 Jian Yang
Jian Yang Yi Chen2†
Yi Chen2† Haoyang Cai
Haoyang Cai- 1Center of Growth, Metabolism, and Aging, Key Laboratory of Bio-Resources and Eco-Environment, College of Life Sciences, Sichuan University, Chengdu, China
- 2Department of Gastrointestinal Surgery, West China Hospital, Sichuan University, Chengdu, China
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common malignancy worldwide. Somatic copy number alterations (CNAs) play a significant role in the development of this lethal cancer. In this study, we present a meta-analysis of CNAs for a total of 1,395 HNSCC samples. Publicly available R packages and in-house scripts were used for genomic array data processing, including normalization, segmentation and CNA calling. We detected 125 regions of significant gains or losses using GISTIC algorithm and found several potential driver genes in these regions. The incidence of chromothripsis in HNSCC was estimated to be 6%, and the chromosome pulverization hotspot regions were detected. We determined 323 genomic locations significantly enriched for breakpoints, which indicate HNSCC-specific genomic instability regions. Unsupervised clustering of genome-wide CNA data revealed a sub-cluster predominantly composed of nasopharynx tumors and presented a large proportion of HPV-positive samples. These results will facilitate the discovery of therapeutic candidates and extend our molecular understanding of HNSCC.
Introduction
Head and neck squamous cell carcinoma (HNSCC) has an incidence of over 600,000 new diagnoses per year worldwide, with a mortality rate of nearly 50% (1, 2). It consists of a heterogeneous group of epithelial tumors arising from oral cavity, nasal cavity, pharynx, larynx, paranasal sinuses and salivary glands. Tobacco use and excessive alcohol consumption are well-established major risk factors for the development of HNSCC. Recently, human papillomavirus (HPV) infection has also been recognized as an increasingly important risk factor for HNSCC (3, 4). Previous studies have concluded that many genes are recurrently mutated in HNSCC but at low frequencies, and functional consequences of these mutations are often unclear. Therefore, a more detailed understanding of the genetic mechanisms of HNSCC is needed to improve prevention and treatment of this cancer.
Genomic instability in the form of copy number alteration (CNA) is a hallmark of cancer cells and a promotor for carcinogenesis (5–8). CNAs have critical roles in activating oncogenes and in inactivating tumor suppressors, and often affect many genes simultaneously (9–13). Like many other cancer types, HNSCCs are characterized by complex patterns of copy number gains and losses throughout the genome (14–17). In 2015, The Cancer Genome Atlas (TCGA) consortium published a robust integrative multiplatform characterization of 279 HNSCCs, including genome-wide profiling of copy number alterations (18). Although many studies have identified multiple regions of CNAs in HNSCC, a comprehensive landscape of copy number changes still remains largely unexplored. The role and implications of these aberrations in HNSCC need to be further evaluated and elucidated.
Recently, a new phenomenon, called chromothripsis, has been described as a new mechanism for cancer initiation and progression (19). It is characterized by the shattering of one or multiple chromosomes followed by a random reassembly of the DNA fragments. All the events are believed to occur in a single catastrophic event rather than a series of subsequent alterations (20–23). This is in contrast to the multistep model of cancer development. It can lead to the simultaneous acquisition of multiple tumor-promoting lesions. Chromothripsis has attracted a great attention in cancer research and has been observed in many tumor types (24–27). However, a comprehensive evaluation of chromothripsis in HNSCC has not been carried out yet. Given the low incidence of chromothripsis, a large number of tumor samples are required to perform such analysis.
Here we present a meta-analysis of genomic copy number alterations for 1,395 HNSCC samples from 19 datasets. In brief, we identified 72 and 53 regions of amplification and deletion, respectively, and detected potential driver genes in these regions. We identified 92 samples with the signs of chromothripsis, giving the incidence of 6%. Furthermore, we found some chromosome pulverization hotspots in HNSCC. The hierarchical clustering analysis identified two major clusters and cluster specific CNA patterns were provided. Our analysis may facilitate further studies on the distinct molecular mechanisms underlying HNSCC.
Materials and Methods
Data Collection and Patient Characteristics
We collected genomic array data from NCBI Gene Expression Omnibus (GEO) (28) and TCGA (29) databases. Our data selection criteria are that (1) the patient was diagnosed with head and neck squamous cell carcinoma, excluding precancerous lesions, metastasis, and recurrence cases, (2) the array platform must be genome-wide, including array comparative genomic hybridization (aCGH) and single nucleotide polymorphisms (SNP) array, and (3) the number of probes of the array platform should be >100K.
Our cohort consists of 873 patients from GEO and 522 patients from TCGA datasets. The primary tumors originated from several sites of the head and neck region, including oral cavity (n = 768, 61%), nasopharynx (n = 73, 5.8%), oropharynx (n = 123, 9.7%), hypopharynx (n = 78, 6.2%), larynx (n = 186, 14.8%), and sinonasal cavity (n = 31, 2.5%). The majority of patients were tobacco users (69.4%), while 55.6% were users of alcohol. Approximately 14% (150 out of 1,065 patients with available HPV test results) of patients were HPV-positive which is consistent with the reported frequency at about 14.17% (85,000 in 600,000) (30). A summary of patients' clinical data is provided in Table 1.
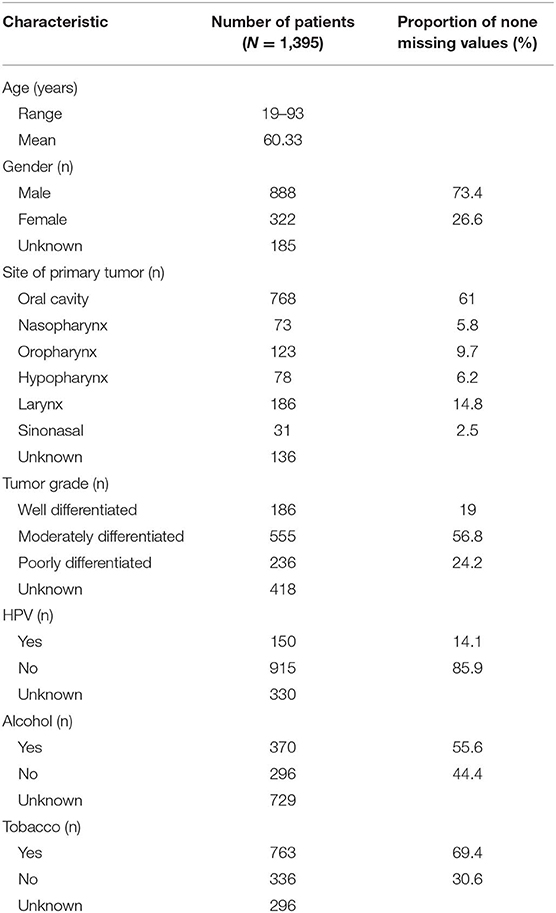
Table 1. Summary of clinical data.
