Biological Sciences, Chinese Academy of Sciences (Shanghai, China). Dulbecco’s modified Eagle’s medium/F-12 (DMEM/F-12) and fetal bovine serum (FBS) were both purchased from Gibco-Invitrogen (Grand Island, NY, United States). The cells were cultured in DMEM/F-12, supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C in a humidified incubator containing 95% air and 5% CO2.
Oxygen Glucose Deprivation (OGD)/Reoxygenation (OGD/R) Model and Drug Treatment
H9c2 cells were incubated with a normal medium in a cell incubator for 24 h. Cells were then exposed to hypoxic conditions (oxygen deprivation, 1% O2) for 24 h in a culture medium with lower glucose and 1% FBS. After hypoxia, the cells were oxygenated under a normal oxygen concentration (reoxygenation) for 24 h in a normal medium. According to the previous study, propofol (Sigma-Aldrich, United States) was added respectively to the cells 1 h before and during the hypoxia-reoxygenation with different concentrations (5, 10, and 20 μM) (Zhao et al., 2015). U0126 (Beyotime, Haimen, China), MEK1/2 inhibitor, was added to the cells during OGD/R at a concentration of 20 μM. After H9c2 cells were incubated with a normal medium in cell incubator for 24 h, propofol and caspase inhibitor Z-VAD-FMK (Beyotime, Haimen, China) were respectively added to the cells for 24 h.
Cell Viability Assay
Cell viability was measured by a cell counting kit-8 (CCK-8), purchased from Tokyo, Japan. Cells were seeded in 96-well cell culture plates at a density of 2 × 104 cells/well. After 24 h in the culture, cells were added with different concentrations of propofol for hypoxia-oxygenation or just were respectively added with different concentrations of propofol and Z-VAD-FMK. Then, the supernatant fluid was removed and a medium containing 10% CCK- 8 reagent was added for 1–3 h incubation at 37°C. Eventually, the absorbance at 450 nm was measured using an AMR-100 automatic enzyme analyzer (Allsheng, Hangzhou, China).
Intracellular ROS Detection
In order to detect intracellular ROS, the cells were preloaded with 10 μM 2, 7-dichlorofluorescin diacetate (DCFH-DA, Beyotime, Haimen, China) for 20 min at 37°C. Afterward, the cells were washed using a culture medium without serum for at least three. A fluorescence microplate reader with an excitation wavelength of 488 nm and an emission wavelength of 525 nm was used to determine the intensity of DCF fluorescence.
Cell Apoptosis Assay
Cells were seeded into a 6-well cell culture plate and an established OGDR model, and treated with propofol. The FITC Annexin V Apoptosis Detection Kit (BD PharmingenTM, United States) was used to detect apoptotic cells according to the manufacturer’s protocol. First, cells were washed twice with cold PBS and then cells were resuspended in a 1X Binding Buffer at a concentration of 1 × 10ˆ6 cells/ml. Second, 100 μl of the solution (1 × 10ˆ5 cells) was transferred to a 5 ml culture tube. Third, 5 μl of FITC Annexin V and 5 μl PI was added. Fourth, the cell were gently vortexed and incubated for 15 min at room temperature (25°C) in the dark. Fifth, 400 μl of the 1X Binding Buffer was added to each tube. Lastly, the cells were analyzed via flow cytometry within 1 h. The proportion of apoptotic cells was calculated by FlowJo software.
Confocal Laser Scanning Fluorescence Microscopy
Confocal fluorescence microscopy was performed using a Laser Scanning Confocal Microscope (TCS SP5; Leica Microsystems, Inc., United States). After cells climbed to the carry sheet glass, we established an OGDR model and treated cells with propofol. The cells were then incubated for 15 min at room temperature with a 100 nM Mito-Tracker Red (Invitrogen, United States) to stain the mitochondrial membrane. Cells were fixed in 4% paraformaldehyde for 15 min and blocked with 5% bovine serum albumin (BSA) for 2 h, and then permeabilized with Triton-X 100 (0.1%) for 15 min. Cells were incubated with the primary mouse anti-Drp1 antibody (1:100) (BD Biosciences, United States) at 4°C overnight, washed with cold PBS, and incubated with Fluorescein-conjugated anti-mouse secondary antibody (1:100) (Proteintech, Wuhan, China) for 2 h at room temperature. The cells were then imaged at x630 magnification. Image J software was used to analyze the mitochondrial fragmentation count.
Mitochondria/Cytosol Fractionation
This assay was conducted using a Mitochondrial Isolation Kit (Pierce, Rockford, IL, United States) according to the manufacturers’ protocol. Cells were homogenized and the homogenates were centrifuged at 750 g for 10 min at 4°C. The pellet was kept as a mitochondrial fraction. The supernatant was centrifuged at 12,000 g for 15 min at 4°C. The supernatant was further centrifuged at 100,000 g for 1 h, and the supernatant was kept as the cytosol fraction. Cytochrome c in cytoplasm or mitochondria was detected with an immunoblot analysis, respectively.
Western Blotting
Cell lysates were separated on 6, 10, 12% SDS-PAGE, and transferred to polyvinylidene difluoride membranes (PVDF, Millipore, Billerica, MA, United States). The membranes were blocked and incubated with primary antibodies against anti-Drp1 (1:1000, BD Biosciences, United States), anti-pDrp1 (1:1000, CST, United States), anti-Fis1 (1:1000, Proteintech, China), anti-Mfn1 (1:1000, Proteintech, China), anti-Mfn2 (1:1000, CST, United States), anti-Opa1 (1:1000, BD Biosciences, United States), anti-ERK (1:1000, CST, United States), anti-pERK (1:1000, CST, United States), anti-Cytochrome c (1:1000, CST, United States), anti-Bax (1:1000, CST, United States), anti-Bak (1:1000, CST, United States), anti-Apaf1 (1:1000, Proteintech, China), anti-Caspase 9 (1:1000, CST, United States), anti-Caspase 6 (1:1000, CST, United States), anti-Caspase 7 (1:1000, CST, United States), anti-Caspase3 (1:1000, CST, United States), anti-COX IV (1:1000, CST, United States) and anti-GAPDH (1:1000, CST, United States) at 4°C overnight. After primary antibody incubation, the membranes were incubated with either goat anti-mouse or goat anti-rabbit horseradish peroxidase-conjugated secondary antibodies, at room temperature for 2 h. Immunoreactive bands were developed with an electrochemiluminescence (ECL) reagent (Thermo-Pierce, Rockford, IL, United States). Images were quantified using Gel-Pro Analyzer software (Media Cybernetics, Silver Spring, MD, United States).
Statistical Analysis
The results were presented as the mean ± standard deviation (SD). Statistical analysis was performed using the software Statistical Package for the Social Science (SPSS, Chicago, IL, United States). The analysis of variance (ANOVA) was used for comparisons among groups and differences with a P < 0.05, were considered statistically significant.
Results
Propofol Enhanced the Survival of H9c2 Cells and Decreased the ROS Level Against OGD/R Injury. Moreover, the H9c2 Cells Proliferation Under Normal Culture Conditions Was Inhibited by Propofol and Caspase Inhibitor Relieved the Effect of Propofol
Compared with the control group, cell viability was significantly decreased in the OGD/R group without propofol. But as the concentration of propofol (5, 10, and 20 μM) went up, H9c2 cells survival increased compared with the OGD/R group without propofol (Figure 1A). In this experiment, OGD/R obviously elevated the intracellular ROS level compared with control group. Interestingly, compared with the OGD/R group without propofol, the ROS level was obviously inhibited with the increasing dose of propofol (5, 10, and 20 μM) (Figure 1B). These results demonstrated that propofol inhibited cell death and decreased the intracellular ROS level induced by OGD/R in the H9c2 cells. Moreover, propofol (10 and 20 μM) inhibited the H9c2 cells proliferation under normal culture conditions and Z-VAD-FMK could relieve the decrease of cell viability induced by 20 μM propofol (Figure 1C). These results demonstrated that high concentrations of propofol could cause damage to the H9c2 cells by activating caspases.
