 Yu Wang
Yu Wang Weixing Yu1
Weixing Yu1- 1Department of Biochemistry and Molecular Biology, School of Basic Medicine, Tongji Medical College, Huazhong University of Science of Technology, Wuhan, China
- 2Department of Neurosurgery, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 3Cell Architecture Research Center, Huazhong University of Science and Technology, Wuhan, China
Acetyl-CoA carboxylases (ACCs) are enzymes that catalyze the carboxylation of acetyl-CoA to produce malonyl-CoA. In mammals, ACC1 and ACC2 are two members of ACCs. ACC1 localizes in the cytosol and acts as the first and rate-limiting enzyme in the de novo fatty acid synthesis pathway. ACC2 localizes on the outer membrane of mitochondria and produces malonyl-CoA to regulate the activity of carnitine palmitoyltransferase 1 (CPT1) that involves in the β-oxidation of fatty acid. Fatty acid synthesis is central in a myriad of physiological and pathological conditions. ACC1 is the major member of ACCs in mammalian, mountains of documents record the roles of ACC1 in various diseases, such as cancer, diabetes, obesity. Besides, acetyl-CoA and malonyl-CoA are cofactors in protein acetylation and malonylation, respectively, so that the manipulation of acetyl-CoA and malonyl-CoA by ACC1 can also markedly influence the profile of protein post-translational modifications, resulting in alternated biological processes in mammalian cells. In the review, we summarize our understandings of ACCs, including their structural features, regulatory mechanisms, and roles in diseases. ACC1 has emerged as a promising target for diseases treatment, so that the specific inhibitors of ACC1 for diseases treatment are also discussed.
Introduction
In mammalian cells, acetyl-CoA is a global currency that can mediate the carbon transactions between metabolic pathways, including glycolysis, tricarboxylic acid cycle, amino acid metabolism, gluconeogenesis, and fatty acid synthesis. Lipid metabolism or fatty acid metabolism is the bank of acetyl-CoA. It can deposit extra acetyl-CoA in the form of fatty acids and regulate the intracellular availability of acetyl-CoA to the global metabolism network by controlling the conversion of acetyl-CoA into fatty acids. As such, fatty acid synthesis is a central pathway in harnessing a myriad of metabolic pathways and related physiologies in cells.
Acetyl-CoA carboxylases (ACCs) are enzymes that catalyze the carboxylation of acetyl-CoA to produce malonyl-CoA, which in turn is utilized by the fatty acid synthase (FASN) to produce long-chain saturated fatty acids (1). There are two members of ACCs in mammalian cells. ACC1 localizes in the cytosol and takes the major responsibility of converting cytoplasmic acetyl-CoA into malonyl-CoA for fatty acid synthesis (2). Despite ACC2 also catalyzing the conversion of acetyl-CoA into malonyl-CoA, it localizes on the outer membrane of mitochondria that makes the downstream pathways of ACC2-produced malonyl-CoA different from ACC1. It is reported that the ACC2-produced malonyl-CoA can allosterically influence the activity of carnitine palmitoyltransferase 1 (CPT1) in the β-oxidation of fatty acid (3). More functional studies about ACC2 are expected in this field.
Fatty acid synthesis controls the storage and expenditure of carbon source and energy, which can regulate the activities of other metabolic pathways, such as amino acid metabolism and glucose metabolism, so that fatty acid synthesis is frequently alternated to harness the intracellular metabolism network to meet the requirement of materials and energy for diseases progressions, such as cancer and metabolic diseases (4–8). ACC1 is the first rate-limiting enzyme in the fatty acid synthesis that plays a central role in fatty acid synthesis, so that ACC1 is the hub of the fatty acid synthesis-related metabolism network. Its dysregulation in diseases is intensively studied, including the roles of ACC1 in regulating tumour cell proliferation, migration, and metabolic disease progression (9–12). In addition, because acetyl-CoA and malonyl-CoA are cofactors in protein acetylation and malonylation, respectively, the emerging non-metabolic functions of ACC1 in diseases are discussed in recent studies (11, 13, 14), which further expand the roles of ACC1 in physiologies and pathophysiologies. ACC1 is therefore considered as a promising therapeutic target for treating diseases, such as cancer and metabolic diseases.
This review summarizes our current knowledge about ACCs, including the structure of ACCs, the regulatory mechanisms, and the roles of ACCs in tumorigenesis and metabolic diseases. Besides, we briefly introduce ACCs inhibitors that are under investigation for cancer and metabolic diseases therapy.
Structure of Acetyl-CoA Carboxylases
In mammals, ACCs have two isoforms: ACC1 and ACC2. Human ACC1 (ACCα, 265 kDa) is encoded by the ACACA gene on chromosome 17q12 while ACC2 (ACCβ, 275 kDa) is encoded by the ACACB gene on chromosome 12q23 (15). ACC1 and ACC2 share 75% identity in amino acid sequence and are composed of conservative domains for enzyme activity (16, 17).
ACC1 and ACC2 have similar structures and molecule bases in catalyzing carboxylation of acetyl-CoA to produce malonyl-CoA. ACC1 is discussed here in terms of ACCs’ structure. ACC1 contains three major functional domains: a biotin carboxylase domain (BC domain), a carboxyl transferase domain (CT domain), and a biotin carboxyl carrier protein domain (BCCP domain) that links the BC domain and CT domain (Figure 1). To perform the catalytic activity, the BC domain of ACC1 firstly consumes ATP and bicarbonate to catalyze the carboxylation of biotin, in which bicarbonate serves as the donor of the carboxyl moiety (18). Then, the BCCP domain transfers the carboxyl moiety from the carboxylated biotin to the CT domain (19), where the carboxyl moiety is transferred to the acetyl-CoA to accomplish the carboxylation of acetyl-CoA, converting acetyl-CoA into malonyl-CoA (20) (Figure 1). Although the BC domain and CT domain are linked by the BCCP domain in a single ACC1 molecule, the spatial dimension of ACC1 is so broad that the functional domains are spatially separated, which makes the carboxylated biotin in the BC domain can hardly reach to the acetyl-CoA that bond by the CT domain of the same molecule of ACC1. To link the cascade reactions of acetyl-CoA carboxylation, ACC1 molecules form homodimers to enable the carboxylated biotin in the BC domain reaching to the acetyl-CoA in the CT domain of the other ACC1 molecule of the homodimer (19–23). Therefore, regulating the formation of ACC1 homodimers is considered as an important mechanism controlling the acetyl-CoA carboxylation activity of ACC1.
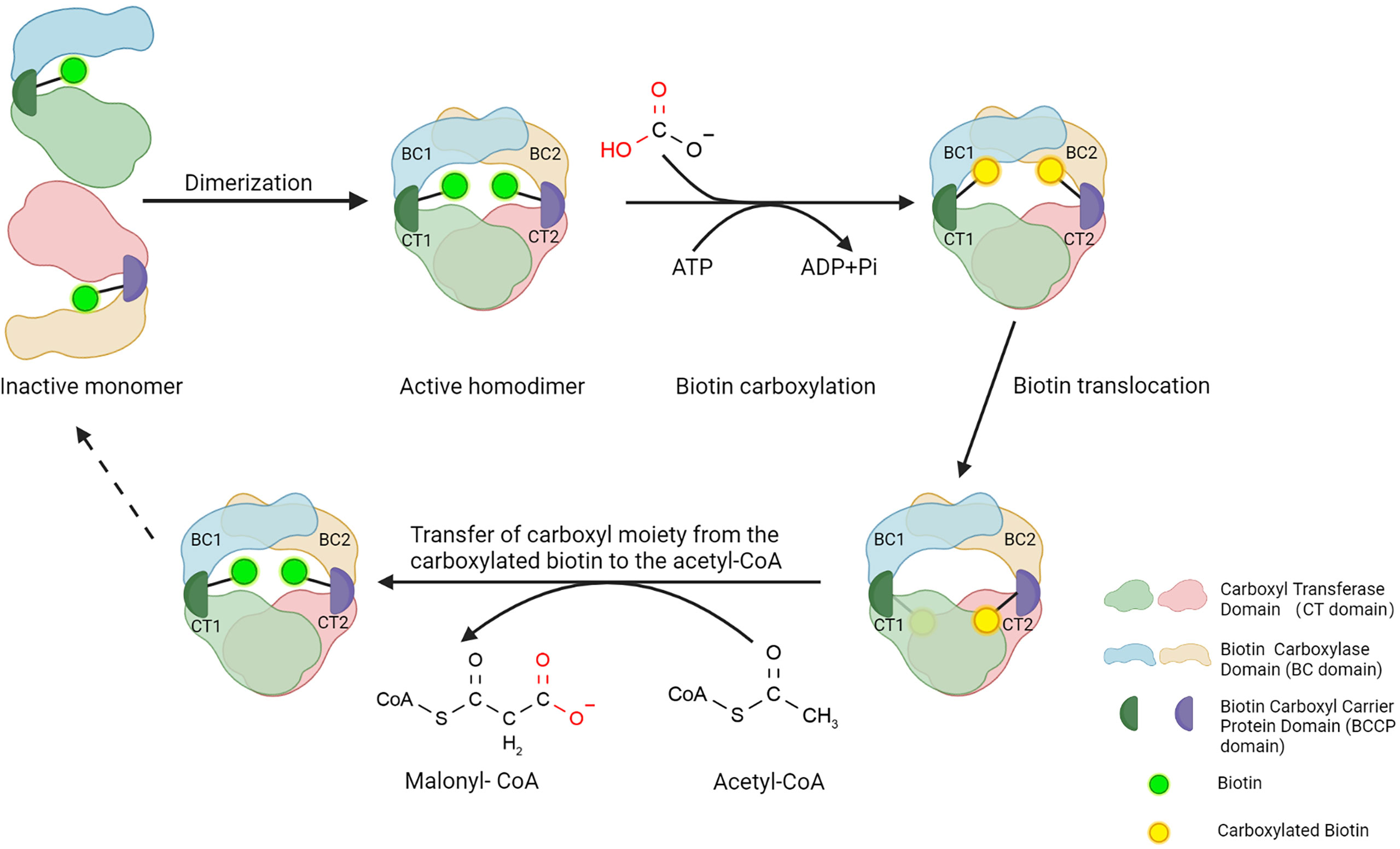
Figure 1 Structure of ACC1 and function of three main domains. Three steps to a functional ACC1: First, the BC domain consumes ATP and catalyzes the carboxylation of biotin, in which bicarbonate serves as the donor of the carboxyl moiety. Subsequently, the BCCP domain transfers the bicarbonate moiety from carboxylated biotin to the CT domain of ACC1. Lastly, the CT domain catalyzes the carboxylation of acetyl-CoA carboxyl moiety, converting acetyl-CoA into malonyl-CoA. Created with BioRender.com.
