 Monica de Gaetano1*
Monica de Gaetano1* Caitriona McEvoy1,2
Caitriona McEvoy1,2 Darrell Andrews1Antonino Cacace1
Darrell Andrews1Antonino Cacace1 Jonathan Hunter1
Jonathan Hunter1 Eoin Brennan1
Eoin Brennan1 Catherine Godson1
Catherine Godson1- 1UCD Diabetes Complications Research Centre, Conway Institute and UCD School of Medicine, University College Dublin, Dublin, Ireland
- 2Renal Transplant Program, University Health Network, Toronto, ON, Canada
Diabetes and its associated chronic complications present a healthcare challenge on a global scale. Despite improvements in the management of chronic complications of the micro-/macro-vasculature, their growing prevalence and incidence highlights the scale of the problem. It is currently estimated that diabetes affects 425 million people globally and it is anticipated that this figure will rise by 2025 to 700 million people. The vascular complications of diabetes including diabetes-associated atherosclerosis and kidney disease present a particular challenge. Diabetes is the leading cause of end stage renal disease, reflecting fibrosis leading to organ failure. Moreover, diabetes associated states of inflammation, neo-vascularization, apoptosis and hypercoagulability contribute to also exacerbate atherosclerosis, from the metabolic syndrome to advanced disease, plaque rupture and coronary thrombosis. Current therapeutic interventions focus on regulating blood glucose, glomerular and peripheral hypertension and can at best slow the progression of diabetes complications. Recently advanced knowledge of the pathogenesis underlying diabetes and associated complications revealed common mechanisms, including the inflammatory response, insulin resistance and hyperglycemia. The major role that inflammation plays in many chronic diseases has led to the development of new strategies aiming to promote the restoration of homeostasis through the “resolution of inflammation.” These strategies aim to mimic the spontaneous activities of the ‘specialized pro-resolving mediators’ (SPMs), including endogenous molecules and their synthetic mimetics. This review aims to discuss the effect of SPMs [with particular attention to lipoxins (LXs) and resolvins (Rvs)] on inflammatory responses in a series of experimental models, as well as evidence from human studies, in the context of cardio- and reno-vascular diabetic complications, with a brief mention to diabetic retinopathy (DR). These data collectively support the hypothesis that endogenously generated SPMs or synthetic mimetics of their activities may represent lead molecules in a new discipline, namely the ‘resolution pharmacology,’ offering hope for new therapeutic strategies to prevent and treat, specifically, diabetes-associated atherosclerosis, nephropathy and retinopathy.
Introduction
Diabetes and its associated complications pose a challenge to human health on a global scale. It is estimated that 425 m people are currently living with diabetes and this is predicted to rise to 700 m by 2025 The Lancet, 2018. The rapid rise in diabetes and its associated complications over the past three decades reflects numerous factors including aging, obesity, urbanization and greater longevity amongst patients (Zimmet et al., 2001). Among NCCDs, diabetes is one of the major global causes of premature mortality. It is frequently underestimated because very often persons with diabetes die from causes related to co-morbidities (Lee I.M. et al., 2012).
The prevalence of diabetes in adults worldwide is predicted to be higher in developed than in developing countries, while, the incidence of diabetes is predicted to be higher in developing countries (Wild et al., 2004). Thus, by the year 2030, the countries with the largest number of people with diabetes are predicted to be India, China and the United States; the countries with increased prevalence of overweight and obese inhabitants, the main drivers of T2D (Yach et al., 2006). The burden on already challenged health care systems is unprecedented.
Diabetes is essentially a disorder of glucose homeostasis. Conventionally, diabetes has been classified as Type-1, Type-2 and gestational diabetes. In T1D, autoimmune destruction of the β-cells of the pancreas creates an insulin-deficient state where patients are dependent on exogenous insulin for survival. The precise mechanisms underlying the pathogenesis remain elusive, but it is likely that genetic and environmental factors converge to drive an autoimmune response. The observed increasing incidence of T1D in developed nations is thought to reflect responses to environmental triggers. Historically, T1D is considered to represent 10% of total number of persons with diabetes (You and Henneberg, 2016). In T2D, peripheral insulin resistance in target tissues (including skeletal muscle, adipose tissue and liver coupled with hypersecretion of insulin) typically precedes eventual β-cell loss. The diabetes epidemic is commonly attributed to T2D (Tuomi, 2005). Gestational diabetes describes insulin resistance observed during pregnancy, which generally resolves in the postnatal period. However, these mothers are at increased risk of T2D in later life (Buchanan and Xiang, 2005).
It is now clear that the above classifications are an over-simplification. It has recently been proposed to re-define diabetes based on six clinical parameters [BMI, age at diagnosis, hemoglobin A1c, glutamate decarboxylase autoantibodies (GADAs) (evidence of autoimmunity); β-cell function and insulin sensitivity]. This has led to the identification of five distinct pathologies associated with different disease progression and risks of complications. Further characterization of the genetic architecture of these subgroups may facilitate identification of patients most at risk of specific complications (Rossing, 2018). According to the proposed classification, the sub-groups can be defined as: (1) SAID: This is the least common subtype (6.4%) and traditionally classified as T1D. These patients had an early onset of disease and were positive for GADAs, had low BMI and were dependent on exogenous insulin (Abegunde et al., 2007). (2) SIDD: these defined as a group of patients who showed insulin deficiency and were GADAs negative. This group was at greatest risk of DR (Jingi et al., 2017). (3) SIRD: This group represented 15.3% of the whole cohort of participants. These patients had a high degree of insulin resistance and were likely to be overweight or obese and showed kidney damage more frequently than other groups. They also had a higher risk of non-alcoholic fatty liver disease (Dutta and Mukhopadhyay, 2018). (4) MOD: Around a fifth of all participants were classified in cluster 4. These patients typically had high BMIs but they did not show insulin resistance (Kahn et al., 2006). (5) Mild age-related diabetes (MARD): Most of the patients (nearly 40%) in the cohort belonged to cluster 5. They were usually older adults with healthier metabolic profiles (including lower BMIs) than the other clusters (Ma et al., 2018).
Diabetes is associated with serious life-threatening and life limiting complications. Acute complications include hyperglycemia-induced ketoacidosis and hypoglycemia. The chronic vascular complications of diabetes have a massive impact on morbidity and mortality. These are classically defined as microvascular and macrovascular complications and reflect responses of susceptible individuals to hyperglycemia, dyslipidemia and hypertension associated with diabetes (Orban et al., 2018). The macrovascular complications include accelerated-CVD and accelerated-atherosclerosis, as discussed below (Duncan et al., 2003).
Complications of the microvasculature include retinopathy, neuropathy and nephropathy. DR is major cause of blindness in the working class (Duh et al., 2017). Diabetic neuropathy develops in almost half of all individuals with diabetes and the lifetime risk of lower limb amputation as much as 15% in certain populations. Diabetic neuropathy is a syndrome encompassing both somatic and autonomic branches of the peripheral nervous system, and, furthermore, contributes to the pathology of other diabetic complications, such as impaired wound healing and erectile dysfunction (Russell and Zilliox, 2014). As will be discussed in more detail below, DKD is the leading cause of ESRD (Piccoli et al., 2015). DKD typically develops over a long period (decades) and, importantly, it is a major risk factor for the development of macrovascular complications, including MI and stroke.
With best medical care the risk of major chronic complications for T1D are cited as 47% for retinopathy, 17% for nephropathy and 14% for CVD. These figures represent a lifetime risk. Figures for T2D are more complex. Although death rates are higher for people with diabetes, relative to age and sex matched cohorts, a recent study has shown that in the United States, whereas death rates for people with and without diabetes have fallen, the greatest decline in mortality was actually seen in those with diabetes, presumably reflecting improved management of glycemia, lipids and hypertension (Gregg et al., 2018). Moreover, in a United Kingdom study, patients with T2D initiated on metformin monotherapy had longer survival than did matched, non-diabetic controls (Bannister et al., 2014). However, the overall mortality in T2D is 60% higher than non-diabetic age and sex matched controls. One consideration on these data is that the lower rates reflect the relatively recent increase in incidence. Mortality is typically associated with chronic complications, such as DKD which develops over decades. The increased incidence may represent a timebomb of diabetes-associated mortality. Indeed, among adults with diabetes, in the United States the prevalence of ESRD has shown the smallest decrease as compared to other diabetic complications (Gregg et al., 2014). To an extent this may reflect the efficacy of preventing atherosclerosis, resulting in increased survival and increased opportunity to develop complications as a consequence of chronic exposure to hyperglycemia. As discussed below, it also reflects the need for therapeutic interventions to specifically target DKD and associated-RF.
This review will focus on describing recent advances in the understanding and elucidation of the underlying mechanisms and in exploring the potential of novel therapeutic approaches for treating diabetes-accelerated atherosclerosis, kidney disease and retinopathy, by using animal and human studies. For more comprehensive reviews of diabetic complications, readers are referred to several excellent recent reviews (Forbes and Cooper, 2013; Papatheodorou et al., 2016; Lotfy et al., 2017).
Diabetes-Associated Atherosclerosis
Definition of DAA
Atherosclerosis is a leading cause of vascular disease worldwide and accounts for about 50% of all deaths in westernized societies and 30% in developing countries (Fuster and Kelly, 2011). Its major clinical manifestations include IHD and ischemic stroke (Lusis, 2000), being, respectively, the world’s first and third causes of death (Barquera et al., 2015).
The strong association between diabetes, low-grade inflammation and atherosclerosis, accounts for one of the major diabetes complications worldwide: DAA (Duncan et al., 2003). Approximately 50% of patients with T2D die prematurely of a cardiovascular cause, and a further 10% die of renal failure (van Dieren S et al., 2010).
Since Ross and Libby redefined atherosclerosis as a progressive, chronic, dyslipidemic and also “inflammatory” disease, advances in basic knowledge of this multifactorial disease defined a key for inflammation in mediating all the phases of athero-progression (Ross, 1999; Libby et al., 2002). Among the numerous markers of high- and low-grade inflammation, C-reactive protein predicts the risk of atherosclerotic complications (see below) (Ross, 1999; Libby et al., 2002). In the recent trial of anti-IL-1β antibody in a large population of high risk atherosclerosis patients (CANTOS), the intervention reduced inflammation and cardiovascular events. Greatest impact was seen in those with highest baseline markers of systemic inflammation. However, its efficacy was similar in those with and without diabetes and, despite decreasing inflammatory markers, did not reduce the incidence of diabetes (Weber and von Hundelshausen, 2017; Everett et al., 2018).
Risk Factors for DAA
The main modifiable risk factors for atherosclerosis have been identified, and they include, but are not limited to, smoking, adiposity, blood pressure, high levels of BMI, high level of LDL, low level of HDL and diabetes (Herrington et al., 2016). T2D is associated with an increased risk of CVD. A role for the lipid-lowering therapy with statins for the primary prevention of CVD in diabetes was demonstrated in CARDS, the first large primary prevention study determining the action of statins in T2D patients, e.g., the efficacy of atorvastatin in preventing disease irrespective of LDL levels (Colhoun et al., 2004). Over the past two decades, developed countries have been able to reduce the contribution of the above mentioned risk factors to mortality, whereas developing countries show an increasing trend due to high BMI and glucose (Barquera et al., 2015).
More recently, the prevalence of coronary atherosclerosis was found to be higher in diabetic than in non-diabetic patients and to be similar for diabetic individuals without clinical CAD and non-diabetics with clinical CAD, implying that prevention measures for asymptomatic diabetic individuals should be similar to secondary preventive approaches among non-diabetic population, as an aggressive prevention measure for atherosclerosis in all diabetic patients, independently of their CAD symptoms (Goraya et al., 2002).
Cellular Pathogenetic Mechanism of DAA
The pathogenesis of atherosclerosis shares several features with other inflammatory diseases, including the infiltration of monocytes and subsequent differentiation to macrophages in response to locally generated signals (Scrivo et al., 2011). At cellular and subcellular levels, inflammatory stimuli or a disturbed blood flow induce endothelial dysfunction (Cunningham and Gotlieb, 2005), altering the homeostatic equilibrium depicted in Figure 1 (left). This vasoreactivity allows lipoproteins apo-B to enter the intima and bind to proteoglycans which trap the LDL particles and increase their susceptibility to oxidation, acetylation and hydrolysis by secretory phospholipases thus amplifying the inflammatory response, characterized by chemokine secretion and adhesion molecules expression on ECs surface. These modifications of lipoprotein induce their aggregation in complexes and subsequent retention, and, additionally, induce monocytes recruitment, a crucial step in early phases of atherogenesis (Lusis, 2000).
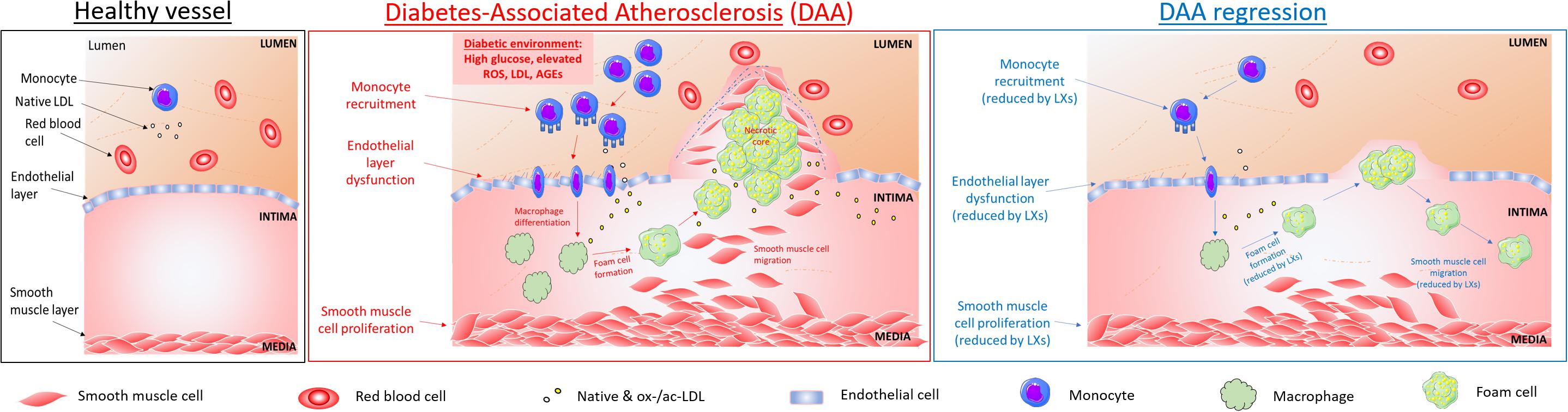
Figure 1. Cellular targets of SPMs in diabetes-associated atherosclerosis. DAA is typified by lesion formation due to inflammatory signals leading to monocyte recruitment into the intima, where they differentiate into macrophages and internalize native and modified lipoproteins, resulting in lipid-loaded foam cells, which accumulate in a necrotic core. Key steps in atherogenesis and progression which may be attenuated by SPMs are highlighted above and include endothelial activation, proliferation and migration (Baker et al., 2009; Brennan et al., 2018a); leukocyte (PMNs and monocytes) recruitment and activation (Colgan et al., 1993; Godson et al., 2000); foam cell transformation and necrotic core formation (Sha et al., 2015); SMCs proliferation and migration (Petri et al., 2015).
Once chemoattracted to the inflammatory injury area, the monocyte undergoes a series of processes that allow cell locomotion (i.e., rolling, adhesion, polarization, crawling) to reach the endothelial transmigration sites, in proximity of low shear stressed athero-prone regions, where blood flow is disturbed, such as bifurcations of arteries (Cunningham and Gotlieb, 2005). Once transmigrated and eventually infiltrated into the intima layer, monocytes differentiate into macrophages in response to locally produced factors, such as M-CSF (Moore and Tabas, 2011). This program of differentiation includes upregulation of class A- macrophage scavenger receptor (SR-A), CD 36 (CD-36) and other cell surface receptors, to facilitate ox-LDL or ac-LDL uptake: in physiologic conditions, this process allows an efficient removal of excessive lipids from the blood circulation (“scavenging” action of macrophage). When homeostasis fails, dysregulation of this phenomenon leads to uncontrolled accumulation of lipids and cholesterol derivatives in macrophages, and their transformation into foam cells in the subintima endothelial layer (Kunjathoor et al., 2002).
Lipid-loaded foam cells, cellular debris, calcium deposits and connective tissue contribute to generate the so-called fatty streak (a hallmark and first sub-clinical sign of atherosclerosis), increasing inflammation and inducing necrosis and foam cell death. As the lesion grows invade both the luminal space and the intima. The necrotic area is confined within a fibrous cap made of connective tissue, composed of SMCs and collagen. Fibrous cap atheromas are the first clinically detectable atherosclerotic lesions (Li and Glass, 2002).
As the lipid core increases in size, the fibrotic cap is invaded by macrophages and lymphocytes, inducing the thinning of the cap. The mechanism by which a sustained macrophage invasion weakens the fibrous cap involves phagocytosis of the ECM and the release of proteolytic enzymes (i.e., plasminogen activators and matrix metalloproteinases, MMPs). The thinned fibrous cap is prone to rupture, exposing the inflammatory and thrombogenic molecules (TF, collagen) of the lipid core, highly increasing the risk of thrombosis (Pepine, 1998).
Molecular Pathogenetic Mechanisms of DAA
As above stated, diabetes accelerates atherosclerosis, contributing to higher rates of mortality and morbidity among diabetic patients. The molecular mechanisms behind this likely reflects increased inflammation and decreased blood flow (Abe and Berk, 2013) but are not fully understood. Several possible triggers have been thus far hypothesized, including hyperglycemia, insulin resistance, increased activation of PDGF-dependent pathways, increased level of TF or decreased level of HDL, and AGEs and their receptors (RAGE) signaling activation (Beckman et al., 2002). Insulin and hyperglycemia play key roles in distinct phases of disease progression, via different mechanisms and differentially affecting the three major cell types: SMCs, macrophages and ECs (Bornfeldt and Tabas, 2011).
It has been hypothesized that, in advanced plaques, insulin resistance may promote apoptosis of PMNs, SMCs, and macrophages. In particular, death of SMCs can lead to the thinning of the fibrotic cap, whereas death of macrophages is associated with a defective phagocytic clearance of the cells (efferocytosis), promoting plaque necrosis (Greenlee-Wacker, 2016). These processes converge to precipitate plaque rupture and acute thrombotic vascular occlusion (Brophy et al., 2017). A relation between diabetes obese-induced adiposity and atherosclerosis in young adults have been observed (McGill et al., 1995). Elevated SFAs has been associated with obesity and insulin resistance (Funaki, 2009) and causes defective efferocytosis of apoptotic macrophage (Li et al., 2009), subsequently causing a secondary cellular necrosis and inflammation amplifying the plaque necrosis (Thorp and Tabas, 2009). The combined pro-apoptotic effect of macrophage insulin resistance and the anti-efferocytic effect of SFAs may create a “perfect storm” for plaque necrosis, as proposed by Bornfeldt and Tabas (2011).
Hyperglycemia accelerates formation of early/mid stage lesions of atherosclerosis by promoting an inflammatory phenotype of which adhesion molecule expression in ECs is a hallmark. Increased flux through the aldose reductase pathway accelerates glucose metabolism and generates ROS. Increased adhesion molecule expression leads to increased monocyte/macrophage accumulation and atherogenesis. In SMCs, a principal effect of increased glucose uptake appears to be increased secretion of the monocyte chemoattractant protein-1, a chemokine which acts in concert with ECs. This leads to an increased production of endothelium-derived contracting factors, which oppose the protective activity of nitric oxide (Meininger et al., 2000; vanDam et al., 2000). Ultimately, this leads to greater recruitment of monocytes into the growing lesion (Bornfeldt and Tabas, 2011), thereby further contributing to an enhanced inflammatory response. Those events have been shown to promote adventitial inflammation and vasa vasorum neovascularization in experimental models of diabetic atherosclerosis. In particular, over the past two decades, the work from Cosentino and Luscher (1998) has established the strong relationship between hyperglycemia, oxidative stress and inflammation, together with an increased risk of CVD in T2D (Beckman et al., 2013; Paneni et al., 2013). Very recently, their studies demonstrated epigenetic regulation of immune-metabolic pathways to increased inflammation, neovascularization and intraplaque hemorrhage in human diabetic atherosclerosis (Guzik and Cosentino, 2018).