For GEO datasets, the raw signal intensity files were downloaded for re-analysis (31, 32). The tumor-paired normal samples were used as reference in data analysis, if available. If one sample was hybridized on two or more array platforms, the sample analyzed by higher resolution array was included in the following analysis. A total of 18 GEO series consist of 873 samples were collected. Supplementary Table 1 shows details of the 18 GEO series. For each patient, clinical data was extracted from GEO website, publication's main text and supplementary file, and was converted to a standardized format. For TCGA dataset, segmented genomic array data (level 3) and clinical information were downloaded from TCGA data portal. There were 522 HNSCC samples, all analyzed by Affymetrix SNP6 arrays. In total, we collected 1,395 HNSCC samples (Supplementary Table 2).
Data Processing and Normalization
Affymetrix raw data files (.CEL) were re-analyzed by the R package aroma.affymetrix with the CRMAv.2 method (33). For non-Affymetrix arrays, probe-level signal intensity and annotation files were processed by in-house Perl scripts. When necessary, genomic coordinates were remapped to hg19/GRCh37 using UCSC's liftOver utility (34). The circular binary segmentation (CBS) (35) algorithm was employed to segment DNA copy number data. The CNA calling cut-off values for genomic gains and losses were set to 0.2 and −0.2, respectively. The X and Y chromosomes were excluded to avoid gender bias.
GISTIC Analysis
GISTIC is a tool used for the detection of peak regions significantly amplified or deleted in a number of samples (36). Default settings were used to run GISTIC 2.0, except that (1) the false discovery rate q-value was set to <0.05 for peak detection, (2) the arm peel method was used to reduce noise, (3) the confidence level used to calculate the region containing a driver was set to 0.95, (4) “Extreme” method was applied for reducing marker-level to the gene-level copy number data, (5) log2 ratios >0.2 and <-0.2 were taken as thresholds for gain and loss detection. Furthermore, for known cancer-related genes, we downloaded 719 cancer consensus genes from the Catalog of Somatic Mutations in Cancer (COSMIC) database (37).
Chromothripsis Detection
We detected chromothripsis-like phenomenon by CTLPScanner web server (http://cgma.scu.edu.cn/CTLPScanner/) (38). The segmented array data were used as input to identify clustering of copy number changes in the genome. The following parameters and thresholds were applied: copy number status switch times ≥20, log10 of Likelihood ratio ≥10, minimum segment size set to 10 Kb and signal value difference between two adjacent segments ≥0.4. For data visualization, signal value for genomic gains and losses were set to 0.2 and −0.2, respectively.
Definition of Copy Number Alteration Breakpoint
The genomic starts and ends of copy number alterations were considered as breakpoints. We used a stringent definition of CNA breakpoints to reduce the bias caused by technical or biological noise. A genomic position was defined as breakpoint if the Log2 signal value alteration between two adjacent genomic segments was >0.4 (39, 40). Copy number segments smaller than 10 Kb were excluded from analysis. Breakpoints located in chromosomal telomeres and centromeres were ignored. To investigate the distribution of breakpoints, the subdivision of the genome and random shuffling of breakpoint positions were performed by in-house Perl scripts. The common fragile sites (CFSs) and non-fragile regions (NFR) of the human genome were obtained from previous publications (41–43). We used the liftOver tool to convert the genome coordinates from assembly hg18/NCBI36 to hg19/ GRCh37 (34).
Results
Genome-Wide CNA Profiles of HNSCC
In our collections, the genomic alterations ranged from whole arm gains and losses to focal high-level amplifications. The mean size of the CNAs was 6.7 Mb, and the average number of CNA events per tumor sample was 443. The CNA frequency plots for each chromosome are shown in Figure 1. It provides a high-resolution view of CNA distribution across the HNSCC genome. The most commonly altered regions in HNSCC included gains of 3q, 5p, 8q, and losses of 3p, 5q, 8p, 13q, 18q, and 21q. The most notable focal CNA was the overrepresentation of 11q13, which may be associated with oncogene CCND1 amplification (44, 45). These results were consistent with previous studies (18, 46–49). We also identified several frequently altered regions that were rarely reported, such as the whole chromosome 4 loss and chromosome 20 gain.
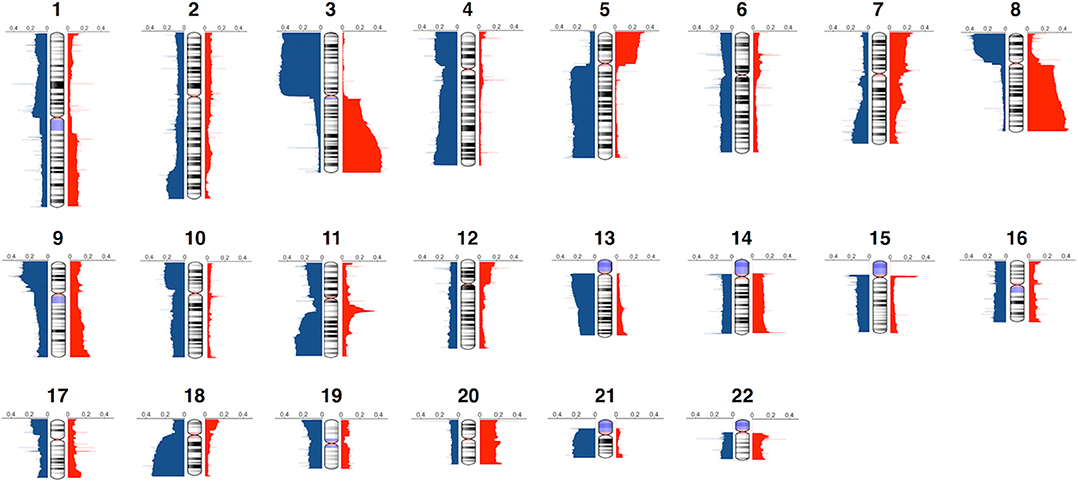
Figure 1. Genome-wide frequency plot of copy number alterations. Copy number gains and losses are represented in red and blue, respectively.