Distribution and Functions of Acetyl-CoA Carboxylases
ACC1 and ACC2 are widely distributed in organs and tissues in mammals. ACC1 is highly enriched in lipogenic tissues, such as liver and adipose tissue, while ACC2 is majorly expressed in oxidative tissues, such as cardiac and skeletal muscle (24), which are consistent with the functions of ACC1 in lipogenesis and ACC2 in regulating fatty acid β-oxidation. In mammalian cells, ACC1 and ACC2 are differently distributed (Figure 2). ACC1 is a cytoplasmic protein that catalyzes the conversion of acetyl-CoA into malonyl-CoA, which is utilized by the fatty acid synthetase (FASN) and subjected to the de novo fatty acid biosynthesis (2). It controls the synthesis of mid-and long-chain fatty acids that serve as building blocks for the cell biology process. Inhibiting ACC1 by 5-tetradecyloxy-2-furoic acid (TOFA) can completely inhibit hepatic de novo lipogenesis (DNL), which is considered a new strategy for non-alcoholic fatty liver disease (NAFLD) treatment (25). Soraphen A, another ACC1 inhibitor, can pharmacologically inhibit fatty acid synthesis in diet-induced obesity mice and significantly suppress weight gain, which sheds new light on controlling diet-induced obesity (26). Liver-specific ACC1 knockout (LACC1 KO) mice can survive but show dysregulated lipid metabolism and deficiency in triglycerides metabolism (27). In cancer cells, inhibition of ACC1 by Soraphen A significantly reduces saturated and mono-unsaturated phospholipid species and increases the proportion of poly-unsaturation, rendering cells vulnerable to oxidative stresses (28). Moreover, activity-impeded ACC1 reduces cytoplasmic membrane fluidity and impairs mobilities of transmembrane receptors, ultimately impairing cell membrane-dependent biological processes (29).
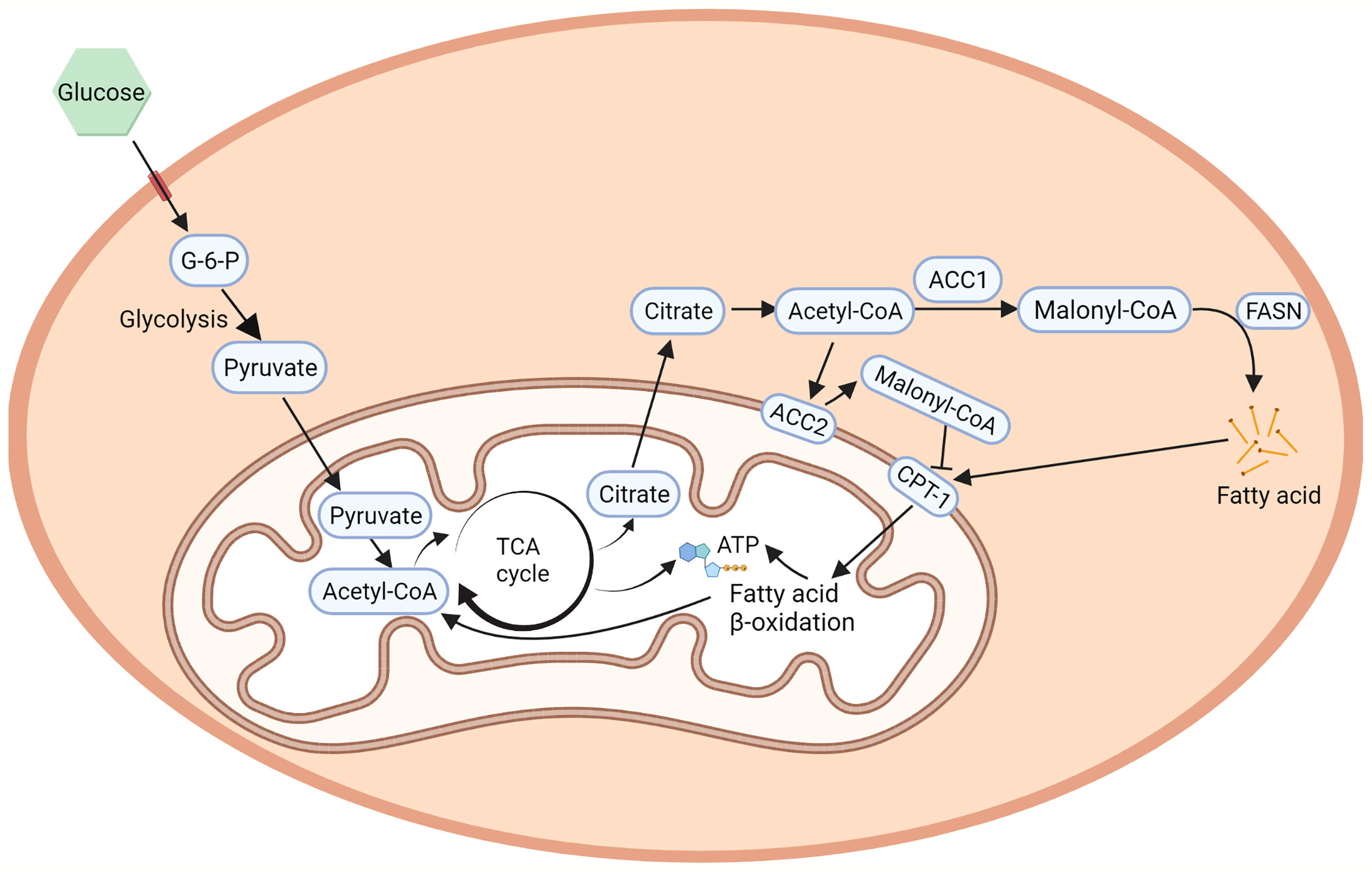
Figure 2 ACCs in fatty acid metabolism. ACC1 is a cytoplasmic protein that catalyzes the conversing of acetyl-CoA to malonyl-CoA in the de novo fatty acid biosynthesis (2). On the other hand, the hydrophobicity of the N-terminal region of ACC2 allows its localization to the outer membrane of the mitochondria, regulates CPT1 which controls fatty acid β-oxidation (25). Created with BioRender.com.
ACC2 contains a hydrophobic N-terminal region that leads ACC2 attaching to the outer membrane of mitochondria (25). The mitochondria-located ACC2 also catalyzes the conversion of acetyl-CoA into malonyl-CoA. However, instead of entering the de novo fatty acid biosynthesis, the ACC2-generated malonyl-CoA locally interacts with carnitine palmitoyltransferase 1 (CPT1) that also localizes on the outer mitochondrial membrane. CPT1 accounts for the first step of long-chain fatty acids β-oxidation in mitochondria. The binding of malonyl-CoA allosterically inhibits the activity of CPT1 and therefore influences the process of fatty acid β-oxidation in mitochondria (30). In animal experiments, inhibition of ACC2 can increase hepatic fat oxidation, reduce hepatic lipids, and improve hepatic insulin sensitivity in mice with NAFLD (31), which is further confirmed in mice with genetic depletion of ACC2 (3, 32, 33). In addition, the fatty acid oxidation rate in the soleus muscle of the ACC2-/- mice is 30% higher than that of wild-type mice and is not affected by the addition of insulin, leading to reduction of body weight under normal food intake and slower weight-gain with high-fat/high-carbohydrate diets (34).
