 Yang He1
Yang He1 Cunlong Wang1Xiulin Zhang1
Cunlong Wang1Xiulin Zhang1 Xuancheng Lu2Jin Xing3Jianyi Lv1Meng Guo1Xueyun Huo1Xin Liu1Jing Lu1Xiaoyan Du1
Xuancheng Lu2Jin Xing3Jianyi Lv1Meng Guo1Xueyun Huo1Xin Liu1Jing Lu1Xiaoyan Du1 Changlong Li1*Zhenwen Chen1*
Changlong Li1*Zhenwen Chen1*- 1School of Basic Medical Sciences, Capital Medical University, Beijing Key Laboratory of Cancer Invasion & Metastasis Research, Beijing, China
- 2Laboratory Animal Center, Chinese Center for Disease Control and Prevention, Beijing, China
- 3Institute for Laboratory Animal Resources, National Institutes for Food and Drug Control, Beijing, China
Helicobacter pylori is designated as a class I carcinogen of human gastric cancer following long-term infection. During this process, H. pylori bacteria persist in proliferation and death, and release bacterial components that come into contact with gastric epithelial cells and regulate host cell function. However, the impact of long-term exposure to H. pylori lysate on the pathological changes of gastric cells is not clear. In this study, we aimed to investigate the regulation and mechanisms involved in gastric cell dysfunction following continuous exposure to H. pylori lysate. We co-cultured gastric cell lines GES-1 and MKN-45 with H. pylori lysate for 30 generations, and we found that sustained exposure to H. pylori lysate inhibited GES-1 cell invasion, migration, autophagy, and apoptosis, while it did not inhibit MKN-45 cell invasion or migration. Furthermore, Mongolian gerbils infected with H. pylori ATCC 43504 strains for 90 weeks confirmed the in vitro results. The clinical and in vitro data indicated that sustained exposure to H. pylori lysate inhibited cell apoptosis and autophagy through the Nod1-NF-κB/MAPK-ERK/FOXO4 signaling pathway. In conclusion, sustained exposure to H. pylori lysate promoted proliferation of gastric epithelial cells and inhibited autophagy and apoptosis via Nod1-NF-κB/MAPK-ERK/FOXO4 signaling pathway. In the process of H. pylori-induced gastric lesions, H. pylori lysate plays as an “accomplice” to carcinogenesis.
Introduction
Helicobacter pylori (H. pylori) is designated as a class I carcinogen of human gastric cancer (1), and it can survive for prolonged periods in the acidic gastric environment. H. pylori infection is closely related to gastritis, peptic ulcer, gastric cancer, gastric mucosa-associated lymphoid tissue (MALT) lymphoma, and even some extragastric diseases (2–5). It is generally believed that the diseases induced by H. pylori infection are caused by living bacteria. H. pylori induces defective autophagy or inhibits autophagy to promote its own colonization (6, 7). Moreover, H. pylori is involved in migration, invasion, autophagy, and apoptosis, eventually leading to gastric cancer (8, 9). H. pylori promotes the malignant transformation of the host cells by transporting cytotoxin-associated gene product A (CagA), an oncoprotein, to cells through the type IV secretion system (T4SS) (10–12). Furthermore, H. pylori secretes vacuolating cytotoxin A (VacA) (13) and destroys the activity of lysosomal calcium channels in host cells, which leads to the formation of dysfunctional enlarged lysosomes and allows H. pylori to colonize in the stomach and, thus, escape from eradication therapy (14). In addition, the outer membrane vesicles (OMVs) released by H. pylori contain a variety of bacterial toxins and antigens (15), which are absorbed by gastric epithelial cells (16) and enhance the carcinogenic potential of H. pylori (17).
During long-term infection by H. pylori, a large number of bacteria persist in proliferation and death. Massive bacterial compositions maintain contact with and stimulate gastric epithelial cells. These bacterial components enter the host cell in multiple ways, such as receptor recognition and OMVs, regulate cell survival and metabolism, and lead to pathological diseases of the gastric mucosal barrier (18–20). During bacterial disintegration in the stomach, various components act on endothelial cells simultaneously. It has been reported that H. pylori lysate promotes hepatocellular carcinoma (HSC) cell proliferation and liver fibrosis (21). Further, Helicobacter suis lysate regulates the apoptosis of gastric epithelial cells (22). To date, most reports have investigated the mechanisms of H. pylori-induced gastric diseases by infecting living bacteria or a single bacterial virulence factor to cell lines for 6 h to 72 h (8, 18, 21, 22). However, these cannot simulate the effects of long-term stimulation of H. pylori on gastric cells. Because H. pylori cannot survive co-cultures with cells for an extended time, long-term co-cultures with gastric epithelial cells using H. pylori lysate instead of living bacteria are used to simulate the regulatory effects of persistent infection on cells. In this process, the effects of H. pylori lysate are also important.
In this study, H. pylori lysate was prepared by ultrasonic lysis and was co-cultured with gastric epithelial cells for 30 consecutive generations to investigate the underlying mechanisms involved in its cellular regulatory activity in vitro and in vivo. Our data demonstrate that sustained exposure of gastric epithelial cells to H. pylori lysate promoted proliferation and inhibited autophagy and apoptosis, and it may further lead to malignant transformation in gastric epithelial cells.
Materials and Methods
Bacterial Culture and Preparation of Bacterial Lysate
The H. pylori strain American Type Culture Collection (ATCC) 43504 (cagA+, vacA+) was obtained from the National Institutes for Food and Drug Control, Beijing. H. pylori was grown on Colombian agar plates (OXOID, UK, CM0331B) containing 5% sterile and defibrated sheep blood (MRC, China, CCS30037.01) at 37°C under microaerophilic conditions for 48 h.
H. pylori was scraped off the plate and washed twice with phosphate buffer saline (PBS) (KeyGen BioTECH, China, KGB5001), then mixed with PBS, and ultrasonic lysis was performed. We used the bicinchoninic acid (BCA) method to detect protein concentration. The lysate was stored at -20°C until use.
Cell Lines and Cell Culture
The human normal gastric epithelial cell line GES-1 and human gastric adenocarcinoma cell line MKN-45 were purchased from Beijing Dingguo Changsheng Biotechnology Co., Ltd. Cells were grown in DMEM (Corning, USA, 10-013-CVR) supplemented with 10% fetal bovine serum (FBS) (PAN, Germany, P30-3302) and 1% penicillin/streptomycin binary antibody solution (KeyGen BioTECH, China, KGY0023) in a humidified environment and under 5% CO2 at 37°C.
GES-1 cells and MKN-45 cells of the experimental group were cultured in medium added with H. pylori lysate for 30 consecutive generations. The other conditions were consistent with those of the control group. The untreated normal cells were labeled as B-GES-1 and B-MKN-45, which were cultured for 30 consecutive generations. The cells co-cultured with H. pylori lysate for 30 generations were labeled as Cul30-GES-1 and Cul30-MKN-45, respectively.
Cell Treatment
A total of 4×105 Cul30-GES-1 and B-GES-1, Cul30-MKN-45, and B-MKN-45 cells were seeded into 6-well plates. After the cells were attached, normal DMEM, DMEM containing H. pylori lysate, or DMEM containing H. pylori (6×106 CFU/mL) (23) was separately added to the 6-well plates for a total of 2 mL per well, and cells were incubated for 24 h.
Determining the Optimum Concentration of H. pylori Lysate to Be Co-Cultured With Cells
The optimum concentration of H. pylori lysate to be co-cultured with cells was determined by MTT. B-GES-1 or B-MKN-45 cells were digested with 0.25% trypsin and washed with PBS. The cell suspension concentration was adjusted to 2.5×104/mL using DMEM medium containing 10% FBS. The cells were inoculated in 96-well plates with a volume of 100 μL per well. The edge wells of the 96-well plate were filled with 200 μL sterile PBS solution, and the culture was continued for 6 h to allow the cells to adhere. After the cells were attached, the medium was discarded, and the cells were washed twice with PBS solution. In the experimental group, medium containing different concentrations of H. pylori lysate was added (0.5 μg/mL, 1 μg/mL, 1.5 μg/mL, 2 μg/mL, 2.5 μg/mL, 3 μg/mL, 3.5 μg/mL, 4 μg/mL, 4.5 μg/mL, 5 μg/mL), in a volume of 200 μL per well, and the control group received 200 μL of normal medium containing an equal amount of PBS solution. Five sub-wells were set for each experimental group and control group. The cells were cultured for another 72 h. After 72 h of culture, the culture medium was discarded. Cells were washed twice with PBS solution. Sterile MTT solution (Solarbio, China, M1020) was added to the well in a final volume of 100 μL. The cells were cultured for 4 h. The solution in the 96-well plate was discarded and 100 μL dimethyl sulfoxide (DMSO) was added to each well. The plate was placed on a shaking table for 10 min to dissolve the methylamine precipitate. Absorbance of each well was read at 490 nm using a microplate reader of a spectrophotometer to measure the cell quantity.
Cell Proliferation Assay
In this study, gastric cells were co-cultured with H. pylori or H. pylori lysate. The cell proliferation of Cul30-GES-1 and B-GES-1, Cul30-MKN-45, and B-MKN-45 were compared by two methods. First, the cells were plated into 96-well plates and then cultured in medium containing H. pylori lysate or H. pylori for 24 h or 48 h. Then MTT colorimetric assay was carried out. Second, plate cloning assay was used. Cul30-GES-1 and B-GES-1 (500 cells/well) were placed in 6-well plates and maintained in DMEM medium containing 10% FBS. After 14 days, the cells were fixed and stained by crystal violet. Visible colonies were then counted by Image J software. Each well was assessed in triplicate.
Wound Healing Assay
B-GES-1 and Cul30-GES-1 were placed in 6-well plates and maintained in DMEM medium containing 10% FBS. The cell concentration was 5×105 cells/mL and 2 mL volume was added per well. When cells reached 90% confluence, a wound was generated, the medium was discarded, and the cells were washed twice with PBS solution to remove any floating cells. In the experimental group, serum free medium containing H. pylori lysate or H. pylori was added at a volume of 2 mL per well, and the control group received 2 mL of serum free medium. The cells were incubated for 24 h. The gap distances were measured to assess the capacity of the cells to migrate.
Transwell Cell Migration and Invasion Assay
Transwell migration chamber was used for the migration assay. After treatment with H. pylori lysate or H. pylori, Cul30-GES-1 and B-GES-1, Cul30-MKN-45, and B-MKN-45 cells (3×104 cells in 200 μL) were placed in the upper chamber in serum-free medium. Medium containing 10% FBS was added to the lower chamber. The cells, following a 24-h incubation, were fixed and stained with 0.1% crystal violet and counted. The assays were performed in triplicate.
For the invasion assay, a total of 3×104 cells were seeded into the upper chamber of a Transwell invasion chamber with serum-free media, while medium containing 10% FBS was added to the lower chamber. The cells, after a 24-h incubation, were fixed and stained with Hoechst (Solarbio, China, B8040) for 10 min in a dark environment. The number of cells that invaded from the upper chamber were counted using Image J software.
Cytokine Level Analysis
Cul30-GES-1, B-GES-1 and Cul30-MKN-45, B-MKN-45 cells were treated with H. pylori lysate or H. pylori for 24 h. Cytokine level analysis was performed by Shanghai Universal Biotech Co., Ltd. The Luminex detection assays were used to measure the expression of each factor in the samples. According to previous reports, eight cytokines in Cul30-GES-1 and B-GES-1 cells related to autophagy, apoptosis, migration, or invasion were selected for detection, including TNF-α, CCL-20, CCL-28, CXCL-2, IFN-γ, TFPI, SLPI, and FAS. Three cytokines, CCL-20, CCL-28, and CXCL-2, in Cul30-MKN-45 and B-MKN-45 cells associated with migration and invasion were also detected.
mCherry-EGFP-LC3 Transfection
Two milliliters of Cul30-GES-1 and B-GES-1 cell suspension with a density of 2.5 × 104 cells/mL was prepared in complete medium and added to a 6-well plate. After the cells were attached, the old medium was discarded, the cells were washed with PBS, and medium containing a 50 μL titer of 108 TU/mL mCherry-EGFP-LC3 lentivirus (purchased from SyngenTech Co., Ltd) and 8 μg/mL Polybrene was added to the culture. After 48 h of infection, the fluorescence expression of cells was observed by fluorescence microscopy. When the efficiency of cell infection reached about 80%, the cells were cultured in medium containing H. pylori lysate or H. pylori for an additional 24 h. The images of mCherry-EGFP-LC3 transfected cells were observed by laser scanning confocal microscopy. The autophagy flux was measured by the color change of mCherry-EGFP.