We further investigated the frequency of arm-level alterations, which were defined as a single CNA that encompasses >50% of a chromosomal arm. Supplementary Figure 1 represents the arm-level alteration frequency measured across all samples. Similar to other cancer types, we observed a negative correlation (r = −0.545, p = 0.0003) between arm-level gain and loss frequencies (5). It reveals that frequently altered chromosomes tend to be either gained or lost, but rarely both.
Driver Gene Identification
GISTIC 2.0 (36) was performed to identify statistically significant recurrent focal CNAs and potential driver genes. We observed 72 and 53 regions of recurrent copy number gain and loss, respectively (q < 0.05) (Supplementary Table 3). The annotation of these regions revealed 1,160 targeted genes. Some of the most significantly altered genes and their significance levels are shown in Figure 2. Thirty-one genes were known cancer genes and listed in COSMIC database (37) (Supplementary Table 4). Among them, 17 genes have been reported to be driver genes in HNSCC, such as ASPSCR1, BIRC3, CBFA2T3, EGFR, ERBB2, FGFR1, NFE2L2, PIK3CA, RECQL4, RNF213, and WHSC1L1 were located in copy-number gain regions, while CDKN2A, FAT1, LRP1B, PTPRD, PTEN, and RB1 were identified in copy-number loss regions. In addition to these known HNSCC driver genes, we also identified novel or recently described genes. There were 39 regions of interest that contained only one candidate driver gene. IRAK1BP1 (6q14.1) is an inhibitory component of TNFR-related pathways (50, 51). Although it can activate NF-kappa-B pathway, which is known as a major regulator of innate and adaptive immune responses, it has not been found associated with cancer. PTPN1 (20q13.13) is a tyrosine-protein phosphatase and acts as a regulator of endoplasmic reticulum unfolded protein response (52, 53). It directly dephosphorylates insulin receptor and insulin receptor substrates, which results in down regulation of insulin signaling. In recent studies, the overexpression of PTPN1 was detected in several cancer types such as colon cancer and breast cancer (54, 55). It has also been reported as an indicator of poor prognosis in gastric cancer (56). These results suggest an essential role for PTPN1 in HNSCC carcinogenesis. The new candidates in these recurrent alterations may direct experimental studies and contribute to molecular mechanism researches of HNSCC.
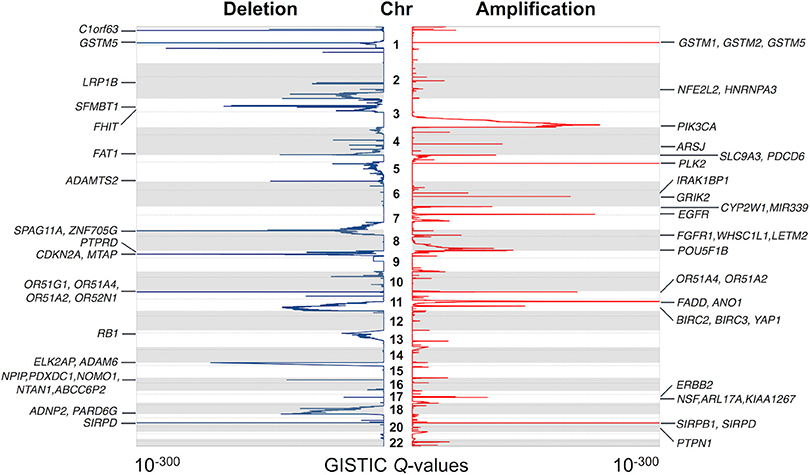
Figure 2. Significantly altered regions and genes identified by GISTIC algorithm. The significance of recurrent amplification and deletion is plotted across the genome. Some of the identified potential cancer driver genes are shown at the corresponding peaks.
Chromothripsis in HNSCC
Using CTLPScanner algorithm (38), we identified 92 chromothripsis cases from 1,395 HNSCC samples, giving an incidence of about 6% (Supplementary Table 5). Compared to other tumor types, the incidence rate of HNSCC is a little bit above the average level (approximately 5% of all cancers) (12, 57, 58). The sizes of affected regions ranged from 30 Mb to the whole chromosome. Supplementary Figure 2 illustrates examples of localized and chromosome-level pulverization. In our dataset, the most frequently shattered chromosome was chromosome 8 (18%, 17 out of 92). Chromosomes 6 and 11 also exhibited a relatively high pulverization rate. We also investigated the number of affected chromosomes per tumor sample, and found that approximately 11% (10 out of 92) of chromothripsis cases carried two or more chromosome shattering events. Figure 3 shows the hotspots of chromothripsis across the genome. These results suggest that HNSCC has a specific pattern of chromothripsis. In our dataset, the follow-up data was available for 772 patients, and the mean follow-up time was 31.4 months. Chromothripsis was detected in 8.2% of this cohort. The Kaplan-Meier analysis indicated that there was no survival difference in patients with chromothripsis patterns compared with those without chromothripsis (log-rank test, p = 0.61) (Supplementary Figure 3).