In addition to the roles in metabolic flow, fatty acids, acetyl-CoA, and malonyl-CoA are effector molecules that participate in signaling pathways in cells (35–37). Correspondingly, ACCs, as the consumer of acetyl-CoA and the producer of malonyl-CoA that function as the rate-limiting enzyme in fatty acid synthesis, play intriguing roles in regulating cellular signaling networks. For example, polyunsaturated fatty acids (PUFAs) are the precursors of various signaling molecules, such as eicosanoids, that regulate the activity of sterol-regulatory-element-binding protein 1c (SREBP1c) in fatty acid metabolism in liver (38). Inhibition of ACCs is considered as a promising strategy for treating liver diseases (39). However, on the other hand, it leads to a decrease in malonyl-CoA and the synthesis of downstream PUFA, which in turn activates SREBP1c and upregulates the expression of glycerol-3-phosphate acyltransferase 1 (GPAT1) that catalyzes triglyceride synthesis, stimulating hepatic VLDL secretion and leading to hypertriglyceridemia (40). As such, hypertriglyceridemia is used to be accompanied with the ACCs-targeting therapies. Acetyl-CoA is another instance. It can regulate gene transcription by donating the acetyl-moiety in the acetyltransferases-mediated histone acetylation (41). Inhibition of ACCs can increase the intracellular acetyl-CoA level and stimulate the influx of calcium into the cells, which lead to the activation and translocation of NFAT (nuclear factor of activated T cells 1) into the nucleus to promote the transcription of adhesion and migration-related genes, promoting adhesion and migration of glioblastoma cells through Ca2+– NFAT signaling. Malonyl-CoA plays roles in regulating dietary behavior and appetite (42). It is shown that mammalian neural tissue was able to rapidly convert administered acetate into acetyl-CoA, which subsequently entered the Krebs cycle to promote ATP production. Excessive ATP level, in turn, down-regulates AMPK activity and secures ACC2’s enzymatic activity. As a result, malonyl-CoA was produced to a great extent, causing the downstream effector molecular pro-opiomelanocortin upregulation and neuropeptide Y downregulation, eventually leading to loss of appetite in mice (43, 44).
In addition to the metabolic functions, ACC1 and ACC2 regulate protein acetylation by manipulating the availability of acetyl-CoA in cells. In liver-specific ACC1 knockout mice, the amount of acetyl-CoA in the extra-mitochondrial space is substantially elevated, which can serve as the substrate cofactor for acetyltransferases and increase the acetylation of proteins to regulate the functionome, including metabolic enzymes that regulate the metabolism network in ACC1 KO mice (13). Another study also shows that attenuated expression of ACC1 leads to a substantial increase in histone acetylation and alters transcriptional regulation, resulting in increased histone acetylation that consequently regulates biological processes in cells via influencing gene transcription (14). While the causal relationship between ACCs’ activities and protein acetylation is confirmed, the detailed mechanism underlying ACC1-related hyperacetylation remains elusive. Besides, ACC1 regulating protein acetylation by controlling the intracellular concentration of acetyl-CoA might also play a role in disease development. A supportive study reported that ACC inhibition regulates smad2 acetylation, which consequently affects the activity of smad2 and breast cancer metastasis (11). Malonyl-CoA is the product of ACCs’ enzymatic reactions. It can also serve as the substrate cofactor in the enzyme-catalyzed protein malonylation. Increased intracellular malonyl-CoA can result in upregulation of protein malonylation, which might affect protein functions and biological activities in cells (9). Despite evidences are supporting the importance of the non-metabolic functions of ACC1 in regulating protein modifications and functions, it is premature to conclude the non-metabolic functions of ACC1 in diseases development and treatment.
Altogether, ACCs regulate the physiologies and pathophysiological processes of cells by executing metabolic and non-metabolic functions. It should sense the alternatives in cells and precisely translate the alternated signals into the responses of cells. As such, sophisticated regulation of ACCs is required to secure the metabolism network matching the physiologies of cells.
Regulation of Acetyl-CoA Carboxylases
The activities of ACCs in cells can be transcriptionally and post-transcriptionally regulated that are tightly associated with the metabolic status of cells. In general, the protein level and enzymatic activities of ACCs are upregulated in nutrient and energy abundant conditions, aiming to store the excess nutrient and energy in the form of fatty acids. Correspondingly, the protein level and enzymatic activities of ACCs are suppressed in nutrient and energy-deficient conditions, aiming to secure the limited energy and nutrients being utilized for survival (45, 46). The AMP-activated protein kinase (AMPK) is the most studied energy sensor that senses the nutrient and energy status of cells and is an important regulator of ACC1. When cells suffering metabolic stresses, such as glucose deprivation or hypoxia, AMPK is activated that can phosphorylate the serine-79 residues in ACC1 (equivalent to ACC2 Ser212) (47). Phosphorylation of the Ser-79 residue effectively blocks the formation of ACC1 homodimer, leaving ACC1 molecules as monomers that are unable to catalyze acetyl-CoA carboxylation (21). The fatty acid synthesis pathway is therefore suppressed. However, when cells return to a nutrient and energy-abundant environment, the phosphorylation of Ser-79 in ACC1 can be removed by the type 2A protein phosphatase (PP2A), allowing the reformation of ACC1 homodimer that is active in catalyzing acetyl-CoA carboxylation (48, 49). Besides nutrient and energy stresses, the Ser-79 residue in ACC1 can be phosphorylated and maintained to prevent lipogenesis in certain pathophysiological processes. For example, the susceptibility gene 1 (BRCA1) C-terminal (BRCT) domain binds to p-ACC1 to from BRCA1/p-ACC1 complex (50), which prevents dephosphorylation of p-ACC1 and constantly suppress the activity of ACC1 to reprogram the metabolism network in breast cancer. Insulin-like growth factor-1 (IGF-1) treatment can disrupt the interaction between BRCA1 and p-ACC1, which leads to the dephosphorylation and reactivation of ACC1 (51).
In addition to phosphorylation, metabolites that are associated with changes in metabolism can allosterically regulate the activities of ACCs. For example, citrate is an intermediate metabolite in the TCA cycle that can allosterically activate ACC1 to drive the fatty acid synthesis pathway in normal condition (52, 53). Intriguingly, opposite effects of citrate on ACCs’ activities were reported (54), the underlying mechanism remains elusive. Glutamate can allosterically activate phosphatase that mediates dephosphorylation and activation of ACCs in cardiomyocytes, which may contribute to the cardioprotective effects of glutamine against lipolysis (55).. Fatty acyl-CoAs can induce the de-dimerization of ACC1 that inhibits the activities of ACC1 and fatty acid synthesis in cells (54). By interplay with metabolites from different metabolic pathways, ACCs mediate the cross-talk between fatty acid synthesis and other metabolic pathways, forming a sophisticated regulation network to secure the metabolic status fit the physiologies of cells.
The protein levels of ACC1 and ACC2 are dynamically regulated in cells. The expression level of ACC1 can be regulated by certain transcription factors. SREBP1c is a well-studied instance. The ACACA gene has two distinct SREBP binding sites, which recruit SREBP1c to initiate RNA Polymerase II-dependent transcription. Carbohydrate response element (ChRE) -binding protein (ChREBP) is another transcription factor that regulates ACC1. It binds to the promoter regions and activates the transcription of ACACA, in response to the high-carbohydrate diet (56, 57). Besides transcription, the protein stability of ACC1 can also be regulated. In breast cancer, small interfering RNA-mediated Aldo-keto reductase family 1B10 (AKR1B10) knockdown induces ACC1 degradation via the ubiquitination-proteasome pathway, resulting in a markedly drop in fatty acid synthesis in RAO-3 cells (58). In prostate cancer, the expression of prolyl isomerase Pin1 positively correlates with the protein level of ACC1. It binds to ACC1 to prevent ACC1 from entering the lysosomal pathway, leading to the stabilization of ACC1 protein and resulting in enhanced activity of ACC1 in cells (59).
ACC1 activity can also be regulated by post-transcriptional and translational mechanisms. There are 64 exons included in the gene ACACA that result in 7 alternatively spliced minor exons (1A, 1B, 1C, 3, 5A’, 5A, and 5B). The exon 5B can lead to transcriptional termination of the upstream exon 5 in two different transcripts, producing a short peptide that leads to the production of truncated ACC1 that affects the transcriptional efficiency and activity of ACC1. These studies suggest that ACC1 activity can be regulated by post-transcriptional and translational mechanisms and consequently result in suppression of fatty acid synthesis (60). The protein level of ACC1 can be post-transcriptionally reduced in calcium/calmodulin-dependent protein kinase kinase2 (CAMKK2) knock out cells, suppressing the proliferation of human prostate cancer cells (61).
Taken together, ACC1 and ACC2 are sophisticatedly regulated in cells to make the process of fatty acid synthesis, as well as its cross-talk metabolic networks, meet the physiologies of cells. There are myriad factors that regulate ACC1’s activities, including nutrients, protein kinases, phosphatases, allosteric regulators, transcriptional factors et al. Dysregulation of these regulatory factors usually serves as causative signaling for the development of cancer and metabolic diseases (10, 54, 61–63). Dysregulation of ACCs in diseases is therefore intensively studied.
Dysregulation of Acetyl-CoA Carboxylases in Diseases
Fatty acid synthesis is central in the cross-talk between multiple biological processes, including membrane biosynthesis, energy storage, and the generation of signaling molecules (64). Lipogenesis is dynamically regulated in response to the physiologies of cells. Correspondingly, dysregulation of fatty acid synthesis can induce or promote the development of diseases. ACCs is the first rate-limiting enzyme in fatty acid synthesis. It is therefore the focus of mountains of studies and be validated as a critical participant in diseases, especially cancer and metabolic diseases (11, 65–71).