Hoechst Staining
After treatment with H. pylori lysate or H. pylori, Cul30-GES-1 and B-GES-1 cells were washed with PBS three times. Next, cells were stained with Hoechst 33342 (10 μg/mL) for 10 min. Nuclear morphologic changes were examined under a fluorescence microscope.
Apoptosis Assay
Flow cytometry was used to detect the effects of H. pylori lysate on the apoptosis of Cul30-GES-1 and B-GES-1 cells using the Annexin V-PE/7-AAD Apoptosis Detection Kit (Vazyme, China, A213-01) according to the manufacturer’s recommendations. The rate of apoptosis was analyzed using LSR Fortessa Flow Cytometer at 488 nm.
Evaluation of Gene Expression via the TCGA Database
The gene mRNA expression data of 132 gastric adenocarcinoma cases (17 cases of H. pylori infection and 115 cases without H. pylori infection) were downloaded from the TCGA database (https://cancergenome.nih.gov/). Log2 transformation and Z-correction were performed to normalize the expression value of each gene.
Reverse Transcriptase Polymerase Chain Reaction
A TRIzol Reagent (Vazyme, China, R401-01) was used to isolate total RNA from Cul30-GES-1 and B-GES-1 cells. Single-stranded DNA was prepared from 1 μg total RNA using reverse transcriptase-bound oligonucleotide (DT) primers. Each cDNA sample (2 μL) was subjected to reverse transcriptase polymerase chain reaction (RT-PCR) amplification using specific primers as detailed in Supplementary Table 1. The data were collected and analyzed. The values were compared with the experimental controls after being normalized to those of GAPDH.
Western Blot
Following treatment with H. pylori lysate or H. pylori, Cul30-GES-1 and B-GES-1, Cul30-MKN-45, and B-MKN-45 cells were lysed by Radio Immunoprecipitation Assay (RIPA) Lysis Buffer (Solarbio, China, R0010) on ice for 30 min. The cell lysate was centrifuged at 13400×g at 4°C for 15 min. The supernatants were collected, and the protein concentration was measured with a BCA protein kit (Thermo Scientific, USA, A53225). The lysate was mixed with PBS and 5× SDS loading buffer (ROBY, China, RBU114-2) and heated at 99°C for 10 min. Western blots were performed on 8% or 10% SDS-polyacrylamide gel electrophoresis (PAGE) gel, and protein samples were transferred onto polyvinylidene fluoride (PVDF) membranes (Merck Millipore, USA, ISEQ00010). PVDF membranes were blocked by 5% skim milk (BD, USA, 232100) and were incubated first with rabbit primary antibodies overnight at 4°C, followed by incubation with a secondary antibody (Solarbio, China, SE134, 1:5000 dilution for western blot) for another 1 h. GAPDH (CST, USA, 5174, 1:1000 dilution for western blot) served as a loading control. The primary antibodies used for western in this study were as follows: LC3B-II (CST, USA 2775, 1:1000 dilution), p62 (CST, USA, 39749, 1:1000 dilution), Caspase-3 (CST, USA, 9662, 1:1000 dilution), Nod1 (CST, USA, 3545S, 1:1000 dilution), RIP2 (Abcam, UK, ab8428, 1:1000 dilution), p-ERK1/2 (CST, USA, 4370S, 1:1000 dilution), ERK1/2 (CST, USA, 4695, 1:1000 dilution), FOXO4 (CST, USA, 9472, 1:1000 dilution), p-IKKA (Abcam, UK, ab38515, 1:1000 dilution), IKKA (Abcam, UK, ab32041, 1:1000 dilution), BCL-2 (Abcam, UK, ab32124, 1:1000 dilution), BNIP3 (Abcam, UK, ab109362, 1:1000 dilution).
Mongolian Gerbil H. pylori Infection Model
Five Mongolian gerbils weighing 60–80 g were used to establish the in vivo H. pylori model and five H. pylori-negative gerbils were used as controls. All gerbils were obtained from the Capital Medical University and were fed at secondary biosafety laboratories at the Chinese Center for Disease Control and Prevention. Gerbils were housed in standard plastic cages in a room with a 12-h light/dark cycle and free access to food and water throughout all experiments. Gerbils 6–8 weeks of age were infected with H. pylori ATCC 43504 strain solution by oral gavage with 0.5 mL 2×109 CFU/mL. Gerbils were fasted for 12 h prior to challenge, and oral gavage was performed 5 times at intervals of 48 h. Before animals were euthanized, the 13C urea breath test and PCR were performed to confirm that H. pylori had colonized the gerbils. Ninety weeks after infection, gerbils were euthanized and the stomach tissue samples and blood were collected.
The animal experiments were conducted in accordance with the Guidelines of the CMU Animal Experiments and Experimental Animals Management Committee under a protocol approved by the Animal Experiments and Experimental Animal Welfare Committee of CMU (Permit number: AEEI-2016-154).
TUNEL Staining
TUNEL staining was performed using the TUNEL Bright Green Apoptosis Detection Assay kit (Vazyme, China, A112) to detect apoptotic cells in the gerbil’s stomach according to the manufacturer’s instructions. Briefly, paraffin sections were dewaxed, hydrated, and treated with proteinase K for 30 min and then incubated with a fluorescently labeled solution of dUTP and TdT enzyme for 80 min at 37°C. Positive controls were incubated with DNase I for 10 min at room temperature prior to the fluorescent labeling procedure, while negative controls were incubated with dUTP for 10 min. The nuclei were then counterstained with Hoechst and the samples were blocked by antifading mounting medium.
Immunohistochemistry
Stomach sections were incubated with rabbit monoclonal anti-LC3 antibody (Abcam, UK, ab128025, 1:100 dilution for immunohistochemistry) overnight at 4°C followed by incubation with corresponding biotinylated secondary antibody. The cell nuclei were counterstained with hematoxylin, and the samples were dehydrated in a gradient series, vitrified with dimethylbenzene, and finally mounted with neutral balsam.
ELISA Analysis
The levels of BCL-2 and BNIP3 in the gerbils’ sera were measured by ELISA using the Gerbil BCL-2 ELISA kit (Enzymatic Biotechnology, China) and Gerbil BNIP3 ELISA kit (Enzymatic Biotechnology, China) following the manufacturer’s instructions. A 47 μL volume of sample dilution solution and 3 μL sample were added to each sample well of the enzyme labeling plate. After mixing, 100 μL enzyme labeling reagent was added into each well and incubated at 37°C for 1 h. The plate was washed, and the color developer was added at 37°C in a dark environment. The reaction was terminated after 15 min. Finally, the absorbance of each well was measured at 450 nm wavelength in a microplate reader.
Statistical Analysis
All statistical analyses were carried out using SPSS v19.0 software. Data are expressed as the mean ± standard deviation. Independent sample t-test and one-way ANOVA analysis were used to determine the significance of the differences between the results. The Mann-Whitney U test was used to analyze gene expression from TCGA databases. Differences were considered statistically significant when the p-value was <0.05.
Results
Optimum Concentration of H. pylori Lysate Co-cultured With Gastric Epithelial Cells
To select a suitable concentration of H. pylori lysate for long-term co-culture with cells, we tested different concentrations of lysate for co-culture with gastric epithelial cells for 72 h. The MTT assay showed that the cell activity decreased as the concentration of H. pylori lysate increased. To ensure the stable growth of cells in long-term co-culture with H. pylori lysate, the concentration of lysate with 70%–80% activity of normal cells was chosen as the optimum concentration, which was determined to be 2 μg/mL for GES-1 cells and 1.5 μg/mL for MKN-45 cells (Figures 1A, B). GES-1 cells and MKN-45 cells of the experimental group were cultured in medium with H. pylori lysate for 30 consecutive generations using the optimum concentration determined above, and they were labeled as Cul30-GES-1 and Cul30-MKN-45, respectively. The untreated normal cells were labeled as B-GES-1 and B-MKN-45.
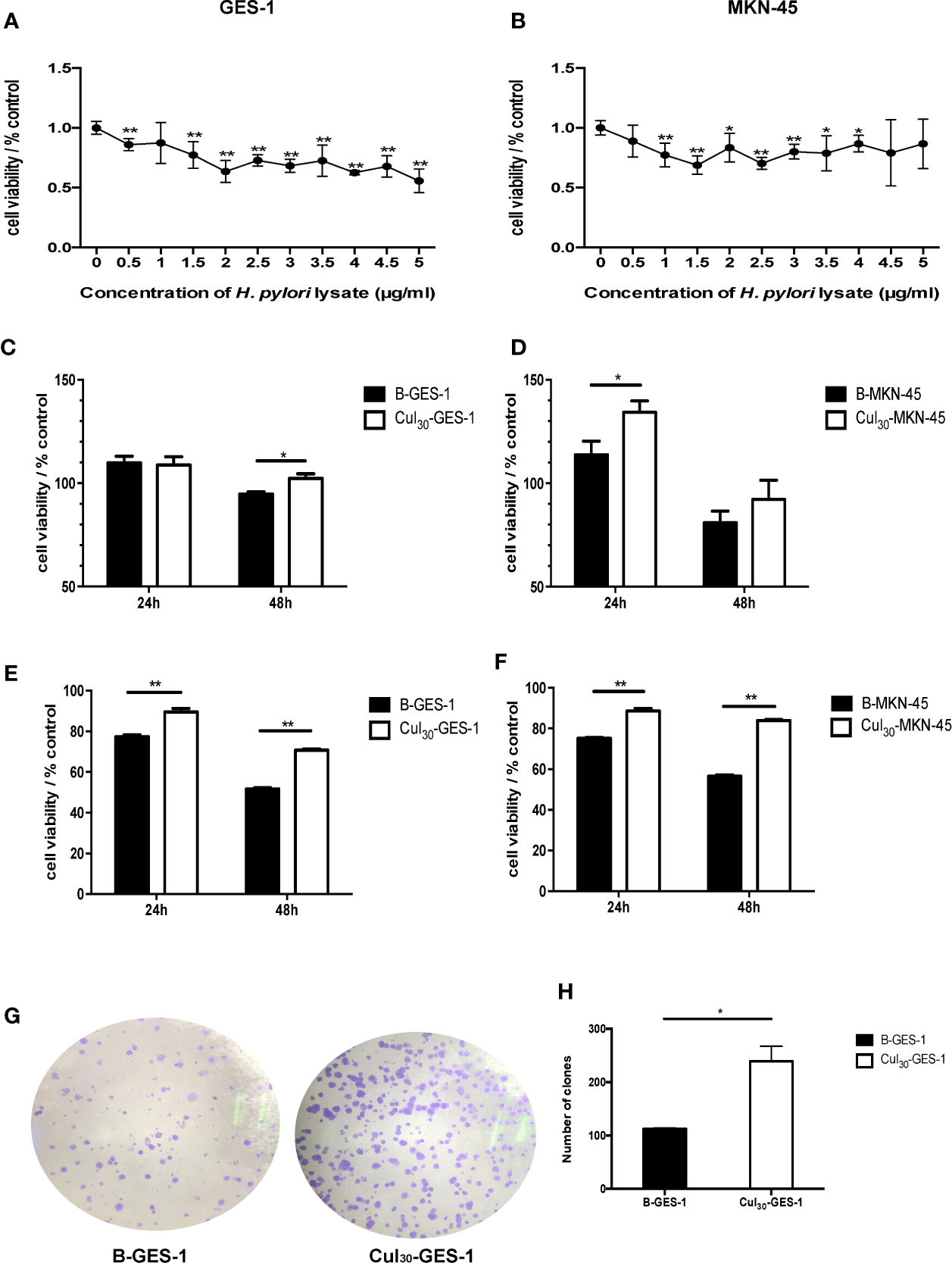
Figure 1 Sustained exposure to H. pylori lysate promotes proliferation of GES-1 and MKN-45 cells. GES-1 (A) and MKN-45 (B) cells were co-cultured with H. pylori lysate at a concentration of 0–5 μg/mL for 72 h, respectively. The concentration of H. pylori lysate exhibiting 70%–80% activity of normal cell activity was determined as the long-term co-culture concentration. Cul30-GES-1, B-GES-1, Cul30-MKN-45, and B-MKN-45 cells (n = 5 experiments) were challenged with H. pylori lysate (2 μg/mL for GES-1 cells and 1.5 μg/mL for MKN-45 cells) (C, D) or H. pylori (6×106 CFU/mL) (E, F) for 24 h or 48 h, respectively. Cell viability was detected by the MTT assay. n = 5. (G, H) The proliferation of Cul30-GES-1 and B-GES-1 cells was detected by the plate cloning assay. Image J software was used to count the visible colonies (n = 3 experiments). *p < 0.05, **p < 0.01.