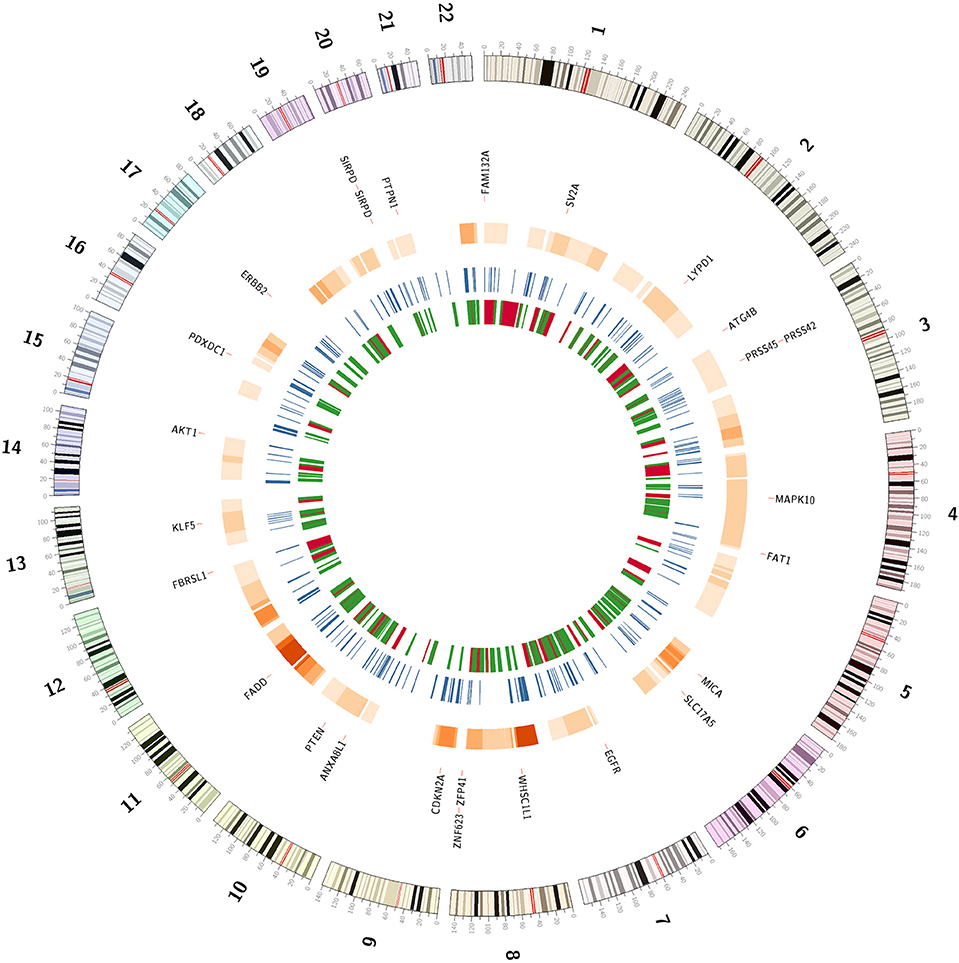
Figure 3. The genomic landscape of chromothripsis and copy number alteration breakpoints. The outermost circle represents potential cancer driver genes located in chromosome pulverization regions or the breakpoint hotspots. The next circle represents hotspots of chromothripsis region. The third circle shows the breakpoint-prone genomic regions. The innermost circle represents common fragile sites and non-fragile regions in red and green, respectively.
Copy Number Alteration Breakpoint Analysis
Chromosome breakage is a hallmark of cancer cells and may contribute to cancer initiation and progression. We identified a total of 193,028 CNA breakpoints in 1,395 HNSCC samples, with the mean breakpoints per sample being 138. We further examined the CNA breakpoint hotspots across the genome. Each chromosome was divided into continuous bins with length of 1 Mb, and the density of breakpoints per bin was calculated. Then, we shuffled the position of copy number alterations 10,000 times to obtain the background distribution of breakpoints. The breakpoint-prone genomic regions were identified through comparing the actual and background distributions. In total, we obtained 323 genomic regions that were significantly enriched for breakpoints of chromosomal rearrangements (Bonferroni corrected p < 0.01) (Supplementary Table 6). Moreover, we compared these regions with known common fragile sites and non-fragile regions described in the literature (41). Among these hotspots, only 58 (18%) were located within CFS, while 103 (32%) overlapped with NFS (Figure 3). These results provide important information about the HNSCC-specific genomic instability regions.
Comprehensive Clustering Analysis
We performed unsupervised hierarchical clustering on the copy number profiles of all HNSCC samples. CNA data were clustered in R based on Euclidean distance using Ward's method, resulting in two major clusters (Figure 4, Supplementary Figure 4). Tumors that were classified in cluster 1 showed a high proportion of the genome affected by CNAs, especially the arm-level events. The most frequent arm-level copy number changes in HNSCC, such as 3q, 8q gain, and 3p, 8p loss, can be found in this cluster. Cluster 2 was characterized by reduced CNAs and tumors were more affected by small focal alterations rather than arm-level events. Furthermore, several additional sub-clusters can be distinguished within both major clusters. For instance, a sub-cluster within cluster 2 was represented by samples harboring few CNAs (M class), a pattern that has been described in previous studies. These genome copy number stable tumors may be primarily driven by mutation rather than CNA. Notably, the hierarchical clustering analysis identified a sub-cluster predominantly composed of nasopharynx tumors and presented a large proportion of HPV-positive samples (Supplementary Table 7). These tumor samples demonstrated a distinctive copy number alteration pattern of 3q gain and lack of 3p loss. These different CNA patterns may be used as markers to cluster HNSCC samples into distinct tumor subtypes, and provide valuable insights into the different molecular mechanisms that underlie HNSCC development.