Signaling regulators of lipid biosynthesis are major downstream targets of oncogenes and tumour suppressor pathways. Alternations of oncogenes and tumour suppressor pathways can manipulate de novo fatty acid synthesis. Dysregulation of fatty acid metabolism, in turn, influences the cellular processes that are linked to diseases, such as cancer. For example, the AMPK pathway is important in regulating cell growth, lipid and glucose metabolism, and autophagy (72). It senses the relative level of ADP to ATP and be activated when the ratio of ADP to ATP increased. When tumour cells suffering metabolic stresses, AMPK can be activated, which then phosphorylates ACCs to suppress the lipid biosynthesis pathway, resulting in metabolism reprogramming that influences the survival and growth of tumour cells. Mutagenic blockage of the AMPK phosphorylation site of ACC (ACC1 Ser76Ala and ACC2 Ser212Ala) increases liver DNL and accelerates the development of hepatocellular carcinoma (HCC). Liver-specific inhibitor ND-654, which mimics ACC phosphorylation, inhibits liver DNL and the progression of HCC, resulting in an improved prognosis for tumour-bearing mice (73). In head and neck squamous cell carcinoma cells (HNSCC), the AMPK activator cetuximab and 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR) can suppress tumour cell growth (74, 75). Abolishing the AMPK phosphorylation sites on ACC1 by mutagenesis can protect HNSCC from cetuximab-induced growth inhibition. Decreased AMPK activity in hereditary leiomyomatosis renal cell cancer (HLRCC) leads to the elevated activity of ACC1, which contributes to the oncogenic growth of HLRCC (76). Metformin is an agonist of AMPK that can promote phosphorylation of ACCs. Metformin treatment can effectively suppress lipogenesis and cancer cell proliferation (10). Because ACC1 can mediate the AMPK-sensed metabolic stress and the downstream of cancer metabolism reprogramming, it is considered a potential target for cancer therapy. However, some studies also show exceptional viewpoints on the AMPK/ACC signaling pathway for tumour growth (77). For example, under energy stress conditions, activated AMPK can phosphorylate and inhibit ACC1, which can suppress the NADPH-consuming fatty acid synthesis and maintain the NADPH homeostasis in tumour cells. Similarly, ACC1 depletion can also suppress the NADPH consumption by fatty acid synthesis, which in turn partially facilitates solid tumour survival under stress conditions (77). Thus, under special conditions, the AMPK/ACC signaling pathway that can alternatively regulate tumour cell proliferation by maintaining NADPH homeostasis.
The phosphatidylinositol-3 kinase (PI3K)/Akt/mammalian target of the rapamycin (mTOR) is another signaling pathway that senses the physiologies of cells and executes important functions by regulating ACC1 activities. In general, receptor tyrosine kinases (RTKs)-mediated activation of PI3K can activate Akt. Hyperactivated Akt then activates mTOR, which processes the upstream signals and forms mTORC1 (78). PI3K/Akt/mTOR signaling pathway regulates tumour metabolism, growth, survival, and metastasis (79, 80). ACC1 is tightly associated with the PI3K/Akt/mTOR signaling pathway in cancer cells. For example, the melanoma antigen ganglioside GD3 is a downstream target of PI3K/Akt/mTOR signaling. In melanoma, GD3 can induce the activation of SREBP-1, which is a transcription factor that regulates the expression of ACC1 (81). In breast cancer, the HER2 oncogene can induce ACC1 expression through translational regulation of the mTOR signaling pathway (82). Correspondingly, inhibition of ACC1 by siRNA or chemical inhibitors can inhibit AKT-related pathways, which is detrimental for cancer, such as human HCC (83). It is therefore concluded that ACC1 protein level and activity can be regulated by various internal alterations, which in turn affects lipid synthesis in tumors. Dysregulated lipid metabolism impacts multiple intracellular processes, such as membrane synthesis and energy metabolism that may influence tumor development ultimately. However, the mechanisms underlying lipid metabolism influencing tumor progressions, such as proliferation and metastasis, have not been fully elucidated. How ACC1 cross-talk with other pathways remains open for discussion.
Metabolic diseases are also tightly associated with the dysregulation of ACCs. In mammals, the accumulation of lipid in tissues, such as muscle and liver, is closely related to insulin resistance that associates with a myriad of metabolic disorders (84, 85). Likewise, dysregulated lipogenesis may lead to metabolic diseases such as obesity, diabetes, and NAFLD (6–8). As a central player of lipogenesis, ACCs promptly participates in the progression of metabolic disease. For example, a high-fat diet leads to increased ACC1 activity and obesity in mice while inhibition of ACC1 antagonizes the high fat diet induced obesity. ACC2 plays roles in controlling diet-induced diabetic nephropathy (DN). High-glucose diets promote lipid deposition and reduce fatty acid β-oxidation in human podocytes. Depletion of ACC2 attenuates the high-glucose diet-promoted lipid deposition and podocyte injury. The expression of glucose transporter 4 (GLUT4) is also restored by ACC2 depletion, which hampers the insulin signaling pathway. Besides, the expression of SIRT1/PGC-1a, an important complex related to the insulin metabolic pathway is also restored in the cells with ACC2 depletion, leading to the reduction of cellular insulin resistance and ultimately alleviating DN-induced cell injury (86). ACCs knockout animal models are powerful tools to understand the roles of ACCs in the progression of metabolic diseases, with which, a study demonstrated that ACC1 is necessary to maintain functional pancreatic β cells and glucose homeostasis in vivo, which indicates that ACC1 might be used to improve insulin secretion during diabetes (71). Liver-specific ACC1-KO mice (LACC1 KO) accumulate 40%-70% lower triglycerides in livers than that of wide-type mice when overnutrition is provided. Similarly, ACC2 knockout (ACC2 KO) mice do not gain weight when fed with high-fat diet (HF) (34). It might be due to the hepatic peroxisome proliferator-activated receptor-γ (PPAR-γ) proteins that are significantly reduced in ACC2 KO mice that are fed with high-fat and high-carbohydrate diet (HFHC). In this case, lipid synthesis-related enzymes such as ACC1, FASN, and ATP citrate lyase (ACL) are decreased, which in turn reduced diet-induced obesity. Besides, ACC2 KO mice are able to alleviate the HFHC diet-induced insulin resistance. ACC2 KO mice with HF diet show reduced AKT level and increased phosphorylation of AKT, which is critical in the insulin signaling pathway that can protect the mice from diabetes (87). The above researches demonstrate that ACCs is responsible for metabolic disorders caused by dietary factors (27). Moreover, hyper-activation of ACC1 can also result in abnormal physiologies in metabolic disease. For instance, the enhanced activity of ACC1 accelerates lipogenesis and lipid accumulation when animal suffering overnutrition and obesity, which leads to the accumulation of triglycerides in hepatocytes and thus causing NAFLD (88).
In general, dysregulated lipogenesis leads to the development of tumorigenesis and metabolic diseases. The roles of ACCs in regulating metabolism reprogramming in cancer and metabolic diseases are revealed in accumulated studies, which shed bright light on diseases treatment. As such, ACCs are becoming a promising therapeutic target for discovering novel therapeutic strategies and therapeutics development.
Acetyl-CoA Carboxylases-Targeting Small Molecules for Therapeutic Proposes
With the evidence of ACCs participating in the progression of diseases and its structural information, countless screenings for ACCs antagonists are performed and several promising leading compounds are confirmed for further validations (65, 83, 89–94). The ACCs inhibitors mainly target its BC domain and CT domain.
The BC domain accounts for the biotin carboxylation and formation of the homodimer of ACCs molecules. The main mechanism of action (MOA) of the BC domain targeting inhibitors is allosterically inhibiting the dimerization of the BC domain, maintaining ACC1 molecules as inactive monomers that are unable to perform the catalytic activity (21). Soraphen A, AMPK activators, and ND-series inhibitors (ND-630, ND-646, ND-654) inhibit ACC1 belong to this category (73, 91, 95–98). These inhibitors can effectively inhibit ACCs activity and affect the process of lipid metabolism and the development of disease (10, 26, 28, 29, 90, 99, 100). It is worth noting that there are studies already confirmed that inhibiting ACCs in the liver by using ND-630 (GS-0976) can significantly reduce 29% liver fat, hepatic steatosis, and markers of liver injury in NAFLD patients (101, 102), which further encourage the finding of ACCs inhibitors for therapeutic proposes.
The CT domain catalyzes the transfer of carboxyl-moiety from the carboxylated biotin to the acetyl-CoA to produce malonyl-CoA. Competitive inhibitors targeting the binding of acetyl-CoA by CT domain are therefore another promising strategy for inhibiting ACCs. TOFA, CP-640186, piperidinyl derived analogs, and spiropiperidine derived compounds are antagonists that belong to this category (103–108). These antagonists can reduce the mice’s appetite and accelerate weight loss (93, 109) and lead to apoptosis in different cancer cell lines (92, 110, 111). Despite no relevant clinical trials of this class of antagonists are found, it keeps recruiting screenings for new leading compounds.
In conclusion, numbers of commercially available ACCs inhibitors have exhibited strong therapeutic effects on disease models in vivo and in vitro, supporting that ACCs are promising therapeutic targets for the treatment of tumour and metabolic diseases. However, no agonist can specifically inhibit one ACCs member and keep another member intact. This might lead to adverse effects, because ACC1 and ACC2, indeed, are different in physiologies and pathophysiologies. To this end, the development of agonists that are specifically against ACC1 or ACC2 might be a promising strategy to target ACCs for diseases treatment.
Limitations and Prospects
Antagonists that target ACCs are intensively studied in clinic but hampered by several side-effects. For example, inhibiting lipogenesis via suppressing the expression of ACCs can reduce hepatic steatosis, but it simultaneously results in hypertriglyceridemia due to the activation of SREBP-1c and increased VLDL secretion (40). Another instance is PF-05175157, the first-in-human clinical trials ACC inhibitor, contributes to DNL reduction in treatment for T2DM but with concomitant reductions in platelet count (112). Recently, an exciting result in phase II clinical trial shows that the co-administration of PF-0522134 (a new ACC1 inhibitor in clinical trial) and PF-6865571 (DGAT2 inhibitor) has a strong effect in treating NASH without the side effect of hypertriglyceridemia (113). However, there are several challenges to address the side-effects of ACCs inhibition in clinical practice.