Insulin and hyperglycemia are not the only possible factors so far correlated to the underlying pathogenetic mechanism of DAA. Hyperglycemia enhances shear stress-induced platelet activation (Gresele et al., 2003). PDGF has been shown to play a major role in the pathology of vascular diseases. Inhibition of PDGF receptor activation attenuates DAA in experimental models (Lassila et al., 2004).
The inflammatory component of microangiopathic processes is independently associated with plaque rupture, leading to coronary thrombosis. TF, the most potent trigger of the coagulation cascade, is increased in diabetic patients with poor glycemic control. Circulating TF microparticles are also associated with apoptosis of plaque macrophages, closing the link among inflammation, plaque rupture and blood thrombogenicity (Fallon et al., 1997; Singh et al., 2012).
AGE/RAGE signaling has been a well-studied cascade in many different disease states, particularly diabetes. It heavily influences both cellular and systemic responses to increase bone matrix proteins through activation of PKC, p38 MAPK, TGFβ, NFκβ and ERK1/2 signaling pathways in both hyperglycemic and calcification conditions. AGE/RAGE signaling has been shown to increase oxidative stress and to promote diabetes-mediated vascular calcification through activation of NADPH oxidase-1 and decreased expression of superoxide dismutase-1. AGE/RAGE signaling in diabetes-mediated vascular calcification is also attributed to increased oxidative stress resulting in the phenotypic switch of SMCs to osteoblast-like cells in AGEs-induced calcification (Kay et al., 2016). HDL, responsible for free cholesterol removal, are reduced in patients with insulin resistance and diabetes, conditions for which the role of obesity is highly detrimental (Rashid and Genest, 2007; Barter, 2011). In addition to their role as lipid lowering agents, via inhibition of 3-hydroxy-3-methylglutaryl coenzyme A reductase, pleiotropic responses to statins may include reduction of SMCs proliferation, as observed in in vitro and ex vivo models (Fleg et al., 2008).
Current Therapies in DAA
There are currently no available therapies for the regression of atherosclerosis (Fuster et al., 1998). Therefore, new therapeutic targets are needed in order to offer an alternative type of intervention to invasive surgery, such as stenting or endarterectomy. Current therapies in DAA adopted antiplatelet/anticoagulant therapy, stabilizing the plaque (Engelberg et al., 1956; Colwell, 1997), including the use of low-dose aspirin (75–162 mg/day) for secondary prevention of cerebrovascular and cardiovascular events in all diabetic patients (Angiolillo, 2009). Evidence that LDL causes CVDs is overwhelming. It has also been proven beyond all doubt that lowering the level of LDL using statins reduces cardiovascular risk. However, many people remain at high risk even when their level of LDL has been reduced by aggressive treatment with statins. One reason for this residual risk can be a low level of HDL, an independent, inverse predictor for CAD. It has therefore been suggested that raising the level of HDL should be considered as a therapeutic strategy for reducing the residual cardiovascular risk that persists in some people, despite aggressive LDL-cholesterol lowering with statins. HDL particles have several functions with the potential to protect against arterial disease, the best known of which relates to their ability to promote cholesterol efflux from macrophages in the artery wall. However, HDLs have several additional protective properties that are independent of their involvement in cholesterol metabolism. For example, they have properties that reduce oxidation, vascular inflammation and thrombosis, improve endothelial function, promote endothelial repair, enhance insulin sensitivity and promote insulin secretion by pancreatic β islet cells (Barter, 2011). These beneficial effects may be responsible for coronary plaque stabilization in patients treated with those molecules which can up regulate HDL expression including Apo-A1 or peroxisomal proliferator-activated receptors agonists, holding great promise in the treatment of diabetic atherosclerosis.
The Regression of DAA
The regression of existing lesions is the holy-grail in management of atherosclerosis. Over the past two decades major advances have been made to this end. Fisher’s lab and his collaborators Young, Hazen, Smith and Moore have firmly established the principle that regression of atherosclerosis is a possible therapeutic goal (Fisher, 2016).
Although monocytes are recruited into the plaque during its growth, they also have the capacity to emigrate from atherosclerotic lesion. Using murine models of regression, including the “transplantation mouse,” a transplant model in which plaque-bearing aortic segments are transferred into normolipidemic mice (Reis et al., 2001); the “reversa mouse,” a genetic “switch” model in which LDL production can be conditionally reduced in LDL-R-/- mice (Feig et al., 2011); and acute treatment models, in which Apo-E-/- mice are injected either with Apo-A1 (Hewing et al., 2014), with a microsomal triglyceride transfer protein inhibitor or with an anti-microRNA (miR) (anti-miR-33) (Rayner et al., 2011; Moore et al., 2013; Distel et al., 2014) a decrease in plaque size and, consequently, regression of pre-existing atherosclerosis was demonstrated (Llodra et al., 2004; Randolph, 2008; Feig et al., 2009). A possible explanation of the reduction in CD68+ macrophage cell content is that monocytes can enter the lymphatic system, reaching the lymph nodes, or they can migrate across the arterial endothelium toward the artery lumen to directly enter the circulating bloodstream (Llodra et al., 2004; Randolph, 2008). The main processes involved in atherogenesis are also the main target for regression, namely, the retention of apo-B-containing lipoproteins in the arterial wall and the reaction of macrophages to these particles (Williams and Tabas, 2005). The resulting foam cells secrete pro-inflammatory cytokines and chemokines, as well as retention factors that amplify the inflammatory response and promote macrophage chemostasis. These accumulating macrophages experience endoplasmic reticulum stress, which, if prolonged, results in apoptosis. This cell death, coupled with defective efferocytosis, due to an uncontrolled lipid accumulation, in which essentially SFAs decrease the fluidity of the plasma membrane, leads to the formation of the necrotic core that is characteristic of advanced plaques (Funaki, 2009; Thorp and Tabas, 2009; Bornfeldt and Tabas, 2011).
The key mechanisms that promote regression are: lipid unload of the foam cell and promotion of RCT, via upregulation of the efflux protein ABCA-1 expression on plaque macrophages and the subsequent cholesterol efflux toward exogenous acceptors (i.e., Apo-E-containing HDL) (Chinetti-Gbaguidi et al., 2011); a decrease in the expression of retention factors (Brodsky and Fisher, 2008); a reduced monocyte recruitment via their transformation in monocyte-derived dendritic cells and subsequent upregulation of CC-chemokine receptor (CC-R)-7 on their surface, which allow emigration to the lymphatic system, restoring permeabilization and reducing lymphatic vessel fibrosis (Ivanov et al., 2016). Finally, the retention/migration factors contributing to macrophage loss from the plaque, through reverse transmigration to the lumen or through trafficking to the adventitial lymphatic (Potteaux et al., 2011).
In the context of diabetes, as depicted in Figure 1 (central panel), regression of atherosclerosis is impaired. High glucose levels modulate LXR-dependent gene expression, by inhibiting the LXR-dependent expression of ABCA1, but not ABCG1 (Hussein et al., 2015) and by inducing miR-33, a key negative regulator of the RCT factors, ABCA1 and HDL (Wijesekara et al., 2012). In mouse models of insulin-deficient diabetes, it has been shown that leukocytosis (monocytosis and neutrophilia) is hyperglycemia-dependent. The myelopoiesis is driven by increased expression of certain DAMPs, specifically, signaling through the pattern recognition AGE/RAGE. The relevance to human health and disease is suggested by the correlation between serum S100A8/S100A9 (the associated DAMPs) and the incidence of CAD in a subset of T1D patients from the Pittsburgh EDC study, highlighting the potential importance of glucose control and lipid-lowering therapy as strategies to promote regression of atherosclerosis in diabetics and also suggesting a number of therapeutic targets, including disruption of the S100A8/S100A9-RAGE signaling axis (Nagareddy et al., 2013).
Diabetic Kidney Disease
Definition of DKD
DKD typically develops over many decades. It is characterized by progressive proteinuria (microalbuminuria 30–299 mg/24 h to macroalbuminuria > 300 mg/24 h) with a subsequent decline in glomerular filtration reflected by increased serum creatinine (National Kidney Foundation, 2002). The pathophysiology of DKD typically reflects the convergence of hemodynamic, metabolic and inflammatory insults in susceptible individuals (Harjutsalo and Groop, 2014). Current interventions focus on tight glycemic control and RAAS blockade by ACE inhibition or angiotensin receptor antagonism to dilate the efferent arteriole and reduce glomerular hypertension. At best, these interventions slow the progress of disease (Forbes and Cooper, 2013). There is a growing appreciation that oxidative stress and inflammation are key drivers of DKD and may be appropriate targets for therapeutic intervention. Circulating inflammatory cytokine levels correlate with albuminuria and elevated levels of soluble TNF-receptor-1 is an independent predictor of decline in renal function (Krolewski et al., 2014).
Cellular Pathogenetic Mechanisms of DKD
The physiological functionality of a healthy glomerulus is outlined in Figure 2 (left). Changes in renal hemodynamics, reflecting glomerular and systemic hypertension, arise early in DKD and lead to glomerular hyperfiltration. RAAS activation leads to increased angiotensin II and endothelin-1 causing efferent arteriolar vasoconstriction and hyperfiltration. Glomerular damage is characterized by podocyte effacement resulting in proteinuria. Renal hypertrophy is also observed in DKD reflecting accumulation of mesangial matrix, glomerular BM thickening and tubular hypertrophy. As matrix expands, it accumulates to form Kimmelstiel–Wilson nodules, a pathological feature of DKD. TIF is considered the major determinant of progression of DKD (Duffield, 2014). The mechanisms underlying TIF have been exhaustively investigated in the context of chronic kidney disease, including DKD (Leaf and Duffield, 2017). At a cellular level, several mechanisms have been proposed including activation of resident fibroblasts to matrix producing myofibroblasts, detachment of pericytes and matrix production, recruitment of fibrocytes from bone marrow and EMT (Kalluri and Weinberg, 2009). Whereas the role of EMT in TIF has been questioned the loss of several epithelial cell markers (de-differentiation) has been observed together with expression of pro-fibrotic mediators such as CTGF and the TGFβ1 activator THBS-1 (Thiery et al., 2009). Experimental evidence suggests that partial EMT and chronic inflammation converge to create a profibrotic milieu facilitating collagen production by fibroblasts and recruited hematopoietic cells in the kidney (Zeisberg and Duffield, 2010; Buchtler et al., 2018). Glomerulosclerosis and TIF lead eventually to organ failure and a requirement for renal replacement therapy (hemodialysis or transplantation). Efforts to directly target inflammation in DKD have included manipulating chemokine and cytokine signals in T2D, such as antagonism of CCR2/CCR5 (Huh et al., 2018).
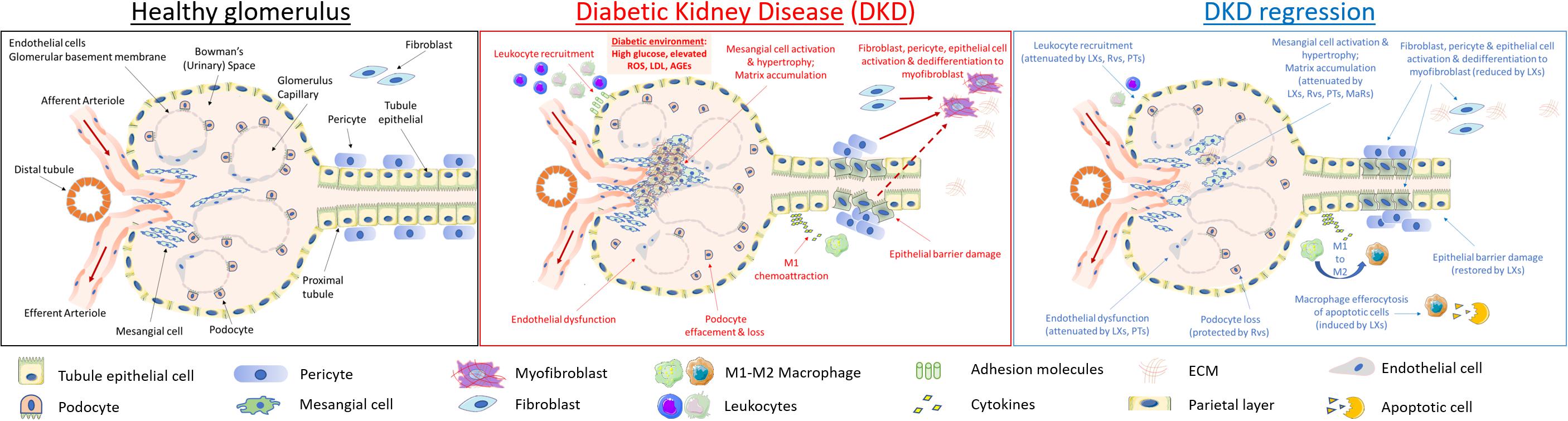
Figure 2. Cellular targets of SPMs in diabetic kidney disease. DKD is typified by mesangial expansion, increased ECM deposition, podocyte loss, renal tubule epithelial cell injury and PMN leukocyte recruitment. Key steps in renal inflammation and fibrosis which may be attenuated by SPMs are highlighted above and include leukocyte recruitment and activation (Godson and Brady, 2000; Leonard et al., 2002; Duffield et al., 2006; Brennan et al., 2018b); renal tubule epithelial cytokine production and dedifferentiation (Kieran et al., 2003; Duffield et al., 2006; Brennan et al., 2013, 2018b); endothelial activation (Baker et al., 2009; Sun et al., 2013); fibroblast to myofibroblast activation (Borgeson et al., 2011; Qu et al., 2012); podocyte effacement (Zhang et al., 2013); macrophage efferocytosis and M1:M2 ratio (Mitchell et al., 2002; Borgeson et al., 2011, 2015); mesangial cell activation and matrix accumulation (McMahon et al., 2002; Rodgers et al., 2005; Wu et al., 2006, 2007, 2009; Tang et al., 2014).
Molecular Pathogenetic Mechanisms of DKD
As depicted in Figure 2 (central panel), high glucose exerts specific toxic effects on the resident cells of the kidney, including specialized parietal epithelial cells (podocytes), mesangial cells, endothelia, fibroblasts and epithelia driving cellular de-differentiation (Eddy and Neilson, 2006; Liu, 2011). Many of these responses are driven by autocrine and paracrine mediators released by target cells and infiltrating monocytes/macrophages, as typified by responses to TGF-β1 and its downstream targets, including CTGF (Murphy et al., 1999; Strutz et al., 2000; Boor and Floege, 2011). Hyperglycemia leads to ROS production and activation of inflammatory responses including NF-kB and janus kinases and signal transducer and activator of transcription proteins (JAK-STAT) activation and subsequent downstream cytokine production (Sifuentes-Franco et al., 2018).
Current Therapies in DKD
Numerous large scale genome-wide association studies have been carried out in DKD over recent years (Sandholm et al., 2014; Ahlqvist et al., 2015; Teumer et al., 2015; Wuttke and Köttgen, 2016; van Zuydam et al., 2018). These studies have frequently implicated inflammatory pathways in the pathogenesis of DKD. Such genetic validation of therapeutic targets includes the JAK-STAT pathway. STAT-1,3 activation is observed in renal biopsies from people with DKD (Berthier et al., 2008). Baricitinib, a small molecule JAK-STAT inhibitor, has shown efficacy in a small scale clinical trial. Treatment with baricitinib was associated with decreased inflammatory biomarkers (e.g., urinary chemokine CCL-2, plasma soluble tumor necrosis factor receptor-1, intracellular adhesion molecule-1 and serum amyloid A). Baricitinib decreased albuminuria in participants with T2D and DKD (Tuttle et al., 2018).
Glucose stimulates inflammasome assembly, caspase-1 activation and IL-1β release (Schroder et al., 2010). IL-1β activation by the NACHT, LRR and PYD domains-containing protein (NLRP) inflammasome is an important component of CKD (Vilaysane et al., 2010). Blockade of IL-1β activity in post MI patients with CKD reduced the risk of adverse cardiovascular events among those with CKD (Ridker et al., 2017). As described above, the CANTOS trial targeted IL-1β in atherosclerosis patients and the intervention reduced inflammation and cardiovascular events (Ridker et al., 2011). Comparable effects were observed among those with baseline albuminuria or diabetes. Canakinumab, however, was without effect on serial measures of eGFR, creatinine, the urinary albumin:creatinine ratio or reported adverse renal events during trial follow-up (Ridker et al., 2018).
Despite the identification of numerous drivers of fibrosis, such as the TGF superfamily, thus far efforts to target RF per se have been unsuccessful. A recent double blind phase II study assessed whether modulating TGF-β1 activity with a TGF-β1-specific, humanized, neutralizing monoclonal antibody was effective in slowing renal function loss in patients with diabetic nephropathy on RAAS inhibition treatment over a 12-month period. No significant impact on disease progression was observed (Voelker et al., 2017). Other approaches have focused on the balance between BMP family agonist-antagonist activities and promoted BMP-7 and or small peptide mimetics, such as THR-123 (Ali and Brazil, 2014; Tampe and Zeisberg, 2014; Brazil et al., 2015). However, some of these data have been controversial (Sugimoto et al., 2012). Systemic administration of BMP-7 protein is problematic due to the low availability in the kidney, explaining the need for a huge amount of BMP-7 for its reno-protective action, which might exert adverse effects elsewhere (Vukicevic et al., 1998; Yanagita, 2012). CTGF/CCN2 has also been proposed as a potential target (Falke et al., 2014, 2017). Other therapeutic approaches which have been proposed in the context of DKD include attenuation of NF-kB signaling (Lee J. et al., 2012), breakdown of AGEs (Rabbani and Thornalley, 2018) or RAGE antagonism (Bongarzone et al., 2017). In this context, bardoxolone methyl is a novel synthetic triterpenoid belonging to the antioxidant inflammation modulator class. Antioxidant inflammation modulators potently induce the antioxidant and cytoprotective transcription factor Nrf2, reduce the pro-inflammatory activity of the IKK-β/NF-κB pathway, increase the production of antioxidant and reductive molecules, and decrease oxidative stress, thereby restoring redox homeostasis in areas of inflammation. Activation of anti-oxidant responses via Nrf2 and inhibition of NF-kB by the triterpenoid bardoxolone methyl reduces oxidative stress, inflammation and promotes mitochondrial function in numerous experimental models of CKD, including DKD (Pergola et al., 2011). Unfortunately, clinical trials of bardoxolone methyl in patients with stage 4 CKD and T2D were prematurely terminated for safety concerns (Tayek and Kalantar-Zadeh, 2013). Bardoxolone methyl treatment was associated with approximately double the risk of heart failure as placebo. Subsequent analysis suggests that these data did not represent toxicity per se and that further development of this compound may be warranted with more careful patient selection (Chin et al., 2014, 2018).
