 Rose N. Njoroge
Rose N. Njoroge Rajita J. Vatapalli
Rajita J. Vatapalli Sarki A. Abdulkadir
Sarki A. Abdulkadir- 1Department of Urology, Northwestern University, Feinberg School of Medicine, Chicago, IL, United States
- 2Robert H. Lurie Comprehensive Cancer Center, Northwestern University Feinberg School of Medicine, Chicago, IL, United States
- 3Department of Pathology, Northwestern University Feinberg School of Medicine, Chicago, IL, United States
Epidemiological and preclinical data suggest that antioxidants are protective against prostate cancer whose pathogenesis has been linked to oxidative stress. However, the selenium and vitamin E Cancer Prevention Trial (SELECT), found no efficacy for selenium in reducing prostate cancer incidence while vitamin E was associated with an increased risk of the disease. These results have called in to question the models used in preclinical chemoprevention efficacy studies and their ability to predict in vivo outcomes. Chemoprevention agents have traditionally been tested on two dimensional monolayer cultures of cell lines derived from advanced prostate cancers. But as SELECT demonstrates, results from advanced disease models were not predictive of the outcome of a primary chemoprevention trial. Additionally, lack of cell-matrix interactions in two dimensional cultures results in loss of biochemical and mechanical cues relevant for native tissue architecture. We use recent findings in three dimensional organoid cultures that recapitulated the SELECT trial results to argue that the organoid model could increase the predictive value of in vitro studies for in vivo outcomes.
Introduction
Prostate cancer (PCa) is the most commonly diagnosed non-cutaneous male malignancy in the United States. One in six men in the US is expected to be diagnosed with PCa in his lifetime. In 2018, an estimated 164,690 men will be diagnosed with PCa and 29,430 men will die from the disease (1). For men with local or regional disease at diagnosis, radical prostatectomy, or radiation therapy are effective treatments with a 100% 5-year survival rate (2). However, treatment related morbidity results in a poorer quality of life (3, 4). Moreover, 20–40% radical prostatectomy and 30–50% radiation therapy patients will experience biochemical recurrence within 10 years (5–7).
Early PCa growth and progression is driven by androgens making androgen deprivation therapy through surgical or medical castration, the standard treatment for advanced and metastatic PCa (8, 9). Eventually, however, there is progression to castration resistant disease, whose treatments confer a median overall survival benefit of less than 5 months (10–16). Moreover, widespread serum prostate-specific antigen screening has been associated with unnecessary biopsies, over diagnosis and overtreatment of indolent PCa (17). Given the drawbacks of screening, treatment associated morbidity and lack of effective treatments for advanced disease, preventing PCa is imperative.
The long natural history of PCa and its stepwise pathogenesis make it particularly amenable to prevention (2, 18, 19). Chemoprevention which uses natural, synthetic, or biological substances to reverse, slow or inhibit the initiation and progression of cancer, is an attractive public health strategy to reduce PCa incidence and treatment related morbidities (20). Androgen suppression using the 5α-reductase inhibitors, finasteride and dutasteride, in the Prostate Cancer Prevention Trial (PCPT) and REduction by DUtasteride of Prostate Cancer Events (REDUCE) randomized trials showed a 23 to 25% reduction in PCa relative risk (21, 22). These inhibitors were however not approved for PCa chemoprevention by the FDA as both were found to increase the risk of high-grade PCa with the possibility of increasing mortality (23). However, a long-term follow-up of the PCPT trial participants found a 43% relative risk reduction in low-grade PCa but no significant differences in overall survival or rates of high-grade disease (24). In spite of these findings, the two inhibitors are still not approved for PCa prevention (25).
Oxidative Stress, a Promising Chemoprevention Target in PCa
Reactive oxygen species (ROS) are reactive molecules resulting from the partial reduction of molecular oxygen. Intracellular ROS comes from exogenous and endogenous sources (26). In small quantities, ROS take part in signal transduction by reversibly oxidizing protein thiol groups affecting numerous physiological processes (27, 28). High levels of ROS however indiscriminately damage cellular macromolecules (29). ROS can modify DNA bases, form DNA adducts, induce DNA cross-linking and cause DNA strand breaks (30). Oxidative DNA lesions that are not removed prior to DNA replication can lead to replication errors, mutations, and genome instability increasing the risk of carcinogenesis (31).
ROS also indirectly increase cancer risk if they cause lesions on tumor suppressor genes or inactivate negative regulators of oncogenes and DNA repair proteins (32, 33). Under physiological conditions, ROS accumulation is regulated by endogenous enzymatic and non-enzymatic antioxidant defense systems (26). Oxidative stress occurs when ROS levels outstrip these cellular antioxidant defenses (34). Oxidative stress has been linked to PCa development and progression. In Nkx3.1−/− mutant mice, dysregulation in the expression of antioxidant and prooxidant enzymes led to oxidative stress and development of HGPIN while Nkx3.1/Pten double mutants which sustained more oxidative damage progressed to adenocarcinoma (35).
Products of oxidative damage; 8-hydroxy-2'-deoxyguanosine, 4-hydroxynonenal-protein-adducts, and nitrotyrosine were also detected in early prostatic tumorigenesis in the transgenic adenocarcinoma of the mouse prostate model (36). In the noble rat hormonal PCa model, administration of testosterone and β-estradiol triggered the expression of prooxidant enzymes and oxidative damage which induced stromal inflammation and dysplasia in the lateral prostate (37). For humans, several PCa risk factors including age, diet, inflammation and androgens are also associated with oxidative stress (38).
Additionally, key genetic and epigenetic changes in PCa have been shown to decrease the expression of genes relevant to prostatic redox homeostasis like GSTP1, Nrf2, NKX3.1, and NADPH oxidases (35, 39–41). Besides, transcription factors important for PCa like NK-κB, AP-1, HIF-1, and p53 are redox sensitive (42). Relative to benign prostate cells, human PCa cell lines display higher oxidative stress which is associated with a more aggressive phenotype (39). Moreover, as PCa progresses, patients display increasing levels of oxidative biomarkers including thiobarbituric acid reactive substances, 8-hydroxy-2'-deoxyguanosine and 4-hydroxynonenal-modified proteins concomitant with loss of antioxidant defenses (43–48).
The Selenium and Vitamin E Cancer Prevention Trial (SELECT)
The links between oxidative stress and PCa pathogenesis are the basis of the notion that use of antioxidants can reduce risk of the disease. The SELECT trial tested the efficacy of the antioxidants selenium and vitamin E in reducing PCa incidence in 35,533 men over the age of 50 with an intended follow up of 12 years (49, 50). As part of the inclusion criteria, the men had to be free of a prior PCa diagnosis, have a non-suspicious digital rectal examination and a serum prostate specific antigen level of ≤ 4 ng/mL (50). SELECT, a phase III clinical trial, randomized participants to a daily dose of 200 μg L-selenomethionine and/or 400 IU α-tocopheryl acetate or placebo (49, 50). After 7 years of follow up, it was found that neither antioxidant reduced PCa incidence and that vitamin E was associated with a 17% increased risk of PCa compared to placebo (51).
In vitro Models of Advanced Disease did not Predict Outcome of a Primary Chemoprevention Clinical Trial
Numerous in vitro studies suggest that vitamin E and selenium are antitumorigenic by inhibiting proliferation, altering redox homeostasis, inducing apoptosis, suppressing androgen receptor signaling, blocking inflammatory signaling among other mechanisms (52–92). SELECT's lack of efficacy and harmful effects however indicate that traditional in vitro models had a low predictive value for the clinical trial outcome (93, 94). In addition, the majority of in vitro chemoprevention studies predominantly use cell lines derived from advanced cancers including LNCaP, DU145, and PC3. These do not model disease initiation and they did not predict the outcome of a primary chemoprevention clinical trial like SELECT (94).
