 Cheng Zhong1,2†
Cheng Zhong1,2† Yirun Li1,2†
Yirun Li1,2† Jing Yang1,2Shengxi Jin1,2
Jing Yang1,2Shengxi Jin1,2 Guoqiao Chen1,2
Guoqiao Chen1,2 Duguang Li1,2
Duguang Li1,2 Xiaoxiao Fan1,2,3*
Xiaoxiao Fan1,2,3* Hui Lin1,2*
Hui Lin1,2*- 1Department of General Surgery, Sir Run Run Shaw Hospital, School of Medicine, Zhejiang University, Hangzhou, China
- 2Biomedical Research Center, Sir Run Run Shaw Hospital, School of Medicine, Zhejiang University, Hangzhou, China
- 3State Key Laboratory of Modern Optical Instrumentations, Centre for Optical and Electromagnetic Research, College of Optical Science and Engineering, International Research Center for Advanced Photonics, Zhejiang University, Hangzhou, China
Although many approaches have been used to treat hepatocellular carcinoma (HCC), the clinical benefits remain limited, particularly for late stage HCC. In recent years, studies have focused on immunotherapy for HCC. Immunotherapies have shown promising clinical outcomes in several types of cancers and potential therapeutic effects for advanced HCC. In this review, we summarize the immune tolerance and immunotherapeutic strategies for HCC as well as the main challenges of current therapeutic approaches. We also present alternative strategies for overcoming these limitations.
Introduction
Liver cancer is the seventh most commonly diagnosed cancer and third leading cause of cancer-related death worldwide. Hepatocellular carcinoma (HCC), the most common form of liver cancer, shows high morbidity and mortality (1). The major clinical risk factor for developing HCC is liver cirrhosis. Chronic infections with hepatitis B virus and hepatitis C virus as well as long-term heavy alcohol consumption are the main causes of cirrhosis development (2).
Surgical resection, liver transplantation, and radiofrequency ablation (RFA) are widely applied in the clinical treatment of early stage HCC (Barcelona Clinic Liver Cancer [BCLC] stage A). For patients with intermediate HCC (BCLC stage B), transarterial chemoembolization is considered as the first-line treatment with a median survival of approximately 40 months (3–5). However, most patients with HCC are first diagnosed in an advanced stage (BCLC stage C). The multi-tyrosine kinase inhibitors sorafenib and regorafenib have been approved by the Food and Drug Administration as first- and second-line treatments for advanced HCC but only increase survival by less than three months (5). Although many treatment approaches have improved the clinical efficacy, patients with HCC suffer from tumor recurrence and show poor survival rates. Thus, novel therapeutic strategies are urgently needed.
Cancer immunotherapy (CIT) has rapidly developed in the past few years and has improved the survival of patients with different tumors. However, only a few patients with specific cancers, such as melanoma or Hodgkin’s lymphoma, exhibit life-altering improvements with CIT. Most patients with solid tumors still respond negatively to immune therapies. In this review, we summarize the immune tolerance and immunotherapeutic strategies for HCC, analyze the limits of current therapeutic approaches, and present alternative strategies which might overcome these limitations.
Immune Tolerance of HCC
The liver is constantly exposed to non-self proteins derived from nutrients or microbiota, which can trigger immune responses. Many mechanisms protect these harmless antigens from being attacked by the hepatic immune system to maintain homeostasis in the hepatic microenvironment (6). In chronic liver disease, continuous inflammation makes the liver an immunosuppressive microenvironment. Chronic hepatitis B virus and hepatitis C virus infection are the most important risk factors for HCC and are associated with 80% of HCC cases globally (7, 8), providing an immunosuppressive milieu for the initiation and progression of HCC (9). Tumor cells and the specific immune system of HCC constitute an immune-resistant microenvironment, allowing tumor tissue to evade the surveillance of the immune system and protecting the tumor tissue from immune system attack.
Tumor Cells Promote Immune Tolerance of HCC
Hepatocytes under chronic pressure gain ‘driver’ mutations (10), leading to growth advantages and gradually transforming them into low-grade dysplastic nodules, high-grade dysplastic nodules, early HCC, and finally advanced HCC (11). The progression of tumor cells under the selective pressure of immune system resulting in the emergence of immune-resistant tumor cells with fewer immunogenic or immunosuppressive factors is named as ‘immunoediting’ (12).
Tumor cells show weakened antigenicity. Tumor-associated antigens (TAAs) are antigens that are either only produced by tumor cells or overexpressed in tumors compared to in normal cells. The most studied TAAs are oncofetal antigens and cancer/testis antigens, including alpha fetoprotein (AFP), glypican-3 (GPC-3), New York esophageal squamous cell carcinoma-1, synovial sarcoma X-2, melanoma antigen gene-A, and human telomerase-reverse transcriptase, which can elicit a defensive immune response in the host. In the progression of a chronically inflamed liver and HCC, genetic and epigenetic alterations under pressure from the microenvironment transform tumor cells and deregulate the expression of TAAs. In addition to decreasing TAAs, HCC cells escape immune attack by releasing immunosuppressive cytokines, such as transforming growth factor-β and indoleamine 2, 3-dioxygenase (13, 14).
Immunosuppressive Cells in HCC
The liver prevents harmless antigens from being attacked by the hepatic immune system and thus maintains homeostasis in the hepatic microenvironment (6). However, long-lasting inflammatory and antigenic stimulation switches the immune system in the liver to an immunosuppressive status, which is exacerbated during the initiation and progression of HCC (15).
Repressive T Cells in HCC
The mechanisms of immunological tolerance for T cells in HCC including inactivation or deletion of effector T cells, mainly refers to CD8+ T cells as well as priming and expansion of regulatory T cells (Treg cells). The presence of tumor-infiltrating lymphocytes (TILs) is associated with a good prognosis and improved overall survival in HCC (16). Cytotoxic infiltrating CD8+ T cells are the major cell type functioning to kill tumor cells. However, persistent exposure to antigens stimulates effector CD8+ T cells to differentiate into exhausted CD8+ T cells (17). Exhausted CD8+ T cells were originally characterized by down-regulated expression of interferon gamma (IFN-γ) during chronic inflammation. Poor expression of tumor necrosis factor-β and interleukin-2 (IL-2) in exhausted CD8+ T cells is also observed, resulting in impaired cytotoxic function (18). In addition to the loss of effector function, exhausted CD8+ T cells express inhibitory receptors (IRs), such as programmed cell death 1 (PD-1), lymphocyte-activation gene 3, T cell immunoglobulin domain and mucin domain-containing protein 3, and cytotoxic T lymphocyte-associated antigen (CTLA)-4 (19–21). IRs are negative regulatory pathways that prevent the immune system from attacking cells indiscriminately. However, in the tumor immune system, IRs protect tumor cells from immune system attack. Persistent and elevated expression of these IRs has been observed in HCC (22, 23).
Treg cells are a subpopulation of T cells that modulate the immune system and play an immune suppressive role in immune tolerance in cancer. Depletion of Treg cells results in severe autoimmunity and allergies (24–26). In HCC, accumulation of intra-tumoral Treg cells correlates with tumor progression and poor prognosis (27, 28). Treg cell depletion can also activate an effective immune response in tumor models in animals (29, 30). Treg cells express the CD4, CD25, and Foxp3 biomarkers. Foxp3 is a key regulatory gene in the development of Treg cells (31). The transcription factor Foxp3 has been proposed to regulate the expression levels of immune-suppressive molecules in Treg cells. Ectopic expression of Foxp3 confers Treg-like suppressive function to CD4+CD25- T cells (32), and various molecules encoded by Foxp3-controlled genes are associated with immune suppression (33).
Myeloid Cells in HCC
There are two types of myeloid cells; marrow-derived suppressor cells (MDSCs) and tumor associated-macrophages (TAMs), which play important roles in the tumor microenvironment. MDSCs are a population of immature myeloid cells with strong immunosuppressive functions and can promote tumoral angiogenesis. MDSCs can differentiate into macrophages, granulocytes, and dendritic cells (DCs) (34). However, in the hypoxic microenvironment of HCC, tumor cells express ectonucleoside triphosphate diphosphohydrolase 2, which can convert extracellular ATP to 5′-AMP and thus prevent the differentiation of MDSCs (35). Arginine is an essential amino acid for the proliferation of CD4+ and CD8+ T cells. MDSCs suppress T-cell proliferation via increased arginase activity, leading to arginine depletion (36). MDSCs also exert an immunosuppressive effect by inducing the differentiation of CD4+ T cells into Treg cells (36).
TAMs are also immunosuppressive myeloid cells. Macrophages can differentiate via two routes, known as macrophage polarization. Classically activated macrophages (M1) produce high levels of IL-12 and low levels of IL-10 and promote tumor initiation, whereas alternatively activated macrophages (M2) are characterized by low IL-12 and high IL-10 production and promote tumor progression. The microenvironment of HCC stimulates macrophages towards M2 polarization, which are named as TAMs (37). A previous study reported that macrophages in the early stage of HCC express high levels of major histocompatibility complex (MHC)-class II and cytokines, such as IL-1β, IL-6, IL-12, and inducible nitric oxide synthase, which suppress tumor progression. However, in advanced HCC, macrophages express M2-like molecules, including macrophage mannose receptor c1, arginase, IL-10, and transforming growth factor-β and low levels of MHC-class II, which promote tumor progression (38).
TAMs promote tumor progression through angiogenesis, tumor cell invasion, and metastasis (39). Infiltrating TAMs contribute to poor prognosis in HCC, and in vivo and in vitro experiments have shown that TAMs in HCC enhance tumor invasion by producing C-C motif chemokine 22 (40). Another study showed that TREM-1+ TAMs in HCC induce immunosuppression by recruiting C-C chemokine receptor type 6-positive Treg cells, releasing CCL20 and producing the immune checkpoint molecule PD-L1 which may endow HCC with anti-PD-L1 therapy resistance (41). Transforming growth factor-β in the HCC environment can promote TAMs to produce T-cell immunoglobulin- and mucin-domain-containing molecule-3, which can promote bone marrow-derived macrophages and peripheral monocytes to differentiate into TAMs (42). After co-culture with tumor cells, TAMs promoted the expansion of CD44+ HCC stem cells by producing IL-6 and signaling via STAT3 (43). The CCR2+ macrophage subset has pro-angiogenic properties in HCC, and inhibition of CCR2+ TAMs in the fibrosis-HCC model significantly suppress angiogenic activities (44).
Hepatic Stellate Cells in HCC
Hepatic stellate cells (HSCs) are the main producers of extracellular matrix in the liver. In liver fibrosis, HSCs are activated towards a myofibroblast-like phenotype and play a key role in fibrogenesis (45). Activated HSCs produce extracellular matrix, cytokines, and growth factors to create a tumor-favoring environment in HCC (46). Activated HSCs in HCC suppress the antitumor immune response by depleting effector T cells and promoting the accumulation of immunosuppressive cells. HSCs can induce apoptosis of activated T cells through PD-L1 signaling (47, 48). Activated HSCs can convert mature peripheral blood monocytes into MDSCs (49). In murine models, HSCs can present antigens to naïve CD4+ T cells and transform activated naïve CD4+ T cells into Foxp3+ Treg cells by producing retinoic acid (50).
