 Abel Tesfaye Anshabo
Abel Tesfaye Anshabo Robert Milne
Robert Milne Shudong Wang
Shudong Wang Hugo Albrecht
Hugo Albrecht- Drug Discovery and Development, Centre for Cancer Diagnostics and Therapeutics, Clinical and Health Sciences, University of South Australia, Adelaide, SA, Australia
Cyclin-dependent kinases (CDKs) are proteins pivotal to a wide range of cellular functions, most importantly cell division and transcription, and their dysregulations have been implicated as prominent drivers of tumorigenesis. Besides the well-established role of cell cycle CDKs in cancer, the involvement of transcriptional CDKs has been confirmed more recently. Most cancers overtly employ CDKs that serve as key regulators of transcription (e.g., CDK9) for a continuous production of short-lived gene products that maintain their survival. As such, dysregulation of the CDK9 pathway has been observed in various hematological and solid malignancies, making it a valuable anticancer target. This therapeutic potential has been utilized for the discovery of CDK9 inhibitors, some of which have entered human clinical trials. This review provides a comprehensive discussion on the structure and biology of CDK9, its role in solid and hematological cancers, and an updated review of the available inhibitors currently being investigated in preclinical and clinical settings.
Introduction
Protein kinases are a large family of enzymes that regulate most eukaryotic cellular processes and signaling pathways through protein phosphorylation. This can activate or inhibit enzymes, increase protein-protein interactions, change cellular localization, or generate a site for the recruitment of proteins (1, 2). Protein kinases are themselves regulated by different transcriptional and post-translational modifications. The overall outcome is the regulation of cellular proliferation, apoptosis, and differentiation (1, 2). The human genome encodes more than 500 protein kinase genes (3). Due to their key functions, deregulation of protein kinase activity as a consequence of genetic mutation or the absence of a negative regulator is associated with a number of pathological disorders (e.g., cancer and inflammatory disorders) (1, 2).
Cyclin-dependent kinases (CDKs) are serine (Ser)/threonine (Thr) protein kinases activated by regulatory cyclin proteins (4). The human genome encodes twenty CDKs (numbered 1-20) belonging to the CDK- and CDK-like branch of the CMGC subfamily of human kinases which include the cyclin-dependent, mitogen-activated, glycogen synthase and CDC-like kinases (4). CDKs together with their cyclin partners play specific roles in numerous cellular processes, such as cell division and transcription, in response to intra- and extra-cellular signals (5). CDK proteins have a two-lobed structure with the active site sandwiched between an amino (N)-terminal lobe comprised mostly of β-sheets and a carboxyl (C)-terminal lobe of α-helices. CDKs are regulated by binding of the cyclin subunits (themselves regulated by formation and degradation) and phosphorylation of conserved residues in the T- and glycine (G) - rich loop structures within the CDK (4). Based on their evolutionary relationships and main functional roles, CDKs may be divided into two main groups: those which regulate the cell cycle (e.g. CDKs 1-7, 14-18) and those regulating transcription (e.g. CDKs 7-13, 18-20) (5). Owing to these central regulatory functions it is perhaps not surprising that dysregulation of CDK activity is closely associated with human malignancies. This is particularly so for CDK9, a key regulator of transcription which is overtly employed by cancer cells for the constant production of short-lived proteins that maintains their survival. In this review, the role of CDK9 in the pathogenesis of hematological and solid cancers will be discussed together with a comprehensive review of its discovery, structure, biological function, regulation, and the available pharmacological inhibitors currently being investigated as anticancer agents.
Discovery of P-TEFb (CDK9-Cyclin T)
Identification of the regulation of gene transcription at the elongation phase began with the observation that cells treated with the nucleoside analogue 5,6-dichloro-1-β-D-ribofuranosyl benzimidazole (DRB), a general inhibitor of transcription, continued to produce short capped ribonucleic acids (RNA) (6). Initially, it was proposed that DRB might cause premature transcription termination (6). Later evidence, however, indicated that it might act at the elongation step as it lacks an inhibitory effect on the transcription efficacy of elongating RNA polymerase II (RNAP II) that has already synthesized RNAs past the transcription elongation checkpoint (7, 8). Parallel to these observations, there was growing evidence that RNAP II pauses transcription shortly after its initiation (9). This was shown by the appearance of long RNA transcripts from a deoxyribonucleic acid (DNA) template in the presence of nuclear extracts at a rate much slower than the appearance of short-length RNA (9). Furthermore, purified RNAP II elongation complexes (cleared of other transcription factors) were only capable of producing short RNAs as compared to incubation of RNAP II with nuclear extracts, indicating the presence of stimulatory transcription elongation factors (9).
Simultaneously, certain agents (e.g., high salt treatment) were shown to initially reduce the elongation rate, but with time caused an increase in the number of long transcripts (8, 9). The two opposite effects were ascribed to dual inhibition of elongation factors and disruption of a transcription pause (9). These findings led to a model suggesting the presence of a block-relief elongation control system directly affected by DRB. The opposing proteins responsible for such block-relief control were subsequently named Positive Transcription Elongation Factor (P-TEF, the one that relieves the pause and is affected by DRB) and Negative Transcription Elongation Factor (N-TEF, the protein preventing transcription elongation) (9).
Following the identification of these two factors, further characterization of the P-TEF complex revealed a component named P-TEFb as the primary target of DRB during transcription elongation (10). The removal of P-TEFb from a nuclear extract incubated with a DNA template totally abolished the production of long RNA transcripts with no effect seen on the level of short-length RNAs (10). The actual mechanism through which P-TEFb relieves the transcriptional pause was not clear until discovery of the importance of phosphorylation of the carboxyl terminal domain (CTD) on the largest subunit (Rpb1) of RNAP II for productive transcription elongation. Notably, hypo-and hyper-phosphorylated states paralleled a pause and productive elongation, respectively (11), and this was accompanied with decreasing long RNA production when RNAP II CTD became increasingly truncated (12). Coincidentally, P-TEFb was shown to have kinase activity specifically directed against the CTD (12).
Prior to the identification of P-TEFb a protein originally named PITALRE (for its characteristic Proline, Isoleucine, Threonine, Alanine, Leucine, Arginine, Glutamic acid motif) has been cloned during efforts to discover Ser/Thr protein kinases with crucial functions during the cell cycle. Its major characteristics were a wide tissue distribution, close similarity with cell cycle kinases, and localization within the nucleus (13). Its real identity and potential role in transcription, however, was later discovered during a search for subunits of P-TEFb. The sequence and function of PITALRE were shown to be identical to that of the small subunit of P-TEFb (14). Like P-TEFb, PITALRE phosphorylated RNAP II CTD and its removal led to inhibition of long RNA production in a DRB-sensitive manner (14). This led to the identification of PITALRE as the kinase component of P-TEFb. Subsequently, the large subunit of P-TEFb was shown to have structural signatures of cyclin proteins (i.e., two sequences of five α-helices) and was required for phosphorylation of the CTD and productive elongation (15).
Consequently, the kinase subunit was renamed CDK9 and its cyclin partner cyclin T (with three subunits identified cyclin T1, T2a, and T2b) (15, 16). Simultaneously, a protein derived from the human immunodeficiency virus 1 (HIV1), known as Trans-Activator of Transcription (TAT), was shown to have a role in viral transcription through the formation of a complex which contained P-TEFb (17). We summarized the timeline for P-TEFb biology key findings in Figure 1.
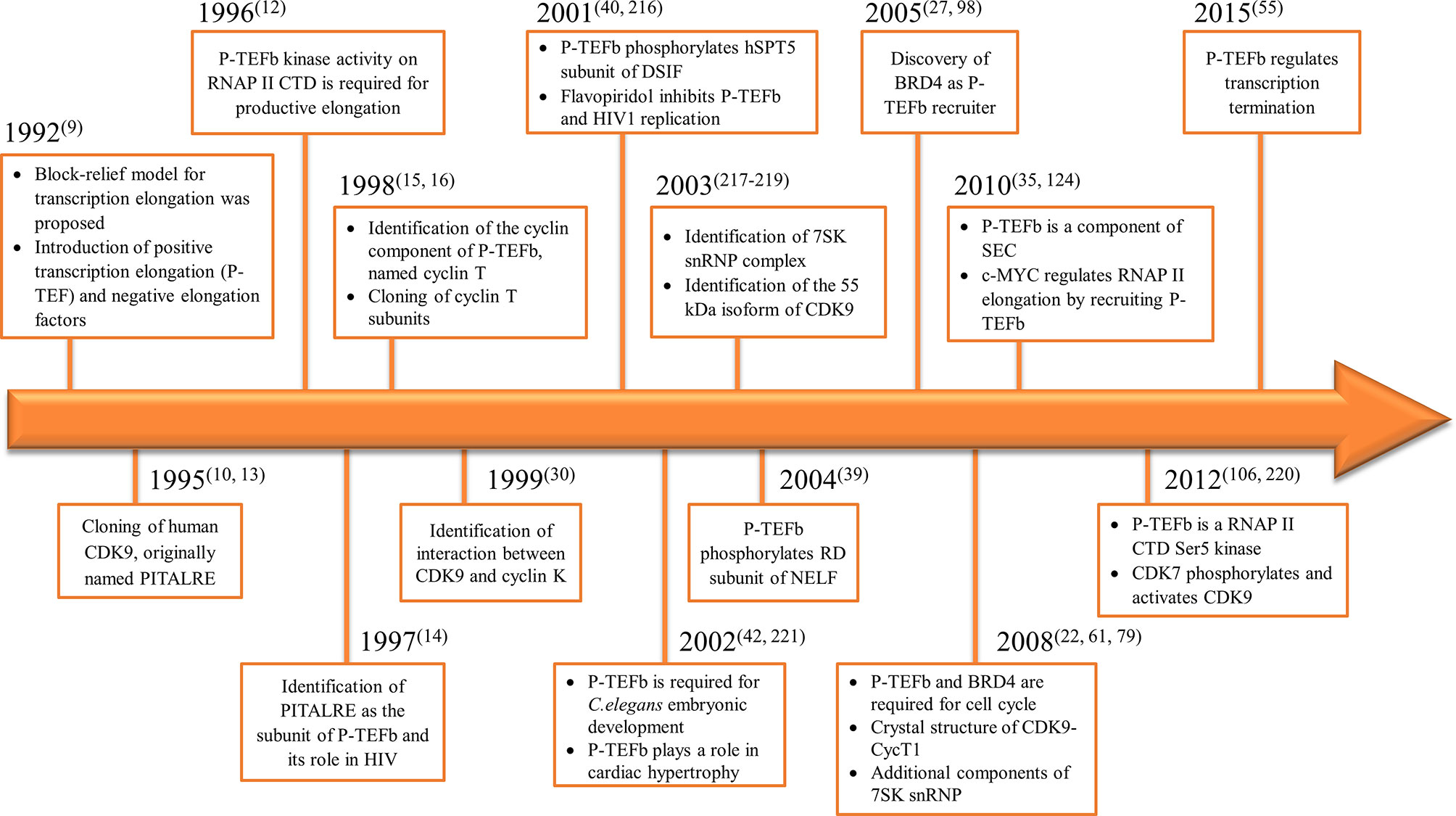
Figure 1 Timeline for the discovery of P-TEFb and its biological roles.
Structure of the CDK9-Cyclin T Complex
The eukaryotic kinases consist of conserved sequences of 250 to 300 amino acids that form a common catalytical core called the kinase domain (18). The kinase domain contains twelve conserved sub-domains that fold into a bilobal tertiary structure with smaller N-terminal and larger C-terminal lobes. The N-terminal region contains mainly β strands while α-helices form a major part of the C-terminal region (19–21).
Structure of CDK9
The N-terminal lobe of CDK9, which spans from amino acid residue 16 to 108, comprises five β structures (β1-5) and one α helix (αC) (Figure 2) (22). The C-terminal lobe (residues 109-330) is composed of seven α-helices (αD-J) and four β strands (β6-9) (22). The interaction between CDK9 and cyclin T1 occurs mainly through the αC helix located on the N-terminal lobe (Figures 2 and 4). This helix contains a peptide sequence highly conserved across CDKs (PITALRE in CDK9) and serves as a site for interaction with cyclin during the activation of CDKs (22).
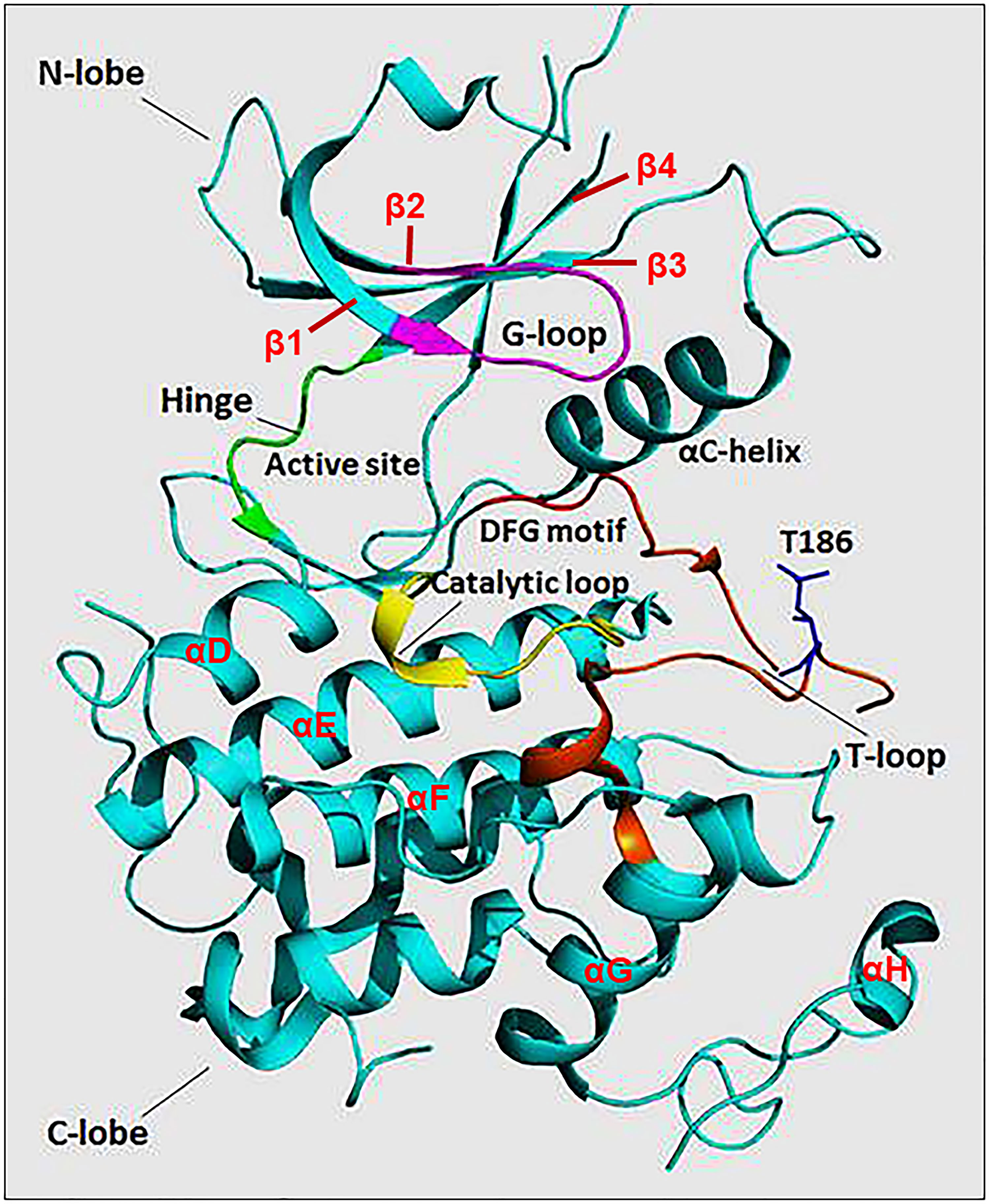
Figure 2 The protein structure of monomeric CDK9 (Protein Data Bank: 3BLQ). The bilobal CDK9 structure is dominated by N-terminal β-sheets (1-4 are shown) and C-terminal α-helices (D-H are shown). The C-terminal also contains β-sheets (6-9, not shown). The two lobes are connected by a hinge region (green) that binds the adenine moiety of adenosine triphosphate (ATP). The N-terminus contains an αC helix and glycine-rich loop (G-loop, purple) which binds cyclin and ATP, respectively. The C-terminus comprises the catalytic loop (yellow), T-loop (brown), and DFG motif that binds Mg+2. The threonine residue (Thr186) involved in CDK9 activation is found in the T-loop structure.
The Adenosine Triphosphate (ATP) Binding Motif forms a cleft between the N- and C-terminal lobes and is highly conserved among CDKs (Figures 2 and 3) (22). In this site, the adenine moiety of ATP is inserted deep into the cleft and the phosphate groups are positioned toward the exterior (18). The hydrophobic pocket harboring the adenine moiety is located between the β-sheets of the N lobe and a hinge region loop which connects the two lobes (20, 22). In this region, the ATP adenine nitrogen atoms, N6 and N1, form hydrogen bonds with the main chain oxygen and nitrogen of Asp104 and Cys106 residues, respectively (22). In addition to hydrogen bonds, multiple interactions of the purine ring with aliphatic and aromatic residues of the hinge region also help in anchoring the adenine moiety (22). The α and β non-transferable phosphates of ATP are held in position through ionic and hydrogen bonds with residues located in the G-loop between β1 and β2 (Figure 2) (20, 22). The β- and γ-phosphates in concert with an aspartate residue and two water molecules form coordination bonds with a cationic Mg+2 cofactor. The aspartate residue involved in this process (Asp167 in CDK9, Asp145 in CDK2) belongs to a ‘DFG’ motif located in a loop between β8 and β9 (Figure 2) (18, 20, 22).

Figure 3 Sequence comparison between CDK9 and CDK2. The sequence identity between the two proteins is 31.9%. Green color indicates residues conserved between CDK9 and CDK2. Red underlined residues indicate the different functional subunits of the kinases. In the T-loop, the phosphorylation of a conserved threonine residue (labelled red) is vital for the activation of both CDK9 (Thr186) and CDK2 (Thr160). The sequence alignment was generated and % sequence similarity determined using UniProt (https://www.uniprot.org/align/) and sequence identifiers were P50750 for CDK9 and P24941 for CDK2.
The Substrate Recognition Motif is located in the cleft between the N- and C- lobes in close proximity to the γ-phosphate of ATP (20). In general, CDKs have a strong preference for substrate motifs which have a proline residue immediately flanking a phospho-Ser or phospho-Thr residue (i.e. Ser/Thr-Pro-X-Arg/Lys) (4). This is ascribed to the presence of a hydrophobic pocket (created by the interaction between Val190 and Arg195 of CDK9) that can only accommodate a proline residue at the +1 position relative to the phosphorylation site (22). While the recognition motif is universal for the CDK family, subtle differences exist between members in their substrate preference, depending on the stringent requirement of a specific residue at the +3 position (4). This has been exemplified with the substrate recognition differences between CDK2 and CDK9, where the latter displayed a strict requirement for the Ser/Thr-Pro-X-Arg/Lys consensus (23).
The residues of the Catalytic Loop are highly conserved among protein kinases suggesting a similar catalytic mechanism (Figures 2 and 3) (24). The main mechanism involves transformation of the hydroxyl group of the Ser or Thr residue on the substrate into a nucleophile capable of attacking the γ-phosphate of ATP (24). A conserved aspartate (Asp149 in CDK9) facilitates this by acting as a general base that helps align the substrate oxygen (22, 24). Two additional residues, namely Lys151 and Thr165, have been suggested to play a secondary role by orientating the substrate (22).
T-Loop: When cyclin is not bound, the catalytic cleft is completely blocked by a C-terminal loop named the T-loop or activation segment (Figures 2 and 3) (20). This conformation hinders critical interactions between different residues and the non-transferable phosphates of ATP vital for locking ATP in a catalytically favorable position. During activation, binding of cyclin physically pulls the T-loop outward from the catalytic cleft and exposes a threonine residue found in the loop (Thr186 in CDK9 and Thr160 in CDK2; Figure 3) (4, 20, 22). The phosphorylation of this residue stabilizes the T-loop in an open position, as phospho-Thr186 coordinates the formation of an intramolecular hydrogen bonding network containing Arg148 and Arg172, resulting in a fully active kinase protein (22).
Structure of Cyclins T and K
In general, cyclins are characterized by the presence of two similar regions, each having five α-helices and a short ending helix (N- or C- terminal helices). Each stalk of helices forms a separate canonical cyclin box, generally composed of approximately 100 conserved amino acid residues each. The two regions are arranged around central helices in an antiparallel fashion, forming a rigid structure that is liable to minor conformational change during binding to a kinase protein (22). Although both the N- and C- terminal regions of cyclins make contact with the kinase protein, interactions leading to activation of the kinase occur mainly through the N-terminal cyclin box region (4, 19). Despite their similarity in structure and sequence, major differences in regions outside of the cyclin box between those cyclins involved in cell cycle control (e.g., cyclins A, B, E) and those in transcription (e.g. cyclins T, H, C, K) are observed. Notably, there is a clear variation in the length and orientation of the short-ending helices (22).
The major cyclin partner of CDK9, cyclin T (‘T’ named after the first letter of HIV TAT), has a close similarity to cyclin C and cyclin H (15, 17). Three cyclin T members are known, namely T1 (726 residues), T2a (663 residues), and T2b (730 residues), which have a high degree of identity (81%) in their cyclin box region (16). HIV TAT interacts only with cyclin T1 in complex with CDK9 to mediate HIV transcription (25, 26). Human cyclin T1 (Figure 7B) contains a recognition motif for TAT-Transactivation Response Element (TAR) complex (TRM, residues 254-272) (26) found downstream of the N-terminal cyclin box (residues 1-263), a putative coiled-coil motif (residues 379–430), histidine-rich motif (residues 506–530) (27, 28), and a C-terminal PEST (Pro-Glu-Ser-Thr) sequence (residues 709–726) (29). CDK9 also forms a complex with cyclin K, which displays CTD kinase activity, despite the fact that cyclin K only shares 29% identity with cyclin T at the amino acid level (30). Nonetheless, the CDK9 interaction site is conserved among cyclin T and K, explaining their similar modes of interaction (31).