Cells at different stages of cancer progression likely respond differently to antioxidants due to differential regulation of prooxidant and antioxidant networks (95). ROS accumulation due to decreased antioxidant activity in premalignant lesions and early stage cancers could promote tumorigenesis through DNA damage and inactivating tumor suppressors (95). Advanced cancers however counter high ROS accumulation by increasing antioxidant activity through several mechanisms like upregulating NRF2 or increasing metabolic generation of reducing equivalents like NADPH mitigating oxidant damage (95).
Increasing the Predictive Value of Preclinical Research by Using Three Dimensional Cell Cultures
In vitro testing of chemoprevention agents has historically utilized conventional two dimensional (2D) culture which does not recapitulate in vivo prostate biology. The human prostate is a multicellular secretory epithelium with a central lumen (96). The prostate's luminal layer is made of polarized columnar luminal epithelial cells which face the lumen and produce prostatic secretions (96). The prostate epithelium sits upon a basement membrane which is surrounded by a stromal compartment (97). Basement membranes are cell adherent sheet-like structured extracellular matrices (ECMs) that include laminins, collagens, proteoglycans, and glycoproteins (98). The ECM provides physical support and serves as a scaffold for tissue organization (99).
In addition, ECM attachment provides biochemical and mechanical cues necessary for anchorage dependent cell growth, proliferation, migration, and differentiation (99). ECM proteins bind growth factors, cytokines and chemokines creating spatial and temporal concentration gradients necessary for spatiotemporal coordination of cellular activity during tissue morphogenesis, wound healing, and chemotaxis (100). Integrins which link the ECM and the actin cytoskeleton also transmit external mechanical and internal actomysin contractile forces from and to the ECM altering cell migration, proliferation, and differentiation (101).
During normal epithelial glandular development, ECM–cell interactions provide contextual cues needed to maintain apico-basal polarity necessary for correct tissue architecture and function (102). The ECM aligns the mitotic spindle perpendicular to the apical–basal axis enabling symmetrical cell division in the plane of the monolayer which maintains tissue structure (102). ECM–cell interactions balance proliferation and cell death for tissue homeostasis and lumen morphogenesis; their disruption contribute to neoplastic transformation (99, 103). In contrast, cells in 2D culture lack physiological cell-cell and cell-matrix interactions leading to loss of native tissue architecture and function (104).
Additionally, attachment of the 2D monolayer to artificial surfaces affects cell morphology and signaling (105, 106). This has been attributed to the high failure rate of drugs screened using 2D in vitro cultures in clinical trials (107). Moreover, 2D cultures have a non-physiologically uniform distribution of oxygen and nutrients (108). Embedding cells in three dimensional (3D) matrices yields more physiological cell-cell and cell-matrix interactions. Organoids are 3D cellular aggregates derived from primary tissues/cells, induced pluripotent stem cells, or embryonic stem cells, which self-renew and self-organize to exhibit similar architecture and functionality as their tissue of origin (109). Organoids can also be derived from established cell lines (110).
Organoid self-renewing property lends itself well to a host of basic and translational research applications. It allows the expansion of cells from minuscule patient samples like solid tumor biopsies and circulating tumor cells for liquid biopsies, facilitating genetic profiling, drug screening, and potentially guiding personalized therapy (111, 112). Organoids can also be harnessed to study tissue development including embryonic development, lineage specification, tissue morphogenesis, and homeostasis and how these processes change during disease (113, 114). For example, organoids have been utilized to investigate the PCa cell of origin which could have prognostic and treatment benefits (115–118). Organoid co-cultures with stromal components will allow research in to the contribution of the tumor microenvironment to malignancy (119).
Prostate organoids phenocopy in vivo glandular morphology; they undergo polarization, lumen formation and produce prostatic secretions (115). Unlike 2D cultures, growth factor, nutrients and oxygen gradients in organoids yield heterogeneous cell populations like in vivo (107). Additionally avascular organoids better model solid tumors which are often poorly vascularized in vivo. Similar to tumors, cells at the center of large organoids are under various stresses including loss of ECM attachment and limits in the diffusion of nutrients and oxygen (120). This in vivo-like physiology of organoid cultures could increase the predictive value of preclinical research (107). Moreover, organoid cultures bridge the gap between 2D and animal models, that are challenging to generate for example the genetically engineered mouse models and which are affected by interspecies differences (121).
Premalignant Organoids Recapitulate Outcome of the Select Trial
Effects of the mean concentrations of the SELECT agents attained in the blood plasma of the SELECT participants was evaluated in prostate organoids derived from normal, premalignant, and malignant prostate epithelial cells (122). The benign organoids were obtained from histologically and genetically normal prostate epithelium isolated from radical prostatectomy tissue (122). The premalignant organoids were derived from the untransformed RWPE-1 cell line which is immortalized with the E7 oncoprotein, modulating the activity of the retinoblastoma tumor suppressor (123, 124). The malignant organoids were cultured from the androgen responsive LNCaP cell line established from a lymph node PCa metastasis (125).
The vehicle-treated malignant organoids were devoid of lumens (filled morphology) a phenotype consistent with the acquisition of anchorage independent survival and loss of glandular differentiation (122). In contrast, normal epithelial cells require ECM attachment for proliferation and survival (99). This is partly because integrin ligation to the ECM regulates growth factor signaling and cell cycle progression (99). In these cells, ECM detachment activates a form of apoptotic cell death termed “anoikis,” which is Greek for homelessness (126). Anoikis clears cells in inappropriate locations preventing dysplastic growth (126). Anoikis also contributes to tissue homeostasis by eliminating cells without ECM contact in the lumen, hollowing glands (127).
Focal loss of ECM and integrin attachment in prostatic intraepithelial neoplasia lesions leads to cells proliferation within the lumen leading to defective glandular structures (128). In addition, PCa progression is marked by a gradual loss of glandular lumina (129). In cancer cells, oncogene activation overrides the requirement for ECM-adhesion leading to anchorage independent growth and resistance to anoikis (126). The SELECT agents decreased proliferation and increased cell death in the ECM distal cells at the center of the malignant organoids (122). This is consistent with reports that show antioxidant efficacy in PCa cell lines (60, 61). In contrast, the agents had no effect on the proliferation of benign organoids (122). Morphologically, the benign organoids had proper glandular structure with well-formed lumens which was not affected by vitamin E and/or selenium treatment (122).
The null effect of antioxidants on the benign organoids is consistent with SELECT where a fraction more subjects on vitamin E developed PCa compared to placebo (51). We posit that these individuals might have harbored initiated cells that progressed to neoplastic transformation with chronic vitamin E exposure. In agreement with this hypothesis, vitamin E alone or in combination with selenium but not selenium alone significantly increased the proliferation of premalignant RWPE-1 cell organoids (122). Therefore only results from the premalignant organoids recapitulated the clinical trial data from SELECT.
Our group has previously demonstrated that the antioxidant NAC causes premalignant prostatic epithelial hyperplasia in mice with prostate specific deletion of the Nkx3.1 tumor suppressor but not in wild type mice (130). In addition, polymorphisms in NKX3.1 were found to modulate PCa risk in men on the interventional arms of the SELECT trial (131). This points to the importance of the underlying genetic background in modifying the response to antioxidant supplementation. However, SELECT's inclusion criteria; prostate-specific antigen levels and a non-suspicious digital rectal exam, could not rule out the existence of initiating molecular aberrations (132).
Additionally, while the vehicle treated premalignant RWPE-1 organoids had more differentiated acinus structures and predominantly hollow lumens those treated with vitamin E had predominantly filled lumens (122). Microarray analysis of RNA extracted from vitamin E treated RWPE-1 organoids displayed significant downregulation of several integrins confirming the loss of matrix attachment (122). Vitamin E therefore increases cell survival in a low matrix environment (122).
