SZT2 variants associated with partial epilepsy or epileptic encephalopathy and the genotype-phenotype correlation
 Sheng Luo1†
Sheng Luo1†  Xing-Guang Ye2†
Xing-Guang Ye2†  Liang Jin1,3†
Liang Jin1,3†  Huan Li1 Yun-Yan He1 Bao-Zhu Guan1
Huan Li1 Yun-Yan He1 Bao-Zhu Guan1  Liang-Di Gao1
Liang-Di Gao1  Xiao-Yu Liang1 Peng-Yu Wang1
Xiao-Yu Liang1 Peng-Yu Wang1  Xin-Guo Lu4 Hong-Jun Yan5
Xin-Guo Lu4 Hong-Jun Yan5  Bing-Mei Li1
Bing-Mei Li1  Yong-Jun Chen3*
Yong-Jun Chen3*  Zhi-Gang Liu2,6* for the China Epilepsy Gene 1.0 Project
Zhi-Gang Liu2,6* for the China Epilepsy Gene 1.0 Project- 1Department of Neurology, Institute of Neuroscience, Key Laboratory of Neurogenetics and Channelopathies of Guangdong Province and the Ministry of Education of China, The Second Affiliated Hospital, Guangzhou Medical University, Guangzhou, China
- 2Department of Pediatrics, Affiliated Foshan Maternity and Child Healthcare Hospital, Southern Medical University, Foshan, China
- 3Department of Neurology, The Affiliated Nanhua Hospital, Hengyang Medical School, University of South China, Hengyang, China
- 4Epilepsy Center and Department of Neurology, Shenzhen Children's Hospital, Shenzhen, China
- 5Epilepsy Center, Guangdong 999 Brain Hospital, Guangzhou, China
- 6The Second School of Clinical Medicine, Southern Medical University, Guangzhou, China
Background: Recessive SZT2 variants are reported to be associated with developmental and epileptic encephalopathy 18 (DEE-18) and occasionally neurodevelopment abnormalities (NDD) without seizures. This study aims to explore the phenotypic spectrum of SZT2 and the genotype-phenotype correlation.
Methods: Trios-based whole-exome sequencing was performed in patients with epilepsy. Previously reported SZT2 mutations were systematically reviewed to analyze the genotype-phenotype correlations.
Results: SZT2 variants were identified in six unrelated cases with heterogeneous epilepsy, including one de novo null variant and five pairs of biallelic variants. These variants had no or low frequencies in controls. All missense variants were predicted to alter the hydrogen bonds with surrounding residues and/or protein stability. The three patients with null variants exhibited DEE. The patients with biallelic null mutations presented severe DEE featured by frequent spasms/tonic seizures and diffuse cortical dysplasia/periventricular nodular heterotopia. The three patients with biallelic missense variants presented mild partial epilepsy with favorable outcomes. Analysis of previously reported cases revealed that patients with biallelic null mutations presented significantly higher frequency of refractory seizures and earlier onset age of seizure than those with biallelic non-null mutations or with biallelic mutations containing one null variant.
Significance: This study suggested that SZT2 variants were potentially associated with partial epilepsy with favorable outcomes without NDD, expanding the phenotypic spectrum of SZT2. The genotype-phenotype correlation helps in understanding the underlying mechanism of phenotypic variation.
Introduction
The SZT2 gene (OMIM* 615463) is expressed in the brain, predominantly in the parietal and frontal cortex (Toutzaris et al., 2010). It encodes seizure threshold 2 protein homolog (SZT2), primarily distributing in the lysosome membrane and peroxisome (Peng et al., 2017). In mice, heterozygous knock-out of Stz2 led to minimal clonic seizures, while the homozygous knock-out causes caused preweaning lethality with incomplete penetrance and maximal tonic hindlimb extension seizures (Frankel et al., 2009). In humans, recessive SZT2 variants were reported to be associated with developmental and epileptic encephalopathy 18 (DEE-18, OMIM* 615463) and occasionally NDD without seizures (Basel-Vanagaite et al., 2013). The SZT2 protein is the core subunit of the KICSTOR complex, with essential roles in regulating the mechanistic target of rapamycin (mTOR) signal transduction (Wolfson et al., 2017). The mTOR signal pathway plays vital roles in multiple cellular functions, including protein synthesis, cell growth and proliferation, and synaptic plasticity, which will influence neuronal excitability (Meng et al., 2013). Previously, a series of mTOR pathway genes have been identified to be associated with epilepsy and neurodevelopment abnormalities (NDD), including TSC1, TSC2, PTEN, STRADA, MTOR, AKT3, PIK3CA, RHEB, DEPDC5, NPRL2, and NPRL3 (Moloney et al., 2021). The majority of these genes are reported to be associated with a similar phenotypic spectrum, ranging from severe DEE or epilepsy with NDD to mild partial epilepsy (Møller et al., 2016; Liu et al., 2018, 2020; Deng et al., 2019; Jiang et al., 2021). However, it is unknown whether the SZT2 variants were associated with partial epilepsy, sharing a similar phenotypic spectrum with other mTOR pathway genes.