Sustained Exposure to H. pylori Lysate Promoted Proliferation of GES-1 and MKN-45 Cells
To study the regulation of H. pylori lysate on cell growth, Cul30-GES-1, B-GES-1, Cul30-MKN-45, and B-MKN-45 cells were cultured with H. pylori lysate (2 μg/mL for GES-1 cells and 1.5 μg/mL for MKN-45 cells) or H. pylori (6×106 CFU/mL) (23) for 24 h or 48 h, respectively. The results of the MTT assay showed that the activities of Cul30-GES-1 and Cul30-MKN-45 cells treated with H. pylori lysate or H. pylori exceeded those of B-GES-1 and B-MKN-45 cells (Figures 1C–F), which was further verified by the colony formation assay of GES-1 cells (Figures 1G, H). These data indicated that long-term treatment of H. pylori lysate promoted the proliferation of GES-1 and MKN-45 cells. Meanwhile, the cells still maintained strong activity under long-term exposure to H. pylori lysate.
Sustained Exposure to H. pylori Lysate Alters Migration and Invasion of Gastric Epithelial Cells
To investigate whether H. pylori lysate would regulate the migratory and invasive ability of GES-1 and MKN-45 cells, Cul30-GES-1, B-GES-1, Cul30-MKN-45, and B-MKN-45 cells were cultured with H. pylori lysate (2 μg/mL for GES-1 cells and 1.5 μg/mL for MKN-45 cells) or H. pylori (6×106 CFU/mL) for 24 h. The results of the wound healing assay showed that the migratory ability of Cul30-GES-1 exposed to H. pylori lysate or H. pylori was significantly inhibited compared with that of B-GES-1 cells (Figure 2A). The transwell migration assay yielded consistent results (Figure 2B). These data revealed that a 24-h exposure to H. pylori lysate had no significant effect on the migratory ability of GES-1 cells, but long-term exposure to H. pylori lysate could significantly inhibit GES-1 cell migration. Nevertheless, in MKN-45 cells, H. pylori lysate or H. pylori exposure only promoted the migratory ability of B-MKN-45 cells with no significant differences (Figure 2C), indicating that long-term exposure of H. pylori lysate did not inhibit migration of gastric cancer cells.
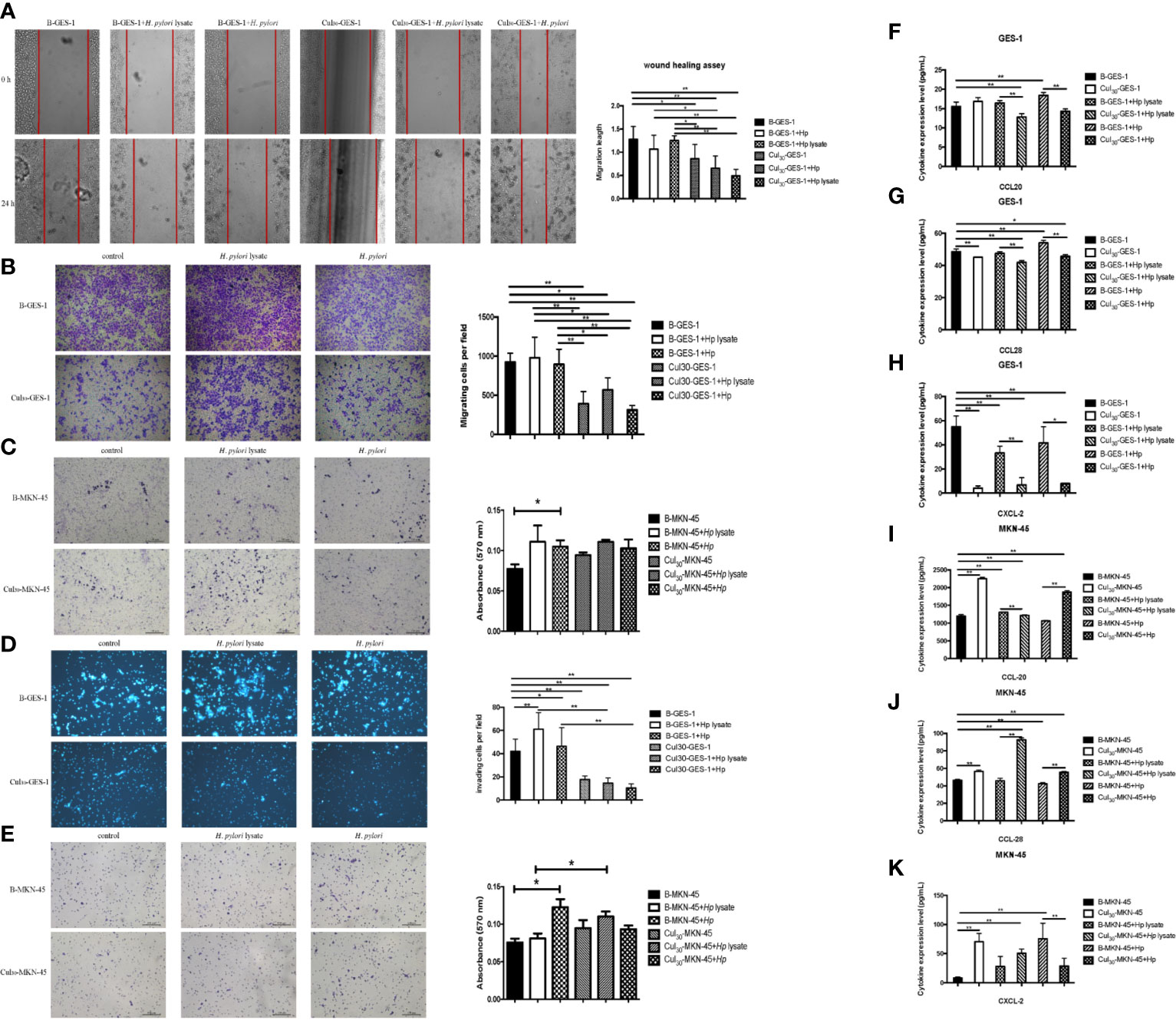
Figure 2 Sustained exposure to H. pylori lysate alters migration and invasion of GES-1 and MKN-45 cells. GES-1 or MKN-45 cells were treated with H. pylori lysate (2 μg/mL for GES-1 cells and 1.5 μg/mL for MKN-45 cells) or H. pylori (6×106 CFU/mL) for 24 h. (A) The wound healing assay was used to determine the migration of Cul30-GES-1 and B-GES-1 cells exposure to H. pylori lysate or H. pylori. Red lines represent the borders of the wounds (n=5 experiments). (B–E) Transwell migration (B, C) and Transwell invasion (D, E) assays were performed to detect the migration and invasion ability of Cul30-GES-1, B-GES-1, Cul30-MKN-45, and B-MKN-45 cells exposed to H. pylori lysate or H. pylori. (F–K) Expression of CCL-20 (F), CCL-28 (G), and CXCL-2 (H) in GES-1 cells, and the expression of CCL-20 (I), CCL-28 (J), and CXCL-2 (K) in MKN-45 cells were measured by Luminex assays. n = 3. *p < 0.05, **p < 0.01.
For the cell invasion assay, short-term exposure to H. pylori lysate or H. pylori promoted the invasive ability of B-GES-1 cells, but it inhibited the invasion of Cul30-GES-1 cells (Figure 2D). In addition, the invasive ability of Cul30-MKN-45 cells challenged by the H. pylori lysate increased significantly (Figure 2E). These results indicated that exposure to H. pylori lysate promoted the invasion of gastric epithelial cells but inhibited the invasion of cells co-cultured with H. pylori lysate. However, treatment of H. pylori lysate did not affect the invasive ability of gastric adenocarcinoma cells, but instead promoted the invasion of MKN-45 cells co-cultured with H. pylori lysate.
Sustained Exposure to H. pylori Lysate Inhibited the Expression of CCL20, CCL28, and CXCL-2 of Gastric Epithelial Cells
The long-term exposure to H. pylori lysate inhibited the migration and invasion of normal gastric epithelial cells, but it had no effect on cancer cells. To investigate the mechanisms that altered migration and invasion of gastric cells, we evaluated the expression of cytokines CCL20, CCL28, and CXCL-2, which promote the migration and invasion of various cancer cells (24–26). Data showed that the expression of CCL20, CCL28, and CXCL-2 in Cul30-GES-1 cells under the treatment of H. pylori (6×106 CFU/mL) or lysate (2 μg/mL for GES-1 cells and 1.5 μg/mL for MKN-45 cells) for 24 h significantly decreased relative to B-GES-1 cells (Figures 2F–H). However, the expression levels of these cytokines in Cul30-MKN-45 cells increased significantly compared with those of B-MKN-45 cells (p<0.01, Figures 2I–K). These results further confirmed that long-term exposure to H. pylori lysate altered migration and invasion of gastric epithelial cells.
Autophagy of Gastric Epithelial Cells Was Inhibited by Persistent Treatment With H. pylori Lysate
After treating Cul30-GES-1 and B-GES-1 cells with H. pylori lysate (2 μg/mL) or H. pylori (6×106 CFU/mL) for 24 h, the expression of an important indicator of autophagy LC3b-II (27) in B-GES-1 cells and Cul30-GES-1 cells treated with H. pylori lysate or H. pylori was upregulated in both cell lines. Further, the level of LC3b-II in H. pylori-treated B-GES-1 cells was significantly higher than that in cells exposed to lysate. Moreover, the expression of LC3b-II in Cul30-GES-1 cells co-cultured with H. pylori lysate or H. pylori was significantly lower than that in B-GES-1 cells (Figure 3A). These results demonstrated that long-term exposure of H. pylori lysate inhibited the autophagy of GES-1 cells. We then detected expression levels of vital autophagy regulator IFN-γ and FAS (28, 29) in B-GES-1 and Cul30-GES-1 cells under different conditions, and further confirmed the above results (Figures 3B, C).