ECM Detached RWPE-1 Cells Have Deficient Glucose Metabolism Which Vitamin E Alleviates by Activating Fatty Acid Oxidation
Despite increased cell growth and survival, the vitamin E treated premalignant organoids had decreased expression of glucose transporters and several glycolytic enzymes implying glucose metabolism dysregulation (122). To further study metabolic changes in RWPE-1 cells under anchorage independent conditions in vitro, Poly-2-hydroxyethyl methacrylate (Poly-HEMA), suspension cultures were used (122). Poly-HEMA, a non-ionic polymer prevents ECM deposition on tissue culture plates and cell adhesion (126, 133). Additionally, growth of human epithelial cancer cells on poly-HEMA coated plates correlates with in vitro growth in soft agar the gold standard for measuring anchorage independent growth and tumorigenicity (134, 135).
Poly-HEMA cultures therefore provide a model for studying the regulation of anchorage-independent cell survival and growth for studies that are difficult to perform in organoids (136). Glucose is catabolized to Acetyl-CoA whose oxidative phosphorylation in the mitochondria produces reducing equivalents which mediate electron transfer in the electron transport chain generating the proton motive force that drives ATP synthesis (137). RWPE-1 poly-HEMA cultures had reduced glucose uptake and ATP levels confirming that loss of attachment jeopardizes cellular metabolism (122). Similar observations have been reported in detached MCF-10A benign breast epithelial cells (138).
Altered metabolism after cell detachment has been attributed to the loss of integrin activation of the PI3K/AKT pathway which is a crucial regulator of glucose and glutamine uptake and metabolism (138). ECM-integrin contact leads to the recruitment of adopter proteins like talin and paxillin as well as signaling molecules like focal adhesion kinase (Fak) and small GTPases to form large macromolecular structures termed focal adhesions connecting the ECM and the actin cytoskeleton (139). Autophosphorylation of FAK (Y397) downstream of integrin signaling activates its kinase function leading to the activation of the SRC/MAPK and PI3K/AKT pathways which are crucial for progression through the G1/S checkpoint, cell survival and proliferation (Figure 1) (140, 141).
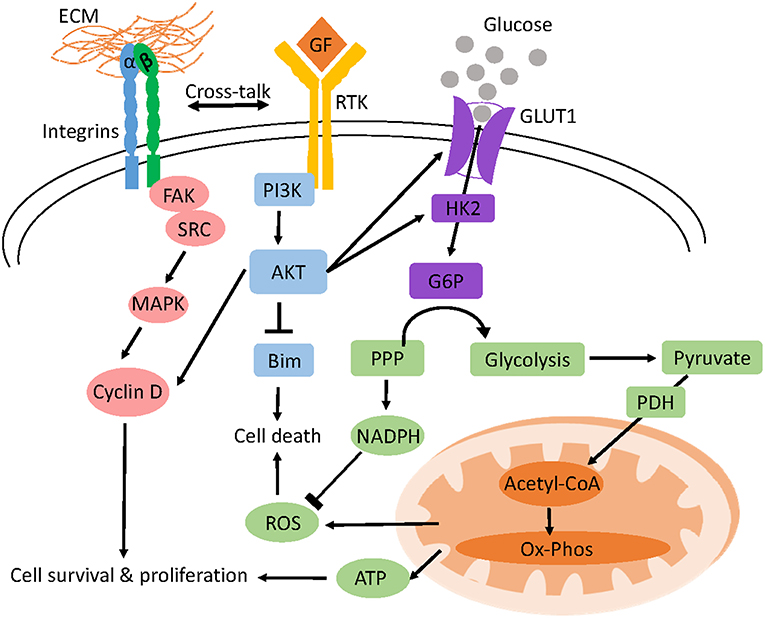
Figure 1. Model of anchorage-dependent regulation of cell survival and glucose metabolism. Adhesion of cells to the ECM activates integrins and receptor tyrosine kinase (RTK) signaling which triggers different pro-survival and proliferation pathways like AKT and MAPK. AKT inhibits BIM, a pro-apoptotic protein preventing anoikis and increases glucose uptake and glycolysis by upregulating the transcription of glucose transporters (GLUT1) and hexokinase 2 (HK2) respectively. Oxidative phosphorylation (OXPHOS) of pyruvate derived from glucose yields ATP for cellular function. Glucose shunted through the PPP pathway generates NADPH which prevents ROS induced cell death.
Cross talk between integrins and receptor protein tyrosine kinases also activates the PI3K/AKT pathway downstream of the epidermal growth factor receptor (142). In contrast, the accumulation of multiple alterations allows cancer cells to circumvent such extracellular regulation enabling them to uptake nutrients constitutively (137). Glucose can also be shunted through the pentose phosphate pathway to generate the reducing equivalent NADPH and the nucleotide structural component, ribose-5-phosphate (137). In detached mammary cells low glucose uptake not only reduces ATP generation but also diminishes pentose phosphate pathway flux and NADPH production leading to ROS accumulation (Figure 2) (138).
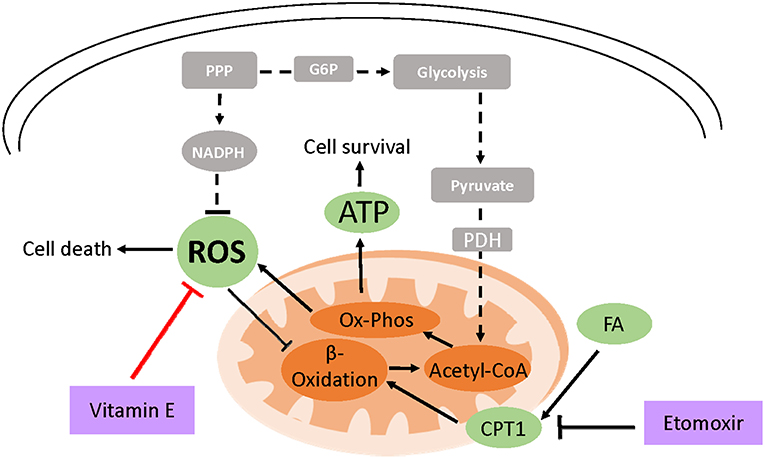
Figure 2. Model of vitamin E-mediated cell survival in ECM detached cells. Due to loss of integrin and PI3K signaling, cell detachment from the ECM results in the reduced expression of glucose transporters and low glucose uptake (shown in broken lines and gray boxes). This decreases ATP production and cell survival. Moreover, low NADPH generation through attenuated PPP flux leads to ROS accumulation which can induce cell death. ROS also inhibits fatty acid beta-oxidation (FAO) an alternative ATP generation pathway under glucose limiting conditions. However, treating detached cells with exogenous antioxidants like vitamin E neutralizes ROS which activates FAO increasing cell survival. Attenuating FAO using etomoxir which inhibits carnitine palmitoyltransferase (CPT1), FAO's rate limiting enzyme, abrogates vitamin E's ATP rescue ultimately diminishing cell survival.
Treatment of detached RWPE-1 cells with vitamin E rescued ATP generation but not glucose uptake (122). Similar findings have been reported in detached mammary cells treated with NAC or trolox, a soluble form of vitamin E (138). When glucose is scarce, cells can catabolize fatty acids for ATP generation. In Akt transformed glioblastoma, cells activate fatty acid oxidation to survive upon glucose withdrawal (143). It has been postulated that ROS inhibits fatty acid oxidation and hydrogen peroxide a form of ROS, has been shown to inhibit peroxisomal FAO (144–146). Pharmacological inhibition of fatty acid oxidation in vitamin E detached RWPE-1 cells abrogated the ATP rescue indicating that the antioxidant activates fatty acid catabolism (122).