Liver Sinusoidal Endothelial Cells in HCC
Liver sinusoidal endothelial cells (LSECs) form a bed in the liver and receive blood from both the hepatic artery and portal veins in the hepatic parenchyma. In addition to functioning as vascular channels, LSECs play a role in the immune system. LSECs function in both pathogen recognition and antigen presentation. LSECs can cross-present antigens to CD8+ T cells by taking up, processing, and transferring antigens to MHC class I. The presentation of antigens produces a tolerogenic response in naïve CD8+ T cells by upregulating PD-L1 on the surface of LSECs, which bind to the PD-1 receptor expressed on naïve CD8+ T cells (51). LSECs also present antigens to the MHC class II complex to activate CD4+ T cells. However, because of the lack of co-stimulatory molecules, LSECs drive naïve CD4+ T cells to develop into Treg cells rather than into T helper cells (52). LSECs express various receptors for angiogenic factors including vascular endothelial growth factor receptors 1 and 2, Tie‑2 (angiopoietin‑1 receptor), and platelet-derived growth factor receptor. The interaction between these receptors and their ligands promotes the proliferation of LSECs and angiogenesis (53, 54).
Immunotherapy for HCC
Immunotherapy for cancer mainly involves three approaches: vaccines, adoptive cell transfer (ACT), and immune checkpoint inhibitors (ICIs) (Table 1). Vaccines or ACT with genetically modified T cells target specific antigens. ICI inhibits the suppressive regulators of T cells and stimulates already present antitumor immune responses to kill tumor cells.
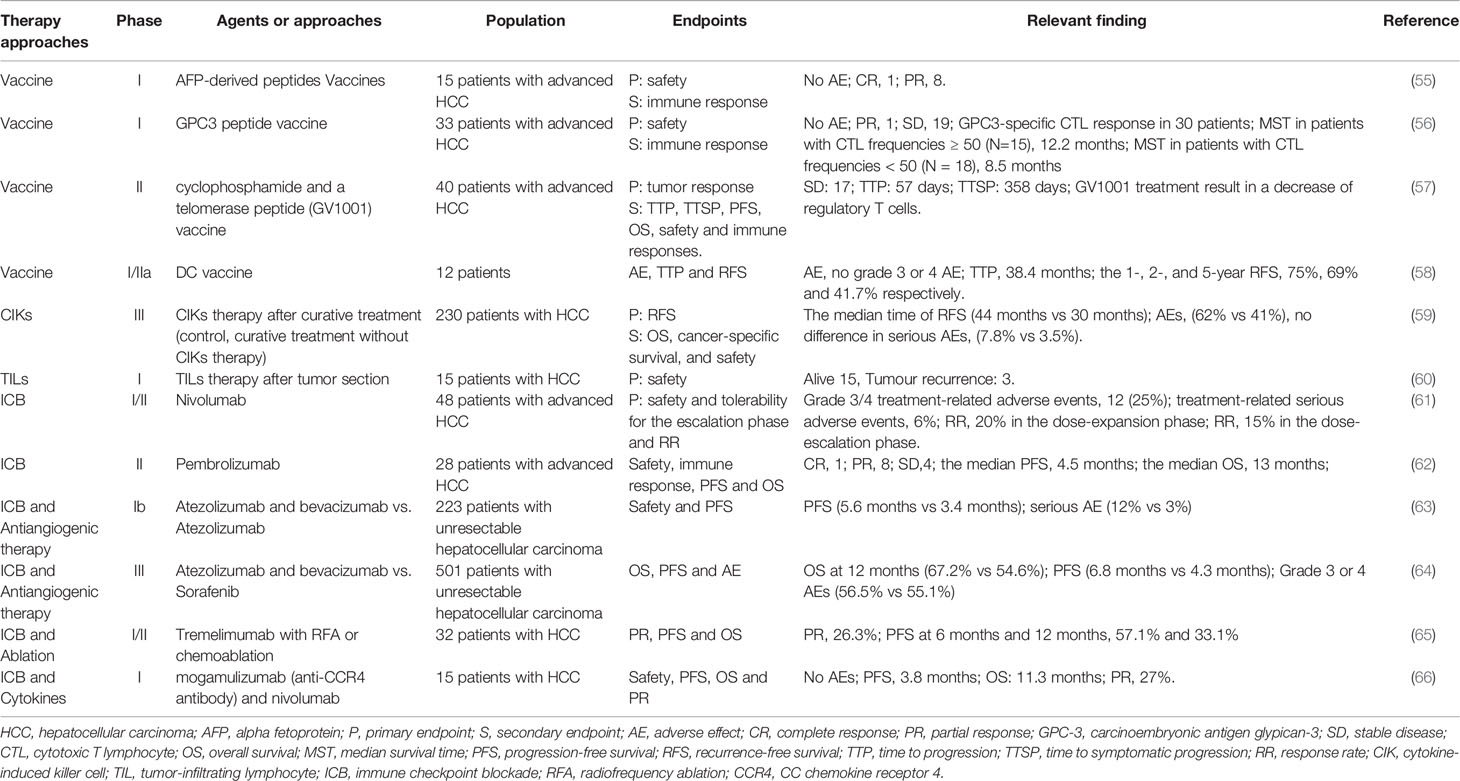
Table 1 Clinical trials of immunotherapy for HCC.
Vaccines
Vaccines have been widely used to prevent various diseases by providing active acquired immunity. Clinical studies using neoantigen peptide, mRNA, or DC vaccines in patients with melanoma have achieved promising results (55, 58, 67, 68). This antigen-based immunotherapy has also been tested for other tumors, such as ovarian cancer, breast cancer (56), and small-cell lung cancer (57). TAAs released from tumor lysates are considered to be optimal vaccines to activate immune response, but the low representation of the TAAs with high immunogenicity limits the clinical effect (69, 70).
Some vaccines are being evaluated for treating HCC. In a phase I trial, administration of AFP-derived peptides as an anti-tumor vaccine was explored in 15 patients with advanced HCC. The results demonstrated that the vaccine was safe and effective. The peptides stimulated the immune system to produce peptide-specific T-cell receptors (TCRs), with one patient showing a complete response and eight patients exhibiting slowing of tumor progression (71). In a phase I trial, a carcinoembryonic antigen glypican-3 (GPC3) peptide vaccine was explored for treating advanced HCC, with 30 of 31 patients (91%) showing a peptide-specific CTL response. For the clinical response among 33 patients, one patient showed a partial response and 19 had stable disease for 2 months (72). A telomerase peptide was also explored as a vaccine target for the treatment of advanced HCC in a phase II trial. No patients showed a complete or partial response, and 17 patients (45.9%) had stable disease for six months (73). Currently, a multi-epitope multi-HLA peptide vaccine is being evaluated in a phase I/II clinical trial for 40 patients with early and intermediate stages of HCC (HepaVac-101-NCT03203005). The results are extremely expected. In addition, lack of high immunogenic vaccines restricts the development of vaccine. A new prediction algorithm is needed for the identification of neoantigens with high immunogenicity, which may have unique homology compared with any human self-antigen and induce vigorous immune response (74–76).
Adoptive Cell Transfer
Patients receiving ACT therapy are directly treated with autologous natural or engineered anti-tumor T cells (77). The transferred cells can divide into three types, including cytokine-induced killer (CIK) cells, TILs, and genetically modified T cells. CIK cells and TILs can enhance the overall immune response by increasing the number of immune cells, whereas the genetically modified T cells target specific antigens.
Cytokine-Induced Killer Cells
CIK cells are a mixture of cytotoxic T cells and natural killer (NK) cells separated from peripheral blood mononuclear cells and are cultured in vitro under treatment with cytokines such as IFN-γ, anti-CD3 antibody and IL-2 to promote their proliferation and anti-tumor activities (78). Reinfusion of expanded and activated CIK cells either alone or as a combined therapeutic strategy has been widely studied to suppress tumor progression, with some impressive results observed in metastatic colorectal cancer, myeloid leukemia, and renal cell carcinoma (79–81). Some studies investigated the efficiency of ACT with CIK cells for HCC treatment. In a Korean phase III clinical trial, CIK cells, including CD3+/CD56+ cells, CD3-/CD56+ NK cells, and CD3+/CD56- cytotoxic T cells (82, 83), were used as an adjuvant treatment for 230 patients with HCC who had been pre-treated with other curative therapies (surgical resection, RFA, or percutaneous ethanol injection). The results showed that the adjuvant immunotherapy group with activated CIK cells had increased overall and recurrence-free survival compared with the control group without adjuvant therapy (median time of recurrence-free survival: 44 vs 30 months) (59).
Tumor-Infiltrating Lymphocytes
The presence of TILs in tumors is associated with good prognosis (60, 84). TILs are obtained from surgical tumor specimens and then cultured in vitro with sequential treatment with IL-2 for expansion and anti-CD3 antibody for activation. These proliferative and activated TILs are then transferred back into patients. ACT with TILs has been studied for the treatment of metastatic human papillomavirus-associated carcinomas, with clinical responses occurring in 5 of 18 (28%) patients in the cervical cancer group and 2 of 11 (18%) patients in the non-cervical cancer group (85). A phase I clinical trial confirmed the safety of ACT using TILs in patients with HCC: The toxicity and immune response of therapy with autologous TIL is being tested in an ongoing phase I clinical trial of patients with advanced HCC (ClinicalTrials.gov number: NCT01462903) (86).
Genetically Modified T Cells
Heterodimeric antibody receptors expressed on the surface of T cells are known to be tumor antigen-specific TCRs that recognize the antigenic peptide-MHC complex. The gene sequence of TCRs that recognize specific TAAs can be analyzed and introduced into autologous T cells by retroviral or lentiviral vectors (87). These proliferative and activated autologous modified TCR-expressing T cells are reinfused into patients. In response to tumor cells, the cells express the target antigen, leading to effective antitumor activity by releasing cytokines such as IFN-γ, granulocyte macrophage colony-stimulating factor, and tumor necrosis factor alpha-α and directly killing tumor cells (88). An AFP TCR with optimal affinity, function, and safety is being evaluated for its clinical efficacy in an early phase clinical trial (ClinicalTrials.gov number: NCT03971747) (89).
Despite the powerful anti-tumor function of immunotherapies based on the interaction between peptide-MHC molecules and TCRs, tumor cells can escape immune surveillance by down-regulating peptide-MHC complex expression (90). ACT with T cells engineered to express a chimeric antigen receptor (CAR) are not limited by the presentation of MHC molecules on the tumor cell surface. CAR can recognize a defined TAA on the surface of tumor cells via the single chain variable fragment region, which is constructed from the variable heavy and variable light sequences of a monoclonal antibody specific for TAAs. Activation signals are transferred into cells by activating the transmembrane adaptor signaling protein CD3ζ and one or more co-stimulatory molecules (CD28, CD137, or OX40). The mechanism of CAR therapy causes tumor variants, which can escape the immune surveillance through deficiencies in antigen presentation, to remain susceptible to CAR therapy (87). Other biomarkers have also been considered as targets for CAR T cell therapy. AFP is a well-known biomarker for HCC, and CAR T-cell therapy targeting the AFP-MHC complex showed robust antitumor activity in AFP-CAR T cells in a mouse xenograft model of liver cancer (91). Another attractive liver cancer-specific target is GPC3 because of its high expression in HCC but low expression in normal tissues (92). GPC3-CAR T cells efficiently eradicated GPC3+ HCC cells rather than GPC3- HCC cells. This approach showed high treatment efficiency in an HCC xenograft model with high levels of GPC3 expression and low treatment efficiency in HCC xenografts with low GPC3 expression (93). Another ACT study of GPC3-CAR T cell transfer into patient-derived HCC xenografts also revealed suppression of tumor cell growth (94).