Interactions Between CDK9 and Cyclin T1
Most of the binding between CDK9 and cyclin T1 involves interactions between the H3, H4, and H5 helices of the cyclin and the αC helix and β4 strand of the CDK (Figures 2 and 4) (22). The H5 helix interacts with the αC helix and enforces an active conformation. While this mechanism is common across CDKs, in contrast to most other CDKs, the N-terminal short helix (HN) in cyclin T1 makes no contact with CDK9, which gives forth to a more solvent-exposed kinase surface.
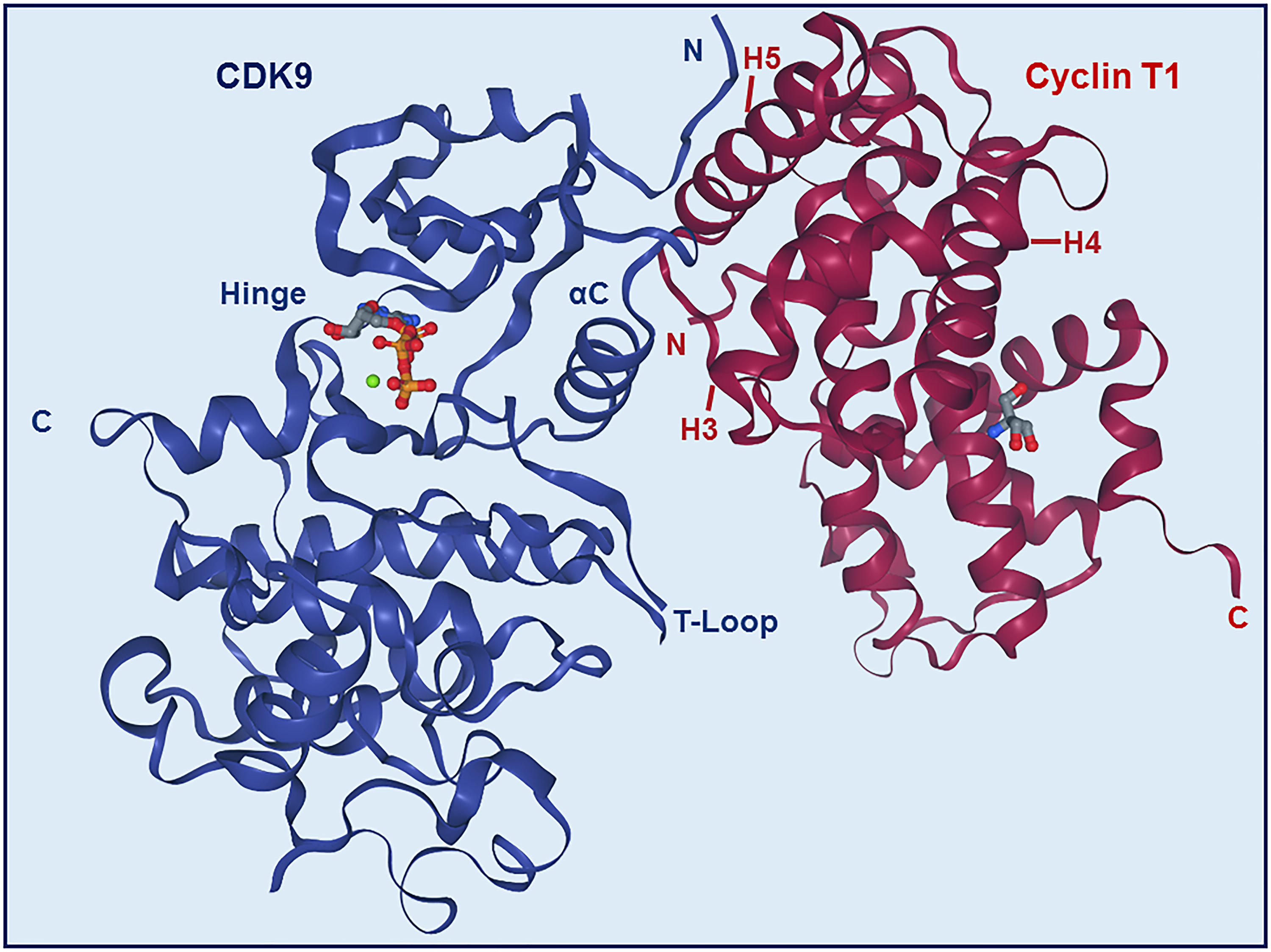
Figure 4 Structure of CDK9-Cyclin T1 complex. Stereo ribbon plot of the 3D structure of CDK9-cyclin T1 in complex with ATP (Protein data bank: 3BLQ). CDK9 (blue) and cyclin T1 (dark red) make contact through the αC helix and β4 strand of CDK9 and H3, H4, and H5 helices of cyclin T1. The CDK9 T-loop and hinge regions are indicated.
Biological Functions of P-TEFb
Control of Transcriptional Elongation and Termination
Normal cellular growth and development are dependent on efficient and intricate regulation of gene expression. This regulation primarily occurs during transcription, which is the initial step of expression and revolves around the interaction of multiple host factors with the CTD of RNAP II. The CTD consists of tandem heptapeptide repeats (52 in mammals) of the consensus sequence Tyr-Ser-Pro-Thr-Ser-Pro-Ser (Y1S2P3T4S5P6S7) (32, 33). These host factors guide RNAP II to gain access to transcription sites, initiate and elongate transcription, and couple transcription of messenger RNA (mRNA) with its processing, including capping, splicing, and polyadenylation (32–34).
Formerly, transcriptional initiation was viewed as the main checkpoint for regulating transcription, while little emphasis was given to transcriptional elongation. This viewpoint, however, has changed considerably as it now becomes apparent that elongation is a highly dynamic and strictly regulated stage of transcription (32). Shortly after RNAP II initiates transcription and synthesizes 20-50 nucleotides of the nascent RNA, it is engaged by factors which significantly hinder its ability to continue elongation (9, 33). In fact, the majority of RNAP II is found paused at the promoter-proximal regions of most mammalian genes, ready to resume transcription elongation (35). While the exact purpose of RNAP II pausing is not clarified, some of the proposed functions include (1): increasing the accessibility of genes that would otherwise have a high chance of being condensed into nucleosomes (2), allowing rapid and synchronous gene activation (3), integrating multiple regulatory signals, and (4) a checkpoint for coupling elongation with 5’ end-capping of nascent RNA (36). Two factors, namely DRB sensitivity-inducing factor (DSIF) and negative elongation factor (NELF), cooperate in pausing RNAP II (37, 38).
The RNAP II requires the kinase activity of P-TEFb to overcome the pause and continue elongation (Figure 5) (14). Upon recruitment to the paused site by bromodomain-containing protein 4 (BRD4), P-TEFb phosphorylates one of the four subunits of NELF (NELF-E or RD) and the human SPT5 (hSPT5) subunit of DSIF (39, 40). These phosphorylation steps liberate NELF from RNAP II, while converting DSIF into a positive elongation factor to track along elongating RNAP II (39, 41). Simultaneously, P-TEFb also phosphorylates the CTD of RNAP II, primarily on the Ser2 residue (Figure 5) (42–44). Phosphorylation of the CTD is vital for the efficient coupling of transcription elongation and pre-mRNA processing (45, 46). Ser2 phosphorylated CTD recruits chromatin-modifying factors, elongation factors, the co-transcriptional splicing machinery, and pre-mRNA 3’ end processing factors (47–50). In addition to P-TEFb, other CTD kinases such as CDK12, CDK13, and BRD4 (an atypical kinase) have been shown to phosphorylate the CTD on the Ser2 residue, although the direct role of this phosphorylation by these kinases, particularly CDKs 12 and 13, remains elusive (51–53).
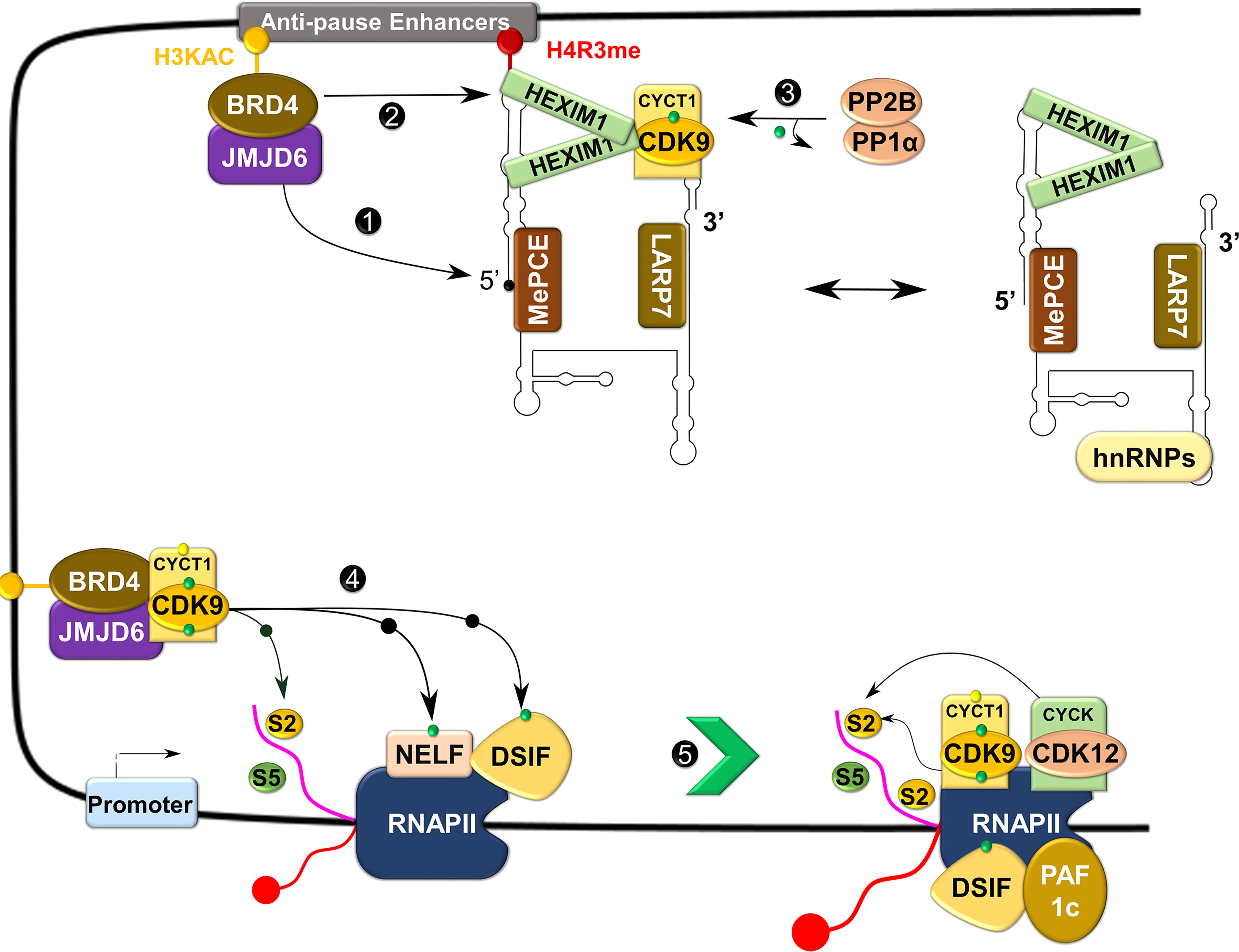
Figure 5 Control of transcriptional elongation by P-TEFb. During active transcription, BRD4 recruits JMJD6 to 7SK snRNP anchored to anti-pause enhancers on chromatin. JMJD6 demethylates both H4R3me and the 5’ hairpin of 7SK RNA, breaking chromatin binding of the former and exposing the latter for degradation (Labelled as 1). Concurrently, acetylated histone (H3KAC)-bound BRD4 interacts with and extracts P-TEFb from 7SK snRNP (2). Protein phosphatases (PP2B and PP1α) also assist in the release of P-TEFb from 7SK snRNP by dephosphorylating CDK9 pThr186 (3). After release, CDK9 is re-phosphorylated on Thr186 by CDK7 and delivered by BRD4-JMJD6 to RNAP II that has been paused in the proximal promoter region. At this site, P-TEFb phosphorylates DSIF, NELF, and RNAP II CTD (4), allowing productive elongation (5).
In addition to the well-known role of P-TEFb in transcriptional elongation, an interdependence between its role in releasing RNAP II from its pause and transcriptional initiation has also been identified (54, 55). Namely, a high degree of RNAP II promoter-proximal pausing, induced by CRISPR-Cas9-based or small-molecule inhibition of CDK9, limits the frequency of any new round of transcriptional initiation (referred to as ‘pause-initiation’ limit), giving an insight into how cells maintain the appropriate quantity of RNA from particular genes (54, 55).
P-TEFb also plays a direct regulatory role in terminating transcription. Analogous to promoter-proximal pausing, an additional major elongation checkpoint that is dependent on P-TEFb has been identified near the terminal poly(A) sites (56). Most RNAP II that escapes promoter-proximal pausing despite the presence of CDK9 inhibitors (e.g., KM05382 and DRB) has the capacity to elongate transcription, but prematurely terminate transcription near poly(A) sites. This termination was directly linked with a loss of association of P-TEFb, DSIF, and poly(A) factors (e.g., Ssu72 and CstF64) to RNAP II (56). Beyond the poly(A) site, CDK9 regulates transcriptional termination by phosphorylating Xrn2, a 5’-3’exoribonuclease, on Thr439 and enhances its cleavage of the RNA transcript from RNAP II (57). Furthermore, CDK9 phosphorylates and inhibits protein phosphatase 1 (PP1) activity on DSIF and RNAP II until the complex reaches transcription termination sites. At these sites, PP1 becomes activated and dephosphorylates DSIF, which leads to the termination of transcription (58).
P-TEFb in the Cell Cycle
Timely progression through each phase of the cell cycle is controlled by spatio-temporal expression of different cyclins (e.g. cyclins D, E, A, and B) which control the kinase activity of their respective cell cycle CDKs (5). Unlike cell cycle CDKs, the expression of CDK9 and its cyclin partner, as well as its kinase activity, does not change in a cell cycle-dependent manner (13, 59). This observation has led the conventional view that P-TEFb is only a transcriptional CDK with a limited role in the cell cycle. Contrary to this view, silencing of Cdk9 by RNA interference (RNAi) induced the arrest of Drosophila cells in the G1 stage of their cycle (60). The missing mechanistic link was provided by BRD4, a mitotic bookmark that remains attached to chromatin during mitosis when all other transcription factors have dissociated (61–64). This “bookmarking” is vital for prompt re-activation of transcription after mitosis (61, 63). Beginning around mid to late anaphase, BRD4 marks many M/G1 genes and in concert with jumonji C-domain-containing protein 6 (JMJD6) induces promoter-proximal pause release, and recruits P-TEFb for RNAPII, NELF and DSIF phosphorylation (Figure 5). Subsequently, this results in the expression of key G1 genes to promote the progression of cells into their S phase (62, 63). Abrogation of this process through BRD4 knockdown reduces the binding of P-TEFb to mitotic chromosomes and the expression of key G1 and G1-associated genes, leading to cell cycle arrest and apoptosis (62).
P-TEFb in Cellular Differentiation
P-TEFb influences many cellular differentiation programs (65–70). For example, CDK9-cyclin T2a interacts directly with myoblast determination protein 1 (MyoD), a basic helix-loop-helix muscle differentiation factor, and promotes MyoD-dependent transcription and activation of myogenic differentiation (66). Similarly, CDK9-cyclin T1 activates muscle differentiation programs by stimulating the transcription program of myocyte enhancer factor 2 [MEF2 (67)], indicating interaction with MyoD or MEFs is dictated by the particular cyclin T.
P-TEFb is also required for the differentiation of monocytes (70), lymphocytes (68), adipocytes (71), and neurons (69, 72). Treatment of monocytes with a potent inducer of differentiation, phorbol 12-myristate 13-acetate, induces increased expression of cyclin T1 and of P-TEFb activity (70). Similarly, the expression of both CDK9 and cyclin T1 is linked to a particular stage of lymphoid differentiation (68). During adipogenesis, P-TEFb (containing CDK955, a minor isoform of CDK9) (73) interacts with, and phosphorylates the peroxisome proliferator-activated receptor gamma (PPAR γ), the master regulator of adipocyte differentiation, to activate transcription of its target genes in pre-adipocytes (71). Furthermore, CDK9-cyclin T1 is required for neuronal differentiation induced by retinoic acid as indicated by increased expression of both CDK9 and cyclin T1 (69, 72). This neuronal differentiation is linked to increased expression of differentiation-associated genes as a result of retinoic acid inducing the interaction of P-TEFb with a transcription factor, called Zinc Finger MYND-Type Containing 8 (72).
Role of P-TEFb in DNA Repair
While the biological role of the CDK9-cyclin T1 complex is well established, the function of CDK9-cyclin K has remained elusive for a long period of time. Although CDK9-cyclin K can phosphorylate RNAP II CTD in vitro (30), this complex can only activate transcription while tethered to RNA but not DNA (74). Insight into the role of CDK9-cyclin K emerged with the identification of cyclin K as a transcription target for p53 in response to DNA damage (75). The depletion of CDK9 and cyclin K, but not cyclin T, with RNAi was shown to impair the cell cycle in response to replication stress and DNA damage (76). Relative to transcriptionally mediated mechanisms, CDK9-cyclin K is presumed to play a direct role in the repair of damaged DNA by interacting with components of the ATM-and Rad3-related (ATR) pathways that respond to DNA damage, such as ATR, ATR interacting protein, and claspin (76). Furthermore, small hairpin RNA (shRNA)-mediated depletion of CDK955 induces double strand DNA breaks and apoptosis (77). CDK955 also interacts with Ku70, a key protein in nonhomologous end-joining, and might play a role in DNA repair. Interestingly, this interaction might involve cyclin K as Ku70 does not associate with either cyclin T1 or T2 (77).
Regulation of P-TEFb Activity
The activity of P-TEFb is highly regulated because its function is important for the efficient expression of most genes (34). Numerous transcriptional, translational, and posttranslational mechanisms are employed to control the protein level and biological activity of P-TEFb (34).
Regulation by Sequestering in an Inactive Complex
P-TEFb can integrate into two functionally opposite complexes, which are in equilibrium. More than half of cellular P-TEFb is reversibly sequestrated in a large inhibitory ribonucleoprotein complex (Figure 5), while the remainder is bound to BRD4 and transcriptionally active (27, 78). Figure 6 shows in more detail the control over the level of inhibitory complex. P-TEFb is sequestrated by hexamethylene bisacetamide-inducible proteins (HEXIM 1 or 2) bound to a small, evolutionarily conserved nuclear RNA called 7SK snRNA (27, 78, 79). In this complex (hereafter named, 7SK snRNP), HEXIM is the main suppressor of P-TEFb function and was initially identified as a protein induced upon treating vascular smooth muscle cells with hexamethylene bisacetamide (78, 80). 7SK snRNA serves as an indispensable scaffold that mediates the interaction between HEXIM 1 or 2 and P-TEFb (Figure 6) (78, 81–83). Due to its vital role, the stability of 7SK snRNA is maintained by two proteins known as La related protein [LARP7 (84, 85)], and methyl phosphate capping enzyme [MePCE (85, 86)], which protect the 7SK snRNA from degradation by exonucleases at the 3’ and 5’ ends, respectively. The reversible sequestration of P-TEFb by 7SK snRNP plays a key role in maintaining a strict balance in the overall cellular gene expression (Figures 5 and 6).
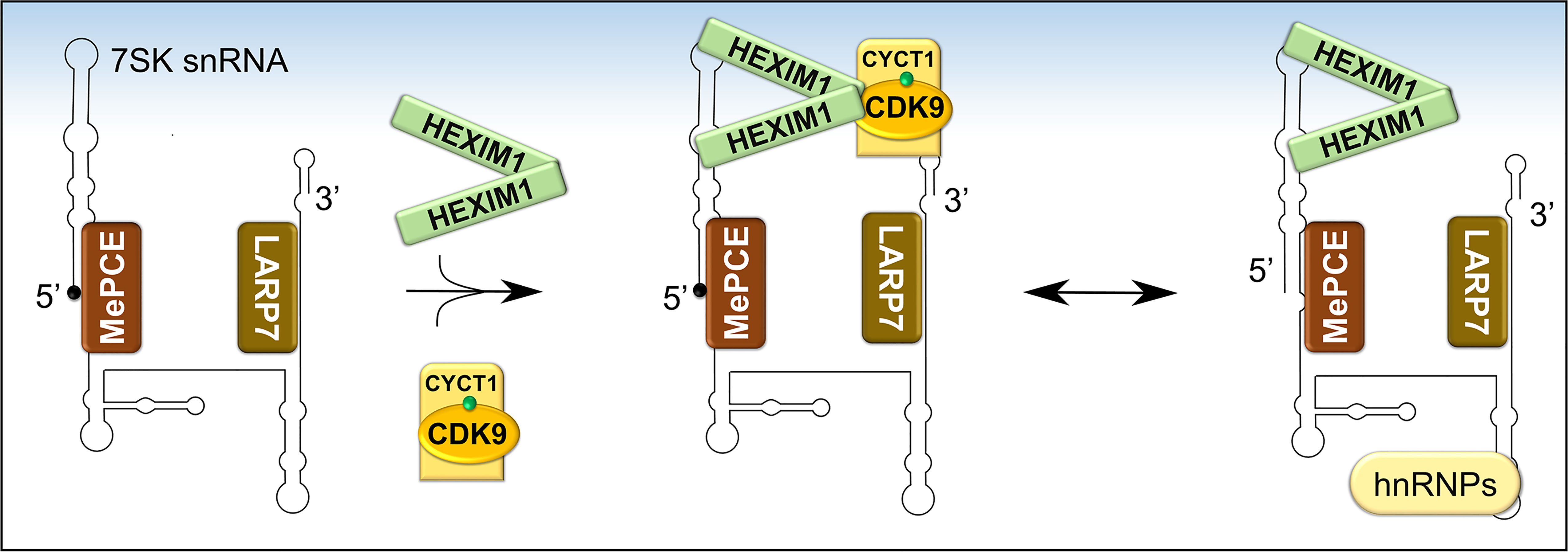
Figure 6 Formation and composition of 7SK snRNP. Following the folding of 7SK RNA into a four-stem loop structure, MePCE and LARP7 bind and protect its 5’ and 3’ ends, respectively, from catalytic degradation. One mechanism of protection involves capping of the 5’ end of 7SK RNA by MePCE (depicted as a black dot). The stable 7SK snRNP core then binds dimers of HEXIM1 which exposes their P-TEFb binding domains. Subsequently, HEXIM1 binds activated P-TEFb (CDK9 phosphorylated on Thr186, green dot) and this inhibits its kinase activity. During transcriptional activation, P-TEFb is released and 7SK snRNP is stabilized by binding to heterogeneous nuclear ribonucleoproteins (hnRNPs).