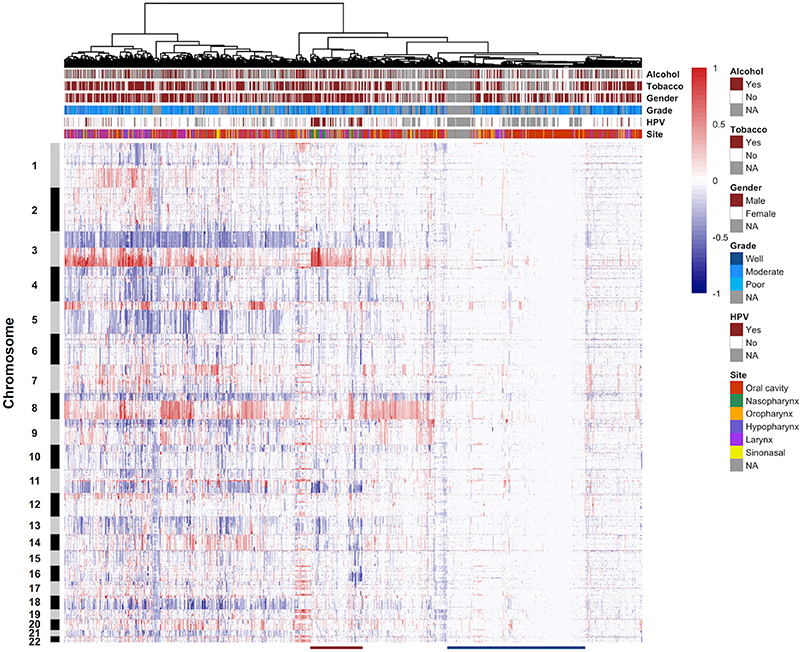
Figure 4. Unsupervised clustering of DNA copy number alteration data. The 1,395 HNSCC samples are arranged along the x-axis and ordered according to their copy number profiles. Chromosome numbers are indicated along the y-axis. Key clinical parameters associated with the samples are displayed in the column header. Red indicates copy number gain, and blue indicates copy number loss. The red line at the bottom of the figure represents a sub-cluster containing a large proportion of HPV-positive samples. The blue line at the bottom of the figure shows a sub-cluster consisted of samples harboring few copy number alterations. NA, not available.
Discussion
The high degree of genomic heterogeneity of HNSCC underscores a challenge to distinguish between driver and passenger alterations from aberrant regions. A large number of tumor samples need to be examined in order to identify driver genes that are not detected when each tumor is analyzed individually. In this study, we characterized genome-wide DNA copy number alterations in HNSCC based on published high resolution aCGH and SNP array data. From the overall CNA profile of 1,395 HNSCC samples, we obtained the most frequent arm-level alterations. Previous studies have indicated that broad and focal copy number alterations may represent different characteristics of cancer cells with independent mechanisms (12, 59). While broad copy number alterations may contribute to immune suppression, focal alterations may activate oncogenes and inactivate tumor suppressors that provide tumor cells with a proliferative advantage (60).
We applied GISTIC algorithm (36) to perform focal CNA analysis and identified a number of candidate genes, including driver genes from other tumor types, as well as genes not previously reported to be associated with any cancer. Over-expression of PTPN1 has been linked to colon cancer and breast cancer development and progression (54, 55). The phosphatase activity of PTPN1 is responsible for the regulation of cell motility and invasion, and may enhance the expression of EGFR. High expression of PTPN1 is strongly associated with poor prognosis in gastric cancer (56). These results support the hypothesis that amplification of PTPN1 is important in the pathogenesis of HNSCC. Another promising candidate gene, IRAK1BP1, can activate NF-kappa-B through the TNFR signaling pathway and acts by enhancing RELA transcriptional activity. This gene is associated with inflammation and several autoimmune diseases (61), but not cancer. Our results indicated that IRAK1BP1 may be important in HNSCC development and involved in immune surveillance.
The single catastrophic event chromothripsis is a novel phenomenon of genomic instability and is distinct from the progressive accumulation of mutations model of cancer development (19, 23). The prevalence of chromothripsis is heterogeneous across various cancer types. According to previous studies, chromothripsis on average can be found in approximately 5% of all cancers, with up to 25% in bone tumors (19, 22, 58, 62). We detected 92 chromothripsis cases out of 1,395 HNSCC samples, giving an incidence about 6%. While the underlying mechanisms resulting chromothripsis are still largely unknown, several hypotheses have been proposed, including micronuclei formation (63, 64), abortive apoptosis (65, 66), premature chromosome condensation (67), breakage-fusion-bridge cycles (68–70), and telomere dysfunction (71). The patterns of chromosomal pulverization may reveal underlying mechanisms of chromothripsis in carcinogenesis. In our cohort, 89% chromothripsis cases affect only a single chromosome, and about 3% cases affect 3 chromosomes simultaneously. Chromothripsis involving multiple chromosomes can result from several chromosomes in a micronucleus or is the consequence of a process of aborted apoptosis. It is likely that more than one mechanism contributes to the generation of chromothripsis in HNSCC. Since chromothripsis was more frequently detected in particular genomic regions, the chromosomal pulverization hotspots may contain critical genes for the regulation of cell cycle control, DNA damage, proliferation and apoptosis. For instance, Rausch et al. (24) have associated chromothripsis with TP53 mutations in subsets of medulloblastoma. In our analysis, the most frequently affected chromosome regions were 8p and 11q (account for 18 and 14% chromothripsis cases, respectively), suggesting that these loci were chromothripsis hotspots. The affected genes in these regions may be crucial for chromothripsis formation in HNSCC.
Genomic rearrangement breakpoints may disrupt tumor suppressor genes or create novel gene fusions with oncogenic potential. We performed genome-wide analysis and identified 323 recurrent breakpoints that were more clustered than would be expected if location of CNAs were randomly distributed throughout the genome. These breakpoint-prone regions revealed HNSCC specific genomic instability regions, by comparing with known common fragile sites and non-fragile regions of human genome. A number of potential driver genes detected by GISTIC were located in these regions. These results expanded our understanding of the impact of genomic rearrangements as a cause of HNSCC.
We performed unsupervised hierarchical clustering analysis based on primary tumor samples from six different sites of head and neck region. The results did not show a clear separation between the samples from different locations, which demonstrated the diversity of genetic alterations in a variety of HNSCC sites, as well as the importance to collect large-scale datasets for analysis. In consistent with previous studies, we observed a sub-cluster with few CNAs, which may represent the M class cancer that predominantly driven by mutations rather than copy number alterations (72). In 2015, the TCGA consortium published a comprehensive systematic evaluation of 279 HNSCCs, and provided an integrated view on the molecular landscape of this cancer type (18). HPV+ and HPV- tumors showed different mutational patterns. In our analysis, most HPV+ tumors displayed specific mutational profiles and were clustered together, suggesting that these tumors reflect a separate genetic subgroup. The distinctive copy number alterations of this group of samples include 3q gain and lack of 3p loss. Human papillomavirus infection has been identified in strong association with oropharyngeal and tonsillar cancers (73–75). However, our cluster analysis showed an enrichment of HPV+ samples in nasopharynx tumors, which was not explicitly noted before. Although the clinical implications of HPV infection in nasopharynx tumors need further investigation, these results are important for patient management and treatment choices.