Immune Checkpoint Inhibitors
The human immune system is in an equilibrium state. Immune checkpoints regulate immune function by suppressing immune activity, interrupting the immune response to avoid overactivation of T cells, and protecting tissues from damage caused by an excessive immune response. Immune checkpoints in tumor tissues promote immune evasion. Most studies of immune checkpoints focused on cytotoxic T-lymphocyte antigen-4 and PD-1 with corresponding PD-L1 ligands. The ICI approach results in nonspecific immune stimulation by targeting negative regulators of T cell signaling pathways.
CTLA-4 Inhibitors
The hepatic microenvironment contains a large number of DCs, which are the major antigen-presenting cells in the liver (95). In a normal hepatic microenvironment, DCs take up foreign peptides and present them to T cells via the TCR (signal 1). In addition to signal 1, activation of T cells requires co-stimulatory molecules from DCs. After stimulation by the peptide-MHC complex, DCs present CD80 and CD86 to T cells and bind to the CD28 receptor on the surface of T cells (signal 2) and further promote maturation, proliferation, activation, and survival of naïve T cells. Signal 2 prevents the recognition of self-antigens, whereas the absence of such a signal leads to T cell anergy. Upon activation, T cells induce CTLA-4 to competitively bind to CD80 and CD86 with higher affinity than CD28, to prevent an excessive immune reaction (96). CTLA-4 inhibitors prevent CTLA-4 from binding to CD80 and CD86, thereby initiating signal 2, which can activate specific T cells in lymphoid organs and promote their migration into the tumor (97).
Two anti-CTLA-4 monoclonal antibodies, ipilimumab and tremelimumab, are being evaluated in clinical trials for the treatment of other tumors. Studies have shown that CTLA-4 inhibitors deplete Treg cells in the tumor, leading to enhanced effector function of antigen-specific T cells in the tumor. Patients with melanoma and administered ipilimumab exhibit Treg cells depletion (98, 99). However, the clinical data associated with application of CTLA-4 inhibitors alone for advanced HCC are limited. A clinical trial of tremelimumab in patients with HCC and chronic hepatitis C revealed a partial response rate of 17.6% and disease control rate of 76.4% (ClinicalTrials.gov number: NCT01008358) (100).
PD-1/PD-L1 Inhibitors
The cell surface receptor PD-1 is expressed on activated T, B, and NK cells and binds PD-L1 and PD-L2 ligands to convey co-inhibitory signals to the TCR. The PD-1 signal terminates immune responses appropriately and maintains self-tolerance by causing apoptosis of antigen‐specific T cells, attenuation of TCR‐mediated activation, and proliferation of T cells (101). The PD-1 signal mediates the function of Treg cells by promoting their differentiation and proliferation (102). These ligands are expressed on leukocytes and tumor cells (103). PD-L1 binding results in phosphorylation of PD-1, inhibiting T cell proliferation and cytokine releasing through SHP2. SHP2 dephosphorylation results in dephosphorylation of key TCR signaling components, most notably CD28 and the ZAP70/CD3zeta signalosome (104, 105). When chronically exposed to antigens, overexpression of PD-1 in T cell induces their exhaustion.
Anti-PD-1 monoclonal antibodies, such as nivolumab and pembrolizumab, and anti-PD-L1 monoclonal antibodies, such as durvalumab and atezolizumab, have been approved for several hematologic and solid malignancies. Many clinical trials for HCC are underway. In a phase I/II of escalation trial, safety was evaluated in 48 patients treated with nivolumab, with grade ≥3 adverse events observed in 31% of patients (15 of 48), which was considered to be a manageable safety profile (61). A phase II study of the efficacy of pembrolizumab in 28 patients showed that one patient achieved a complete response and eight patients achieved partial responses (62).
Limits of Current Immunotherapies
Although immunotherapy has shown promising clinical outcomes in some tumors, including melanoma, non‐small cell lung carcinoma, and urothelial carcinoma, the application in HCC faces some limitations (Figure 1) (106–108).
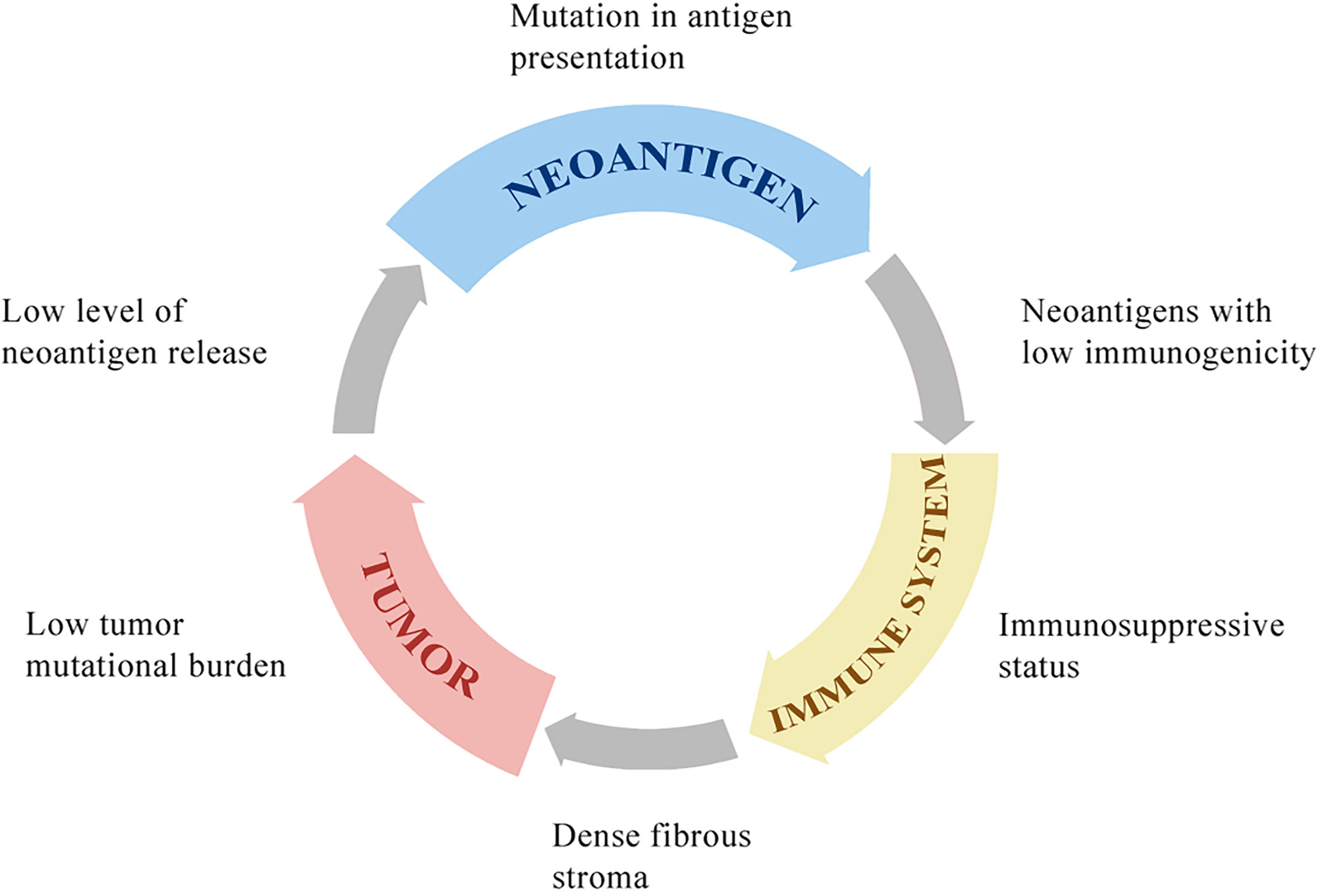
Figure 1 The potential mechanisms of resistance to immunotherapies. HCC with low tumor mutational burden releases few neoantigens. The mutation in antigen presentation pathways also inhibits tumor-specific peptide presentation. Most of these neoantigens cannot drive effective anti-tumor immunity because of low immunogenicity. The immune system in the tumor microenvironment is under immunosuppressive status, with few effector CD8+ T cells, many regulatory CD4+ T cells, and other immunosuppressive cells, which is associated with poor clinical response to immunotherapy. The dense fibrous stroma around tumor islets inhibits immune cells’ access to the tumor.
Tumor Mutational Burden
Immunotherapies are ineffective for HCC because of the low tumor mutational burden of HCC compared to that of melanoma or non‐small cell lung carcinoma (109). Neoantigens are tumor-specific peptides that result from somatic mutations in cancer cells. A larger number of somatic mutations is associated with higher levels of neoantigens, and the tumor mutational burden is used to evaluate somatic mutations in cancer to give a useful estimation of the tumor neoantigen load (110, 111). Neoantigens are newly expressed antigens on the surface of tumor cells and can be recognized and presented to T cells to result in adaptive immune response activation. However, even in tumors with a high tumor mutational burden, such as those with deficiencies in DNA damage repair pathways resulting in the accumulation of DNA mutations, a high mutational load is not related to high levels of neoantigens. In fact, only a minority of mutations generates peptides that bind to MHC molecules and present on the surface of tumor cells, and fewer can be recognized by T cells (67, 112). The antigen presentation pathway in tumor cells can be inhibited by mutations in antigen presentation genes. For example, in metastatic melanomas, the loss of b2-microglobulin may result in defects in antigen presentation and escape from immune recognition (113). Not all neoantigens presented on the surface of tumor cells can drive effective antitumor immunity. A study reported that neoantigens could be expressed on either all tumor cells (clonal) or a subpopulation of tumor cells (subclonal). Tumors with a high load of clonal neoantigens show an excellent response to ICI therapy, whereas tumors with a high load of subclonal neoantigens evade immunotherapy (114) (Figure 2).