Furthermore, fatty acid oxidation inhibition decreased cell survival and enhanced luminal clearance in vitamin E treated premalignant organoids indicating that antioxidants support anchorage independent cell survival (122). Moreover, other antioxidant-driven metabolic rescue mechanisms following loss of matrix attachment have also been described (147). In lung cancer spheroids, loss of matrix attachment upregulates glutamine reductive metabolism by cytosolic isocitrate dehydrogenase-1 to generate NADPH which is shuttled to inhibit mitochondrial ROS, enhancing cell growth (147). However, despite lowering ROS levels, selenium was not protumorigenic on the premalignant organoids (122). The effect of selenium on PCa risk has been shown to depend on baseline selenium status (148).
Conclusions
The data showing antitumorigenic effects of antioxidants on malignant organoids replicates numerous two dimensional in vitro studies. However, 3D organoids starkly reveal differential effects of antioxidants along the prostate cancer evolution spectrum. In this system, vitamin E had a null effect in benign organoids and a pro-tumorigenic effect in premalignant organoids in a manner highly reminiscent of the SELECT trial results. These findings demonstrate that the use of preclinical models that better mimic in vivo conditions and disease stage yield data that is more relevant for clinical translation.
More broadly, organoid data in several cancers now show that neutralizing ROS promotes anchorage independent cell growth and survival implying that ROS accumulation can imperil detached cells (122, 138, 147). Anchorage independence facilitates cell growth and survival in non-native environments for example in metastasis (149). Therefore, identifying mechanisms that enable anchorage independence could offer clues on how to impede cancer metastasis (149). Finally, given the role that metabolism plays in carcinogenesis, preclinical studies ought to include metabolic endpoints when assessing potential chemopreventive agents.
Author Contributions
SA and RN contributed to conception and design of the manuscript. RN researched supporting evidence and wrote the manuscript. RV and SA contributed to critical revision of content and figure design. SA secured grant funding.
Funding
This work was supported by NCI grants 1P50CA180995, R01CA123484, and R01 CA167966.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. (2018) 68:7–30. doi: 10.3322/caac.21442
2. Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. (2016) 66:271–89. doi: 10.3322/caac.21349
3. Potosky AL, Legler J, Albertsen PC, Stanford JL, Gilliland FD, Hamilton AS, et al. Health outcomes after prostatectomy or radiotherapy for prostate cancer: results from the Prostate Cancer Outcomes Study. J Natl Cancer Inst. (2000) 92:1582–92. doi: 10.1093/jnci/92.19.1582
4. Stanford JL, Feng Z, Hamilton AS, Gilliland FD, Stephenson RA, Eley JW, et al. Urinary and sexual function after radical prostatectomy for clinically localized prostate cancer: the Prostate Cancer Outcomes Study. JAMA (2000) 283:354–60. doi: 10.1001/jama.283.3.354
5. Roehl KA, Han M, Ramos CG, Antenor JA, Catalona WJ. Cancer progression and survival rates following anatomical radical retropubic prostatectomy in 3,478 consecutive patients: long-term results. J Urol. (2004) 172:910–4. doi: 10.1097/01.ju.0000134888.22332.bb
6. Freedland SJ, Humphreys EB, Mangold LA, Eisenberger M, Dorey FJ, Walsh PC, et al. Risk of prostate cancer-specific mortality following biochemical recurrence after radical prostatectomy. JAMA (2005) 294:433–9. doi: 10.1001/jama.294.4.433
7. Kupelian PA, Mahadevan A, Reddy CA, Reuther AM, Klein EA. Use of different definitions of biochemical failure after external beam radiotherapy changes conclusions about relative treatment efficacy for localized prostate cancer. Urology (2006) 68:593–8. doi: 10.1016/j.urology.2006.03.075
9. Eisenberger MA, Blumenstein BA, Crawford ED, Miller G, McLeod DG, Loehrer PJ, et al. Bilateral orchiectomy with or without flutamide for metastatic prostate cancer. N Engl J Med. (1998) 339:1036–42. doi: 10.1056/NEJM199810083391504
10. Fizazi K, Scher HI, Molina A, Logothetis CJ, Chi KN, Jones RJ, et al. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. (2012) 13:983–92. doi: 10.1016/S1470-2045(12)70379-0
11. Ryan CJ, Smith MR, Fizazi K, Saad F, Mulders PF, Sternberg CN, et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. (2015) 16:152–60. doi: 10.1016/S1470-2045(14)71205-7
12. Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. (2012) 367:1187–97. doi: 10.1056/NEJMoa1207506
13. Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. (2010) 363:411–22. doi: 10.1056/NEJMoa1001294
14. Berthold DR, Pond GR, Soban F, de Wit R, Eisenberger M, Tannock IF. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer: updated survival in the TAX 327 study. J Clin Oncol. (2008) 26:242–5. doi: 10.1200/JCO.2007.12.4008
15. de Bono JS, Oudard S, Ozguroglu M, Hansen S, Machiels JP, Kocak I, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet (2010) 376:1147–54. doi: 10.1016/S0140-6736(10)61389-X
16. Parker C, Nilsson S, Heinrich D, O'Sullivan JM, Fossa SD, Chodacki A, et al. Updated analysis of the phase III, double-blind, randomized, multinational study of radium-223 chloride in castration-resistant prostate cancer (CRPC) patients with bone metastases (ALSYMPCA). J Clin Oncol. (2012) 30. doi: 10.1200/jco.2012.30.18_suppl.lba4512
17. Duffy MJ. Prostate-specific antigen: does the current evidence support its use in prostate cancer screening? Ann Clin Biochem. (2011) 48(Pt 4):310–6. doi: 10.1258/acb.2011.010273
18. De Marzo AM, Haffner MC, Lotan TL, Yegnasubramanian S, Nelson WG. Premalignancy in Prostate Cancer: Rethinking What we Know. Cancer Prev Res. (2016) 9:648–56. doi: 10.1158/1940-6207.CAPR-15-0431
19. Sakr WA, Partin AW. Histological markers of risk and the role of high-grade prostatic intraepithelial neoplasia. Urology (2001) 57(4 Suppl. 1):115–20. doi: 10.1016/S0090-4295(00)00953-5
20. Sporn MB, Dunlop NM, Newton DL, Smith JM. Prevention of chemical carcinogenesis by vitamin A and its synthetic analogs (retinoids). Fed Proc. (1976) 35:1332–8.