In summary, we performed a comprehensive characterization of somatic genomic alterations based on a large cohort of HNSCC samples. Several newly identified driver genes in focal alterations, such as IRAK1BP1 and PTPN1, may facilitate molecular mechanism studies of HNSCC. Analysis of chromothripsis regions and breakpoint hotspots directed 8p and 11q as commonly affected regions in HNSCC that may related to disease development. Clustering of patients may provide evidence for patient stratification and differential treatment. These results provide valuable new insights into the mechanisms of genomic instability in HNSCC, and will facilitate the discovery of therapeutic and diagnostic candidates.
Data Availability Statement
Publicly available datasets were analyzed in this study. This data can be found in TCGA data portal (https://portal.gdc.cancer.gov/) and NCBI GEO database (https://www.ncbi.nlm.nih.gov/geo/). We integrated the following datasets: TCGA-HNSCC, GSE11938, GSE20306, GSE20939, GSE23831, GSE25103, GSE31984, GSE33229, GSE33983, GSE34507, GSE36790, GSE39367, GSE40777,GSE47443, GSE51265, GSE57201, GSE66136, GSE68717, and GSE85514.
Author Contributions
HC conceived the study. JY and YC collected raw data and performed data analysis. HL participated in interpretation of results and prepared figures. HC and JY drafted the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos. 31771394, 31571314, and U1603120).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Yang Cao for his valuable comments and suggestions.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.00321/full#supplementary-material
References
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. (2015) 136:E359–86. doi: 10.1002/ijc.29210
2. Leemans CR, Braakhuis BJM, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer. (2011) 11:9–22. doi: 10.1038/nrc2982
3. Braakhuis BJM, Snijders PJF, Keune WJH, Meijer CJLM, Ruijter-Schippers HJ, Leemans CR, et al. Genetic patterns in head and neck cancers that contain or lack transcriptionally active human papillomavirus. J Natl Cancer Inst. (2004) 96:998–1006. doi: 10.1093/jnci/djh183
4. Castellsagué X, Alemany L, Quer M, Halec G, Quirós B, Tous S, et al. HPV involvement in head and neck cancers: comprehensive assessment of biomarkers in 3680 patients. J Natl Cancer Inst. (2016) 108:djv403. doi: 10.1093/jnci/djv403
5. Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature. (2010) 463:899–905. doi: 10.1038/nature08822
6. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. (2009) 458:719–24. doi: 10.1038/nature07943
7. Baudis M. Genomic imbalances in 5918 malignant epithelial tumors: an explorative meta-analysis of chromosomal CGH data. BMC Cancer. (2007) 7:226. doi: 10.1186/1471-2407-7-226
8. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
9. Stephens PJ, McBride DJ, Lin ML, Varela I, Pleasance ED, Simpson JT, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. (2009) 462:1005–10. doi: 10.1038/nature08645
10. Xue W, Kitzing T, Roessler S, Zuber J, Krasnitz A, Schultz N, et al. A cluster of cooperating tumor-suppressor gene candidates in chromosomal deletions. Proc Natl Acad Sci USA. (2012) 109:8212–7. doi: 10.1073/pnas.1206062109
11. Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. (2012) 489:519–25. doi: 10.1038/nature11404
12. Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. (2013) 45:1134–40. doi: 10.1038/ng.2760
13. Kim T-M, Xi R, Luquette LJ, Park RW, Johnson MD, Park PJ. Functional genomic analysis of chromosomal aberrations in a compendium of 8000 cancer genomes. Genome Res. (2013) 23:217–27. doi: 10.1101/gr.140301.112
14. Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. (2011) 333:1157–60. doi: 10.1126/science.1208130
15. Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. (2011) 333:1154–7. doi: 10.1126/science.1206923
16. Hedberg ML, Goh G, Chiosea SI, Bauman JE, Freilino ML, Zeng Y, et al. Genetic landscape of metastatic and recurrent head and neck squamous cell carcinoma. J Clin Invest. (2016) 126:169–80. doi: 10.1172/JCI82066
17. Seiwert TY, Zuo Z, Keck MK, Khattri A, Pedamallu CS, Stricker T, et al. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin Cancer Res. (2015) 21:632–41. doi: 10.1158/1078-0432.CCR-13-3310
18. Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. (2015) 517:576–82. doi: 10.1038/nature14129
19. Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. (2011) 144:27–40. doi: 10.1016/j.cell.2010.11.055
20. Liu P, Erez A, Nagamani SCS, Dhar SU, Kołodziejska KE, Dharmadhikari AV, et al. Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell. (2011) 146:889–903. doi: 10.1016/j.cell.2011.07.042
21. Kloosterman WP, Tavakoli-Yaraki M, van Roosmalen MJ, van Binsbergen E, Renkens I, Duran K, et al. Constitutional chromothripsis rearrangements involve clustered double-stranded DNA breaks and nonhomologous repair mechanisms. Cell Rep. (2012) 1:648–55. doi: 10.1016/j.celrep.2012.05.009
22. Forment JV, Kaidi A, Jackson SP. Chromothripsis and cancer: causes and consequences of chromosome shattering. Nat Rev Cancer. (2012) 12:663–70. doi: 10.1038/nrc3352
23. Korbel JO, Campbell PJ. Criteria for inference of chromothripsis in cancer genomes. Cell. (2013) 152:1226–36. doi: 10.1016/j.cell.2013.02.023