Prior to sequestering P-TEFb, HEXIM1 is found as a dimer formed through its two C-terminal coiled-coil (CR) regions (residues 279-352) and is incapable of interacting with and/or inhibiting P-TEFb (Figures 6 and 7A) (87, 88). Such inability emanates from an autoinhibitory electrostatic interaction between a highly basic region (BR, residues 150-177, KHRR motif) and two acidic regions (AR1 and 2, residues 211-249) of HEXIM1, conferring a conformation that opposes its interaction with P-TEFb (Figure 7A) (78, 89). Binding of the 5’ terminal hairpin of 7SK snRNA with the BR of HEXIM1 causes a conformational change in HEXIM1, unmasking its C-terminal P-TEFb binding domain (residues 181-359) for interaction with the cyclin box of cyclin T1 (88, 89). In this complex, cyclin T1 also makes contact with the 3’ hairpin of 7SK RNA (Figure 7B) (90). HEXIM1 inactivates CDK9 through its PYNT motif (residues 202-205), which masks the catalytic site of CDK9 and occupies its substrate recognition motif (Figures 6 and 7A) (88, 91). Interestingly, phosphorylation of CDK9 on its Thr186 residue is required for the sequestration of P-TEFb in the inhibitory complex (88, 92). This suggests that the inhibitory complex serves as a pool for transcriptionally active kinase, as Thr186 phosphorylation is critical for P-TEFb kinase activity. Because HEXIM homodimerizes, it is proposed that the 7SK snRNP complex comprises two HEXIM1 molecules, binding to 7SK snRNA and P-TEFb (88).
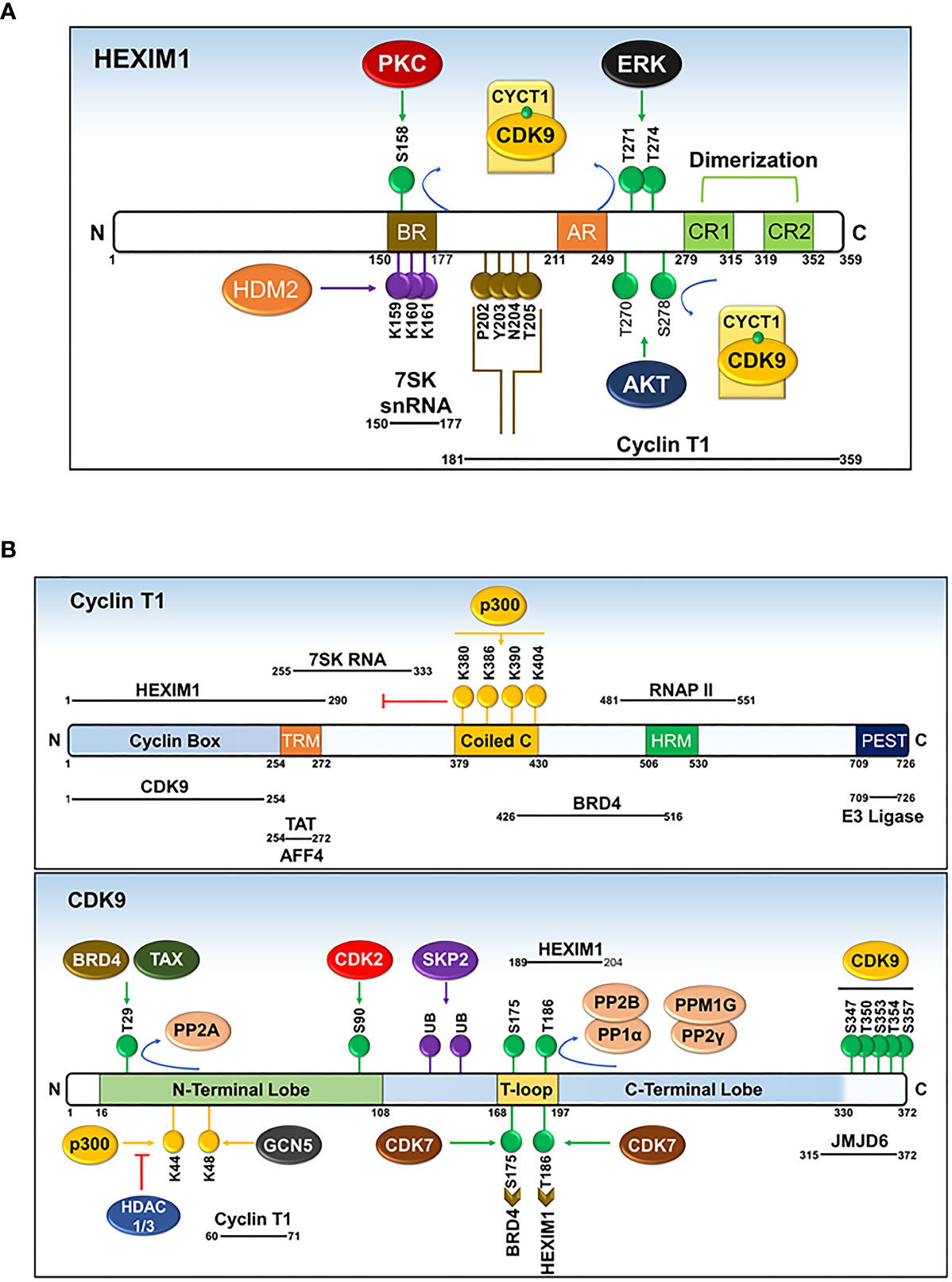
Figure 7 Domains and post-translational modifications of HEXIM1 (A), cyclin T1 (B), and CDK9 (B). Phosphorylated (green), ubiquitinated (purple), acetylated (yellow), and PYNT (brown) motifs are shown. Numbers indicate the positions of amino acid residues. BR, basic region; AR, acidic region, encompassing AR1 and AR2; CR, coiled-coil region. Solid black lines with indicated spans of amino acid residues indicate the regions responsible for interactions with other binding partners. Modified from Cho, Schroeder and Ott, Cell Cycle, 9 (9), 1697-1705 (2010).
Orchestrating the function of P-TEFb to activate paused RNAP II complexes on some genes while sparing others requires specific delivery of P-TEFb. Previously, simple diffusion of 7SK snRNP was proposed as the mechanism for transporting P-TEFb to activated genes based on the finding that 7SK snRNP is readily extracted from the nuclei under non-harsh conditions (e.g., under low salt treatment) (93). Separate reports, however, have indicated the presence of 7SK snRNP on chromatin. This is based on the existence of inactive P-TEFb and components of the inhibitory complex (e.g. HEXIM1, LARP7, and 7SK snRNA) with the non-phosphorylated form of RNAP II in the pre-initiation complex (94). Coincidentally, subsequent evidence has shown the co-occupancy of HEXIM1, LARP7, and 7SK snRNA with RNAP II on the transcribed loci of a wide number of active protein-encoding genes and further identified a chromatin anchoring mechanism for 7SK snRNP (e.g., methylated histone (H4R3me) on specific enhancers, termed anti-pause enhancers) (95, 96). These observations indicate that 7SK snRNP is tethered to chromatin to selectively guide the function of a transcription-ready P-TEFb (Figure 5).
The precise molecular mechanism for the liberation of P-TEFb from 7SK snRNP in response to cellular signals or stress conditions (e.g., UV, actinomycin D) remains to be elucidated but might involve post-translational modifications of the components of 7SK snRNP or direct recruitment by transcription regulators (e.g. BRD4, HIV TAT) (Figures 7A, B and 8). Multiple modifications directed towards HEXIM1, CDK9, and cyclin T1 induce the release of P-TEFb. For instance, T-cell receptor signaling disrupts 7SK snRNP and activates P-TEFb function via phosphorylation of HEXIM1 mediated by protein kinase C (PKC, on Ser158) and extracellular-signal-regulated kinase (ERK, on Tyr271, 274) (Figure 7A) (97, 98). In a different cellular context (e.g. HIV infected cells), hexamethylene bisacetamide (HMBA) disrupts 7SK snRNP by activating the Phosphatidylinositol 3-kinase (PI3K)/Protein Kinase B (AKT) pathway, which inhibits the interaction between HEXIM1 and P-TEFb by phosphorylating HEXIM1 on Thr270 and Ser278 (Figures 7A and 8) (99). In addition, HMBA and UV light activate Ca+2-dependent phosphatases (PP2B and PP1α) which cause dephosphorylation of CDK9 on Thr186, releasing P-TEFb (100). Besides these phosphorylation-dephosphorylation mechanisms, acetylation of cyclin T1 on its C-terminal lysine residues (Lys, K380, 386, 390 and 404) by p300 further contribute to the release of P-TEFb (Figures 7B and 8) (101).
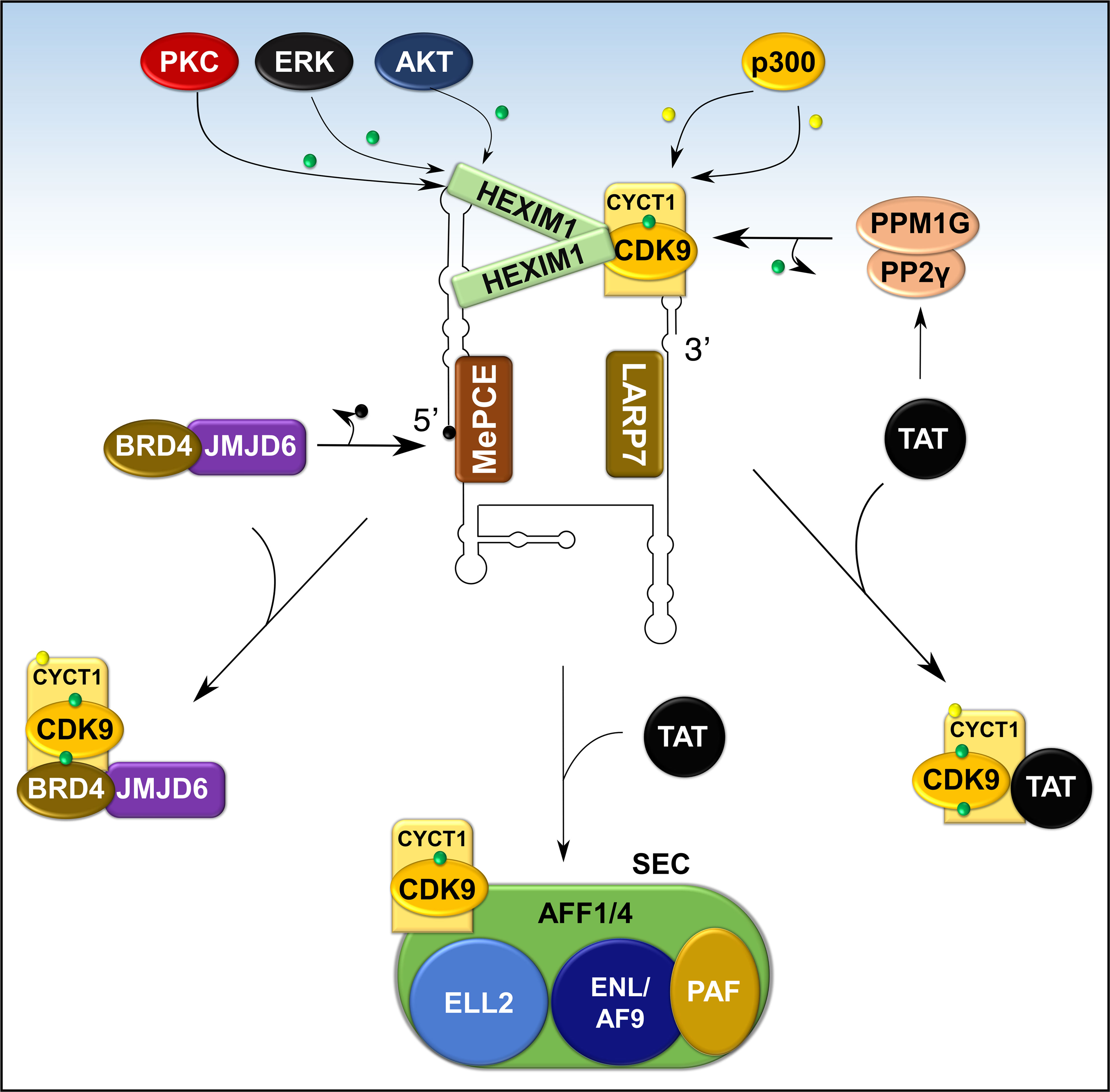
Figure 8 Recruitment of P-TEFb from 7SK snRNP. Various signaling pathways and stress conditions liberate P-TEFb from 7SK snRNP through PTMs of the components of 7SK snRNP (HEXIM1 and cyclin T1) or direct recruitment (BRD4, TAT, or super elongation complex, SEC). The major PTM involves phosphorylation (green dot) of HEXIM1 in various residues and acetylation (yellow dot) of cyclin T1. TAT recruits P-TEFb by collaborating with phosphatases (PPM1G and PP2γ) which dephosphorylate CDK9 on Thr186. Meanwhile, BRD4 interacts with JMJD6, a histone demethylase, which demethylates (black dot) the 5’ end of 7SK RNA and destabilizes the inhibitory core. AFF1/4, ALL-fused gene from chromosome 1/4 family member; ELL2, Eleven-nineteen lysine-rich in leukemia; ENL, Eleven nineteen leukemia; AF9, ALL-fused gene from chromosome 9.
In addition to intracellular signaling, transcriptional factors can also directly release P-TEFb from 7SK snRNP. The HIV1 TAT activates HIV transcription by hijacking P-TEFb from 7SK snRNA through a high-affinity competition with HEXIM1 for interaction with cyclin T1 (102). To complement this direct effect, TAT also recruits protein phosphatases PPM1G/PP2Cγ to the HIV promoter to dephosphorylate CDK9 on Thr186 to enhance the release of P-TEFb (Figure 8) (96). In normal cellular transcription, the release of P-TEFb from 7SK snRNP depends on BRD4 (27, 103). BRD4 is a ubiquitously expressed nuclear protein that recognizes acetylated histone during active transcription (Figure 5) and serves as an adaptor for recruiting key transcription factors (104). During active transcription, acetylated histone-bound BRD4 recruits an arginine histone demethylase, JMJD6 (105), to chromatin-anchored 7SK snRNP on anti-pause enhancers (A-PE). Here, JMJD6 releases P-TEFb from 7SK snRNP by demethylating both H4R3me and the 5’ hairpin of 7SK RNA, which dissociates 7SK snRNP from chromatin and exposes 7SK RNA for degradation (Figure 8) (95). Once released, both BRD4 and JMJD6 interact with respective cyclin T1 and CDK9 (Figures 7A, B), through their P-TEFb interacting domains [PID; BRD4, residues 1209-1362 and JMJD6, residues 1-305 (95, 106)], and deliver P-TEFb to poised RNAP II.
A small fraction of active P-TEFb can also be found in the Super Elongation Complex (SEC), a multicomponent, potent transcription activator (Figure 8). The components of SEC are known fusion partners for Mixed Lineage Leukemia (MLL) and this multiplex is actively recruited by TAT during HIV1 replication (107, 108). Components of SEC such as AF9, AFF1/4, ENL, ELL interact directly with P-TEFb (also see section on hematological malignancies below) and this interaction is increased by TAT. Besides BRD4 or SEC, other transcription factors such as NFκB might directly or indirectly deliver P-TEFb to target genes (96, 107).
Once P-TEFb is extracted by BRD4/JMJD6 or other transcription factors, 7SK snRNA is stabilized by binding to heterogeneous nuclear ribonucleoproteins (hnRNPs A1, A2/B1, R, and Q; Figure 6) (109, 110). The exact mechanism for the reassembly of the 7SK snRNP is not known but it is proposed that upon termination of transcription, hnRNPs are recruited by the nascent mRNA, thereby releasing 7SK snRNA to reassemble with P-TEFb and HEXIM1 (109, 110).
Regulation of P-TEFb by Post-Translational Modifications
Besides sequestration in an inhibitor complex, the biological activity of P-TEFb is further controlled by post-translational modifications directed toward CDK9 and cyclin T1. These modifications, which include phosphorylation, acetylation, and ubiquitination of CDK9 and/or cyclin T1, increase or decrease the activity of P-TEFb.
Regulation by Phosphorylation
Among the various modifications documented, phosphorylation of several Ser and Thr residues of CDK9 and its cyclin T1 partner plays a key regulatory function (Figure 7B). Phosphorylation of a conserved Thr186 residue in the T-loop structure of CDK9 (Figure 2) is necessary for its enzymatic activity (88, 92). This phosphorylation triggers a conformational change in the CDK9-cyclin T1 heterodimer exposing the ATP and substrate binding sites (22). Moreover, as described above, binding of P-TEFb to the components of 7SK snRNP is also dependent on the phosphorylation of CDK9 at Thr186 (pThr186), indicating that the inhibitory complex serves as a pool for efficient and prompt release of active P-TEFb in response to stress conditions (92, 100). Initially, autophosphorylation was described as the main mechanism for the formation of pThr186, based on an in vitro kinase assay employing purified CDK9-cyclin T1 complex (22). Recent evidence, however, revealed that CDK7, a CDK-activating kinase (CAK) for various cell cycle CDKs, is responsible for forming pThr186 (111). Besides CDK7, a global search for other kinases responsible for forming pThr186 indicated that siRNA knockdown of Ca2+/calmodulin-dependent kinase 1D decreases the level of pThr186, although a direct link for this role was not established (112).
Once P-TEFb dissociates from the inhibitory complex, CDK9 is phosphorylated on a second highly conserved T-loop residue, Ser175 (Figure 7B) (103). CDK9 carrying pSer175 is found exclusively outside of the 7SK snRNP complex and this phosphorylation step promotes the binding of P-TEFb with BRD4 and/or TAT (103, 113). It was proposed that pSer175 induces a conformational change in P-TEFb favoring the interaction of cyclin T1 with BRD4 (103). Recently, an in vitro kinase assay and cellular experiments employing THZ1, a highly selective covalent CDK7 inhibitor, have identified CDK7 (as part of CAK) as the kinase that phosphorylates CDK9 on Ser175 (114). The direct contribution of pSer175 to the kinase activity of CDK9 is not clearly established. This comes from various findings showing that a mutation of Ser175 to alanine renders CDK9 inactive in an in vitro kinase assay (88), while at the same time, it increased TAT-dependent HIV1 transcription at the cellular level (113). Furthermore, mutation of Ser175 to aspartic acid, mimicking the phosphorylated Ser, failed to activate both CDK9 kinase activity in vitro (88) and TAT-dependent HIV1 transcription in vivo (113).
The activity of CDK9 is reduced by phosphorylation of an N-terminal Thr29 (115), a modification that is homologous to inhibitory phosphorylation of CDK2 on Thr15 (116). Surprisingly, BRD4 and TAX [transactivator of Human T-lymphotropic Virus Type 1 (HTLV-1)], which recruit P-TEFb for basal and HTLV-1 transcriptions, respectively, induced phosphorylation of CDK9 on Thr29 (Figure 7B) (115, 117). This phosphorylation mainly occurs in the pre-initiation complex, following recruitment of CDK9 by either BRD4 or TAX, and is necessary for limiting the function of CDK9 during initiation of transcription (117). Following succession from transcription initiation to elongation, PP2A dephosphorylates pThr29 (Figure 7B) and activates the positive transcription elongation activity of CDK9 (117).
Additional phosphorylation of CDK9, vital mainly for the activation of HIV1 replication, includes phosphorylation of a cluster of C-terminal Ser/Thr residues (Ser347, Ser353, Ser357; Thr350, Thr354) and an N-terminal Ser90 residue (Figure 7B) (118–120). Autophosphorylation of the C-terminal residues of CDK9 and cyclin T1 increases the binding of the TAT-P-TEFb complex to the TAR RNA (118, 119). In addition, phosphorylation of CDK9 on Ser90 by CDK2 positively contributes to HIV1 replication (120). Besides the above-mentioned residues, several other phosphorylated residues of CDK9 with unknown function, were identified by mass spectrometry (121).
Regulation by Acetylation
Acetylation of both CDK9 and cyclin T1 is another important post-translational modification that regulates the biological function of P-TEFb (Figure 7B). Namely, acetylation of four lysine residues within the coiled-coil region of cyclin T1 (Lys380, Lys386, Lys390, and Lys404) induces the dissociation of P-TEFb from 7SK snRNP and is found exclusively in the active P-TEFb complex (101). Acetylation of these residues is mediated by the histone acetyl-transferase (HAT), p300, which liberates P-TEFb from HEXIM1 and 7SK snRNA (Figures 7B and 8) (101). Consequently, acetylated cyclin T1 binds the second bromodomain (BDII) of BRD4 and supports the recruitment and transcriptional activity of P-TEFb (122). In contrast to normal cellular transcription, the HIV1 TAT protein has evolved the ability to recruit the non-acetylated cyclin T1 directly from the inhibitory complex to activate HIV1 transcription (102).