In this study, trio-based whole-exome sequencing (WES) was performed in patients with epilepsy. A total of 11 SZT2 variants were identified in six unrelated patients with heterogeneous epilepsy, including DEE in the patients with null variants and partial epilepsy in the patients with compound heterozygous missense variants. To explore the underlying mechanism of phenotypic heterogeneity, we systematically reviewed the previously reported SZT2 variants and analyzed the correlation between genotype and phenotype.
Materials and methods
Patients
The patients were recruited at the Epilepsy Center of the Second Affiliated Hospital of Guangzhou Medical University, Shenzhen Child Hospital, Guangdong 999 Brain Hospital, and Foshan Maternity and Child Healthcare Hospital, through the platform of China Epilepsy Project 1.0. Patients with acquired causes were excluded. Clinical phenotypes of epileptic seizures and epilepsy syndromes were assessed following the criteria of the Commission on Classification and Terminology of the International League Against Epilepsy (1981, 1989, 2001, 2010, 2017). Detailed clinical features were collected, including gender, current age, seizure onset age, seizure type and frequency, outcome, response to antiepileptic drugs, family history, and results from general and neurological examinations. Brain MRI scans were performed to detect brain structure abnormalities. Long-term video electroencephalograms (EEG), including open-close eyes test, hyperventilation, intermittent photic stimulation, and sleep monitoring, were performed. All patients were followed up for at least 1 year.
This study adhered to the principles of the International Committee of Medical Journal Editors concerning patient consent for research or participation and received approval from the ethics committee of the Foshan Maternity and Child Healthcare Hospital (FSFY-MEC-2022-069). Written informed consent was provided by the patient's legal guardians.
Whole exon sequencing
Blood samples of the probands, their parents, and other available family members were collected. Genomic DNAs were extracted from blood samples using the Qiagen Flexi Gene DNA kit (Qiagen, Hilden, Germany). WES was performed using a NextSeq500 sequencing instrument (Illumina, San Diego, California, USA) following the standard procedures previously described (Wang et al., 2018). The sequencing data were generated by massively parallel sequencing with an average depth of >125x and >98% coverage of the capture region on the chip for obtaining high-quality reads that were mapped to the Genome Reference Consortium Human genome build 37 by Burrows-Wheeler alignment. Variants were called and qualified with the Genome Analysis Toolkit (DePristo et al., 2011). Sanger sequences were used to validate candidate variants.
Genetic analysis
To derive potentially pathogenic variants, a case-by-case analytical approach was adopted in each case, as described in our previous study (Li et al., 2021; Wang et al., 2021). Initially, the polymorphic variants with a minor allele frequency ≥ 0.005 in the gnomAD database were removed. Potentially disease-causing variants were retained, including frameshift, non-sense, canonical splice site, initiation codon, in-frame, and missense variants. We screened SZT2 mutations by inheritance origin, including de novo mutations, co-segregated mutations, homozygous mutations, and compound heterozygous mutations, which present the genetic difference between the affected child and the parents and thus explain the occurrence of phenotype in a given family (trio). All SZT2 mutations identified in this study were annotated into the reference transcript NM_015284.
Analysis of genotype-phenotype correlation
The SZT2 mutations and related phenotypes were systematically reviewed from the PubMed database (http://www.ncbi.nlm.nih.gov/pubmed/) and the Human Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk/ac/index.php) up to November 2022. Null variants were used to define the variant that causes gross malformation of the protein and leads to loss of function (LOF) or haploinsufficiency, such as non-sense, frameshifting, initiation codon, and canonical splice site variants (Richards et al., 2015). The missense and in-frame indel/insert variants were treated as non-null variants.
Bioinformatic analyses
To evaluate the damaging effect of candidate missense variants, protein modeling was performed by using the Iterative Threading ASSEmbly Refinement software (I-TASSER, https://zhanglab.ccmb.med.umich.edu/I-TASSER/) (Yang and Zhang, 2015). Protein structure changes were visualized and analyzed by PyMOL Molecular Graphics System (Version 2.3.2; Schrödinger, LLC; New York, USA). Protein stability changes of each variant were predicted by the I-Mutant Suite server (gpcr2.biocomp.unibo.it/cgi/predictors/I-Mutant3.0/I-Mutant3.0.cgi) (Capriotti et al., 2005), which was indicated by free energy change (DDG). Negative DDG values indicate abnormally reduced mutant protein stability.
Statistical analysis
Statistical analyses were performed in R (version 4.0.3). The two-tailed Fisher's exact test was used to compare the difference between groups. P-value < 0.05 was considered statistically significant.
Results
Identification of SZT2 variants
A total of 11 SZT2 variants were identified in six unrelated cases, including one heterozygous mutation (c.6707-2A>G), one pair of biallelic mutations containing one null variant (c.6113A>C/p.Tyr2038Ser & c.8584C>T/p.Gln2862Ter), one pair of biallelic null mutations (c.6226-1G>A & c.7855C>T/p.Arg2619Ter), and three pairs of biallelic missense mutations. (c.5018C>T/p.Ser1673Phe & c.3667G>A/p.Ala1223Thr, c.8773C>T/p.His2925Tyr & c.5380C>T/p.Arg1794Trp, and c.9629G>A/p.Arg3210Gln & c.8470G>C/p.Glu2824Gln) (Figures 1A, B; Table 1). The heterozygous mutation was of de novo. The five pairs of compound heterozygous mutations originated from their asymptomatic parents, consistent with Mendelian recessive heredity patterns.