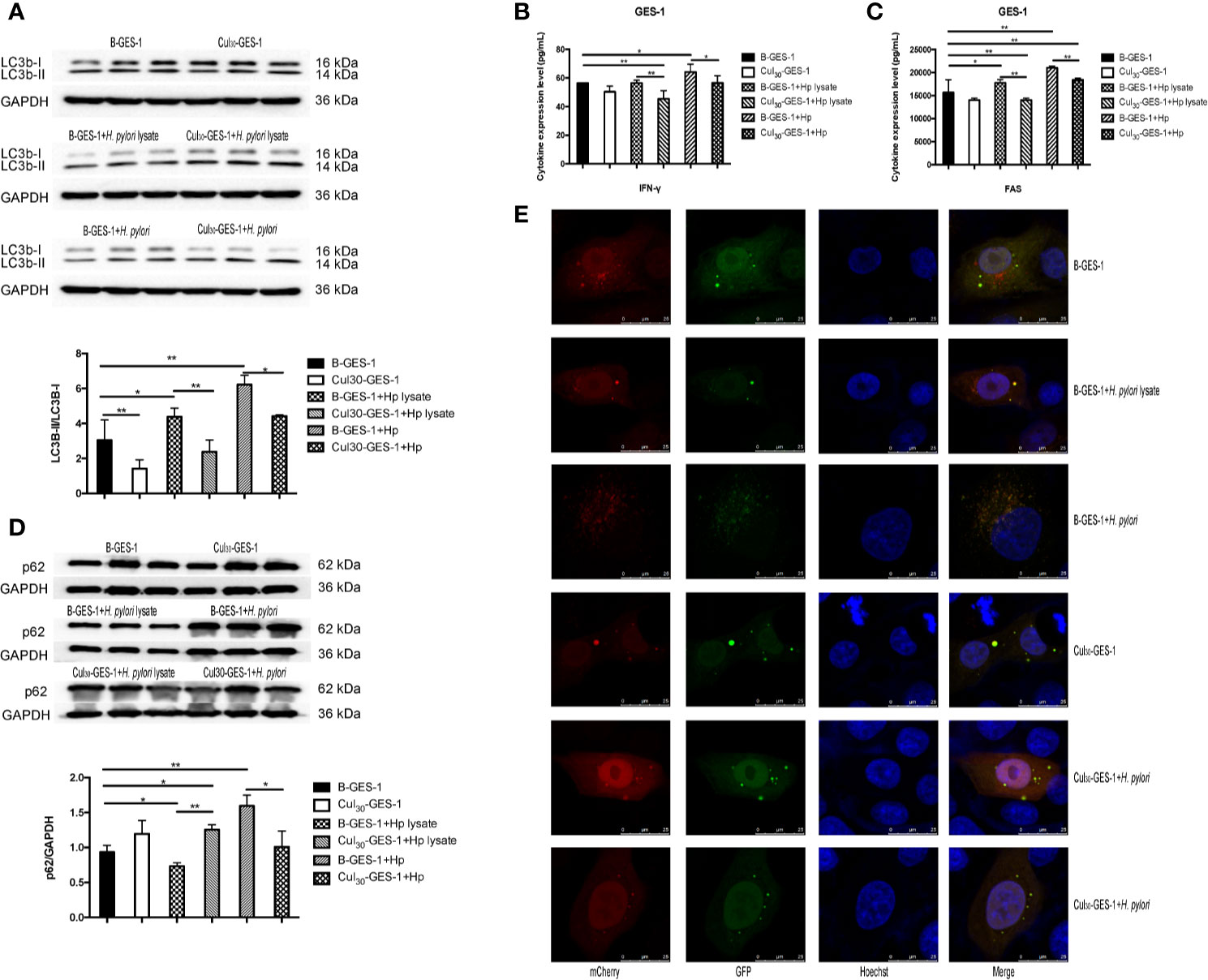
Figure 3 The treatment of H. pylori lysate inhibits autophagy of GES-1 cells. Cul30-GES-1 and B-GES-1 cells were challenged with H. pylori lysate (2 μg/mL) or H. pylori (6×106 CFU/mL) for 24 h, and the expression of LC3b-II/LC3b-I (A) and p62 (D) in Cul30-GES-1 and B-GES-1 cells exposed to H. pylori lysate or H. pylori were detected by western blot. The expression of IFN-γ (B) and FAS (C) were measured by Luminex assays. (E) Representative fluorescence images of autophagosomes and autolysosomes in GES-1cells treated with H. pylori lysate using the tandem mCherry-EGFP-LC3 fusion protein assay. The autophagy flux was evaluated by the ratio of red spots to yellow spots. The yellow spots indicate autophagosomes, while the red spots indicate autolysosomes. If the phagosome and lysosome fuses normally, then the red fluorescence is greater than the yellow fluorescence. If downstream autophagy is blocked, the phagosome and lysosome cannot fuse normally, and then yellow fluorescence is the main color visualized. n=3. *p < 0.05, **p < 0.01.
The expression of LC3b-II and SQSTM1/p62 is regulated by the production and removal of autophagosomes (27). We next evaluated the expression of p62 in lysate-treated GES-1 cells. Western blot assays showed that the p62 level was inhibited in B-GES-1 cells after 24-h treatment of H. pylori lysate, while it was enhanced in Cul30-GES-1 cells (Figure 3D). Furthermore, we transfected B-GES-1 and Cul30-GES-1 cells with mCherry-EGFP-LC3 lentiviral vector. The EGFP signal in the mCherry-EGFP-LC3 fusion protein was quenched under acidic pH in autophagolysosomes, which allowed easier detection of autophagolysosomes (GFP-negative/RFP-positive; red dots) and autophagosomes (GFP positive/RFP positive; yellow dots) (Figure 3E). In B-GES-1 and lysate-treated B-GES-1 cells, the red puncta were more numerous than the yellow puncta, which was in contrast with Cul30-GES-1 cells (Figure 3E). These observations suggested that long-term treatment of H. pylori lysate might inhibit autophagy flux of GES-1 cells.
Constant Treatment of H. pylori Lysate Inhibited Apoptosis of GES-1 Cells and Contributed to a Tendency for Cell Canceration
We next identified whether H. pylori lysate would regulate the apoptosis of GES-1 cells. After being challenged by H. pylori lysate (2 μg/mL) or H. pylori (6×106 CFU/mL) for 24 h, the expression of caspase-3 in Cul30-GES-1 cells decreased significantly, compared with that of B-GES-1 cells (Figure 4A). We also stained the nucleus of GES-1 cells with Hoechst and performed flow cytometry analysis, and the results further supported the above results (Figures 4B, C). In addition, we evaluated the expression of TNF-α, SLPI, and TFPI, which promote cell death (30–32). The expression levels of SLPI in Cul30-GES-1 cells treated with H. pylori or H. pylori lysate were significantly decreased compared to the control group (Figure 4D). After continuous co-culture with H. pylori lysate, the expression of TNF-α in Cul30-GES-1 cells showed a decreased trend (Figure 4E). However, there were no significant differences in the expression of TFPI between the two groups (Figure 4F).
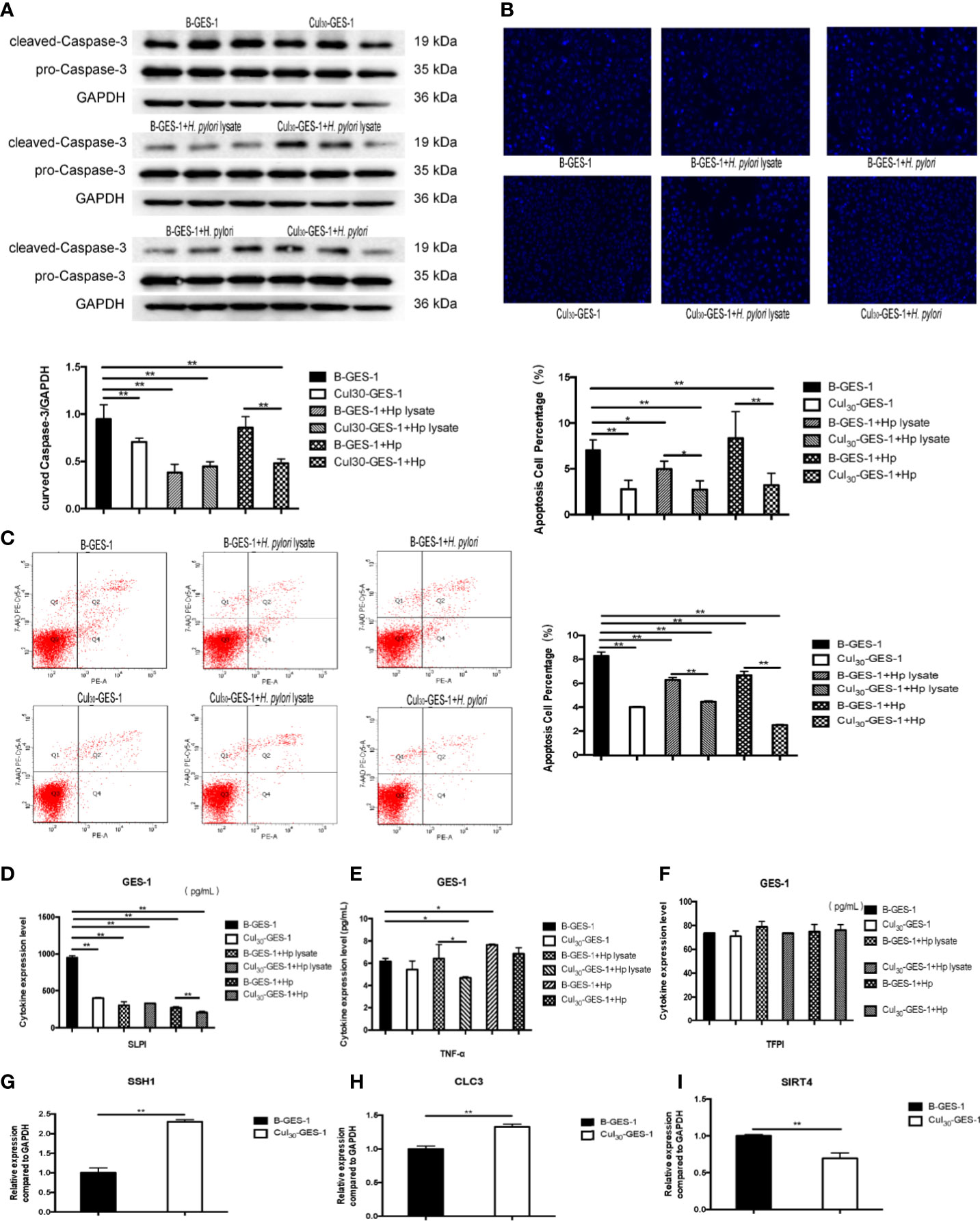
Figure 4 Continuous treatment of H. pylori lysate inhibits apoptosis of GES-1. Cul30-GES-1 and B-GES-1 cells were challenged with H. pylori lysate (2 μg/mL) or H. pylori (6×106 CFU/mL) for 24 h and (A) the expression of cleaved Caspase-3 in Cul30-GES-1 and B-GES-1 cells exposed to H. pylori lysate or H. pylori were analyzed by Western blot. (B) Cells treated with H. pylori lysate or H. pylori stained with Hoechst 33342 (10 μg/mL) for 10 min. Nuclear morphologic changes were examined under a fluorescence microscope. (C) Cell apoptosis detected by flow cytometry. (D–F) The expression of SLPI (D), TNF-α (E), and TFPI (F) of B-GES-1 and Cul30-GES-1 cells were measured by Luminex assays. (G–I) Real-time qPCR results showing mRNA levels of SSH1 (G), CLC3 (H), and SIRT4 (I) of Cul30-GES-1, and normalized to control cells. n=3. *p < 0.05, **p < 0.01.
Given that persistent stimulation by H. pylori lysate exposure promoted proliferation and inhibited autophagy and apoptosis of GES-1 cells, we hypothesized that the treatment could promote the tendency for malignant progression. We detected the mRNA expression level of three gastric cancer biomarkers, chloride channel-3 (CLC-3), slingshot protein phosphatase 1 (SSH1), and sirtuin 4 (SIRT4) (33–35) in Cul30-GES-1 and B-GES-1 cells. We found that the mRNA levels of SSH1 and CLC3 increased significantly, while SIRT4 decreased, compared with the control group (Figures 4G–I). These data suggested that long-term stimulation by H. pylori lysate may contribute to the tendency of malignant transformation of gastric epithelial cells.