Novel Therapeutic Approaches in DKD
It is important to note some recent advances that suggest renoprotection in response to newer therapeutics which regulate blood glucose and reduce cardiovascular risk in T2D. Intriguingly these reno-protective responses may be independent of glucose lowering. Such interventions include the incretin-based therapeutics (GLP-1-RAs, e.g., liraglutide or DPP-4 inhibitors and SGLT-2 inhibitors) enhancing glycemic control with a low risk of hypoglycemia. However, the use of these agents is limited in those with significant renal impairment. A recent trial treatment with liraglutide, a GLP-1 analog, was associated with a 22% lower incidence of doubling serum creatinine, persistent macroalbuminuria, development of ESRD or death from renal disease relative to controls (Mann et al., 2017). Similar data have been reported for other GLP-1 receptor agonists and for DPP-4 inhibitors which inhibit breakdown of endogenous GLP-1. SGLT-2 inhibitors suppress glucose reabsorption by the proximal tubule and therefore increase glucose excretion. The SGLT-2 inhibitors target reabsorption of both glucose and sodium as a result there is increased sodium delivery to the macula densa activating tubule-glomerular feedback afferent arteriolar vaso-modulation, resulting in increased renal blood flow and decreased glomerular hyperfiltration. SGLT-2 inhibition is associated with lower rates of albuminuria and lowering rates of eGFR decline (Tomkin, 2014). Intriguingly, bariatric surgery in T2D appears to have specific reno-protective effects which may relate to enhanced GLP-1 responsiveness (Docherty and le Roux, 2014). Miras et al. (2015) reported that, 1-year post-bariatric surgery, a decrease urinary albumin/creatinine ratio was observed whereas no benefit was seen on other microvascular complications, i.e., retinopathy or neuropathy. SGLT-2 inhibitors and GLP-1 targeting drugs attenuate inflammatory responses in DKD. As we will discuss below, we propose that exploiting the bioactivity of endogenous lipid modulators that promote the resolution of inflammation and suppress fibrosis is a novel therapeutic paradigm worthy of consideration as adjuvant therapy in DKD.
The Role of Macrophage in Diabetes Complications
As described, macrophages are key players in atherosclerotic lesions, regulating the local inflammatory milieu and plaque stability by the secretion of many inflammatory molecules, growth factors and cytokines (Wolfs et al., 2011). The macrophage paradigm classically reflects the heterogeneity of their monocyte progenitor: alternative crawling monocytes continuously patrol the endothelium of blood vessels in the steady state. The patrolling monocytes rarely extravasate in the steady state. In contrast, during inflammation or infection, classical monocytes are the first to extravasate when inflammation signaling occurs, and, within few hours they differentiate in M1 “pro-inflammatory macrophages,” induced by INFγ + LPS or by TNFa, characterized by a high phagocytic profile. At later stages of inflammation, non-classical monocytes (or non-phlogistic monocytes) trans-migrate and initiate a differentiation program into ‘M2’-like macrophages, which play a role in resolving of inflammation and tissue repair (Geissmann et al., 2008). M2 macrophages can polarize toward different phenotypes according to various stimuli present in their surrounding micro-environment (Mosser and Edwards, 2008) and to their distinct gene expression profiles (Mantovani et al., 2004). In particular, “M2a” or “alternative” macrophage is the product of Th2 activation (by IL4 and IL13 cytokines or fungal and helminth infections) and is responsible for a type II inflammatory response (consisting in killing parasites and inducing a Th2 response to allergy). “M2b” or “type II” macrophage is elicited by IL-1 receptor ligands, immune complexes and LPS, triggering the activation of Th2 system. “M2c” or “deactivated” macrophage is induced by IL10, TGF-β and glucocorticoids and is mainly immunoregulatory, through matrix deposition and tissue re-modeling (Martinez and Gordon, 2014). A fourth type, “M2d,” or “angiogenic” macrophage is elicited by IL-6 and adenosine and is mainly involved in wound healing (Ferrante and Leibovich, 2012). However, recent findings provide evidence for proliferation of local macrophages or trans-differentiation from other vascular cells as alternative sources (Nagenborg et al., 2017). In particular, it has been shown that cholesterol-loading induces the trans-differentiation of SMCs to macrophage-like cells (Rong et al., 2001) and more recently, it has been shown that approximately 50% of foam cells might have a SMC origin (Allahverdian et al., 2014). Linear tracing studies from Randolph’s lab have shown that tissue-specific factors drive highly specialized macrophage functions irrespective of their ontological origin, suggesting tremendous plasticity and redundancy in the mononuclear phagocyte system. Whether embryonic and adult macrophages possess specialized roles has yet to be formally tested. However, the conceptual understanding and genetic tools are now sufficiently developed to precisely follow both embryonic and adult macrophage subsets in health and disease, which should allow important and unanswered questions in the field to be addressed. In order to develop novel therapies, a critical future goal is to harness this new found understanding that different macrophage lineages exist within tissues and clarify whether these distinct lineages differentially contribute to tissue damage and repair (Epelman et al., 2014).
Role of Macrophage in DAA
In the context of atherosclerosis, macrophages uniquely possess a dual functionality, regulating lipid accumulation and metabolism and sustaining the chronic inflammatory response, two well-documented pathways associated with the pathogenesis of the disease (Moore and Tabas, 2011).
Established atherosclerotic plaques from patients with existing CAD undergoing carotid endarterectomy classified as symptomatic (where the patient has experienced previous ischemic events but without any CVD diagnosis) or asymptomatic (where a patient has no history of ischemic events or CVD) have been recently comprehensive histologically and immunohistochemically characterized for their cellular content and macrophage subsets of atherosclerotic lesion. Symptomatic plaques were defined as highly hemorrhagically active and the internal carotid was the most diseased segment, based on the predominant prevalence of fibrotic and necrotic tissue, calcifications, and hemorrhagic events. Immunohistochemical analysis showed that both M1 and M2 macrophages are present in human plaques. However, M2 macrophages were localized to more stable locations within the lesion. Importantly, M1 markers and Th 1-associated cytokines were highly expressed in symptomatic plaques, whereas expression of the M2 markers, mannose receptor and CD163 and Th2 cytokines were inversely related with disease progression (de Gaetano et al., 2016). A strong relation between macrophage, mitochondria and glucose dysregulation has recently emerged in a number of studies from Fredman and Tabas (2017). Clearance of ACs by phagocytes (efferocytosis) prevents post-apoptotic necrosis and dampens inflammation. Mitochondrial fission in response to AC uptake is a critical process that enables macrophages to clear multiple ACs and to avoid the pathologic consequences of defective efferocytosis in vivo (Yurdagul et al., 2017).
Role of Macrophage in DKD
In a renal context, macrophages constitute a major subset of the infiltrating inflammatory cells and their contribution to renal fibrogenesis is well established (Duffield, 2010). Macrophage infiltration has been found to correlate with TIF on kidney biopsies (Young et al., 1995), and to correlate negatively with outcome in CKD of diverse etiologies (Tinckam et al., 2005; Duffield, 2010). However, the role of macrophages in this context is not entirely clear-cut. M1 macrophages are recruited to the kidney at early time points in a murine IRI model, whereas at later time points, M2 macrophages predominate. Additionally, in this model, depletion of macrophages prior to IRI has been found to attenuate inflammation and TIF, whereas macrophage depletion after 3–5 days is shown to slow tubular cell proliferation and repair (Lee et al., 2011). Macrophages exhibit some plasticity, and may not remain committed to a single phenotype. As a component of the programmed resolution of inflammation, a phenotypic change is triggered by altered cytokine and lipid mediator profiles in the microenvironment, and M1 phenotype macrophages thus ‘switch’ to a pro-resolving M2 phenotype (Nathan and Ding, 2010; Lee et al., 2011). In the context of chronic inflammation, or repeated injuries, the factors that determine if macrophages are predominantly reparative versus predominantly pro-inflammatory remain unclear. Directing more of the macrophage population toward a pro-resolving phenotype may provide a novel therapeutic approach in CKD. Although much of our current understanding of the ontogeny and functional plasticity of macrophages has been derived from murine models, it is important to note that, together with the above mentioned contribution from Randolph on macrophage ontology, a recent study in human heart reveals two populations of macrophages with different origins and functions: CCR2 expressing macrophages are recruited from bone marrow and proliferate and are functionally proinflammatory and abundant in regions of scarring, whereas macrophages lacking CCR2 are maintained by local proliferation and express genes associated with tissue repair (Bajpai et al., 2018).
Diabetic Retinopathy: A Brief Overview
Definition, Pathogenesis, and Current Therapies
Diabetic retinopathy (DR) is a microvascular complication of diabetes, clinically characterized by progressive alterations in the microvasculature that lead to retinal ischemia, neovascularization, altered retinal permeability and macular edema. It is currently the leading cause of blindness in the adult working population (Congdon et al., 2004; Yau et al., 2012).
The disease can be divided into two main stages – non-proliferative retinopathy and proliferative retinopathy distinguished by the absence or presence of abnormal neovascularization, respectively. The final stage “proliferative retinopathy” is characterized by neovascularization of the disk or iris or vitreous hemorrhage or retinal detachment (Wilkinson et al., 2003). Macular edema or “diabetic maculopathy” can occur at both the non-proliferative and proliferative stages as a result of fluid accumulation under the macula.
The pathological and morphological alterations associated with DR were long considered to be primarily microvascular in nature, as a result of hyperglycemia and the metabolic pathways it activates. The onset of clinically detectable DR is characterized by changes in the micro-vessels of the eye which includes thickening of the BM, loss of vascular permeability, loss of pericytes, capillary occlusions and microaneurysms (Xu et al., 2014).
However, recent studies have demonstrated that retinal neurodegeneration is a critical feature associated with the progression of the disease and may in fact precede the development of clinically detectable microvascular damage (Lieth et al., 2000; Puro, 2002).
Under pathological DR conditions, break-down of the BRB occurs as a result of the presence of increased levels of vascular permeability factors, such as VEGF in the vitreous of the eye. The resulting “leaky” vasculature leads to increased albumin flux into the retina and fluid accumulation resulting in macular edema and possible vessel hemorrhage (Klaassen et al., 2013).
Thickening of the vascular BM occurs early in the disease and represents one of the first histologically detectable structural alterations. Several biochemical alterations contribute to BM thickening in vivo. Increased expression of the matrix components of the BM, including fibronectin (Roy et al., 1996), collagen IV (Roy et al., 1994) and laminin (Ljubimov et al., 1996) can be detected long before the formation of diabetic lesions. BM turnover is tightly regulated by the delicate balance of synthesis and degradation of BM components by MMPs, urokinases and their inhibitors. This balance is disturbed during DR (Kowluru et al., 2012).
Several interconnected biochemical pathways associated with hyperglycemia have been implicated in the pathogenesis of DR, including increased polyol pathway flux, increased hexosamine pathway flux and activation of protein kinase C. A crucial role is played by hyperglycemia-induced ROS production and AGEs formation (Forbes and Cooper, 2013). The retina is the most metabolically active tissue in the body, rendering it particularly susceptible to oxidative stress (Wu et al., 2014). Although all retinal cells express RAGE ubiquitously, retinal pericytes, in particular, have been shown to accumulate AGEs, contributing to BRB breakdown, which is in part accredited to pericyte loss, but also to AGE-induced leukocyte adherence to retinal ECs (Moore et al., 2003).
Growing consensus is emerging in the predominant role of inflammation in the pathogenesis of DR (Rubsam et al., 2018). The formation of AGEs and the activation of PKC have been implicated in the activation of pro-inflammatory mediators, such as NF-κB, connecting hyperglycemic-induced oxidative stress to inflammation. An increase in a number of pro-inflammatory cytokines and chemokines has been demonstrated in both diabetic patients and models of experimental retinopathy (Doganay et al., 2002; Sato et al., 2009). Blocking the activity of pro-inflammatory cytokines (such as TNF-α, IL-6, and IL-1) has shown beneficial effects in models of retinopathy. An IL-1 receptor antagonist reduces inflammatory responses in a rodent model of T2D (Vallejo et al., 2014) while breakdown of the BRB was completely ablated in a TNF-α knockout diabetic mouse (Huang et al., 2011). Chemokines, such as MCP-1 and IL-8, are also elevated in diabetic eye disease and contributed to neovascularization and fibrosis (Yoshida et al., 2003). However, their expression was reduced by inhibitors of VEGF, suggesting that the action of both MCP-1 and IL-8 are mediated through pathways involving VEGF. Hyperglycemic conditions also drive increased expression of a number of growth factors (including VEGF and TGFβ) mediating the retinal damage associated with DR, such as BM thickening, vascular permeability and neovascularization.
TNF-a and VEGF have received particular attention for their role in the vascular lesion and neovascularization associated with late stage retinopathy. Therefore, anti-TNFa (i.e., Infliximab) (Sfikakis et al., 2005) and anti-VEGF (i.e., Avastin) (Haritoglou et al., 2006) intravitreal therapies are standard clinical therapeutic options for the treatment of DR.
Many of the agents developed to target the various biochemical pathways driven by hyperglycemia have had limited effect clinically, pointing to a need for new therapeutics targets.
The Role of SPMs in Resolving Inflammation
The inflammatory response consists of two phases: initiation and resolution. The initiation phase is characterized by the site-specific accumulation and coordinated activation of a host of immune effector cells in an inflammatory cytokine and pro-inflammatory lipid mediator rich environment. Inflammation is critical in the host response to infection and injury, however, timely resolution is necessary for the restoration of tissue homeostasis, thereby limiting excessive tissue injury, preventing the development of a chronic inflammatory state (Serhan et al., 2007). Non-resolving inflammation is a major driver of disease. Multiple mechanisms ensure physiological resolution toward tissue homeostasis. Cells like macrophages switch phenotypes by secreting molecules like reactive oxygen intermediates, lipids and proteins which impact a cell from displaying pro- or an anti-inflammatory behaviors (Buckley et al., 2014).
Whereas inflammation and its effective outcome, i.e., a return to homeostasis, were typically considered a manifestation of the passive dissipation of pro-inflammatory stimuli, including lipids, such as prostaglandins and leukotrienes, it is now clear that the resolution of inflammation is an active and dynamically regulated process reflecting responses to endogenously generated mediators, including cytokine and lipids (Godson and Brady, 2000; Maderna and Godson, 2009; Serhan, 2014).
The specialized SPMs are a family of endogenously produced pro-resolving lipid mediators derived from the metabolism of PUFAs, which include LXs, resolvins (Rvs), protectins (PDs) and MaRs. They were discovered by Serhan et al. (1984). LXs (Lipoxygenase interaction products) were firstly isolated in a human leukocyte (Serhan et al., 1984) and classified as derivatives of the ω6 fatty acid arachidonic acid (20:4, n-6). Rvs (Resolution phase interaction products) were firstly identified in a resolving inflammatory exudate in 2000 (Serhan et al., 2000), PDs (termed neuroprotectin D1 if generated in neural tissue for its protection in neurons, glial cells, and brain stroke; or protectin D1 for other tissue in 2004 (Bazan, 2005) and MaRs (Macrophage mediator in Resolving Inflammation) in 2009 (Serhan et al., 2009). Rvs, PDs and MaRs are classified as derivatives of ω3 fatty acids: specifically, Rvs can either form from the EPA (20:5, n-3) [RVs E-series] or from the DHA (22:6, n-3) [RVs D-series]; while, PDs and MaRs only derive from DHA. As their precursors, all these derivatives are classified as PUFAs and they demonstrated potent anti-inflammatory and immunoregulatory actions (Serhan et al., 2008).
Within a few hours from barrier break, tissue injury or trauma, eicosanoids are crucial in initiating the cardinal signs of inflammation (redness, heat, pain and swelling). As part of the vascular response, leukocytes traffic to the site of injury. The prostaglandins PGE2 and PGI2 (involved in vasodilation) and the leukotriene LTB4 (involved in chemotaxis and adhesion) stimulate the migration of PMN to the tissue. In parallel to the PMN–monocyte sequence, lipid mediator composition of the inflammatory exudate switches class, from eicosanoids to SPMs, marking the beginning of the end of the acute inflammatory response. LXs are the first SPM to be locally produced, highlighting its role as “stop” signal to eicosanoid production (exudate switch), as firstly described by the work of Levy et al. (2001). LXs and Rvs also stimulate the recruitment of monocytes. The resolving macrophages then clear apoptotic PMNs, inflammatory debris by efferocytosis (stimulated by LXs, Rvs, PDs). After this has taken place, normal structure and homeostasis can be restored. “Resolution” is defined as the period between peak inflammatory cell influx and the clearance of these cells from the tissue site and the restoration of functional homeostasis. Subsequent post-resolution events involves activation of adaptive immunity B- and T-lymphocytes (Fullerton and Gilroy, 2016).
Failed resolution can lead to increased levels of prostaglandins and leukotrienes, chronic inflammation and fibrosis. Ultimately SPMs reduce the magnitude and duration of inflammation (Arita et al., 2007), stimulate re-epithelialization (Hellmann et al., 2018), wound healing (Dalli, 2017) and tissue regeneration (Dalli, 2017).
While most of the studies involving SPMs have been conducted on rodents models, major and recent advances have been represented by the work of Motwani et al. (2016) in humans, where a new translational model of self-resolving acute inflammatory response triggered by the intradermal injection of UV-killed Escherichia coli into the forearm of healthy volunteers was described. For the first time SPMs endogenous production have been identified in humans over the course of the inflammatory response. It has also been shown that resolution is an active process accelerated by addition of exogenous SPMs.
The molecular mechanisms through which SPMs exert their responses include activation of distinct GPCRs and regulation of gene expression. The binding, and consequent activation, of the LX/N-formyl peptide receptor-2 (ALX/FPR2) GPCR by lipids, such as LXA4 and RvD1 as well as Annexin-1 peptide (Krishnamoorthy et al., 2010; Maderna et al., 2010; Bena et al., 2012), and the RvE1 agonism at the ChemR23 GPCR (Arita et al., 2007) are key to reduce PMN infiltration and subsequently stimulate efferocytosis by macrophages, heralding the initiation of pro-resolving cascade of events.
SPMs in Acute Injuries
The anti-inflammatory and pro-resolving properties of SPMs, including LXs, Rvs and their mimetics, particularly 15(R/S)-methyl-LXA4 (Wu et al., 2013), benzo-LXA4 (Sun et al., 2009), BDA-RvD1 (Orr et al., 2015) and have been demonstrated in several types of experimental acute renal and peritoneal injury (see below).
Moreover, SPMs have recently been shown to play a key role in dampening both sterile inflammation and infection (or non-sterile inflammation). In this context, a recent advance is represented by the above mentioned study on the self-resolving properties of SPMs in an acute and local E. coli-induced translational skin-blisters model (Motwani et al., 2016).
Biosynthesis and Functions of LXs in Acute Injuries
Native LXs, LXA4 and LXB4, are endogenous eicosanoids, transcellularly biosynthesized by 5- and 15-LO interaction of activated leukocytes with epithelium, endothelium or platelets (Serhan et al., 1984; Serhan, 1989; Serhan and Sheppard, 1990) Acetylation of cyclooxygenase-2 by aspirin can trigger the biosynthesis of their 15R-carbon epimers, 15-epi-LXA4 and 15-epi-LXB4 [15-epi-LXs or aspirin-triggered LXs (ATLs)] (Serhan, 2005). Although native LXs have demonstrated potent anti-inflammatory and pro-resolution bioactions (Claria et al., 1996; Fierro and Serhan, 2001; Chandrasekharan and Sharma-Walia, 2015), their therapeutic potential is compromised for by their chemical instability and for by their rapid metabolic inactivation by prostaglandin dehydrogenase-mediated metabolic inactivation in vivo (Clish et al., 2000), with the growing need to synthesize their mimetics.
First-generation synthetic LXA4 analogs were designed in 1995–1998 by Serhan, Petasis and colleagues to minimize metabolism of the molecule (Parkinson, 2006). These relatively stable pharmacological agents, together with myeloid-specific ALX-R-expressing transgenic mice, have provided powerful tools to explore LX functions in vivo. Among those, pharmacokinetic analysis of ATLa, such as methyl (5R,6R,7E,9E,11Z,13E,15S)-16-(4-fluorophenoxy)-5,6,15-trihydroxy-7,9,11,13-hexadecatetraenoate, revealed β-oxidation as a novel route for LXA4 metabolism, prompting the development of second-generation 3-oxa-LXA4 analogs with improved pharmacokinetic disposition (Parkinson, 2006).
Second-generation 3-oxa-LXA4 analogs, such as (5R,6R,7E,9E,11Z,13E,15S)-16-(4-fluorophenoxy)-3-oxa-5,6,15-trihydroxy-7,9,11,13-hexadecatetraenoic acid, have shown potency and efficacy comparable to ATLa in diverse animal models after topical, intravenous or oral delivery (Guilford and Parkinson, 2005).