21. Andriole GL, Bostwick DG, Brawley OW, Gomella LG, Marberger M, Montorsi F, et al. Effect of dutasteride on the risk of prostate cancer. N Engl J Med. (2010) 362:1192–202. doi: 10.1056/NEJMoa0908127
22. Thompson IM, Goodman PJ, Tangen CM, Lucia MS, Miller GJ, Ford LG, et al. The influence of finasteride on the development of prostate cancer. N Engl J Med. (2003) 349:215–24. doi: 10.1056/NEJMoa030660
23. Hamilton RJ, Freedland SJ. 5-alpha reductase inhibitors and prostate cancer prevention: where do we turn now? BMC Med. (2011) 9:105. doi: 10.1186/1741-7015-9-105
24. Thompson IM Jr, Goodman PJ, Tangen CM, Parnes HL, Minasian LM, Godley PA, et al. Long-term survival of participants in the prostate cancer prevention trial. N Engl J Med. (2013) 369:603–10. doi: 10.1056/NEJMoa1215932
25. Liss MA, Thompson IM. Prostate cancer prevention with 5-alpha reductase inhibitors: concepts and controversies. Curr Opin Urol. (2018) 28:42–5. doi: 10.1097/MOU.0000000000000464
26. Ye ZW, Zhang J, Townsend DM, Tew KD. Oxidative stress, redox regulation and diseases of cellular differentiation. Bba Gen Subj. (2015) 1850:1607–21. doi: 10.1016/j.bbagen.2014.11.010
27. Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol. (2014) 24:R453–62. doi: 10.1016/j.cub.2014.03.034
28. Holmstrom KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol. Cell Biol. (2014) 15:411–21. doi: 10.1038/nrm3801
29. Kalyanaraman B. Teaching the basics of redox biology to medical and graduate students: Oxidants, antioxidants and disease mechanisms. Redox Biol. (2013) 1:244–57. doi: 10.1016/j.redox.2013.01.014
30. Cadet J, Wagner JR. DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harbor Perspect Biol. (2013) 5:a012559. doi: 10.1101/cshperspect.a012559
31. Klaunig JE, Kamendulis LM, Hocevar BA. Oxidative stress and oxidative damage in carcinogenesis. Toxicol Pathol. (2010) 38:96–109. doi: 10.1177/0192623309356453
32. Wei H. Activation of oncogenes and/or inactivation of anti-oncogenes by reactive oxygen species. Med Hypotheses (1992) 39:267–70. doi: 10.1016/0306-9877(92)90120-2
33. Cerutti PA. Oxy-radicals and cancer. Lancet (1994) 344:862–3. doi: 10.1016/S0140-6736(94)92832-0
34. Halliwell B. Free radicals, antioxidants, and human disease: curiosity, cause, or consequence? Lancet (1994) 344:721–4. doi: 10.1016/S0140-6736(94)92211-X
35. Ouyang X, DeWeese TL, Nelson WG, Abate-Shen C. Loss-of-function of Nkx3.1 promotes increased oxidative damage in prostate carcinogenesis. Cancer Res. (2005) 65:6773–9. doi: 10.1158/0008-5472.CAN-05-1948
36. Tam NN, Nyska A, Maronpot RR, Kissling G, Lomnitski L, Suttie A, et al. Differential attenuation of oxidative/nitrosative injuries in early prostatic neoplastic lesions in TRAMP mice by dietary antioxidants. Prostate (2006) 66:57–69. doi: 10.1002/pros.20313
37. Tam NN, Leav I, Ho SM. Sex hormones induce direct epithelial and inflammation-mediated oxidative/nitrosative stress that favors prostatic carcinogenesis in the noble rat. Am J Pathol. (2007) 171:1334–41. doi: 10.2353/ajpath.2007.070199
38. Fleshner NE, Klotz LH. Diet, androgens, oxidative stress and prostate cancer susceptibility. Cancer Metast Rev. (1998)17:325–30. doi: 10.1023/A:1006118628183
39. Kumar B, Koul S, Khandrika L, Meacham RB, Koul HK. Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res. (2008) 68:1777–85. doi: 10.1158/0008-5472.CAN-07-5259
40. Tam NN, Gao Y, Leung YK, Ho SM. Androgenic regulation of oxidative stress in the rat prostate: involvement of NAD(P)H oxidases and antioxidant defense machinery during prostatic involution and regrowth. Am J Pathol. (2003) 163:2513–22. doi: 10.1016/S0002-9440(10)63606-1
41. Lin X, Asgari K, Putzi MJ, Gage WR, Yu X, Cornblatt BS, et al. Reversal of GSTP1 CpG island hypermethylation and reactivation of pi-class glutathione S-transferase (GSTP1) expression in human prostate cancer cells by treatment with procainamide. Cancer Res. (2001) 61:8611–6.
42. Turpaev KT. Reactive oxygen species and regulation of gene expression. Biochem Biokhim. (2002) 67:281–92. doi: 10.1023/A:1014819832003
43. Bostwick DG, Alexander EE, Singh R, Shan A, Qian J, Santella RM, et al. Antioxidant enzyme expression and reactive oxygen species damage in prostatic intraepithelial neoplasia and cancer. Cancer (2000) 89:123–34. doi: 10.1002/1097-0142(20000701)89:1<123::AID-CNCR17>3.0.CO;2-9
44. Oberley TD, Zhong W, Szweda LI, Oberley LW. Localization of antioxidant enzymes and oxidative damage products in normal and malignant prostate epithelium. Prostate (2000) 44:144–55. doi: 10.1002/1097-0045(20000701)44:2<144::AID-PROS7>3.0.CO;2-G
45. Yossepowitch O, Pinchuk I, Gur U, Neumann A, Lichtenberg D, Baniel J. Advanced but not localized prostate cancer is associated with increased oxidative stress. J Urol. (2007) 178(4 Pt 1):1238–43; discussion 43–4. doi: 10.1016/j.juro.2007.05.145
46. Aydin A, Arsova-Sarafinovska Z, Sayal A, Eken A, Erdem O, Erten K, et al. Oxidative stress and antioxidant status in non-metastatic prostate cancer and benign prostatic hyperplasia. Clin Biochem. (2006) 39:176–9. doi: 10.1016/j.clinbiochem.2005.11.018
47. Arsova-Sarafinovska Z, Eken A, Matevska N, Erdem O, Sayal A, Savaser A, et al. Increased oxidative/nitrosative stress and decreased antioxidant enzyme activities in prostate cancer. Clin Biochem. (2009) 42:1228–35. doi: 10.1016/j.clinbiochem.2009.05.009
48. Kaya E, Ozgok Y, Zor M, Eken A, Bedir S, Erdem O, et al. Oxidative stress parameters in patients with prostate cancer, benign prostatic hyperplasia and asymptomatic inflammatory prostatitis: a prospective controlled study. Adv Clin Exp Med. (2017) 26:1095–9. doi: 10.17219/acem/66837
49. Lippman SM, Klein EA, Goodman PJ, Lucia MS, Thompson IM, Ford LG, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA (2009) 301:39–51. doi: 10.1001/jama.2008.864
50. Lippman SM, Goodman PJ, Klein EA, Parnes HL, Thompson IM Jr, Kristal AR, et al. Designing the Selenium and Vitamin E Cancer Prevention Trial (SELECT). J Natl Cancer Inst. (2005) 97:94–102. doi: 10.1093/jnci/dji009
51. Klein EA, Thompson IM Jr, Tangen CM, Crowley JJ, Lucia MS, Goodman PJ, et al. Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA (2011) 306:1549–56. doi: 10.1001/jama.2011.1437