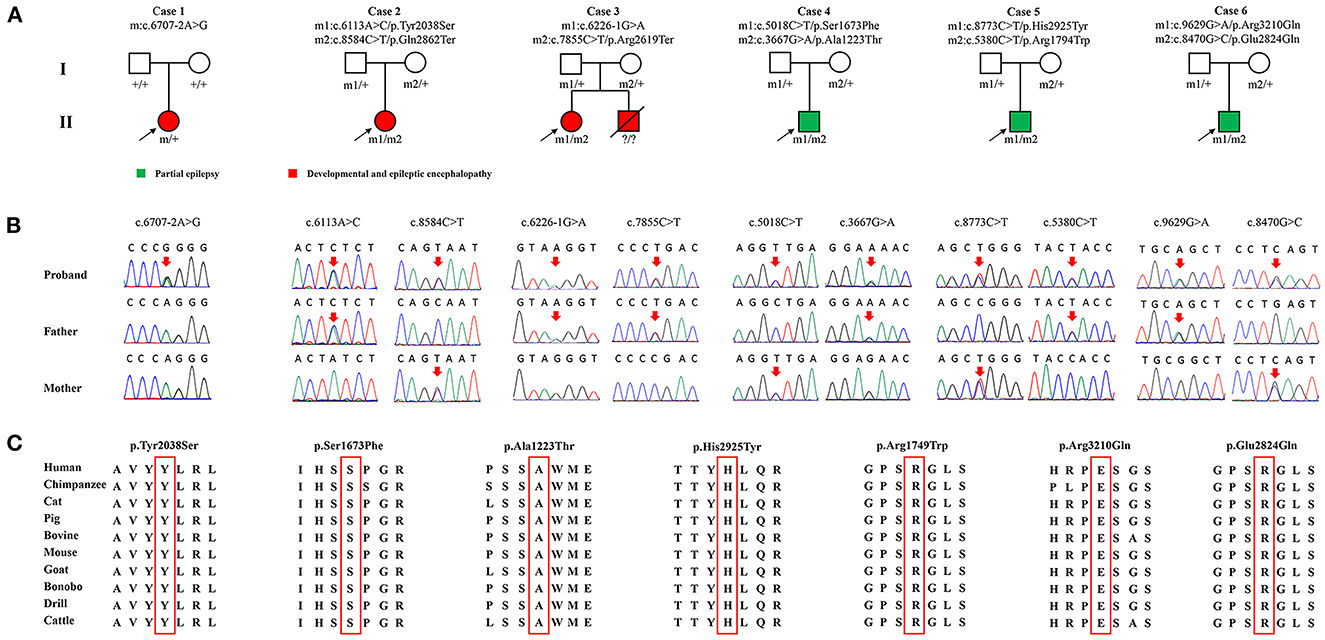
Figure 1. Genetic characterization of the probands. (A) Pedigrees of the cases with SZT2 mutations and their corresponding phenotypes. (B) DNA sequence chromatogram of the SZT2 mutations. Arrows indicated the positions of the mutations. (C) The amino acid sequence alignment showed that all seven missense variants were located in residues that are highly conserved across mammals.
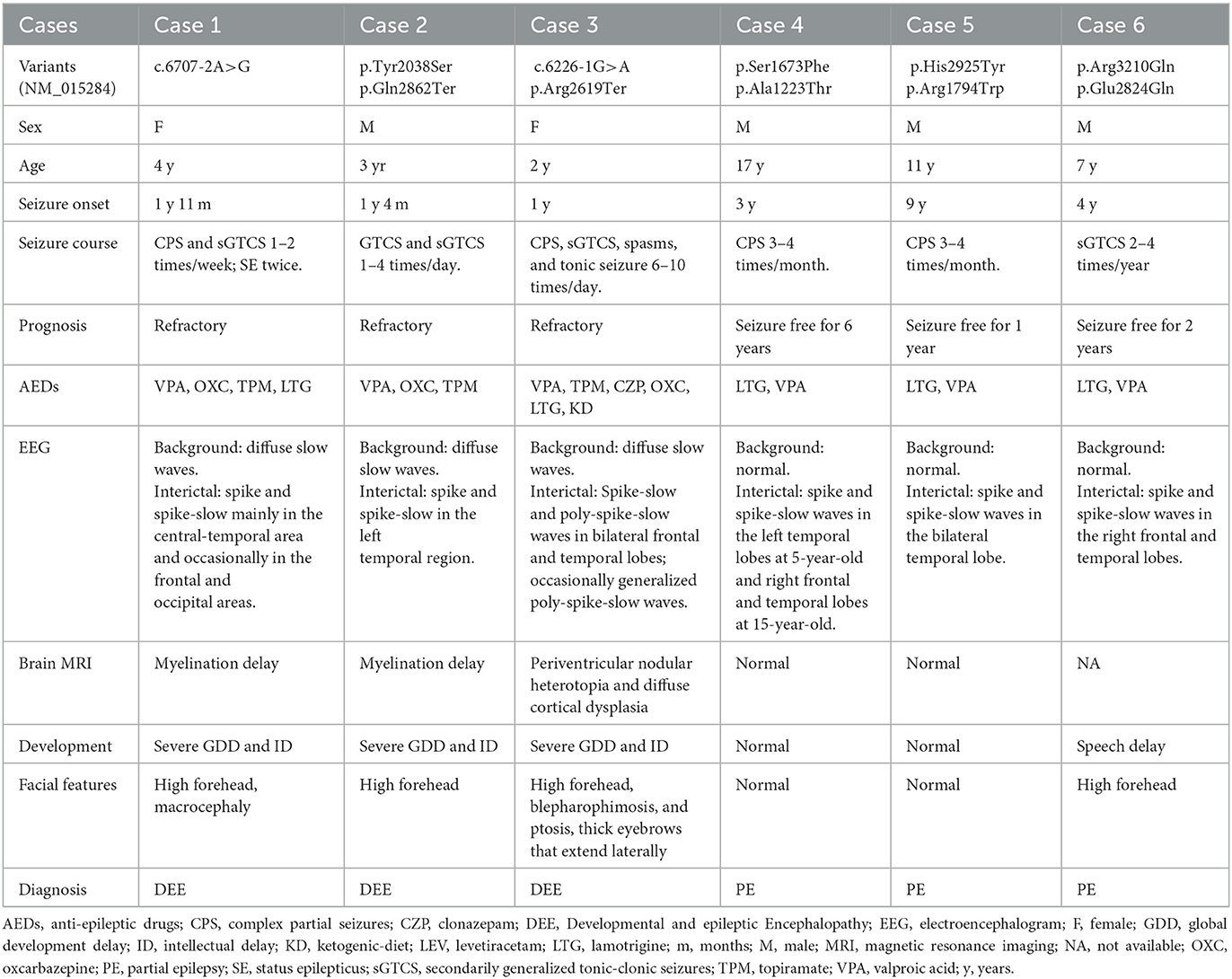
Table 1. Clinical characteristics of patients with SZT2 variants.