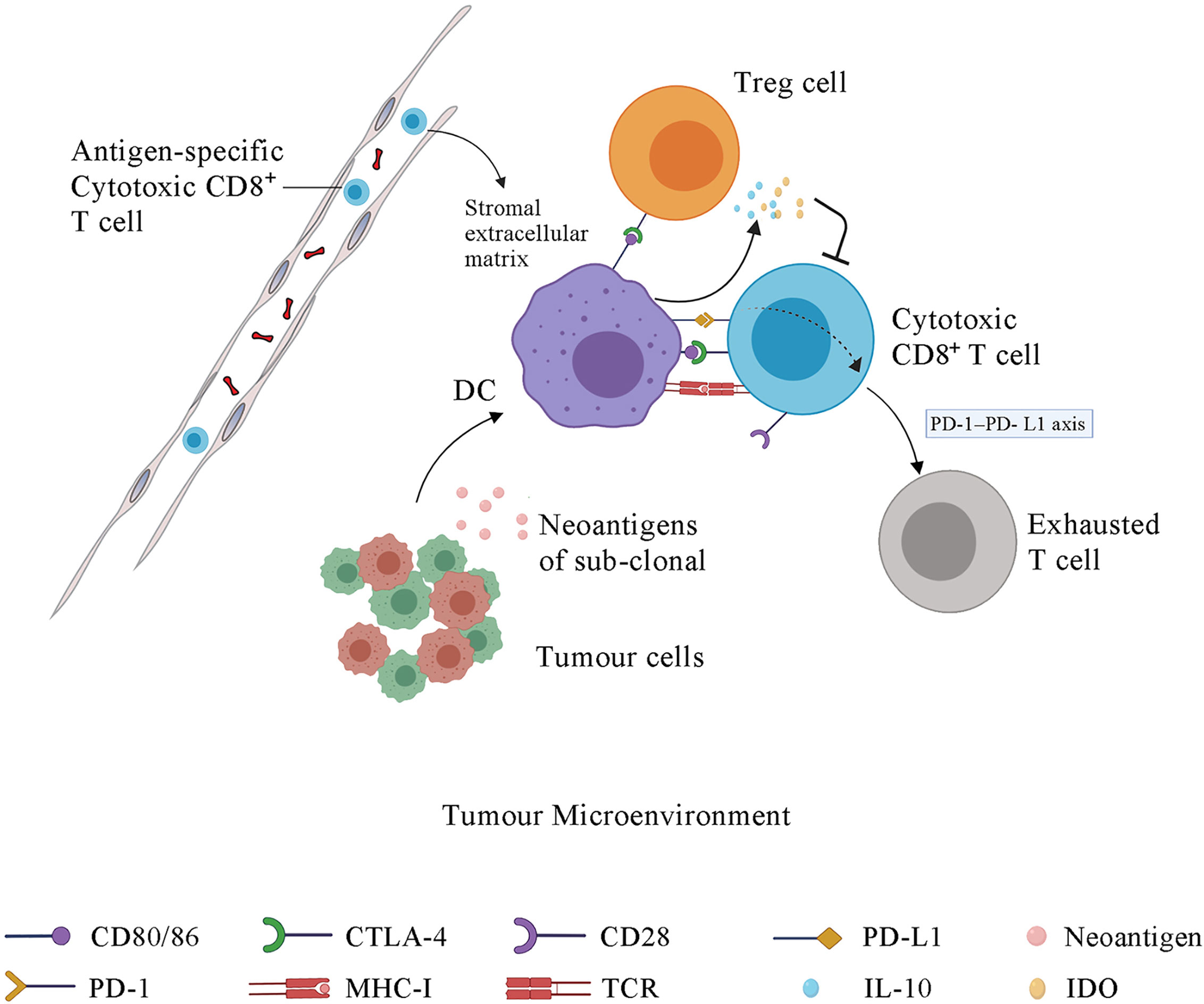
Figure 2 The immune response in tumor microenvironment and the function of immune checkpoints. Some sub-clonal tumor cells release neoantigens while others do not, contributing to the immune response to only part of tumor cells and thus leading to the failure of tumor immunotherapy. Upon antigen recognition, DCs present the antigen-MHC molecules, bind to the TCR on T cell membrane and stimulate the proliferation and activation of CD4+ T cells and CD8+ T cells in lymph node. Then the antigen-specific cytotoxic T cells migrate to tumor microenvironment via blood system. The stromal extracellular matrix in tumor may prevents T cell infiltration. CTLA-4, which is the membrane receptor of activated T cells, outcompetes CD28 for binding to the CD80/86 expressed on the DC membrane, further inhibiting the signal 2, which is essential for the maturation, proliferation, activation and survival of T cells. The interaction of PD-1 and PD-L1 promotes the differentiation and proliferation of Treg cells and induces the cytotoxic CD8+ T cells into an exhausted state. DCs under the influence of CLTA-4 signal and PD-1 signal release some immunosuppressive molecules, such as IL-10 and IDO, which suppress T cells activation. IDO, indoleamine 2,3-dioxygenase; PD-1, programmed cell death protein 1; PD-L1, programmed cell death 1 ligand 1; CTLA-4, cytotoxic T-lymphocyte protein 4; DC, dendritic cell; Treg cell, regulatory T cell.
Tumor Microenvironment
The tumor microenvironment (TME) contains many components, including bone marrow inflammatory cells, lymphocytes, blood vessels, fibroblastic cells, and the extracellular-derived matrix composed of collagen and proteoglycans. The clinical efficacy of ICI depends on three tumor immune status characteristics. First, antigen-specific CD8+ T cells must be present within the TME. Second, the resident immune cell populations in the TME must be polarized towards an immune permissive state. Third, tumor cells must have MHC class I-mediated antigen presentation and PD-1 signaling as the dominant mechanism of immune tolerance. A tumor with these characteristics is vulnerable to ICIs and named as an immune “hot” tumor. Immune “cold” tumors lack these characteristics and are associated with poor clinical response to ICI therapy (115). The absence of CD8+ T cells in the TME in several tumor types has been associated with poor clinical outcomes of ICI therapy (116–118). A study of the stroma of human lung tumors showed that the stromal extracellular matrix influences the migration and positioning of T cells (119) (Figure 2).
Efforts to Enhance Immunotherapy for HCC
Although the immunotherapy approaches discussed previously have achieved impressive clinical efficacy in other tumors, they have failed to benefit patients with advanced HCC. The limitations of these approaches are discussed above. Here, we summarize some potential combinatorial strategies for enhancing the effects of immunotherapy for HCC.
Epigenetic Modulation
Epigenetic modification plays an important role in tumor progression, causing transcriptional aberrations in gene expression and immune function changes, which may result in a favorable TME (120). In contrast, epigenetic therapy has the potential to enhance immunotherapy for HCC by converting an immune “cold” tumor into an immune “hot” tumor (121). Epigenetic therapy can promote the expression of immunogenic antigens on the tumor surface such as cancer testis antigens (122–125). Cancer testis antigens are a group of proteins expressed on male germ cells but not in healthy adult somatic tissues and can serve as target antigens for antitumor immunotherapy (126, 127).
Epigenetic modification can regulate the composition of immune cell populations. Methylation of DNA represses genes related to effector function, proliferation, metabolic activity, and tissue homing of exhausted T cells. Chronic antigen stimulation drives CD8+ effector T cells towards the exhausted phenotype, which is characterized by a series of changes in gene expression associated with alterations in methylation, leading to increased PD-1 expression and decreased CXCR3 expression (128). De novo DNA methylation is essential for establishing exhaustion in T cells, whereas treatment with ICI contributes to rejuvenation of exhausted T cells (128) (Figure 3). Azacitidine and histone deacetylase inhibitors have been shown to suppress MYC signaling, activate interferon responsiveness, and potentiate the recruitment of T cells in mouse models of non‐small cell lung carcinoma (129). An EZH2 inhibitor (DZNep) and DNMT1 c (5-azacytidine) can augment anti-PD-L1 immunotherapy for HCC by increasing the release of the chemokines CXCL9 and CXCL10, which stimulate T cell trafficking into the TME. This combination therapy strategy can also upregulate the expression of cancer testis antigens New York esophageal squamous cell carcinoma-1 and L antigen family member, which are normally expressed at low levels, as neoantigens to stimulate the adaptive immune response (130). The potential therapeutic strategy combined with epigenetic modulation has emerged in recent years and is promising for treating HCC.
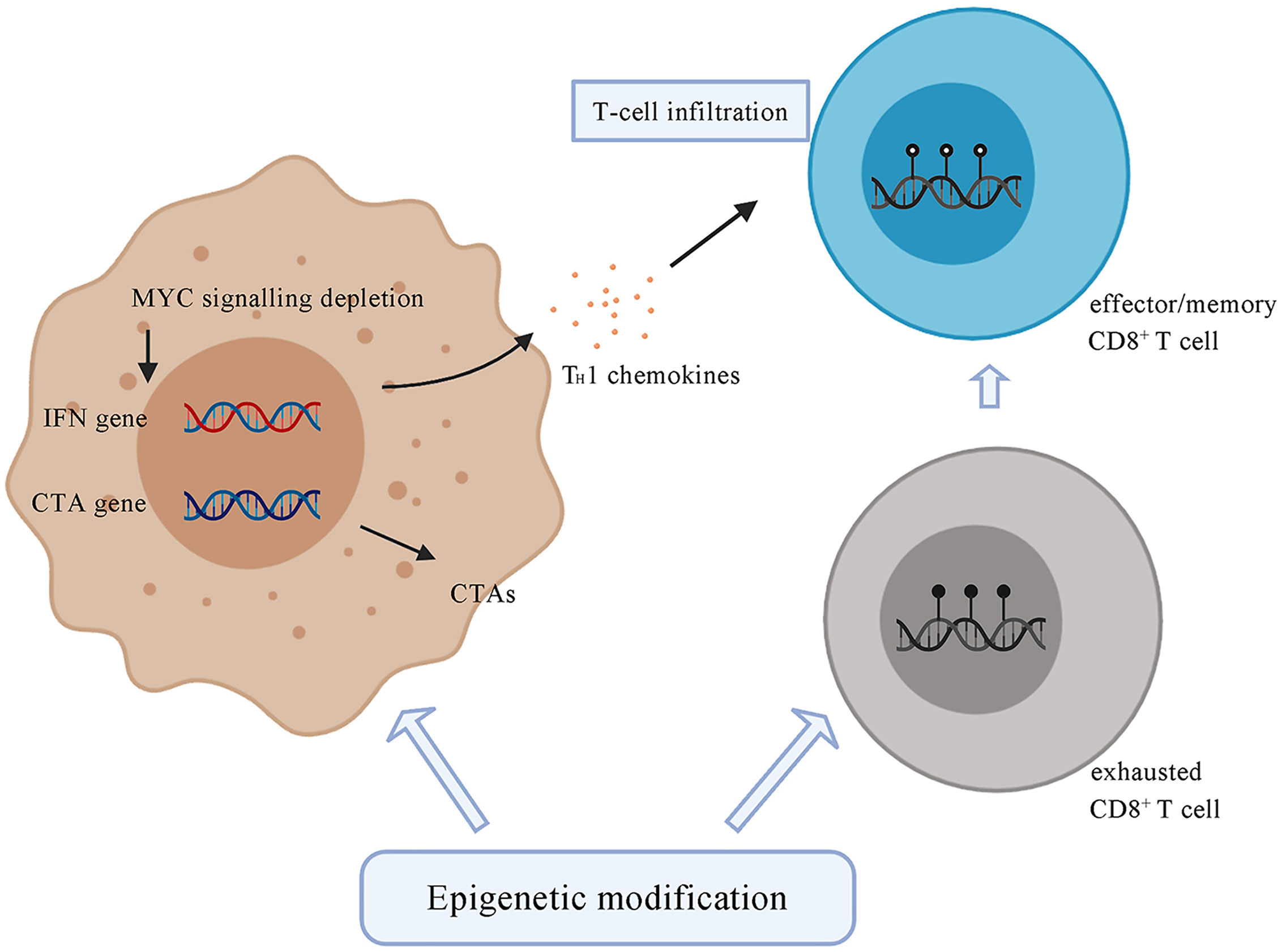
Figure 3 Tumor cells under the treatment of epigenetic drugs upregulate the expression of CTAs, such as NY-ESO-1 and LAGE. Epigenetic modification contributes to the depletion of MYC signalling, activates type I interferon signalling and potentiates the recruitment of T cells. Epigenetic agents can modulate the state of CD8+ T cells by transforming exhausted CD8+ T cells, which are characterized by a series of changes in effector genes associated with alterations in methylation, into effector or memory CD8+ T cells. CTA, cancer testis antigens; NY-ESO-1, New York Esophageal Squamous Cell Carcinoma-1; LAGE, L antigen family member.
Antiangiogenic Therapy
The hypoxia microenvironment stimulates tumor angiogenesis and promotes HCC development. Drugs target angiogenic pathways, including vascular endothelial growth factor (VEGF), are approved for the treatment of advanced HCC (131). Anti-VEGF therapy are widely used in HCC treatment (132). Sorafenib, a tyrosine kinase inhibitor (TKI), can disturb VEGF signaling pathway and approved for HCC treatment (133, 134). Despite survival benefits observed, the high rate of acquired resistance to sorafenib limits its use for advanced HCC treatment.