52. Ledesma MC, Jung-Hynes B, Schmit TL, Kumar R, Mukhtar H, Ahmad N. Selenium and vitamin E for prostate cancer: post-SELECT (Selenium and Vitamin E Cancer Prevention Trial) status. Mol Med. (2011) 17:134–43. doi: 10.2119/molmed.2010.00136
53. Combs GF Jr, Gray WP. Chemopreventive agents: selenium. Pharmacol Ther. (1998) 79:179–92. doi: 10.1016/S0163-7258(98)00014-X
54. Kim YS, Milner J. Molecular targets for selenium in cancer prevention. Nutr Cancer (2001) 40:50–4. doi: 10.1207/S15327914NC401_10
55. Abdulah R, Kobayashi K, Yamazaki C, Koyama H. Molecular targets of selenium in prostate cancer prevention (Review). Int J Oncol. (2011) 39:301–9. doi: 10.3892/ijo.2011.1035
56. Whanger PD. Selenium and its relationship to cancer: an update. Br J Nutr. (2004) 91:11–28. doi: 10.1079/BJN20031015
57. Constantinou C, Papas A, Constantinou AI. Vitamin E and cancer: an insight into the anticancer activities of vitamin E isomers and analogs. Int J Cancer (2008) 123:739–52. doi: 10.1002/ijc.23689
58. Gunawardena K, Murray DK, Meikle AW. Vitamin E and other antioxidants inhibit human prostate cancer cells through apoptosis. Prostate (2000) 44:287–95. doi: 10.1002/1097-0045(20000901)44:4<287::AID-PROS5>3.0.CO;2-Z
59. Crispen PL, Uzzo RG, Golovine K, Makhov P, Pollack A, Horwitz EM, et al. Vitamin E succinate inhibits NF-kappa B and prevents the development of a metastatic phenotype in prostate cancer cells: implications for chemoprevention. Prostate. (2007) 67:582–90. doi: 10.1002/pros.20468
60. Shiau CW, Huang JW, Wang DS, Weng JR, Yang CC, Lin CH, et al. alpha-Tocopheryl succinate induces apoptosis in prostate cancer cells in part through inhibition of Bcl-xL/Bcl-2 function. J Biol Chem. (2006) 281:11819–25. doi: 10.1074/jbc.M511015200
61. Ni J, Chen M, Zhang Y, Li R, Huang J, Yeh S. Vitamin E succinate inhibits human prostate cancer cell growth via modulating cell cycle regulatory machinery. Biochem Biophys Res Commun. (2003) 300:357–63. doi: 10.1016/S0006-291X(02)02851-6
62. Morley S, Thakur V, Danielpour D, Parker R, Arai H, Atkinson J, et al. Tocopherol transfer protein sensitizes prostate cancer cells to vitamin E. J Biol Chem. (2010) 285:35578–89. doi: 10.1074/jbc.M110.169664
63. Chang E, Ni J, Yin Y, Lin CC, Chang P, James NS, et al. Alpha-vitamin E derivative, RRR-alpha-tocopheryloxybutyric acid inhibits the proliferation of prostate cancer cells. Asian J Androl. (2007) 9:31–9. doi: 10.1111/j.1745-7262.2007.00246.x
64. Zhang Y, Ni J, Messing EM, Chang E, Yang CR, Yeh S. Vitamin E succinate inhibits the function of androgen receptor and the expression of prostate-specific antigen in prostate cancer cells. Proc Natl Acad Sci USA. (2002) 99:7408–13. doi: 10.1073/pnas.102014399
65. Israel K, Yu W, Sanders BG, Kline K. Vitamin E succinate induces apoptosis in human prostate cancer cells: role for Fas in vitamin E succinate-triggered apoptosis. Nutr Cancer (2000) 36:90–100. doi: 10.1207/S15327914NC3601_13
66. Jia L, Yu W, Wang P, Li J, Sanders BG, Kline K. Critical roles for JNK, c-Jun, and Fas/FasL-Signaling in vitamin E analog-induced apoptosis in human prostate cancer cells. Prostate (2008) 68:427–41. doi: 10.1002/pros.20716
67. Redman C, Scott JA, Baines AT, Basye JL, Clark LC, Calley C, et al. Inhibitory effect of selenomethionine on the growth of three selected human tumor cell lines. Cancer Lett. (1998) 125:103–10. doi: 10.1016/S0304-3835(97)00497-7
68. Zhong W, Oberley TD. Redox-mediated effects of selenium on apoptosis and cell cycle in the LNCaP human prostate cancer cell line. Cancer Res. (2001) 61:7071–8.
69. Webber MM, Perez-Ripoll EA, James GT. Inhibitory effects of selenium on the growth of DU-145 human prostate carcinoma cells in vitro. Biochem Biophys Res Commun. (1985) 130:603–9. doi: 10.1016/0006-291X(85)90459-0
70. He Q, Rashid A, Rong R, Hillman MJ, Huang Y, Sheikh MS. Death receptor 5 regulation during selenium-mediated apoptosis in human prostate cancer cells. Cancer Biol Ther. (2002) 1:287–90. doi: 10.4161/cbt.83
71. Gundimeda U, Schiffman JE, Chhabra D, Wong J, Wu A, Gopalakrishna R. Locally generated methylseleninic acid induces specific inactivation of protein kinase C isoenzymes: relevance to selenium-induced apoptosis in prostate cancer cells. J Biol Chem. (2008) 283:34519–31. doi: 10.1074/jbc.M807007200
72. Gasparian AV, Yao YJ, Lu JX, Yemelyanov AY, Lyakh LA, Slaga TJ, et al. Selenium compounds inhibit I kappa B kinase (IKK) and nuclear factor-kappa B (NF-kappa B) in prostate cancer cells. Mol Cancer Ther. (2002) 1:1079–87.
73. Christensen MJ, Nartey ET, Hada AL, Legg RL, Barzee BR. High selenium reduces NF-kappaB-regulated gene expression in uninduced human prostate cancer cells. Nutr Cancer (2007) 58:197–204. doi: 10.1080/01635580701328701
74. Lu Z, Qi L, Li GX, Bo XJ, Liu GD, Wang JM. Se-methylselenocysteine suppresses the growth of prostate cancer cell DU145 through connexin 43-induced apoptosis. J Cancer Res Ther. (2015) 11:840–5. doi: 10.4103/0973-1482.139265
75. Jiang C, Wang Z, Ganther H, Lu JX. Caspases as key executors of methyl selenium-induced apoptosis (Anoikis) of DU-145 prostate cancer cells. Cancer Res. (2001) 61:3062–70.
76. Jiang C, Wang Z, Ganther H, Lu J. Distinct effects of methylseleninic acid versus selenite on apoptosis, cell cycle, and protein kinase pathways in DU145 human prostate cancer cells. Mol Cancer Ther. (2002) 1:1059–66.
77. Wang Z, Jiang C, Lu J. Induction of caspase-mediated apoptosis and cell-cycle G1 arrest by selenium metabolite methylselenol. Mol Carcinogen. (2002) 34:113–20. doi: 10.1002/mc.10056
78. Liu SA, Zhang HT, Zhu LY, Zhao LJ, Dong Y. Kruppel-like factor 4 is a novel mediator of selenium in growth inhibition. Mol Cancer Res. (2008) 6:306–13. doi: 10.1158/1541-7786.MCR-07-0159
79. Venkateswaran V, Klotz LH, Fleshner NE. Selenium modulation of cell proliferation and cell cycle biomarkers in human prostate carcinoma cell lines. Cancer Res. (2002) 62:2540–5.
80. Dong Y, Lee SO, Zhang HT, Marshall J, Gao AC, Ip C. Prostate specific antigen expression is down-regulated by selenium through disruption of androgen receptor signaling. Cancer Res. (2004) 64:19–22. doi: 10.1158/0008-5472.CAN-03-2789
81. Cho SD, Jiang C, Malewicz B, Dong Y, Young CY, Kang KS, et al. Methyl selenium metabolites decrease prostate-specific antigen expression by inducing protein degradation and suppressing androgen-stimulated transcription. Mol Cancer Ther. (2004) 3:605–11.