The principal challenge is that the inhibitors can hardly distinguish ACC1 from ACC2. As described above, ACC1 and ACC2 share 75% identity in amino acid sequence and are similar in structures that are composed of conservative domains for enzyme activity. However, the ACCs antagonists, such as Soraphen A and TOFA, can target and influence the activity of both ACC1 and ACC2, which might lead to the side-effects caused by the inhibition of unwanted ACC isoform. In nutrient-abundant condition, fatty acid synthesis and breakdown are coordinately controlled, avoiding a wasteful cycle of metabolism. However, in cancer cells, both fatty acid synthesis and breakdown are boosted to support cancer growth. To this end, coordinately antagonizing the dysregulation of ACC1 and ACC2 in cancer cells would be a promising strategy for cancer treatment. So far, none of ACCs inhibitors is approved useful in clinic. This might be due to the fact that ACC inhibitors that are not isoform-specific only partially reverse cancer’s preferences. Moreover, it is shown that the selectively inhibition of ACC2 may be ineffective in treating some metabolic diseases (114). A selective inhibitor targeting ACC1 that shows anti-NAFLD/NASH effects in pre-clinical models is reported in a recent study (115), which is expected to strengthen the efficacy.
Accumulating studies indicate the importance of ACCs in tumour cell growth which shows the great potential of ACCs in the treatment of cancer. However, studies on the role of ACCs in cancer have been attributed to their roles in fatty acid synthesis, the exact mechanism of which remains to be investigated. The role of fatty acid metabolism in cancer biology is not fully understood (116). More in-depth research about fatty acid metabolism in cancer will help examine and detail the roles of the ACCs, in cancer initiation, progression, and development.
Author Contributions
YuW: draft the manuscript and iconography. WY, SL, DG, and JH: proofread the manuscript and iconography. YugW: conceptualized, supervised, and finalized the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (31970577 and 91957110).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Menendez JA, Lupu R. Fatty Acid Synthase and the Lipogenic Phenotype in Cancer Pathogenesis. Nat Rev Cancer (2007) 7:763–77. doi: 10.1038/nrc2222
2. Abu-Elheiga L, Matzuk MM, Kordari P, Oh W, Shaikenov T, Gu Z, et al. Mutant Mice Lacking Acetyl-CoA Carboxylase 1 Are Embryonically Lethal. Proc Natl Acad Sci USA (2005) 102:12011–6. doi: 10.1073/pnas.0505714102
3. Schreurs M, Kuipers F, van der Leij FR. Regulatory Enzymes of Mitochondrial Beta-Oxidation as Targets for Treatment of the Metabolic Syndrome. Obes Rev (2010) 11:380–8. doi: 10.1111/j.1467-789X.2009.00642.x
4. Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr. Cellular Fatty Acid Metabolism and Cancer. Cell Metab (2013) 18:153–61. doi: 10.1016/j.cmet.2013.05.017
5. Hodson L, Gunn PJ. The Regulation of Hepatic Fatty Acid Synthesis and Partitioning: The Effect of Nutritional State. Nat Rev Endocrinol (2019) 15:689–700. doi: 10.1038/s41574-019-0256-9
6. Postic C, Girard J. Contribution of De Novo Fatty Acid Synthesis to Hepatic Steatosis and Insulin Resistance: Lessons From Genetically Engineered Mice. J Clin Invest (2008) 118:829–38. doi: 10.1172/JCI34275
7. Ronnett GV, Kim E-K, Landree LE, Tu Y. Fatty Acid Metabolism as a Target for Obesity Treatment. Physiol Behav (2005) 85:25–35. doi: 10.1016/j.physbeh.2005.04.014
8. Imamura F, Fretts AM, Marklund M, Ardisson Korat AV, Yang W-S, Lankinen M, et al. Fatty Acids in the De Novo Lipogenesis Pathway and Incidence of Type 2 Diabetes: A Pooled Analysis of Prospective Cohort Studies. PloS Med (2020) 17:e1003102. doi: 10.1371/journal.pmed.1003102
9. Bruning U, Morales-Rodriguez F, Kalucka J, Goveia J, Taverna F, Queiroz KC, et al. Impairment of Angiogenesis by Fatty Acid Synthase Inhibition Involves mTOR Malonylation. Cell Metab (2018) 28:866–80.e15. doi: 10.1016/j.cmet.2018.07.019
10. Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinilkunnil T, Chen ZP, et al. Single Phosphorylation Sites in Acc1 and Acc2 Regulate Lipid Homeostasis and the Insulin-Sensitizing Effects of Metformin. Nat Med (2013) 19:1649–54. doi: 10.1038/nm.3372
11. Rios Garcia M, Steinbauer B, Srivastava K, Singhal M, Mattijssen F, Maida A, et al. Acetyl-CoA Carboxylase 1-Dependent Protein Acetylation Controls Breast Cancer Metastasis and Recurrence. Cell Metab (2017) 26:842–55.e5. doi: 10.1016/j.cmet.2017.09.018
12. Wakil SJ, Abu-Elheiga LA. Fatty Acid Metabolism: Target for Metabolic Syndrome. J Lipid Res (2009) 50:S138–S43. doi: 10.1194/jlr.R800079-JLR200
13. Chow JD, Lawrence RT, Healy ME, Dominy JE, Liao JA, Breen DS, et al. Genetic Inhibition of Hepatic Acetyl-CoA Carboxylase Activity Increases Liver Fat and Alters Global Protein Acetylation. Mol Metab (2014) 3:419–31. doi: 10.1016/j.molmet.2014.02.004
14. Galdieri L, Vancura A. Acetyl-CoA Carboxylase Regulates Global Histone Acetylation. J Biol Chem (2012) 287:23865–76. doi: 10.1074/jbc.M112.380519
15. Tong L. Acetyl-Coenzyme A Carboxylase: Crucial Metabolic Enzyme and Attractive Target for Drug Discovery. Cell Mol Life Sci (2005) 62:1784–803. doi: 10.1007/s00018-005-5121-4
16. Abu-Elheiga L, Almarza-Ortega DB, Baldini A, Wakil SJ. Human Acetyl-CoA Carboxylase 2. Molecular Cloning, Characterization, Chromosomal Mapping, and Evidence for Two Isoforms. J Biol Chem (1997) 272:10669–77. doi: 10.1074/jbc.272.16.10669
17. Madauss KP, Burkhart WA, Consler TG, Cowan DJ, Gottschalk WK, Miller AB, et al. The Human ACC2 CT-Domain C-Terminus Is Required for Full Functionality and Has a Novel Twist. Acta Crystallogr D Biol Crystallogr (2009) 65:449–61. doi: 10.1107/s0907444909008014
18. Lee CK, Cheong HK, Ryu KS, Lee JI, Lee W, Jeon YH, et al. Biotinoyl Domain of Human Acetyl-CoA Carboxylase: Structural Insights Into the Carboxyl Transfer Mechanism. Proteins (2008) 72:613–24. doi: 10.1002/prot.21952
19. Chen Y, Elizondo-Noriega A, Cantu DC, Reilly PJ. Structural Classification of Biotin Carboxyl Carrier Proteins. Biotechnol Lett (2012) 34:1869–75. doi: 10.1007/s10529-012-0978-4
20. Zhang H, Yang Z, Shen Y, Tong L. Crystal Structure of the Carboxyltransferase Domain of Acetyl-Coenzyme A Carboxylase. Science (2003) 299:2064–7. doi: 10.1126/science.1081366
21. Wei J, Tong L. Crystal Structure of the 500-kDa Yeast Acetyl-CoA Carboxylase Holoenzyme Dimer. Nature (2015) 526:723–7. doi: 10.1038/nature15375