Despite of high rate of resistance to anti-VEGF drugs for HCC patients, some studies have reported that these drugs can enhance immune response. Drugs targeting VEGF-A/VEGFR-2 axis inhibited Treg cells accumulation in colorectal cancer (135). A VEGFR-2 inhibitor (DC101) promoted tumor-specific CD8+ T cells infiltration (136). These findings promote the combined strategies of anti-VEGF drugs and ICBs for HCC treatment. Bevacizumab is an anti-VEGF agent approved to treat metastatic colorectal cancer, glioblastoma, renal cell cancer and cervical cancer (137–139). However, the clinical efficacy of Bevacizumab for HCC treatment was less, with 13% response rates in a phase II study (140). Recent studies focus on the combination of anti-VEGF therapy and immunotherapy. In an open-label, multicenter, multiarm, phase Ib study, atezolizumab plus bevacizumab shows optional results, with longer progression-free survival compare with atezolizumab alone for patients with unresectable HCC (63). A similar result is showed in another clinical trial, the combination strategy of atezolizumab and bevacizumab for HCC treatment showed better overall and progression-free survival outcomes than sorafenib in 501 patients with unresectable HCC in a global, multicenter, open-label, phase III trial (ClinicalTrials.gov number: NCT03434379) (64). Other anti-VEGF drugs are also being evaluated in the combined therapy with immunotherapy for HCC treatment (ClinicalTrials.gov number: NCT03170960 (Cabozantinib and Atezolizumab) and NCT03006926 (Lenvatinib and Pembrolizumab)).
Inducing the Formation of Tertiary Lymphoid Structures
Although intra-tumoral infiltration by immune cells is a predictor of sensitivity to ICI treatment and many studies have focused on the role of T cell in antitumor responses, other immune cells have not been widely examined. Recently, studies revealed that the presence of intra-tumoral tertiary lymphoid structures (TLSs) improves ICI treatment of melanoma (141). TLSs are ectopic lymphoid aggregates that reflect lymphoid neogenesis occurring in non-lymphoid tissues in response to chronic inflammation, characterized by mature DCs in a T-cell zone adjacent to B-cell follicles including a germinal center (142, 143). TLSs are found in most types of cancer, with high TLS densities associated with improved clinical outcomes (144). In HCC, intra-tumoral TLSs are correlated with a decreased risk of early HCC recurrence after surgical resection, which may reflect ongoing, effective antitumor immunity (145). Therapeutic strategies to induce the formation of TLSs may enhance the antitumor immunotherapy of HCC (145). A reagent targeting LIGHT, a member of tumor necrosis factor superfamily of cytokines, can induce the formation of TLSs and can be combined with ICI to increase the number of TILs, conferring a survival benefit in mice with insulinomas (146). Other strategies aimed at stromal cells, which participate in the establishment of TLSs (147). Stromal cells derived from lymph nodes and induce TLSs cause infiltration of host immune cell subsets to suppress tumor growth in vivo (148).
Locoregional Therapy
Locoregional therapies such as RFA can be as efficient as surgical resection of HCC nodules (149) but patients treated with this therapy frequently experience cancer recurrence. Although it is not effective as monotherapy, locoregional therapy causes tumor cell death via the release of tumor antigens and stimulation antitumor immunity (150), named as immunogenic cell death (ICD). ICD may enhance the anti-tumor immune reaction through the antigens and adjuvants released during this process. ICD of tumor cells results in the release of neoantigens which may be recognized by DCs followed by activation of the adaptive immune response (151). Moreover, heat shock proteins induced by RFA have been shown to enhance the immune response by activating the natural immune response and augmenting the antigen-specific cytotoxic T-cell response (152–154). Although the effect of immune activation by locoregional therapy alone is not sufficient for treating HCC, it may be an effective adjuvant for immunotherapy (150).
Tremelimumab combined with RFA or chemoablation for advanced HCC resulted a partial response in 26.3% of patients (5 of 19), with a clear increase in CD8+ T cells. Progression-free survival rates at 6 and 12 months were 57.1% and 33.1%, respectively, and the median overall survival was 12.3 months (65).
Chemotherapy
Chemotherapeutic drugs alone for treating HCC, such as oxaliplatin, have shown limited effects on the overall survival of patients with advanced HCC. These cytotoxic drugs induce tumor cell death, which may also stimulate anti-tumor immunity by induced ICD. The cytotoxic effect also induces a decrease in the immunosuppressive cell population, such as MDSCs and Treg cells (155, 156). High-dose chemotherapy, which is the proper strategy for the treatment of HCC, leads to the death of both tumor and immune cells. The suppressed immune system then loses its function and no longer targets therapy-resistant tumor cells.
Low dose metronomically administered chemotherapy can increase the ablation of immunosuppressive Treg cells (156, 157), promote the maturation and activation of DCs (158, 159), and improve the activation and functionality of cytotoxic NK and CD8+ T cells (160). Treatment with metronomic cyclophosphamide affected gliomas by activating anti-tumor CD8+ T cell responses and immune memory in an immune-competent mouse model with implanted GL261 glioma (160). Pre-treatment with metronomic chemotherapy for HCC may enhance the effect of ICI and avoid unacceptable toxicity (161).
Cytokines
Although cytokines have multiple functions in the formation of the immune system, cytokine treatment alone as an immunotherapy for HCC is limited. IFN-α was the first immunotherapy tested in many clinical trials. Although IFN-α has anti-proliferative, immunostimulatory, and anti-angiogenesis properties (162), most trials failed to show clinical benefits (163, 164).
Overexpression of cytokine CCL5 in CTNNB1-mutant HCC cells led to the recruitment of CD103+ DCs and antigen-specific CD8+ T cells, which may enhance the clinical outcome of ICI therapy (165). CCR4 expressed by Treg cells can suppress anti-tumor immune response. In a phase I study, the safety and efficacy of combined mogamulizumab (anti-CCR4 antibody) and nivolumab are evaluated for patients with HCC, with four (27%) tumor responses among 15 patients. During treatment, the immune system activated with population of Treg cells decreased and effector CD8+ cells increased (66). Although immunotherapy using cytokines alone is limited for treating HCC, the potential advantage of cytokines as adjuvants to enhance the clinical efficacy of immunotherapy is promising.
Conclusion
Immunotherapy as monotherapy or combined with other therapeutic strategies has demonstrated clinical efficacy. Although some patients benefit from these therapeutic approaches, most patients suffering from advanced HCC do not. Novel immunotherapy strategies are currently being evaluated.
Author Contributions
Conceptualization, XF and HL. Resources, CZ. Writing – Original Draft Preparation, CZ and YL. Writing – Review and Editing, CZ and SJ. Visualization, GC and DL. Supervision, JY, XF and HL. Funding Acquisition, HL. All authors contributed to the article and approved the submitted version.
Funding
We are greatly indebted to the subjects enrolled in our study. This work was supported by the National Key Research and Development Program (2016YFC0906400), National Natural Science Foundation of China (81400656, 81872297, and 81874059), Zhejiang province analysis and test technology project (2018C37062), the Fundamental Research Funds for the Central Universities(2016XZZX002-05).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2018) 68(6):394–424. doi: 10.3322/caac.21492
2. Parikh S, Hyman D. Hepatocellular cancer: a guide for the internist. Am J Med (2007) 120(3):194–202. doi: 10.1016/j.amjmed.2006.11.020
3. Burrel M, Reig M, Forner A, Barrufet M, de Lope CR, Tremosini S, et al. Survival of patients with hepatocellular carcinoma treated by transarterial chemoembolisation (TACE) using Drug Eluting Beads. Implications for clinical practice and trial design. J Hepatol (2012) 56(6):1330–5. doi: 10.1016/j.jhep.2012.01.008
4. EASL. Clinical Practice Guidelines: Management of hepatocellular carcinoma. J Hepatol (2018) 69(1):182–236. doi: 10.1016/j.jhep.2018.03.019
5. Malagari K, Pomoni M, Moschouris H, Bouma E, Koskinas J, Stefaniotou A, et al. Chemoembolization with doxorubicin-eluting beads for unresectable hepatocellular carcinoma: five-year survival analysis. Cardiovasc Intervent Radiol (2012) 35(5):1119–28. doi: 10.1007/s00270-012-0394-0
6. Heymann F, Tacke F. Immunology in the liver–from homeostasis to disease. Nat Rev Gastroenterol Hepatol (2016) 13(2):88–110. doi: 10.1038/nrgastro.2015.200
7. El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology (2012) 142(6):1264–73.e1. doi: 10.1053/j.gastro.2011.12.061
8. Yang JD, Roberts LR. Hepatocellular carcinoma: A global view. Nat Rev Gastroenterol Hepatol (2010) 7(8):448–58. doi: 10.1038/nrgastro.2010.100
9. Zongyi Y, Xiaowu L. Immunotherapy for hepatocellular carcinoma. Cancer Lett (2020) 470:8–17. doi: 10.1016/j.canlet.2019.12.002
10. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Sci (New York NY) (2013) 339(6127):1546–58. doi: 10.1126/science.1235122
11. Kudo M. Multistep human hepatocarcinogenesis: correlation of imaging with pathology. J Gastroenterol (2009) 44 Suppl 19:112–8. doi: 10.1007/s00535-008-2274-6
12. Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases–elimination, equilibrium and escape. Curr Opin Immunol (2014) 27:16–25. doi: 10.1016/j.coi.2014.01.004
13. Pan K, Wang H, Chen M-s, Zhang H-k, Weng D-s, Zhou J, et al. Expression and prognosis role of indoleamine 2,3-dioxygenase in hepatocellular carcinoma. J Cancer Res Clin Oncol (2008) 134(11):1247–53. doi: 10.1007/s00432-008-0395-1
14. Yan W, Liu X, Ma H, Zhang H, Song X, Gao L, et al. Tim-3 fosters HCC development by enhancing TGF-β-mediated alternative activation of macrophages. Gut (2015) 64(10):1593–604. doi: 10.1136/gutjnl-2014-307671
15. Tagliamonte M, Mauriello A, Cavalluzzo B, Ragone C, Manolio C, Petrizzo A, et al. Tackling hepatocellular carcinoma with individual or combinatorial immunotherapy approaches. Cancer Lett (2020) 473:25–32. doi: 10.1016/j.canlet.2019.12.029
16. Ding W, Xu X, Qian Y, Xue W, Wang Y, Du J, et al. Prognostic value of tumor-infiltrating lymphocytes in hepatocellular carcinoma: A meta-analysis. Medicine (2018) 97(50):e13301. doi: 10.1097/MD.0000000000013301
18. Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, et al. TOX transcriptionally and epigenetically programs CD8 T cell exhaustion. Nature (2019) 571(7764):211–8. doi: 10.1038/s41586-019-1325-x
19. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol (2015) 15(8):486–99. doi: 10.1038/nri3862
20. Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol (2009) 10(1):29–37. doi: 10.1038/ni.1679
21. McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu Rev Immunol (2019) 37:457–95. doi: 10.1146/annurev-immunol-041015-055318
22. Wu K, Kryczek I, Chen L, Zou W, Welling TH. Kupffer cell suppression of CD8+ T cells in human hepatocellular carcinoma is mediated by B7-H1/programmed death-1 interactions. Cancer Res (2009) 69(20):8067–75. doi: 10.1158/0008-5472.CAN-09-0901
23. Zhou G, Sprengers D, Boor PPC, Doukas M, Schutz H, Mancham S, et al. Antibodies Against Immune Checkpoint Molecules Restore Functions of Tumor-Infiltrating T Cells in Hepatocellular Carcinomas. Gastroenterology (2017) 153(4):1107–19.e10. doi: 10.1053/j.gastro.2017.06.017
24. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell (2008) 133(5):775–87. doi: 10.1016/j.cell.2008.05.009
25. Vignali DAA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol (2008) 8(7):523–32. doi: 10.1038/nri2343