More recently, a new class of LX-analogs featuring a benzo-fused ring system have been designed and proved to be as potent as native LXA4 in a series of in vitro and in vivo studies (O’Sullivan et al., 2007; Petasis et al., 2008). In particular, it was found to stimulate phagocytosis of apoptotic PMN by macrophages, in a zymosan-induced peritonitis murine model of acute inflammation (O’Sullivan et al., 2007). Further exploration of the mechanism of action through which PMN phagocytosis by bone marrow-derived macrophage was elicited revealed that expression, activation and internalization of ALX/FPR2 by LXA4 and the glucocorticoid-derived Annexin A1 peptide (Ac2-26) were essential (Maderna et al., 2010).
In early 2000, the work from Leonard et al. (2002) suggested a framework for understanding SPMs bioactions in renal IRI and the molecular basis for renoprotection by LXs in this setting. They firstly demonstrated, in a murine renal IRI, that the stable synthetic LXA4 analog 15-epi-16-(FPhO)-LXA4-Me is reno-protective, as gauged by lower serum creatinine, attenuated leukocyte infiltration and reduced morphologic tubule injury. Subsequently, they employed complementary oligonucleotide microarray and bioinformatic analyses to probe the transcriptomic events that underpin LX renoprotection and found that epi-LXA4 modified the expression of many differentially expressed pathogenic mediators, including cytokines, growth factors, adhesion molecules and proteases. Importantly, this LX-modulated transcriptomic response included many genes expressed by renal parenchymal cells (such as the Claudin family epithelial tight junctions) (Kieran et al., 2003).
Biosynthesis and Functions of Rvs in Acute Injuries
Rvs are produced by 12/15-LO, p450, and/or 5-LO, in trans-cellular or intracellular biosynthetic systems of leukocytes or leukocytes plus endothelia/epithelia (Serhan et al., 2000). The novel lipid mediators produced from EPA were first isolated from resolving exudates that proved to contain 18R-HEPE as well as several other related bioactive compounds and were therefore collectively named 18R-E series (Serhan et al., 2000). The first bioactive product isolated from exudates, coined RvE1, reduced inflammation and blocked human PMN trans-endothelial migration.
RvDs are derived from DHA. During inflammation, endogenous DHA is converted to 17S-HEPE which are then converted in 17S-hydroxyl-containing RvDs (RvD1–RvD6) and docosa-conjugated triene-containing PD1/NPD1, via 15-LO (15S-lipoxygenation)-initiated biochemical pathways (Serhan et al., 2002; Hong et al., 2003; Marcheselli et al., 2003) or to 14S hydroxyl-containing MaRs via 12-LO (12S-lipoxygenation)-initiated biochemical pathways. 5-LO catalyzes sequentially with 15-LO or 12/15-LO, generating RvDs (Hong et al., 2003) and some MaRs (Serhan et al., 2009).
RvD1 is converted by eicosanoid oxidoreductases to 17-oxo-RvD1 and 8-oxo-RvD1. The former is an inactive metabolite, while the latter is still effective in suppressing PMN infiltration (Sun et al., 2007). RvE1 is metabolized to 12-oxo-RvE, 18-oxo-RvE1, 10,11-dihydroxy RvE, 19-hydroxy RvE1, 20-hydroxy RvE1 in tissue or cells, of which the first four metabolites are inactive partially or completely in inflammation resolution, and thus are representative of RvE1 metabolic deactivation (Arita et al., 2006; Hong et al., 2008). Human PMNs convert PD1 to its omega-22 hydroxy product (Serhan and Petasis, 2011). The metabolic deactivation of Rvs dysregulated in pathological conditions, may result in their deficiency, or in diminishing the pharmacological efficacy of administered resolvins. Therefore, a series of stable analogs have been successfully synthesized, such as a p-fluorophenoxyl added to RvE1 and RvD1 ω-terminal, which blocks the critical metabolic inactivation of RvE1 or RvD1 without attenuating the anti-inflammatory pro-resolving activities (Arita et al., 2006; Hong et al., 2008; Tang et al., 2014) In particular, the RvE1 analog 19-(p-fluorophenoxy)-RvE1 was synthesized to resist rapid metabolic inactivation and proved to retain biological activity reducing PMN infiltration and pro-inflammatory cytokine/chemokine production in vivo. These results established the structure of a novel RvE1 initial metabolite, indicating that conversion of RvE1 to the oxo product represents a mode of RvE1 inactivation. Moreover, the designed RvE1 analog, which resisted further metabolism/inactivation, could be a useful tool to evaluate the actions of RvE1 in complex disease models (Arita et al., 2006).
These lipids act as paracrine and autocrine mediators of leukocytes to promote resolution of acute injuries, including AKI-initiated inflammation and fibrosis and rescue of kidney functions (Zhao et al., 2016), by shortening PMN life span and promoting macrophage efferocytosis of ACs and the subsequent exit of the phagocytes from inflammatory tissue.
RvD1 and RvE1 also switch macrophage to the phenotype that produces pro-resolving interleukin-10. RvDs or protectin/neuroprotectin D1 (PD1/NPD1) inhibits PMN infiltration into injured kidney, blocks TLR-mediated inflammatory activation of macrophage and mitigates renal dysfunction. RvDs also repress renal interstitial fibrosis, and PD1 promotes reno-protective heme-oxygenase-1 expression. These findings provide novel approaches for targeting inflammation resolution and LMs or modulation of LM-associated pathways for developing better clinical treatments for AKI. Moreover, in LPS-induced AKI, RvD1 could decrease TNFα level, ameliorate kidney pathological injury, protect kidney function, and improve animal survival by down-regulating NFκB inflammatory signal as well as inhibiting renal cell apoptosis (Zhao et al., 2016). Intriguingly, RvE1 counter-regulates leukocytes partially via increased LXA4 biosynthesis (Levy et al., 2011). Since AKI is the major complication of renal allograft transplantation (Bellomo et al., 2012), these results further demonstrate the effectiveness of LXA4 or RvE1 in reducing AKI. LX actions converge with the pro-resolving characteristics of RvD1, as LXA4 and RvD1 both activate the same GPCRs ALXR/FPR2 and GPR32.
SPMs in Chronic Diabetes Complications
Unresolved inflammation drives the development of clinically relevant chronic diseases. Here, we focus the attention on the role of SPMs, particularly LXs and Rvs, on DAA, CKD and, briefly, on DR.
As discussed previously, sustained, non-resolved low-grade inflammation, over decades, promotes formation of atherosclerotic lesions characterized by large necrotic cores, thin fibrous caps and thrombosis. In advanced atherosclerosis, there is an imbalance between levels of SPMs and proinflammatory lipid mediators, which results in sustained leukocyte influx into lesions, inflammatory macrophage polarization, and impaired efferocytosis. In animal models of advanced atherosclerosis, restoration of SPMs limits plaque progression by suppressing inflammation, enhancing efferocytosis, and promoting an increase in collagen cap thickness (Fredman and Tabas, 2017).
From a CKD-perspective, there is a clear mechanistic link between non-resolving inflammation and fibrosis. Non-resolving inflammation results in sustained secretion of pro-fibrotic cytokines and other inflammatory mediators from both resident and infiltrating cells, eliciting fibroblast proliferation and epithelial cell de-differentiation. Sustained or unresolved inflammation is recognized to be an underlying component of many chronic disease states in diverse organ systems, including CKD (Serhan, 2014; Brennan E.P. et al., 2017).
The Role of LXs and Rvs in Atherosclerosis and DAA
It is now established that the local LO-induced biosynthesis of lipid mediators, including LXA4, RvD1 and PD1, protects against atherosclerosis. These mediators exert potent agonist actions on macrophages and vascular ECs that can control the magnitude of the local inflammatory response (Merched et al., 2008), as depicted in Figure 1 (left).
Enhanced biosynthesis of LXA4 in transgenic mice is associated with decreased lesion formation in models of atherosclerosis (Merched et al., 2008). Atheroprotective responses of macrophages and ECs to SPMs include enhanced efferocytosis of apoptotic debris and modulation of adhesion molecules expression (VCAM-1, ICAM-1, P-Sel). It has been shown that LXA4 increases ABCA1 expression and promotes cholesterol efflux through LXRα pathway in THP-1 macrophage-derived foam cells (Sha et al., 2015). Moreover, it has been recently demonstrated that ATL signals through FPR2/ALX in vascular SMCs and protects against intimal hyperplasia after carotid ligation (Petri et al., 2015).
Over the past few years, Brennan’s work focused on the role of miR in both DKD (see details below) and DAA. The let-7 miRNA family plays a key role in modulating inflammatory responses. Vascular SMC proliferation and EC dysfunction are critical in the pathogenesis of atherosclerosis, including in the setting of diabetes. The therapeutic potential of LXA4-induced restoration of let-7 mimic levels was observed in vitro in SMCs, in vivo via tail vein injection in a 24 h murine model, and ex vivo, where significant changes to the secretome in response to let-7 therapy were seen. It has been proposed that restoration of let-7 expression, a mimic of response to LXA4, could provide a new target for an anti-inflammatory approach in diabetic vascular disease (Brennan E. et al., 2017). Very recently, LXA4 and the synthetic LX mimic benzo-LXA4 have also been shown to be athero-protective in murine model of DAA (STZ-induced diabetic ApoE-/- mouse). Here there was significant reduction in plaque area. The authors also demonstrated that these SPMs could attenuate vascular SMCs migration and proliferation, EC-monocytes interactions, as well as modulate the pro-inflammatory secretome signature in human carotid plaque explants. Of particular note was the finding that LX treatment reduced pre-existing plaque burden in diabetic mice (Brennan et al., 2018a).
Oxidation of native LDLs plays an important role in the development of atherosclerosis. A very recent work showed that although ox-LDLs are known to be pro-inflammatory and deleterious in the context of atherosclerosis, they are also able to induce a pro-resolution effect by self-induction of RvD1 from HMEC (Dufour et al., 2018). Moreover, circulating inflammation-resolving lipid mediators RvD1 and DHA are decreased in patients with acutely symptomatic carotid disease (Bazan et al., 2017). Similarly, RvE1 and ATL plasma levels were found to be significantly lower in symptomatic peripheral arteries disease than in healthy controls (Ho et al., 2010).
In addition to lipid agonists, the ALX/FPR2 can also bind peptides, such as Annexin-1 (Maderna et al., 2010). In an advanced model of atherosclerosis, the Annexin-1 derivative acetylated peptide (Ac2-26), was delivered using Collagen IV-targeted nanoparticles and it showed therapeutic effect in fat-fed LDL-R-/- mice, including an increase in the protective collagen layer overlying lesions, suppression of oxidative stress and a decrease in plaque necrosis, thus, suggesting a new form of therapy (Fredman et al., 2015).
The Role of LXs in CKD and DKD
Advances in understanding the effects of LXs in the context of RF arose from investigating their actions on the main cell types involved in kidney failure (mesangial cells, fibroblasts, epithelia, adipocytes) (see details below). As outlined in Figure 2 (right), work from Rodgers, McMahon and Mitchell investigated the potential of LXA4 to regulate PDGF-induced gene expression and the associated autocrine TGFβ1 production in human renal mesangial cells, and found that LXA4 is a potent modulator of matrix accumulation and pro-fibrotic change, thus suggesting a potential protective role in progressive renal disease (McMahon et al., 2002; Mitchell et al., 2004; Rodgers et al., 2005). In an experimental model of RF, i.e., unilateral ureteric obstruction (UUO), LXA4 and its synthetic benzo-analog attenuated injury by inhibiting TGFβ1-induced fibroblast activation, proliferation and gene expression (Borgeson et al., 2011).
Aging, defined as a state of chronic, low-grade, sterile inflammation (inflamm-aging) (Franceschi et al., 2017) and adiposity, have recently been proposed as one of the major risk factors underlying the pathophysiological development of obesity-associated complications, including T2D, and its complications DAA and DKD (Todd et al., 2015). Therefore, of particular relevance in the diabetes context is the work that Borgeson et al. (2011) subsequently carried out, in 2012 and 2015, on the effect of the native LXA4 on obesity-induced adipose tissue inflammation and related diseases. Firstly, using a model of age-associated adipose inflammation, inflamm-aging it was shown that LXA4 attenuates adipose inflammation, decreasing IL-6 and increasing IL-10 expression. The altered cytokine milieu correlated with increased the insulin-regulated glucose transporter-4 and the insulin receptor substrate-1 expression, suggesting improved insulin sensitivity. Further investigations revealed the ability of LXA4 to rescue macrophage-induced desensitization to insulin-stimulated signaling and glucose uptake in cultured adipocytes, thus suggesting that LXA4 may represent a potentially useful and novel therapeutic strategy to subvert adipose inflammation and insulin resistance, key components of T2D (Borgeson et al., 2012). Later on, the role of LXs in obesity-related pathologies was further explored by investigating their impact on impaired glucose tolerance, adipose inflammation, fatty liver and CKD. In particular, LXs attenuated obesity-induced CKD, reducing glomerular expansion, mesangial matrix and urinary H2O2. These data suggested a protective role for LXs against obesity-induced systemic disease, and supported a novel therapeutic paradigm for treating obesity and associated pathologies, such as TD2 and its related complications (Borgeson et al., 2015). A role in the context of aging-related pathologies (including obesity, atherosclerosis, renal disease and diabetes) for SPMs has been also recently reviewed by Doyle et al. (2018).
Certain miRs have been implicated in fibrosis (both renal and cystic). In cultured HK-2 cells, LXA4 suppresses TGF- 1-induced RF through a mechanism involving upregulation of the miR let-7c and downregulation of TGF R1. Expression of let-7c targets is dysregulated in human RF (Brennan et al., 2013). The effects of let-7 on TGF 1-mediated responses of renal epithelia have also been shown by others, including Cooper and Kantharidis, leading to the proposal that let-7b miR represents a potential new target for the treatment of RF in diabetic and non-diabetic nephropathy (Wang et al., 2014; Kantharidis et al., 2015; Brennan E. et al., 2017). Interestingly, LXA4 demonstrated to attenuate TGF- 1-induced fibrotic responses whereby epithelial cells express mesenchymal markers (Brennan et al., 2013). In cultured renal epithelia upregulation of thrombospondin and CTGF is a well-documented fibrotic response (Liu et al., 2013). While, in cystic fibrosis, miR181b is indeed downregulated by LXA4 and RvD1, through ALX/FPR2 activation (Pierdomenico et al., 2015). Moreover, very recent interesting observations showed that LXs can also reverse established atherosclerosis (Brennan et al., 2018a) and DKD (Brennan et al., 2018a).
Very recently, in a DKD murine model, Brennan has also identified a series of transcripts regulated by LXA4 and Benzo-LXA4, modulating well established (TGF-β1, PDGF, TNF-α, NF-κβ) and novel (early growth response-1) networks in DKD, demonstrating that LXs can reverse established diabetic complications and supporting a therapeutic paradigm to promote the resolution of inflammation (Brennan et al., 2018b). Interestingly, a recent study from Goicoechea measured circulating level of ATL in patients with diabetic and non-diabetic kidney disease and found that diabetes was associated with lower levels of the SPMs and that this could be restored by 12-month low dose aspirin treatment (Goicoechea et al., 2017).
The Role of Rvs in Diabetic Wound Healing
The work from Spite greatly deepened the knowledge around the SPMs properties of re-epithelialization and/or re-vascularization post ischemia, particularly focussing on Rvs bioactions. RvD2 stimulates arteriogenic revascularization in a murine model of hind limb ischemia suggesting that resolvins may be a novel class of mediators that both resolve inflammation and promote arteriogenesis (Zhang et al., 2016), a mechanism which can provide protection against nephropathy and atherosclerosis.
Altered resolution of acute inflammation in the context of obesity and diabetes, in which PMN apoptosis is delayed and macrophage efferocytosis is defective, cause persistent leukocyte and AC accumulation and defective wound closure (Baltzis et al., 2014). Wound healing in diabetes is enhanced by RvD1 and RvE1 via the promotion of macrophage-mediated AC clearance and re-epithelialization (Bannenberg et al., 2005; Spite et al., 2014). Moreover, RvD1 decreases adipose tissue macrophage accumulation and improves insulin sensitivity in obese-diabetic mice, suggesting that RvD1 could provide a novel therapeutic strategy for treating obesity-induced diabetes (Hellmann et al., 2011).
The Role of LXs and Rvs in DR
Although the anti-inflammatory (anti-TNFα and anti-VEGF) approach is still the standard therapy for DR, recent in vitro and in vivo models are shifting the attention toward a pro-resolving novel strategy (Das, 2013; Wang and Daggy, 2017).
Since corneal, retinal neuronal degeneration (Srinivasan et al., 2017), conjunctivitis (Stuebiger et al., 2015) and uveitis (Sivaraj et al., 2009) have been associated with DR, the effects of LXs (Gronert, 2005; Biteman et al., 2007; He et al., 2011; Hodges et al., 2017) and Rvs (Tian et al., 2009; Settimio et al., 2012; Li et al., 2013; Lee et al., 2015) in dampening DR are of relevance.
In a well established in vivo model of STZ-induced Diabetes, hyperglycemia induces persistent inflammation and tissue damage, due to decreased expression of heme-oxygenase (HO) in the ciliar body (Rossi et al., 2006). Recently, the effect of RvD1on STZ-induced DR has been explored. RvD1 regulates the NLRP3 inflammasome and NFκB signaling pathway (Yin et al., 2017).
Moreover, by using an in vivo deletion of 12/15-LOX model, associated with exacerbated inflammation and impaired wound healing, due to a failure of HO-1 induction, it has been demonstrated that LXA4, restored the HO synthesis and activity, rescuing the wound healing phenotype (Biteman et al., 2007).
Overall, the therapeutic potential of SPMs in the treatment of DR are promising.
Realizing Therapeutic Potential
The therapeutic challenges presented by diabetes-associated complications such as DAA and DKD are well documented, and experimental evidence, as outlined above, suggests a role for SPM-based mimetics as adjuvants to current therapies. Clinical trials specifically investigating the therapeutic potentials of LXs and Rvs have been limited.
In a randomized controlled trial, AT-LXA4 and a comparatively stable analog of LXB4, 15R/S-methyl-LXB4, reduced the severity of eczema in a study of 60 infants (Wu et al., 2013).
A synthetic analog of RvE1 is in clinical phase III testing for the treatment of the inflammation-based dry eye syndrome; along with this study, other clinical trials using an RvE1 analog to treat various conditions are underway, such as in a single study where inhaled LXA4 decreased LTC4-initiated bronchoprovocation in patients with asthma (Basil and Levy, 2016). RvE1, Mar1 and NPD1 are in clinical development studies for the treatment of neurodegenerative diseases and hearing loss (Serhan et al., 2015; Basil and Levy, 2016).
A clinical trial phase-I evaluating the effects of n-3 fatty acid supplementation on plasma SPMs in patients with CKD showed that endogenous production of SPMs was increased after 8-weeks n-3 fatty acid supplementation in patients with CKD, potentially impacting also patient risk of CVD complications (Mas et al., 2016).
More recently, Gilroy introduced the above mentioned first translational cantharidin-induced skin blister model in healthy male volunteers, providing insights into the mechanisms of self-resolving infections in humans, identifying cells and soluble mediators that may control the resolution phase. Further use of this model will improve our understanding of the evolution and resolution of inflammation in humans, how defects in these over-lapping pathways may contribute to the variability in disease longevity/chronicity, and lends itself to the screen of putative anti-inflammatory or pro-resolution therapies (Motwani et al., 2016).
Summary, Conclusions, and Future Perspectives
Aging populations, increasing urbanization and widening social inequalities are all contributing factors to the rapid rise in diabetes prevalence seen over the past 40 years worldwide. Reducing premature mortality from non-communicable diseases, including diabetes, has become a global priority. For people with either T1D or T2D, advances in clinical care, such as development of better glucose-lowering drugs and structured education programs promoting life-style changes, have led to considerable increases in life expectancy. Effectively, more people are living with diabetes for longer. Understanding the disease course, onset of complications, and comorbid conditions is critical to improving specialized care for people with diabetes.