22. Broussard TC, Kobe MJ, Pakhomova S, Neau DB, Price AE, Champion TS, et al. The Three-Dimensional Structure of the Biotin Carboxylase-Biotin Carboxyl Carrier Protein Complex of E. Coli Acetyl-CoA Carboxylase. Structure (2013) 21:650–7. doi: 10.1016/j.str.2013.02.001
23. Hunkeler M, Hagmann A, Stuttfeld E, Chami M, Guri Y, Stahlberg H, et al. Structural Basis for Regulation of Human Acetyl-CoA Carboxylase. Nature (2018) 558:470–4. doi: 10.1038/s41586-018-0201-4
24. Munday MR. Regulation of Mammalian Acetyl-CoA Carboxylase. Biochem Soc Trans (2002) 30:1059–64. doi: 10.1042/bst0301059
25. Harada N, Oda Z, Hara Y, Fujinami K, Okawa M, Ohbuchi K, et al. Hepatic De Novo Lipogenesis Is Present in Liver-Specific ACC1-Deficient Mice. Mol Cell Biol (2007) 27:1881–8. doi: 10.1128/mcb.01122-06
26. Schreurs M, van Dijk TH, Gerding A, Havinga R, Reijngoud DJ, Kuipers F. Soraphen, an Inhibitor of the Acetyl-CoA Carboxylase System, Improves Peripheral Insulin Sensitivity in Mice Fed a High-Fat Diet. Diabetes Obes Metab (2009) 11:987–91. doi: 10.1111/j.1463-1326.2009.01078.x
27. Mao J, DeMayo FJ, Li H, Abu-Elheiga L, Gu Z, Shaikenov TE, et al. Liver-Specific Deletion of Acetyl-CoA Carboxylase 1 Reduces Hepatic Triglyceride Accumulation Without Affecting Glucose Homeostasis. Proc Natl Acad Sci USA (2006) 103:8552–7. doi: 10.1073/pnas.0603115103
28. Rysman E, Brusselmans K, Scheys K, Timmermans L, Derua R, Munck S, et al. De Novo Lipogenesis Protects Cancer Cells From Free Radicals and Chemotherapeutics by Promoting Membrane Lipid Saturation. Cancer Res (2010) 70:8117–26. doi: 10.1158/0008-5472.Can-09-3871
29. Stoiber K, Nagło O, Pernpeintner C, Zhang S, Koeberle A, Ulrich M, et al. Targeting De Novo Lipogenesis as a Novel Approach in Anti-Cancer Therapy. Br J Cancer (2018) 118:43–51. doi: 10.1038/bjc.2017.374
30. Abu-Elheiga L, Brinkley WR, Zhong L, Chirala SS, Woldegiorgis G, Wakil SJ. The Subcellular Localization of Acetyl-CoA Carboxylase 2. Proc Natl Acad Sci USA (2000) 97:1444–9. doi: 10.1073/pnas.97.4.1444
31. Savage DB, Choi CS, Samuel VT, Liu ZX, Zhang D, Wang A, et al. Reversal of Diet-Induced Hepatic Steatosis and Hepatic Insulin Resistance by Antisense Oligonucleotide Inhibitors of Acetyl-CoA Carboxylases 1 and 2. J Clin Invest (2006) 116:817–24. doi: 10.1172/jci27300
32. Essop MF, Camp HS, Choi CS, Sharma S, Fryer RM, Reinhart GA, et al. Reduced Heart Size and Increased Myocardial Fuel Substrate Oxidation in ACC2 Mutant Mice. Am J Physiol Heart Circ Physiol (2008) 295:H256–65. doi: 10.1152/ajpheart.91489.2007
33. Eaton S. Control of Mitochondrial Beta-Oxidation Flux. Prog Lipid Res (2002) 41:197–239. doi: 10.1016/s0163-7827(01)00024-8
34. Abu-Elheiga L, Matzuk MM, Abo-Hashema KA, Wakil SJ. Continuous Fatty Acid Oxidation and Reduced Fat Storage in Mice Lacking Acetyl-CoA Carboxylase 2. Science (2001) 291:2613–6. doi: 10.1126/science.1056843
35. Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G. Acetyl Coenzyme A: A Central Metabolite and Second Messenger. Cell Metab (2015) 21:805–21. doi: 10.1016/j.cmet.2015.05.014
36. Snaebjornsson MT, Janaki-Raman S, Schulze A. Greasing the Wheels of the Cancer Machine: The Role of Lipid Metabolism in Cancer. Cell Metab (2020) 31:62–76. doi: 10.1016/j.cmet.2019.11.010
37. Saggerson D. Malonyl-CoA, a Key Signaling Molecule in Mammalian Cells. Annu Rev Nutr (2008) 28:253–72. doi: 10.1146/annurev.nutr.28.061807.155434
38. Wang M, Ma LJ, Yang Y, Xiao Z, Wan JB. N-3 Polyunsaturated Fatty Acids for the Management of Alcoholic Liver Disease: A Critical Review. Crit Rev Food Sci Nutr (2019) 59:S116–s29. doi: 10.1080/10408398.2018.1544542
39. Zhang XJ, Cai J, Li H. Targeting ACC for NASH Resolution. Trends Mol Med (2022) 28:5–7. doi: 10.1016/j.molmed.2021.11.002
40. Kim CW, Addy C, Kusunoki J, Anderson NN, Deja S, Fu X, et al. Acetyl CoA Carboxylase Inhibition Reduces Hepatic Steatosis But Elevates Plasma Triglycerides in Mice and Humans: A Bedside to Bench Investigation. Cell Metab (2017) 26:394–406.e6. doi: 10.1016/j.cmet.2017.07.009
41. Lee JV, Berry CT, Kim K, Sen P, Kim T, Carrer A, et al. Acetyl-CoA Promotes Glioblastoma Cell Adhesion and Migration Through Ca(2+)-NFAT Signaling. Genes Dev (2018) 32:497–511. doi: 10.1101/gad.311027.117
42. Fadó R, Rodríguez-Rodríguez R, Casals N. The Return of Malonyl-CoA to the Brain: Cognition and Other Stories. Prog Lipid Res (2021) 81:101071. doi: 10.1016/j.plipres.2020.101071
43. Frost G, Sleeth ML, Sahuri-Arisoylu M, Lizarbe B, Cerdan S, Brody L, et al. The Short-Chain Fatty Acid Acetate Reduces Appetite via a Central Homeostatic Mechanism. Nat Commun (2014) 5:3611. doi: 10.1038/ncomms4611
44. Wyss MT, Magistretti PJ, Buck A, Weber B. Labeled Acetate as a Marker of Astrocytic Metabolism. J Cereb Blood Flow Metab (2011) 31:1668–74. doi: 10.1038/jcbfm.2011.84
45. Swinnen JV, Ulrix W, Heyns W, Verhoeven G. Coordinate Regulation of Lipogenic Gene Expression by Androgens: Evidence for a Cascade Mechanism Involving Sterol Regulatory Element Binding Proteins. Proc Natl Acad Sci USA (1997) 94:12975–80. doi: 10.1073/pnas.94.24.12975
46. Atkinson LL, Fischer MA, Lopaschuk GD. Leptin Activates Cardiac Fatty Acid Oxidation Independent of Changes in the AMP-Activated Protein Kinase-Acetyl-CoA Carboxylase-Malonyl-CoA Axis. J Biol Chem (2002) 277:29424–30. doi: 10.1074/jbc.M203813200
47. Steinberg GR, Kemp BE. AMPK in Health and Disease. Physiol Rev (2009) 89:1025–78. doi: 10.1152/physrev.00011.2008
48. Sanders FW, Griffin JL. De Novo Lipogenesis in the Liver in Health and Disease: More Than Just a Shunting Yard for Glucose. Biol Rev (2016) 91:452–68. doi: 10.1111/brv.12178
49. Auger C, Knuth CM, Abdullahi A, Samadi O, Parousis A, Jeschke MG. Metformin Prevents the Pathological Browning of Subcutaneous White Adipose Tissue. Mol Metab (2019) 29:12–23. doi: 10.1016/j.molmet.2019.08.011
50. Moreau K, Dizin E, Ray H, Luquain C, Lefai E, Foufelle F, et al. BRCA1 Affects Lipid Synthesis Through Its Interaction With Acetyl-CoA Carboxylase. J Biol Chem (2006) 281:3172–81. doi: 10.1074/jbc.M504652200
51. Koobotse M, Holly J, Perks C. Elucidating the Novel BRCA1 Function as a Non-Genomic Metabolic Restraint in ER-Positive Breast Cancer Cell Lines. Oncotarget (2018) 9:33562–76. doi: 10.18632/oncotarget.26093
52. Beaty NB, Lane MD. Kinetics of Activation of Acetyl-CoA Carboxylase by Citrate. Relationship to the Rate of Polymerization of the Enzyme. J Biol Chem (1983) 258:13043–50. doi: 10.1016/S0021-9258(17)44077-4
53. Vagelos PR, Alberts AW, Martin DB. Studies on the Mechnism of Activation of Acetyl Coenzyme A Carboxylase by Citrate. J Biol Chem (1963) 238:533–40 https://pubmed.ncbi.nlm.nih.gov/13995702/.