26. Josefowicz SZ, Lu L-F, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol (2012) 30:531–64. doi: 10.1146/annurev.immunol.25.022106.141623
27. Gao Q, Qiu SJ, Fan J, Zhou J, Wang XY, Xiao YS, et al. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol (2007) 25(18):2586–93. doi: 10.1200/jco.2006.09.4565
28. Chen KJ, Lin SZ, Zhou L, Xie HY, Zhou WH, Taki-Eldin A, et al. Selective recruitment of regulatory T cell through CCR6-CCL20 in hepatocellular carcinoma fosters tumor progression and predicts poor prognosis. PloS One (2011) 6(9):e24671. doi: 10.1371/journal.pone.0024671
29. Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell (2018) 174(5):1293–308.e36. doi: 10.1016/j.cell.2018.05.060
30. De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, et al. Transcriptional Landscape of Human Tissue Lymphocytes Unveils Uniqueness of Tumor-Infiltrating T Regulatory Cells. Immunity (2016) 45(5):1135–47. doi: 10.1016/j.immuni.2016.10.021
31. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299(5609):1057–61. doi: 10.1126/science.1079490
32. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4(4):330–6. doi: 10.1038/ni904
33. Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity (2009) 30(5):636–45. doi: 10.1016/j.immuni.2009.04.010
34. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol (2016) 37(3):208–20. doi: 10.1016/j.it.2016.01.004
35. Chiu DK-C, Tse AP-W, Xu IM-J, Di Cui J, Lai RK-H, Li LL, et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat Commun (2017) 8(1):517. doi: 10.1038/s41467-017-00530-7
36. Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Krüger C, Manns MP, et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology (2008) 135(1):234–43. doi: 10.1053/j.gastro.2008.03.020
37. Prieto J, Melero I, Sangro B. Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol (2015) 12(12):681–700. doi: 10.1038/nrgastro.2015.173
38. Wang B, Li Q, Qin L, Zhao S, Wang J, Chen X. Transition of tumor-associated macrophages from MHC class II(hi) to MHC class II(low) mediates tumor progression in mice. BMC Immunol (2011) 12:43. doi: 10.1186/1471-2172-12-43
39. Qian B, Deng Y, Im JH, Muschel RJ, Zou Y, Li J, et al. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PloS One (2009) 4(8):e6562. doi: 10.1371/journal.pone.0006562
40. Yeung OW, Lo CM, Ling CC, Qi X, Geng W, Li CX, et al. Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma. J Hepatol (2015) 62(3):607–16. doi: 10.1016/j.jhep.2014.10.029
41. Wu Q, Zhou W, Yin S, Zhou Y, Chen T, Qian J, et al. Blocking Triggering Receptor Expressed on Myeloid Cells-1-Positive Tumor-Associated Macrophages Induced by Hypoxia Reverses Immunosuppression and Anti-Programmed Cell Death Ligand 1 Resistance in Liver Cancer. Hepatology (2019) 70(1):198–214. doi: 10.1002/hep.30593
42. Liu F, Liu Y, Chen Z. Tim-3 expression and its role in hepatocellular carcinoma. J Hematol Oncol (2018) 11(1):126. doi: 10.1186/s13045-018-0667-4
43. Wan S, Zhao E, Kryczek I, Vatan L, Sadovskaya A, Ludema G, et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology (2014) 147(6):1393–404. doi: 10.1053/j.gastro.2014.08.039
44. Bartneck M, Schrammen PL, Möckel D, Govaere O, Liepelt A, Krenkel O, et al. The CCR2 Macrophage Subset Promotes Pathogenic Angiogenesis for Tumor Vascularization in Fibrotic Livers. Cell Mol Gastroenterol Hepatol (2019) 7(2):371–90. doi: 10.1016/j.jcmgh.2018.10.007
45. Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev (2008) 88(1):125–72. doi: 10.1152/physrev.00013.2007
46. Coulouarn C, Clément B. Stellate cells and the development of liver cancer: therapeutic potential of targeting the stroma. J Hepatol (2014) 60(6):1306–9. doi: 10.1016/j.jhep.2014.02.003
47. Charles R, Chou H-S, Wang L, Fung JJ, Lu L, Qian S. Human hepatic stellate cells inhibit T-cell response through B7-H1 pathway. Transplantation (2013) 96(1):17–24. doi: 10.1097/TP.0b013e318294caae
48. Zhao W, Zhang L, Yin Z, Su W, Ren G, Zhou C, et al. Activated hepatic stellate cells promote hepatocellular carcinoma development in immunocompetent mice. Int J Cancer (2011) 129(11):2651–61. doi: 10.1002/ijc.25920
49. Höchst B, Schildberg FA, Sauerborn P, Gäbel YA, Gevensleben H, Goltz D, et al. Activated human hepatic stellate cells induce myeloid derived suppressor cells from peripheral blood monocytes in a CD44-dependent fashion. J Hepatol (2013) 59(3):528–35. doi: 10.1016/j.jhep.2013.04.033
50. Dunham RM, Thapa M, Velazquez VM, Elrod EJ, Denning TL, Pulendran B, et al. Hepatic stellate cells preferentially induce Foxp3+ regulatory T cells by production of retinoic acid. J Immunol (Baltimore Md 1950) (2013) 190(5):2009–16. doi: 10.4049/jimmunol.1201937
51. Limmer A, Ohl J, Kurts C, Ljunggren HG, Reiss Y, Groettrup M, et al. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T-cell tolerance. Nat Med (2000) 6(12):1348–54. doi: 10.1038/82161
52. Knolle PA, Schmitt E, Jin S, Germann T, Duchmann R, Hegenbarth S, et al. Induction of cytokine production in naive CD4(+) T cells by antigen-presenting murine liver sinusoidal endothelial cells but failure to induce differentiation toward Th1 cells. Gastroenterology (1999) 116(6):1428–40. doi: 10.1016/s0016-5085(99)70508-1
53. Wu S-D, Ma Y-S, Fang Y, Liu L-L, Fu D, Shen X-Z. Role of the microenvironment in hepatocellular carcinoma development and progression. Cancer Treat Rev (2012) 38(3):218–25. doi: 10.1016/j.ctrv.2011.06.010
54. Hernandez-Gea V, Toffanin S, Friedman SL, Llovet JM. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology (2013) 144(3):512–27. doi: 10.1053/j.gastro.2013.01.002
55. Sahin U, Derhovanessian E, Miller M, Kloke B-P, Simon P, Löwer M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature (2017) 547(7662):222–6. doi: 10.1038/nature23003
56. Kalli KR, Block MS, Kasi PM, Erskine CL, Hobday TJ, Dietz A, et al. Folate Receptor Alpha Peptide Vaccine Generates Immunity in Breast and Ovarian Cancer Patients. Clin Cancer Res an Off J Am Assoc Cancer Res (2018) 24(13):3014–25. doi: 10.1158/1078-0432.CCR-17-2499
57. Chiappori AA, Williams CC, Gray JE, Tanvetyanon T, Haura EB, Creelan BC, et al. Randomized-controlled phase II trial of salvage chemotherapy after immunization with a TP53-transfected dendritic cell-based vaccine (Ad.p53-DC) in patients with recurrent small cell lung cancer. Cancer Immunol Immunother (2019) 68(3):517–27. doi: 10.1007/s00262-018-2287-9
58. Lee JH, Lee Y, Lee M, Heo MK, Song JS, Kim KH, et al. A phase I/IIa study of adjuvant immunotherapy with tumour antigen-pulsed dendritic cells in patients with hepatocellular carcinoma. Br J Cancer (2015) 113(12):1666–76. doi: 10.1038/bjc.2015.430
59. Lee JH, Lee J-H, Lim Y-S, Yeon JE, Song T-J, Yu SJ, et al. Adjuvant immunotherapy with autologous cytokine-induced killer cells for hepatocellular carcinoma. Gastroenterology (2015) 148(7):1383–91.e6. doi: 10.1053/j.gastro.2015.02.055
60. Adams S, Gray RJ, Demaria S, Goldstein L, Perez EA, Shulman LN, et al. Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J Clin Oncol (2014) 32(27):2959–66. doi: 10.1200/jco.2013.55.0491
61. El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet (2017) 389(10088):2492–502. doi: 10.1016/S0140-6736(17)31046-2
62. Feun LG, Li Y-Y, Wu C, Wangpaichitr M, Jones PD, Richman SP, et al. Phase 2 study of pembrolizumab and circulating biomarkers to predict anticancer response in advanced, unresectable hepatocellular carcinoma. Cancer (2019) 125(20):3603–14. doi: 10.1002/cncr.32339
63. Lee MS, Ryoo BY, Hsu CH, Numata K, Stein S, Verret W, et al. Atezolizumab with or without bevacizumab in unresectable hepatocellular carcinoma (GO30140): an open-label, multicentre, phase 1b study. Lancet Oncol (2020) 21(6):808–20. doi: 10.1016/s1470-2045(20)30156-x
64. Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med (2020) 382(20):1894–905. doi: 10.1056/NEJMoa1915745
65. Duffy AG, Ulahannan SV, Makorova-Rusher O, Rahma O, Wedemeyer H, Pratt D, et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J Hepatol (2017) 66(3):545–51. doi: 10.1016/j.jhep.2016.10.029
66. Doi T, Muro K, Ishii H, Kato T, Tsushima T, Takenoyama M, et al. A Phase I Study of the Anti-CC Chemokine Receptor 4 Antibody, Mogamulizumab, in Combination with Nivolumab in Patients with Advanced or Metastatic Solid Tumors. Clin Cancer Res (2019) 25(22):6614–22. doi: 10.1158/1078-0432.Ccr-19-1090
67. Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science (2015) 348(6236):803–8. doi: 10.1126/science.aaa3828
68. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature (2017) 547(7662):217–21. doi: 10.1038/nature22991
69. Chiang CL, Coukos G, Kandalaft LE. Whole Tumor Antigen Vaccines: Where Are We? Vaccines (Basel) (2015) 3(2):344–72. doi: 10.3390/vaccines3020344
70. Buonaguro L. New vaccination strategies in liver cancer. Cytokine Growth Factor Rev (2017) 36:125–9. doi: 10.1016/j.cytogfr.2017.06.010
71. Nakagawa H, Mizukoshi E, Kobayashi E, Tamai T, Hamana H, Ozawa T, et al. Association Between High-Avidity T-Cell Receptors, Induced by α-Fetoprotein-Derived Peptides, and Anti-Tumor Effects in Patients With Hepatocellular Carcinoma. Gastroenterology (2017) 152(6):1395–406.e10. doi: 10.1053/j.gastro.2017.02.001
72. Sawada Y, Yoshikawa T, Nobuoka D, Shirakawa H, Kuronuma T, Motomura Y, et al. Phase I trial of a glypican-3-derived peptide vaccine for advanced hepatocellular carcinoma: immunologic evidence and potential for improving overall survival. Clin Cancer Res an Off J Am Assoc Cancer Res (2012) 18(13):3686–96. doi: 10.1158/1078-0432.CCR-11-3044
73. Greten TF, Forner A, Korangy F, N’Kontchou G, Barget N, Ayuso C, et al. A phase II open label trial evaluating safety and efficacy of a telomerase peptide vaccination in patients with advanced hepatocellular carcinoma. BMC Cancer (2010) 10:209. doi: 10.1186/1471-2407-10-209
74. Buonaguro L, Petrizzo A, Tagliamonte M, Tornesello ML, Buonaguro FM. Challenges in cancer vaccine development for hepatocellular carcinoma. J Hepatol (2013) 59(4):897–903. doi: 10.1016/j.jhep.2013.05.031
75. Petrizzo A, Tagliamonte M, Mauriello A, Costa V, Aprile M, Esposito R, et al. Unique true predicted neoantigens (TPNAs) correlates with anti-tumor immune control in HCC patients. J Transl Med (2018) 16(1):286. doi: 10.1186/s12967-018-1662-9
76. Buonaguro L, Mauriello A, Cavalluzzo B, Petrizzo A, Tagliamonte M. Immunotherapy in hepatocellular carcinoma. Ann Hepatol (2019) 18(2):291–7. doi: 10.1016/j.aohep.2019.04.003
77. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Sci (New York NY) (2015) 348(6230):62–8. doi: 10.1126/science.aaa4967
78. Zoll B, Lefterova P, Csipai M, Finke S, Trojaneck B, Ebert O, et al. Generation of cytokine-induced killer cells using exogenous interleukin-2, -7 or -12. Cancer Immunol Immunother (1998) 47(4):221–6. doi: 10.1007/s002620050524
79. Zhao H, Wang Y, Yu J, Wei F, Cao S, Zhang X, et al. Autologous Cytokine-Induced Killer Cells Improves Overall Survival of Metastatic Colorectal Cancer Patients: Results From a Phase II Clinical Trial. Clin Colorectal Cancer (2016) 15(3):228–35. doi: 10.1016/j.clcc.2016.02.005
80. Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med (2016) 8(357):357ra123. doi: 10.1126/scitranslmed.aaf2341
81. Zhao X, Zhang Z, Li H, Huang J, Yang S, Xie T, et al. Cytokine induced killer cell-based immunotherapies in patients with different stages of renal cell carcinoma. Cancer Lett (2015) 362(2):192–8. doi: 10.1016/j.canlet.2015.03.043
82. Verneris MR, Ito M, Baker J, Arshi A, Negrin RS, Shizuru JA. Engineering hematopoietic grafts: purified allogeneic hematopoietic stem cells plus expanded CD8+ NK-T cells in the treatment of lymphoma. Biol Blood Marrow Transpl (2001) 7(10):532–42. doi: 10.1016/s1083-8791(01)70014-6
83. Ochoa AC, Gromo G, Alter BJ, Sondel PM, Bach FH. Long-term growth of lymphokine-activated killer (LAK) cells: role of anti-CD3, beta-IL 1, interferon-gamma and -beta. J Immunol (1987) 138(8):2728–33.