Screening of Genes Involved in H. pylori-Induced Gastric Cancer Based on TCGA Database
To determine potential pathways affected by persistence stimulation by H. pylori lysate, we downloaded a dataset from a patient cohort with gastric adenocarcinoma from the TCGA database (https://cancergenome.nih.gov/). We divided the data into two groups according to patients with (n=17) or without H. pylori infection (n=115). We found that compared with H. pylori negative group, H. pylori infection induced the downregulation of the forkhead box O4 (FOXO4) gene (Figure 5A), upregulation of B-cell lymphoma-2 (BCL2), and upregulation of growth arrest and DNA-damage-inducible Beta (GADD45B) genes (Figures 5B, C). Moreover, H. pylori infection resulted in upregulation of RIPK2, TNF receptor-associated factor (TRAF)1 and TRAF2 genes, and downregulation of autophagy related 12 homolog (ATG12) genes, although these differences were not statistically significant (Figures 5D–G).
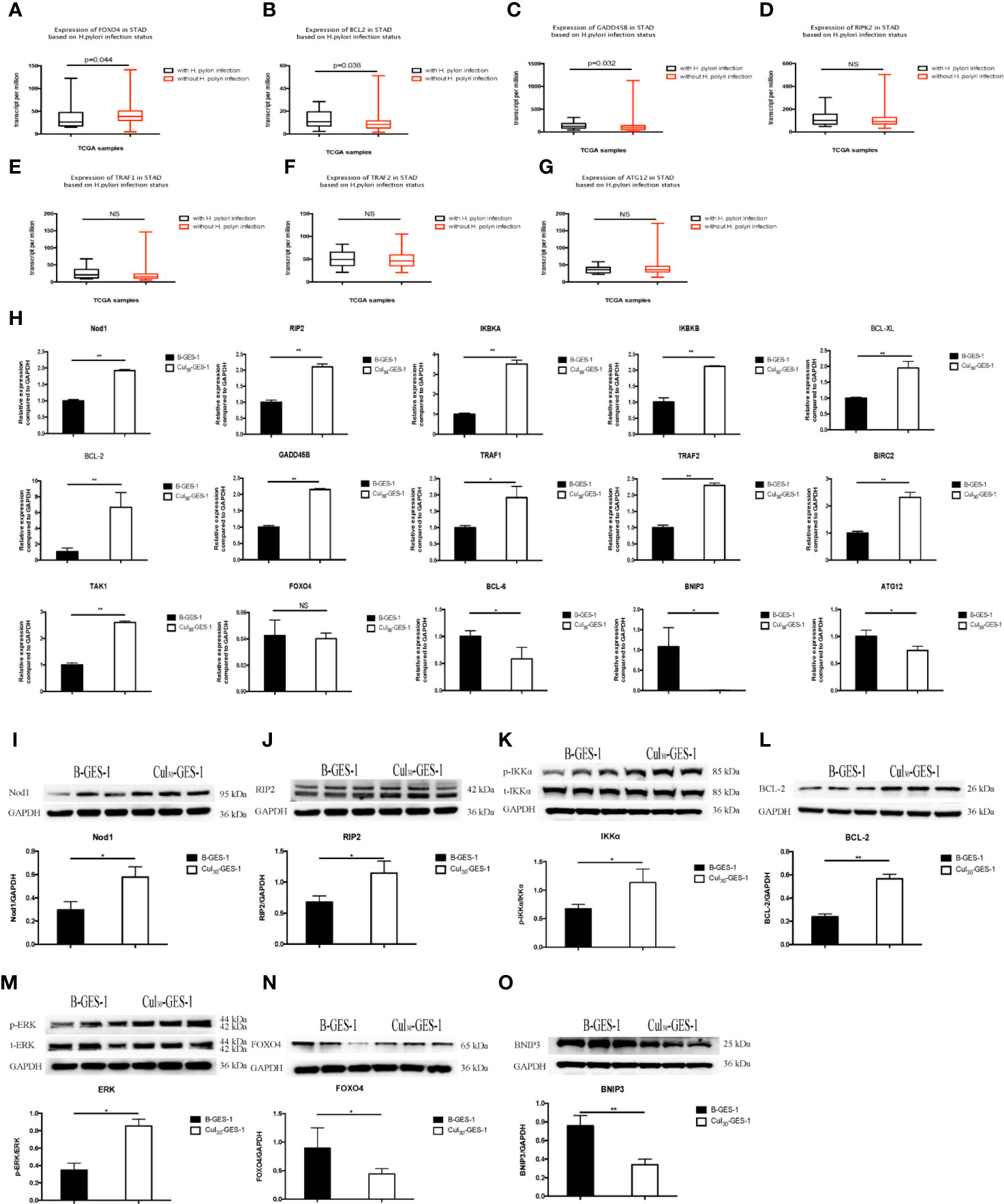
Figure 5 Pathways related to the inhibition of autophagy and apoptosis of gastric epithelial cells sustained exposure to H. pylori lysate. (A–F) Gene expression among H. pylori-positive (n=17) and H. pylori-negative (n=115) patients with gastric adenocarcinoma analyzed from the TCGA database. Down-regulation of FOXO4 (A) and ATG12 (G) is shown in H. pylori-positive samples, while BCL2 (B), GADD45B (C), RIP2 (D), TRAF1 (E), and TRAF2 (F) are upregulated. (H) mRNA expression of related genes in B-GES-1 and Cul30-GES-1 cells tested by Real-time qPCR. (I–O) Expression of Nod1, RIP2, p-IKKA/IKKA, BCL-2, p-ERK/ERK, FOXO4, and BNIP3 measured by Western blot. n=3. *p < 0.05, **p < 0.01. NS, No Significance.
The Nod Receptor Pathway Was Related to the Inhibition of Autophagy and Apoptosis of Gastric Epithelial Cells Under Persistent Treatment of H. pylori Lysate
Previous studies have indicated that Nod1-RIP2 regulates apoptosis through the NF-κB pathway and also regulates autophagy (36). Therefore, we evaluated the mRNA expression levels of Nod1, receptor-interacting protein 2 (RIP2), IκB kinase-α (IKBKA), and IκB kinase-β (IKBKB). All genes were significantly upregulated in Cul30-GES-1 cells, compared with B-GES-1 cells (Figure 5H). We then explored the mRNA levels of apoptosis-related downstream genes. B-cell lymphoma-XL (BCL-XL), BCL-2, GADD45B, TRAF1, TRAF2, and baculoviral IAP repeat-containing protein 2 (BIRC2) were upregulated in Cul30-GES-1 cells (Figure 5H). Moreover, we verified the protein levels of these genes. In accordance with the results above, the protein levels of Nod1, RIP2, IKKα, and BCL-2 increased significantly (Figures 5I–L). These results suggested that long-term exposure to H. pylori lysate may regulate the apoptosis of gastric epithelial cells through the Nod1-RIP2-NF-κB pathway.
Nod1/RIP2 inhibits FOXO4 expression through the mitogen-activated protein kinase (MAPK)/ERK pathway (37). Thus, we verified the mRNA and protein levels of related pathway and vital genes. The results indicated that the mRNA level of TAK1 was significantly higher than that of the control group, while the mRNA level of FOXO4 was inhibited, although there were no statistically significant differences (Figure 5H). The mRNA levels of B-cell lymphoma-6 (BCL-6), BNIP3, and ATG12 were also downregulated (Figure 5H). Western blot showed that the phosphorylation level of extracellular regulated protein kinases (ERK) increased (Figure 5M), while the expression of FOXO4 and BNIP3 decreased significantly (Figures 5N, O). These results suggest that long-term exposure of H. pylori lysate may regulate apoptosis and autophagy of gastric epithelial cells through the Nod1-RIP2-MAPK/ERK-FOXO4 pathway.
Long-Term Infection of H. pylori Inhibited Autophagy and Apoptosis of Gastric Epithelial Cells In vivo
We presumed that H. pylori proliferates continuously in the course of chronic infection. H. pylori and its lysate are in constant contact with gastric epithelial cells in vivo. Autophagy and apoptosis of cells may be inhibited, which is conducive to the sustained colonization of H. pylori, and this may promote a tendency toward progression to gastric cancer. We then carried out in vivo experiments to verify the results of the above in vitro results. Mongolian gerbils were infected with the H. pylori 43504 strain for 90 weeks and then the gastric tissues were collected. The TUNEL staining assay and immunohistochemistry were performed to determine the degree of apoptosis and autophagy induced in gastric epithelial cells. Compared to the control group, the apoptosis and autophagy of gerbils continuously exposed to H. pylori infection were remarkably inhibited (Figures 6A, B). Bcl-2 and BNIP3 are two key regulators of autophagy and apoptosis (38). The expression of BNIP3 has also been reported to be absent in gastric cancer (39). The serum levels of Bcl-2 and BNIP3 in gerbils were also tested, and the results were consistent with the data above (Figure 6C). Furthermore, hyperplastic lesions were identified in the stomach tissue of gerbils infected with H. pylori (Figures 6D, F), showing a tendency of gastric lesions to transform to cancer.
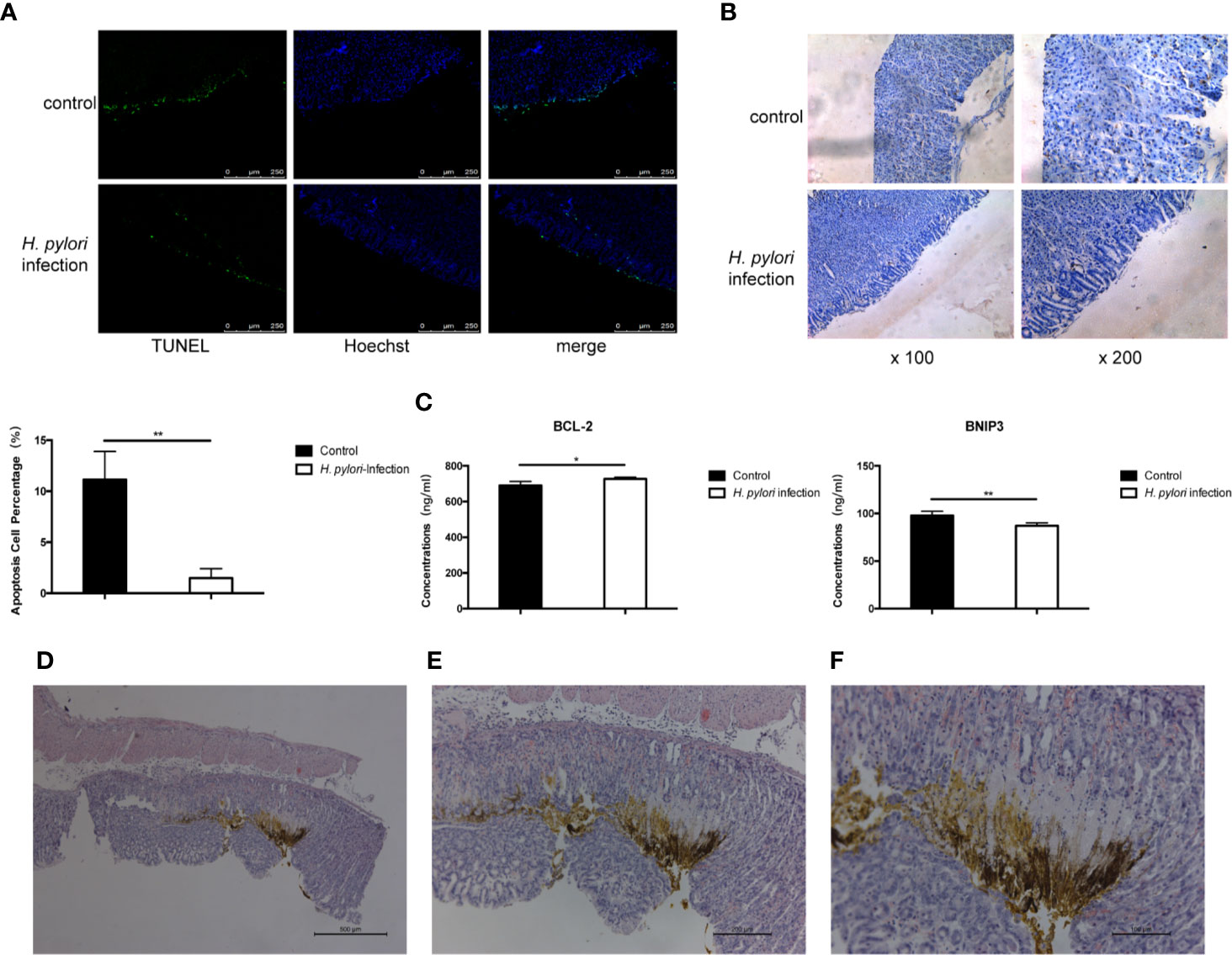
Figure 6 Long-term infection of H. pylori inhibits autophagy and apoptosis of gastric epithelial cells in vivo. Mongolian gerbils were infected by H. pylori for 90 weeks. Serum and gastric tissue were collected. (A) TUNEL staining assay and immunohistochemistry (B) of LC3 was performed to determine the apoptosis and autophagy of gerbil gastric epithelial cells. (C) The serum levels of BCL-2 and BNIP3 in gerbils (tested by ELISA; n=5). *p < 0.05, **p < 0.01. (D–F) HE staining revealing the pathology of Mongolian gerbil stomach samples. Representative histologic images from H. pylori-infected gerbils at original magnification ×50 (D), ×100 (E), and ×200 (F).