54. Wakil SJ, Stoops JK, Joshi VC. Fatty Acid Synthesis and Its Regulation. Annu Rev Biochem (1983) 52:537–79. doi: 10.1146/annurev.bi.52.070183.002541
55. Boone AN, Chan A, Kulpa JE, Brownsey RW. Bimodal Activation of Acetyl-CoA Carboxylase by Glutamate. J Biol Chem (2000) 275:10819–25. doi: 10.1074/jbc.275.15.10819
56. Ishii S, Iizuka K, Miller BC, Uyeda K. Carbohydrate Response Element Binding Protein Directly Promotes Lipogenic Enzyme Gene Transcription. Proc Natl Acad Sci USA (2004) 101:15597–602. doi: 10.1073/pnas.0405238101
57. Iizuka K, Horikawa Y. ChREBP: A Glucose-Activated Transcription Factor Involved in the Development of Metabolic Syndrome. Endocr J (2008) 55:617–24. doi: 10.1507/endocrj.k07e-110
58. Ma J, Yan R, Zu X, Cheng JM, Rao K, Liao DF, et al. Aldo-Keto Reductase Family 1 B10 Affects Fatty Acid Synthesis by Regulating the Stability of Acetyl-CoA Carboxylase-Alpha in Breast Cancer Cells. J Biol Chem (2008) 283:3418–23. doi: 10.1074/jbc.M707650200
59. Ueda K, Nakatsu Y, Yamamotoya T, Ono H, Inoue Y, Inoue MK, et al. Prolyl Isomerase Pin1 Binds to and Stabilizes Acetyl CoA Carboxylase 1 Protein, Thereby Supporting Cancer Cell Proliferation. Oncotarget (2019) 10:1637–48. doi: 10.18632/oncotarget.26691
60. Mao J, Chirala SS, Wakil SJ. Human Acetyl-CoA Carboxylase 1 Gene: Presence of Three Promoters and Heterogeneity at the 5’-Untranslated mRNA Region. Proc Natl Acad Sci USA (2003) 100:7515–20. doi: 10.1073/pnas.1332670100
61. Penfold L, Woods A, Muckett P, Nikitin AY, Kent TR, Zhang S, et al. CAMKK2 Promotes Prostate Cancer Independently of AMPK via Increased Lipogenesis. Cancer Res (2018) 78:6747–61. doi: 10.1158/0008-5472.Can-18-0585
62. Karami KJ, Coppola J, Krishnamurthy K, Llanos DJ, Mukherjee A, Venkatachalam KV. Effect of Food Deprivation and Hormones of Glucose Homeostasis on the Acetyl CoA Carboxylase Activity in Mouse Brain: A Potential Role of Acc in the Regulation of Energy Balance. Nutr Metab (Lond) (2006) 3:15. doi: 10.1186/1743-7075-3-15
63. Kaulage MH, Bhattacharya S, Muniyappa K. Structural Characterization of I-Motif Structure in the Human Acetyl-CoA Carboxylase 1 Gene Promoters and Their Role in the Regulation of Gene Expression. Chembiochem (2018) 19:1078–87. doi: 10.1002/cbic.201800021
64. Röhrig F, Schulze A. The Multifaceted Roles of Fatty Acid Synthesis in Cancer. Nat Rev Cancer (2016) 16:732–49. doi: 10.1038/nrc.2016.89
65. Luo DX, Tong DJ, Rajput S, Wang C, Liao DF, Cao D, et al. Targeting Acetyl-CoA Carboxylases: Small Molecular Inhibitors and Their Therapeutic Potential. Recent Pat Anticancer Drug Discov (2012) 7:168–84. doi: 10.2174/157489212799972918
66. Wang MD, Wu H, Fu GB, Zhang HL, Zhou X, Tang L, et al. Acetyl-Coenzyme A Carboxylase Alpha Promotion of Glucose-Mediated Fatty Acid Synthesis Enhances Survival of Hepatocellular Carcinoma in Mice and Patients. Hepatology (2016) 63:1272–86. doi: 10.1002/hep.28415
67. Milgraum LZ, Witters LA, Pasternack GR, Kuhajda FP. Enzymes of the Fatty Acid Synthesis Pathway Are Highly Expressed in In Situ Breast Carcinoma. Clin Cancer Res (1997) 3:2115–20 https://pubmed.ncbi.nlm.nih.gov/9815604/.
68. Yang JH, Kim NH, Yun JS, Cho ES, Cha YH, Cho SB, et al. Snail Augments Fatty Acid Oxidation by Suppression of Mitochondrial ACC2 During Cancer Progression. Life Sci Alliance (2020) 3:e202000683. doi: 10.26508/lsa.202000683
69. Brusselmans K, De Schrijver E, Verhoeven G, Swinnen JV. RNA Interference-Mediated Silencing of the Acetyl-CoA-Carboxylase-Alpha Gene Induces Growth Inhibition and Apoptosis of Prostate Cancer Cells. Cancer Res (2005) 65:6719–25. doi: 10.1158/0008-5472.Can-05-0571
70. Li K, Zhang C, Chen L, Wang P, Fang Y, Zhu J, et al. The Role of acetyl-coA Carboxylase2 in Head and Neck Squamous Cell Carcinoma. PeerJ (2019) 7:e7037. doi: 10.7717/peerj.7037
71. Cantley J, Davenport A, Vetterli L, Nemes NJ, Whitworth PT, Boslem E, et al. Disruption of Beta Cell Acetyl-CoA Carboxylase-1 in Mice Impairs Insulin Secretion and Beta Cell Mass. Diabetologia (2019) 62:99–111. doi: 10.1007/s00125-018-4743-7
72. Herzig S, Shaw RJ. AMPK: Guardian of Metabolism and Mitochondrial Homeostasis. Nat Rev Mol Cell Biol (2018) 19:121–35. doi: 10.1038/nrm.2017.95
73. Lally JSV, Ghoshal S, DePeralta DK, Moaven O, Wei L, Masia R, et al. Inhibition of Acetyl-CoA Carboxylase by Phosphorylation or the Inhibitor ND-654 Suppresses Lipogenesis and Hepatocellular Carcinoma. Cell Metab (2019) 29:174–82.e5. doi: 10.1016/j.cmet.2018.08.020
74. Luo J, Hong Y, Lu Y, Qiu S, Chaganty BK, Zhang L, et al. Acetyl-CoA Carboxylase Rewires Cancer Metabolism to Allow Cancer Cells to Survive Inhibition of the Warburg Effect by Cetuximab. Cancer Lett (2017) 384:39–49. doi: 10.1016/j.canlet.2016.09.020
75. Li K, Chen L, Lin Z, Zhu J, Fang Y, Du J, et al. Role of the AMPK/ACC Signaling Pathway in TRPP2-Mediated Head and Neck Cancer Cell Proliferation. BioMed Res Int (2020) 2020:4375075. doi: 10.1155/2020/4375075
76. Tong WH, Sourbier C, Kovtunovych G, Jeong SY, Vira M, Ghosh M, et al. The Glycolytic Shift in Fumarate-Hydratase-Deficient Kidney Cancer Lowers AMPK Levels, Increases Anabolic Propensities and Lowers Cellular Iron Levels. Cancer Cell (2011) 20:315–27. doi: 10.1016/j.ccr.2011.07.018
77. Jeon SM, Chandel NS, Hay N. AMPK Regulates NADPH Homeostasis to Promote Tumour Cell Survival During Energy Stress. Nature (2012) 485:661–5. doi: 10.1038/nature11066
78. Schlessinger J. Cell Signaling by Receptor Tyrosine Kinases. Cell (2000) 103:211–25. doi: 10.1016/s0092-8674(00)00114-8
79. Alzahrani AS. PI3K/Akt/mTOR Inhibitors in Cancer: At the Bench and Bedside. Semin Cancer Biol (2019) 59:125–32. doi: 10.1016/j.semcancer.2019.07.009
80. Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front Oncol (2014) 4:64. doi: 10.3389/fonc.2014.00064
81. Yamauchi Y, Furukawa K, Hamamura K, Furukawa K. Positive Feedback Loop Between PI3K-Akt-Mtorc1 Signaling and the Lipogenic Pathway Boosts Akt Signaling: Induction of the Lipogenic Pathway by a Melanoma Antigen. Cancer Res (2011) 71:4989–97. doi: 10.1158/0008-5472.Can-10-4108
82. Yoon S, Lee MY, Park SW, Moon JS, Koh YK, Ahn YH, et al. Up-Regulation of Acetyl-CoA Carboxylase Alpha and Fatty Acid Synthase by Human Epidermal Growth Factor Receptor 2 at the Translational Level in Breast Cancer Cells. J Biol Chem (2007) 282:26122–31. doi: 10.1074/jbc.M702854200
83. Calvisi DF, Wang C, Ho C, Ladu S, Lee SA, Mattu S, et al. Increased Lipogenesis, Induced by AKT-Mtorc1-RPS6 Signaling, Promotes Development of Human Hepatocellular Carcinoma. Gastroenterology (2011) 140:1071–83. doi: 10.1053/j.gastro.2010.12.006
84. Engin A. The Definition and Prevalence of Obesity and Metabolic Syndrome. Adv Exp Med Biol (2017) 960:1–17. doi: 10.1007/978-3-319-48382-5_1
85. Santoleri D, Titchenell PM. Resolving the Paradox of Hepatic Insulin Resistance. Cell Mol Gastroenterol Hepatol (2019) 7:447–56. doi: 10.1016/j.jcmgh.2018.10.016
86. Wang Q, Zhao B, Zhang J, Sun J, Wang S, Zhang X, et al. Faster Lipid β-Oxidation Rate by Acetyl-CoA Carboxylase 2 Inhibition Alleviates High-Glucose-Induced Insulin Resistance via SIRT1/PGC-1α in Human Podocytes. J Biochem Mol Toxicol (2021) 35:e22797. doi: 10.1002/jbt.22797
87. Abu-Elheiga L, Wu H, Gu Z, Bressler R, Wakil SJ. Acetyl-CoA Carboxylase 2-/- Mutant Mice are Protected Against Fatty Liver Under High-Fat, High-Carbohydrate Dietary and De Novo Lipogenic Conditions. J Biol Chem (2012) 287:12578–88. doi: 10.1074/jbc.M111.309559
88. Alves-Bezerra M, Cohen DE. Triglyceride Metabolism in the Liver. Compr Physiol (2017) 8:1–8. doi: 10.1002/cphy.c170012