24. Rausch T, Jones DTW, Zapatka M, Stütz AM, Zichner T, Weischenfeldt J, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell. (2012) 148:59–71. doi: 10.1016/j.cell.2011.12.013
25. Kloosterman WP, Hoogstraat M, Paling O, Tavakoli-Yaraki M, Renkens I, Vermaat JS, et al. Chromothripsis is a common mechanism driving genomic rearrangements in primary and metastatic colorectal cancer. Genome Biol. (2011) 12:R103. doi: 10.1186/gb-2011-12-10-r103
26. Molenaar JJ, Koster J, Zwijnenburg DA, van Sluis P, Valentijn LJ, van der Ploeg I, et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature. (2012) 483:589–93. doi: 10.1038/nature10910
27. Bochtler T, Granzow M, Stölzel F, Kunz C, Mohr B, Kartal-Kaess M, et al. Marker chromosomes can arise from chromothripsis and predict adverse prognosis in acute myeloid leukemia. Blood. (2017) 129:1333–42. doi: 10.1182/blood-2016-09-738161
28. Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. (2013) 41:D991–5. doi: 10.1093/nar/gks1193
29. Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA, Mills GB, Shaw KRM, Ozenberger BA, et al. The cancer genome atlas pan-cancer analysis project. Nat Genet. (2013) 45:1113–20. doi: 10.1038/ng.2764
30. Dok R, Nuyts S. HPV positive head and neck cancers: molecular pathogenesis and evolving treatment strategies. Cancers (Basel). (2016) 8:41. doi: 10.3390/cancers8040041
31. Cai H, Kumar N, Ai N, Gupta S, Rath P, Baudis M. Progenetix: 12 years of oncogenomic data curation. Nucleic Acids Res. (2014) 42:D1055–62. doi: 10.1093/nar/gkt1108
32. Cai H, Gupta S, Rath P, Ai N, Baudis M. arrayMap 2014: an updated cancer genome resource. Nucleic Acids Res. (2015) 43:D825–30. doi: 10.1093/nar/gku1123
33. Bengtsson H, Wirapati P, Speed TP. A single-array preprocessing method for estimating full-resolution raw copy numbers from all Affymetrix genotyping arrays including GenomeWideSNP 5 & 6. Bioinformatics. (2009) 25:2149–56. doi: 10.1093/bioinformatics/btp371
34. Karolchik D, Barber GP, Casper J, Clawson H, Cline MS, Diekhans M, et al. The UCSC genome browser database: 2014 update. Nucleic Acids Res. (2014) 42:D764–70. doi: 10.1093/nar/gkt1168
35. Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. (2004) 5:557–72. doi: 10.1093/biostatistics/kxh008
36. Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. (2011) 12:R41. doi: 10.1186/gb-2011-12-4-r41
37. Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. (2015) 43:D805–11. doi: 10.1093/nar/gku1075
38. Yang J, Liu J, Ouyang L, Chen Y, Liu B, Cai H. CTLPScanner: a web server for chromothripsis-like pattern detection. Nucleic Acids Res. (2016) 44:W252–8. doi: 10.1093/nar/gkw434
39. Zheng S, Fu J, Vegesna R, Mao Y, Heathcock LE, Torres-Garcia W, et al. A survey of intragenic breakpoints in glioblastoma identifies a distinct subset associated with poor survival. Genes Dev. (2013) 27:1462–72. doi: 10.1101/gad.213686.113
40. Smida J, Xu H, Zhang Y, Baumhoer D, Ribi S, Kovac M, et al. Genome-wide analysis of somatic copy number alterations and chromosomal breakages in osteosarcoma. Int J Cancer. (2017) 141:816–28. doi: 10.1002/ijc.30778
41. Fungtammasan A, Walsh E, Chiaromonte F, Eckert KA, Makova KD. A genome-wide analysis of common fragile sites: what features determine chromosomal instability in the human genome? Genome Res. (2012) 22:993–1005. doi: 10.1101/gr.134395.111
42. Durkin SG, Glover TW. Chromosome fragile sites. Annu Rev Genet. (2007) 41:169–92. doi: 10.1146/annurev.genet.41.042007.165900
43. Sarni D, Kerem B. The complex nature of fragile site plasticity and its importance in cancer. Curr Opin Cell Biol. (2016) 40:131–6. doi: 10.1016/j.ceb.2016.03.017
44. Noorlag R, van Kempen PMW, Stegeman I, Koole R, van Es RJJ, Willems SM. The diagnostic value of 11q13 amplification and protein expression in the detection of nodal metastasis from oral squamous cell carcinoma: a systematic review and meta-analysis. Virchows Arch. (2015) 466:363–73. doi: 10.1007/s00428-015-1719-6
45. Jarmuz-Szymczak M, Pelinska K, Kostrzewska-Poczekaj M, Bembnista E, Giefing M, Brauze D, et al. Heterogeneity of 11q13 region rearrangements in laryngeal squamous cell carcinoma analyzed by microarray platforms and fluorescence in situ hybridization. Mol Biol Rep. (2013) 40:4161–71. doi: 10.1007/s11033-013-2496-4
46. Hayes DN, Van Waes C, Seiwert TY. Genetic landscape of human papillomavirus-associated head and neck cancer and comparison to tobacco-related tumors. J Clin Oncol. (2015) 33:3227–34. doi: 10.1200/JCO.2015.62.1086
47. Leemans CR, Snijders PJF, Brakenhoff RH. The molecular landscape of head and neck cancer. Nat Rev Cancer. (2018) 18:269–82. doi: 10.1038/nrc.2018.11
48. Califano J, van der Riet P, Westra W, Nawroz H, Clayman G, Piantadosi S, et al. Genetic progression model for head and neck cancer: implications for field cancerization. Cancer Res. (1996) 56:2488–92. doi: 10.1016/S0194-5998(96)80631-0
49. Gross AM, Orosco RK, Shen JP, Egloff AM, Carter H, Hofree M, et al. Multi-tiered genomic analysis of head and neck cancer ties TP53 mutation to 3p loss. Nat Genet. (2014) 46:939–43. doi: 10.1038/ng.3051
50. Conner JR, Smirnova II, Moseman AP, Poltorak A. IRAK1BP1 inhibits inflammation by promoting nuclear translocation of NF-kappaB p50. Proc Natl Acad Sci USA. (2010) 107:11477–82. doi: 10.1073/pnas.1006894107