The de novo heterozygous mutation (c.6707-2A>G) was absent in the gnomAD database. Among variants of biallelic mutations, three null variants and two missense variants (c.6113A>C/p.Tyr2038Ser and c.8470G>C/p.Glu2824Gln) were not presented in controls, while the remaining five missense variants presented low frequencies (MAF < 0.005) in controls of the gnomAD database (Table 2).
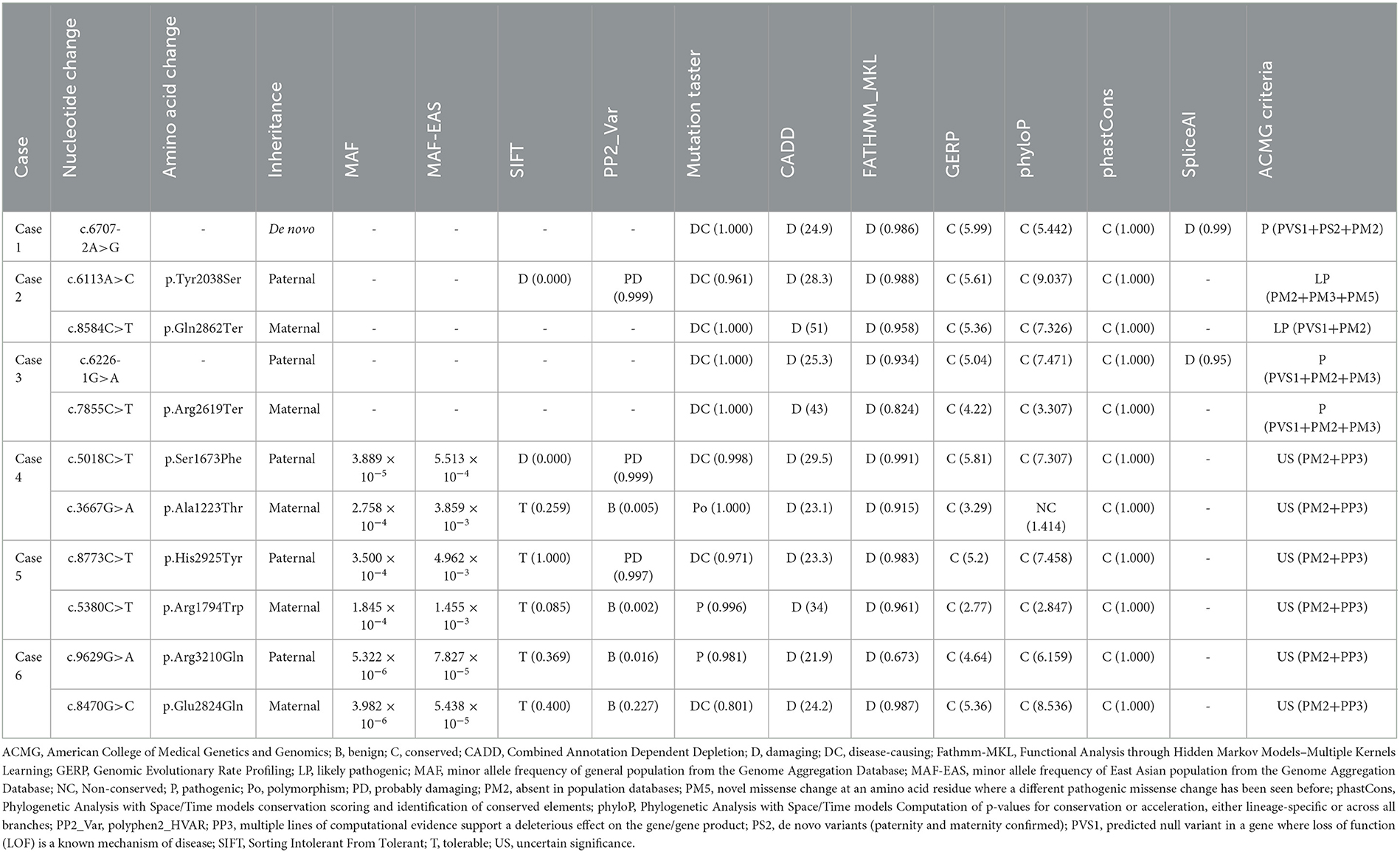
Table 2. Genetics characteristics of SZT2 mutations identified in this study.