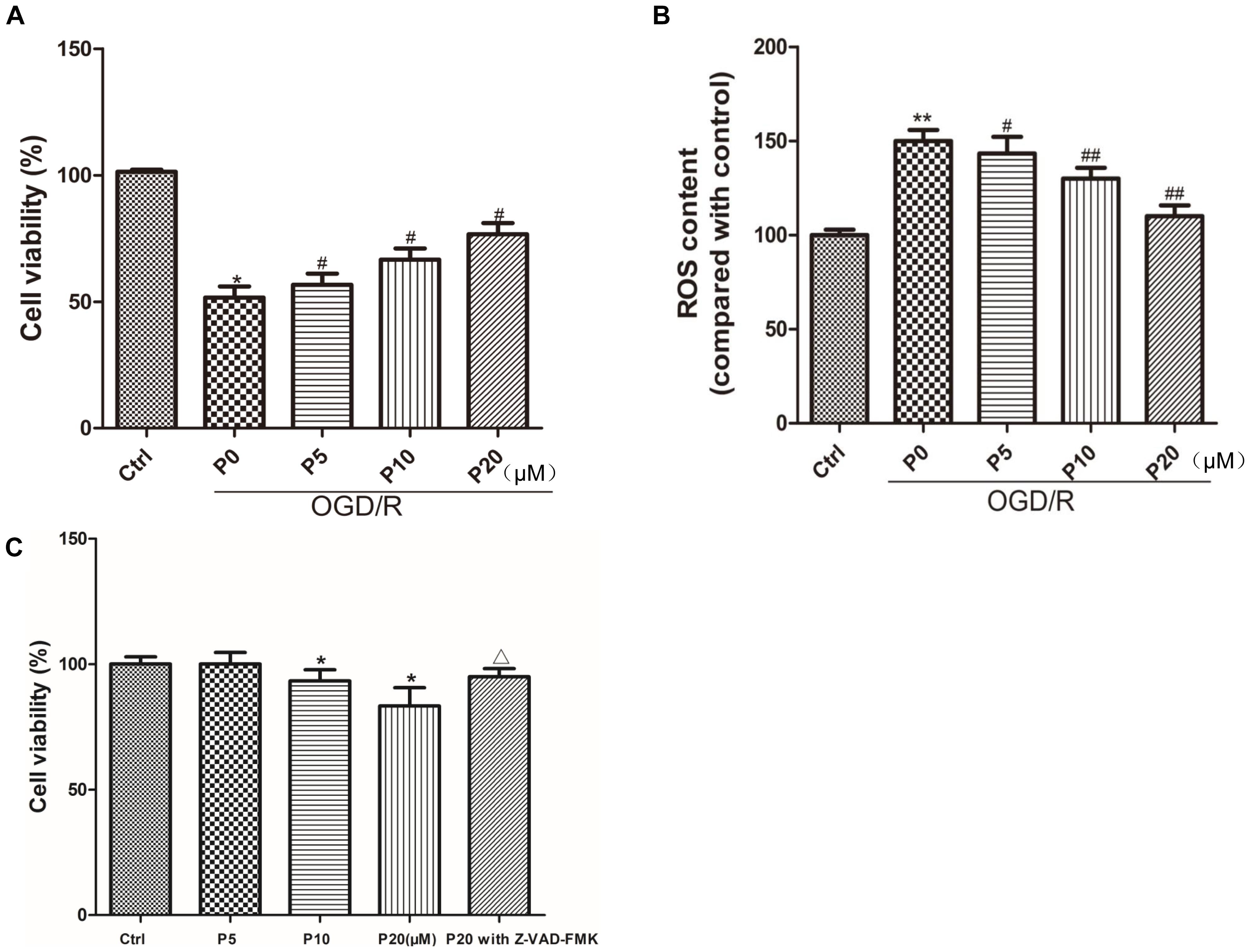
Figure 1. Propofol enhanced the survival of H9c2 cells and decreased ROS level against OGD/R injury. Moreover, H9c2 cells proliferation under normal culture conditions was inhibited by propofol and caspase inhibitor relieved the effect of propofol. (A,C) Cell viability was assessed by measuring the CCK8. The viability of control group was defined as 100%. (B) Effects of propofol (5, 10, and 20 μM) on increased intracellular ROS induced by OGD/R in H9c2 cells. The ROS contents of control group was defined as 100. The results were shown as mean ± SD from at least three independent experiments. ∗P < 0.05, ∗∗P < 0.01 versus control, #P < 0.05, ##P < 0.01 versus OGD/R group without propofol, ΔP < 0.05 versus 20 μM propofol group.
Propofol Inhibited Cell Apoptosis Induced by OGD/R in H9c2 Cells
Cardiomyocyte apoptosis in acute myocardial ischemia-reperfusion injury could be one of the main mechanisms. The results of the present study demonstrated that H9c2 cell apoptosis in the OGD/R group without propofol, was significantly increased compared with the control group. Compared with the OGD/R group without propofol, propofol alleviated H9c2 cell apoptosis induced by OGD/R, in a dose-dependent manner, at the range of 5∼20 μM (Figure 2). These results were consistent with a previous report that propofol could attenuate cardiomyocytes apoptosis induced by H2O2 (Liu et al., 2017).
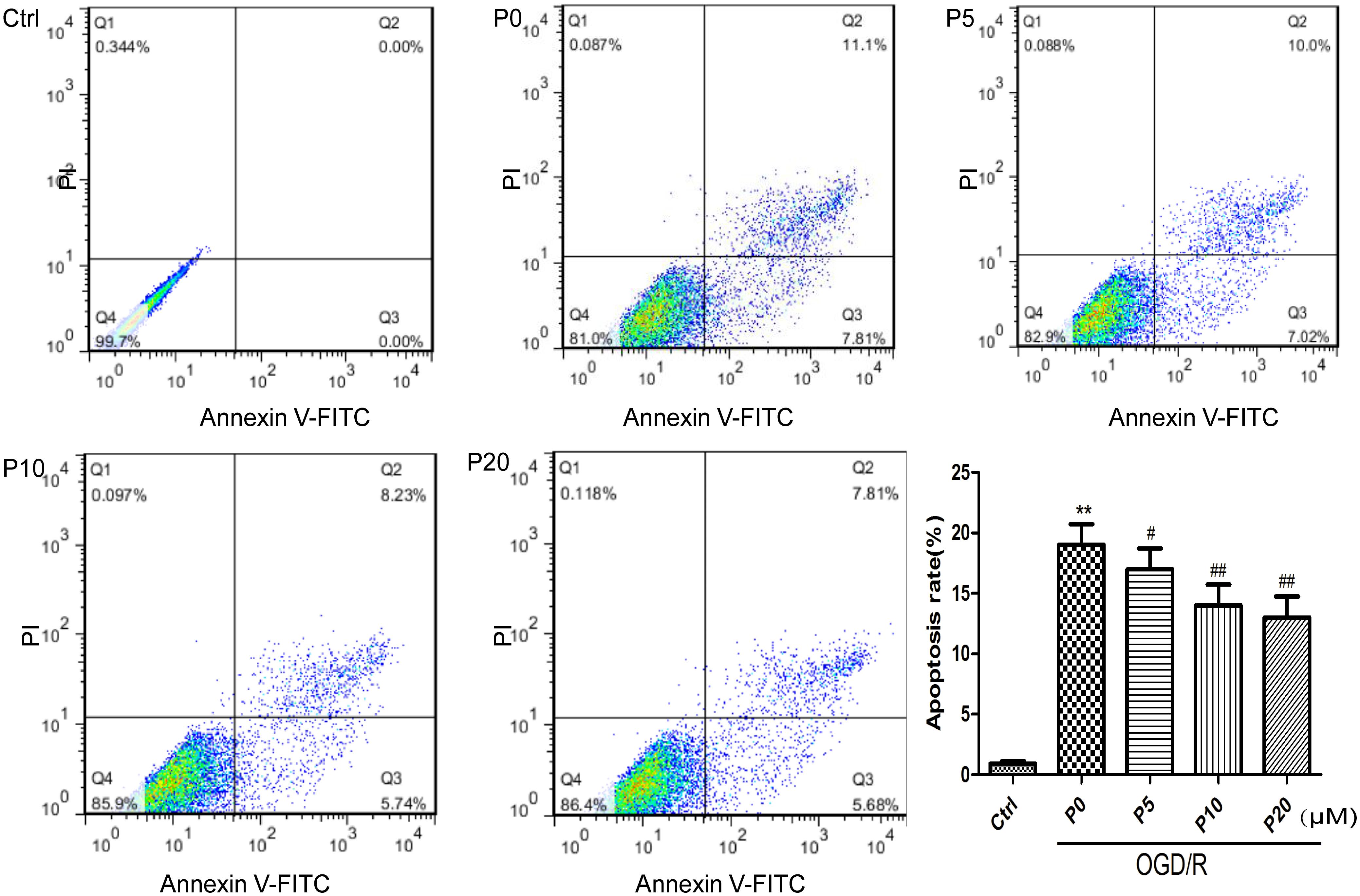
Figure 2. Propofol significantly inhibited H9c2 cells apoptosis induced by OGD/R injury. The cells were analyzed by flow cytometry. The data are presented as the mean ± SD of at least three independent experiments. ∗∗P < 0.01 versus control, #P < 0.05, ##P < 0.01 versus OGD/R group without propofol.
Propofol Inhibited Mitochondrial Fission in H9c2 Cells During OGD Injury
When oxidative stress occurs during acute I/R injury, mitochondrial fission can be caused in HL-1 cardiac cells (Ong et al., 2010). Compared with the control group, mitochondrial fragmentation was obvious, and the quantity of mitochondria increased in H9c2 cells during OGD injury (Figures 3A,B,D,E). Compared with the OGD group without propofol, propofol (20 μM) significantly inhibited mitochondrial fission and reduced the quantity of mitochondria (Figures 3B–E).