The most prevalent complications are affecting the microvascular (DKD, DR) and the macrovascular (DAA) systems. As mortality from cardiovascular complications continues to decline, attention must be turned to identifying, preventing, and treating other diabetes complications. In this context, advances in research in the molecular biology of such complications unveiled novel players and novel unified mechanisms driving different diabetes related complications. As highlighted here, inflammation is central to these processes. Evidence is accumulating that agonism of resolution of inflammation is a rational and tractable target that may be an attractive adjuvant in the context of chronic complications of diabetes applying a novel therapeutic paradigm to a vast and growing unmet need.
Author Contributions
MdG, EB, and CG conceived and designed the review article. MdG prepared the first draft of the manuscript. CM, DA, AC, JH, EB, and CG contributed to the final version of the manuscript. All authors read and approved the final manuscript.
Funding
Work in the author’s lab is funded by Science Foundation Ireland (SFI 15/1A/3152; SFI 15/US/B3130), The Health Research Board, the European Union’s Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie grant agreement no. 675111, JDRF strategic research award. MdG was supported by an IRC Government of Ireland postdoctoral fellowship (IRC GOIPD/2017/1060).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
18R-/17S-HEPE, 18R-/17S-hydroeicosapentaenoic acid; ABC-A/G, ATP-binding cassette-subfamily A/G; ACs, apoptotic cells; ACE, angiotensin-converting enzyme; AGE, advanced glycation end-product; AKI, acute kidney injury; ALX/FPR2, lipoxin/N-formyl peptide receptor-2; Apo, apolipoprotein; ATL, aspirin-triggered LX; ATLa, aspirin-triggered LX analog; BM, basement membrane; BMI, body mass index; BMP, bone morphogenetic protein; BRB, blood-retinal barrier; CAD, coronary artery disease; CANTOS, Canakinumab Anti-inflammatory Thrombosis Outcome Study; CARDS, Collaborative Atorvastatin Diabetes Study; CCR, CC-chemokine receptor; CD, cluster of differentiation; ChemR23, chemerin-like-1; CTGF, connective tissue growth factor; CVD, cardiovascular disease; DAA, diabetes-associated atherosclerosis; DAMPs, damage-associated molecular patterns; DHA, docosahexaenoic acid; DKD, diabetic kidney disease; DPP-4, di-peptidyl peptidase-4; ECs, endothelial cells; ECM, extracellular matrix; eGRF, estimated glomerular filtration rate; EMT, epithelial-mesenchymal transition; EPA, eicosapentaenoic acid; ESRD, end stage renal disease; GADA, GAD autoantibody; GLP-1-RA, glucagon-like peptide-1 receptor agonist; GPCRs, G-protein coupled receptors; HDL, high density lipoprotein; HK-2, human proximal tubular epithelial; HO, heme-oxygenase; IHD, ischemic heart disease; IKK-β, inhibitor of nuclear factor kappa-β; IRI, ischemia reperfusion injury; JAK/STAT, janus kinase/signal transducers and activators of transcription; LDL, low density lipoprotein; LDL-R, LDL-receptor; LO, lipoxygenase; LX, lipoxin; MaR, maresin; MARDs, mild age-related diabetes; M-CSF, macrophage colony-stimulating factor; MI, myocardial infarction; MMP, matrix metalloproteinase; MOD, mild obesity-related diabetes; NCCD, non-communicable chronic disease; NF-kB, nuclear factor kappa beta; NLRP, NACHT, LRR and PYD domains-containing protein; Nrf2, nuclear factor (erythroid-derived 2)-like 2; ox-/ac-LDL, oxidized-/acetylated-LDL; PD, protectin; PDGF, platelet-derived growth factor; PMNs, polymorphonuclear neutrophils; PUFA, polyunsaturated fatty acid; RAAS, renin–angiotensin–aldosterone system; RAGE, receptor for advanced glycation end-product; RCT, reverse cholesterol transport; RF, renal fibrosis; ROS, reactive oxygen species; Rv, resolvin; SAID, severe autoimmune diabetes; SFA, saturated fatty acid; SGLT-2, sodium glucose cotransporter-2; SIDD, severe insulin-deficient diabetes; SIRD, severe insulin-resistant diabetes; SMC, smooth muscle cell; SPM, specialized pro-resolving mediator; SR-A, class A-macrophage scavenger receptor; STZ, streptozotocin; T1D/T2D, type-1/type-2 diabetes; TF, tissue factor; TGF-β1, transforming growth factor-β1; Th, T helper; THBS-1, thrombospondin-1; TIF, tubulointerstitial fibrosis; TLR, toll-like receptor; VEGF, vascular endothelial growth factor.
References
Abe, J. I., and Berk, B. C. (2013). Athero-prone flow activation of the SREBP2-NLRP3 inflammasome mediates focal atherosclerosis. Circulation 128, 579–582. doi: 10.1161/CIRCULATIONAHA.113.004390
Abegunde, D. O., Mathers, C. D., Adam, T., Ortegon, M., and Strong, K. (2007). The burden and costs of chronic diseases in low-income and middle-income countries. Lancet 370, 1929–1938. doi: 10.1016/S0140-6736(07)61696-1
Ahlqvist, E., Van Zuydam, N. R., Groop, L. C., and McCarthy, M. I. (2015). The genetics of diabetic complications. Nat. Rev. Nephrol. 11:277. doi: 10.1038/nrneph.2015.37
Ali, I. H. A., and Brazil, D. P. (2014). Bone morphogenetic proteins and their antagonists: current and emerging clinical uses. Br. J. Pharmacol. 171, 3620–3632. doi: 10.1111/bph.12724
Allahverdian, S., Chehroudi, A. C., McManus, B. M., Abraham, T., and Francis, G. A. (2014). Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation 129, 1551–1559. doi: 10.1161/CIRCULATIONAHA.113.005015
Angiolillo, D. J. (2009). Antiplatelet therapy in diabetes: efficacy and limitations of current treatment strategies and future directions. Diabetes Care 32, 531–540. doi: 10.2337/dc08-2064
Arita, M., Oh, S. F., Chonan, T., Hong, S., Elangovan, S., Sun, Y. P., et al. (2006). Metabolic inactivation of resolvin E1 and stabilization of its anti-inflammatory actions. J. Biol. Chem. 281, 22847–22854. doi: 10.1074/jbc.M603766200
Arita, M., Ohira, T., Sun, Y. P., Elangovan, S., Chiang, N., and Serhan, C. N. (2007). Resolvin E1 selectively interacts with leukotriene B4 receptor BLT1 and ChemR23 to regulate inflammation. J. Immunol. 178, 3912–3917. doi: 10.4049/jimmunol.178.6.3912
Bajpai, G., Schneider, C., Wong, N., Bredemeyer, A., and Hulsmans, M. (2018). The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med. 24, 1234–1245. doi: 10.1038/s41591-018-0059-x
Baker, N., O’Meara, S. J., Scannell, M., Maderna, P., and Godson, C. (2009). Lipoxin A4: anti-inflammatory and anti-angiogenic impact on endothelial cells. J. Immunol. 182, 3819–3826. doi: 10.4049/jimmunol.0803175
Baltzis, D., Eleftheriadou, I., and Veves, A. (2014). Pathogenesis and treatment of impaired wound healing in diabetes mellitus: new insights. Adv. Ther. 31, 817–836. doi: 10.1007/s12325-014-0140-x
Bannenberg, G. L., Chiang, N., Ariel, A., Arita, M., Tjonahen, E., Gotlinger, K. H., et al. (2005). Molecular circuits of resolution: formation and actions of resolvins and protectins. J. Immunol. 174, 4345–4355. doi: 10.4049/jimmunol.174.7.4345
Bannister, C. A., Holden, S. E., Jenkins-Jones, S., Morgan, C. L., Halcox, J. P., Schernthaner, G., et al. (2014). Can people with type 2 diabetes live longer than those without? A comparison of mortality in people initiated with metformin or sulphonylurea monotherapy and matched, non-diabetic controls. Diabetes. Obes. Metab. 16, 1165–1173. doi: 10.1111/dom.12354
Barquera, S., Pedroza-Tobías, A., Medina, C., Hernández-Barrera, L., Bibbins-Domingo, K., Lozano, R., et al. (2015). Global overview of the epidemiology of atherosclerotic cardiovascular disease. Arch. Med. Res. 46, 328–338. doi: 10.1016/j.arcmed.2015.06.006
Barter, P. J. (2011). The causes and consequences of low levels of high density lipoproteins in patients with diabetes. Diabetes Metab J. 35, 101–106. doi: 10.4093/dmj.2011.35.2.101
Basil, M. C., and Levy, B. D. (2016). Specialized pro-resolving mediators: endogenous regulators of infection and inflammation. Nat. Rev. Immunol. 16, 51–67. doi: 10.1038/nri.2015.4
Bazan, H. A., Lu, Y., Jun, B., Fang, Z., Woods, T. C., and Hong, S. (2017). Circulating inflammation-resolving lipid mediators RvD1 and DHA are decreased in patients with acutely symptomatic carotid disease. Prostaglandins Leukot. Essent. Fatty Acids 125, 43–47. doi: 10.1016/j.plefa.2017.08.007
Bazan, N. G. (2005). Neuroprotectin D1 (n.d.): a DHA-derived mediator that protects brain and retina against cell injury-induced oxidative stress. Brain Pathol. 15, 159–166. doi: 10.1111/j.1750-3639.2005.tb00513.x
Beckman, J. A., Creager, M. A., and Libby, P. (2002). Diabetes and atherosclerosis: epidemiology, pathophysiology, and management. JAMA 287, 2570–2581. doi: 10.1001/jama.287.19.2570
Beckman, J. A., Paneni, F., Cosentino, F., and Creager, M. A. (2013). Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part II. Eur. Heart J. 34, 2444–2452. doi: 10.1093/eurheartj/eht142
Bellomo, R., Kellum, J. A., and Ronco, C. (2012). Acute kidney injury. Lancet 380, 756–766. doi: 10.1016/S0140-6736(11)61454-2
Bena, S., Brancaleone, V., Wang, J. M., Perretti, M., and Flower, R. J. (2012). Annexin A1 interaction with the FPR2/ALX receptor: identification of distinct domains and downstream associated signaling. J. Biol. Chem. 287, 24690–24697. doi: 10.1074/jbc.M112.377101
Berthier, C. C., Zhang, H., Schin, M., Henger, A., Nelson, R. G., Yee, B., et al. (2008). Enhanced expression of JAK-STAT pathway members in human diabetic nephropathy. Diabetes 58, 469–477. doi: 10.2337/db08-1328
Biteman, B., Hassan, I. R., Walker, E., Leedom, A. J., Dunn, M., Seta, F., et al. (2007). Interdependence of lipoxin A4 and heme-oxygenase in counter-regulating inflammation during corneal wound healing. FASEB J. 21, 2257–2266. doi: 10.1096/fj.06-7918com
Bongarzone, S., Savickas, V., Luzi, F., and Gee, A. D. (2017). Targeting the receptor for advanced glycation endproducts (RAGE): a medicinal chemistry perspective. J. Med. Chem. 60, 7213–7232. doi: 10.1021/acs.jmedchem.7b00058
Boor, P., and Floege, J. (2011). Chronic kidney disease growth factors in renal fibrosis. Clin. Exp. Pharmacol. Physiol. 38, 441–450. doi: 10.1111/j.1440-1681.2011.05487.x
Borgeson, E., Docherty, N. G., Murphy, M., Rodgers, K., Ryan, A., O’Sullivan, T. P., et al. (2011). Lipoxin A(4) and benzo-lipoxin A(4) attenuate experimental renal fibrosis. FASEB J. 25, 2967–2979. doi: 10.1096/fj.11-185017
Borgeson, E., Johnson, A. M., Lee, Y. S., Till, A., Syed, G. H., Ali-Shah, S. T., et al. (2015). Lipoxin A4 attenuates obesity-induced adipose inflammation and associated liver and kidney disease. Cell Metab. 22, 125–137. doi: 10.1016/j.cmet.2015.05.003
Borgeson, E., McGillicuddy, F. C., Harford, K. A., Corrigan, N., Higgins, D. F., Maderna, P., et al. (2012). Lipoxin A4 attenuates adipose inflammation. FASEB J. 26, 4287–4294. doi: 10.1096/fj.12-208249
Bornfeldt, K. E., and Tabas, I. (2011). Insulin resistance, hyperglycemia, and atherosclerosis. Cell Metab. 14, 575–585. doi: 10.1016/j.cmet.2011.07.015
Brazil, D. P., Church, R. H., Surae, S., Godson, C., and Martin, F. (2015). BMP signalling: agony and antagony in the family. Trends Cell Biol. 25, 249–264. doi: 10.1016/j.tcb.2014.12.004
Brennan, E., Wang, B., McClelland, A., Mohan, M., Marai, M., Beuscart, O., et al. (2017). Protective effect of let-7 miRNA family in regulating inflammation in diabetes-associated atherosclerosis. Diabetes Metab. Res. Rev. 66, 2266–2277. doi: 10.2337/db16-1405
Brennan, E. P., Cacace, A., and Godson, C. (2017). Specialized pro-resolving mediators in renal fibrosis. Mol. Aspects Med. 58, 102–113. doi: 10.1016/j.mam.2017.05.001
Brennan, E. P., Mohan, M., McClelland, A., de Gaetano, M., Tikellis, C., Marai, M., et al. (2018a). Lipoxins protect against inflammation in diabetes-associated atherosclerosis. Diabetes 67, 2657–2667. doi: 10.2337/db17-1317
Brennan, E. P., Mohan, M., McClelland, A., Tikellis, C., Ziemann, M., Kaspi, A., et al. (2018b). Lipoxins regulate the early growth response-1 network and reverse diabetic kidney disease. J. Am. Soc. Nephrol. 29, 1437–1448. doi: 10.1681/ASN.2017101112
Brennan, E. P., Nolan, K. A., Borgeson, E., Gough, O. S., McEvoy, C. M., Docherty, N. G., et al. (2013). Lipoxins attenuate renal fibrosis by inducing let-7c and suppressing TGFbetaR1. J. Am. Soc. Nephrol. 24, 627–637. doi: 10.1681/ASN.2012060550
Brodsky, J. L., and Fisher, E. A. (2008). The many intersecting pathways underlying apolipoprotein B secretion and degradation. Trends Endocrinol. Metab. 19, 254–259. doi: 10.1016/j.tem.2008.07.002
Brophy, M. L., Dong, Y., Wu, H., Rahman, H. N. A., Song, K., and Chen, H. (2017). Eating the dead to keep atherosclerosis at bay. Front. Cardiovasc. Med. 4:2. doi: 10.3389/fcvm.2017.00002
Buchanan, T. A., and Xiang, A. H. (2005). Gestational diabetes mellitus. J. Clin. Invest. 115, 485–491. doi: 10.1172/JCI200524531
Buchtler, S., Grill, A., Hofmarksrichter, S., Stockert, P., Schiechl-Brachner, G., Rodriguez Gomez, M., et al. (2018). Cellular origin and functional relevance of collagen I production in the kidney. J. Am. Soc. Nephrol. 29, 1859–1873. doi: 10.1681/ASN.2018020138
Buckley, C. D., Gilroy, D. W., and Serhan, C. N. (2014). Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity 40, 315–327. doi: 10.1016/j.immuni.2014.02.009
Chandrasekharan, J. A., and Sharma-Walia, N. (2015). Lipoxins: nature’s way to resolve inflammation. J. Inflamm. Res. 8, 181–192.
Chin, M. P., Bakris, G. L., Block, G. A., Chertow, G. M., Goldsberry, A., Inker, L. A., et al. (2018). Bardoxolone methyl improves kidney function in patients with chronic kidney disease stage 4 and type 2 diabetes: post-hoc analyses from bardoxolone methyl evaluation in patients with chronic kidney disease and type 2 diabetes study. Am. J. Nephrol. 47, 40–47. doi: 10.1159/000486398
Chin, M. P., Reisman, S. A., Bakris, G. L., O’Grady, M., Linde, P. G., McCullough, P. A., et al. (2014). Mechanisms contributing to adverse cardiovascular events in patients with type 2 diabetes mellitus and stage 4 chronic kidney disease treated with bardoxolone methyl. Am. J. Nephrol. 39, 499–508. doi: 10.1159/000362906
Chinetti-Gbaguidi, G., Baron, M., Bouhlel, M. A., Vanhoutte, J., Copin, C., Sebti, Y., et al. (2011). Human atherosclerotic plaque alternative macrophages display low cholesterol handling but high phagocytosis because of distinct activities of the PPARG and LXRα pathways. Circ. Res. 108, 985–995. doi: 10.1161/CIRCRESAHA.110.233775
Claria, J., Lee, M. H., and Serhan, C. N. (1996). Aspirin-triggered lipoxins (15-epi-LX) are generated by the human lung adenocarcinoma cell line (A549)-neutrophil interactions and are potent inhibitors of cell proliferation. Mol. Med. 2, 583–596. doi: 10.1007/BF03401642
Clish, C. B., Levy, B. D., Chiang, N., Tai, H. H., and Serhan, C. N. (2000). Oxidoreductases in lipoxin A4 metabolic inactivation: a novel role for 15-onoprostaglandin 13-reductase/leukotriene B4 12-hydroxydehydrogenase in inflammation. J. Biol. Chem. 275, 25372–25380. doi: 10.1074/jbc.M002863200
Colgan, S. P., Serhan, C. N., Parkos, C. A., Delp-Archer, C., and Madara, J. L. (1993). Lipoxin A4 modulates transmigration of human neutrophils across intestinal epithelial monolayers. J. Clin. Invest. 92, 75–82. doi: 10.1172/JCI116601
Colhoun, H. M., Betteridge, D. J., Durrington, P. N., Hitman, G. A., Neil, H. A., Livingstone, S. J., et al. (2004). Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet 364, 685–696. doi: 10.1016/S0140-6736(04)16895-5
Colwell, J. A. (1997). Aspirin therapy in diabetes. Diabetes Care 20:1767. doi: 10.2337/diacare.20.11.1767
Congdon, N., O’Colmain, B., Klaver, C. C., Klein, R., Munoz, B., Friedman, D. S., et al. (2004). Causes and prevalence of visual impairment among adults in the United States. Arch. Ophthalmol. 122, 477–485. doi: 10.1001/archopht.122.4.477
Cosentino, F., and Luscher, T. F. (1998). Endothelial dysfunction in diabetes mellitus. J. Cardiovasc. Pharmacol. 32(Suppl 3), S54–S61.
Cunningham, K. S., and Gotlieb, A. I. (2005). The role of shear stress in the pathogenesis of atherosclerosis. Lab. Invest. 85, 9–23. doi: 10.1038/labinvest.3700215
Dalli, J. (2017). Does promoting resolution instead of inhibiting inflammation represent the new paradigm in treating infections? Mol. Aspects Med. 58, 12–20. doi: 10.1016/j.mam.2017.03.007
Das, U. N. (2013). Lipoxins, resolvins, and protectins in the prevention and treatment of diabetic macular edema and retinopathy. Nutrition 29, 1–7. doi: 10.1016/j.nut.2012.02.003
de Gaetano, M., Crean, D., Barry, M., and Belton, O. (2016). M1- and M2-type macrophage responses are predictive of adverse outcomes in human atherosclerosis. Front. Immunol. 7:275. doi: 10.3389/fimmu.2016.00275
Distel, E., Barrett, T. J., Chung, K., Girgis, N. M., Parathath, S., Essau, C. C., et al. (2014). mir33 inhibition overcomes deleterious effects of diabetes on atherosclerosis plaque regression in mice. Circ. Res. 115:759. doi: 10.1161/CIRCRESAHA.115.304164
Docherty, N. G., and le Roux, C. W. (2014). Improvements in the metabolic milieu following Roux-en-Y gastric bypass and the arrest of diabetic kidney disease. Exp. Physiol. 99, 1146–1153. doi: 10.1113/expphysiol.2014.078790
Doganay, S., Evereklioglu, C., Er, H., Turkoz, Y., Sevinc, A., Mehmet, N., et al. (2002). Comparison of serum NO, TNF-alpha, IL-1β, sIL-2R, IL-6 and IL-8 levels with grades of retinopathy in patients with diabetes mellitus. Eye 16, 163–170. doi: 10.1038/sj/eye/6700095
Doyle, R., Sadlier, D. M., and Godson, C. (2018). Pro-resolving lipid mediators: agents of anti-ageing? Semin Immunol. doi: 10.1016/j.smim.2018.09.002 [Epub ahead of print].