82. Dong Y, Zhang HT, Gao AC, Marshall JR, Ip C. Androgen receptor signaling intensity is a key factor in determining the sensitivity of prostate cancer cells to selenium inhibition of growth and cancer-specific biomarkers. Mol Cancer Ther. (2005) 4:1047–55. doi: 10.1158/1535-7163.MCT-05-0124
83. Chun JY, Nadiminty N, Lee SO, Onate SA, Lou W, Gao AC. Mechanisms of selenium down-regulation of androgen receptor signaling in prostate cancer. Mol Cancer Ther. (2006) 5:913–8. doi: 10.1158/1535-7163.MCT-05-0389
84. Gazi MH, Gong A, Donkena KV, Young CY. Sodium selenite inhibits interleukin-6-mediated androgen receptor activation in prostate cancer cells via upregulation of c-Jun. Clin Chim Acta. (2007) 380:145–50. doi: 10.1016/j.cca.2007.01.031
85. Morris JD, Pramanik R, Zhang X, Carey AM, Ragavan N, Martin FL, et al. Selenium- or quercetin-induced retardation of DNA synthesis in primary prostate cells occurs in the presence of a concomitant reduction in androgen-receptor activity. Cancer Lett. (2006) 239:111–22. doi: 10.1016/j.canlet.2005.07.037
86. Hu H, Jiang C, Li G, Lu J. PKB/AKT and ERK regulation of caspase-mediated apoptosis by methylseleninic acid in LNCaP prostate cancer cells. Carcinogenesis (2005) 26:1374–81. doi: 10.1093/carcin/bgi094
87. Zu K, Bihani T, Lin A, Park YM, Mori K, Ip C. Enhanced selenium effect on growth arrest by BiP/GRP78 knockdown in p53-null human prostate cancer cells. Oncogene (2006) 25:546–54. doi: 10.1038/sj.onc.1209071
88. Xiang N, Zhao R, Zhong W. Sodium selenite induces apoptosis by generation of superoxide via the mitochondrial-dependent pathway in human prostate cancer cells. Cancer Chemother Pharmacol. (2009) 63:351–62. doi: 10.1007/s00280-008-0745-3
89. Li GX, Hu H, Jiang C, Schuster T, Lu J. Differential involvement of reactive oxygen species in apoptosis induced by two classes of selenium compounds in human prostate cancer cells. Int J Cancer (2007) 120:2034–43. doi: 10.1002/ijc.22480
90. Berggren M, Sittadjody S, Song Z, Samira JL, Burd R, Meuillet EJ. Sodium selenite increases the activity of the tumor suppressor protein, PTEN, in DU-145 prostate cancer cells. Nutr Cancer (2009) 61:322–31. doi: 10.1080/01635580802521338
91. Xiang N, Zhao R, Song G, Zhong W. Selenite reactivates silenced genes by modifying DNA methylation and histones in prostate cancer cells. Carcinogenesis (2008) 29:2175–81. doi: 10.1093/carcin/bgn179
92. Hurst R, Elliott RM, Goldson AJ, Fairweather-Tait SJ. Se-methylselenocysteine alters collagen gene and protein expression in human prostate cells. Cancer Lett. (2008) 269:117–26. doi: 10.1016/j.canlet.2008.04.025
93. Bosland MC, Ozten N, Eskra JN, Mahmoud AM. A perspective on prostate carcinogenesis and chemoprevention. Curr Pharmacol Rep. (2015) 1:258–65. doi: 10.1007/s40495-015-0031-0
94. Bosland MC. Is There a Future for Chemoprevention of Prostate Cancer? Cancer Prev Res. (2016) 9:642–7. doi: 10.1158/1940-6207.CAPR-16-0088
95. Assi M. The differential role of reactive oxygen species in early and late stages of cancer. Am J Physiol Regul Integr Compar Physiol. (2017) 313:R646–53. doi: 10.1152/ajpregu.00247.2017
96. Shen MM, Abate-Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev. (2010) 24:1967–2000. doi: 10.1101/gad.1965810
97. Knudsen BS, Vasioukhin V. Mechanisms of prostate cancer initiation and progression. Adv Cancer Res. (2010) 109:1–50. doi: 10.1016/B978-0-12-380890-5.00001-6
98. Jayadev R, Sherwood DR. Basement membranes. Curr Biol. (2017) 27:R207–11. doi: 10.1016/j.cub.2017.02.006
99. Pickup MW, Mouw JK, Weaver VM. The extracellular matrix modulates the hallmarks of cancer. Embo Rep. (2014) 15:1243–53. doi: 10.15252/embr.201439246
100. Macri L, Silverstein D, Clark RAF. Growth factor binding to the pericellular matrix and its importance in tissue engineering. Adv Drug Deliv Rev. (2007) 59:1366–81. doi: 10.1016/j.addr.2007.08.015
101. Wang N, Suo Z. Long-distance propagation of forces in a cell. Biochem Biophys Res Commun. (2005) 328:1133–8. doi: 10.1016/j.bbrc.2005.01.070
102. Roignot J, Peng X, Mostov K. Polarity in mammalian epithelial morphogenesis. Cold Spring Harbor Perspect Biol. (2013) 5:a013789. doi: 10.1101/cshperspect.a013789
103. Xia J, Swiercz JM, Banon-Rodriguez I, Matkovic I, Federico G, Sun T, et al. Semaphorin-plexin signaling controls mitotic spindle orientation during epithelial morphogenesis and repair. Dev Cell (2015) 33:299–313. doi: 10.1016/j.devcel.2015.02.001
104. Xu R, Boudreau A, Bissell MJ. Tissue architecture and function: dynamic reciprocity via extra- and intra-cellular matrices. Cancer Metast Rev. (2009) 28:167–76. doi: 10.1007/s10555-008-9178-z
105. Hoarau-Vechot J, Rafii A, Touboul C, Pasquier J. Halfway between 2D and animal models: are 3D cultures the ideal tool to study cancer-microenvironment interactions? Int J Mol Sci. (2018) 19:E181. doi: 10.3390/ijms19010181
106. Simian M, Bissell MJ. Organoids: a historical perspective of thinking in three dimensions. J Cell Biol. (2017) 216:31–40. doi: 10.1083/jcb.201610056
107. Edmondson R, Broglie JJ, Adcock AF, Yang L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev Technol. (2014) 12:207–18. doi: 10.1089/adt.2014.573
108. Antoni D, Burckel H, Josset E, Noel G. Three-dimensional cell culture: a breakthrough in vivo. Int J Mol Sci. (2015) 16:5517–27. doi: 10.3390/ijms16035517
109. Lancaster MA, Knoblich JA. Organogenesis in a dish: modeling development and disease using organoid technologies. Science (2014) 345:1247125. doi: 10.1126/science.1247125
110. Tyson DR, Inokuchi J, Tsunoda T, Lau A, Ornstein DK. Culture requirements of prostatic epithelial cell lines for acinar morphogenesis and lumen formation in vitro: role of extracellular calcium. Prostate (2007) 67:1601–13. doi: 10.1002/pros.20628
111. Praharaj PP, Bhutia SK, Nagrath S, Bitting RL, Deep G. Circulating tumor cell-derived organoids: Current challenges and promises in medical research and precision medicine. Biochim Biophys Acta Rev Cancer (2018) 1869:117–27. doi: 10.1016/j.bbcan.2017.12.005
112. Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell (2014) 159:176–87. doi: 10.1016/j.cell.2014.08.016
113. Fatehullah A, Tan SH, Barker N. Organoids as an in vitro model of human development and disease. Nat Cell Biol. (2016) 18:246–54. doi: 10.1038/ncb3312
114. Debnath J, Brugge JS. Modelling glandular epithelial cancers in three-dimensional cultures. Nat Rev Cancer (2005) 5:675–88. doi: 10.1038/nrc1695
115. Karthaus WR, Iaquinta PJ, Drost J, Gracanin A, van Boxtel R, Wongvipat J, et al. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell (2014) 159:163–75. doi: 10.1016/j.cell.2014.08.017
116. Park JW, Lee JK, Phillips JW, Huang P, Cheng D, Huang J, et al. Prostate epithelial cell of origin determines cancer differentiation state in an organoid transformation assay. Proc Natl Acad Sci USA. (2016) 113:4482–7. doi: 10.1073/pnas.1603645113
117. Agarwal S, Hynes PG, Tillman HS, Lake R, Abou-Kheir WG, Fang L, et al. Identification of different classes of luminal progenitor cells within prostate tumors. Cell Rep. (2015) 13:2147–58. doi: 10.1016/j.celrep.2015.10.077
118. Liu X, Grogan TR, Hieronymus H, Hashimoto T, Mottahedeh J, Cheng DH, et al. Low CD38 identifies progenitor-like inflammation-associated luminal cells that can initiate human prostate cancer and predict poor outcome. Cell Rep. (2016) 17:2596–606. doi: 10.1016/j.celrep.2016.11.010
119. Wang S, Gao D, Chen Y. The potential of organoids in urological cancer research. Nat Rev Urol. (2017) 14:401–14. doi: 10.1038/nrurol.2017.65
120. Achilli TM, Meyer J, Morgan JR. Advances in the formation, use and understanding of multi-cellular spheroids. Expert Opin Biol Ther. (2012) 12:1347–60. doi: 10.1517/14712598.2012.707181
121. Yamada KM, Cukierman E. Modeling tissue morphogenesis and cancer in 3D. Cell (2007) 130:601–10. doi: 10.1016/j.cell.2007.08.006
122. Njoroge RN, Unno K, Zhao JC, Naseem AF, Anker JF, McGee WA, et al. Organoids model distinct Vitamin E effects at different stages of prostate cancer evolution. Sci Rep. (2017) 7:16285. doi: 10.1038/s41598-017-16459-2
123. Dyson N, Howley PM, Munger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science (1989) 243:934–7. doi: 10.1126/science.2537532
124. Bello D, Webber MM, Kleinman HK, Wartinger DD, Rhim JS. Androgen responsive adult human prostatic epithelial cell lines immortalized by human papillomavirus 18. Carcinogenesis (1997) 18:1215–23. doi: 10.1093/carcin/18.6.1215
125. Horoszewicz JS, Leong SS, Kawinski E, Karr JP, Rosenthal H, Chu TM, et al. LNCaP model of human prostatic carcinoma. Cancer Res. (1983) 43:1809–18.
126. Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol. (1994) 124:619–26. doi: 10.1083/jcb.124.4.619
127. Mailleux AA, Overholtzer M, Schmelzle T, Bouillet P, Strasser A, Brugge JS. BIM regulates apoptosis during mammary ductal morphogenesis, and its absence reveals alternative cell death mechanisms. Dev Cell (2007) 12:221–34. doi: 10.1016/j.devcel.2006.12.003
128. Nagle RB, Cress AE. Metastasis Update: human prostate carcinoma invasion via tubulogenesis. Prostate Cancer (2011) 2011:249290. doi: 10.1155/2011/249290
129. Zhang H, Eisenried A, Zimmermann W, Shively JE. Role of CEACAM1 and CEACAM20 in an in vitro model of prostate morphogenesis. PLoS ONE (2013) 8:e53359. doi: 10.1371/journal.pone.0053359
130. Martinez EE, Anderson PD, Logan M, Abdulkadir SA. Antioxidant treatment promotes prostate epithelial proliferation in Nkx3.1 mutant mice. PLoS ONE (2012) 7:e46792. doi: 10.1371/journal.pone.0046792
131. Martinez EE, Darke AK, Tangen CM, Goodman PJ, Fowke JH, Klein EA, et al. A functional variant in NKX3.1 associated with prostate cancer risk in the Selenium and Vitamin E Cancer Prevention Trial (SELECT). Cancer Prev Res. (2014) 7:950–7. doi: 10.1158/1940-6207.CAPR-14-0075
132. Klein EA, Thompson IM, Lippman SM, Goodman PJ, Albanes D, Taylor PR, et al. SELECT: the selenium and vitamin E cancer prevention trial. Urol Oncol Semin Ori (2003) 21:59–65. doi: 10.1016/S1078-1439(02)00301-0
133. Folkman J, Moscona A. Role of cell shape in growth control. Nature (1978) 273:345–9. doi: 10.1038/273345a0
134. Rotem A, Janzer A, Izar B, Ji Z, Doench JG, Garraway LA, et al. Alternative to the soft-agar assay that permits high-throughput drug and genetic screens for cellular transformation. Proc Natl Acad Sci USA. (2015) 112:5708–13. doi: 10.1073/pnas.1505979112
135. Fukazawa H, Nakano S, Mizuno S, Uehara Y. Inhibitors of anchorage-independent growth affect the growth of transformed cells on poly(2-hydroxyethyl methacrylate)-coated surfaces. Int J Cancer (1996) 67:876–82. doi: 10.1002/(SICI)1097-0215(19960917)67:6<876::AID-IJC19>3.0.CO;2-#
136. Grassian AR, Coloff JL, Brugge JS. Extracellular matrix regulation of metabolism and implications for tumorigenesis. Cold Spring Harbor Symp Quant Biol. (2011) 76:313–24. doi: 10.1101/sqb.2011.76.010967
137. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. (2016) 23:27–47. doi: 10.1016/j.cmet.2015.12.006
138. Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature (2009) 461:109–13. doi: 10.1038/nature08268
139. Geiger B, Bershadsky A, Pankov R, Yamada KM. Transmembrane crosstalk between the extracellular matrix–cytoskeleton crosstalk. Nat Rev Mol Cell Biol. (2001) 2:793–805. doi: 10.1038/35099066
140. Xia H, Nho RS, Kahm J, Kleidon J, Henke CA. Focal adhesion kinase is upstream of phosphatidylinositol 3-kinase/Akt in regulating fibroblast survival in response to contraction of type I collagen matrices via a beta(1) integrin viability signaling pathway. J Biol. Chem. (2004) 279:33024–34. doi: 10.1074/jbc.M313265200
141. Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. (2005) 6:56–68. doi: 10.1038/nrm1549
142. Cabodi S, Moro L, Bergatto E, Boeri Erba E, Di Stefano P, Turco E, et al. Integrin regulation of epidermal growth factor (EGF) receptor and of EGF-dependent responses. Biochem Soc Trans. (2004) 32(Pt3):438–42. doi: 10.1042/bst0320438
143. Buzzai M, Bauer DE, Jones RG, DeBerardinis RJ, Hatzivassiliou G, Elstrom RL, et al. The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid beta-oxidation. Oncogene (2005) 24:4165–73. doi: 10.1038/sj.onc.1208622
144. Sheikh FG, Pahan K, Khan M, Barbosa E, Singh I. Abnormality in catalase import into peroxisomes leads to severe neurological disorder. Proc Natl Acad Sci USA. (1998) 95:2961–6. doi: 10.1073/pnas.95.6.2961
145. Gulati S, Ainol L, Orak J, Singh AK, Singh I. Alterations of peroxisomal function in ischemia-reperfusion injury of rat kidney. Biochim Biophys Acta (1993) 1182:291–8. doi: 10.1016/0925-4439(93)90071-8
146. Hashimoto F, Hayashi H. Significance of catalase in peroxisomal fatty acyl-CoA beta-oxidation: NADH oxidation by acetoacetyl-CoA and H2O2. J Biochem. (1990) 108:426–31. doi: 10.1093/oxfordjournals.jbchem.a123217
147. Jiang L, Shestov AA, Swain P, Yang C, Parker SJ, Wang QA, et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature (2016) 532:255–8. doi: 10.1038/nature17393
148. Algotar AM, Stratton MS, Ahmann FR, Ranger-Moore J, Nagle RB, Thompson PA, et al. Phase 3 clinical trial investigating the effect of selenium supplementation in men at high-risk for prostate cancer. Prostate (2013) 73:328–35. doi: 10.1002/pros.22573
Keywords: organoids, vitamin E, selenium, select, prostate cancer, anoikis, metabolism, extracellular matrix (ECM)
Citation: Njoroge RN, Vatapalli RJ and Abdulkadir SA (2019) Organoids Increase the Predictive Value of in vitro Cancer Chemoprevention Studies for in vivo Outcome. Front. Oncol. 9:77. doi: 10.3389/fonc.2019.00077
Received: 21 September 2018; Accepted: 29 January 2019;
Published: 20 February 2019.
Edited by:
Imtiaz Ahmad Siddiqui, University of Colorado Anschutz Medical Campus, United StatesReviewed by:
Guangwen Cao, Second Military Medical University, ChinaChandra Singh, University of Wisconsin-Madison, United States
Copyright © 2019 Njoroge, Vatapalli and Abdulkadir. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sarki A. Abdulkadir, sarki.abdulkadir@northwestern.edu