In addition to cyclin T1, CDK9 is also acetylated on two N-terminal lysine residues, namely Lys44 and Lys48, by p300 and GCN5 (Figure 7B) (123, 124). While p300 and GCN5 acetylate both lysine residues, Lys44 is the preferred site for p300 while GCN5 mainly targets Lys48 (123, 124). Contrary findings have been reported regarding the outcome of CDK9 acetylation on its kinase and transcriptional activity (123, 124). On the one hand, Fu et al. reported that acetylation of CDK9 on Lys44 increases its activity as the mutation of Lys44 to Arginine or overexpression of histone deacetylases (HDAC1 and 3, directed towards Lys44) markedly impaired CDK9’s kinase and transcriptional activities (123). On the other hand, Sabo et al. showed that acetylation of CDK9 inhibits the binding of ATP to CDK9 and thus hinders its kinase activity (124). Recently, the latter finding was supported by a study showing that by counteracting GCN5-mediated Lys48 acetylation with sirtuin7, a nicotinamide adenine dinucleotide (NAD)-dependent deacetylase, there was an increased transcriptional activity of P-TEFb (125).
Regulation by Ubiquitination
Polyubiquitination of CDK9 and its negative regulator HEXIM1 provides an additional mechanism for the regulation of P-TEFb (Figures 7A, B) (29, 126). Recruitment of the SCF E3 ubiquitin ligase core components (S-phase kinase-associated protein 1 (SKP1), cul-1, and p45SKP2) by cyclin T1 through its C-terminal PEST domain (residues 709-726) mediates polyubiquitination and subsequent degradation of CDK9 by proteasomes (29). Since the protein level of CDK9 does not change in a manner akin to kinases regulating the cell cycle (59), the functional relevance of proteolytic degradation of CDK9 to its regulation is not clear. Interestingly, contrary to the well-known function of ubiquitination, HIV1 transactivation by TAT is increased by the ubiquitination of CDK9 which facilitates the formation of a ternary complex between P-TEFb, TAT, and TAR RNA (127). TAT also recruits the UBE2O ubiquitin ligase in the cytoplasm to ubiquitinate HEXIM1 in a non-degradative manner (128). This ubiquitination step releases HEXIM1 from 7SK snRNP and liberates P-TEFb for transport from the cytoplasmic pool to the nucleus (128). Similarly, ubiquitination of HEXIM1 by human double minute-2 protein (HDM2, Figure 7A), a p53-specific E3 ubiquitin ligase, does not lead to proteasome-mediated degradation, but instead increases its sequestering and thus inhibition of P-TEFb, suggesting a role of ubiquitination beyond proteasome-mediated degradation (126).
The Role of P-TEFb in Cancer
A plethora of genetic aberrations have been discovered as underlying causes for blood and solid cancers. Despite the overwhelming amount of known cancer-causing mutations, most tumors are reliant on continuously activated gene expression. Therefore, it does not come as a surprise that many studies have been conducted to highlight the link between P-TEFb and most known types of cancer. Here, we first focus comprehensively on blood cancers since the rationale for the use of CDK9 inhibitors can be best supported with known genetic aberrations and gene mutations underlaying these diseases. We also highlight the potential role of P-TEFb in solid tumors using breast, prostate, and hepatocellular cancers as examples. Table 1 provides a summary of the studies providing links between P-TEFb and a range of other cancers as well as those discussed in more detail below.
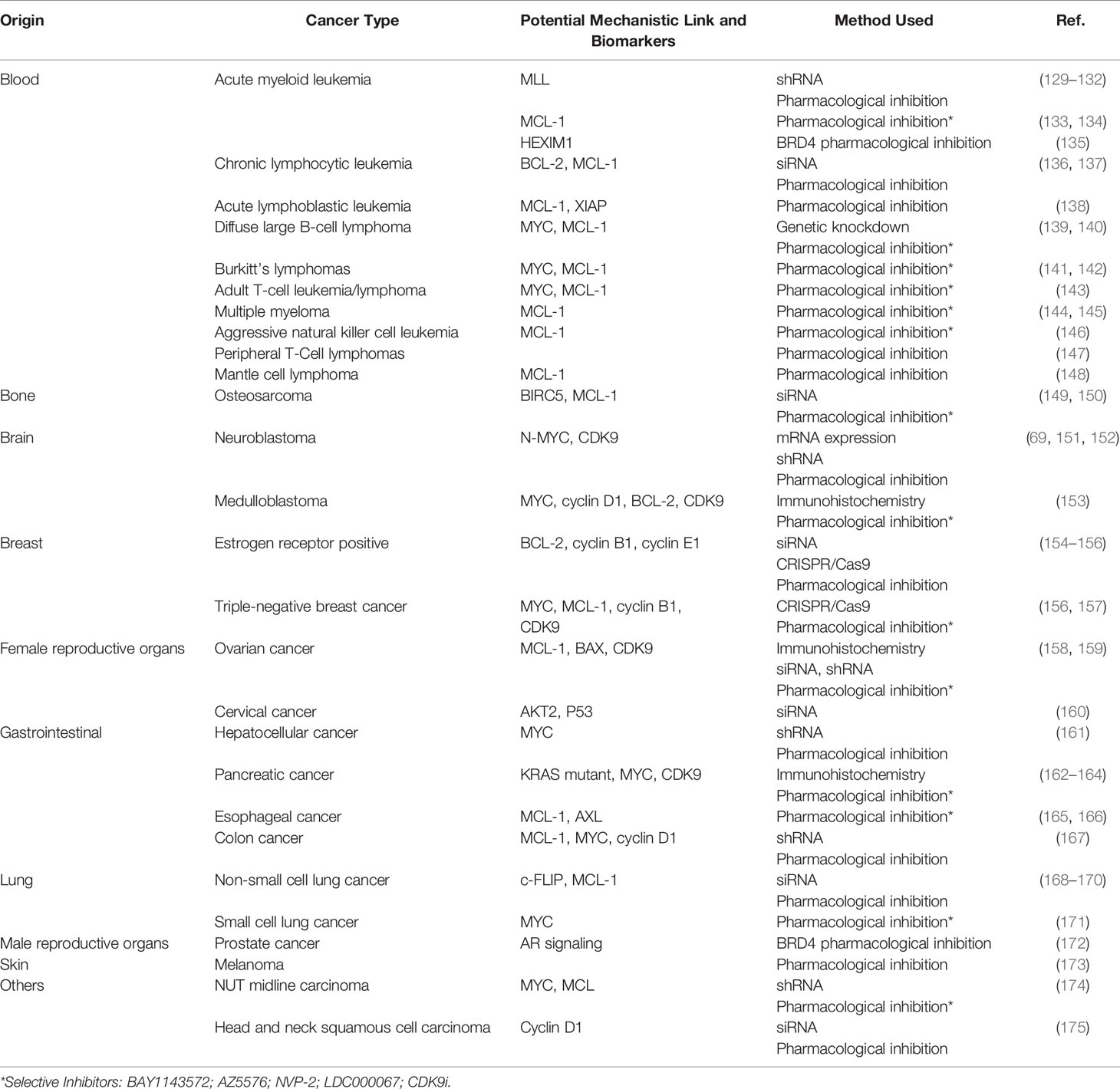
Table 1 Studies that have described links between P-TEFb and different cancers.
Hematological Malignancies
P-TEFb in Leukemia
P-TEFb plays a well-recognized role in the pathogenesis of many hematological malignancies, such as leukemia. This is particularly true in leukemia harboring a chromosomal translocationw mutation on chromosome 11q23 (176). This loci encodes for a histone 3 lysine 4 methyltransferase protein, called mixed-lineage leukemia (MLL) (177). MLL is ubiquitously expressed in myeloid and lymphoid progenitor cells and increases the expression of a cluster of HOXA homeobox genes (e.g. HOXA7 and 9) and the gene for the HOX-dimerization partner, MEIS1 (177). These genes control self-renewal of hematopoietic stem cells and are downregulated during hematopoietic differentiation (178). For reasons not clearly defined, chromosome 11 frequently undergoes an in-frame translocation mutation at the locus 11q23, where the 5’ end of MLL (containing its target gene binding motifs) is fused with the 3’ end of a wide variety of unrelated partner genes, generating chimeric MLL fusion proteins (Figure 9) (179). These fusion proteins are aberrant transcription factors that increase the expression of HOXA and MEIS1 genes, leading to a pre-leukemic state by blocking hematopoietic differentiation (177). The actively proliferating pre-leukemic progenitor cells are highly susceptible to secondary mutations (e.g., mutation in the fms-like tyrosine kinase (FLT3) receptor) which aids in their transformation into acute leukemia (180).
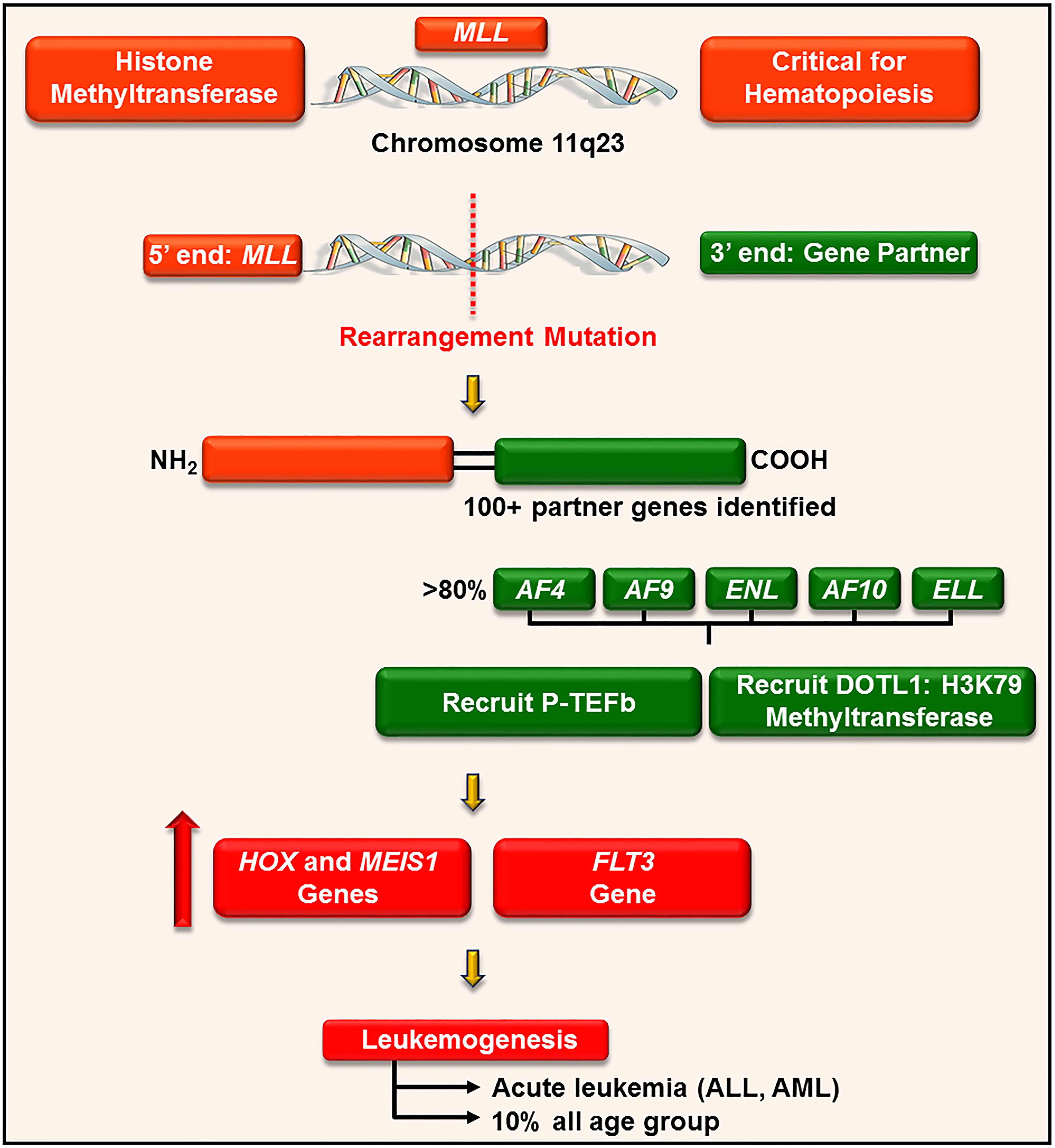
Figure 9 P-TEFb is required for the MLL transcription program and leukemogenesis. MLL is a histone methyltransferase ubiquitously expressed in hematopoietic progenitor cells and plays a key role in their self-renewal. For unknown reasons, MLL gene (on chromosome 11q23) undergoes a trans-locational mutation where its 5’ end is fused with the 3’ end of numerous genes. The majority of these partner genes are nuclear transcription factors that recruit P-TEFb and DOTL-1 leading to upregulated expression of HOX, MEIS1, and FLT3. These proteins drive leukemogenesis by blocking differentiation and driving active proliferation.
MLL rearrangement leukemia has a poor prognosis and accounts for approximately 10% of all cases of acute leukemia in humans, irrespective of age. Specifically, MLL fusion is associated with > 70% of infantile acute lymphoblastic leukemia (ALL), 35 – 50% of infant acute myeloid leukemia (AML), and 1-15% of therapy-related leukemia (e.g. patients treated with topoisomerase II inhibitors) (177).
MLL is involved in more than 100 different rearrangements and 64 translocation partner genes have been identified (Figure 9) (179). Despite a large number of partners, only nine proteins account for more than 90% of MLL rearrangements, namely AF4, AF9, ENL, AF6, AF10, ELL, AF1p, AF17, and SEPT6 (179). The majority of these translocation partners collectively associate to form a large macromolecule called ENL-interacting proteins or elongation-assisting proteins (EAP) (181). EAP consist of three major components (1): SEC (e.g. AF4, AF9, ENL, and AFF4) which recruits P-TEFb and other elongation factors (e.g. ELL1-3, Figure 8) to activate transcription elongation (2, 176), Dot1-containing complex (DotCom) which methylates lysine 79 on histone 3 through the activity of disruptor of telomeric silencing 1-like (DOT1L) (182), and (3) MLL fusion partner ENL, which forms a scaffold for the multiprotein complex and inhibits negative regulators of MLL (e.g. polycomb proteins) (183). These combined effects lead to continuous ectopic expression of HOXA genes and finally contribute to leukemogenesis. In line with these roles, pharmacological inhibition of CDK9 (by flavopiridol, dinaciclib, and CDKI-73) significantly delay disease progression and improve survival in mouse models of MLL-ENL and MLL-AF9, respectively (129, 130, 184).
Leukemia harboring MLL fusions possess a high level of expression and frequent activating mutations in the FLT3 gene (185, 186). FLT3 is a membrane-bound receptor tyrosine kinase that regulates the survival, proliferation, and differentiation of hematopoietic stem cells (187). Stimulation of FLT3 by its cognate ligand activates signal transduction networks, mainly through PI3K/AKT and mitogen-activated protein kinase (MAPK) pathways (187). FLT3 is mutated and constitutively active in 30% of AML, and the most common mutation, known as internal tandem duplication (ITD), involves an in-frame duplication of short sequences in the juxtamembrane domain of the receptor. ITD thwarts an auto-inhibitory mechanism built into the FLT3 wild type (WT) receptor leading to dimerization and constitutive activation of its signaling despite the absence of a ligand (187). Moreover, the co-occurrence of MLL fusion (MLL-AF9) and FLT3-ITD was found to accelerate the onset of AML in a mouse model, suggesting cooperation of the two oncogenes in leukemogenesis (188). This highlights the role played by P-TEFb for the transcription of MLL-fusion genes in leukemic cells harboring both MLL fusion and FLT3-ITD.
Besides its role in MLL, P-TEFb is also involved in the pathogenesis of AML and chronic lymphocytic leukemia (CLL) through its direct role in myeloid cell leukemia 1 (MCL-1) transcription. High expression of MCL-1 is responsible for the development and survival of AML cells (189). Evaluation of clinical AML samples (e.g. leukemic blasts and primary human hematopoietic subsets) indicated high MCL-1 expression (190). In addition, MCL-1 is also upregulated in 50% of recurrent AML cases and is linked with a poor prognosis and/or response to chemotherapy (191). The continuous survival state of leukemic blasts mediated by high MCL-1 expression requires constant activation of P-TEFb due to the short half-life of MCL-1 (133, 134). Therefore, CDK9 inhibition dramatically reduces MCL-1 expression in AML and produces strong anti-leukemic activity in AML animal models (134, 192). These observations have led to the current clinical trials investigating the value of CDK9 inhibitors (e.g., BAY1143572 and CDKI-73) for AML treatment (193).
CLL is the most common leukemia in elderly patients and is characterized by the accumulation of mature but functionally impotent B-lymphocytes in blood, bone marrow, and lymph nodes (194). Relative to other hematological malignancies, CLL is a non-proliferative form of leukemia associated with constitutive activation of the B-cell receptor signaling pathway and overexpression of the B-cell lymphoma 2 (BCL-2) family of anti-apoptotic proteins (e.g., BCL-2, MCL-1) (195, 196). The dependence of CLL on P-TEFb-mediated transcription was confirmed by the apoptotic effects on CLL cells of inhibiting CDK9 by pharmacological (e.g., flavopiridol, dinaciclib, and CDKI-73) and biological means (siRNA) (136, 197, 198). This reliance was further established by the strong anticancer effect of both flavopiridol and dinaciclib in refractory CLL patients in phase II/III clinical trials (137, 199–201).
P-TEFb in Other Hematological Malignancies
P-TEFb has also been implicated in the development of lymphoma, particularly diffuse large B-cell lymphoma. This is a non-Hodgkin lymphoma characterized by malignant and diffuse proliferation of large B lymphocytes (202). Dysregulation of c-MYC is essential in its pathogenesis and involves either a c-MYC rearrangement mutation, most commonly involving its translocation into heavy- or light-chain immunoglobulin loci (5-15%), or gene amplification (30-50%). These changes often confer aggressive clinical courses (202). As a general transcription factor, MYC directly interacts and recruits P-TEFb to its promoter and other target genes to mediate RNAP II pause release (35). Inhibition of CDK9 by selective inhibitors (e.g., AZ5576) or by genetic knockdown negatively regulated MYC and MCL-1 expression, and induced apoptosis in primary and transformed cells from this lymphoma, providing an attractive therapeutic strategy (139, 140).
Furthermore, dysregulation of MYC and MCL-1 transcription is associated with the development of adult T-cell leukemia/lymphoma, an aggressive proliferation of mature T lymphocytes transformed by HTLV-1 (143). Similar to diffuse large B-cell lymphoma, cells from this leukemia/lymphoma are highly susceptible to the apoptotic effect of selective pharmacological inhibition of CDK9 (143). Other rare hematological malignancies such as multiple myeloma, aggressive natural killer cell leukemia, and peripheral T-Cell lymphomas have also been found responsive to CDK9 inhibitors (144, 146, 147).
Solid Cancers
P-TEFb in Breast Cancer
There are three major subtypes of breast cancer, based on the presence or absence of receptors for estrogen (ER+/-), progesterone (PR+/-) and human epidermal growth factor receptor 2 (HER2+/-). The majority of patients (70%) are diagnosed with ER+/PR+/HER2- cancers, followed by HER2+ (15-20%), and ER-/PR-/HER2- triple-negative (15%) cancers (203). These subtypes have different risk profiles, treatment strategies, and clinical outcomes (203). Besides this classification, genetic profiling has been used to identify different breast cancer subtypes having diverse clinical courses (204). Regardless of these genetic heterogeneities, breast cancers are reliant on continuously activated gene expression programs that are dependent on P-TEFb. For instance, the survival and proliferation of ER+ breast cancers are dependent on overexpression of the MYB proto-oncogene, a transcription factor which is a direct target for ER signaling, and regulator of BCL-2, cyclin B1, and cyclin E1 (154, 205). MYB transcription is regulated by ER-mediated recruitment of P-TEFb to transcription pause sites (205). In addition, breast cancers have evolved the capability of downregulating a tumor-suppressive microRNA (miRNA), miR-874, which suppresses proliferation by downregulating CDK9 expression (155). Apart from a direct mechanistic role, P-TEFb-mediated overexpression of MYC is associated with ER-independent growth in breast cancers resistant to hormone therapy (206). These roles are reinforced by the induction of apoptosis and inhibition of cell growth in both hormone therapy-sensitive and -resistant ER+ breast cancer cell lines (e.g., MCF-7) as a consequence of CDK9 inhibition by biological or pharmacological means (154, 155, 206).
Relative to ER/PR+ and HER2+ breast cancers, triple-negative breast cancers (TNBCs) are more aggressive subtypes with a higher frequency of relapses (207). The high level of inherent genetic heterogeneity in TNBCs and consequent lack of unifying molecular alterations has created a challenge for targeted therapy (207). Despite this heterogeneity, however, the gene expression profiles of TNBCs display a uniform trait of activated transcription of a cluster of TNBC-specific genes (156). These clusters are of transcriptional regulators and signal transducers, such as epidermal growth factor receptor (EGFR), Fos-related antigen 1, Forkhead box C1, MYC, and SOX9. Most are associated with large enhancer regions occupied by multiple transcriptional factors, called super-enhancers. As a result, these genes are exceptionally active and sensitive to inhibitors targeting key regulators of transcription (e.g., CDK7 by THZ1 and CDK9 by BAY1143572 and dinaciclib) (156, 157, 208). Clinically, high CDK9 expression in TNBC patients renders comparatively poor overall survival (157).
P-TEFb in Prostate Cancer
Androgen receptor (AR) signaling regulates the expression of genes vital for the growth, differentiation, and survival of prostate cells (209). The AR interacts directly with P-TEFb or its recruiter protein, BRD4, to mediate expression of its target genes (e.g., prostate-specific antigen, PSA) (172, 210). Moreover, P-TEFb phosphorylates AR on Ser81 and influences its chromatin binding, nuclear localization, and transcriptional activity (211, 212). These functions are overtly activated in prostate cancer cell lines and primary clinical samples and abrogated by non-selective CDK9 inhibitors (e.g., flavopiridol, roscovitine) (212–214).