Duffield, J. S. (2010). Macrophages and Immunologic Inflammation of the Kidney. Seminars in Nephrology. Amsterdam: Elsevier.
Duffield, J. S. (2014). Cellular and molecular mechanisms in kidney fibrosis. J. Clin. Invest. 124, 2299–2306. doi: 10.1172/JCI72267
Duffield, J. S., Hong, S., Vaidya, V. S., Lu, Y., Fredman, G., Serhan, C. N., et al. (2006). Resolvin D series and protectin D1 mitigate acute kidney injury. J. Immunol. 177, 5902–5911. doi: 10.4049/jimmunol.177.9.5902
Dufour, D., Khalil, A., Nuyens, V., Rousseau, A., Delporte, C., Noyon, C., et al. (2018). Native and myeloperoxidase-oxidized low-density lipoproteins act in synergy to induce release of resolvin-D1 from endothelial cells. Atherosclerosis 272, 108–117. doi: 10.1016/j.atherosclerosis.2018.03.012
Duh, E. J., Sun, J. K., and Stitt, A. W. (2017). Diabetic retinopathy: current understanding, mechanisms, and treatment strategies. JCI Insight 2:e93751. doi: 10.1172/jci.insight.93751
Duncan, B. B., Schmidt, M. I., Pankow, J. S., Ballantyne, C. M., Couper, D., Vigo, A., et al. (2003). Low-grade systemic inflammation and the development of type 2 diabetes: the atherosclerosis risk in communities study. Diabetes Metab. Res. Rev. 52, 1799–1805. doi: 10.2337/diabetes.52.7.1799
Dutta, D., and Mukhopadhyay, S. (2018). Novel diabetes subgroups. Lancet Diabetes Endocrinol. 6:438. doi: 10.1016/S2213-8587(18)30129-3
Eddy, A. A., and Neilson, E. G. (2006). Chronic kidney disease progression. J. Am. Soc. Nephrol. 17, 2964–2966. doi: 10.1681/ASN.2006070704
Engelberg, H., Kuhn, R., and Steinman, M. (1956). A controlled study of the effect of intermittent heparin therapy on the course of human coronary atherosclerosis. Circulation 13, 489–498. doi: 10.1161/01.CIR.13.4.489
Epelman, S., Lavine, K. J., and Randolph, G. J. (2014). Origin and functions of tissue macrophages. Immunity 41, 21–35. doi: 10.1016/j.immuni.2014.06.013
Everett, B. M., Donath, M. Y., Pradhan, A. D., Thuren, T., Pais, P., Nicolau, J. C., et al. (2018). Anti-inflammatory therapy with canakinumab for the prevention and management of diabetes. J. Am. Coll. Cardiol. 71, 2392–2401. doi: 10.1016/j.jacc.2018.03.002
Falke, L. L., Goldschmeding, R., and Nguyen, T. Q. (2014). A perspective on anti-CCN2 therapy for chronic kidney disease. Nephrol. Dial. Transpl. 29(suppl 1), i30–i37. doi: 10.1093/ndt/gft430
Falke, L. L., Leeuwis, J. W., Lyons, K. M., Mummery, C. L., Nguyen, T. Q., and Goldschmeding, R. (2017). CCN2 reduction mediates protective effects of BMP7 treatment in obstructive nephropathy. J. Cell Commun. Signal. 11, 39–48. doi: 10.1007/s12079-016-0358-2
Fallon, J. T., DavidGertz, S., Chesebro, J. H., and Badimon, J. J. (1997). Tissue factor modulates the thrombogenicity of human atherosclerotic plaques. Circulation 95, 594–599. doi: 10.1161/01.CIR.95.3.594
Feig, J. E., Parathath, S., Rong, J. X., Mick, S. L., Vengrenyuk, Y., Grauer, L., et al. (2011). Reversal of hyperlipidemia with a genetic switch favorably affects the content and inflammatory state of macrophages in atherosclerotic plaques. Circulation 123, 989–998. doi: 10.1161/CIRCULATIONAHA.110.984146
Feig, J. E., Quick, J. S., and Fisher, E. A. (2009). The role of a murine transplantation model of atherosclerosis regression in drug discovery. Curr. Opin. Investig. Drugs 10, 232–238.
Ferrante, C. J., and Leibovich, S. J. (2012). Regulation of macrophage polarization and wound healing. Adv. Wound Care 1, 10–16. doi: 10.1089/wound.2011.0307
Fierro, I. M., and Serhan, C. N. (2001). Mechanisms in anti-inflammation and resolution: the role of lipoxins and aspirin-triggered lipoxins. Braz. J. Med. Biol. Res. 34, 555–566. doi: 10.1590/S0100-879X2001000500002
Fisher, E. A. (2016). Regression of atherosclerosis: the journey from the liver to the plaque and back. Arterioscler. Thromb. Vasc. Biol. 36, 226–235. doi: 10.1161/ATVBAHA.115.301926
Fleg, J. L., Mete, M., Howard, B. V., Umans, J. G., Roman, M. J., Ratner, R. E., et al. (2008). Effect of statins alone versus statins plus ezetimibe on carotid atherosclerosis in type 2 diabetes: the SANDS (stop atherosclerosis in native diabetics study) trial. J. Am. Coll. Cardiol. 52, 2198–2205. doi: 10.1016/j.jacc.2008.10.031
Forbes, J. M., and Cooper, M. E. (2013). Mechanisms of diabetic complications. Physiol. Rev. 93, 137–188. doi: 10.1152/physrev.00045.2011
Franceschi, C., Garagnani, P., Vitale, G., Capri, M., and Salvioli, S. (2017). Inflammaging and ‘Garb-aging’. Trends Endocrinol. Metab. 28, 199–212. doi: 10.1016/j.tem.2016.09.005
Fredman, G., Kamaly, N., Spolitu, S., Milton, J., Ghorpade, D., Chiasson, R., et al. (2015). Targeted nanoparticles containing the proresolving peptide Ac2-26 protect against advanced atherosclerosis in hypercholesterolemic mice. Sci. Transl. Med. 7:275ra220. doi: 10.1126/scitranslmed.aaa1065
Fredman, G., and Tabas, I. (2017). Boosting inflammation resolution in atherosclerosis: the next frontier for therapy. Am. J. Pathol. 187, 1211–1221. doi: 10.1016/j.ajpath.2017.01.018
Fullerton, J. N., and Gilroy, D. W. (2016). Resolution of inflammation: a new therapeutic frontier. Nat. Rev. Drug Discov. 15, 551–567. doi: 10.1038/nrd.2016.39
Funaki, M. (2009). Saturated fatty acids and insulin resistance. J. Med. Invest. 56, 88–92. doi: 10.2152/jmi.56.88
Fuster, V., Badimon, J. J., and Chesebro, J. H. (1998). Atherothrombosis: mechanisms and clinical therapeutic approaches. Vasc. Med. 3, 231–239. doi: 10.1177/1358836X9800300310
Fuster, V., and Kelly, B. (2011). Summary of the institute of medicine report promoting cardiovascular health in the developing world. Glob. Heart 6, 133–142. doi: 10.1016/j.gheart.2011.08.002
Geissmann, F., Auffray, C., Palframan, R., Wirrig, C., Ciocca, A., Campisi, L., et al. (2008). Blood monocytes: distinct subsets, how they relate to dendritic cells, and their possible roles in the regulation of T-cell responses. Immunol. Cell Biol. 86, 398–408. doi: 10.1038/icb.2008.19
Godson, C., and Brady, H. R. (2000). Lipoxins: novel anti-inflammatory therapeutics? Curr. Opin. Invest. Drugs 1, 380–385.
Godson, C., Mitchell, S., Harvey, K., Petasis, N. A., Hogg, N., and Brady, H. R. (2000). Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J. Immunol. 164, 1663–1667. doi: 10.4049/jimmunol.164.4.1663
Goicoechea, M., Sanchez-Nino, M. D., Ortiz, A., Garcia de Vinuesa, S., Quiroga, B., Bernis, C., et al. (2017). Low dose aspirin increases 15-epi-lipoxin A4 levels in diabetic chronic kidney disease patients. Prostaglandins Leukot. Essent. Fatty Acids 125, 8–13. doi: 10.1016/j.plefa.2017.08.009
Goraya, T. Y., Leibson, C. L., Palumbo, P. J., Weston, S. A., Killian, J. M., Pfeifer, E. A., et al. (2002). Coronary atherosclerosis in diabetes mellitus. A population-based autopsy study. J. Am. Coll. Cardiol. 40, 946–953. doi: 10.1016/S0735-1097(02)02065-X
Greenlee-Wacker, M. C. (2016). Clearance of apoptotic neutrophils and resolution of inflammation. Immunol. Rev. 273, 357–370. doi: 10.1111/imr.12453
Gregg, E. W., Cheng, Y. J., Srinivasan, M., Lin, J., Geiss, L. S., Albright, A. L., et al. (2018). Trends in cause-specific mortality among adults with and without diagnosed diabetes in the USA: an epidemiological analysis of linked national survey and vital statistics data. Lancet 391, 2430–2440. doi: 10.1016/S0140-6736(18)30314-3
Gregg, E. W., Li, Y., Wang, J., Rios Burrows, N., Ali, M. K., Rolka, D., et al. (2014). Changes in diabetes-related complications in the United States, 1990–2010. New Engl. J. Med. 370, 1514–1523. doi: 10.1056/NEJMoa1310799
Gresele, P., Guglielmini, G., De Angelis, M., Ciferri, S., Ciofetta, M., Falcinelli, E., et al. (2003). Acute, short-term hyperglycemia enhances shear stress-induced platelet activation in patients with type II diabetes mellitus. J. Am. Coll. Cardiol. 41, 1013–1020. doi: 10.1016/S0735-1097(02)02972-8
Gronert, K. (2005). Lipoxins in the eye and their role in wound healing. Prostaglandins Leukot. Essent. Fatty Acids 73, 221–229. doi: 10.1016/j.plefa.2005.05.009
Guilford, W. J., and Parkinson, J. F. (2005). Second-generation beta-oxidation resistant 3-oxa-lipoxin A4 analogs. Prostaglandins Leukot. Essent. Fatty Acids 73, 245–250. doi: 10.1016/j.plefa.2005.05.012
Guzik, T. J., and Cosentino, F. (2018). Epigenetics and immunometabolism in diabetes and aging. Antioxid. Redox Signal. 29, 257–274. doi: 10.1089/ars.2017.7299
Haritoglou, C., Kook, D., Neubauer, A., Wolf, A., Priglinger, S., Strauss, R., et al. (2006). Intravitreal bevacizumab (Avastin) therapy for persistent diffuse diabetic macular edema. Retina 26, 999–1005. doi: 10.1097/01.iae.0000247165.38655.bf
Harjutsalo, V., and Groop, P. H. (2014). Epidemiology and risk factors for diabetic kidney disease. Adv. Chronic Kidney Dis. 21, 260–266. doi: 10.1053/j.ackd.2014.03.009
He, J., Kakazu, A. H., Bazan, N. G., and Bazan, H. E. (2011). Aspirin-triggered lipoxin A4 (15-epi-LXA4) increases the endothelial viability of human corneas storage in Optisol-GS. J. Ocul. Pharmacol. Ther. 27, 235–241. doi: 10.1089/jop.2010.0187
Hellmann, J., Sansbury, B. E., Wong, B., Li, X., Singh, M., Nuutila, K., et al. (2018). Biosynthesis of D-series resolvins in skin provides insights into their role in tissue repair. J. Invest. Dermatol. 138, 2051–2060. doi: 10.1016/j.jid.2018.03.1498
Hellmann, J., Tang, Y., Kosuri, M., Bhatnagar, A., and Spite, M. (2011). Resolvin D1 decreases adipose tissue macrophage accumulation and improves insulin sensitivity in obese-diabetic mice. FASEB J. 25, 2399–2407. doi: 10.1096/fj.10-178657
Herrington, W., Lacey, B., Sherliker, P., Armitage, J., and Lewington, S. (2016). Epidemiology of atherosclerosis and the potential to reduce the global burden of atherothrombotic disease. Circ. Res. 118, 535–546. doi: 10.1161/CIRCRESAHA.115.307611
Hewing, B., Parathath, S., Barrett, T., Chung, W. K., Astudillo, Y. M., Hamada, T., et al. (2014). Effects of native and myeloperoxidase-modified apolipoprotein a-I on reverse cholesterol transport and atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 34, 779–789. doi: 10.1161/ATVBAHA.113.303044
Ho, K. J., Spite, M., Owens, C. D., Lancero, H., Kroemer, A. H., Pande, R., et al. (2010). Aspirin-triggered lipoxin and resolvin E1 modulate vascular smooth muscle phenotype and correlate with peripheral atherosclerosis. Am. J. Pathol. 177, 2116–2123. doi: 10.2353/ajpath.2010.091082
Hodges, R. R., Li, D., Shatos, M. A., Bair, J. A., Lippestad, M., Serhan, C. N., et al. (2017). Lipoxin A4 activates ALX/FPR2 receptor to regulate conjunctival goblet cell secretion. Mucosal Immunol. 10, 46–57. doi: 10.1038/mi.2016.33
Hong, S., Gronert, K., Devchand, P. R., Moussignac, R. L., and Serhan, C. N. (2003). Novel docosatrienes and 17S-resolvins generated from docosahexaenoic acid in murine brain, human blood, and glial cells. Autacoids in anti-inflammation. J. Biol. Chem. 278, 14677–14687. doi: 10.1074/jbc.M300218200
Hong, S., Porter, T. F., Lu, Y., Oh, S. F., Pillai, P. S., and Serhan, C. N. (2008). Resolvin E1 metabolome in local inactivation during inflammation-resolution. J. Immunol. 180, 3512–3519. doi: 10.4049/jimmunol.180.5.3512
Huang, H., Gandhi, J. K., Zhong, X., Wei, Y., Gong, J., Duh, E. J., et al. (2011). TNFalpha is required for late BRB breakdown in diabetic retinopathy, and its inhibition prevents leukostasis and protects vessels and neurons from apoptosis. Invest. Ophthalmol. Vis. Sci. 52, 1336–1344. doi: 10.1167/iovs.10-5768
Huh, J. H., Kim, H. M., Lee, E. S., Kwon, M. H., Lee, B. R., Ko, H. J., et al. (2018). Dual CCR2/5 antagonist attenuates obesity-induced insulin resistance by regulating macrophage recruitment and M1/M2 status. Obesity 26, 378–386. doi: 10.1002/oby.22103
Hussein, M. A., Shrestha, E., Ouimet, M., Barrett, T. J., Leone, S., Moore, K. J., et al. (2015). LXR-mediated ABCA1 expression and function are modulated by high glucose and PRMT2. PLoS One 10:e0135218. doi: 10.1371/journal.pone.0135218
Ivanov, S., Scallan, J. P., Kim, K. W., Werth, K., Johnson, M. W., Saunders, B. T., et al. (2016). CCR7 and IRF4-dependent dendritic cells regulate lymphatic collecting vessel permeability. J. Clin. Invest. 126, 1581–1591. doi: 10.1172/JCI84518
Jingi, A. M., Tankeu, A. T., Ateba, N. A., and Noubiap, J. J. (2017). Mechanism of worsening diabetic retinopathy with rapid lowering of blood glucose: the synergistic hypothesis. BMC Endocrine Disord. 17:63. doi: 10.1186/s12902-017-0213-3
Kahn, S. E., Hull, R. L., and Utzschneider, K. M. (2006). Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444):840.
Kalluri, R., and Weinberg, R. A. (2009). The basics of epithelial-mesenchymal transition. J. Clin. Invest. 119, 1420–1428. doi: 10.1172/JCI39104
Kantharidis, P., Hagiwara, S., Brennan, E., and McClelland, A. D. (2015). Study of microRNA in diabetic nephropathy: isolation, quantification and biological function. Nephrology 20, 132–139. doi: 10.1111/nep.12374
Kay, A. M., Simpson, C. L., and Stewart, J. A. (2016). The role of AGE/RAGE signaling in diabetes-mediated vascular calcification. J. Diabetes Res. 2016:6809703. doi: 10.1155/2016/6809703
Kieran, N. E., Doran, P. P., Connolly, S. B., Greenan, M. C., Higgins, D. F., Leonard, M., et al. (2003). Modification of the transcriptomic response to renal ischemia/reperfusion injury by lipoxin analog. Kidney Int. 64, 480–492. doi: 10.1046/j.1523-1755.2003.00106.x
Klaassen, I., Van Noorden, C. J., and Schlingemann, R. O. (2013). Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog. Retin. Eye Res. 34, 19–48. doi: 10.1016/j.preteyeres.2013.02.001
Kowluru, R. A., Zhong, Q., and Santos, J. M. (2012). Matrix metalloproteinases in diabetic retinopathy: potential role of MMP-9. Expert. Opin. Investig. Drugs 21, 797–805. doi: 10.1517/13543784.2012.681043
Krishnamoorthy, S., Recchiuti, A., Chiang, N., Yacoubian, S., Lee, C.-H., Yang, R., et al. (2010). Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc. Natl. Acad. Sci. U.S.A. 107, 1660–1665.