Discussion
The global infection rate of H. pylori is about 44% (40). H. pylori is the most important risk factor for the development of gastric cancer (41). In the process of colonization, large amounts of H. pylori bacteria die and breakdown naturally, releasing lysate components, which influence the host cell in several ways. For example, its OMVs bind with pattern recognition receptors (PRR) on the surface of the host cell and regulate important cytological functions, including migration, invasion, apoptosis, autophagy, and carcinogenesis (22, 42, 43). However, there are some limitations that may have influenced the results of these studies. One is that the exposure time to bacterial lysate is generally too short to simulate an effective H. pylori infection in vivo. In addition, notably, when the components of the H. pylori lysate were isolated, they were antagonistic to each other in regulating some of the functions of the host cells. For instance, VacA induced apoptosis, while CagA blocked this effect and inhibited apoptosis. On the contrary, VacA also inhibited cytoskeleton deformity induced by CagA (27, 28). A single bacterial toxin cannot completely replace the regulatory effects of H. pylori on host cells. Therefore, it is necessary to use a H. pylori lysate instead of live bacteria to study the regulation of the host cell function by the continuous exposure to H. pylori infection. Considering that gastric epithelial cells are known to be initial contact points of bacteria in the gastric mucosa during H. pylori infection (44), human gastric epithelial cells GES-1 were used in our study. We co-cultured H. pylori lysate with GES-1 and MKN-45 cells for 30 generations and established a cell model of chronic stimulation of H. pylori lysate able to simulate the long-term symbiosis of H. pylori and host cells more closely, and to explore any relevant pathogenesis.
Cell proliferation is related to apoptosis, autophagy, and carcinogenesis. However, the effects of H. pylori and H. pylori lysate on the proliferation of gastric epithelial cells are not consistent. In vitro experiments showed that H. pylori promoted proliferation of gastric epithelial cells at low concentrations but promoted apoptosis at higher concentrations (45). Nonetheless, the effects of H. pylori lysate were consistent with those of H. pylori (46). It is generally believed that the contrasting results are due to the various types of H. pylori strains used and the different gastric epithelial cells with which they interacted. In this study, we found that the lysate of the H. pylori ATCC 43504 strain inhibited proliferation of B-GES-1 and B-MKN-45 cells dose-dependently. To ensure the survival of cells and achieve the goal of long-term co-culture of cells and H. pylori lysate, we chose a concentration of H. pylori lysate when the cell value-added rate was about 70%–80% as the optimum concentration. After continuous co-culture with H. pylori lysate, proliferation of Cul30-GES-1 cells significantly increased, which may be due to the tolerance of cells to H. pylori lysate.
Cell invasion and migration are two important characteristics in the process of cell carcinogenesis. H. pylori infection promotes migration and invasion of gastric epithelial cells in a CagA-dependent manner (47). CagA is translocated into the host cell mainly by the T4SS of H. pylori (48) and interacts with E-cadherin (49), resulting in the increase of movement and elongation of the host cell (50). In this study, differently from previous studies, we found that constant exposure to H. pylori lysate inhibited the migration and invasion of GES-1 cells. This may be due to the brief H. pylori stimulation, only 24 h, while we challenged gastric cells for 30 generations. Furthermore, ultrasonic lysis was used to prepare H. pylori lysate, and this process destroyed the T4SS, allowing only a small amount of CagA to enter the host cells through OMVs, and thus its promoting effect on cell migration and invasion may have been antagonized by other components, like lipopolysaccharide (LPS) (43). It is worth noting that differently from the effects on GES-1 cells, constant stimulation by H. pylori lysate showed less inhibition on the migration and invasion potential of MKN-45 cells, which may be due to the malignant properties of the cells. Although originating from normal cells, cancer cells have unique biological characteristics and behavior. For example, studies found that the mRNA and protein levels of HER2 in MKN-45 cells are significantly higher than that in GES-1 cells, and HER2 is subsequently proved to promote the migration and invasion of gastric cancer cells by upregulating CXCR4 (51). The higher migration and invasion characteristics of cancer cells may explain the reduced inhibitory effects of H. pylori lysate on the migration and invasion of MKN-45 cells.
Autophagy disorders interfere with health and disease (52). Many studies have shown that the inflammatory pathway promotes tumor progression by regulating autophagy (53). When H. pylori infects gastric epithelial cells for a short time, VacA and urease induce autophagy (54), but after a prolonged co-culture, H. pylori destroys the autophagy pathway and accumulates cells due to dysfunctional autophagy (7). We found that sustained exposure to H. pylori lysate blocked the autophagy flux of Cul30-GES-1 cells, resulting in a failure of fusion of autophagosomes and autolysosomes, and the accumulation of cells with defective autophagy. H. pylori can invade gastric epithelial cells and are isolated by lysosomal acidified autophagy (55) to promote survival and colonization (56, 57). Our results suggest that H. pylori lysates play vital roles in the process of chronic infection and inhibit the autophagy of host cells, which contributes to the survival and colonization of H. pylori.
The dysregulation of autophagy and apoptosis has adverse effects on the body and may even lead to cancer. It is believed that autophagy induced by H. pylori is associated with apoptosis, while autophagy occurs earlier than apoptosis (58). It has been reported that H. pylori infection inhibits apoptosis of the gastric epithelial cell (59, 60). We found that long-term stimulation of H. pylori lysate also inhibited the apoptosis of gastric epithelial cells, and we assumed that long-term exposure to H. pylori lysate may contribute to the initiation of the malignant transformation of the cell. To further evaluate the potential carcinogenic effects of exposure to H. pylori lysate, we detected mRNA levels of CLC-3, SSH1, and SIRT4, and the results confirmed our hypothesis. Sustained exposure to H. pylori lysate promoted the proliferation of gastric epithelial cells, inhibited autophagy and apoptosis, and facilitated the survival and colonization of bacteria, which may further promote the malignant transformation of cells. This was also confirmed by results in vivo. We infected Mongolian gerbils with H. pylori 43504 strain for 90 weeks. Although no carcinogenesis was evidenced in the stomach, dysplasia was present. Gastric epithelial dysplasia is a crucial pathology stage of the Correa cascade leading to gastric cancer (61). The observed dysplasia in model indicates that the pathological changes in the stomach of infected gerbils were indicative of transformation into cancer. These data suggest that H. pylori lysate acts as an “accomplice” in the process of H. pylori-induced gastric diseases.
Subsequently, we explored the underlying pathways involved in the long-term exposure cell model. Through the screening of clinical data and cell experiments, we found that continuous stimulation of H. pylori lysate upregulated mRNA and protein levels of Nod1-RIP2-NF-κB and of downstream genes BCL-2 and GADD45B, which is consistent with previous reports. The NF-κB pathway regulates cell apoptosis (60) and the regulation of BCL-2 by NF-κB plays an important role in host cell apoptosis induced by H. pylori infection (62). H. pylori activates NF-κB, inflammation and gastric cancer via Nod1-dependent activation (63). Furthermore, Nod1 is a member of the Nod-like receptor (NLR) family, a cytoplasmic recognition receptor in cells, which recognizes a variety of ligands, including peptidoglycan (PGN) and flagellin from bacterial pathogens and viral and bacterial RNA (19). The Nod1 mRNA expression level was also shown to be upregulated in gastric cancer tissues (64). These results suggested that H. pylori lysate may regulate apoptosis of gastric epithelial cells via the Nod1-RIP2- NF-κB pathway. In addition, we found that long-term stimulation of H. pylori lysate promoted the phosphorylation of ERK, and then inhibited the levels of FOXO4 and its downstream genes, BCL-6, BNIP3, and ATG12, which is consistent with reports indicating that the FOXO pathway regulates cell autophagy and apoptosis (65, 66).
In conclusion, we established a long-term gastric epithelial cell line model co-culture with H. pylori lysate to explore the effects of sustained exposure to H. pylori lysate on gastric cells, and we found that continuous treatment of H. pylori lysate promoted gastric epithelial cell proliferation and inhibited cell autophagy and apoptosis via the Nod1-NF-κB/MAPK-ERK/FOXO4 pathway (Figure 7). In the process of H. pylori-induced gastric lesions, H. pylori lysate acts as an “accomplice.”