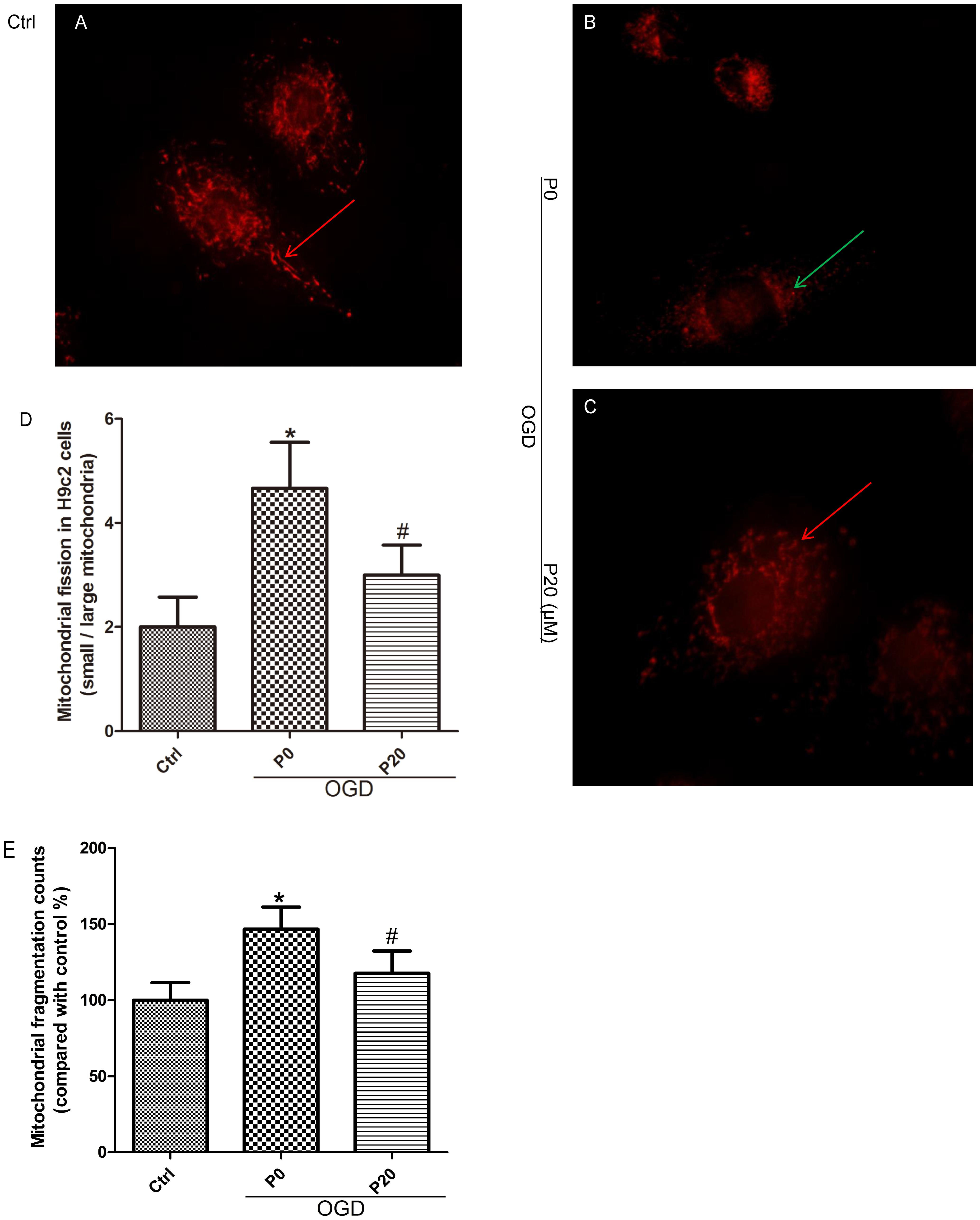
Figure 3. Propofol inhibited mitochondrial fission in H9c2 cells during OGD injury. (A,B,D,E) Compared with control group, small mitochondria (green arrows) increased in H9c2 cells during OGD injury. Scale bar: 10 μm. (B–E) Propofol increased the quantity and proportion of large mitochondria (red arrows) in H9c2 cells during OGD injury. The data are presented as the mean ± SD of at least three independent experiments. ∗P < 0.05 versus control, #P < 0.05, versus OGD group without propofol.
Propofol Alleviated High Levels of Mitochondrial Fusion and Fission During OGD/R in H9c2 Cells by Regulating Mitochondrial Membrane Remodeling Dynamin Family Proteins
It is well-accepted that Drp1 and Fis1 proteins promote mitochondrial fission, whereas Mfn1, Mfn2 and Opa1 proteins mediate mitochondrial fusion. Furthermore, high expression of Drp1, Mfn1 and Mfn2 may lead to cell death in the heart (Whelan et al., 2012; Park et al., 2014). The experimental results demonstrated that in the OGD/R group without propofol, the levels of Drp1 and Fis1 proteins were significantly elevated compared with the control group. Moreover, with the increase of propofol concentration, the levels of Drp1 and Fis1 proteins were significantly decreased during OGD/R injury (Figures 4A–C). These results may suggest that propofol could inhibit the high expression of mitochondrial fission proteins induced by OGD/R injury. Compared with the control group, the levels of Mfn1 and Mfn2 proteins were also significantly elevated during OGD/R injury. Additionally, with the increase of propofol concentration, the high levels of Mfn1 and Mfn2 proteins during OGD/R injury were significantly decreased (Figures 4A,D,E). However, there was no significant difference in the levels of Opa1 between the control and OGD/R without propofol group. Propofol did not influence the levels of Opa1 during OGD/R injury (Figures 4A,F). Propofol may also inhibit the high levels of some mitochondrial fusion proteins induced by OGD/R injury. The experimental results demonstrated that mitochondrial fission and mitochondrial fusion were significantly elevated by OGD/R injury and were alleviated by propofol.
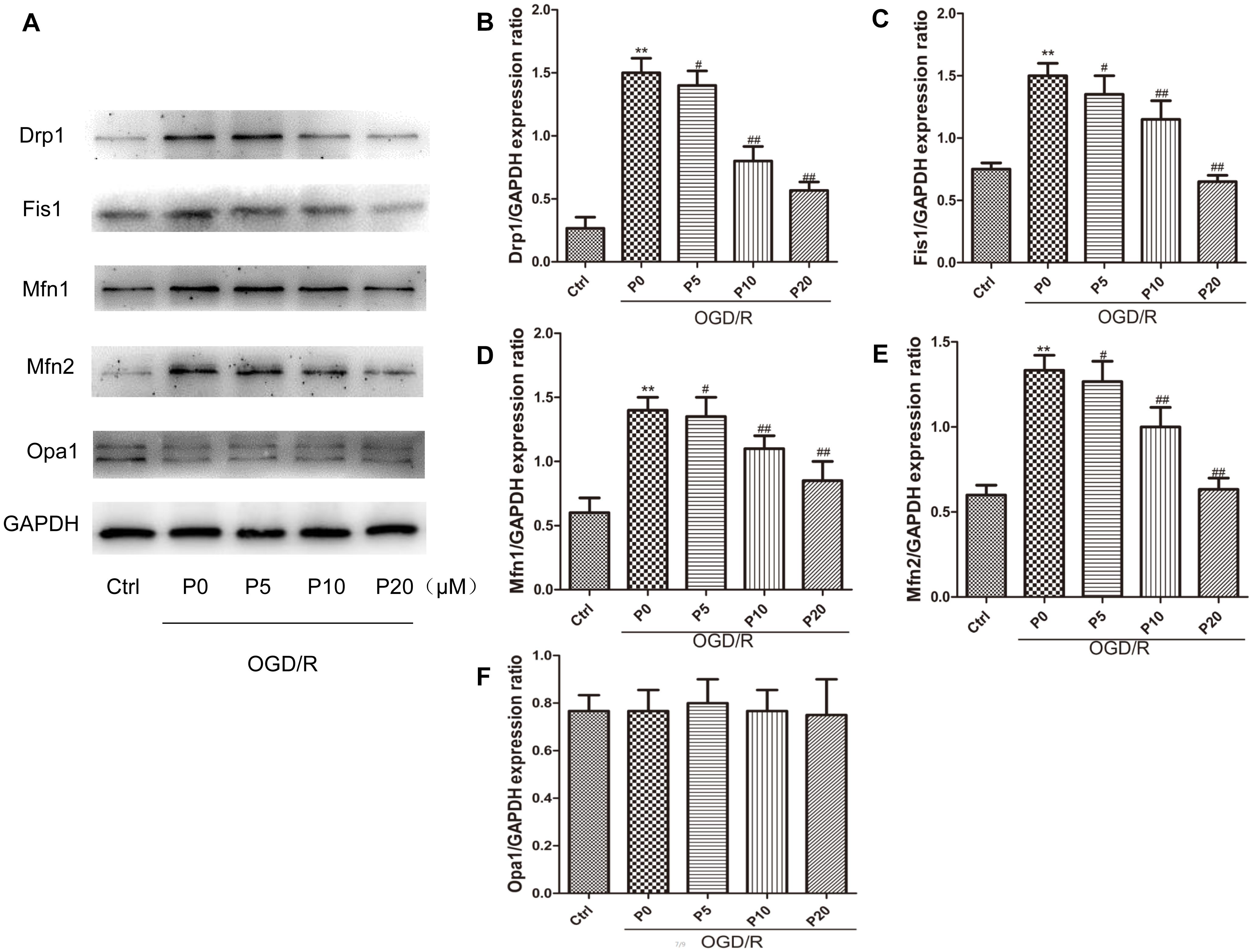
Figure 4. Propofol inhibited high expression of some mitochondrial dynamin family proteins induced by OGD/R in H9c2 cells. (A) Representative bands of Drp1, Fis1, Mfn1, Mfn2 and Opa1 by western blotting. (B–E) With the increase of propofol concentration (5, 10, 20 μM), the levels of Drp1, Fis1, Mfn1 and Mfn2 protein were significantly decreased during OGD/R injury. (F) However, there was no significant difference in the levels of Opa1 between control group and OGD/R group with or without propofol. The data are presented as the mean ± SD of at least three independent experiments. ∗∗P < 0.01 versus control, #P < 0.05, ##P < 0.01 versus OGD/R group without propofol.