Androgen deprivation has been the mainstay of treatment for advanced prostate cancers, but inevitably the disease relapses leading to a castration-resistant form (CRPC). In these phenotypes, AR signaling is still maintained via diverse mechanisms such as AR amplification or constitutively active AR splicing variants (215). Several lines of evidence point to P-TEFb involvement in AR signaling of CRPC. A group of AR-regulated enhancer RNAs (small non-encoding RNAs) are upregulated and interact with P-TEFb to promote the growth of CRPC (216). In addition, BRD4 interacts directly with and recruits the AR to target genetic loci that drive the proliferation of CRPC (172). Correspondingly, MYC, a critical downstream transcriptional target of BRD4/P-TEFb, was shown to be responsible for the overexpression of full-length AR and AR splice variants in CRPC patient samples (217). Besides MYC, the upregulation of MCL-1 in CRPC is responsible for the androgen-independent survival of CRPC (218). As an outcome, inhibition of the dysregulated AR transcription in CRPC through specific BRD4 inhibitors (e.g., JQ1) produces strong suppression of cellular proliferation (172, 219), although the direct consequence of P-TEFb inhibition in CRPC has not been established yet.
P-TEFb in MYC-Dependent Hepatocellular Cancer
Hepatocellular cancer is an aggressive, highly lethal (< 1-year survival rate in the advanced stage), and frequent type of primary liver cancer which originates from a series of genetic and epigenetic events following chronic liver diseases (220). Genomic studies identified MYC amplification and TP53 inactivation as frequent genetic alterations (221, 222). Gene silencing using a shRNA library identified the requirement of CDK9 for the sustained proliferation of hepatocellular cancer cells and their dependence on MYC (161). This finding was reinforced by the antitumor effects arising from shRNA-mediated and pharmacological (e.g., PHA-767491) inhibition of CDK9 in murine and human cell lines driven by MYC. Furthermore, the silencing of CDK9 inhibited MYC-dependent liver tumorigenesis in a mouse model and suppressed the proliferation of xenografts of murine and human hepatocellular cancer cells (161). A high level of CDK9 expression concurrent with a downregulation of miRNA-206, an inhibitor of translation from CDK9 mRNA, was noted in hepatocellular cancer cell lines (223).
Inhibitors of CDK9 as Therapeutic Agents for Cancer
The discovery of flavopiridol as the first clinical CDK inhibitor, launched a race for the discovery of alternative small molecules with more potent and selective CDK9 inhibition, and some have entered clinical trials for treating solid and hematological malignancies. These inhibitors are competitive at the highly conserved catalytic ATP binding site (224), and as a consequence, they tend to target multiple CDKs and/or protein kinases, rendering them less attractive for use as therapeutic agents and chemical probes (225). Nevertheless, several inhibitors have been developed with improved selectivity towards CDK9, but only limited data are available regarding their broader selectivity profile and pharmacological properties.
First Generation CDK9 Inhibitors
Flavopiridol (Alvocidib) was the first pan-CDK inhibitor to enter clinical trials with half maximum inhibitory values (IC50) below 400 nM against CDKs 1, 2, 4, 6, 7, and 9 (136, 226, 227). Initially, the anticancer mechanisms were attributed to the arrest of cells at the G1 and G2/M phases of their cycle through inhibition of CDK4/6 and CDK1, respectively. Later, the primary mechanism of action was ascribed to downregulation of cell cycle- and apoptosis-related genes via inhibition of CDKs 7 and 9 (136, 228). Flavopiridol showed inadequate efficacy relative to its toxicity when tested clinically against various solid and hematological malignancies, either as a single agent or in combination with other anticancer agents. Regardless of these outcomes, timed sequential combinations with cytarabine and mitoxantrone reached complete remission (CR) rates of 36-68% in relapsed/refractory (R/R) AML and newly diagnosed poor-risk AML (229). As a result, orphan drug designation has been assigned for the treatment of AML patients.
Seliciclib (roscovitine/CYC202) was the second pan-CDK inhibitor to enter clinical trials. It inhibited CDK9 with lower potency compared to flavopiridol (IC50 = 950 vs. 7 nM, respectively), but with improved selectivity (230). Seliciclib was more potent against CDKs 2, 5, and 7 (IC50 = 100, 160, and 490 nM, respectively) than CDK9 (230), and demonstrated anticancer activities in numerous preclinical cancer models by inducing cell cycle arrest and apoptosis (231, 232). Unfortunately, these effects could not be translated into clinical use as a kinase inhibitor mono-therapy, due to limited efficacy and a myriad of toxicities (233). Currently, combination trials of seliciclib with sapacitabine in BRCA-mutant solid tumors are ongoing.
Dinaciclib (SCH 727965) inhibits CDKs 1, 2, 5, and 9 with a similar potency (IC50 = 3, 1, 1, and 4 nM, respectively), and displays better selectivity for CDKs relative to other protein kinases (234). The compound induces cell cycle arrest and apoptosis in multiple cancer cell lines representing a broad range of cancer types and showed in vivo antitumor efficacy after intraperitoneal administration in tumor xenograft models (130, 136, 141, 151, 162, 208). Dinaciclib was well tolerated in phase I clinical trials when administered weekly for 3 weeks, leading to its progression into phase II trials against solid cancers (235). In subsequent breast and lung cancer trials, dinaciclib did not perform better than comparator agents, resulting in premature termination (236, 237). Encouraged by the positive outcome of flavopiridol in CLL patients (199), dinaciclib was investigated in relapsed and refractory CLL and 54% of patients showed a partial response with limited side-effects (e.g. cytopenia and tumor lysis syndrome) (137). Hence, dinaciclib progressed to a phase III trial with the anti-CD20 monoclonal antibody ofatumumab as comparator (200). Although dinaciclib did demonstrate efficacy and was tolerated, the phase III trial was terminated early (due to reasons unrelated to safety and efficacy), precluding definitive conclusions (200). Besides CLL, dinaciclib has also been trialed as a single agent in patients with relapsed multiple myeloma having a partial response rate of 11% (238). Currently, clinical trials are investigating the efficacy of dinaciclib in combination with a BCL-2 inhibitor (venetoclax) for R/R AML (NCT03484520), immunotherapy (pembrolizumab) for R/R hematological malignancies (NCT02684617), and a Poly (ADP-ribose) polymerase (PARP) inhibitor (veliparib) against solid cancers (NCT01434316).
Over the years, numerous other small molecule CDK9 inhibitors have been discovered (e.g., CDKI-73, TG02) (198, 239). These compounds are currently either in preclinical development or in the early stages of clinical trials. Supplementary Table 1 provides detailed information on the development stages of various CDK9 inhibitors.
Second Generation CDK9 Inhibitors
BAY1143572 (atuveciclib), a benzyl sulfoximine, is one of the most selective and potent CDK9 inhibitors (IC50 = 6 nM) currently being evaluated in clinical trials (192). The compound inhibited the proliferation of cancer cell lines at sub-micromolar concentrations and suppressed the growth of subcutaneous xenograft models of AML (134, 192), TNBC (157), lymphoma (143, 146), and esophageal (165) cancer. BAY1143572 inhibited the phosphorylation of RNAP II CTD on Ser2, downregulated MCL-1 and MYC, and induced apoptosis. Based on these findings, BAY1143572 is being evaluated in two phase I clinical trials involving patients with advanced cancers (gastric cancer, TNBC, and DLBCL; NCT01938638) and acute leukemias (NCT02345382), with results yet to be reported. In a follow-up lead optimization, a related compound, BAY1251152, was identified with increased potency (IC50 for CDK9 = 3 nM) and solubility to allow intravenous (IV) administration (240). BAY1251152 demonstrated antitumor efficacy in AML xenograft models and progressed to phase I clinical evaluation (240). BAY1251152 was administered once weekly for 3 weeks as a 30 minute IV infusion to patients with metastatic solid cancers or aggressive non-Hodgkin lymphoma and caused the disease to stabilize in 12 out of 31 patients with a manageable safety profile (NCT02635672) (241). This compound is also currently under investigation in patients with advanced hematological malignancies (phase I, NCT02745743).
AZD4573 is a potent inhibitor of CDK9 (IC50 < 4 nM) having more than ten-fold selectivity for CDK9 over CDKs 1 – 7 (242). It downregulated MCL-1 and induced rapid apoptosis in a large panel of hematologic cancer cell lines after a short exposure (i.e., 6 h) (242). The compound led to the regression of subcutaneous tumor xenografts and disseminated models of AML after twice weekly dosing through the intraperitoneal route alone as single agent or in combination with venetoclax (242). Consequently, AZD4573 is currently being evaluated in a phase I clinical trial for patients with hematological malignancies (NCT03263637).
i-CDK9 is a compound of pico-molar potency with a 600-fold selectivity for CDK9 over other CDKs (243). Besides CDKs, i-CDK9 inhibits dual-specificity tyrosine-phosphorylation-regulated kinases (DYRK) 1A and 1B, although at a lower potency compared to CDK9. i-CDK9 reduced the phosphorylation of CTD on Ser2 and SPT5 of DSIF on Thr775, downregulated MCL-1, and induced apoptosis in HeLa cells. Consistent with CDK9 inhibition, chromatin immunoprecipitation followed by parallel DNA sequencing (ChIP-seq) indicated that greater than 50% of genes in HeLa cells contained RNAP II paused in their promoter-proximal regions following incubation with i-CDK9 (243). Of interest was a small group of genes including MYC, which displayed a rebound expression in the presence of i-CDK9 prior to complete suppression of CTD Ser2 phosphorylation. This paradox was ascribed to a compensatory release of CDK9 from 7SK snRNP by BRD4 (Figure 5), which was abrogated by a combined inhibition of CDK9 and BRD4 (using JQ1). Due to its poor pharmacological properties, i-CDK9 has not progressed into clinical testing.
NVP-2 is an aminopyrimidine based inhibitor and a chemical analogue of i-CDK9 which potently and selectively inhibits CDK9 (IC50 = 0.5 nM) (244). NVP-2 displayed antiproliferative activity against numerous leukemia cell lines, associated with downregulation of MCL-1 and induction of apoptosis. RNA sequencing and ChIP-seq analysis have shown that NVP-2 downregulated a large percentage of total mRNA in MOLT4 cells and increased the localization of RNAP II near promoter-proximal regions (244). Concurrently, the same research team also described a novel method of achieving selectivity by linking SNS032 (a non-selective CDK9 inhibitor) to a thalidomide (THAL) moiety that recruits E3 ligase cereblon (CRBN) to catalyze the proteasomal degradation of CDK9. Through such a method, the specific degradation of CDK9 by THAL-SNS-032 was achieved, which resulted in prolonged antiproliferative activity and apoptosis compared to ATP-competitive inhibition (e.g., NVP-2, SNS032). The pharmacokinetic and pharmacodynamic characteristics of these compounds have not been reported yet.
Discussion
In summary, CDK9 along with cyclin T1 (constituting the P-TEFb complex) plays a key role in transcription by allowing RNAPII to facilitate the productive elongation of transcripts. Its roles extend beyond transcriptional elongation with functions in the cell cycle, differentiation, DNA repair, and transcriptional initiation and termination. Detailed structural characterization has revealed a conserved cyclin-dependent phosphor-transfer mechanism across CDKs, with some subtle differences in substrate recognition and cyclin binding for CDK9. P-TEFb activity is regulated by sequestering into an inactive complex and various post-translational modifications. Given its pivotal functions, when CDK9 becomes overactive in many hematological and solid cancers there is a continuous production of short-lived proteins that maintain the survival of cancer cells. This addiction to transcription makes cancer cells highly susceptible to the inhibition of CDK9 relative to non-transformed cells. Understanding the biology and function of CDK9 has advanced dramatically since its discovery in 1994 (13) and this has had a positive impact on the design and the use of specific inhibitors as a potential strategy for the treatment of several diseases. In line with this, several first generation CDK9 inhibitors have been developed and tested in clinical trials mostly in combination with conventional chemotherapeutic agents. Unfortunately, these investigational new drug entities have been hampered by severe adverse effects and to date none of them have made it to clinical approval (Supplementary Table 1).
Nevertheless, the authors predict that future development will be guided by insights into the molecular structure and function of CDK9, which will serve as the driving force for further improvements in the potency and specificity of novel inhibitors. Meanwhile, a more advanced understanding of its biology is likely to pave the way for establishing a sounder basis for the future value of CDK9 inhibitors for cancer therapy. One missing piece of knowledge in the CDK9 puzzle is a full validation of this target for cancer treatment. Experimental in vivo validation would be best assessed with CDK9-deficient mice. Unfortunately, knockout of CDK9 or its binding partner cyclin T2, is embryonically fatal to the mouse (245, 246). Designs for future studies might use conditional genetic knockout of the CDK9 or cyclin T1/2 genes in various established cancer models to provide more information about the role of the P-TEFb complex in tumor formation. The use of specific inhibitors of CDK9 as chemical probes may well be applied to finally confirm the outcomes from these sophisticated models. To this end, new hope has arisen in recent years from second generation inhibitors, with their much-improved specificity for CDK9 inhibition (Supplementary Table 1). Overall, a holistic understanding of the underlying CDK9 biology will be a prerequisite to optimizing the use of novel kinase inhibitors as mono and/or adjuvant therapies for the future treatment of various neoplastic disorders.
Author Contributions
AA: wrote the manuscript. HA, RM, and SW: conceptualized and critically revised the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.678559/full#supplementary-material
References
1. Cicenas J, Valius M. The CDK Inhibitors in Cancer Research and Therapy. J Cancer Res Clin Oncol (2011) 137(10):1409–18. doi: 10.1007/s00432-011-1039-4
2. Endicott JA, Noble ME, Johnson LN. The Structural Basis for Control of Eukaryotic Protein Kinases. Annu Rev Biochem (2012) 81:587–613. doi: 10.1146/annurev-biochem-052410-090317
3. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The Protein Kinase Complement of the Human Genome. Science (2002) 298(5600):1912–34. doi: 10.1126/science.1075762
5. Malumbres M, Barbacid M. Cell Cycle, CDKs and Cancer: A Changing Paradigm. Nat Rev Cancer (2009) 9(3):153–66. doi: 10.1038/nrc2602
6. Laub O, Jakobovits EB, Aloni Y. 5,6-dichloro-1-beta-ribofuranosylbenzimidazole Enhances Premature Termination of Late Transcription of Simian Virus 40 DNA. Proc Natl Acad Sci USA (1980) 77(6):3297–301. doi: 10.1073/pnas.77.6.3297
7. Chodosh LA, Fire A, Samuels M, Sharp PA. 5,6-Dichloro-1-beta-D-ribofuranosylbenzimidazole Inhibits Transcription Elongation by RNA Polymerase II In Vitro. J Biol Chem (1989) 264(4):2250–7. doi: 10.1016/S0021-9258(18)94169-4
8. Kephart DD, Marshall NF, Price DH. Stability of Drosophila RNA Polymerase II Elongation Complexes In Vitro. Mol Cell Biol (1992) 12(5):2067–77. doi: 10.1128/mcb.12.5.2067
9. Marshall NF, Price DH. Control of Formation of Two Distinct Classes of RNA Polymerase II Elongation Complexes. Mol Cell Biol (1992) 12(5):2078–90. doi: 10.1128/mcb.12.5.2078
10. Marshall NF, Price DH. Purification of P-TEFb, a Transcription Factor Required for the Transition Into Productive Elongation. J Biol Chem (1995) 270(21):12335–8. doi: 10.1074/jbc.270.21.12335
11. O’Brien T, Hardin S, Greenleaf A, Lis JT. Phosphorylation of RNA Polymerase II C-Terminal Domain and Transcriptional Elongation. Nature (1994) 370(6484):75–7. doi: 10.1038/370075a0
12. Marshall NF, Peng J, Xie Z, Price DH. Control of RNA Polymerase II Elongation Potential by a Novel Carboxyl-Terminal Domain Kinase. J Biol Chem (1996) 271(43):27176–83. doi: 10.1074/jbc.271.43.27176
13. Grana X, De Luca A, Sang N, Fu Y, Claudio PP, Rosenblatt J, et al. PITALRE, a Nuclear CDC2-related Protein Kinase That Phosphorylates the Retinoblastoma Protein In Vitro. Proc Natl Acad Sci USA (1994) 91(9):3834–8. doi: 10.1073/pnas.91.9.3834
14. Zhu Y, Pe’ery T, Peng J, Ramanathan Y, Marshall N, Marshall T, et al. Transcription Elongation Factor P-TEFb is Required for HIV-1 Tat Transactivation In Vitro. Genes Dev (1997) 11(20):2622–32. doi: 10.1101/gad.11.20.2622
15. Peng J, Marshall NF, Price DH. Identification of a Cyclin Subunit Required for the Function of Drosophila P-Tefb. J Biol Chem (1998) 273(22):13855–60. doi: 10.1074/jbc.273.22.13855
16. Peng J, Zhu Y, Milton JT, Price DH. Identification of Multiple Cyclin Subunits of Human P-Tefb. Genes Dev (1998) 12(5):755–62. doi: 10.1101/gad.12.5.755