According to ACMG guidelines, one missense that constituted biallelic mutation with a null variant and all four null variants were evaluated to be “pathogenic” or “likely pathogenic,” while the other six missense variants that constituted three pairs of compound heterozygous mutations were rated as “uncertain significance.” All missense variants identified in this study were predicted to be “damaging” by at least four in silico tools (Table 2). Amino acid sequence alignment indicated that they were located in highly conservated residues across mammals (Figure 1C).
The SZT2 gene contains 71 exons and encodes a protein that is constituted by a structural domain (1083aa-1189aa), with the function of mediating the interaction with the GATOR1 complex, and other parts with unknown functions. The detailed location of variants in the protein were visualized in Figure 2A.
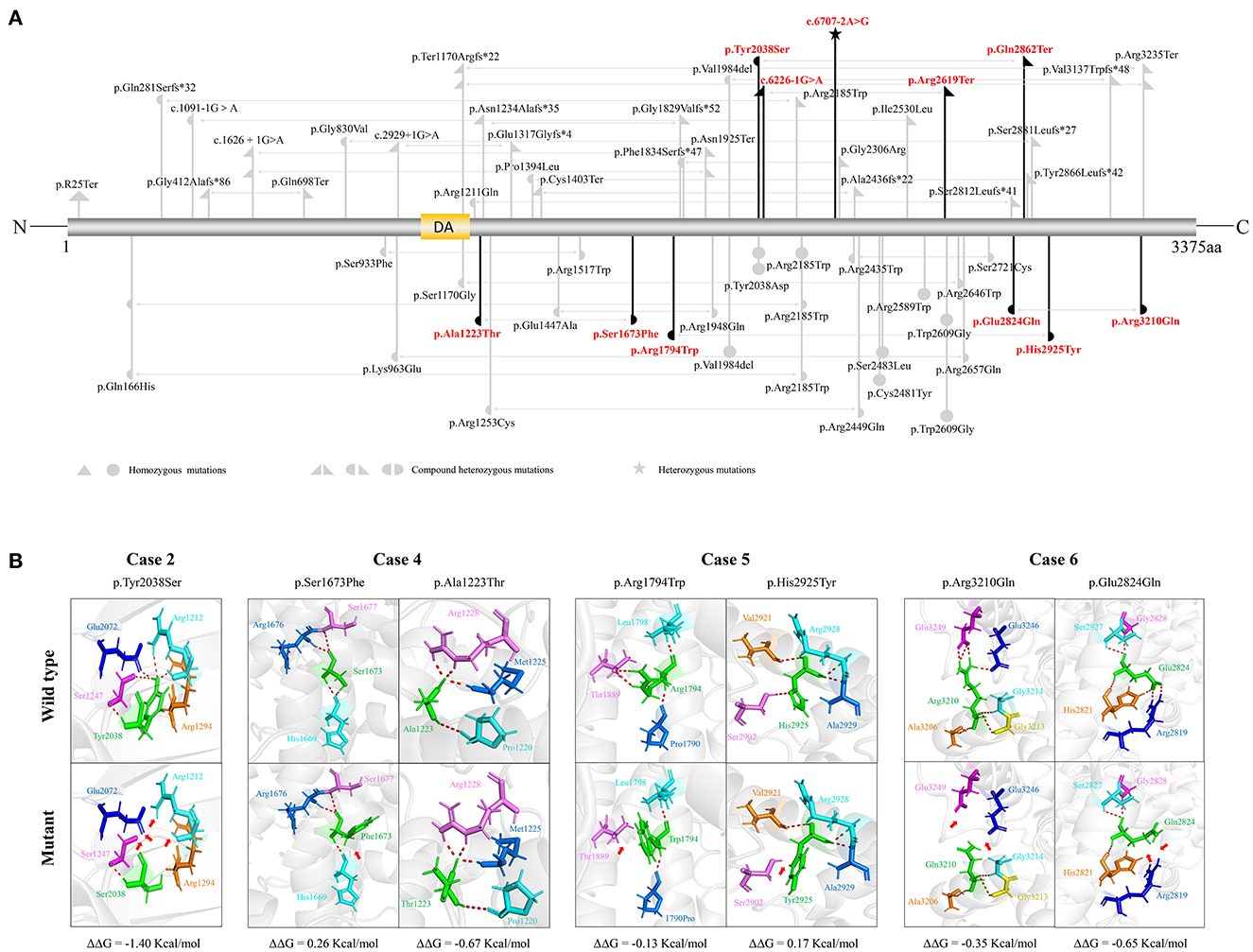
Figure 2. Schematic illustration of variants location, hydrogen bond changes, and protein stability prediction. (A) Schematic diagram of SZT2 protein and the localization of the SZT2 variants identified in this study (red) and reported previously (gray). (B) Hydrogen bond changes and DDG values of SZT2 variants. The red dotted line represented hydrogen bonds. Arrows indicated the positions with hydrogen bond changes. Six of seven missense variants were predicted to alter hydrogen bonds with surrounding residues. The variant p.Ala1223Thr was not predicted to alter hydrogen bonds with surrounding residues but was predicted to decrease the protein stability.