84. Balermpas P, Michel Y, Wagenblast J, Seitz O, Weiss C, Rödel F, et al. Tumour-infiltrating lymphocytes predict response to definitive chemoradiotherapy in head and neck cancer. Br J Cancer (2014) 110(2):501–9. doi: 10.1038/bjc.2013.640
85. Stevanović S, Helman SR, Wunderlich JR, Langhan MM, Doran SL, Kwong MLM, et al. A Phase II Study of Tumor-infiltrating Lymphocyte Therapy for Human Papillomavirus-associated Epithelial Cancers. Clin Cancer Res an Off J Am Assoc Cancer Res (2019) 25(5):1486–93. doi: 10.1158/1078-0432.CCR-18-2722
86. Jiang S-S, Tang Y, Zhang Y-J, Weng DS, Zhou Z-G, Pan K, et al. A phase I clinical trial utilizing autologous tumor-infiltrating lymphocytes in patients with primary hepatocellular carcinoma. Oncotarget (2015) 6(38):41339–49. doi: 10.18632/oncotarget.5463
87. Jensen MC, Riddell SR. Design and implementation of adoptive therapy with chimeric antigen receptor-modified T cells. Immunol Rev (2014) 257(1):127–44. doi: 10.1111/imr.12139
88. Morgan RA, Dudley ME, Yu YY, Zheng Z, Robbins PF, Theoret MR, et al. High efficiency TCR gene transfer into primary human lymphocytes affords avid recognition of melanoma tumor antigen glycoprotein 100 and does not alter the recognition of autologous melanoma antigens. J Immunol (2003) 171(6):3287–95. doi: 10.4049/jimmunol.171.6.3287
89. Luo X, Cui H, Cai L, Zhu W, Yang W-C, Patrick M, et al. Selection of a Clinical Lead TCR Targeting Alpha-Fetoprotein-Positive Liver Cancer Based on a Balance of Risk and Benefit. Front Immunol (2020) 11:623. doi: 10.3389/fimmu.2020.00623
90. Garrido F, Ruiz-Cabello F, Cabrera T, Pérez-Villar JJ, López-Botet M, Duggan-Keen M, et al. Implications for immunosurveillance of altered HLA class I phenotypes in human tumours. Immunol Today (1997) 18(2):89–95. doi: 10.1016/s0167-5699(96)10075-x
91. Liu H, Xu Y, Xiang J, Long L, Green S, Yang Z, et al. Targeting Alpha-Fetoprotein (AFP)-MHC Complex with CAR T-Cell Therapy for Liver Cancer. Clin Cancer Res (2017) 23(2):478–88. doi: 10.1158/1078-0432.CCR-16-1203
92. Baumhoer D, Tornillo L, Stadlmann S, Roncalli M, Diamantis EK, Terracciano LM. Glypican 3 expression in human nonneoplastic, preneoplastic, and neoplastic tissues: a tissue microarray analysis of 4,387 tissue samples. Am J Clin Pathol (2008) 129(6):899–906. doi: 10.1309/HCQWPWD50XHD2DW6
93. Gao H, Li K, Tu H, Pan X, Jiang H, Shi B, et al. Development of T cells redirected to glypican-3 for the treatment of hepatocellular carcinoma. Clin Cancer Res (2014) 20(24):6418–28. doi: 10.1158/1078-0432.CCR-14-1170
94. Jiang Z, Jiang X, Chen S, Lai Y, Wei X, Li B, et al. Anti-GPC3-CAR T Cells Suppress the Growth of Tumor Cells in Patient-Derived Xenografts of Hepatocellular Carcinoma. Front Immunol (2016) 7:690. doi: 10.3389/fimmu.2016.00690
95. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature (1998) 392(6673):245–52. doi: 10.1038/32588
96. Darvin P, Toor SM, Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med (2018) 50(12):1–11. doi: 10.1038/s12276-018-0191-1
97. Liu Y, Zheng P. Preserving the CTLA-4 Checkpoint for Safer and More Effective Cancer Immunotherapy. Trends Pharmacol Sci (2020) 41(1):4–12. doi: 10.1016/j.tips.2019.11.003
98. Tarhini AA, Edington H, Butterfield LH, Lin Y, Shuai Y, Tawbi H, et al. Immune monitoring of the circulation and the tumor microenvironment in patients with regionally advanced melanoma receiving neoadjuvant ipilimumab. PloS One (2014) 9(2):e87705. doi: 10.1371/journal.pone.0087705
99. Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, et al. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci USA (2015) 112(19):6140–5. doi: 10.1073/pnas.1417320112
100. Sangro B, Gomez-Martin C, de la Mata M, Iñarrairaegui M, Garralda E, Barrera P, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol (2013) 59(1):81–8. doi: 10.1016/j.jhep.2013.02.022
101. Pardoll D, Drake C. Immunotherapy earns its spot in the ranks of cancer therapy. J Exp Med (2012) 209(2):201–9. doi: 10.1084/jem.20112275
102. Nikolova M, Lelievre J-D, Carriere M, Bensussan A, Lévy Y. Regulatory T cells differentially modulate the maturation and apoptosis of human CD8+ T-cell subsets. Blood (2009) 113(19):4556–65. doi: 10.1182/blood-2008-04-151407
103. Taube JM, Young GD, McMiller TL, Chen S, Salas JT, Pritchard TS, et al. Differential Expression of Immune-Regulatory Genes Associated with PD-L1 Display in Melanoma: Implications for PD-1 Pathway Blockade. Clin Cancer Res (2015) 21(17):3969–76. doi: 10.1158/1078-0432.CCR-15-0244
104. Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett (2004) 574(1-3):37–41. doi: 10.1016/j.febslet.2004.07.083
105. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science (2017) 355(6332):1428–33. doi: 10.1126/science.aaf1292
106. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi: 10.1056/NEJMoa1003466
107. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Sci (New York NY) (2015) 348(6230):124–8. doi: 10.1126/science.aaa1348
108. Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet (2016) 387(10031):1909–20. doi: 10.1016/S0140-6736(16)00561-4
109. Yarchoan M, Hopkins A, Jaffee EM. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N Engl J Med (2017) 377(25):2500–1. doi: 10.1056/NEJMc1713444
110. Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, Stenzinger A, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol Off J Eur Soc Med Oncol (2019) 30(1):44–56. doi: 10.1093/annonc/mdy495
111. Tran L, Theodorescu D. Determinants of Resistance to Checkpoint Inhibitors. Int J Mol Sci (2020) 21(5):1594. doi: 10.3390/ijms21051594
112. Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer (2014) 14(2):135–46. doi: 10.1038/nrc3670
113. Restifo NP, Marincola FM, Kawakami Y, Taubenberger J, Yannelli JR, Rosenberg SA. Loss of functional beta 2-microglobulin in metastatic melanomas from five patients receiving immunotherapy. J Natl Cancer Inst (1996) 88(2):100–8. doi: 10.1093/jnci/88.2.100
114. McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Sci (New York NY) (2016) 351(6280):1463–9. doi: 10.1126/science.aaf1490
115. Topper MJ, Vaz M, Marrone KA, Brahmer JR, Baylin SB. The emerging role of epigenetic therapeutics in immuno-oncology. Nat Rev Clin Oncol (2020) 17(2):75–90. doi: 10.1038/s41571-019-0266-5
116. Pagès F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med (2005) 353(25):2654–66. doi: 10.1056/NEJMoa051424
117. Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA (2005) 102(51):18538–43. doi: 10.1073/pnas.0509182102
118. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (2006) 313(5795):1960–4. doi: 10.1126/science.1129139
119. Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean M-C, Validire P, Trautmann A, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest (2012) 122(3):899–910. doi: 10.1172/JCI45817
120. Yin J, Leavenworth JW, Li Y, Luo Q, Xie H, Liu X, et al. Ezh2 regulates differentiation and function of natural killer cells through histone methyltransferase activity. Proc Natl Acad Sci USA (2015) 112(52):15988–93. doi: 10.1073/pnas.1521740112
121. Jones PA, Ohtani H, Chakravarthy A, De Carvalho DD. Epigenetic therapy in immune-oncology. Nat Rev Cancer (2019) 19(3):151–61. doi: 10.1038/s41568-019-0109-9
122. Natsume A, Wakabayashi T, Tsujimura K, Shimato S, Ito M, Kuzushima K, et al. The DNA demethylating agent 5-aza-2’-deoxycytidine activates NY-ESO-1 antigenicity in orthotopic human glioma. Int J Cancer (2008) 122(11):2542–53. doi: 10.1002/ijc.23407
123. Moreno-Bost A, Szmania S, Stone K, Garg T, Hoerring A, Szymonifka J, et al. Epigenetic modulation of MAGE-A3 antigen expression in multiple myeloma following treatment with the demethylation agent 5-azacitidine and the histone deacetlyase inhibitor MGCD0103. Cytotherapy (2011) 13(5):618–28. doi: 10.3109/14653249.2010.529893
124. Goodyear O, Agathanggelou A, Novitzky-Basso I, Siddique S, McSkeane T, Ryan G, et al. Induction of a CD8+ T-cell response to the MAGE cancer testis antigen by combined treatment with azacitidine and sodium valproate in patients with acute myeloid leukemia and myelodysplasia. Blood (2010) 116(11):1908–18. doi: 10.1182/blood-2009-11-249474
125. Almstedt M, Blagitko-Dorfs N, Duque-Afonso J, Karbach J, Pfeifer D, Jäger E, et al. The DNA demethylating agent 5-aza-2’-deoxycytidine induces expression of NY-ESO-1 and other cancer/testis antigens in myeloid leukemia cells. Leuk Res (2010) 34(7):899–905. doi: 10.1016/j.leukres.2010.02.004
126. Scanlan MJ, Gure AO, Jungbluth AA, Old LJ, Chen YT. Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunol Rev (2002) 188:22–32. doi: 10.1034/j.1600-065x.2002.18803.x
127. Loriot A, Boon T, De Smet C. Five new human cancer-germline genes identified among 12 genes expressed in spermatogonia. Int J Cancer (2003) 105(3):371–6. doi: 10.1002/ijc.11104
128. Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P, et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell (2017) 170(1):142–57.e19. doi: 10.1016/j.cell.2017.06.007
129. Topper MJ, Vaz M, Chiappinelli KB, DeStefano Shields CE, Niknafs N, Yen RC, et al. Epigenetic Therapy Ties MYC Depletion to Reversing Immune Evasion and Treating Lung Cancer. Cell (2017) 171(6):1284–300.e21. doi: 10.1016/j.cell.2017.10.022
130. Hong YK, Li Y, Pandit H, Li S, Pulliam Z, Zheng Q, et al. Epigenetic modulation enhances immunotherapy for hepatocellular carcinoma. Cell Immunol (2019) 336:66–74. doi: 10.1016/j.cellimm.2018.12.010
131. Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol (2009) 10(1):25–34. doi: 10.1016/s1470-2045(08)70285-7
132. Morse MA, Sun W, Kim R, He AR, Abada PB, Mynderse M, et al. The Role of Angiogenesis in Hepatocellular Carcinoma. Clin Cancer Res (2019) 25(3):912–20. doi: 10.1158/1078-0432.Ccr-18-1254
133. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med (2008) 359(4):378–90. doi: 10.1056/NEJMoa0708857
134. European Association For The Study Of The Liver, European Organisation For Research And Treatment Of Cancer. EASL-EORTC clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol (2012) 56(4):908–43. doi: 10.1016/j.jhep.2011.12.001
135. Terme M, Pernot S, Marcheteau E, Sandoval F, Benhamouda N, Colussi O, et al. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res (2013) 73(2):539–49. doi: 10.1158/0008-5472.Can-12-2325
136. Manning EA, Ullman JG, Leatherman JM, Asquith JM, Hansen TR, Armstrong TD, et al. A vascular endothelial growth factor receptor-2 inhibitor enhances antitumor immunity through an immune-based mechanism. Clin Cancer Res (2007) 13(13):3951–9. doi: 10.1158/1078-0432.Ccr-07-0374
137. Escudier B, Pluzanska A, Koralewski P, Ravaud A, Bracarda S, Szczylik C, et al. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet (2007) 370(9605):2103–11. doi: 10.1016/s0140-6736(07)61904-7
138. Bennouna J, Sastre J, Arnold D, Österlund P, Greil R, Van Cutsem E, et al. Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): a randomised phase 3 trial. Lancet Oncol (2013) 14(1):29–37. doi: 10.1016/s1470-2045(12)70477-1
139. Tewari KS, Sill MW, Long HJ,3, Penson RT, Huang H, Ramondetta LM, et al. Improved survival with bevacizumab in advanced cervical cancer. N Engl J Med (2014) 370(8):734–43. doi: 10.1056/NEJMoa1309748
140. Siegel AB, Cohen EI, Ocean A, Lehrer D, Goldenberg A, Knox JJ, et al. Phase II trial evaluating the clinical and biologic effects of bevacizumab in unresectable hepatocellular carcinoma. J Clin Oncol (2008) 26(18):2992–8. doi: 10.1200/jco.2007.15.9947
141. Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature (2020) 577(7791):561–5. doi: 10.1038/s41586-019-1914-8
142. Dieu-Nosjean M-C, Giraldo NA, Kaplon H, Germain C, Fridman WH, Sautès-Fridman C. Tertiary lymphoid structures, drivers of the anti-tumor responses in human cancers. Immunol Rev (2016) 271(1):260–75. doi: 10.1111/imr.12405
143. Sautès-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer (2019) 19(6):307–25. doi: 10.1038/s41568-019-0144-6
144. Sautès-Fridman C, Lawand M, Giraldo NA, Kaplon H, Germain C, Fridman WH, et al. Tertiary Lymphoid Structures in Cancers: Prognostic Value, Regulation, and Manipulation for Therapeutic Intervention. Front Immunol (2016) 7:407. doi: 10.3389/fimmu.2016.00407
145. Calderaro J, Petitprez F, Becht E, Laurent A, Hirsch TZ, Rousseau B, et al. Intra-tumoral tertiary lymphoid structures are associated with a low risk of early recurrence of hepatocellular carcinoma. J Hepatol (2019) 70(1):58–65. doi: 10.1016/j.jhep.2018.09.003
146. Johansson-Percival A, He B, Li Z-J, Kjellén A, Russell K, Li J, et al. De novo induction of intratumoral lymphoid structures and vessel normalization enhances immunotherapy in resistant tumors. Nat Immunol (2017) 18(11):1207–17. doi: 10.1038/ni.3836
147. Nayar S, Campos J, Smith CG, Iannizzotto V, Gardner DH, Mourcin F, et al. Immunofibroblasts are pivotal drivers of tertiary lymphoid structure formation and local pathology. Proc Natl Acad Sci USA (2019) 116(27):13490–7. doi: 10.1073/pnas.1905301116
148. Zhu G, Nemoto S, Mailloux AW, Perez-Villarroel P, Nakagawa R, Falahat R, et al. Induction of Tertiary Lymphoid Structures With Antitumor Function by a Lymph Node-Derived Stromal Cell Line. Front Immunol (2018) 9:1609:1609. doi: 10.3389/fimmu.2018.01609
149. Lam CM, Ng KK, Poon RT, Ai V, Yuen J, Fan ST. Impact of radiofrequency ablation on the management of patients with hepatocellular carcinoma in a specialized centre. Br J Surg (2004) 91(3):334–8. doi: 10.1002/bjs.4448
150. Zerbini A, Pilli M, Penna A, Pelosi G, Schianchi C, Molinari A, et al. Radiofrequency thermal ablation of hepatocellular carcinoma liver nodules can activate and enhance tumor-specific T-cell responses. Cancer Res (2006) 66(2):1139–46. doi: 10.1158/0008-5472.Can-05-2244
151. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol (2013) 31:51–72. doi: 10.1146/annurev-immunol-032712-100008
152. Schueller G, Stift A, Friedl J, Dubsky P, Bachleitner-Hofmann T, Benkoe T, et al. Hyperthermia improves cellular immune response to human hepatocellular carcinoma subsequent to co-culture with tumor lysate pulsed dendritic cells. Int J Oncol (2003) 22(6):1397–402. doi: 10.3892/ijo.22.6.1397
153. Schueller G, Kettenbach J, Sedivy R, Bergmeister H, Stift A, Fried J, et al. Expression of heat shock proteins in human hepatocellular carcinoma after radiofrequency ablation in an animal model. Oncol Rep (2004) 12(3):495–9. doi: 10.3892/or.12.3.495
154. Schueller G, Kettenbach J, Sedivy R, Stift A, Friedl J, Gnant M, et al. Heat shock protein expression induced by percutaneous radiofrequency ablation of hepatocellular carcinoma in vivo. Int J Oncol (2004) 24(3):609–13. doi: 10.3892/ijo.24.3.609
155. Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol (2008) 8(1):59–73. doi: 10.1038/nri2216
156. Banissi C, Ghiringhelli F, Chen L, Carpentier AF. Treg depletion with a low-dose metronomic temozolomide regimen in a rat glioma model. Cancer Immunol Immunother (2009) 58(10):1627–34. doi: 10.1007/s00262-009-0671-1
157. Generali D, Bates G, Berruti A, Brizzi MP, Campo L, Bonardi S, et al. Immunomodulation of FOXP3+ regulatory T cells by the aromatase inhibitor letrozole in breast cancer patients. Clin Cancer Res (2009) 15(3):1046–51. doi: 10.1158/1078-0432.CCR-08-1507
158. Tanaka H, Matsushima H, Mizumoto N, Takashima A. Classification of chemotherapeutic agents based on their differential in vitro effects on dendritic cells. Cancer Res (2009) 69(17):6978–86. doi: 10.1158/0008-5472.Can-09-1101
159. Nars MS, Kaneno R. Immunomodulatory effects of low dose chemotherapy and perspectives of its combination with immunotherapy. Int J Cancer (2013) 132(11):2471–8. doi: 10.1002/ijc.27801
160. Wu J, Waxman DJ. Metronomic cyclophosphamide eradicates large implanted GL261 gliomas by activating antitumor Cd8(+) T-cell responses and immune memory. Oncoimmunology (2015) 4(4):e1005521. doi: 10.1080/2162402x.2015.1005521
161. Kareva I. A Combination of Immune Checkpoint Inhibition with Metronomic Chemotherapy as a Way of Targeting Therapy-Resistant Cancer Cells. Int J Mol Sci (2017) 18(10):2134. doi: 10.3390/ijms18102134
162. Dunk AA, Ikeda T, Pignatelli M, Thomas HC. Human lymphoblastoid interferon. In vitro and in vivo studies in hepatocellular carcinoma. J Hepatol (1986) 2(3):419–29. doi: 10.1016/s0168-8278(86)80053-8
163. Llovet JM, Sala M, Castells L, Suarez Y, Vilana R, Bianchi L, et al. Randomized controlled trial of interferon treatment for advanced hepatocellular carcinoma. Hepatology (2000) 31(1):54–8. doi: 10.1002/hep.510310111
164. Chen LT, Chen MF, Li LA, Lee PH, Jeng LB, Lin DY, et al. Long-term results of a randomized, observation-controlled, phase III trial of adjuvant interferon Alfa-2b in hepatocellular carcinoma after curative resection. Ann Surg (2012) 255(1):8–17. doi: 10.1097/SLA.0b013e3182363ff9
Keywords: immunotherapy, hepatocellular carcinoma, immune tolerance, tumor mutational burden, tumor microenvironment, epigenetic modification, tertiary lymphoid structure
Citation: Zhong C, Li Y, Yang J, Jin S, Chen G, Li D, Fan X and Lin H (2021) Immunotherapy for Hepatocellular Carcinoma: Current Limits and Prospects. Front. Oncol. 11:589680. doi: 10.3389/fonc.2021.589680
Received: 31 July 2020; Accepted: 10 March 2021;
Published: 29 March 2021.
Edited by:
Victor Ho Fun Lee, The University of Hong Kong, Hong KongReviewed by:
Muhammad A. Rizvi, Lehigh Valley Health Network, United StatesLuigi Buonaguro, Istituto Nazionale Tumori Fondazione G. Pascale (IRCCS), Italy
Copyright © 2021 Zhong, Li, Yang, Jin, Chen, Li, Fan and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Lin, 369369@zju.edu.cn; Xiaoxiao Fan, fanxx_gs@zju.edu.cn
†These authors have contributed equally to this work