Mitochondrial Morphology and Quantity Kept Stable and Propofol Inhibited Drp1 Colocalization With Mitochondria in H9c2 Cells During OGD/R Injury
From the experimental results, we observed that the mitochondrial morphology and quantity of H9c2 cells remained stable among the control group, the OGD/R without propofol group and the OGD/R with 20 μM propofol group (Figures 5A,D,G,J,K). The result was consistent with the levels of mitochondrial membrane remodeling dynamin family proteins. Previous research has shown that Drp1 in other normal cells distributed throughout the cytoplasm, with lower-levels of colocalization with the mitochondria (Wang et al., 2015). However, surprisingly we found that most Drp1 was collocated with the mitochondria, or surrounded them in the H9c2 cells, whether in the control or OGD/R groups (Figures 5C,F,I). But there was a lower fluorescence signal for Drp1 in the control group (Figure 5B), which was consistent with lower levels of Drp1. Using 20 μM propofol treatment, there was more Drp1 in the cytoplasm of the H9c2 cells during OGD/R injury, compared with the group that received no propofol treatment (Figures 5E,F,H,I). The results suggested that plenty of Drp1 colocalizing with mitochondria or by surrounding them, worked in mitochondrial fission and propofol could significantly prevent Drp1 from cytoplasm to mitochondria.
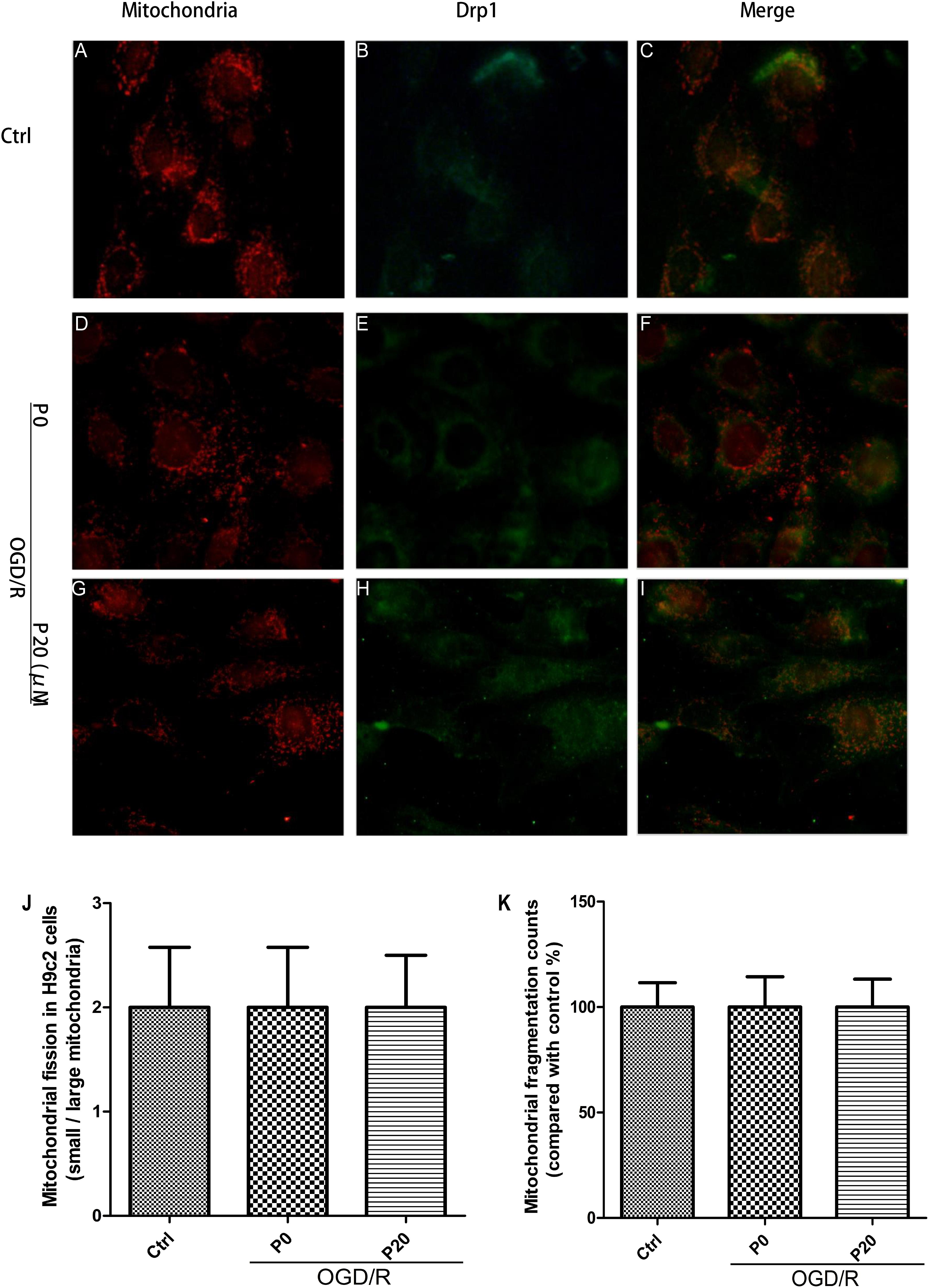
Figure 5. Mitochondrial morphology and quantity kept stable and propofol inhibited Drp1 colocalization with mitochondria in H9c2 cells during OGD/R injury. (A,D,G,J,K) Mitochondrial (red) morphology and quantity of H9c2 cells kept stable among control group, OGD/R group without propofol and OGD/R group with 20 μM propofol. Scale bar: 25 μm. (B,C) There was a lower fluorescence signal for Drp1 (green) and most Drp1 was collocated with the mitochondria or surrounded them in control group. (E,H) The distribution of Drp1 in OGD/R group with 20 μM propofol was more diffuse than the distribution of it in OGD/R group without propofol. (F) Plenty of Drp1 was collocated with mitochondria or surrounded them in OGD/R group without propofol. (I) Using 20 μM propofol treatment, Drp1 (green) was significantly prevented from cytoplasm to mitochondria during OGD/R injury. The data are presented as the mean ± SD of at least three independent experiments.
Propofol Could Inhibit Drp1 Phosphorylation Through Decreasing ERK Activation During OGD/R
As previously reported, ERK is phosphorylated when oxidative stress occurs, relating to apoptosis, but its mechanism remains unclear (Kodiha et al., 2009; Rasola et al., 2010). The experimental results demonstrated that during OGD/R, the levels of pERK were significantly decreased by propofol in a dose-dependent manner at the range of 10∼20 μM (Figures 6A,B). Furthermore, U0126 can markedly inhibit ERK phosphorylation (Figures 6A,C). ERK1/2 have been reported to phosphorylate Drp1 at serine 616, resulting in increased mitochondrial fission (Qi et al., 2011; Yu et al., 2011; Strack et al., 2013). The levels of pDrp1 (S616) during OGD/R were significantly decreased by 20 μM propofol and U0126 (Figures 6A,D,F). Mfn1 was also found to be phosphorylated by ERK, which favored Bak oligomerization contributing to apoptosis (Pyakurel et al., 2015). U0126 could not influence the higher levels of Mfn1 in H9c2 cells induced by OGD/R (Figures 6A,E). These results demonstrated that propofol could inhibit Drp1 phosphorylation in H9c2 cells by decreasing ERK activation during OGD/R.