89. Sugimoto Y, Naniwa Y, Nakamura T, Kato H, Yamamoto M, Tanabe H, et al. A Novel Acetyl-CoA Carboxylase Inhibitor Reduces De Novo Fatty Acid Synthesis in HepG2 Cells and Rat Primary Hepatocytes. Arch Biochem Biophys (2007) 468:44–8. doi: 10.1016/j.abb.2007.09.012
90. Corominas-Faja B, Cuyàs E, Gumuzio J, Bosch-Barrera J, Leis O, Martin ÁG, et al. Chemical Inhibition of Acetyl-CoA Carboxylase Suppresses Self-Renewal Growth of Cancer Stem Cells. Oncotarget (2014) 5:8306–16. doi: 10.18632/oncotarget.2059
91. Svensson RU, Parker SJ, Eichner LJ, Kolar MJ, Wallace M, Brun SN, et al. Inhibition of Acetyl-CoA Carboxylase Suppresses Fatty Acid Synthesis and Tumor Growth of Non-Small-Cell Lung Cancer in Preclinical Models. Nat Med (2016) 22:1108–19. doi: 10.1038/nm.4181
92. He D, Sun X, Yang H, Li X, Yang D. TOFA Induces Cell Cycle Arrest and Apoptosis in ACHN and 786-O Cells Through Inhibiting PI3K/Akt/mTOR Pathway. J Cancer (2018) 9:2734–42. doi: 10.7150/jca.26374
93. Loftus TM, Jaworsky DE, Frehywot GL, Townsend CA, Ronnett GV, Lane MD, et al. Reduced Food Intake and Body Weight in Mice Treated With Fatty Acid Synthase Inhibitors. Science (2000) 288:2379–81. doi: 10.1126/science.288.5475.2379
94. Gao YS, Qian MY, Wei QQ, Duan XB, Wang SL, Hu HY, et al. WZ66, a Novel Acetyl-CoA Carboxylase Inhibitor, Alleviates Nonalcoholic Steatohepatitis (NASH) in Mice. Acta Pharmacol Sin (2020) 41:336–47. doi: 10.1038/s41401-019-0310-0
95. Shen Y, Volrath SL, Weatherly SC, Elich TD, Tong L. A Mechanism for the Potent Inhibition of Eukaryotic Acetyl-Coenzyme A Carboxylase by Soraphen A, a Macrocyclic Polyketide Natural Product. Mol Cell (2004) 16:881–91. doi: 10.1016/j.molcel.2004.11.034
96. Jang S, Gornicki P, Marjanovic J, Bass E, PI T, Rodriguez P, et al. Activity and Structure of Human Acetyl-CoA Carboxylase Targeted by a Specific Inhibitor. FEBS Lett (2018) 592:2048–58. doi: 10.1002/1873-3468.13097
97. Cho YS, Lee JI, Shin D, Kim HT, Jung HY, Lee TG, et al. Molecular Mechanism for the Regulation of Human ACC2 Through Phosphorylation by AMPK. Biochem Biophys Res Commun (2010) 391:187–92. doi: 10.1016/j.bbrc.2009.11.029
98. Harriman G, Greenwood J, Bhat S, Huang X, Wang R, Paul D, et al. Acetyl-CoA Carboxylase Inhibition by ND-630 Reduces Hepatic Steatosis, Improves Insulin Sensitivity, and Modulates Dyslipidemia in Rats. Proc Natl Acad Sci USA (2016) 113:E1796–805. doi: 10.1073/pnas.1520686113
99. Beckers A, Organe S, Timmermans L, Scheys K, Peeters A, Brusselmans K, et al. Chemical Inhibition of Acetyl-CoA Carboxylase Induces Growth Arrest and Cytotoxicity Selectively in Cancer Cells. Cancer Res (2007) 67:8180–7. doi: 10.1158/0008-5472.Can-07-0389
100. Scaglia N, Chisholm JW, Igal RA. Inhibition of Stearoylcoa Desaturase-1 Inactivates Acetyl-CoA Carboxylase and Impairs Proliferation in Cancer Cells: Role of AMPK. PloS One (2009) 4:e6812. doi: 10.1371/journal.pone.0006812
101. Lawitz EJ, Coste A, Poordad F, Alkhouri N, Loo N, McColgan BJ, et al. Acetyl-CoA Carboxylase Inhibitor GS-0976 for 12 Weeks Reduces Hepatic De Novo Lipogenesis and Steatosis in Patients With Nonalcoholic Steatohepatitis. Clin Gastroenterol Hepatol (2018) 16:1983–91.e3. doi: 10.1016/j.cgh.2018.04.042
102. Alkhouri N, Lawitz E, Noureddin M, DeFronzo R, Shulman GI. GS-0976 (Firsocostat): An Investigational Liver-Directed Acetyl-CoA Carboxylase (ACC) Inhibitor for the Treatment of Non-Alcoholic Steatohepatitis (NASH). Expert Opin Investig Drugs (2020) 29:135–41. doi: 10.1080/13543784.2020.1668374
103. Chen L, Duan Y, Wei H, Ning H, Bi C, Zhao Y, et al. Acetyl-CoA Carboxylase (ACC) as a Therapeutic Target for Metabolic Syndrome and Recent Developments in ACC1/2 Inhibitors. Expert Opin Investig Drugs (2019) 28:917–30. doi: 10.1080/13543784.2019.1657825
104. McCune SA, Harris RA. Mechanism Responsible for 5-(Tetradecyloxy)-2-Furoic Acid Inhibition of Hepatic Lipogenesis. J Biol Chem (1979) 254:10095–101.
105. Halvorson DL, McCune SA. Inhibition of Fatty Acid Synthesis in Isolated Adipocytes by 5-(Tetradecyloxy)-2-Furoic Acid. Lipids (1984) 19:851–6. doi: 10.1007/bf02534514
106. Zhang H, Tweel B, Li J, Tong L. Crystal Structure of the Carboxyltransferase Domain of Acetyl-Coenzyme A Carboxylase in Complex With CP-640186. Structure (2004) 12:1683–91. doi: 10.1016/j.str.2004.07.009
107. Harwood HJ Jr., Petras SF, Shelly LD, Zaccaro LM, Perry DA, Makowski MR, et al. Isozyme-Nonselective N-Substituted Bipiperidylcarboxamide Acetyl-CoA Carboxylase Inhibitors Reduce Tissue Malonyl-CoA Concentrations, Inhibit Fatty Acid Synthesis, and Increase Fatty Acid Oxidation in Cultured Cells and in Experimental Animals. J Biol Chem (2003) 278:37099–111. doi: 10.1074/jbc.M304481200
108. Chonan T, Oi T, Yamamoto D, Yashiro M, Wakasugi D, Tanaka H, et al. (4-Piperidinyl)-Piperazine: A New Platform for Acetyl-CoA Carboxylase Inhibitors. Bioorg Med Chem Lett (2009) 19:6645–8. doi: 10.1016/j.bmcl.2009.10.012
109. Bengtsson C, Blaho S, Saitton DB, Brickmann K, Broddefalk J, Davidsson O, et al. Design of Small Molecule Inhibitors of Acetyl-CoA Carboxylase 1 and 2 Showing Reduction of Hepatic Malonyl-CoA Levels In Vivo in Obese Zucker Rats. Bioorg Med Chem (2011) 19:3039–53. doi: 10.1016/j.bmc.2011.04.014
110. Nishi K, Suzuki K, Sawamoto J, Tokizawa Y, Iwase Y, Yumita N, et al. Inhibition of Fatty Acid Synthesis Induces Apoptosis of Human Pancreatic Cancer Cells. Anticancer Res (2016) 36:4655–60. doi: 10.21873/anticanres.11016
111. Guseva NV, Rokhlin OW, Glover RA, Cohen MB. TOFA (5-Tetradecyl-Oxy-2-Furoic Acid) Reduces Fatty Acid Synthesis, Inhibits Expression of AR, Neuropilin-1 and Mcl-1 and Kills Prostate Cancer Cells Independent of P53 Status. Cancer Biol Ther (2011) 12:80–5. doi: 10.4161/cbt.12.1.15721
112. Griffith DA, Kung DW, Esler WP, Amor PA, Bagley SW, Beysen C, et al. Decreasing the Rate of Metabolic Ketone Reduction in the Discovery of a Clinical Acetyl-CoA Carboxylase Inhibitor for the Treatment of Diabetes. J Med Chem (2014) 57:10512–26. doi: 10.1021/jm5016022
113. Calle RA, Amin NB, Carvajal-Gonzalez S, Ross TT, Bergman A, Aggarwal S, et al. ACC Inhibitor Alone or Co-Administered With a DGAT2 Inhibitor in Patients With Non-Alcoholic Fatty Liver Disease: Two Parallel, Placebo-Controlled, Randomized Phase 2a Trials. Nat Med (2021) 27(10):1836–48. doi: 10.1038/s41591-021-01489-1
114. Olson DP, Pulinilkunnil T, Cline GW, Shulman GI, Lowell BB. Gene Knockout of Acc2 has Little Effect on Body Weight, Fat Mass, or Food Intake. Proc Natl Acad Sci USA (2010) 107:7598–603. doi: 10.1073/pnas.0913492107
115. Tamura Y, Sugama J, Iwasaki S, Sasaki M, Yasuno H, Aoyama K, et al. Selective Acetyl-CoA Carboxylase 1 Inhibitor Improves Hepatic Steatosis and Hepatic Fibrosis in a Pre-Clinical NASH Model. J Pharmacol Exp Ther (2021) 379(3):280–9. doi: 10.1124/jpet.121.000786
Keywords: acetyl-CoA carboxylase, lipogenesis, cancer metabolism, tumorigenesis, metabolic diseases
Citation: Wang Y, Yu W, Li S, Guo D, He J and Wang Y (2022) Acetyl-CoA Carboxylases and Diseases. Front. Oncol. 12:836058. doi: 10.3389/fonc.2022.836058
Received: 15 December 2021; Accepted: 10 February 2022;
Published: 11 March 2022.
Edited by:
Xuefei Li, Institute of Synthetic Biology, Shenzhen Institutes of Advanced Technology (CAS), ChinaReviewed by:
Kumar Pichumani, Houston Methodist Research Institute, United StatesDe Huang, Duke University, United States
Copyright © 2022 Wang, Yu, Li, Guo, He and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yugang Wang, yugangw@hust.edu.cn