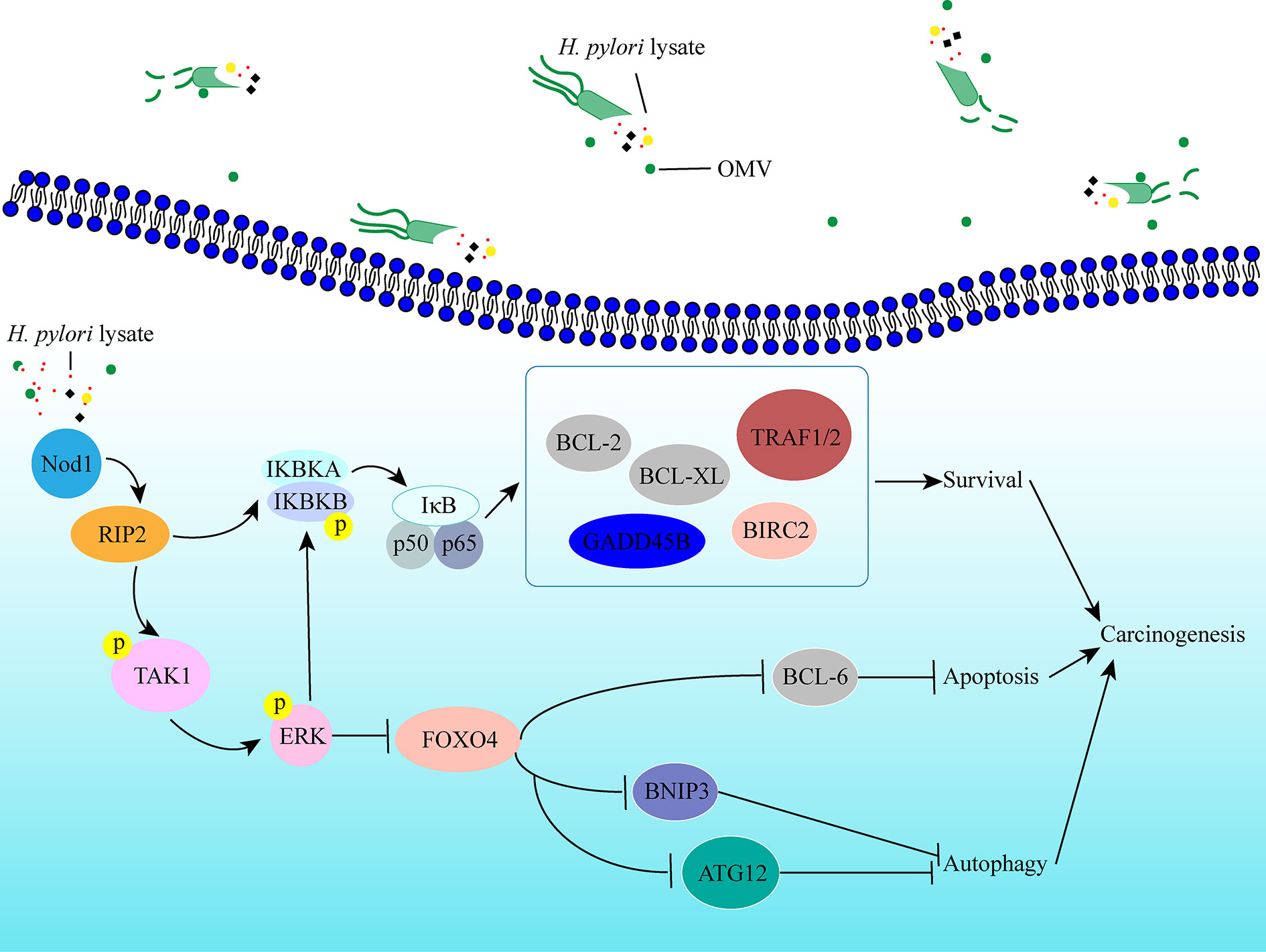
Figure 7 Schematic representation of the signaling pathways induced by prolonged exposure to H. pylori. Sustained exposure to H. pylori lysate inhibits apoptosis and autophagy of gastric epithelial cells via the Nod1-NF-κB/MAPK-ERK/FOXO4 pathway, which promotes cells survival, and may contribute to the tendency toward cell malignant transformation.
Data Availability Statement
Publicly available datasets were analyzed in this study. This data can be found here: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga.
Ethics Statement
The animal study was reviewed and approved by the Animal Experiments and Experimental Animal Welfare Committee of CMU (Permit number: AEEI-2016-154), Capital Medical University.
Author Contributions
All authors contributed to the study conception and design. ZC and CL designed the study. YH and CW conducted the experiments. YH created the figures and wrote the manuscript. XZ conducted the animal experiments. XLu mainly provided technical and material support. JX cultivated H. pylori and prepared H. pylori lysate. JLv evaluated the gene expression via the TCGA database. MG, XH, XLi, JLu, and XD did analysis and interpretation of data. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (No. 32070537, 31772545, 31970512, 31872308, 83902332), High-level Teachers in Beijing Municipal Universities in the Period of 13th Five Plan (No. IDHT20170516), National Key Research and Development Plan of China (No. 2017YFD0501602), and Beijing Science and Technology Program (D181100000518002).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.581364/full#supplementary-material
Abbreviations
H. pylori, Helicobacter pylori; T4SS, type IV secretion system; CagA, cytotoxin associated gene product A; VacA, vacuolating cytotoxin A; OMV, outer membrane vesicles; LPS, lipopolysaccharide; PGN, peptidoglycan; Ure, urease; STAT3, signal transducer and activator of transcription 3; ATCC, American type culture collection; PBS, phosphate buffer saline; BCA, bicinchoninic acid; DMEM, dulbecco’s modified eagle medium; FBS, fetal bovine serum; MTT, 3-(4, 5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide; DMSO, dimethyl sulfoxide; EGFP, enhanced green fluorescent protein; 7-AAD, 7-Aminoactinomycin D; TCGA database, The Cancer Genome Atlas database; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; RIP2, receptor-interacting protein 2; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor kappa B; ERK, extracellular regulated protein kinases; p-ERK, phosphorylated extracellular regulated protein kinase; FOXO4, forkhead box O4; IKKA/IKBKA, IκB kinase-α; p-IKKA, phosphorylated IκB kinase-α; BCL-2, B-cell lymphoma-2; BNIP3, BCL2 interacting protein 3; BCL-XL, B-cell lymphoma-XL; GADD45B, growth arrest and DNA-damage-inducible Beta; TRAF1/2, TNF receptor-associated factor ½; BIRC2, baculoviral IAP repeat-containing protein 2; ATG12, autophagy related 12 homolog; dUTP, 2’-deoxyuridine 5’-triphosphate; TdT enzyme, terminal deoxynucleotidyl transferase; PRR, pattern recognition receptor; NLR, Nod like receptor; CCL20, C-C motif chemokine ligand 20; CCL28, C-C motif chemokine ligand 28; CXCL-2, C-X-C motif chemokine ligand 2; IFN-γ, interferon- γ; FAS, cell surface death receptor; TNF-α, tumor necrosis factor- α; SLPI, secretory leukocyte peptidase inhibitor; TFPI, tissue factor pathway inhibitor; CLC-3, chloride channel-3; SSH1, slingshot protein phosphatase 1; SIRT4, sirtuin 4.
References
1. Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, et al. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med (2001) 345:784–9. doi: 10.1056/NEJMoa001999
2. Schulz C, Schütte K, Malfertheiner P. Helicobacter pylori and other gastric microbiota in gastroduodenal pathologies. Dig Dis (2016) 34:210–6. doi: 10.1159/000443353
3. Ahn HJ, Lee DS. Helicobacter pylori in gastric carcinogenesis. World J Gastrointest Oncol (2015) 7:455–65. doi: 10.4251/wjgo.v7.i12.455
4. Plummer M. Global burden of gastric cancer attributable to Helicobacter pylori. Int J Cancer (2015) 136:487–90. doi: 10.1002/ijc.28999
5. Möller H, Heseltine E, Vainio H. Working group report on schistosomes, liver flukes and Helicobacter pylori. Int J Cancer (1995) 60:587–9. doi: 10.1002/ijc.2910600502
6. Eslami M, Yousefi B, Kokhaei P, Arabkari V, Ghasemian A. Current information on the association of Helicobacter pylori with autophagy and gastric cancer. J Cell Physiol (2019) 234:14800–11. doi: 10.1002/jcp.28279
7. Hu W, Zhang L, Li MX, Shen J, Liu XD, Xiao ZG, et al. Vitamin D3 activates the autolysosomal degradation function against Helicobacter pylori through the PDIA3 receptor in gastric epithelial cells. Autophagy (2019) 15:707–25. doi: 10.1080/15548627.2018.1557835
8. Xie J, Lin Z, Xian Y, Xian Y, Kong S, Lai Z, et al. Patchouli alcohol protects against Helicobacter pylori urease-induced apoptosis, oxidative stress and inflammatory response in human gastric epithelial cells. Int Immunopharmacol (2016) 35:43–52. doi: 10.1016/j.intimp.2016.02.022
9. Wang J, Yao Y, Zhang Q, Li S, Tang L. Inflammatory responses induced by Helicobacter pylori on the carcinogenesis of gastric epithelial GES−1 cells. Int J Oncol (2019) 54:2200–10. doi: 10.3892/ijo.2019.4775
10. Takahashi KA, Knight CT, Hatakeyama M. Molecular anatomy and pathogenic actions of Helicobacter pylori CagA that underpin gastric carcinogenesis. Cell Mol Immunol (2020) 17:50–63. doi: 10.1038/s41423-019-0339-5
11. Neel BG, Gu H, Pao L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci (2003) 28:284–93. doi: 10.1016/S0968-0004(03)00091-4
12. Easton JB, Royer AR, Middlemas DS. The protein tyrosine phosphatase, Shp2, is required for the complete activation of the RAS/MAPK pathway by brain-derived neurotrophic factor. J Neurochem (2006) 97:834–45. doi: 10.1111/j.1471-4159.2006.03789.x
13. Gauthier NC, Monzo P, Kaddai V, Doye A, Ricci V, Boquet P. Helicobacter pylori VacA cytotoxin: a probe for a clathrin-independent and Cdc42-dependent pinocytic pathway routed to late endosomes. Mol Biol Cell (2005) 16:4852–66. doi: 10.1091/mbc.e05-05-0398
14. Capurro MI, Prashar A, Jones NL. Helicobacter pylori MCOLN1/TRPML1 inhibition - a novel strategy used by to escape autophagic killing and antibiotic eradication therapy. Autophagy (2019) 16:169–70. doi: 10.1080/15548627.2019.1677322
15. Keenan J, Day T, Neal S, Cook B, Bagshaw P. A role for the bacterial outer membrane in the pathogenesis of Helicobacter pylori infection. FEMS Microbiol Lett (2000) 182:259–64. doi: 10.1111/j.1574-6968.2000.tb08905.x
16. Parker H, Chitcholtan K, Hampton MB, Keenan JI. Uptake of Helicobacter pylori outer membrane vesicles by gastric epithelial cells. Infect Immun (2010) 78:5054–61. doi: 10.1128/IAI.00299-10
17. Chitcholtan K, Hampton MB, Keenan JI. Outer membrane vesicles enhance the carcinogenic potential of Helicobacter pylori. Carcinogenesis (2008) 29:2400–5. doi: 10.1093/carcin/bgn218
18. Mnich E, Kowalewicz KM, Sicińska P, Hinc K, Obuchowski M, Gajewski A, et al. Impact of Helicobacter pylori on the healing process of the gastric barrier. World J Gastroenterol (2016) 22:7536–58. doi: 10.3748/wjg.v22.i33.7536
19. Geddes K, Magalhães JG, Girardin SE. Unleashing the therapeutic potential of NOD-like receptors. Nat Rev Drug Discovery (2009) 8:465–79. doi: 10.1038/nrd2783
20. Allison CC, Kufer TA, Kremmer E, Kaparakis M, Ferrero RL. Helicobacter pylori induces MAPK phosphorylation and AP-1 activation via a NOD1-dependent mechanism. J Immunol (2009) 183:8099–109. doi: 10.4049/jimmunol.0900664
21. Ki MR, Goo MJ, Park JK, Hong IH, Ji AR, Han SY, et al. Helicobacter pylori accelerates hepatic fibrosis by sensitizing transforming growth factor-β1-induced inflammatory signaling. Lab Invest (2010) 90:1507–16. doi: 10.1038/labinvest.2010.109
22. Flahou B, Haesebrouck F, Chiers K, Van DK, De SL, Devreese B, et al. Gastric epithelial cell death caused by Helicobacter suis and Helicobacter pylori γ-glutamyl transpeptidase is mainly glutathione degradation-dependent. Cell Microbiol (2011) 13:1933–55. doi: 10.1111/j.1462-5822.2011.01682.x
23. He Q, Li G, Wang X, Wang S, Hu J, Yang L, et al. A decrease of histone deacetylase 6 expression caused by Helicobacter pylori infection is associated with oncogenic transformation in gastric cancer. Cell Physiol Biochem (2017) 42:1326–35. doi: 10.1159/000478961
24. Guo W, Li H, Liu H, Ma X, Yang S, Wang Z. DEPDC1 drives hepatocellular carcinoma cell proliferation, invasion and angiogenesis by regulating the CCL20/CCR6 signaling pathway. Oncol Rep (2019) 42:1075–89. doi: 10.3892/or.2019.7221
25. Yang XL, Liu KY, Lin FJ, Shi HM, Ou ZL. CCL28 promotes breast cancer growth and metastasis through MAPK-mediated cellular anti-apoptosis and pro-metastasis. Oncol Rep (2017) 38:1393–401. doi: 10.3892/or.2017.5798
26. Zheng Z, Zhao F, Zhu D, Han J, Chen H, Cai Y, et al. Long Non-Coding RNA LUCAT1 Promotes Proliferation and Invasion in Clear Cell Renal Cell Carcinoma Through AKT/GSK-3β Signaling Pathway. Cell Physiol Biochem (2018) 48:891–904. doi: 10.1159/000491957
27. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Arozena AA. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy (2016) 12:1–222. doi: 10.1080/15548627.2015.1100356
28. Zhang Y, Wu Y, Cheng Y, Zhao Z, Tashiro S, Onodera S, et al. Fas-mediated autophagy requires JNK activation in HeLa cells. Biochem Biophys Res Commun (2008) 377:1205–10. doi: 10.1016/j.bbrc.2008.10.151
29. Chen Y, Fang Y, Chang C Lin C, Hsu L, Wu S, Chui Y, et al. S100A10 Regulates ULK1 Localization to ER-Mitochondria Contact Sites in IFN-γ-Triggered Autophagy. J Mol Biol (2017) 429:142–57. doi: 10.1016/j.jmb.2016.11.009
30. Li S, Yang X, Feng Z, Wang P, Zhu W, Cui S. Catalase enhances viability of human chondrocytes in culture by reducing reactive oxygen species and counteracting tumor necrosis factor-α-induced apoptosis. Cell Physiol Biochem (2018) 49:2427–42. doi: 10.1159/000493841
31. Rosso M, Lapyckyj L, Amiano N, Besso MJ, Sánchez M, Chuluyan E, et al. Secretory Leukocyte Protease Inhibitor (SLPI) expression downregulates E-cadherin, induces β-catenin re-localisation and triggers apoptosis-related events in breast cancer cells. Biol Cell (2014) 106:308–22. doi: 10.1111/boc.201300075
32. Feng C, Ho Y, Sun C, Xia G, Ding Q, Gu B. TFPI-2 expression is decreased in bladder cancer and is related to apoptosis. J BUON (2016) 21:1518–23.