17. Wei P, Garber ME, Fang SM, Fischer WH, Jones KA. A Novel CDK9-associated C-Type Cyclin Interacts Directly With HIV-1 Tat and Mediates its High-Affinity, Loop-Specific Binding to TAR Rna. Cell (1998) 92(4):451–62. doi: 10.1016/s0092-8674(00)80939-3
18. Hanks SK, Hunter T. Protein Kinases 6. The Eukaryotic Protein Kinase Superfamily: Kinase (Catalytic) Domain Structure and Classification. FASEB J (1995) 9(8):576–96. doi: 10.1096/fasebj.9.8.7768349
19. Jeffrey PD, Russo AA, Polyak K, Gibbs E, Hurwitz J, Massague J, et al. Mechanism of CDK Activation Revealed by the Structure of a cyclinA-CDK2 Complex. Nature (1995) 376(6538):313–20. doi: 10.1038/376313a0
20. De Bondt HL, Rosenblatt J, Jancarik J, Jones HD, Morgan DO, Kim SH. Crystal Structure of Cyclin-Dependent Kinase 2. Nature (1993) 363(6430):595–602. doi: 10.1038/363595a0
21. Russo AA, Jeffrey PD, Patten AK, Massague J, Pavletich NP. Crystal Structure of the p27Kip1 Cyclin-Dependent-Kinase Inhibitor Bound to the Cyclin A-Cdk2 Complex. Nature (1996) 382(6589):325–31. doi: 10.1038/382325a0
22. Baumli S, Lolli G, Lowe ED, Troiani S, Rusconi L, Bullock AN, et al. The Structure of P-TEFb (CDK9/Cyclin T1), its Complex With Flavopiridol and Regulation by Phosphorylation. EMBO J (2008) 27(13):1907–18. doi: 10.1038/emboj.2008.121
23. Echalier A, Endicott JA, Noble ME. Recent Developments in Cyclin-Dependent Kinase Biochemical and Structural Studies. Biochim Biophys Acta (2010) 1804(3):511–9. doi: 10.1016/j.bbapap.2009.10.002
24. Smith GK, Ke Z, Guo H, Hengge AC. Insights Into the Phosphoryl Transfer Mechanism of Cyclin-Dependent Protein Kinases From Ab Initio QM/MM Free-Energy Studies. J Phys Chem B (2011) 115(46):13713–22. doi: 10.1021/jp207532s
25. Napolitano G, Licciardo P, Gallo P, Majello B, Giordano A, Lania L. The CDK9-associated Cyclins T1 and T2 Exert Opposite Effects on HIV-1 Tat Activity. AIDS (1999) 13(12):1453–9. doi: 10.1097/00002030-199908200-00003
26. Garber ME, Wei P, KewalRamani VN, Mayall TP, Herrmann CH, Rice AP, et al. The Interaction Between HIV-1 Tat and Human Cyclin T1 Requires Zinc and a Critical Cysteine Residue That is Not Conserved in the Murine CycT1 Protein. Genes Dev (1998) 12(22):3512–27. doi: 10.1101/gad.12.22.3512
27. Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The Bromodomain Protein Brd4 is a Positive Regulatory Component of P-TEFb and Stimulates RNA Polymerase II-dependent Transcription. Mol Cell (2005) 19(4):523–34. doi: 10.1016/j.molcel.2005.06.027
28. Taube R, Lin X, Irwin D, Fujinaga K, Peterlin BM. Interaction Between P-TEFb and the C-terminal Domain of RNA Polymerase II Activates Transcriptional Elongation From Sites Upstream or Downstream of Target Genes. Mol Cell Biol (2002) 22(1):321–31. doi: 10.1128/mcb.22.1.321-331.2002
29. Kiernan RE, Emiliani S, Nakayama K, Castro A, Labbe JC, Lorca T, et al. Interaction Between Cyclin T1 and SCF(SKP2) Targets CDK9 for Ubiquitination and Degradation by the Proteasome. Mol Cell Biol (2001) 21(23):7956–70. doi: 10.1128/MCB.21.23.7956-7970.2001
30. Fu TJ, Peng J, Lee G, Price DH, Flores O. Cyclin K Functions as a CDK9 Regulatory Subunit and Participates in RNA Polymerase II Transcription. J Biol Chem (1999) 274(49):34527–30. doi: 10.1074/jbc.274.49.34527
31. Baumli S, Hole AJ, Noble ME, Endicott JA. The CDK9 C-Helix Exhibits Conformational Plasticity That may Explain the Selectivity of CAN508. ACS Chem Biol (2012) 7(5):811–6. doi: 10.1021/cb2004516
32. Sims RJ,3, Belotserkovskaya R, Reinberg D. Elongation by RNA Polymerase II: The Short and Long of it. Genes Dev (2004) 18(20):2437–68. doi: 10.1101/gad.1235904
33. Saunders A, Core LJ, Lis JT. Breaking Barriers to Transcription Elongation. Nat Rev Mol Cell Biol (2006) 7(8):557–67. doi: 10.1038/nrm1981
34. Zhou Q, Li T, Price DH. RNA Polymerase II Elongation Control. Annu Rev Biochem (2012) 81:119–43. doi: 10.1146/annurev-biochem-052610-095910
35. Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, et al. c-Myc Regulates Transcriptional Pause Release. Cell (2010) 141(3):432–45. doi: 10.1016/j.cell.2010.03.030
36. Adelman K, Lis JT. Promoter-Proximal Pausing of RNA Polymerase II: Emerging Roles in Metazoans. Nat Rev Genet (2012) 13(10):720–31. doi: 10.1038/nrg3293
37. Wada T, Takagi T, Yamaguchi Y, Watanabe D, Handa H. Evidence That P-TEFb Alleviates the Negative Effect of DSIF on RNA Polymerase II-dependent Transcription In Vitro. EMBO J (1998) 17(24):7395–403. doi: 10.1093/emboj/17.24.7395
38. Yamaguchi Y, Takagi T, Wada T, Yano K, Furuya A, Sugimoto S, et al. NELF, a Multisubunit Complex Containing RD, Cooperates With DSIF to Repress RNA Polymerase II Elongation. Cell (1999) 97(1):41–51. doi: 10.1016/s0092-8674(00)80713-8
39. Fujinaga K, Irwin D, Huang Y, Taube R, Kurosu T, Peterlin BM. Dynamics of Human Immunodeficiency Virus Transcription: P-TEFb Phosphorylates RD and Dissociates Negative Effectors From the Transactivation Response Element. Mol Cell Biol (2004) 24(2):787–95. doi: 10.1128/mcb.24.2.787-795.2004
40. Kim JB, Sharp PA. Positive Transcription Elongation Factor B Phosphorylates hSPT5 and RNA Polymerase II Carboxyl-Terminal Domain Independently of Cyclin-Dependent Kinase-Activating Kinase. J Biol Chem (2001) 276(15):12317–23. doi: 10.1074/jbc.M010908200
41. Yamada T, Yamaguchi Y, Inukai N, Okamoto S, Mura T, Handa H. P-TEFb-mediated Phosphorylation of Hspt5 C-terminal Repeats is Critical for Processive Transcription Elongation. Mol Cell (2006) 21(2):227–37. doi: 10.1016/j.molcel.2005.11.024
42. Shim EY, Walker AK, Shi Y, Blackwell TK. CDK-9/Cyclin T (P-TEFb) is Required in Two Postinitiation Pathways for Transcription in the C. Elegans Embryo. Genes Dev (2002) 16(16):2135–46. doi: 10.1101/gad.999002
43. Ni Z, Schwartz BE, Werner J, Suarez JR, Lis JT. Coordination of Transcription, RNA Processing, and Surveillance by P-TEFb Kinase on Heat Shock Genes. Mol Cell (2004) 13(1):55–65. doi: 10.1016/s1097-2765(03)00526-4
44. Czudnochowski N, Bosken CA, Geyer M. Serine-7 But Not Serine-5 Phosphorylation Primes RNA Polymerase II CTD for P-TEFb Recognition. Nat Commun (2012) 3:842. doi: 10.1038/ncomms1846
45. McCracken S, Fong N, Yankulov K, Ballantyne S, Pan G, Greenblatt J, et al. The C-terminal Domain of RNA Polymerase II Couples mRNA Processing to Transcription. Nature (1997) 385(6614):357–61. doi: 10.1038/385357a0
46. Harlen KM, Churchman LS. The Code and Beyond: Transcription Regulation by the RNA Polymerase II Carboxy-Terminal Domain. Nat Rev Mol Cell Biol (2017) 18(4):263–73. doi: 10.1038/nrm.2017.10
47. Gu B, Eick D, Bensaude O. CTD Serine-2 Plays a Critical Role in Splicing and Termination Factor Recruitment to RNA Polymerase II In Vivo. Nucleic Acids Res (2013) 41(3):1591–603. doi: 10.1093/nar/gks1327
48. Krogan NJ, Kim M, Tong A, Golshani A, Cagney G, Canadien V, et al. Methylation of Histone H3 by Set2 in Saccharomyces Cerevisiae is Linked to Transcriptional Elongation by RNA Polymerase II. Mol Cell Biol (2003) 23(12):4207–18. doi: 10.1128/mcb.23.12.4207-4218.2003
49. Li B, Howe L, Anderson S, Yates JR,3, Workman JL. The Set2 Histone Methyltransferase Functions Through the Phosphorylated Carboxyl-Terminal Domain of RNA Polymerase II. J Biol Chem (2003) 278(11):8897–903. doi: 10.1074/jbc.M212134200
50. Qiu H, Hu C, Gaur NA, Hinnebusch AG. Pol II CTD Kinases Bur1 and Kin28 Promote Spt5 CTR-Independent Recruitment of Paf1 Complex. EMBO J (2012) 31(16):3494–505. doi: 10.1038/emboj.2012.188
51. Devaiah BN, Lewis BA, Cherman N, Hewitt MC, Albrecht BK, Robey PG, et al. BRD4 is an Atypical Kinase That Phosphorylates Serine2 of the RNA Polymerase II Carboxy-Terminal Domain. Proc Natl Acad Sci USA (2012) 109(18):6927–32. doi: 10.1073/pnas.1120422109
52. Bartkowiak B, Liu P, Phatnani HP, Fuda NJ, Cooper JJ, Price DH, et al. CDK12 is a Transcription Elongation-Associated CTD Kinase, the Metazoan Ortholog of Yeast Ctk1. Genes Dev (2010) 24(20):2303–16. doi: 10.1101/gad.1968210
53. Liang K, Gao X, Gilmore JM, Florens L, Washburn MP, Smith E, et al. Characterization of Human Cyclin-Dependent Kinase 12 (CDK12) and CDK13 Complexes in C-terminal Domain Phosphorylation, Gene Transcription, and RNA Processing. Mol Cell Biol (2015) 35(6):928–38. doi: 10.1128/MCB.01426-14
54. Shao W, Zeitlinger J. Paused RNA Polymerase II Inhibits New Transcriptional Initiation. Nat Genet (2017) 49(7):1045–51. doi: 10.1038/ng.3867
55. Gressel S, Schwalb B, Decker TM, Qin W, Leonhardt H, Eick D, et al. CDK9-Dependent RNA Polymerase II Pausing Controls Transcription Initiation. Elife (2017) 6:1–24. doi: 10.7554/eLife.29736
56. Laitem C, Zaborowska J, Isa NF, Kufs J, Dienstbier M, Murphy S. CDK9 Inhibitors Define Elongation Checkpoints At Both Ends of RNA Polymerase II-transcribed Genes. Nat Struct Mol Biol (2015) 22(5):396–403. doi: 10.1038/nsmb.3000
57. Sanso M, Levin RS, Lipp JJ, Wang VY, Greifenberg AK, Quezada EM, et al. P-TEFb Regulation of Transcription Termination Factor Xrn2 Revealed by a Chemical Genetic Screen for Cdk9 Substrates. Genes Dev (2016) 30(1):117–31. doi: 10.1101/gad.269589.115
58. Parua PK, Booth GT, Sanso M, Benjamin B, Tanny JC, Lis JT, et al. A Cdk9-PP1 Switch Regulates the Elongation-Termination Transition of RNA Polymerase II. Nature (2018) 558(7710):460–4. doi: 10.1038/s41586-018-0214-z
59. Garriga J, Bhattacharya S, Calbo J, Marshall RM, Truongcao M, Haines DS, et al. CDK9 is Constitutively Expressed Throughout the Cell Cycle, and its Steady-State Expression is Independent of SKP2. Mol Cell Biol (2003) 23(15):5165–73. doi: 10.1128/mcb.23.15.5165-5173.2003
60. Bettencourt-Dias M, Giet R, Sinka R, Mazumdar A, Lock WG, Balloux F, et al. Genome-Wide Survey of Protein Kinases Required for Cell Cycle Progression. Nature (2004) 432(7020):980–7. doi: 10.1038/nature03160
61. Zhao R, Nakamura T, Fu Y, Lazar Z, Spector DL. Gene Bookmarking Accelerates the Kinetics of Post-Mitotic Transcriptional Re-Activation. Nat Cell Biol (2011) 13(11):1295–304. doi: 10.1038/ncb2341
62. Yang Z, He N, Zhou Q. Brd4 Recruits P-TEFb to Chromosomes At Late Mitosis to Promote G1 Gene Expression and Cell Cycle Progression. Mol Cell Biol (2008) 28(3):967–76. doi: 10.1128/MCB.01020-07
63. Dey A, Nishiyama A, Karpova T, McNally J, Ozato K. Brd4 Marks Select Genes on Mitotic Chromatin and Directs Postmitotic Transcription. Mol Biol Cell (2009) 20(23):4899–909. doi: 10.1091/mbc.E09-05-0380
64. Mochizuki K, Nishiyama A, Jang MK, Dey A, Ghosh A, Tamura T, et al. The Bromodomain Protein Brd4 Stimulates G1 Gene Transcription and Promotes Progression to S Phase. J Biol Chem (2008) 283(14):9040–8. doi: 10.1074/jbc.M707603200
65. Foskett SM, Ghose R, Tang DN, Lewis DE, Rice AP. Antiapoptotic Function of Cdk9 (Tak/P-TEFb) in U937 Promonocytic Cells. J Virol (2001) 75(3):1220–8. doi: 10.1128/JVI.75.3.1220-1228.2001
66. Simone C, Stiegler P, Bagella L, Pucci B, Bellan C, De Falco G, et al. Activation of MyoD-dependent Transcription by cdk9/cyclin T2. Oncogene (2002) 21(26):4137–48. doi: 10.1038/sj.onc.1205493
67. Nojima M, Huang Y, Tyagi M, Kao HY, Fujinaga K. The Positive Transcription Elongation Factor B is an Essential Cofactor for the Activation of Transcription by Myocyte Enhancer Factor 2. J Mol Biol (2008) 382(2):275–87. doi: 10.1016/j.jmb.2008.07.017
68. Bellan C, De Falco G, Lazzi S, Micheli P, Vicidomini S, Schurfeld K, et al. Cdk9/Cyclin T1 Expression During Normal Lymphoid Differentiation and Malignant Transformation. J Pathol (2004) 203(4):946–52. doi: 10.1002/path.1588
69. De Falco G, Bellan C, D’Amuri A, Angeloni G, Leucci E, Giordano A, et al. Cdk9 Regulates Neural Differentiation and its Expression Correlates With the Differentiation Grade of Neuroblastoma and PNET Tumors. Cancer Biol Ther (2005) 4(3):277–81. doi: 10.4161/cbt.4.3.1497
70. Herrmann CH, Carroll RG, Wei P, Jones KA, Rice AP. Tat-Associated Kinase, TAK, Activity is Regulated by Distinct Mechanisms in Peripheral Blood Lymphocytes and Promonocytic Cell Lines. J Virol (1998) 72(12):9881–8. doi: 10.1128/JVI.72.12.9881-9888.1998
71. Iankova I, Petersen RK, Annicotte JS, Chavey C, Hansen JB, Kratchmarova I, et al. Peroxisome Proliferator-Activated Receptor Gamma Recruits the Positive Transcription Elongation Factor B Complex to Activate Transcription and Promote Adipogenesis. Mol Endocrinol (2006) 20(7):1494–505. doi: 10.1210/me.2005-0222
72. Ghosh K, Tang M, Kumari N, Nandy A, Basu S, Mall DP, et al. Positive Regulation of Transcription by Human ZMYND8 Through Its Association With P-TEFb Complex. Cell Rep (2018) 24(8):2141–54.e6. doi: 10.1016/j.celrep.2018.07.064
73. Shore SM, Byers SA, Maury W, Price DH. Identification of a Novel Isoform of Cdk9. Gene (2003) 307:175–82. doi: 10.1016/s0378-1119(03)00466-9
74. Lin X, Taube R, Fujinaga K, Peterlin BM. P-TEFb Containing Cyclin K and Cdk9 can Activate Transcription Via RNA. J Biol Chem (2002) 277(19):16873–8. doi: 10.1074/jbc.M200117200
75. Mori T, Anazawa Y, Matsui K, Fukuda S, Nakamura Y, Arakawa H. Cyclin K as a Direct Transcriptional Target of the p53 Tumor Suppressor. Neoplasia (2002) 4(3):268–74. doi: 10.1038/sj.neo.7900235
76. Yu DS, Zhao R, Hsu EL, Cayer J, Ye F, Guo Y, et al. Cyclin-Dependent Kinase 9-Cyclin K Functions in the Replication Stress Response. EMBO Rep (2010) 11(11):876–82. doi: 10.1038/embor.2010.153
77. Liu H, Herrmann CH, Chiang K, Sung TL, Moon SH, Donehower LA, et al. 55K Isoform of CDK9 Associates With Ku70 and is Involved in DNA Repair. Biochem Biophys Res Commun (2010) 397(2):245–50. doi: 10.1016/j.bbrc.2010.05.092
78. Michels AA, Fraldi A, Li Q, Adamson TE, Bonnet F, Nguyen VT, et al. Binding of the 7SK snRNA Turns the HEXIM1 Protein Into a P-TEFb (CDK9/Cyclin T) Inhibitor. EMBO J (2004) 23(13):2608–19. doi: 10.1038/sj.emboj.7600275
79. Nguyen VT, Kiss T, Michels AA, Bensaude O. 7SK Small Nuclear RNA Binds to and Inhibits the Activity of CDK9/Cyclin T Complexes. Nature (2001) 414(6861):322–5. doi: 10.1038/35104581
80. Turano M, Napolitano G, Dulac C, Majello B, Bensaude O, Lania L. Increased HEXIM1 Expression During Erythroleukemia and Neuroblastoma Cell Differentiation. J Cell Physiol (2006) 206(3):603–10. doi: 10.1002/jcp.20502
81. Michels AA, Nguyen VT, Fraldi A, Labas V, Edwards M, Bonnet F, et al. MAQ1 and 7SK RNA Interact With CDK9/Cyclin T Complexes in a Transcription-Dependent Manner. Mol Cell Biol (2003) 23(14):4859–69. doi: 10.1128/mcb.23.14.4859-4869.2003
82. Yik JH, Chen R, Nishimura R, Jennings JL, Link AJ, Zhou Q. Inhibition of P-TEFb (Cdk9/Cyclin T) Kinase and RNA Polymerase II Transcription by the Coordinated Actions of HEXIM1 and 7SK Snrna. Mol Cell (2003) 12(4):971–82. doi: 10.1016/s1097-2765(03)00388-5
83. Sano M, Abdellatif M, Oh H, Xie M, Bagella L, Giordano A, et al. Activation and Function of Cyclin T-Cdk9 (Positive Transcription Elongation Factor-B) in Cardiac Muscle-Cell Hypertrophy. Nat Med (2002) 8(11):1310–7. doi: 10.1038/nm778
84. He N, Jahchan NS, Hong E, Li Q, Bayfield MA, Maraia RJ, et al. A La-related Protein Modulates 7SK snRNP Integrity to Suppress P-TEFb-dependent Transcriptional Elongation and Tumorigenesis. Mol Cell (2008) 29(5):588–99. doi: 10.1016/j.molcel.2008.01.003
85. Barboric M, Lenasi T, Chen H, Johansen EB, Guo S, Peterlin BM. 7sk snRNP/P-TEFb Couples Transcription Elongation With Alternative Splicing and is Essential for Vertebrate Development. Proc Natl Acad Sci USA (2009) 106(19):7798–803. doi: 10.1073/pnas.0903188106
86. Jeronimo C, Forget D, Bouchard A, Li Q, Chua G, Poitras C, et al. Systematic Analysis of the Protein Interaction Network for the Human Transcription Machinery Reveals the Identity of the 7SK Capping Enzyme. Mol Cell (2007) 27(2):262–74. doi: 10.1016/j.molcel.2007.06.027
87. Blazek D, Barboric M, Kohoutek J, Oven I, Peterlin BM. Oligomerization of HEXIM1 Via 7SK snRNA and Coiled-Coil Region Directs the Inhibition of P-Tefb. Nucleic Acids Res (2005) 33(22):7000–10. doi: 10.1093/nar/gki997
88. Li Q, Price JP, Byers SA, Cheng D, Peng J, Price DH. Analysis of the Large Inactive P-TEFb Complex Indicates That it Contains One 7SK Molecule, a Dimer of HEXIM1 or HEXIM2, and Two P-TEFb Molecules Containing Cdk9 Phosphorylated At Threonine 186. J Biol Chem (2005) 280(31):28819–26. doi: 10.1074/jbc.M502712200
89. Barboric M, Kohoutek J, Price JP, Blazek D, Price DH, Peterlin BM. Interplay Between 7SK snRNA and Oppositely Charged Regions in HEXIM1 Direct the Inhibition of P-Tefb. EMBO J (2005) 24(24):4291–303. doi: 10.1038/sj.emboj.7600883
90. Egloff S, Van Herreweghe E, Kiss T. Regulation of Polymerase II Transcription by 7SK snRNA: Two Distinct RNA Elements Direct P-TEFb and HEXIM1 Binding. Mol Cell Biol (2006) 26(2):630–42. doi: 10.1128/MCB.26.2.630-642.2006
91. Kobbi L, Demey-Thomas E, Braye F, Proux F, Kolesnikova O, Vinh J, et al. An Evolutionary Conserved Hexim1 Peptide Binds to the Cdk9 Catalytic Site to Inhibit P-Tefb. Proc Natl Acad Sci USA (2016) 113(45):12721–6. doi: 10.1073/pnas.1612331113
92. Chen R, Yang Z, Zhou Q. Phosphorylated Positive Transcription Elongation Factor B (P-TEFb) is Tagged for Inhibition Through Association With 7SK Snrna. J Biol Chem (2004) 279(6):4153–60. doi: 10.1074/jbc.M310044200
93. Biglione S, Byers SA, Price JP, Nguyen VT, Bensaude O, Price DH, et al. Inhibition of HIV-1 Replication by P-TEFb Inhibitors DRB, Seliciclib and Flavopiridol Correlates With Release of Free P-TEFb From the Large, Inactive Form of the Complex. Retrovirology (2007) 4:47. doi: 10.1186/1742-4690-4-47
94. D’Orso I, Frankel AD. RNA-Mediated Displacement of an Inhibitory snRNP Complex Activates Transcription Elongation. Nat Struct Mol Biol (2010) 17(7):815–21. doi: 10.1038/nsmb.1827
95. Liu W, Ma Q, Wong K, Li W, Ohgi K, Zhang J, et al. Brd4 and JMJD6-associated Anti-Pause Enhancers in Regulation of Transcriptional Pause Release. Cell (2013) 155(7):1581–95. doi: 10.1016/j.cell.2013.10.056