Krolewski, A. S., Niewczas, M. A., Skupien, J., Gohda, T., Smiles, A., Eckfeldt, J. H., et al. (2014). Early progressive renal decline precedes the onset of microalbuminuria and its progression to macroalbuminuria. Diabetes Care 37, 226–234. doi: 10.2337/dc13-0985
Kunjathoor, V. V., Febbraio, M., Podrez, E. A., Moore, K. J., Andersson, L., Koehn, S., et al. (2002). Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J. Biol. Chem. 277, 49982–49988. doi: 10.1074/jbc.M209649200
Lassila, M., Allen, T. J., Cao, Z., Thallas, V., Jandeleit-Dahm, K. A., Candido, R., et al. (2004). Imatinib attenuates diabetes-associated atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 24, 935–942. doi: 10.1161/01.ATV.0000124105.39900.db
Leaf, I. A., and Duffield, J. S. (2017). What can target kidney fibrosis? Nephrol. Dial. Transplant. 32(suppl 1), i89–i97. doi: 10.1093/ndt/gfw388
Lee, I. M., Shiroma, E. J., Lobelo, F., Puska, P., Blair, S. N., Katzmarzyk, P. T., et al. (2012). Effect of physical inactivity on major non-communicable diseases worldwide: an analysis of burden of disease and life expectancy. Lancet 380, 219–229. doi: 10.1016/S0140-6736(12)61031-9
Lee, J., Rhee, M. H., Kim, E., and Cho, J. Y. (2012). BAY 11-7082 is a broad-spectrum inhibitor with anti-inflammatory activity against multiple targets. Mediat. Inflamm. 2012:416036. doi: 10.1155/2012/416036
Lee, J. E., Sun, Y., Gjorstrup, P., and Pearlman, E. (2015). Inhibition of corneal inflammation by the resolvin E1. Invest. Ophthalmol. Vis. Sci. 56, 2728–2736. doi: 10.1167/iovs.14-15982
Lee, S., Huen, S., Nishio, H., Nishio, S., Lee, H. K., Choi, B.-S., et al. (2011). Distinct macrophage phenotypes contribute to kidney injury and repair. J. Am. Soc. Nephrol. 22, 317–326. doi: 10.1681/ASN.2009060615
Leonard, M. O., Hannan, K., Burne, M. J., Lappin, D. W., Doran, P., Coleman, P., et al. (2002). 15-Epi-16-(para-fluorophenoxy)-lipoxin A(4)-methyl ester, a synthetic analogue of 15-epi-lipoxin A(4), is protective in experimental ischemic acute renal failure. J. Am. Soc. Nephrol. 13, 1657–1662. doi: 10.1097/01.ASN.0000015795.74094.91
Levy, B. D., Clish, C. B., Schmidt, B., Gronert, K., and Serhan, C. N. (2001). Lipid mediator class switching during acute inflammation: signals in resolution. Nat. Immunol. 2, 612–619. doi: 10.1038/89759
Levy, B. D., Zhang, Q. Y., Bonnans, C., Primo, V., Reilly, J. J., Perkins, D. L., et al. (2011). The endogenous pro-resolving mediators lipoxin A4 and resolvin E1 preserve organ function in allograft rejection. Prostaglandins Leukot Essent Fatty Acids 84, 43–50. doi: 10.1016/j.plefa.2010.09.002
Li, A. C., and Glass, C. K. (2002). The macrophage foam cell as a target for therapeutic intervention. Nat. Med. 8, 1235–1242. doi: 10.1038/nm1102-1235
Li, D., Hodges, R. R., Jiao, J., Carozza, R. B., Shatos, M. A., Chiang, N., et al. (2013). Resolvin D1 and aspirin-triggered resolvin D1 regulate histamine-stimulated conjunctival goblet cell secretion. Mucosal Immunol. 6, 1119–1130. doi: 10.1038/mi.2013.7
Li, S., Sun, Y., Liang, C.-P., Thorp, E. B., Han, S., Jehle, A. W., et al. (2009). Defective phagocytosis of apoptotic cells by macrophages in atherosclerotic lesions of ob/ob mice and reversal by a fish oil diet. Circ. Res. 105:1072. doi: 10.1161/CIRCRESAHA.109.199570
Libby, P., Ridker, P. M., and Maseri, A. (2002). Inflammation and atherosclerosis. Circulation 105, 1135–1143. doi: 10.1161/hc0902.104353
Lieth, E., Gardner, T. W., Barber, A. J., Antonetti, D. A., et al. (2000). Retinal neurodegeneration: early pathology in diabetes. Clin. Exp. Ophthalmol. 28, 3–8. doi: 10.1046/j.1442-9071.2000.00222.x
Liu, Y. (2011). Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 7:684. doi: 10.1038/nrneph.2011.149
Liu, Y., Liu, H., Meyer, C., Li, J., Nadalin, S., Knigsrainer, A., et al. (2013). Transforming growth factor-β (TGF-β)-mediated connective tissue growth factor (CTGF) expression in hepatic stellate cells requires Stat3 signaling activation. J. Biol. Chem. 288, 30708–30719. doi: 10.1074/jbc.M113.478685
Ljubimov, A. V., Burgeson, R. E., Butkowski, R. J., Couchman, J. R., Wu, R. R., Ninomiya, Y., et al. (1996). Extracellular matrix alterations in human corneas with bullous keratopathy. Invest. Ophthalmol. Vis. Sci. 37, 997–1007.
Llodra, J., Angeli, V., Liu, J., Trogan, E., Fisher, E. A., and Randolph, G. J. (2004). Emigration of monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc. Natl. Acad. Sci. U.S.A. 101, 11779–11784. doi: 10.1073/pnas.0403259101
Lotfy, M., Adeghate, J., Kalasz, H., Singh, J., and Adeghate, E. (2017). Chronic complications of diabetes mellitus: a mini review. Curr. Diabetes Rev. 13, 3–10. doi: 10.2174/1573399812666151016101622
Ma, J., Yue, J., Huang, R., Liao, Y., Li, S., and Liu, W. (2018). Reversion of aging-related DHEAS decline in mouse plasma alleviates aging-related glucose tolerance impairment by potentiation of glucose-stimulated insulin secretion of acute phase. Biochem. Biophys. Res. Commun. 500, 671–675. doi: 10.1016/j.bbrc.2018.04.134
Maderna, P., Cottell, D. C., Toivonen, T., Dufton, N., Dalli, J., Perretti, M., et al. (2010). FPR2/ALX receptor expression and internalization are critical for lipoxin A(4) and annexin-derived peptide-stimulated phagocytosis. FASEB J. 24, 4240–4249. doi: 10.1096/fj.10-159913
Maderna, P., and Godson, C. (2009). Lipoxins: resolutionary road. Br. J. Pharmacol. 158, 947–959. doi: 10.1111/j.1476-5381.2009.00386.x
Mann, J. F. E., Orsted, D. D., Brown-Frandsen, K., Marso, S. P., Poulter, N. R., Rasmussen, S., et al. (2017). Liraglutide and renal outcomes in type 2 diabetes. N. Engl. J. Med. 377, 839–848. doi: 10.1056/NEJMoa1616011
Mantovani, A., Sica, A., Sozzani, S., Allavena, P., Vecchi, A., and Locati, M. (2004). The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 25, 677–686. doi: 10.1016/j.it.2004.09.015
Marcheselli, V. L., Hong, S., Lukiw, W. J., Tian, X. H., Gronert, K., Musto, A., et al. (2003). Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J. Biol. Chem. 278, 43807–43817. doi: 10.1074/jbc.M305841200
Martinez, F. O., and Gordon, S. (2014). The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000prime Rep. 6, 13–13. doi: 10.12703/P6-13
Mas, E., Barden, A., Burke, V., Beilin, L. J., Watts, G. F., Huang, R. C., et al. (2016). A randomized controlled trial of the effects of n-3 fatty acids on resolvins in chronic kidney disease. Clin. Nutr. 35, 331–336. doi: 10.1016/j.clnu.2015.04.004
McGill, H. C. Jr., McMahan, C. A., Malcom, G. T., Oalmann, M. C., and Strong, J. P. (1995). Relation of glycohemoglobin and adiposity to atherosclerosis in youth. Pathobiological Determinants of Atherosclerosis in Youth (PDAY) research group. Arterioscler Thromb Vasc. Biol. 15, 431–440. doi: 10.1161/01.ATV.15.4.431
McMahon, B., Mitchell, D., Shattock, R., Martin, F., Brady, H. R., and Godson, C. (2002). Lipoxin, leukotriene, and PDGF receptors cross-talk to regulate mesangial cell proliferation. FASEB J. 16, 1817–1819. doi: 10.1096/fj.02-0416fje
Meininger, C. J., Marinos, R. S., Hatakeyama, K., Martinez-Zaguilan, R., Rojas, J. D., Kelly, K. A., et al. (2000). Impaired nitric oxide production in coronary endothelial cells of the spontaneously diabetic BB rat is due to tetrahydrobiopterin deficiency. Biochem. J. 349(Pt 1): 353–356. doi: 10.1042/bj3490353
Merched, A. J., Ko, K., Gotlinger, K. H., Serhan, C. N., and Chan, L. (2008). Atherosclerosis: evidence for impairment of resolution of vascular inflammation governed by specific lipid mediators. FASEB J. 22, 3595–3606. doi: 10.1096/fj.08-112201
Mitchell, D., Rodgers, K., Hanly, J., McMahon, B., Brady, H. R., Martin, F., et al. (2004). Lipoxins inhibit Akt/PKB activation and cell cycle progression in human mesangial cells. Am. J. Pathol. 164, 937–946. doi: 10.1016/S0002-9440(10)63181-1
Mitchell, S., Thomas, G., Harvey, K., Cottell, D., Reville, K., Berlasconi, G., et al. (2002). Lipoxins, aspirin-triggered epi-lipoxins, lipoxin stable analogues, and the resolution of inflammation: stimulation of macrophage phagocytosis of apoptotic neutrophils in vivo. J. Am. Soc. Nephrol. 13, 2497–2507. doi: 10.1097/01.ASN.0000032417.73640.72
Moore, K. J., Sheedy, F. J., and Fisher, E. A. (2013). Macrophages in atherosclerosis: a dynamic balance. Nat. Rev. Immunol. 13, 709–721. doi: 10.1038/nri3520
Moore, K. J., and Tabas, I. (2011). Macrophages in the pathogenesis of atherosclerosis. Cell 145, 341–355. doi: 10.1016/j.cell.2011.04.005
Moore, T. C., Moore, J. E., Kaji, Y., Frizzell, N., Usui, T., Poulaki, V., et al. (2003). The role of advanced glycation end products in retinal microvascular leukostasis. Invest. Ophthalmol. Vis. Sci. 44, 4457–4464. doi: 10.1167/iovs.02-1063
Mosser, D. M., and Edwards, J. P. (2008). Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969. doi: 10.1038/nri2448
Motwani, M. P., Flint, J. D., De Maeyer, R. P. H., Fullerton, J. N., Smith, A. M., Marks, D. J. B., et al. (2016). Novel translational model of resolving inflammation triggered by UV-killed E. coli. J. Pathol. 2, 154–165.
Murphy, M., Godson, C., Cannon, S., Kato, S., Mackenzie, H. S., Martin, F., et al. (1999). Suppression subtractive hybridization identifies high glucose levels as a stimulus for expression of connective tissue growth factor and other genes in human mesangial cells. J. Biol. Chem. 274, 5830–5834. doi: 10.1074/jbc.274.9.5830
Nagareddy, P. R., Murphy, A. J., Stirzaker, R. A., Hu, Y., Yu, S., Miller, R. G., et al. (2013). Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 17, 695–708. doi: 10.1016/j.cmet.2013.04.001
Nagenborg, J., Goossens, P., Biessen, E. A. L., and Donners, M. M. P. C. (2017). Heterogeneity of atherosclerotic plaque macrophage origin, phenotype and functions: implications for treatment. Eur. J. Pharmacol. 816, 14–24. doi: 10.1016/j.ejphar.2017.10.005
National Kidney Foundation (2002). K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am. J. Kidney Dis. 39, S1–S266.
Nathan, C., and Ding, A. (2010). Nonresolving inflammation. Cell 140, 871–882. doi: 10.1016/j.cell.2010.02.029
O’Sullivan, T. P., Vallin, K. S., Shah, S. T., Fakhry, J., Maderna, P., Scannell, M., et al. (2007). Aromatic lipoxin A4 and lipoxin B4 analogues display potent biological activities. J. Med. Chem. 50, 5894–5902. doi: 10.1021/jm060270d
Orr, S. K., Colas, R. A., Dalli, J., Chiang, N., and Serhan, C. N. (2015). Proresolving actions of a new resolvin D1 analog mimetic qualifies as an immunoresolvent. Am. J. Physiol. Lung. Cell. Mol. Physiol. 308, L904–L911. doi: 10.1152/ajplung.00370.2014
Orban, J.-C., Van Obberghen, E., and Ichai, C. (2018). Acute Complications of Diabetes. Metabolic Disorders and Critically Ill Patients, Berlin: Springer, 341–363. doi: 10.1007/978-3-319-64010-5_15
Paneni, F., Beckman, J. A., Creager, M. A., and Cosentino, F. (2013). Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part I. Eur. Heart J. 34, 2436–2443. doi: 10.1093/eurheartj/eht149
Papatheodorou, K., Papanas, N., Banach, M., Papazoglou, D., and Edmonds, M. (2016). Complications of diabetes 2016. J. Diabetes Res. 2016:6989453. doi: 10.1155/2016/6989453
Parkinson, J. F. (2006). Lipoxin and synthetic lipoxin analogs: an overview of anti-inflammatory functions and new concepts in immunomodulation. Inflamm. Allergy Drug Targets 5, 91–106. doi: 10.2174/187152806776383125
Pepine, C. J. (1998). The effects of angiotensin-converting enzyme inhibition on endothelial dysfunction: potential role in myocardial ischemia. Am. J. Cardiol. 82, 23S–27S. doi: 10.1016/S0002-9149(98)90427-1
Pergola, P. E., Krauth, M., Huff, J. W., Ferguson, D. A., Ruiz, S., Meyer, C. J., et al. (2011). Effect of bardoxolone methyl on kidney function in patients with T2D and stage 3b–4 CKD. Am. J. Nephrol. 33, 469–476. doi: 10.1159/000327599
Petasis, N. A., Keledjian, R., Sun, Y. P., Nagulapalli, K. C., Tjonahen, E., Yang, R., et al. (2008). Design and synthesis of benzo-lipoxin A4 analogs with enhanced stability and potent anti-inflammatory properties. Bioorg. Med. Chem. Lett. 18, 1382–1387. doi: 10.1016/j.bmcl.2008.01.013
Petri, M. H., Laguna-Fernandez, A., Tseng, C. N., Hedin, U., Perretti, M., and Back, M. (2015). Aspirin-triggered 15-epi-lipoxin A(4) signals through FPR2/ALX in vascular smooth muscle cells and protects against intimal hyperplasia after carotid ligation. Int. J. Cardiol. 179, 370–372. doi: 10.1016/j.ijcard.2014.11.010
Piccoli, G. B., Grassi, G., Cabiddu, G., Nazha, M., Roggero, S., Capizzi, I., et al. (2015). Diabetic kidney disease: a syndrome rather than a single disease. Rev. Diabetic Stud. 12, 87–109. doi: 10.1900/RDS.2015.12.87
Pierdomenico, A. M., Recchiuti, A., Simiele, F., Codagnone, M., Mari, V. C., Davì, G., et al. (2015). MicroRNA-181b regulates ALX/FPR2 receptor expression and proresolution signaling in human macrophages. J. Biol. Chem. 290, 3592–3600. doi: 10.1074/jbc.M114.592352
Potteaux, S., Gautier, E. L., Hutchison, S. B., van Rooijen, N., Rader, D. J., Thomas, M. J., et al. (2011). Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe–/– mice during disease regression. J. Clin. Invest. 121, 2025–2036. doi: 10.1172/JCI43802
Puro, D. G. (2002). Diabetes-induced dysfunction of retinal Muller cells. Trans. Am. Ophthalmol. Soc. 100, 339–352.
Qu, X., Zhang, X., Yao, J., Song, J., Nikolic-Paterson, D. J., and Li, J. (2012). Resolvins E1 and D1 inhibit interstitial fibrosis in the obstructed kidney via inhibition of local fibroblast proliferation. J. Pathol. 228, 506–519. doi: 10.1002/path.4050
Rabbani, N., and Thornalley, P. J. (2018). Advanced glycation end products in the pathogenesis of chronic kidney disease. Kidney Int. 93, 803–813. doi: 10.1016/j.kint.2017.11.034
Randolph, G. J. (2008). Emigration of monocyte-derived cells to lymph nodes during resolution of inflammation and its failure in atherosclerosis. Curr. Opin. Lipidol. 19, 462–468. doi: 10.1097/MOL.0b013e32830d5f09
Rashid, S., and Genest, J. (2007). Effect of obesity on high-density lipoprotein metabolism. Obesity 15, 2875–2888. doi: 10.1038/oby.2007.342
Rayner, K. J., Sheedy, F.J., Esau, C. C., Hussain, F. N., Temel, R. E., Parathath, S., et al. (2011). Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J. Clin. Invest. 121, 2921–2931. doi: 10.1172/JCI57275
Reis, E. D., Li, J., Fayad, Z. A., Rong, J. X., Hansoty, D., Aguinaldo, J.-G., et al. (2001). Dramatic remodeling of advanced atherosclerotic plaques of the apolipoprotein E–deficient mouse in a novel transplantation model. J. Vasc. Surg. 34, 541A–542A. doi: 10.1067/mva.2001.115963
Ridker, P. M., Everett, B. M., Thuren, T., MacFadyen, J. G., Chang, W. H., Ballantyne, C., et al. (2017). Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 377, 1119–1131. doi: 10.1056/NEJMoa1707914
Ridker, P. M., MacFadyen, J. G., Glynn, R. J., Koenig, W., Libby, P., Everett, B. M., et al. (2018). Inhibition of interleukin-1β by canakinumab and cardiovascular outcomes in patients with chronic kidney disease. J. Am. Coll. Cardiol. 71, 2405–2414. doi: 10.1016/j.jacc.2018.03.490
Ridker, P. M., Thuren, T., Zalewski, A., and Libby, P. (2011). Interleukin-1β inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS). Am. Heart. J. 162, 597–605. doi: 10.1016/j.ahj.2011.06.012
Rodgers, K., McMahon, B., Mitchell, D., Sadlier, D., and Godson, C. (2005). Lipoxin A4 modifies platelet-derived growth factor-induced pro-fibrotic gene expression in human renal mesangial cells. Am. J. Pathol. 167, 683–694. doi: 10.1016/S0002-9440(10)62043-3
Rong, J. X., Li, J., Reis, E. D., Choudhury, R. P., Dansky, H. M., Elmalem, V. I., et al. (2001). Elevating high-density lipoprotein cholesterol in apolipoprotein E-deficient mice remodels advanced atherosclerotic lesions by decreasing macrophage and increasing smooth muscle cell content. Circulation 104, 2447–2452. doi: 10.1161/hc4501.098952
Ross, R. (1999). Atherosclerosis–an inflammatory disease. N. Engl. J. Med. 340, 115–126. doi: 10.1056/NEJM199901143400207
Rossi, S., D’Amico, M., Capuano, A., Romano, M., Petronella, P., and Di Filippo, C. (2006). Hyperglycemia in streptozotocin-induced diabetes leads to persistent inflammation and tissue damage following uveitis due to reduced levels of ciliary body heme oxygenase-1. Mediat. Inflamm. 2006:60285. doi: 10.1155/MI/2006/60285
Rossing, P. (2018). Subclassification of diabetes based on quantitative traits. Nat. Rev. Nephrol. 14, 355–356. doi: 10.1038/s41581-018-0011-9
Roy, S., Cagliero, E., and Lorenzi, M. (1996). Fibronectin overexpression in retinal microvessels of patients with diabetes. Invest. Ophthalmol. Vis. Sci. 37, 258–266.