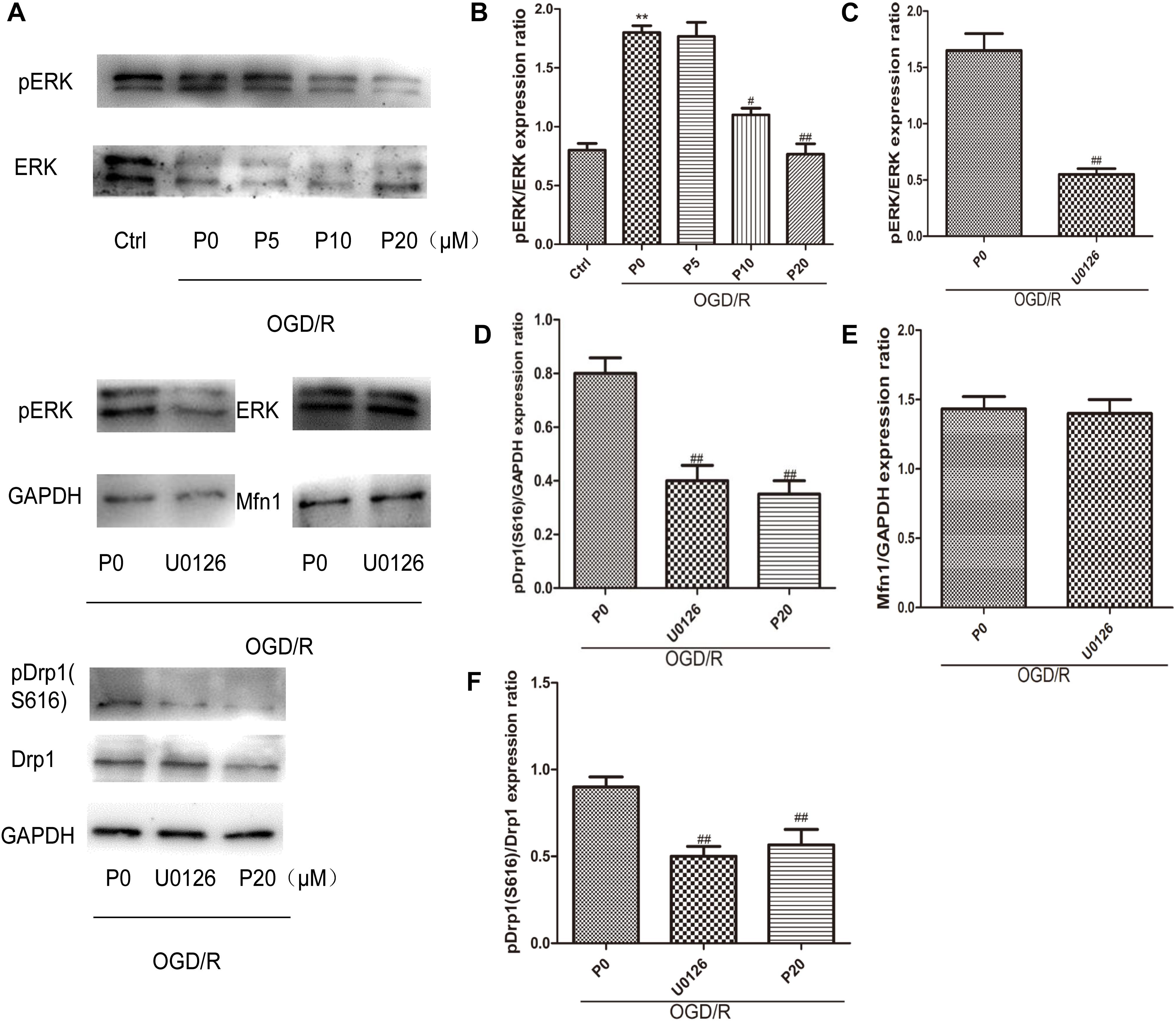
Figure 6. Propofol could inhibit Drp1 phosphorylation through decreasing ERK activation in H9c2 cells during OGD/R. (A) Representative bands of pERK, ERK, pDrp1, Drp1 and Mfn1 by western blotting. (B) With the increase of propofol concentration (10, 20 μM), the levels of pERK were significantly decreased during OGD/R injury. (C) U0126 can markedly inhibit ERK phosphorylation. (D,F) The levels of pDrp1 (S616) during OGD/R were significantly decreased by 20 μM propofol and U0126. (E) There was no significant difference in the levels of Mfn1 between OGD/R group without propofol and OGD/R group with U0126. The data are presented as the mean ± SD of at least three independent experiments. ∗∗P < 0.01 versus control, #P < 0.05, ##P < 0.01 versus OGD/R group without propofol.
Propofol Ameliorated H9c2 Cells Apoptosis During OGD/R via Inhibiting Mitochondrial Cytochrome c Release, Caspase-9, Caspase-6, Caspase-7, and Caspase-3 Activation
As is the case for most cells, the mitochondrial pathway (intrinsic pathway) is one of upstream apoptotic signaling. It involves the sequential release of cytochrome c, the recruitment of Apaf-1 (apoptotic protease-activating factor-1), and the activation of caspase-9 and downstream executioner caspases. The Bcl-2 family proapoptotic members Bax and Bak can mediate MOMP, releasing cytochrome c, along with other proapoptotic factors, into the cytosol (Frank et al., 2001). In the control group the levels of Bax, Bak, cytosol cytochrome c, Apaf-1, Cleaved caspase-9, Cleaved caspase-6, Cleaved caspase-7 and Cleaved caspase-3 were lower. Conversely, the levels of Bax, Bak, cytosol cytochrome c, Apaf-1, Cleaved caspase-9, Cleaved caspase-6, Cleaved caspase-7 and Cleaved caspase-3, were higher in the OGD/R without propofol group. In addition, with the increase of the propofol concentration (10, 20 μM), the levels of Bax, Bak, cytosol cytochrome c, Apaf-1, Cleaved caspase-9, Cleaved caspase-6, Cleaved caspase-7 and Cleaved caspase-3, were markedly reduced during OGD/R injury (Figures 7A–F,H–K). In the OGD/R without propofol group, the level of mitochondrial cytochrome c protein was significantly reduced compared with the control group. But with the increase of the propofol concentration (10, 20 μM), the level of mitochondrial cytochrome c protein was obviously elevated during OGD/R injury (Figures 7A,G). The experimental results demonstrated that propofol could ameliorate H9c2 cells apoptosis during OGD/R, via inhibiting the intrinsic pathway.
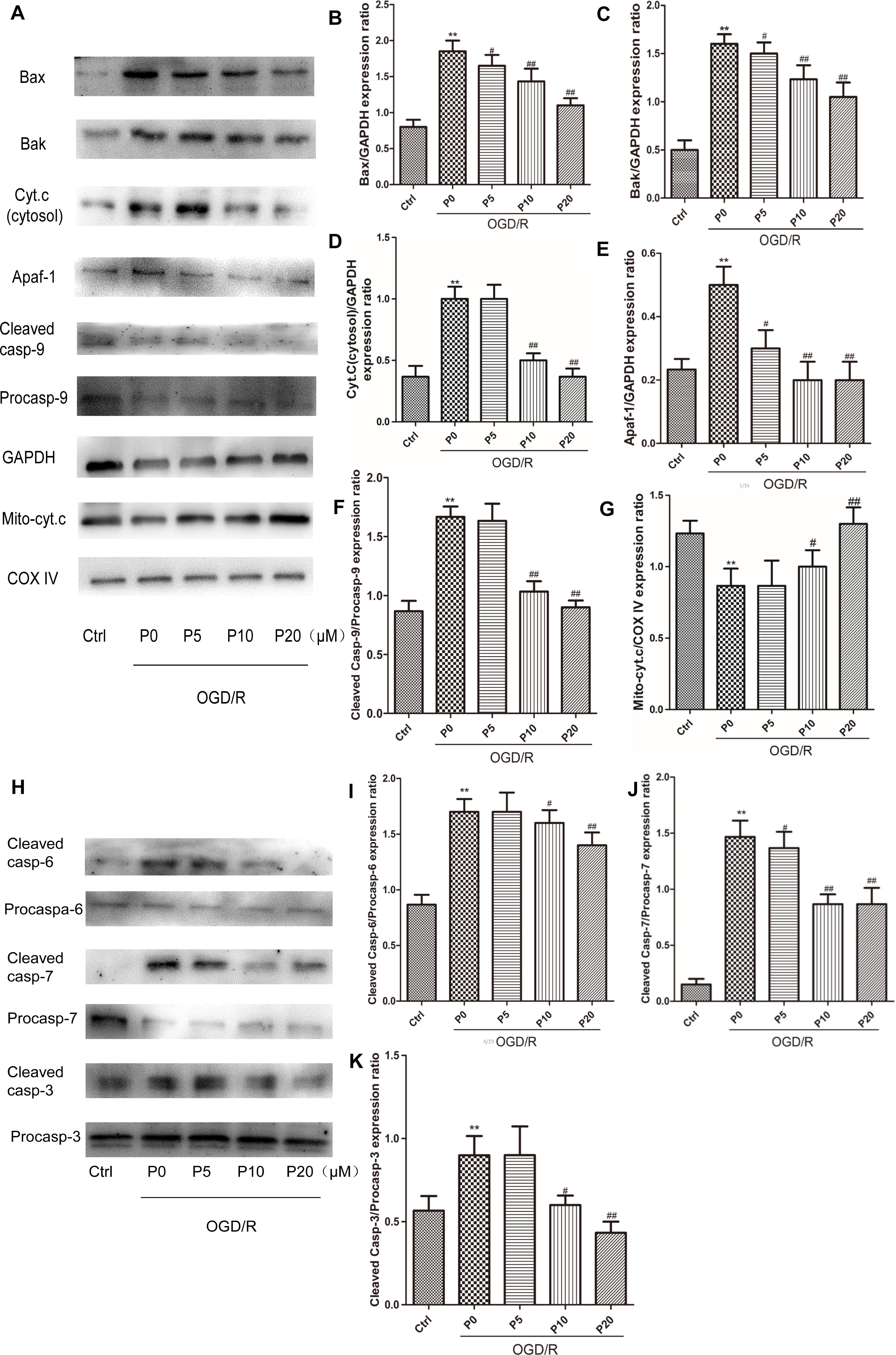
Figure 7. Propofol ameliorated H9c2 cells apoptosis during OGD/R via inhibiting intrinsic apoptosis pathway. (A,H) Representative bands of Bax, Bak, cytosol cytochrome c, Apaf-1, caspase-9, mitochondrial cytochrome c, caspase-6, caspase-7, and caspase-3 by western blotting. (B,C,E,J) With the increase of propofol concentration (5, 10, 20 μM), the levels of Bax, Bak, Apaf-1, and Cleaved caspase-7 were gradually reduced during OGD/R injury. (D,F,I,K) While the levels of cytosol cytochrome c, Cleaved caspase-9, Cleaved caspase-6 and Cleaved caspase-3 were markedly inhibited by 10 and 20 μM propofol during OGD/R injury. (G) And with the increase of propofol concentration (10, 20 μM), the level of mitochondrial cytochrome c protein was obviously elevated during OGD/R injury. The data are presented as the mean ± SD of at least three independent experiments. ∗∗P < 0.01 versus control, #P < 0.05, ##P < 0.01 versus OGD/R group without propofol.