51. Kim JM, Cho HH, Lee SY, Hong CP, Yang JW, Kim YS, et al. Role of IRAK1 on TNF-induced proliferation and NF-κB activation in human bone marrow mesenchymal stem cells. Cell Physiol Biochem. (2012) 30:49–60. doi: 10.1159/000339045
52. Sangwan V, Paliouras GN, Abella JV, Dubé N, Monast A, Tremblay ML, et al. Regulation of the Met receptor-tyrosine kinase by the protein-tyrosine phosphatase 1B and T-cell phosphatase. J Biol Chem. (2008) 283:34374–83. doi: 10.1074/jbc.M805916200
53. Krishnan N, Fu C, Pappin DJ, Tonks NK. H2S-Induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci Signal. (2011) 4:ra86. doi: 10.1126/scisignal.2002329
54. Hoekstra E, Das AM, Swets M, Cao W, van der Woude CJ, Bruno MJ, et al. Increased PTP1B expression and phosphatase activity in colorectal cancer results in a more invasive phenotype and worse patient outcome. Oncotarget. (2016) 7:21922–38. doi: 10.18632/oncotarget.7829
55. Hilmarsdottir B, Briem E, Halldorsson S, Kricker J, Ingthorsson S, Gustafsdottir S, et al. Inhibition of PTP1B disrupts cell-cell adhesion and induces anoikis in breast epithelial cells. Cell Death Dis. (2017) 8:e2769. doi: 10.1038/cddis.2017.177
56. Wang N, She J, Liu W, Shi J, Yang Q, Shi B, et al. Frequent amplification of PTP1B is associated with poor survival of gastric cancer patients. Cell Cycle. (2015) 14:732–43. doi: 10.1080/15384101.2014.998047
57. Zhang C-Z, Leibowitz ML, Pellman D. Chromothripsis and beyond: rapid genome evolution from complex chromosomal rearrangements. Genes Dev. (2013) 27:2513–30. doi: 10.1101/gad.229559.113
58. Cai H, Kumar N, Bagheri HC, von Mering C, Robinson MD, Baudis M. Chromothripsis-like patterns are recurring but heterogeneously distributed features in a survey of 22,347 cancer genome screens. BMC Genomics. (2014) 15:82. doi: 10.1186/1471-2164-15-82
59. Beroukhim R, Getz G, Nghiemphu L, Barretina J, Hsueh T, Linhart D, et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci USA. (2007) 104:20007–12. doi: 10.1073/pnas.0710052104
60. Davoli T, Uno H, Wooten EC, Elledge SJ. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science. (2017) 355:eaaf8399. doi: 10.1126/science.aaf8399
61. Lessard CJ, Li H, Adrianto I, Ice JA, Rasmussen A, Grundahl KM, et al. Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjögren's syndrome. Nat Genet. (2013) 45:1284–92. doi: 10.1038/ng.2792
62. Yang J, Deng G, Cai H. ChromothripsisDB: a curated database of chromothripsis. Bioinformatics. (2016) 32:1433–5. doi: 10.1093/bioinformatics/btv757
63. Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, et al. DNA breaks and chromosome pulverization from errors in mitosis. Nature. (2012) 482:53–8. doi: 10.1038/nature10802
64. Zhang C-Z, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, et al. Chromothripsis from DNA damage in micronuclei. Nature. (2015) 522:179–84. doi: 10.1038/nature14493
65. Tubio JMC, Estivill X. Cancer: When catastrophe strikes a cell. Nature. (2011) 470:476–7. doi: 10.1038/470476a
66. Ichim G, Lopez J, Ahmed SU, Muthalagu N, Giampazolias E, Delgado ME, et al. Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol Cell. (2015) 57:860–72. doi: 10.1016/j.molcel.2015.01.018
67. Meyerson M, Pellman D. Cancer genomes evolve by pulverizing single chromosomes. Cell. (2011) 144:9–10. doi: 10.1016/j.cell.2010.12.025
68. Nones K, Waddell N, Wayte N, Patch A-M, Bailey P, Newell F, et al. Genomic catastrophes frequently arise in esophageal adenocarcinoma and drive tumorigenesis. Nat Commun. (2014) 5:5224. doi: 10.1038/ncomms6224
69. Sorzano COS, Pascual-Montano A, Sánchez de Diego A, Martínez-AC, van Wely KHM. Chromothripsis: breakage-fusion-bridge over and over again. Cell Cycle. (2013) 12:2016–23. doi: 10.4161/cc.25266
70. Li Y, Schwab C, Ryan S, Papaemmanuil E, Robinson HM, Jacobs P, et al. Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature. (2014) 508:98–102. doi: 10.1038/nature13115
71. Maciejowski J, Li Y, Bosco N, Campbell PJ, de Lange T. Chromothripsis and kataegis induced by telomere crisis. Cell. (2015) 163:1641–54. doi: 10.1016/j.cell.2015.11.054
72. Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. (2013) 45:1127–33. doi: 10.1038/ng.2762
73. Massarelli E, Ferrarotto R, Glisson BS. New strategies in human papillomavirus-related oropharynx cancer: effecting advances in treatment for a growing epidemic. Clin Cancer Res. (2015) 21:3821–8. doi: 10.1158/1078-0432.CCR-14-1329
74. Letsolo BT, Faust H, Ekblad L, Wennerberg J, Forslund O. Establishment and characterization of a human papillomavirus type 16-positive tonsillar carcinoma xenograft in BALB/c nude mice. Head Neck. (2016) 38:417–25. doi: 10.1002/hed.23918
Keywords: head and neck squamous cell carcinoma, meta-analysis, copy number alteration, chromothripsis, genomic array
Citation: Yang J, Chen Y, Luo H and Cai H (2020) The Landscape of Somatic Copy Number Alterations in Head and Neck Squamous Cell Carcinoma. Front. Oncol. 10:321. doi: 10.3389/fonc.2020.00321
Received: 16 July 2019; Accepted: 24 February 2020;
Published: 12 March 2020.
Edited by:
Pawel Golusinski, University of Zielona Góra, PolandReviewed by:
Jozsef Dudas, Innsbruck Medical University, AustriaMichal Masternak, University of Central Florida, United States
Copyright © 2020 Yang, Chen, Luo and Cai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haoyang Cai, haoyang.cai@scu.edu.cn
†These authors have contributed equally to this work