The molecular effects of the missense variants were analyzed by using the I-TASSER web tool for protein modeling and PyMOL software for visualization. Six of seven missense variants were predicted to alter hydrogen bonds with surrounding residues. The variant p.Ala1223Thr was not predicted to alter hydrogen bonds with surrounding residues but was predicted to decrease the protein stability (Figure 2B).
No pathogenic or likely pathogenic variants in the other epilepsy-related gene were identified in the six patients (Wang et al., 2017).
Clinical features of the cases with SZT2 variants
The detailed clinical features of the patients with SZT2 variants were summarized in Table 1.
The patients with null SZT2 variants were all diagnosed with DEE (case 1 case 2, and case 3). They presented daily or weekly seizures with onset ages before 2-year-old. Focal seizures and secondarily generalized tonic-clonic seizures (sGTCS) were the common seizure type. The patients with biallelic null variants (case 3) presented also frequent spasms and tonic seizures. Seizures of the three cases were refractory. The interictal EEG recorded focal or multifocal discharge with backgrounds of diffuse slow waves (Figures 3A, B). The MRI scans detected abnormalities of brain structure, including myelination delay in cases 1 and 2 and diffuse cortical dysplasia/periventricular nodular heterotopia in case 3 (Figures 3C, D). The three patients also exhibited global developmental delays and intellectual disabilities.
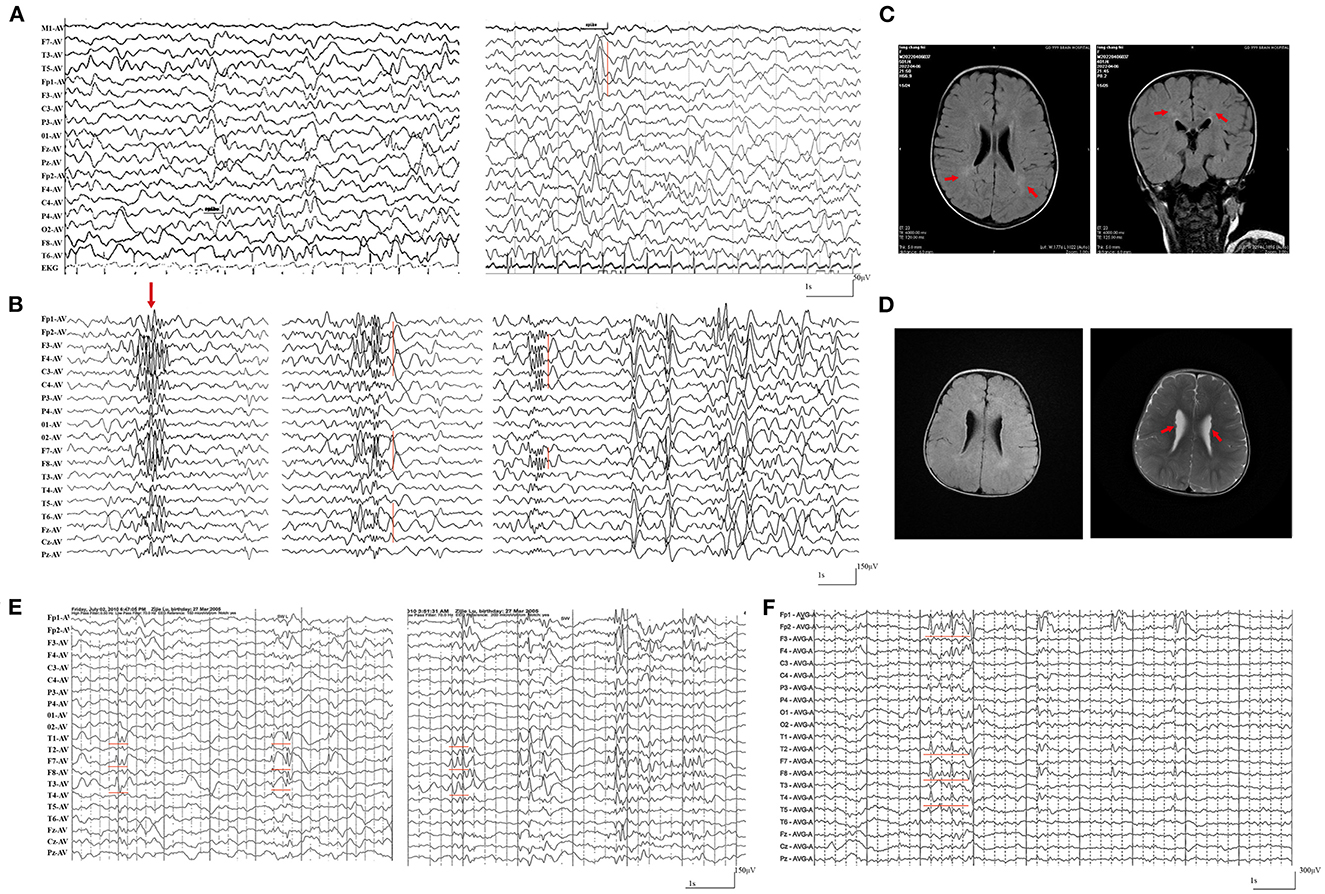
Figure 3. Representative electroencephalography (EEG) and magnetic resonance imaging (MRI) of the cases with SZT2 mutations. (A) Interictal EEG of case 2 at the age of 2-year-old showed general slowing of background (left) and focal sharp waves in the left temporal lobe. (B) Interictal EEG of case 3 at the age of 1.5-year-old showed generalized poly-spike waves (left panel) and multifocal poly-spikes/poly-spike-slow waves in the (middle and right panel). (C) The MRI of case 2 at the age of 2-year-old showed hyperintensities in the white matter of bilateral periventricular, indicating myelination delay. (D) The MRI of case 3 at the age of 1.5-years-old showed diffuse cortical dysplasia and periventricular nodular heterotopia (right panel). (E) Interictal EEG of case 4 at 5-year-old showed spike-slow waves in the left temporal lobe. (F) Interictal EEG of case 4 at 15-year-old showed spike-slow waves in the right temporal and frontal lobes.