Discussion
In the present study, we found that propofol could inhibit high levels of mitochondrial fission and fusion of the H9c2 cells, by regulating membrane remodeling dynamin family proteins during OGD/R injury and then ameliorate H9c2 cells apoptosis by the mitochondrial pathway. The concentration of propofol was consistent with those of previous investigations (Wang et al., 2009; Zhao et al., 2015). In our study, we observed that propofol at a concentration of 5 to 20 μM, significantly reduced cell death in a dose-dependent way and attenuated the level of ROS during OGD/R injury, which indicated that propofol had a strong protective effect against oxidative stress induced by I/R injury in cardiac myocytes. But when we treated H9c2 cells with propofol directly in a normal cell culture for 24 h, cell proliferation was obviously inhibited. And Z-VAD-FMK could relieve the decrease of cell viability induced by 20 μM propofol. This suggested that the protective effect of propofol was indeed due to the reduction of OGD/R damage, rather than the higher proliferation of the H9c2 cells during recovery. At the same time, it also demonstrated that high concentrations of propofol could cause damage to the H9c2 cells by activating caspases. As the concentration of propofol went up, propofol alleviated cell apoptosis induced by OGD/R in a dose-dependent manner, at the range of 5∼20 μM. Therefore, we further explored the mechanism of propofol inhibiting H9c2 cells apoptosis induced by OGD/R injury.
Previous studies showed that altering mitochondrial morphology affected cell apoptosis. Studies indicated that mitochondria underwent fission when HL-1 cardiac cells were subjected to simulated ischemia, generating fragmented mitochondria (Brady et al., 2006). We also observed that mitochondrial fission was obvious in H9c2 cells during OGD injury and propofol could significantly inhibited it. Mitochondrial ROS, which is generated during acute I/R injury is known to induce mitochondrial fission (Plotnikov et al., 2008). In our study, the level of ROS was higher in H9c2 cells during OGD/R injury. In the OGD/R without propofol group, the levels of Drp1, Fis1, Mfn1 and Mfn2 protein were significantly elevated compared with the control group. With the increase of propofol concentration (5, 10, 20 μM), the high levels of Drp1, Fis1, Mfn1 and Mfn2 protein during OGD/R injury were significantly decreased. The experimental results might demonstrate that mitochondrial fission and mitochondrial fusion were significantly elevated by OGD/R injury and were alleviated by propofol. In mitochondrial staining we observed that mitochondrial morphology and the quantity of H9c2 cells, remained stable in the control, OGD/R without propofol and OGD/R groups with 20 μM propofol. The result was consistent with the levels of mitochondrial membrane remodeling dynamin family proteins. Though mitochondrial fission was obvious in H9c2 cells during OGD injury, mitochondrial morphology and the quantity of H9c2 cells remained stable during OGD/R injury. This difference may be due to a high level of mitochondrial fusion during reperfusion.
It has been reported that propofol increases both the activities and protein expressions of superoxide dismutase and catalase to reduce intracellular ROS (Zhao et al., 2015). Additionally, it has been shown that mitochondrial fission induced by Drp1 could reduce inner membrane proton leak/uncoupling in other cell types, contributing to ROS production from mitochondria (Galloway et al., 2012a; Wang et al., 2015). Thus, propofol may reduce ROS production in H9c2 cells by decreasing the levels of Drp1 during OGD/R injury. ERK1/2 can be phosphorylated when oxidative stress occurs, which further phosphorylates Drp1 at serine 616, resulting in increased mitochondrial fission (Kodiha et al., 2009; Yu et al., 2011). Our results demonstrated that in immunofluorescence, plenty of Drp1 colocalizing with mitochondria or by surrounding them, worked in mitochondrial fission. Moreover, propofol could inhibit Drp1 phosphorylation at serine 616 in H9c2 cells by decreasing ERK activation during OGD/R, thus resulting in inhibiting mitochondrial fission.
A previous study also showed that genetic inhibition of Drp1 in HL-1 cardiac cells, not only inhibited the mitochondrial fission induced by acute I/R injury but also reduced cell death in this setting (Ong et al., 2010). Drp1 has been demonstrated to co-localize with Bax at the outer mitochondrial membrane in response to apoptotic stimuli (Karbowski et al., 2002). Bax assembles with Bak and Drp1 in the form of a ring, to delineate the area required for MOMP (Grosse et al., 2016). Interestingly, we found that propofol could inhibit the high expression of Drp1, Fis1, Bax and Bak proteins induced by OGD/R injury. Fis1 has been demonstrated to promote the migration of Drp1 to mitochondria in the cytoplasm (Stojanovski et al., 2004). We observed that in immunofluorescence, propofol can significantly prevent Drp1 from cytoplasm to mitochondria during OGD/R injury. Therefore, propofol could reduce Drp1 colocalization with mitochondria by lowering levels of Drp1 and Fis1. Propofol may inhibit outer membrane permeabilization by reducing the high levels of Drp1, Bax and Bak during OGD/R injury.
When Mfn1 is phosphorylated by ERK, mitochondria fragment and Bak oligomerization is favored, therefore cytochrome c release and cell death (Pyakurel et al., 2015). Our results demonstrated that propofol could significantly decrease ERK activation in H9c2 cells during OGD/R injury. U0126 did not influence the higher levels of non-phosphorylated Mfn1 in H9c2 cells induced by OGD/R. We speculate that propofol could still inhibit Mfn1 phosphorylation in H9c2 cells, by decreasing ERK activation during OGD/R injury. In the heart, acceleration of Mfn1 degradation by membrane-associated RING-CH (MARCH5) under stress, remains an important quality control system that inhibits cell death (Park et al., 2014). Oxidative stress with H2O2 leads to concurrent increases in Mfn2 expression and apoptosis in cultured neonatal rat cardiomyocytes, and Mfn2 silencing inhibits oxidative stress-induced apoptosis in H9c2 cells. Mfn2 also suppresses phosphatidylinositol 3 kinase-protein kinase B (PI3K-Akt) cell survival signaling pathways (Shen et al., 2007). Mfn2 has been demonstrated to co-localize with both Bax and Bak in the outer mitochondrial membrane (Neuspiel et al., 2005). Our data indicated that propofol could inhibit the high expression of the Mfn1 and Mfn2 protein induced by OGD/R injury. OPA1 mediates mitochondrial inner membrane fusion and regulates mitochondrial cristae morphology (Olichon et al., 2003; Cipolat et al., 2004). Anti-apoptotic effects of Opa1 have been attributed to its role in regulating mitochondrial cristae morphology and cytochrome c distribution (Korwitz et al., 2016). However, in our study, propofol did not influence the levels of Opa1 during OGD/R injury and there were no significant changes of Opa1 in OGD/R injury. This could be because the H9c2 cells were less damaged, which did not damage the mitochondrial inner membrane, but affected the mitochondrial outer membrane.
To further test that propofol inhibits higher outer mitochondrial membrane permeabilization induced by OGD/R injury, we measured the levels of cytochrome c in the cytoplasm or mitochondria. Our results indicated that cytochrome c was released into the cytoplasm and the process was inhibited by propofol during OGD/R injury. Cytochrome c, Apaf-1 and procaspase-9 in the cytoplasm combine to form the apoptosome, which leads to procaspase-9 activation (Srinivasula et al., 1998; Zhang et al., 2002). Active caspase-9 then activates the executioner caspase-3, -6, and -7 to complete apoptosis. We found that the expression levels of Apaf-1 and the activities of caspase-9, -3, -6, and -7 were higher in H9c2 cells during OGD/R injury, which could be reversed by propofol. This demonstrated that propofol could reduce H9c2 cells apoptosis during OGD/R injury by inhibiting the mitochondria pathway, which was due to reducing high levels of mitochondrial fusion and fission induced by OGD/R injury (Figure 8).