Roy, S., Maiello, M., and Lorenzi, M. (1994). Increased expression of basement membrane collagen in human diabetic retinopathy. J. Clin. Invest. 93, 438–442. doi: 10.1172/JCI116979
Rubsam, A., Parikh, S., and Fort, P. E. (2018). Role of Inflammation in Diabetic Retinopathy. Int. J. Mol. Sci. 19: E942. doi: 10.3390/ijms19040942
Russell, J. W., and Zilliox, L. A. (2014). Diabetic neuropathies. Continuum 20, 1226–1240. doi: 10.1212/01.CON.0000455884.29545.d2
Sandholm, N., Forsblom, C., Mäkinen, V.-P., McKnight, A. J., Österholm, A.-M., He, B., et al. (2014). Genome-wide association study of urinary albumin excretion rate in patients with type 1 diabetes. Diabetologia 57, 1143–1153. doi: 10.1007/s00125-014-3202-3
Sato, T., Kusaka, S., Shimojo, H., and Fujikado, T. (2009). Simultaneous analyses of vitreous levels of 27 cytokines in eyes with retinopathy of prematurity. Ophthalmology 116, 2165–2169. doi: 10.1016/j.ophtha.2009.04.026
Schroder, K., Zhou, R., and Tschopp, J. (2010). The NLRP3 Inflammasome: a sensor for metabolic danger? Science 327, 296–300. doi: 10.1126/science.1184003
Scrivo, R., Vasile, M., Bartosiewicz, I., and Valesini, G. (2011). Inflammation as common soil of the multifactorial diseases. Autoimmun. Rev. 10, 369–374. doi: 10.1016/j.autrev.2010.12.006
Serhan, C. N. (1989). On the relationship between leukotriene and lipoxin production by human neutrophils: evidence for differential metabolism of 15-HETE and 5-HETE. Biochim. Biophys. Acta 1004, 158–168. doi: 10.1016/0005-2760(89)90264-6
Serhan, C. N. (2005). Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot. Essent. Fatty Acids 73, 141–162. doi: 10.1016/j.plefa.2005.05.002
Serhan, C. N. (2014). Novel pro-resolving lipid mediators in inflammation are leads for resolution physiology. Nature 510, 92–101. doi: 10.1038/nature13479
Serhan, C. N., Brain, S. D., Buckley, C. D., Gilroy, D. W., Haslett, C., O’Neill, L. A. J., et al. (2007). Resolution of inflammation: state of the art, definitions and terms. FASEB J. 21, 325–332. doi: 10.1096/fj.06-7227rev
Serhan, C. N., Chiang, N., and Dalli, J. (2015). The resolution code of acute inflammation: novel pro-resolving lipid mediators in resolution. Semin. Immunol. 27, 200–215. doi: 10.1016/j.smim.2015.03.004
Serhan, C. N., Clish, C. B., Brannon, J., Colgan, S. P., Chiang, N., and Gronert, K. (2000). Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J. Exp. Med. 192, 1197–1204. doi: 10.1084/jem.192.8.1197
Serhan, C. N., Hamberg, M., and Samuelsson, B. (1984). Lipoxins: novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc. Natl. Acad. Sci. U.S.A. 81, 5335–5339. doi: 10.1073/pnas.81.17.5335
Serhan, C. N., Hong, S., Gronert, K., Colgan, S. P., Devchand, P. R., and Mirick, G. (2002). Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 196, 1025–1037. doi: 10.1084/jem.20020760
Serhan, C. N., and Petasis, N. A. (2011). Resolvins and protectins in inflammation resolution. Chem. Rev. 111, 5922–5943. doi: 10.1021/cr100396c
Serhan, C. N., and Sheppard, K. A. (1990). Lipoxin formation during human neutrophil-platelet interactions. Evidence for the transformation of leukotriene A4 by platelet 12-lipoxygenase in vitro. J. Clin. Invest. 85, 772–780. doi: 10.1172/JCI114503
Serhan, C. N., Yacoubian, S., and Yang, R. (2008). Anti-inflammatory and proresolving lipid mediators. Annu. Rev. Pathol. 3, 279–312. doi: 10.1146/annurev.pathmechdis.3.121806.151409
Serhan, C. N., Yang, R., Martinod, K., Kasuga, K., Pillai, P. S., Porter, T. F., et al. (2009). Maresins: novel macrophage mediators with potent antiinflammatory and proresolving actions. J. Exp. Med. 206, 15–23. doi: 10.1084/jem.20081880
Settimio, R., Clara, D. F., Franca, F., Francesca, S., and Michele, D. (2012). Resolvin D1 reduces the immunoinflammatory response of the rat eye following uveitis. Mediat. Inflamm. 2012:318621. doi: 10.1155/2012/318621
Sfikakis, P. P., Markomichelakis, N., Theodossiadis, G. P., Grigoropoulos, V., Katsilambros, N., and Theodossiadis, P. G. (2005). Regression of sight-threatening macular edema in type 2 diabetes following treatment with the anti-tumor necrosis factor monoclonal antibody infliximab. Diabetes Care 28, 445–447. doi: 10.2337/diacare.28.2.445
Sha, Y. H., Hu, Y. W., Gao, J. J., Wang, Y. C., Ma, X., Qiu, Y. R., et al. (2015). Lipoxin A4 promotes ABCA1 expression and cholesterol efflux through the LXRalpha signaling pathway in THP-1 macrophage-derived foam cells. Int. J. Clin. Exp. Pathol. 8, 6708–6715.
Sifuentes-Franco, S., Padilla-Tejeda, D. E., and Carrillo-Ibarra, S. (2018). Oxidative stress, apoptosis, and mitochondrial function in diabetic nephropathy. Int. J. Endocrinol. 2018:1875870. doi: 10.1155/2018/1875870
Singh, A., Boden, G., Homko, C., Gunawardana, J., and Rao, A. K. (2012). Whole-blood tissue factor procoagulant activity is elevated in type 1 diabetes: effects of hyperglycemia and hyperinsulinemia. Diabetes Care 35, 1322–1327. doi: 10.2337/dc11-2114
Sivaraj, R. R., Oswal, K., Stavrou, P., and Murray, P. I. (2009). Diabetes mellitus and uveitis: a longitudinal study. Invest. Ophthalmol. Vis. Sci. 50, 296–296.
Spite, M., Clària, J., and Serhan, C. N. (2014). Resolvins, specialized pro-resolving lipid mediators and their potential roles in metabolic diseases. Cell Metab. 19, 21–36. doi: 10.1016/j.cmet.2013.10.006
Srinivasan, S., Dehghani, C., Pritchard, N., Edwards, K., Russell, A. W., Malik, R. A., et al. (2017). Corneal and retinal neuronal degeneration in early stages of diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 58, 6365–6373. doi: 10.1167/iovs.17-22736
Strutz, F., Zeisberg, M., Hemmerlein, B., Sattler, B., Hummel, K., Becker, V., et al. 2000). Basic fibroblast growth factor expression is increased in human renal fibrogenesis and may mediate autocrine fibroblast proliferation. Kidney Int. 57, 1521–1538. doi: 10.1046/j.1523-1755.2000.00997.x
Stuebiger, N., Smiddy, W., Wang, J., Jiang, H., and DeBuc, D. C. (2015). Assesment of conjunctival microangiopathy in a patient with diabetes mellitus using the retinal function imager. J. Clin. Exp. Ophthalmol. 6:400. doi: 10.4172/2155-9570.1000400
Sugimoto, H., LeBleu, V. S., Bosukonda, D., Keck, P., Taduri, G., Bechtel, W., et al. (2012). Activin-like kinase 3 is important for kidney regeneration and reversal of fibrosis. Nat. Med. 18:396. doi: 10.1038/nm.2629
Sun, Y.-P., Tjonahen, E., Keledjian, R., Zhu, M., Yang, R., Recchiuti, A., et al. (2009). Anti-inflammatory and pro-resolving properties of benzo-lipoxin A(4) analogs. Prostaglandins Leukot. Essent. Fatty Acids 81, 357–366. doi: 10.1016/j.plefa.2009.09.004
Sun, Y. B., Qu, X., Li, X., Nikolic-Paterson, D. J., and Li, J. (2013). Endothelial dysfunction exacerbates renal interstitial fibrosis through enhancing fibroblast Smad3 linker phosphorylation in the mouse obstructed kidney. PLoS One 8:e84063. doi: 10.1371/journal.pone.0084063
Sun, Y. P., Oh, S. F., Uddin, J., Yang, R., Gotlinger, K., Campbell, E., et al. (2007). Resolvin D1 and its aspirin-triggered 17R epimer. Stereochemical assignments, anti-inflammatory properties, and enzymatic inactivation. J. Biol. Chem. 282, 9323–9334. doi: 10.1074/jbc.M609212200
Tampe, D., and Zeisberg, M. (2014). Potential approaches to reverse or repair renal fibrosis. Nat. Rev. Nephrol. 10:226. doi: 10.1038/nrneph.2014.14
Tang, H., Liu, Y., Yan, C., Petasis, N. A., Serhan, C. N., and Gao, H. (2014). Protective actions of aspirin-triggered (17R) resolvin D1 and its analogue, 17R-hydroxy-19-para-fluorophenoxy-resolvin D1 methyl ester, in C5a-dependent IgG immune complex-induced inflammation and lung injury. J. Immunol. 193, 3769–3778. doi: 10.4049/jimmunol.1400942
Tayek, J. A., and Kalantar-Zadeh, K. (2013). The extinguished beacon of bardoxolone: not a monday morning quarterback story. Am. J. Nephrol. 37, 208–211. doi: 10.1159/000346950
Teumer, A., Tin, A., Sorice, R., Gorski, M., Yeo, N. C., Chu, A. Y., et al. (2015). Genome-wide association studies identify genetic loci associated with albuminuria in diabetes. Diabetes, 65, 803–817. doi: 10.2337/db15-1313
Thiery, J. P., Acloque, H., Huang, R. Y., and Nieto, M. A. (2009). Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890. doi: 10.1016/j.cell.2009.11.007
Thorp, E., and Tabas, I. (2009). Mechanisms and consequences of efferocytosis in advanced atherosclerosis. J. Leukoc. Biol. 86, 1089–1095. doi: 10.1189/jlb.0209115
Tian, H., Lu, Y., Sherwood, A. M., Hongqian, D., and Hong, S. (2009). Resolvins E1 and D1 in choroid-retinal endothelial cells and leukocytes: biosynthesis and mechanisms of anti-inflammatory actions. Invest. Ophthalmol. Vis. Sci. 50, 3613–3620. doi: 10.1167/iovs.08-3146
Tinckam, K. J., Djurdjev, O., and Magil, A. B. (2005). Glomerular monocytes predict worse outcomes after acute renal allograft rejection independent of C4d status. Kidney Int. 68, 1866–1874. doi: 10.1111/j.1523-1755.2005.00606.x
Todd, J. N., Dahlstrom, E. H., Salem, R. M., Sandholm, N., Forsblom, C., McKnight, A. J., et al. (2015). Genetic evidence for a causal role of obesity in diabetic kidney disease. Diabetes Metab. Res. Rev. 64, 4238–4246. doi: 10.2337/db15-0254
Tomkin, G. H. (2014). Treatment of type 2 diabetes, lifestyle, GLP1 agonists and DPP4 inhibitors. World J. Diabetes 5, 636–650. doi: 10.4239/wjd.v5.i5.636
Tuomi, T. (2005). Type 1 and type 2 diabetes. What Do They Have in Common? Diabetes 54(Suppl 2), S40–S45. doi: 10.2337/diabetes.54.suppl_2.S40
Tuttle, K. R., Brosius, F. C., Adler, S. G., Kretzler, M., Mehta, R. L., Tumlin, J. A., et al. (2018). JAK1/JAK2 inhibition by baricitinib in diabetic kidney disease: results from a Phase 2 randomized controlled clinical trial. Nephrol. Dial. Transpl. 33, 1–10. doi: 10.1093/ndt/gfx377
Vallejo, S., Palacios, E., Romacho, T., Villalobos, L., Peiro, C., and Sanchez-Ferrer, C. F. (2014). The interleukin-1 receptor antagonist anakinra improves endothelial dysfunction in streptozotocin-induced diabetic rats. Cardiovasc. Diabetol. 13:158. doi: 10.1186/s12933-014-0158-z
van Zuydam, N. R., Ahlqvist, E., and Sandholm, N. (2018). A genome-wide association study of diabetic kidney disease in subjects with type 2 diabetes. Diabetes 67, 1414–1427. doi: 10.2337/db17-0914
vanDam, B., Demirci, C., Reitsma, H. J., van Lambalgen, A. A., van den Bos, G. C., Tangelder, G. J., et al. (2000). Alterations in nitric oxide activity and sensitivity in early streptozotocin-induced diabetes depend on arteriolar size. Int. J. Exp. Diabetes Res. 1, 221–232. doi: 10.1155/EDR.2000.221
van Dieren S., Beulens, J. W., van der Schouw, Y. T., Grobbee, D. E., Neal, B. (2010). The global burden of diabetes and its complications: an emerging pandemic. Eur. J. Cardiovasc. Prev. Rehabil. 17(1 Suppl.), s3–s8.
Vilaysane, A., Chun, J., Seamone, M. E., Wang, W., Chin, R., Hirota, S., et al. (2010). The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J. Am. Soc. Nephrol. 21, 1732–44. doi: 10.1681/ASN.2010020143
Voelker, J., Berg, P. H., Sheetz, M., Duffin, K., Shen, T., Moser, B., et al. (2017). Anti-TGF-beta1 antibody therapy in patients with diabetic nephropathy. J. Am. Soc. Nephrol. 28, 953–962. doi: 10.1681/ASN.2015111230
Vukicevic, S., Basic, V., Rogic, D., Basic, N., Shih, M. S., Shepard, A., et al. (1998). Osteogenic protein-1 (bone morphogenetic protein-7) reduces severity of injury after ischemic acute renal failure in rat. J. Clin. Invest. 102, 202–214. doi: 10.1172/JCI2237
Wang, B., Jha, J. C., Hagiwara, S., McClelland, A. D., Jandeleit-Dahm, K., Thomas, M. C., et al. (2014). Transforming growth factor-beta1-mediated renal fibrosis is dependent on the regulation of transforming growth factor receptor 1 expression by let-7b. Kidney Int. 85, 352–361. doi: 10.1038/ki.2013.372
Wang, H., and Daggy, B. P. (2017). The role of fish oil in inflammatory eye diseases. Biomed. Hub. 2, 6–6. doi: 10.1159/000455818
Weber, C., and von Hundelshausen, P. (2017). CANTOS Trial validates the inflammatory pathogenesis of atherosclerosis: setting the stage for a new chapter in therapeutic targeting. Circ. Res. 121, 1119–1121. doi: 10.1161/CIRCRESAHA.117.311984
Wijesekara, N., Zhang, L.-H., Kang, M. H., Abraham, T., Bhattacharjee, A., Warnock, G. L., et al. (2012). miR-33a modulates ABCA1 expression, cholesterol accumulation, and insulin secretion in pancreatic islets. Diabetes 61, 653–658. doi: 10.2337/db11-0944
Wild, S., Roglic, G., Green, A., Sicree, R., and King, H. (2004). Global prevalence of diabetes. Estimates for the year 2000 and projections for 2030. Diabetes Care 27, 1047–1053. doi: 10.2337/diacare.27.5.1047
Wilkinson, C. P., Ferris, F. L. III, Klein, R. E., Lee, P. P., Agardh, C. D., Davis, M., et al. (2003). Proposed international clinical diabetic retinopathy and diabetic macular edema disease severity scales. Ophthalmology 110, 1677–1682. doi: 10.1016/S0161-6420(03)00475-5
Williams, K. J., and Tabas, I. (2005). Lipoprotein retention–and clues for atheroma regression. Arterioscler. Thromb. Vasc. Biol. 25, 1536–1540. doi: 10.1161/01.ATV.0000174795.62387.d3
Wolfs, I. M., Donners, M. M., and de Winther, M. P. (2011). Differentiation factors and cytokines in the atherosclerotic plaque micro-environment as a trigger for macrophage polarisation. Thromb. Haemost. 106, 763–771. doi: 10.1160/TH11-05-0320
Wu, S.-H., Chen, X.-Q., Liu, B., Wu, H.-J., and Dong, L. (2013). Efficacy and safety of 15(R/S)-methyl-lipoxin A4 in topical treatment of infantile eczema. Br. J. Dermatol. 168, 172–178. doi: 10.1111/j.1365-2133.2012.11177.x
Wu, S. H., Liao, P. Y., Dong, L., and Jiang, X. Y. (2006). Protective effects of 15-methyl-lipoxin A4 on mesangioproliferative nephritis in rats. Zhongguo Dang Dai Er Ke Za Zhi 8, 225–230.
Wu, S. H., Liao, P. Y., Yin, P. L., Zhang, Y. M., and Dong, L. (2009). Elevated expressions of 15-lipoxygenase and lipoxin A4 in children with acute poststreptococcal glomerulonephritis. Am. J. Pathol. 174, 115–122. doi: 10.2353/ajpath.2009.080671
Wu, S. H., Wu, X. H., Liao, P. Y., and Dong, L. (2007). Signal transduction involved in protective effects of 15(R/S)-methyl- lipoxin A(4) on mesangioproliferative nephritis in rats. Prostaglandins Leukot. Essent. Fatty Acids 76, 173–180. doi: 10.1016/j.plefa.2006.12.006
Wu, Y., Tang, L., and Chen, B. (2014). Oxidative stress: implications for the development of diabetic retinopathy and antioxidant therapeutic perspectives. Oxid. Med. Cell. Longev. 2014:752387. doi: 10.1155/2014/752387
Wuttke, M., and Köttgen, A. (2016). Insights into kidney diseases from genome-wide association studies. Nat. Rev. Nephrol. 12:549. doi: 10.1038/nrneph.2016.107
Xu, H., Curtis, T., and Stitt, A. (2014). Pathophysiology and Pathogenesis of Diabetic Retinopathy. Amsterdam: Diapedia, The Living Textbook of Diabetes.
Yach, D., Stuckler, D., and Brownell, K. D. (2006). Epidemiologic and economic consequences of the global epidemics of obesity and diabetes. Nat. Med. 12: 62. doi: 10.1038/nm0106-62
Yanagita, M. (2012). Inhibitors/antagonists of TGF-β system in kidney fibrosis. Nephrol. Dial. Transpl. 27, 3686–3691. doi: 10.1093/ndt/gfs381
Yau, J. W., Rogers, S. L., Kawasaki, R., Lamoureux, E. L., Kowalski, J. W., Bek, T., et al. (2012). Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care 35, 556–564. doi: 10.2337/dc11-1909
Yin, Y., Chen, F., Wang, W., Wang, H., and Zhang, X. (2017). Resolvin D1 inhibits inflammatory response in STZ-induced diabetic retinopathy rats: possible involvement of NLRP3 inflammasome and NF-kappaB signaling pathway. Mol. Vis. 23, 242–250.
Yoshida, S., Yoshida, A., Ishibashi, T., Elner, S. G., and Elner, V. M. (2003). Role of MCP-1 and MIP-1alpha in retinal neovascularization during postischemic inflammation in a mouse model of retinal neovascularization. J. Leukoc. Biol. 73, 137–144. doi: 10.1189/jlb.0302117
You, W.-P., and Henneberg, M. (2016). Type 1 diabetes prevalence increasing globally and regionally: the role of natural selection and life expectancy at birth. BMJ Open Diabetes Res. Care 4:e000161. doi: 10.1136/bmjdrc-2015-000161
Young, B. A., Burdmann, E. A., Johnson, R. J., Alpers, C. E., Giachelli, C. M., Eng, E., et al. (1995). Cellular proliferation and macrophage influx precede interstitial fibrosis in cyclosporine nephrotoxicity. Kidney Int. 48, 439–448. doi: 10.1038/ki.1995.312
Yurdagul, A., Doran, A. C., Cai, B., Fredman, G., and Tabas, I. A. (2017). Mechanisms and consequences of defective efferocytosis in atherosclerosis. Front. Cardiovasc. Med. 4:86. doi: 10.3389/fcvm.2017.00086
Zeisberg, M., and Duffield, J. S. (2010). Resolved: EMT produces fibroblasts in the kidney. J. Am. Soc. Nephrol. 21, 1247–1253. doi: 10.1681/ASN.2010060616
Zhang, J., Pippin, J. W., Krofft, R. D., Naito, S., Liu, Z. H., and Shankland, S. J. (2013). Podocyte repopulation by renal progenitor cells following glucocorticoids treatment in experimental FSGS. Am. J. Physiol. Renal Physiol. 304, F1375–F1389. doi: 10.1152/ajprenal.00020.2013
Zhang, M. J., Sansbury, B. E., Hellmann, J., Baker, J. F., Guo, L., Parmer, C. M., et al. (2016). Resolvin D2 enhances post-ischemic revascularization while resolving inflammation. Circulation 134, 666–680. doi: 10.1161/CIRCULATIONAHA.116.021894
Zhao, Y.-L., Zhang, L., Yang, Y.-Y., Tang, Y., Zhou, J.-J., Feng, Y.-Y., et al. (2016). Resolvin D1 protects lipopolysaccharide-induced acute kidney injury by down-regulating nuclear factor-kappa b signal and inhibiting Apoptosis. Chin. Med. J. 129, 1100–1107. doi: 10.4103/0366-6999.180517
Keywords: diabetic kidney disease, diabetes-associated atherosclerosis, diabetic retinopathy, lipoxins, resolvins
Citation: de Gaetano M, McEvoy C, Andrews D, Cacace A, Hunter J, Brennan E and Godson C (2018) Specialized Pro-resolving Lipid Mediators: Modulation of Diabetes-Associated Cardio-, Reno-, and Retino-Vascular Complications. Front. Pharmacol. 9:1488. doi: 10.3389/fphar.2018.01488
Received: 04 September 2018; Accepted: 05 December 2018;
Published: 19 December 2018.
Edited by:
Lucy V. Norling, Queen Mary University of London, United KingdomReviewed by:
Joan Clària, Hospital Clínic de Barcelona, SpainAntonio Recchiuti, Università degli Studi G. d’Annunzio Chieti e Pescara, Italy
Copyright © 2018 de Gaetano, McEvoy, Andrews, Cacace, Hunter, Brennan and Godson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Monica de Gaetano, monica.degaetano@ucd.ie