96. McNamara RP, McCann JL, Gudipaty SA, D’Orso I. Transcription Factors Mediate the Enzymatic Disassembly of Promoter-Bound 7SK snRNP to Locally Recruit P-TEFb for Transcription Elongation. Cell Rep (2013) 5(5):1256–68. doi: 10.1016/j.celrep.2013.11.003
97. Fujinaga K, Barboric M, Li Q, Luo Z, Price DH, Peterlin BM. PKC Phosphorylates HEXIM1 and Regulates P-TEFb Activity. Nucleic Acids Res (2012) 40(18):9160–70. doi: 10.1093/nar/gks682
98. Kim YK, Mbonye U, Hokello J, Karn J. T-Cell Receptor Signaling Enhances Transcriptional Elongation From Latent HIV Proviruses by Activating P-TEFb Through an ERK-dependent Pathway. J Mol Biol (2011) 410(5):896–916. doi: 10.1016/j.jmb.2011.03.054
99. Contreras X, Barboric M, Lenasi T, Peterlin BM. HMBA Releases P-TEFb From HEXIM1 and 7SK snRNA Via PI3K/Akt and Activates HIV Transcription. PloS Pathog (2007) 3(10):1459–69. doi: 10.1371/journal.ppat.0030146
100. Chen R, Liu M, Li H, Xue Y, Ramey WN, He N, et al. PP2B and PP1alpha Cooperatively Disrupt 7SK snRNP to Release P-TEFb for Transcription in Response to Ca2+ Signaling. Genes Dev (2008) 22(10):1356–68. doi: 10.1101/gad.1636008
101. Cho S, Schroeder S, Kaehlcke K, Kwon HS, Pedal A, Herker E, et al. Acetylation of Cyclin T1 Regulates the Equilibrium Between Active and Inactive P-TEFb in Cells. EMBO J (2009) 28(10):1407–17. doi: 10.1038/emboj.2009.99
102. Barboric M, Yik JH, Czudnochowski N, Yang Z, Chen R, Contreras X, et al. Tat Competes With HEXIM1 to Increase the Active Pool of P-TEFb for HIV-1 Transcription. Nucleic Acids Res (2007) 35(6):2003–12. doi: 10.1093/nar/gkm063
103. Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, et al. Recruitment of P-TEFb for Stimulation of Transcriptional Elongation by the Bromodomain Protein Brd4. Mol Cell (2005) 19(4):535–45. doi: 10.1016/j.molcel.2005.06.029
104. Filippakopoulos P, Picaud S, Fedorov O, Keller M, Wrobel M, Morgenstern O, et al. Benzodiazepines and Benzotriazepines as Protein Interaction Inhibitors Targeting Bromodomains of the BET Family. Bioorg Med Chem (2012) 20(6):1878–86. doi: 10.1016/j.bmc.2011.10.080
105. Chang B, Chen Y, Zhao Y, Bruick RK. JMJD6 is a Histone Arginine Demethylase. Science (2007) 318(5849):444–7. doi: 10.1126/science.1145801
106. Bisgrove DA, Mahmoudi T, Henklein P, Verdin E. Conserved P-TEFb-interacting Domain of BRD4 Inhibits HIV Transcription. Proc Natl Acad Sci USA (2007) 104(34):13690–5. doi: 10.1073/pnas.0705053104
107. He N, Liu M, Hsu J, Xue Y, Chou S, Burlingame A, et al. Hiv-1 Tat and Host AFF4 Recruit Two Transcription Elongation Factors Into a Bifunctional Complex for Coordinated Activation of HIV-1 Transcription. Mol Cell (2010) 38(3):428–38. doi: 10.1016/j.molcel.2010.04.013
108. Sobhian B, Laguette N, Yatim A, Nakamura M, Levy Y, Kiernan R, et al. Hiv-1 Tat Assembles a Multifunctional Transcription Elongation Complex and Stably Associates With the 7SK Snrnp. Mol Cell (2010) 38(3):439–51. doi: 10.1016/j.molcel.2010.04.012
109. Barrandon C, Bonnet F, Nguyen VT, Labas V, Bensaude O. The Transcription-Dependent Dissociation of P-TEFb-HEXIM1-7SK RNA Relies Upon Formation of Hnrnp-7SK RNA Complexes. Mol Cell Biol (2007) 27(20):6996–7006. doi: 10.1128/MCB.00975-07
110. Van Herreweghe E, Egloff S, Goiffon I, Jady BE, Froment C, Monsarrat B, et al. Dynamic Remodelling of Human 7SK snRNP Controls the Nuclear Level of Active P-Tefb. EMBO J (2007) 26(15):3570–80. doi: 10.1038/sj.emboj.7601783
111. Larochelle S, Amat R, Glover-Cutter K, Sanso M, Zhang C, Allen JJ, et al. Cyclin-Dependent Kinase Control of the Initiation-to-Elongation Switch of RNA Polymerase II. Nat Struct Mol Biol (2012) 19(11):1108–15. doi: 10.1038/nsmb.2399
112. Ramakrishnan R, Rice AP. Cdk9 T-loop Phosphorylation is Regulated by the Calcium Signaling Pathway. J Cell Physiol (2012) 227(2):609–17. doi: 10.1002/jcp.22760
113. Mbonye UR, Gokulrangan G, Datt M, Dobrowolski C, Cooper M, Chance MR, et al. Phosphorylation of CDK9 At Ser175 Enhances HIV Transcription and is a Marker of Activated P-TEFb in CD4(+) T Lymphocytes. PLoS Pathog (2013) 9(5):e1003338. doi: 10.1371/journal.ppat.1003338
114. Mbonye U, Wang B, Gokulrangan G, Shi W, Yang S, Karn J. Cyclin-Dependent Kinase 7 (CDK7)-Mediated Phosphorylation of the CDK9 Activation Loop Promotes P-TEFb Assembly With Tat and Proviral HIV Reactivation. J Biol Chem (2018) 293(26):10009–25. doi: 10.1074/jbc.RA117.001347
115. Zhou M, Lu H, Park H, Wilson-Chiru J, Linton R, Brady JN. Tax Interacts With P-TEFb in a Novel Manner to Stimulate Human T-lymphotropic Virus Type 1 Transcription. J Virol (2006) 80(10):4781–91. doi: 10.1128/JVI.80.10.4781-4791.2006
116. Gu Y, Rosenblatt J, Morgan DO. Cell Cycle Regulation of CDK2 Activity by Phosphorylation of Thr160 and Tyr15. EMBO J (1992) 11(11):3995–4005. doi: 10.1002/j.1460-2075.1992.tb05493.x
117. Zhou M, Huang K, Jung KJ, Cho WK, Klase Z, Kashanchi F, et al. Bromodomain Protein Brd4 Regulates Human Immunodeficiency Virus Transcription Through Phosphorylation of CDK9 At Threonine 29. J Virol (2009) 83(2):1036–44. doi: 10.1128/JVI.01316-08
118. Garber ME, Mayall TP, Suess EM, Meisenhelder J, Thompson NE, Jones KA. CDK9 Autophosphorylation Regulates High-Affinity Binding of the Human Immunodeficiency Virus Type 1 tat-P-TEFb Complex to TAR Rna. Mol Cell Biol (2000) 20(18):6958–69. doi: 10.1128/mcb.20.18.6958-6969.2000
119. Fong YW, Zhou Q. Relief of Two built-In Autoinhibitory Mechanisms in P-TEFb is Required for Assembly of a Multicomponent Transcription Elongation Complex At the Human Immunodeficiency Virus Type 1 Promoter. Mol Cell Biol (2000) 20(16):5897–907. doi: 10.1128/mcb.20.16.5897-5907.2000
120. Breuer D, Kotelkin A, Ammosova T, Kumari N, Ivanov A, Ilatovskiy AV, et al. CDK2 Regulates HIV-1 Transcription by Phosphorylation of CDK9 on Serine 90. Retrovirolog (2012) 9:94. doi: 10.1186/1742-4690-9-94
121. Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, et al. Global, In Vivo, and Site-Specific Phosphorylation Dynamics in Signaling Networks. Cell (2006) 127(3):635–48. doi: 10.1016/j.cell.2006.09.026
122. Schroder S, Cho S, Zeng L, Zhang Q, Kaehlcke K, Mak L, et al. Two-Pronged Binding With Bromodomain-Containing Protein 4 Liberates Positive Transcription Elongation Factor B From Inactive Ribonucleoprotein Complexes. J Biol Chem (2012) 287(2):1090–9. doi: 10.1074/jbc.M111.282855
123. Fu J, Yoon HG, Qin J, Wong J. Regulation of P-TEFb Elongation Complex Activity by CDK9 Acetylation. Mol Cell Biol (2007) 27(13):4641–51. doi: 10.1128/MCB.00857-06
124. Sabo A, Lusic M, Cereseto A, Giacca M. Acetylation of Conserved Lysines in the Catalytic Core of Cyclin-Dependent Kinase 9 Inhibits Kinase Activity and Regulates Transcription. Mol Cell Biol (2008) 28(7):2201–12. doi: 10.1128/MCB.01557-07
125. Blank MF, Chen S, Poetz F, Schnolzer M, Voit R, Grummt I. SIRT7-Dependent Deacetylation of CDK9 Activates RNA Polymerase II Transcription. Nucleic Acids Res (2017) 45(5):2675–86. doi: 10.1093/nar/gkx053
126. Lau J, Lew QJ, Diribarne G, Michels AA, Dey A, Bensaude O, et al. Ubiquitination of HEXIM1 by HDM2. Cell Cycle (2009) 8(14):2247–54. doi: 10.4161/cc.8.14.9015
127. Barboric M, Zhang F, Besenicar M, Plemenitas A, Peterlin BM. Ubiquitylation of Cdk9 by Skp2 Facilitates Optimal Tat Transactivation. J Virol (2005) 79(17):11135–41. doi: 10.1128/JVI.79.17.11135-11141.2005
128. Faust TB, Li Y, Bacon CW, Jang GM, Weiss A, Jayaraman B, et al. The HIV-1 Tat Protein Recruits a Ubiquitin Ligase to Reorganize the 7SK snRNP for Transcriptional Activation. Elife (2018) 7:1–30. doi: 10.7554/eLife.31879
129. Garcia-Cuellar MP, Fuller E, Mathner E, Breitinger C, Hetzner K, Zeitlmann L, et al. Efficacy of Cyclin-Dependent-Kinase 9 Inhibitors in a Murine Model of Mixed-Lineage Leukemia. Leukemia (2014) 28(7):1427–35. doi: 10.1038/leu.2014.40
130. Baker A, Gregory GP, Verbrugge I, Kats L, Hilton JJ, Vidacs E, et al. The CDK9 Inhibitor Dinaciclib Exerts Potent Apoptotic and Antitumor Effects in Preclinical Models of MLL-Rearranged Acute Myeloid Leukemia. Cancer Res (2016) 76(5):1158–69. doi: 10.1158/0008-5472.CAN-15-1070
131. Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. Rnai Screen Identifies Brd4 as a Therapeutic Target in Acute Myeloid Leukaemia. Nature (2011) 478(7370):524–8. doi: 10.1038/nature10334
132. Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, et al. Inhibition of BET Recruitment to Chromatin as an Effective Treatment for MLL-fusion Leukaemia. Nature (2011) 478(7370):529–33. doi: 10.1038/nature10509
133. Yin T, Lallena MJ, Kreklau EL, Fales KR, Carballares S, Torrres R, et al. A Novel CDK9 Inhibitor Shows Potent Antitumor Efficacy in Preclinical Hematologic Tumor Models. Mol Cancer Ther (2014) 13(6):1442–56. doi: 10.1158/1535-7163.MCT-13-0849
134. Scholz A, Oellerich T, Hussain A, Lindner S, Luecking U, Walter AO, et al. BAY 1143572, a First-in-Class, Highly Selective, Potent and Orally Available Inhibitor of PTEFb/CDK9 Currently in Phase I, Shows Convincing Anti-Tumor Activity in Preclinical Models of Acute Myeloid Leukemia (AML). Cancer Res (2016) 76:Abstract 3022. doi: 10.1158/1538-7445.Am2016-3022
135. Devaraj SG, Fiskus W, Shah B, Qi J, Sun B, Iyer SP, et al. HEXIM1 Induction is Mechanistically Involved in Mediating anti-AML Activity of BET Protein Bromodomain Antagonist. Leukemia (2016) 30(2):504–8. doi: 10.1038/leu.2015.142
136. Chen R, Keating MJ, Gandhi V, Plunkett W. Transcription Inhibition by Flavopiridol: Mechanism of Chronic Lymphocytic Leukemia Cell Death. Blood (2005) 106(7):2513–9. doi: 10.1182/blood-2005-04-1678
137. Flynn J, Jones J, Johnson AJ, Andritsos L, Maddocks K, Jaglowski S, et al. Dinaciclib is a Novel Cyclin-Dependent Kinase Inhibitor With Significant Clinical Activity in Relapsed and Refractory Chronic Lymphocytic Leukemia. Leukemia (2015) 29(7):1524–9. doi: 10.1038/leu.2015.31
138. Xie S, Jiang H, Zhai XW, Wei F, Wang SD, Ding J, et al. Antitumor Action of CDK Inhibitor LS-007 as a Single Agent and in Combination With ABT-199 Against Human Acute Leukemia Cells. Acta Pharmacol Sin (2016) 37(11):1481–9. doi: 10.1038/aps.2016.49
139. Hashiguchi T, Bruss N, Best S, Lam V, Danilova O, Paiva CJ, et al. Cyclin-Dependent Kinase-9 is a Therapeutic Target in MYC-Expressing Diffuse Large B-Cell Lymphoma. Mol Cancer Ther (2019) 18(9):1520–32. doi: 10.1158/1535-7163.MCT-18-1023
140. Rowland T, Paiva C, Rowley J, Chen A, Drew L, Hurlin P, et al. Selective Targeting Cyclin-Dependent Kinase-9 (Cdk9) Downmodulates c-MYC and Induces Apoptosis in Diffuse Large B-Cell Lymphoma (Dlbcl) Cells. Blood. (2016) 128(22):289. doi: 10.1182/blood.V128.22.289.289
141. Gregory GP, Hogg SJ, Kats LM, Vidacs E, Baker AJ, Gilan O, et al. CDK9 Inhibition by Dinaciclib Potently Suppresses Mcl-1 to Induce Durable Apoptotic Responses in Aggressive MYC-driven B-Cell Lymphoma In Vivo. Leukemia (2015) 29(6):1437–41. doi: 10.1038/leu.2015.10
142. Cidado J, Proia T, Gregory G, Todorovski I, Boiko S, San Martin M, et al. Az’5576, a Selective CDK9 Inhibitor, Demonstrates In Vitro and In Vivo Activity in Diverse Preclinical Models of nonHodgkin Lymphoma. Cancer Res (2017) 77:Abstract 4295. doi: 10.1158/1538-7445.Am2017-4295
143. Narita T, Ishida T, Ito A, Masaki A, Kinoshita S, Suzuki S, et al. Cyclin-Dependent Kinase 9 is a Novel Specific Molecular Target in Adult T-cell Leukemia/Lymphoma. Blood (2017) 130(9):1114–24. doi: 10.1182/blood-2016-09-741983
144. Manohar SM, Rathos MJ, Sonawane V, Rao SV, Joshi KS. Cyclin-Dependent Kinase Inhibitor, P276-00 Induces Apoptosis in Multiple Myeloma Cells by Inhibition of Cdk9-T1 and RNA Polymerase II-dependent Transcription. Leuk Res (2011) 35(6):821–30. doi: 10.1016/j.leukres.2010.12.010
145. Dai Y, Chen S, Pei XY, Ramakrishnan V, Wang M, Orlowski R, et al. Targeting CDK9 Dramatically Potentiates Abt-737-Induced Apoptosis in Human Multiple Myeloma Cells Through a Bim-Dependent Mechanism. Blood (2009) 114(22):125–. doi: 10.1182/blood.V114.22.297.297
146. Kinoshita S, Ishida T, Ito A, Narita T, Masaki A, Suzuki S, et al. Cyclin-Dependent Kinase 9 as a Potential Specific Molecular Target in NK-cell Leukemia/Lymphoma. Haematologica (2018) 103(12):2059–68. doi: 10.3324/haematol.2018.191395
147. Franco LC, Morales F, Boffo S, Giordano A. Cdk9: A Key Player in Cancer and Other Diseases. J Cell Biochem (2018) 119(2):1273–84. doi: 10.1002/jcb.26293
148. Horing E, Montraveta A, Heine S, Kleih M, Schaaf L, Vohringer MC, et al. Dual Targeting of MCL1 and NOXA as Effective Strategy for Treatment of Mantle Cell Lymphoma. Br J Haematol (2017) 177(4):557–61. doi: 10.1111/bjh.14571
149. Ma H, Seebacher NA, Hornicek FJ, Duan Z. Cyclin-Dependent Kinase 9 (CDK9) is a Novel Prognostic Marker and Therapeutic Target in Osteosarcoma. EBioMedicine (2019) 39:182–93. doi: 10.1016/j.ebiom.2018.12.022
150. Tang W, Wang W, Zhao Y, Zhao Z. MicroRNA-874 Inhibits Cell Proliferation and Invasion by Targeting Cyclin-Dependent Kinase 9 in Osteosarcoma. Oncol Lett (2018) 15(5):7649–54. doi: 10.3892/ol.2018.8294
151. Chen Z, Wang Z, Pang JC, Yu Y, Bieerkehazhi S, Lu J, et al. Multiple CDK Inhibitor Dinaciclib Suppresses Neuroblastoma Growth Via Inhibiting CDK2 and CDK9 Activity. Sci Rep (2016) 6:29090. doi: 10.1038/srep29090
152. Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM, et al. CDK7 Inhibition Suppresses Super-Enhancer-Linked Oncogenic Transcription in MYCN-driven Cancer. Cell (2014) 159(5):1126–39. doi: 10.1016/j.cell.2014.10.024
153. Song H, Bhakat R, Kling MJ, Coulter DW, Chaturvedi NK, Ray S, et al. Targeting Cyclin-Dependent Kinase 9 Sensitizes Medulloblastoma Cells to Chemotherapy. Biochem Biophys Res Commun (2019) 520(2):250–6. doi: 10.1016/j.bbrc.2019.09.118
154. Mitra P, Yang RM, Sutton J, Ramsay RG, Gonda TJ. CDK9 Inhibitors Selectively Target Estrogen Receptor-Positive Breast Cancer Cells Through Combined Inhibition of MYB and MCL-1 Expression. Oncotarget (2016) 7(8):9069–83. doi: 10.18632/oncotarget.6997
155. Wang L, Gao W, Hu F, Xu Z, Wang F. MicroRNA-874 Inhibits Cell Proliferation and Induces Apoptosis in Human Breast Cancer by Targeting CDK9. FEBS Lett (2014) 588(24):4527–35. doi: 10.1016/j.febslet.2014.09.035
156. Wang Y, Zhang T, Kwiatkowski N, Abraham BJ, Lee TI, Xie S, et al. CDK7-Dependent Transcriptional Addiction in Triple-Negative Breast Cancer. Cell (2015) 163(1):174–86. doi: 10.1016/j.cell.2015.08.063
157. Brisard D, Eckerdt F, Marsh LA, Blyth GT, Jain S, Cristofanilli M, et al. Antineoplastic Effects of Selective CDK9 Inhibition With Atuveciclib on Cancer Stem-Like Cells in Triple-Negative Breast Cancer. Oncotarget (2018) 9(99):37305–18. doi: 10.18632/oncotarget.26468
158. Wang J, Dean DC, Hornicek FJ, Shi H, Duan Z. Cyclin-Dependent Kinase 9 (CDK9) is a Novel Prognostic Marker and Therapeutic Target in Ovarian Cancer. FASEB J (2019) 33(5):5990–6000. doi: 10.1096/fj.201801789RR
159. Lam F, Abbas AY, Shao H, Teo T, Adams J, Li P, et al. Targeting RNA Transcription and Translation in Ovarian Cancer Cells With Pharmacological Inhibitor CDKI-73. Oncotarget (2014) 5(17):7691–704. doi: 10.18632/oncotarget.2296
160. Xu J, Xu S, Fang Y, Chen T, Xie X, Lu W. Cyclin-Dependent Kinase 9 Promotes Cervical Cancer Development Via AKT2/p53 Pathway. IUBMB Life (2019) 71(3):347–56. doi: 10.1002/iub.1983
161. Huang CH, Lujambio A, Zuber J, Tschaharganeh DF, Doran MG, Evans MJ, et al. CDK9-Mediated Transcription Elongation is Required for MYC Addiction in Hepatocellular Carcinoma. Genes Dev (2014) 28(16):1800–14. doi: 10.1101/gad.244368.114
162. Allaway RJ, Fischer DA, de Abreu FB, Gardner TB, Gordon SR, Barth RJ, et al. Genomic Characterization of Patient-Derived Xenograft Models Established From Fine Needle Aspirate Biopsies of a Primary Pancreatic Ductal Adenocarcinoma and From Patient-Matched Metastatic Sites. Oncotarget (2016) 7(13):17087–102. doi: 10.18632/oncotarget.7718
163. Blake DR, Vaseva AV, Hodge RG, Kline MP, Gilbert TSK, Tyagi V, et al. Application of a MYC Degradation Screen Identifies Sensitivity to CDK9 Inhibitors in KRAS-mutant Pancreatic Cancer. Sci Signal (2019) 12(590):1–15. doi: 10.1126/scisignal.aav7259
164. Kretz AL, Schaum M, Richter J, Kitzig EF, Engler CC, Leithauser F, et al. CDK9 is a Prognostic Marker and Therapeutic Target in Pancreatic Cancer. Tumour Biol (2017) 39(2):1010428317694304. doi: 10.1177/1010428317694304
165. Veeranki OL, Tong Z, Dokey R, Mejia A, Zhang J, Qiao Y, et al. Targeting Cyclin-Dependent Kinase 9 by a Novel Inhibitor Enhances Radiosensitization and Identifies Axl as a Novel Downstream Target in Esophageal Adenocarcinoma. Oncotarget (2019) 10(45):4703–18. doi: 10.18632/oncotarget.27095
166. Tong Z, Mejia A, Veeranki O, Verma A, Correa A, Patel V, et al. Abstract 3859: Targeting CDK9 and MCL-1 by a New CDK9/P-TEFB Inhibitor With and Without 5-Fluorouracil in Esophageal Adenocarcinoma. Cancer Res (2019) 79(13):3859–. doi: 10.1158/1538-7445.am2019-3859
167. Rahaman MH, Lam F, Zhong L, Teo T, Adams J, Yu M, et al. Targeting CDK9 for Treatment of Colorectal Cancer. Mol Oncol (2019) 13(10):2178–93. doi: 10.1002/1878-0261.12559
168. Wang X, Yu C, Wang C, Ma Y, Wang T, Li Y, et al. Novel Cyclin-Dependent Kinase 9 (CDK9) Inhibitor With Suppression of Cancer Stemness Activity Against non-Small-Cell Lung Cancer. Eur J Med Chem (2019) 181:111535. doi: 10.1016/j.ejmech.2019.07.038
169. Shan B, Zhuo Y, Chin D, Morris CA, Morris GF, Lasky JA. Cyclin-Dependent Kinase 9 is Required for Tumor Necrosis Factor-Alpha-Stimulated Matrix Metalloproteinase-9 Expression in Human Lung Adenocarcinoma Cells. J Biol Chem (2005) 280(2):1103–11. doi: 10.1074/jbc.M406293200
170. Lemke J, von Karstedt S, Abd El Hay M, Conti A, Arce F, Montinaro A, et al. Selective CDK9 Inhibition Overcomes TRAIL Resistance by Concomitant Suppression of cFlip and Mcl-1. Cell Death Differ (2014) 21(3):491–502. doi: 10.1038/cdd.2013.179