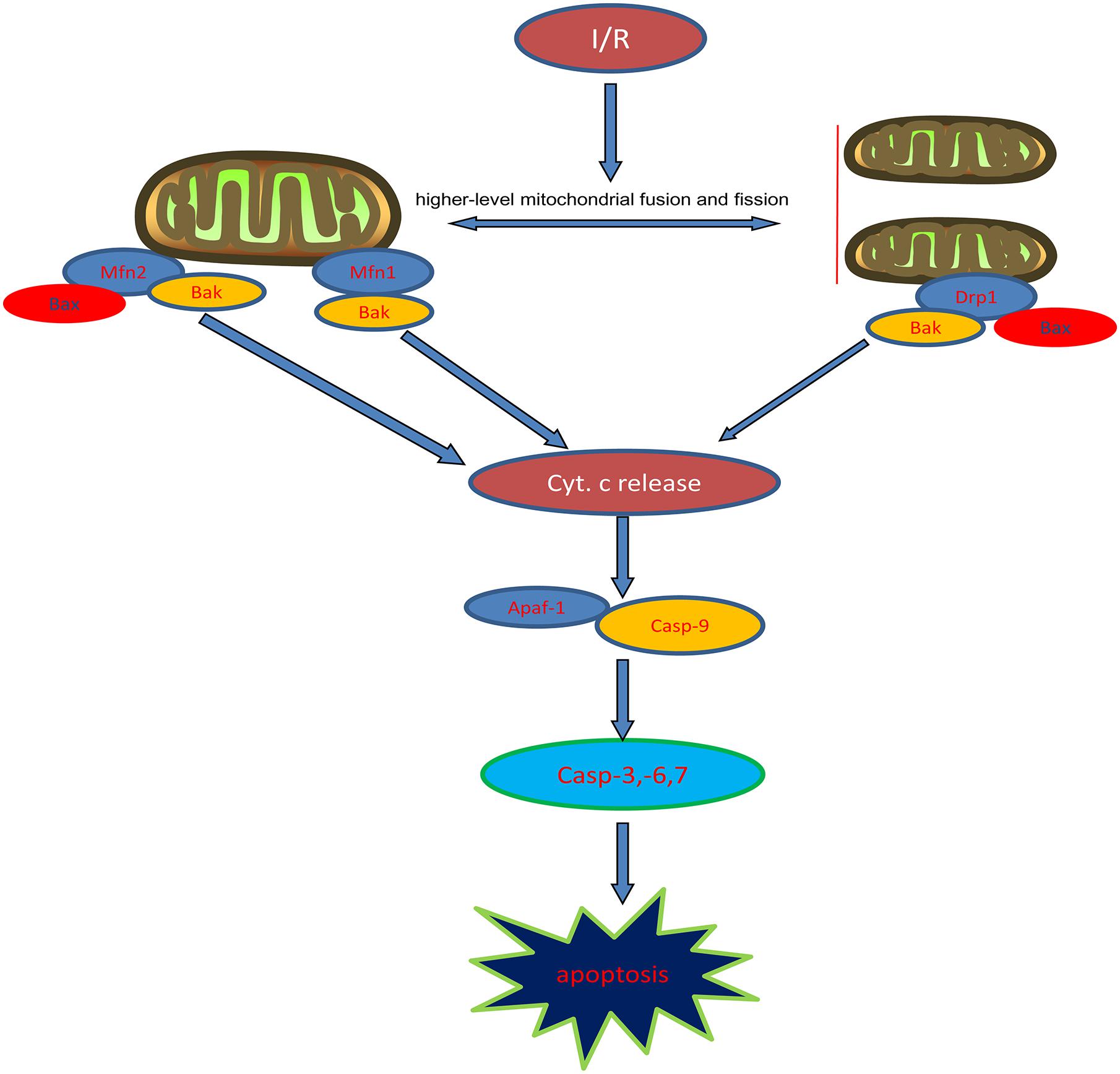
Figure 8. Diagram of mitochondrial dynamics regulating intrinsic apoptosis pathway.
However, all our experimental results were limited to the level of cells in vitro, and the study on the regulation of propofol on mitochondrial morphology was limited to the regulation of the protein level. Further animal experiments need to be conducted to verify our experimental results, and the effect of propofol on mitochondrial fusion and fission may be studied from other regulatory levels. In short, our study may reveal a new cardioprotective mechanism of propofol during acute ischemia/reperfusion injury.
Author Contributions
LZ wrote the manuscript and conceived and conducted the entire study. ZW, JH, and YW wrote the manuscript and amended the references. JZ, DZha, and DZho edited the whole article and offered technical support. HZ, WZ, and MY offered technical assistance and reviewed the text. SZ and JP performed the experiments and analyzed the data. All authors have read and approved the manuscript.
Funding
This project was supported by the National Natural Science Foundation of China (Grant Nos. 81570293 and 81471232), the Scientific Research Foundation of Shanghai Jiao Tong University School of Medicine (Grant No. 12XJ3008), the Shanghai Health Committee Key Weak Discipline Construction Project (Grant No. 2016ZB0205), and the Scientific Research Foundation of Shanghai Songjiang District Committee of Science and Technology (Grant No. 2017sjkjgg31).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Brady, N. R., Hamacher-Brady, A., and Gottlieb, R. A. (2006). Proapoptotic BCL-2 family members and mitochondrial dysfunction during ischemia/reperfusion injury, a study employing cardiac HL-1 cells and GFP biosensors. Biochim. Biophys. Acta Bioenerget. 1757, 667–678. doi: 10.1016/j.bbabio.2006.04.011
Cipolat, S., de Brito, O. M., Dal Zilio, B., and Scorrano, L. (2004). OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. U.S.A. 101, 15927–15932. doi: 10.1073/pnas.0407043101
Frank, S., Gaume, B., Bergmann-Leitner, E. S., Leitner, W. W., Robert, E. G., Catez, F., et al. (2001). The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell 1, 515–525. doi: 10.1016/S1534-5807(01)00055-7
Galloway, C. A., Lee, H., Nejjar, S., Jhun, B. S., Yu, T. Z., Hsu, W., et al. (2012a). Transgenic control of mitochondrial fission induces mitochondrial uncoupling and relieves diabetic oxidative stress. Diabetes Metab. Res. Rev. 61, 2093–2104. doi: 10.2337/db11-1640
Galloway, C. A., Lee, H., and Yoon, Y. (2012b). Mitochondrial morphology-emerging role in bioenergetics. Free Radic. Biol. Med. 53, 2218–2228. doi: 10.1016/j.freeradbiomed.2012.09.035
Grosse, L., Wurm, C. A., Bruser, C., Neumann, D., Jans, D. C., and Jakobs, S. (2016). Bax assembles into large ring-like structures remodeling the mitochondrial outer membrane in apoptosis. EMBO J. 35, 402–413. doi: 10.15252/embj.201592789
Hanouz, J. L., Yvon, A., Flais, F., Rouet, R., Ducouret, P., Bricard, H., et al. (2003). Propofol decreases reperfusion-induced arrhythmias in a model of “border zone” between normal and ischemic-reperfused guinea pig myocardium. Anesth. Analg. 97, 1230–1238. doi: 10.1213/01.Ane.0000086731.87098.99
Haunstetter, A., and Izumo, S. (1998). Apoptosis – basic mechanisms and implications for cardiovascular disease. Circ. Res. 82, 1111–1129. doi: 10.1161/01.RES.82.11.1111
Ishihara, N., Nomura, M., Jofuku, A., Kato, H., Suzuki, S. O., Masuda, K., et al. (2009). Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 11, 958–966. doi: 10.1038/ncb1907
Jin, Y. C., Kim, W., Ha, Y. M., Shin, I. W., Sohn, J. T., Kim, H. J., et al. (2009). Propofol limits rat myocardial ischemia and reperfusion injury with an associated reduction in apoptotic cell death in vivo. Vasc. Pharmacol. 50, 71–77. doi: 10.1016/j.vph.2008.10.002
Karbowski, M., Lee, Y. J., Gaume, B., Jeong, S. Y., Frank, S., Nechushtan, A., et al. (2002). Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J. Cell Biol. 159, 931–938. doi: 10.1083/jcb.200209124
Ko, S. H., Yu, C. W., Lee, S. K., Choe, H., Chung, M. J., Kwak, Y. G., et al. (1997). Propofol attenuates ischemia-reperfusion injury in the isolated rat heart. Anesth. Analg. 85, 719–724. doi: 10.1097/00000539-199710000-00002
Kodiha, M., Banski, P., and Stochaj, U. (2009). Interplay between MEK and PI3 kinase signaling regulates the subcellular localization of protein kinases ERK1/2 and Akt upon oxidative stress. FEBS Lett. 583, 1987–1993. doi: 10.1016/j.febslet.2009.05.011
Kokita, N., and Hara, A. (1996). Propofol attenuates hydrogen peroxide-induced mechanical and metabolic derangements in the isolated rat heart. Anesthesiology 84, 117–127. doi: 10.1097/00000542-199601000-00014
Korwitz, A., Merkwirth, C., Richter-Dennerlein, R., Troder, S. E., Sprenger, H. G., Quiros, P. M., et al. (2016). Loss of OMA1 delays neurodegeneration by preventing stress-induced OPA1 processing in mitochondria. J. Cell Biol. 212, 157–166. doi: 10.1083/jcb.201507022
Li, W. G., Zhang, Y., Liu, Y. R., Yue, F., Lu, Y. M., Qiu, H. R., et al. (2012). In vitro kinetic evaluation of the free radical scavenging ability of propofol. Anesthesiology 116, 1258–1266. doi: 10.1097/ALN.0b013e3182567dcc
Liu, X. R., Cao, L., Li, T., Chen, L. L., Yu, Y. Y., Huang, W. J., et al. (2017). Propofol attenuates H2O2-induced oxidative stress and apoptosis via the mitochondria- and ER-medicated pathways in neonatal rat cardiomyocytes. Apoptosis 22, 639–646. doi: 10.1007/s10495-017-1349-3
Montessuit, S., Somasekharan, S. P., Terrones, O., Lucken-Ardjomande, S., Herzig, S., Schwarzenbacher, R., et al. (2010). Membrane remodeling induced by the dynamin-related protein Drp1 stimulates bax oligomerization. Cell 142, 889–901. doi: 10.1016/j.cell.2010.08.017
Neuspiel, M., Zunino, R., Gangaraju, S., Rippstein, P., and McBride, H. (2005). Activated mitofusin 2 signals mitochondrial fusion, interferes with Bax activation, and reduces susceptibility to radical induced depolarization. J. Biol. Chem. 280, 25060–25070. doi: 10.1074/jbc.M501599200