The cases with biallelic missense variants were diagnosed with childhood partial epilepsy (cases 4, 5, and 6). They suffered from monthly or yearly focal seizures and/or sGTCS. Seizure-free was achieved with the combined treatment of lamotrigine (LTG) and valproic acid (VPA). The interictal EEG detected focal abnormalities with features of idiopathic epilepsies, including shifting, bilateral, multiple focal discharges with normal backgrounds (Figures 3E, F). The MRI scans detected no brain structural abnormalities. The three patients all exhibited normal neurodevelopment, except case 6 with mild speech delays.
Genotype-phenotype correlation
Previously, a total of 41 pairs of recessive mutations were identified in 50 patients, including 3 homozygous null mutations, 7 pairs of compound heterozygous null mutations, 12 pairs of compound heterozygous mutations containing 1 null variant, 8 homozygous missense mutations, 9 pairs of compound heterozygous missense mutations, 2 homozygous in-frame indel mutations (Figure 4A; Supplementary Table S1). To analyze the relationship between genotype and phenotype, the genotype was classified into: (1) biallelic null mutations, (2) biallelic mutations containing one null variant, (3) and biallelic non-null mutations. The patients with biallelic null mutations had earlier onset ages of seizure with median of 5 months, comparing the patients with biallelic mutations with one null variant (12 months) and biallelic non-null mutations (36 months) (Figure 4B). The patients with biallelic null mutations all presented DEE, with most (8/9) having refractory seizures. Patients with biallelic mutations containing one null variant also exhibited DEE, but with fewer refractory seizures (2/7). Patients with biallelic non-null mutations also showed fewer refractory seizures (7/20), and three patients did not have seizures (3/20) (Figure 4C). Patients with biallelic null mutations presented higher percentages of refractory seizures than those with biallelic mutations containing one null variant (8/9 vs. 2/7; p = 0.035) and biallelic non-null variants (8/9 vs. 7/17; p = 0.049) (Figure 4D).