171. Christensen CL, Kwiatkowski N, Abraham BJ, Carretero J, Al-Shahrour F, Zhang T, et al. Targeting Transcriptional Addictions in Small Cell Lung Cancer With a Covalent CDK7 Inhibitor. Cancer Cell (2014) 26(6):909–22. doi: 10.1016/j.ccell.2014.10.019
172. Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, et al. Therapeutic Targeting of BET Bromodomain Proteins in Castration-Resistant Prostate Cancer. Nature (2014) 510(7504):278–82. doi: 10.1038/nature13229
173. Abdullah C, Wang X, Becker D. Expression Analysis and Molecular Targeting of Cyclin-Dependent Kinases in Advanced Melanoma. Cell Cycle (2011) 10(6):977–88. doi: 10.4161/cc.10.6.15079
174. Bragelmann J, Dammert MA, Dietlein F, Heuckmann JM, Choidas A, Bohm S, et al. Systematic Kinase Inhibitor Profiling Identifies CDK9 as a Synthetic Lethal Target in NUT Midline Carcinoma. Cell Rep (2017) 20(12):2833–45. doi: 10.1016/j.celrep.2017.08.082
175. Storch K, Cordes N. The Impact of CDK9 on Radiosensitivity, DNA Damage Repair and Cell Cycling of HNSCC Cancer Cells. Int J Oncol (2016) 48(1):191–8. doi: 10.3892/ijo.2015.3246
176. Lin C, Smith ER, Takahashi H, Lai KC, Martin-Brown S, Florens L, et al. AFF4, a Component of the ELL/P-TEFb Elongation Complex and a Shared Subunit of MLL Chimeras, can Link Transcription Elongation to Leukemia. Mol Cell (2010) 37(3):429–37. doi: 10.1016/j.molcel.2010.01.026
177. Muntean AG, Hess JL. The Pathogenesis of Mixed-Lineage Leukemia. Annu Rev Pathol (2012) 7:283–301. doi: 10.1146/annurev-pathol-011811-132434
178. Argiropoulos B, Humphries RK. Hox Genes in Hematopoiesis and Leukemogenesis. Oncogene (2007) 26(47):6766–76. doi: 10.1038/sj.onc.1210760
179. Meyer C, Kowarz E, Hofmann J, Renneville A, Zuna J, Trka J, et al. New Insights to the MLL Recombinome of Acute Leukemias. Leukemia (2009) 23(8):1490–9. doi: 10.1038/leu.2009.33
180. Brown P, Levis M, Shurtleff S, Campana D, Downing J, Small D. FLT3 Inhibition Selectively Kills Childhood Acute Lymphoblastic Leukemia Cells With High Levels of FLT3 Expression. Blood (2005) 105(2):812–20. doi: 10.1182/blood-2004-06-2498
181. Mueller D, Bach C, Zeisig D, Garcia-Cuellar MP, Monroe S, Sreekumar A, et al. A Role for the MLL Fusion Partner ENL in Transcriptional Elongation and Chromatin Modification. Blood (2007) 110(13):4445–54. doi: 10.1182/blood-2007-05-090514
182. Mohan M, Herz HM, Takahashi YH, Lin C, Lai KC, Zhang Y, et al. Linking H3K79 Trimethylation to Wnt Signaling Through a Novel Dot1-containing Complex (Dotcom). Genes Dev (2010) 24(6):574–89. doi: 10.1101/gad.1898410
183. Maethner E, Garcia-Cuellar MP, Breitinger C, Takacova S, Divoky V, Hess JL, et al. Mll-ENL Inhibits Polycomb Repressive Complex 1 to Achieve Efficient Transformation of Hematopoietic Cells. Cell Rep (2013) 3(5):1553–66. doi: 10.1016/j.celrep.2013.03.038
184. McCalmont H, Li KL, Jones L, Toubia J, Bray SC, Casolari DA, et al. Efficacy of Combined CDK9/BET Inhibition in Preclinical Models of MLL-rearranged Acute Leukemia. Blood Adv (2020) 4(2):296–300. doi: 10.1182/bloodadvances.2019000586
185. Libura M, Asnafi V, Tu A, Delabesse E, Tigaud I, Cymbalista F, et al. FLT3 and MLL Intragenic Abnormalities in AML Reflect a Common Category of Genotoxic Stress. Blood (2003) 102(6):2198–204. doi: 10.1182/blood-2003-01-0162
186. Armstrong SA, Kung AL, Mabon ME, Silverman LB, Stam RW, Den Boer ML, et al. Inhibition of FLT3 in MLL. Validation of a Therapeutic Target Identified by Gene Expression Based Classification. Cancer Cell (2003) 3(2):173–83. doi: 10.1016/s1535-6108(03)00003-5
187. Grafone T, Palmisano M, Nicci C, Storti S. An Overview on the Role of FLT3-tyrosine Kinase Receptor in Acute Myeloid Leukemia: Biology and Treatment. Oncol Rev (2012) 6(1):e8. doi: 10.4081/oncol.2012.e8
188. Stubbs MC, Kim YM, Krivtsov AV, Wright RD, Feng Z, Agarwal J, et al. Mll-AF9 and FLT3 Cooperation in Acute Myelogenous Leukemia: Development of a Model for Rapid Therapeutic Assessment. Leukemia (2008) 22(1):66–77. doi: 10.1038/sj.leu.2404951
189. Glaser SP, Lee EF, Trounson E, Bouillet P, Wei A, Fairlie WD, et al. Anti-Apoptotic Mcl-1 is Essential for the Development and Sustained Growth of Acute Myeloid Leukemia. Genes Dev (2012) 26(2):120–5. doi: 10.1101/gad.182980.111
190. Xiang Z, Luo H, Payton JE, Cain J, Ley TJ, Opferman JT, et al. Mcl1 Haploinsufficiency Protects Mice From Myc-induced Acute Myeloid Leukemia. J Clin Invest (2010) 120(6):2109–18. doi: 10.1172/JCI39964
191. Kaufmann SH, Karp JE, Svingen PA, Krajewski S, Burke PJ, Gore SD, et al. Elevated Expression of the Apoptotic Regulator Mcl-1 At the Time of Leukemic Relapse. Blood (1998) 91(3):991–1000. doi: 10.1182/blood.V91.3.991
192. Lucking U, Scholz A, Lienau P, Siemeister G, Kosemund D, Bohlmann R, et al. Identification of Atuveciclib (BAY 1143572), the First Highly Selective, Clinical PTEFb/CDK9 Inhibitor for the Treatment of Cancer. ChemMedChe (2017) 12(21):1776–93. doi: 10.1002/cmdc.201700447
193. Rahaman MH, Yu Y, Zhong L, Adams J, Lam F, Li P, et al. Cdki-73: An Orally Bioavailable and Highly Efficacious CDK9 Inhibitor Against Acute Myeloid Leukemia. Invest New Drugs (2019) 37(4):625–35. doi: 10.1007/s10637-018-0661-2
194. O’Reilly A, Murphy J, Rawe S, Garvey M. Chronic Lymphocytic Leukemia: A Review of Front-line Treatment Options, With a Focus on Elderly Cll Patients. Clin Lymphoma Myeloma Leuk (2018) 18(4):249–56. doi: 10.1016/j.clml.2018.02.003
195. Pepper C, Lin TT, Pratt G, Hewamana S, Brennan P, Hiller L, et al. Mcl-1 Expression has In Vitro and In Vivo Significance in Chronic Lymphocytic Leukemia and is Associated With Other Poor Prognostic Markers. Blood (2008) 112(9):3807–17. doi: 10.1182/blood-2008-05-157131
196. Stevenson FK, Krysov S, Davies AJ, Steele AJ, Packham G. B-Cell Receptor Signaling in Chronic Lymphocytic Leukemia. Blood (2011) 118(16):4313–20. doi: 10.1182/blood-2011-06-338855
197. Johnson AJ, Yeh YY, Smith LL, Wagner AJ, Hessler J, Gupta S, et al. The Novel Cyclin-Dependent Kinase Inhibitor Dinaciclib (SCH727965) Promotes Apoptosis and Abrogates Microenvironmental Cytokine Protection in Chronic Lymphocytic Leukemia Cells. Leukemia (2012) 26(12):2554–7. doi: 10.1038/leu.2012.144
198. Walsby E, Pratt G, Shao H, Abbas AY, Fischer PM, Bradshaw TD, et al. A Novel Cdk9 Inhibitor Preferentially Targets Tumor Cells and Synergizes With Fludarabine. Oncotarget (2014) 5(2):375–85. doi: 10.18632/oncotarget.1568
199. Byrd JC, Lin TS, Dalton JT, Wu D, Phelps MA, Fischer B, et al. Flavopiridol Administered Using a Pharmacologically Derived Schedule is Associated With Marked Clinical Efficacy in Refractory, Genetically High-Risk Chronic Lymphocytic Leukemia. Blood (2007) 109(2):399–404. doi: 10.1182/blood-2006-05-020735
200. Ghia P, Scarfo L, Perez S, Pathiraja K, Derosier M, Small K, et al. Efficacy and Safety of Dinaciclib vs Ofatumumab in Patients With Relapsed/Refractory Chronic Lymphocytic Leukemia. Blood (2017) 129(13):1876–8. doi: 10.1182/blood-2016-10-748210
201. Wang S, Fischer PM. Cyclin-Dependent Kinase 9: A Key Transcriptional Regulator and Potential Drug Target in Oncology, Virology and Cardiology. Trends Pharmacol Sci (2008) 29(6):302–13. doi: 10.1016/j.tips.2008.03.003
202. Nguyen L, Papenhausen P, Shao H. The Role of c-MYC in B-Cell Lymphomas: Diagnostic and Molecular Aspects. Genes (Basel) (2017) 8(4):1–23. doi: 10.3390/genes8040116
203. Waks AG, Winer EP. Breast Cancer Treatment: A Review. JAMA (2019) 321(3):288–300. doi: 10.1001/jama.2018.19323
204. Baird RD, Caldas C. Genetic Heterogeneity in Breast Cancer: The Road to Personalized Medicine? BMC Med (2013) 11:151. doi: 10.1186/1741-7015-11-151
205. Mitra P, Pereira LA, Drabsch Y, Ramsay RG, Gonda TJ. Estrogen Receptor-Alpha Recruits P-TEFb to Overcome Transcriptional Pausing in Intron 1 of the MYB Gene. Nucleic Acids Res (2012) 40(13):5988–6000. doi: 10.1093/nar/gks286
206. Sengupta S, Biarnes MC, Jordan VC. Cyclin Dependent Kinase-9 Mediated Transcriptional De-Regulation of cMYC as a Critical Determinant of Endocrine-Therapy Resistance in Breast Cancers. Breast Cancer Res Treat (2014) 143(1):113–24. doi: 10.1007/s10549-013-2789-2
207. Abramson VG, Mayer IA. Molecular Heterogeneity of Triple Negative Breast Cancer. Curr Breast Cancer Rep (2014) 6(3):154–8. doi: 10.1007/s12609-014-0152-1
208. Rajput S, Khera N, Guo Z, Hoog J, Li S, Ma CX. Inhibition of Cyclin Dependent Kinase 9 by Dinaciclib Suppresses Cyclin B1 Expression and Tumor Growth in Triple Negative Breast Cancer. Oncotarget (2016) 7(35):56864–75. doi: 10.18632/oncotarget.10870
209. Gelmann EP. Molecular Biology of the Androgen Receptor. J Clin Oncol (2002) 20(13):3001–15. doi: 10.1200/JCO.2002.10.018
210. Lee DK, Duan HO, Chang C. Androgen Receptor Interacts With the Positive Elongation Factor P-TEFb and Enhances the Efficiency of Transcriptional Elongation. J Biol Chem (2001) 276(13):9978–84. doi: 10.1074/jbc.M002285200
211. Gordon V, Bhadel S, Wunderlich W, Zhang J, Ficarro SB, Mollah SA, et al. CDK9 Regulates AR Promoter Selectivity and Cell Growth Through Serine 81 Phosphorylation. Mol Endocrinol (2010) 24(12):2267–80. doi: 10.1210/me.2010-0238
212. Chen S, Gulla S, Cai C, Balk SP. Androgen Receptor Serine 81 Phosphorylation Mediates Chromatin Binding and Transcriptional Activation. J Biol Chem (2012) 287(11):8571–83. doi: 10.1074/jbc.M111.325290
213. Russo JW, Liu X, Ye H, Calagua C, Chen S, Voznesensky O, et al. Phosphorylation of Androgen Receptor Serine 81 is Associated With its Reactivation in Castration-Resistant Prostate Cancer. Cancer Lett (2018) 438:97–104. doi: 10.1016/j.canlet.2018.09.014
214. Arisan ED, Obakan P, Coker-Gurkan A, Calcabrini A, Agostinelli E, Unsal NP. CDK Inhibitors Induce Mitochondria-Mediated Apoptosis Through the Activation of Polyamine Catabolic Pathway in LNCaP, DU145 and PC3 Prostate Cancer Cells. Curr Pharm Des (2014) 20(2):180–8. doi: 10.2174/13816128113199990029
215. Feldman BJ, Feldman D. The Development of Androgen-Independent Prostate Cancer. Nat Rev Cancer (2001) 1(1):34–45. doi: 10.1038/35094009
216. Zhao Y, Wang L, Ren S, Wang L, Blackburn PR, McNulty MS, et al. Activation of P-TEFb by Androgen Receptor-Regulated Enhancer Rnas in Castration-Resistant Prostate Cancer. Cell Rep (2016) 15(3):599–610. doi: 10.1016/j.celrep.2016.03.038
217. Bai S, Cao S, Jin L, Kobelski M, Schouest B, Wang X, et al. A Positive Role of c-Myc in Regulating Androgen Receptor and its Splice Variants in Prostate Cancer. Oncogene (2019) 38(25):4977–89. doi: 10.1038/s41388-019-0768-8
218. Santer FR, Erb HH, Oh SJ, Handle F, Feiersinger GE, Luef B, et al. Mechanistic Rationale for MCL1 Inhibition During Androgen Deprivation Therapy. Oncotarget (2015) 6(8):6105–22. doi: 10.18632/oncotarget.3368
219. Asangani IA, Wilder-Romans K, Dommeti VL, Krishnamurthy PM, Apel IJ, Escara-Wilke J, et al. Bet Bromodomain Inhibitors Enhance Efficacy and Disrupt Resistance to AR Antagonists in the Treatment of Prostate Cancer. Mol Cancer Res (2016) 14(4):324–31. doi: 10.1158/1541-7786.MCR-15-0472
220. Villanueva A. Hepatocellular Carcinoma. N Engl J Med (2019) 380(15):1450–62. doi: 10.1056/NEJMra1713263
221. Peng SY, Lai PL, Hsu HC. Amplification of the C-Myc Gene in Human Hepatocellular Carcinoma: Biologic Significance. J Formos Med Assoc (1993) 92(10):866–70.
222. Hussain SP, Schwank J, Staib F, Wang XW, Harris CC. TP53 Mutations and Hepatocellular Carcinoma: Insights Into the Etiology and Pathogenesis of Liver Cancer. Oncogene (2007) 26(15):2166–76. doi: 10.1038/sj.onc.1210279
223. Pang C, Huang G, Luo K, Dong Y, He F, Du G, et al. miR-206 Inhibits the Growth of Hepatocellular Carcinoma Cells Via Targeting CDK9. Cancer Med (2017) 6(10):2398–409. doi: 10.1002/cam4.1188
224. Sonawane YA, Taylor MA, Napoleon JV, Rana S, Contreras JI, Natarajan A. Cyclin Dependent Kinase 9 Inhibitors for Cancer Therapy. J Med Chem (2016) 59(19):8667–84. doi: 10.1021/acs.jmedchem.6b00150
225. Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The History and Future of Targeting Cyclin-Dependent Kinases in Cancer Therapy. Nat Rev Drug Discov (2015) 14(2):130–46. doi: 10.1038/nrd4504
226. Kim KS, Sack JS, Tokarski JS, Qian L, Chao ST, Leith L, et al. Thio- and Oxoflavopiridols, Cyclin-Dependent Kinase 1-Selective Inhibitors: Synthesis and Biological Effects. J Med Chem (2000) 43(22):4126–34. doi: 10.1021/jm000231g
227. Chao SH, Price DH. Flavopiridol Inactivates P-TEFb and Blocks Most RNA Polymerase II Transcription In Vivo. J Biol Chem (2001) 276(34):31793–9. doi: 10.1074/jbc.M102306200
228. Chao SH, Fujinaga K, Marion JE, Taube R, Sausville EA, Senderowicz AM, et al. Flavopiridol Inhibits P-TEFb and Blocks HIV-1 Replication. J Biol Chem (2000) 275(37):28345–8. doi: 10.1074/jbc.C000446200
229. Zeidner JF, Karp JE. Clinical Activity of Alvocidib (Flavopiridol) in Acute Myeloid Leukemia. Leuk Res (2015) 39(12):1312–8. doi: 10.1016/j.leukres.2015.10.010
230. McClue SJ, Blake D, Clarke R, Cowan A, Cummings L, Fischer PM, et al. In Vitro and In Vivo Antitumor Properties of the Cyclin Dependent Kinase Inhibitor CYC202 (R-Roscovitine). Int J Cancer (2002) 102(5):463–8. doi: 10.1002/ijc.10738
231. Meijer L, Borgne A, Mulner O, Chong JP, Blow JJ, Inagaki N, et al. Biochemical and Cellular Effects of Roscovitine, a Potent and Selective Inhibitor of the Cyclin-Dependent Kinases cdc2, Cdk2 and Cdk5. Eur J Biochem (1997) 243(1-2):527–36. doi: 10.1111/j.1432-1033.1997.t01-2-00527.x
232. Mohapatra S, Chu B, Zhao X, Pledger WJ. Accumulation of p53 and Reductions in XIAP Abundance Promote the Apoptosis of Prostate Cancer Cells. Cancer Res (2005) 65(17):7717–23. doi: 10.1158/0008-5472.CAN-05-0347
233. Whittaker SR, Mallinger A, Workman P, Clarke PA. Inhibitors of Cyclin-Dependent Kinases as Cancer Therapeutics. Pharmacol Ther (2017) 173:83–105. doi: 10.1016/j.pharmthera.2017.02.008
234. Parry D, Guzi T, Shanahan F, Davis N, Prabhavalkar D, Wiswell D, et al. Dinaciclib (SCH 727965), a Novel and Potent Cyclin-Dependent Kinase Inhibitor. Mol Cancer Ther (2010) 9(8):2344–53. doi: 10.1158/1535-7163.MCT-10-0324
235. Nemunaitis JJ, Small KA, Kirschmeier P, Zhang D, Zhu Y, Jou YM, et al. A First-in-Human, Phase 1, Dose-Escalation Study of Dinaciclib, a Novel Cyclin-Dependent Kinase Inhibitor, Administered Weekly in Subjects With Advanced Malignancies. J Transl Med (2013) 11:259. doi: 10.1186/1479-5876-11-259
236. Mita MM, Joy AA, Mita A, Sankhala K, Jou YM, Zhang D, et al. Randomized Phase II Trial of the Cyclin-Dependent Kinase Inhibitor Dinaciclib (MK-7965) Versus Capecitabine in Patients With Advanced Breast Cancer. Clin Breast Cancer (2014) 14(3):169–76. doi: 10.1016/j.clbc.2013.10.016
237. Stephenson JJ, Nemunaitis J, Joy AA, Martin JC, Jou YM, Zhang D, et al. Randomized Phase 2 Study of the Cyclin-Dependent Kinase Inhibitor Dinaciclib (MK-7965) Versus Erlotinib in Patients With non-Small Cell Lung Cancer. Lung Cancer (2014) 83(2):219–23. doi: 10.1016/j.lungcan.2013.11.020
238. Kumar SK, LaPlant B, Chng WJ, Zonder J, Callander N, Fonseca R, et al. Dinaciclib, a Novel CDK Inhibitor, Demonstrates Encouraging Single-Agent Activity in Patients With Relapsed Multiple Myeloma. Blood (2015) 125(3):443–8. doi: 10.1182/blood-2014-05-573741
239. Goh KC, Novotny-Diermayr V, Hart S, Ong LC, Loh YK, Cheong A, et al. TG02, a Novel Oral Multi-Kinase Inhibitor of CDKs, JAK2 and FLT3 With Potent Anti-Leukemic Properties. Leukemia (2012) 26(2):236–43. doi: 10.1038/leu.2011.218
240. Luecking UT, Scholz A, Kosemund D, Bohlmann R, Briem H, Lienau P, et al. Identification of Potent and Highly Selective PTEFb Inhibitor BAY 1251152 for the Treatment of Cancer: From P.O. to I.V. Application Via Scaffold Hops. Cancer Res (2017) 77:Abstract 984. doi: 10.1158/1538-7445.Am2017-984
241. Diamond JR, Moreno V, Lim EA, Cordoba R, Cai C, Ince SJ. Phase I Dose Escalation Study of the First-in-Class Selective PTEFb Inhibitor BAY 1251152 in Patients With Advanced Cancer: Novel Target Validation and Early Evidence of Clinical Activity. J Clin Oncol (2018) 36(15):Abstract 2507. doi: 10.1200/JCO.2018.36.15_suppl.2507
242. Cidado J, Boiko S, Proia T, Ferguson D, Criscione SW, San Martin M, et al. Azd4573 Is a Highly Selective CDK9 Inhibitor That Suppresses MCL-1 and Induces Apoptosis in Hematologic Cancer Cells. Clin Cancer Res (2020) 26(4):922–34. doi: 10.1158/1078-0432.CCR-19-1853
243. Lu H, Xue Y, Yu GK, Arias C, Lin J, Fong S, et al. Compensatory Induction of MYC Expression by Sustained CDK9 Inhibition Via a BRD4-dependent Mechanism. Elife (2015) 4:e06535. doi: 10.7554/eLife.06535
244. Olson CM, Jiang B, Erb MA, Liang Y, Doctor ZM, Zhang Z, et al. Pharmacological Perturbation of CDK9 Using Selective CDK9 Inhibition or Degradation. Nat Chem Biol (2018) 14(2):163–70. doi: 10.1038/nchembio.2538
245. Kohoutek J, Li Q, Blazek D, Luo Z, Jiang H, Peterlin BM. Cyclin T2 is Essential for Mouse Embryogenesis. Mol Cell Biol (2009) 29(12):3280–5. doi: 10.1128/MCB.00172-09
Keywords: cancer, CDKs, transcription, P-TEFb, CDK9 inhibitors
Citation: Anshabo AT, Milne R, Wang S and Albrecht H (2021) CDK9: A Comprehensive Review of Its Biology, and Its Role as a Potential Target for Anti-Cancer Agents. Front. Oncol. 11:678559. doi: 10.3389/fonc.2021.678559
Received: 09 March 2021; Accepted: 16 April 2021;
Published: 10 May 2021.
Edited by:
Paolo Armando Gagliardi, University of Bern, SwitzerlandReviewed by:
Emanuele Middonti, University of Turin, ItalyBishajit Sarkar, Jahangirnagar University, Bangladesh
Md. Asad Ullah, Jahangirnagar University, Bangladesh
Copyright © 2021 Anshabo, Milne, Wang and Albrecht. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hugo Albrecht, hugo.albrecht@unisa.edu.au; Shudong Wang, shudong.wang@unisa.edu.au