33. Gu Z, Li Y, Yang X, Yu M, Chen Z, Zhan C, et al. Overexpression of CLC-3 is regulated by XRCC5 and is a poor prognostic biomarker for gastric cancer. J Hematol Oncol (2018) 11:115. doi: 10.1186/s13045-018-0660-y
34. Daryabari SS, Safaralizadeh R, Hosseinpourfeizi M, Moaddab Y, Shokouhi B. Overexpression of SSH1 in gastric adenocarcinoma and its correlation with clinicopathological features. J Gastrointest Oncol (2018) 9:728–33. doi: 10.21037/jgo.2018.03.09
35. Sun H, Huang D, Liu G, Jian F, Zhu J, Zhang L. SIRT4 acts as a tumor suppressor in gastric cancer by inhibiting cell proliferation, migration, and invasion. Onco Targets Ther (2018) 11:3959–68. doi: 10.2147/OTT.S156143
36. Velloso FJ, Trombetta LM, Anschau V, Sogayar MC, Correa RG. NOD-like receptors: major players (and targets) in the interface between innate immunity and cancer. Biosci Rep (2019) 39:1–21. doi: 10.1042/BSR20181709
37. Roy SK. Inhibition of PI3K/AKT and MAPK/ERK pathways causes activation of FOXO transcription factor, leading to cell cycle arrest and apoptosis in pancreatic cancer. J Mol Signal (2010) 5:10. doi: 10.1186/1750-2187-5-10
38. Almasi S, Kennedy BE, El AM, Sterea AM, Gujar S, Partida SS, et al. TRPM2 channel-mediated regulation of autophagy maintains mitochondrial function and promotes gastric cancer cell survival via the JNK-signaling pathway. J Biol Chem (2018) 293:3637–50. doi: 10.1074/jbc.M117.817635
39. Murai M, Toyota M, Suzuki H, Satoh A, Sasaki Y, Akino K, et al. Aberrant methylation and silencing of the BNIP3 gene in colorectal and gastric cancer. Clin Cancer Res (2005) 11:1021–7.
40. Zamani M, Ebrahimtabar F, Zamani V, Miller WH, Alizadeh NR, Shokri SJ, et al. Systematic review with meta-analysis: the worldwide prevalence of Helicobacter pylori infection. Aliment Pharmacol Ther (2018) 47:868–76. doi: 10.1111/apt.14561
41. Ferlay J, Soerjomataram I, Dikshit RE, Mathers C, Rebelo M, Parkin DM, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer (2015) 136:E359–86. doi: 10.1002/ijc.29210
42. Zhang XS, Tegtmeyer N, Traube L, Jindal S, Perez PG, Sticht H, et al. The effect of Helicobacter pylori infection and different H. pylori components on the proliferation and apoptosis of gastric epithelial cells and fibroblasts. PloS One (2019) 14:e0220636. doi: 10.1371/journal.pone.0220636
43. Greenfield LK, Jones NL. Modulation of autophagy by Helicobacter pylori and its role in gastric carcinogenesis. Trends Microbiol (2013) 21:602–12. doi: 10.1016/j.tim.2013.09.004
44. Hooi JKY, Lai WY, Ng WK, Suen MMY, Underwood FE, Tanyingoh D, et al. Global prevalence of Helicobacter pylori Infection: Systematic review and meta-analysis. Gastroenterology (2017) 153:420–9. doi: 10.1053/j.gastro.2017.04.022
45. Hirabayashi K, Fujimori T, Sakuma K, Terano A. Effect of Helicobacter pylori eradication from patients with gastritis evaluated by the pathological grading and cell proliferation related factors. Nihon Rinsho (1999) 57:179–84.
46. Wang L. The study of effects of Helicobacter pylori infection on gastric carcinoma SGC-7901 cell proliferation and apoptosis. Master Thesis. Chinese: TianJin Medical university, China (2011).
47. Lee YS, Lee DY, Yu DY, Kim S, Lee C. Helicobacter pylori induces cell migration and invasion through casein kinase 2 in gastric epithelial cells. Helicobacter (2014) 19:465–75. doi: 10.1111/hel.12144
48. Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. Disruption of the epithelial apical junctional complex by Helicobacter pylori CagA. Science (2003) 300:1430–4. doi: 10.1126/science.1081919
49. Murata KN, Kurashima Y, Teishikata Y, Yamahashi Y, Saito Y, Higashi H, et al. Helicobacter pylori CagA interacts with E-cadherin and deregulates the beta-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene (2007) 26:4617–26. doi: 10.1038/sj.onc.1210251
50. Kikuchi K, Murata KN, Kondo S, Hatakeyama M. Helicobacter pylori stimulates epithelial cell migration via CagA-mediated perturbation of host cell signaling. Microbes Infect (2012) 14:470–6. doi: 10.1016/j.micinf.2011.12.003
51. Bao W, Fu H-J, Xie Q-S, Wang L, Zhang R, Guo Z-Y, et al. HER2 interacts with CD44 to up-regulate CXCR4 via epigenetic silencing of microRNA-139 in gastric cancer cells. Gastroenterology (2011) 141:2076–87.e6. doi: 10.1053/j.gastro
52. Deretic V, Jiang S, Dupont N. Autophagy intersections with conventional and unconventional secretion in tissue development, remodeling and inflammation. Trends Cell Biol (2012) 22:397–406. doi: 10.1016/j.tcb.2012.04.008
53. Monkkonen T, Debnath J. Inflammatory signaling cascades and autophagy in cancer. Autophagy (2018) 14:190–8. doi: 10.1080/15548627.2017.1345412
54. Terebiznik MR, Raju D, Vazquez CL, Torbricki K, Kulkarni R, Blanke SR, et al. Effect of Helicobacter pylori’s vacuolating cytotoxin on the autophagy pathway in gastric epithelial cells. Autophagy (2009) 5:370–9. doi: 10.4161/auto.5.3.7663
55. Zhang L, Hu W, Cho CH, Chan FK, Yu J, Fitzgerald JR, et al. Reduced lysosomal clearance of autophagosomes promotes survival and colonization of Helicobacter pylori. J Pathol (2018) 244:432–44. doi: 10.1002/path.5033
56. Yang X, Si R, Liang Y, Ma B, Jiang Z, Wang B, et al. Mir-30d increases intracellular survival of Helicobacter pylori through inhibition of autophagy pathway. World J Gastroenterol (2016) 22:3978–91. doi: 10.3748/wjg.v22.i15.3978
57. Zhu P, Xue J, Zhang ZJ, Jia YP, Tong YN, Han D, et al. Helicobacter pylori VacA induces autophagic cell death in gastric epithelial cells via the endoplasmic reticulum stress pathway. Cell Death Dis (2017) 8:3207. doi: 10.1038/s41419-017-0011-x
58. Al-Maleki AR, Loke MF, Lui SY, Ramli NSK, Khosravi Y, Ng CG, et al. Helicobacter pylori outer inflammatory protein A (OipA) suppresses apoptosis of AGS gastric cells in vitro. Cell Microbiol (2017) 19:e12771. doi: 10.1111/cmi.12771
59. Posselt G, Wiesauer M, Chichirau BE, Engler D, Krisch LM, Gadermaier G, et al. Helicobacter pylori-controlled c-Abl localization promotes cell migration and limits apoptosis. Cell Commun Signal (2019) 17:10. doi: 10.1186/s12964-019-0323-9
60. Wan X, Yuan S, Wang Y, Tao H, Jiang W, Guan Z, et al. Helicobacter pylori inhibits the cleavage of TRAF1 a CagA-dependent mechanism. World J Gastroenterol (2016) 22:10566–74. doi: 10.3748/wjg.v22.i48.10566
61. Correa P. Human gastric carcinogenesis: A multistep and multifactorial process-first American cancer society award lecture on cancer epidemiology and prevention. Cancer Res (1992) 52:6735–40.
62. Chu SH, Lim JW, Kim DG, Lee E, Kim KH, Kim H. Down-regulation of Bcl-2 is mediated by NF-κB activation in Helicobacter pylori-induced apoptosis of gastric epithelial cells. Scand J Gastroenterol (2011) 46:148–55. doi: 10.3109/00365521.2010.525255
63. Suarez G, Romero GJ, Piazuelo MB, Wang G, Maier RJ, Forsberg LS, et al. Modification of Helicobacter pylori Peptidoglycan Enhances NOD1 Activation and Promotes Cancer of the Stomach. Cancer Res (2015) 75:1749–59. doi: 10.1158/0008-5472.CAN-14-2291
64. Mohammadian MR, Tehrani M, Taghizadeh S, Shokri SJ, Fakheri H, Ajami A. Association of Nucleotide-binding Oligomerization Domain Receptors with Peptic Ulcer and Gastric Cancer. Iran J Allergy Asthma Immunol (2016) 15:355–62.
65. Hou T, Li Z, Zhao Y, Zhu W. Mechanisms controlling the anti-neoplastic functions of FoxO proteins. Semin Cancer Biol (2018) 50:101–14. doi: 10.1016/j.semcancer.2017.11.007
Keywords: Helicobacter pylori, apoptosis, autophagy, gastric epithelial cell, carcinogenesis
Citation: He Y, Wang C, Zhang X, Lu X, Xing J, Lv J, Guo M, Huo X, Liu X, Lu J, Du X, Li C and Chen Z (2020) Sustained Exposure to Helicobacter pylori Lysate Inhibits Apoptosis and Autophagy of Gastric Epithelial Cells. Front. Oncol. 10:581364. doi: 10.3389/fonc.2020.581364
Received: 08 July 2020; Accepted: 30 September 2020;
Published: 29 October 2020.
Edited by:
Aamir Ahmad, University of Alabama at Birmingham, United StatesReviewed by:
Ravi Manoharan, University of Madras, IndiaAlex C. Kornke, University of York, United Kingdom
Copyright © 2020 He, Wang, Zhang, Lu, Xing, Lv, Guo, Huo, Liu, Lu, Du, Li and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Changlong Li, licl@ccmu.edu.cn; Zhenwen Chen, czwen@ccmu.edu.cn