Olichon, A., Baricault, L., Gas, N., Guillou, E., Valette, A., Belenguer, P., et al. (2003). Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 278, 7743–7746. doi: 10.1074/jbc.C200677200
Ong, S. B., Kalkhoran, S. B., Hernandez-Resendiz, S., Samangouei, P., Ong, S. G., and Hausenloy, D. J. (2017). Mitochondrial-shaping proteins in cardiac health and disease – the long and the short of It! Cardiovasc. Drugs Ther. 31, 87–107. doi: 10.1007/s10557-016-6710-1
Ong, S. B., Subrayan, S., Lim, S. Y., Yellon, D. M., Davidson, S. M., and Hausenloy, D. J. (2010). Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 121, 2012–2022. doi: 10.1161/Circulationaha.109.906610
Park, Y. Y., Nguyen, O. T. K., Kang, H., and Cho, H. (2014). MARCH5-mediated quality control on acetylated Mfn1 facilitates mitochondrial homeostasis and cell survival. Cell Death Dis. 5:e1172. doi: 10.1038/Cddis.2014.142
Plotnikov, E. Y., Vasileva, A. K., Arkhangelskaya, A. A., Pevzner, I. B., Skulachev, V. P., and Zorov, D. B. (2008). Interrelations of mitochondrial fragmentation and cell death under ischemia/reoxygenation and UV-irradiation: protective effects of SkQ1, lithium ions and insulin. FEBS Lett. 582, 3117–3124. doi: 10.1016/j.febslet.2008.08.002
Pyakurel, A., Savoia, C., Hess, D., and Scorrano, L. (2015). Extracellular regulated kinase phosphorylates mitofusin 1 to control mitochondrial morphology and apoptosis. Mol. Cell. 58, 244–254. doi: 10.1016/j.molcel.2015.02.021
Qi, X., Disatnik, M. H., Shen, N., Sobel, R. A., and Mochly-Rosen, D. (2011). Aberrant mitochondrial fission in neurons induced by protein kinase C delta under oxidative stress conditions in vivo. Mol. Biol. Cell 22, 256–265. doi: 10.1091/mbc.E10-06-0551
Rasola, A., Sciacovelli, M., Chiara, F., Pantic, B., Brusilow, W. S., and Bernardi, P. (2010). Activation of mitochondrial ERK protects cancer cells from death through inhibition of the permeability transition. Proc. Natl. Acad. Sci. U.S.A. 107, 726–731. doi: 10.1073/pnas.0912742107
Schaper, J., Meiser, E., and Stammler, G. (1985). Ultrastructural morphometric analysis of myocardium from dogs, rats, hamsters, mice, and from human hearts. Circ. Res. 56, 377–391. doi: 10.1161/01.RES.56.3.377
Shen, T., Zheng, M., Cao, C. M., Chen, C. L., Tang, J., Zhang, W. R., et al. (2007). Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis. J. Biol. Chem. 282, 23354–23361. doi: 10.1074/jbc.M702657200
Srinivasula, S. M., Ahmad, M., Fernandes-Alnemri, T., and Alnemri, E. S. (1998). Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol. Cell. 1, 949–957. doi: 10.1016/S1097-2765(00)80095-7
Stojanovski, D., Koutsopoulos, O. S., Okamoto, K., and Ryan, M. T. (2004). Levels of human Fis1 at the mitochondrial outer membrane regulate mitochondrial morphology. J. Cell Sci. 117, 1201–1210. doi: 10.1242/jc.01058
Strack, S., Wilson, T. J., and Cribbs, J. T. (2013). Cyclin-dependent kinases regulate splice-specific targeting of dynamin-related protein 1 to microtubules. J. Cell Biol. 201, 1037–1051. doi: 10.1083/jcb.201210045
Vasileiou, I., Xanthos, T., Koudouna, E., Perrea, D., Klonaris, C., Katsargyris, A., et al. (2009). Propofol: a review of its non-anaesthetic effects. Eur. J. Pharmacol. 605, 1–8. doi: 10.1016/j.ejphar.2009.01.007
Wahr, J., Vender, J., Gilbert, H. C., Spiess, B., Horrow, J. C., and Maddi, R. (2000). Effect of propofol with and without EDTA on haemodynamics and calcium and magnesium homeostasis during and after cardiac surgery. Intensive Care Med. 26, S443–S451. doi: 10.1007/Pl00003788
Wang, B. H., Shravah, J., Luo, H. L., Raedschelders, K., Chen, D. D. Y., and Ansley, D. M. (2009). Propofol protects against hydrogen peroxide-induced injury in cardiac H9c2 cells via Akt activation and Bcl-2 up-regulation. Biochem. Biophys. Res. Commun. 389, 105–111. doi: 10.1016/j.bbrc.2009.08.097
Wang, L., Yu, T. Z., Lee, H., O’Brien, D. K., Sesaki, H., and Yoon, Y. (2015). Decreasing mitochondrial fission diminishes vascular smooth muscle cell migration and ameliorates intimal hyperplasia. Cardiovasc. Res. 106, 272–283. doi: 10.1093/cvr/cvv005
Whelan, R. S., Konstantinidis, K., Wei, A. C., Chen, Y., Reyna, D. E., Jha, S., et al. (2012). Bax regulates primary necrosis through mitochondrial dynamics. Proc. Natl. Acad. Sci. U.S.A. 109, 6566–6571. doi: 10.1073/pnas.1201608109
Xia, Z. Y., Godin, D. V., Chang, T. K. H., and Ansley, D. M. (2003). Dose-dependent protection of cardiac function by propofol during ischemia and early reperfusion in rats: effects on 15-F-2t-isoprostane formation. Can. J. Physiol. Pharm. 81, 14–21. doi: 10.1139/Y02-170
Yu, T. Z., Jhun, B. S., and Yoon, Y. (2011). High-glucose stimulation increases reactive oxygen species production through the calcium and mitogen-activated protein kinase-mediated activation of mitochondrial fission. Antiox. Redox Signal. 14, 425–437. doi: 10.1089/ars.2010.3284
Zhang, Y. P., Chong, E., and Herman, B. (2002). Age-associated increases in the activity of multiple caspases in Fisher 344 rat organs. Exp. Gerontol. 37, 777–789. doi: 10.1016/S0531-5565(02)00013-X
Keywords: propofol, ischemia, reperfusion, mitochondrial dynamics, apoptosis
Citation: Zhao L, Zhuang J, Wang Y, Zhou D, Zhao D, Zhu S, Pu J, Zhang H, Yin M, Zhao W, Wang Z and Hong J (2019) Propofol Ameliorates H9c2 Cells Apoptosis Induced by Oxygen Glucose Deprivation and Reperfusion Injury via Inhibiting High Levels of Mitochondrial Fusion and Fission. Front. Pharmacol. 10:61. doi: 10.3389/fphar.2019.00061
Received: 25 September 2018; Accepted: 18 January 2019;
Published: 12 February 2019.
Edited by:
Changhua Wang, Wuhan University, ChinaReviewed by:
Klara Komici, University of Molise, ItalyMilica S. Prostran, University of Belgrade, Serbia
Copyright © 2019 Zhao, Zhuang, Wang, Zhou, Zhao, Zhu, Pu, Zhang, Yin, Zhao, Wang and Hong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zejian Wang, wangzejian@sjtu.edu.cn Jiang Hong, jhong.pku@163.com