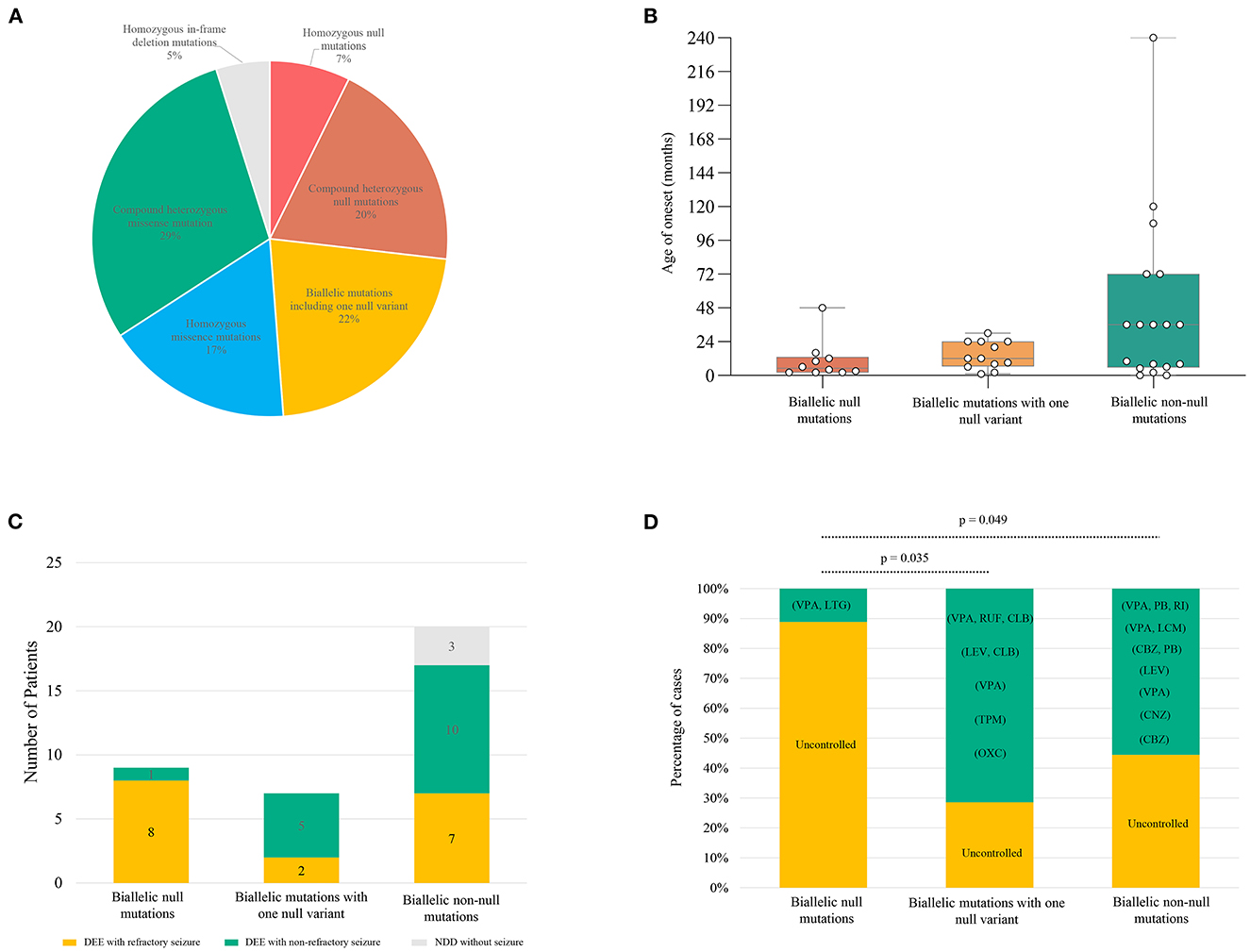
Figure 4. Genotype–phenotype correlation of previously reported SZT2 mutations. (A) The pie chart of the genotype distribution of SZT2 variants. (B) The boxplot of onset ages of the patients with different genotypes. (C) The stacked bar chart of phenotypes of the patients with different genotypes. Patients with detailed clinical information were taken into the analysis (n = 36). (D) The stacked bar chart of drug response of the patients with different genotypes. Patients with biallelic null mutations presented higher percentages of refractory seizures than those with biallelic mutations containing one null variant (8/9 vs. 2/7; p = 0.035) and biallelic non-null variants (8/9 vs. 7/17; p = 0.049). CBZ, carbamazepine; CLB, clobazam; CNZ, clonazepam; LCM, lacosamide; LEV, levetiracetam; LTG, lamotrigine; OXC, oxcarbazepine; PB, phenobarbital; RUF, rufinamide; TPM, topiramate; VPA, valproic acid.
Discussion
In this study, we identified novel SZT2 variants in six unrelated patients with heterogeneous epilepsy, including one de novo null variant and five pairs of biallelic variants. These variants had no or low frequencies in controls. All missense variants were predicted to alter the hydrogen bonding with surrounding residue and/or protein stability. The patients with monoallelic or biallelic null mutations presented severe DEE, while the three patients with biallelic missense mutations exhibited mild partial epilepsy. Analysis of previously reported cases revealed that patients with biallelic null mutations presented significantly higher percentages of refractory seizures and earlier onset ages of seizure than those with other genotypes. This study suggested that SZT2 variants were potentially associated with partial epilepsy with favorable outcomes without NDD. The genotype-phenotype correlation helps in understanding the underlying mechanism of phenotypic variation.
The SZT2 protein is the core subunit of the KICSTOR complex (consisting of KPTN, ITFG2, C12orf66, and SZT2), negatively regulating mTORC1 signaling (Figure 5) (Wolfson et al., 2017). LOF of SZT2 causes overactivation of mTORC1 signaling, which is one of the hallmarks of epilepsy and brain malformations (Marsan and Baulac, 2018). Experimentally, Szt2 knockout mice presented spontaneous seizures (Frankel et al., 2009). The variants identified in this study included one heterozygous null variant, one pair of biallelic null mutations, and one pair of biallelic mutations containing one null variant, which were possibly associated with LOF. The six missense variants in the three pairs of biallelic variants were predicted to alter the hydrogen bonding/protein stability and be “damaged” by diverse in silico tools, thus being considered to be potentially deleterious. These evidence suggested that LOF may be the pathogenic mechanism for SZT2.
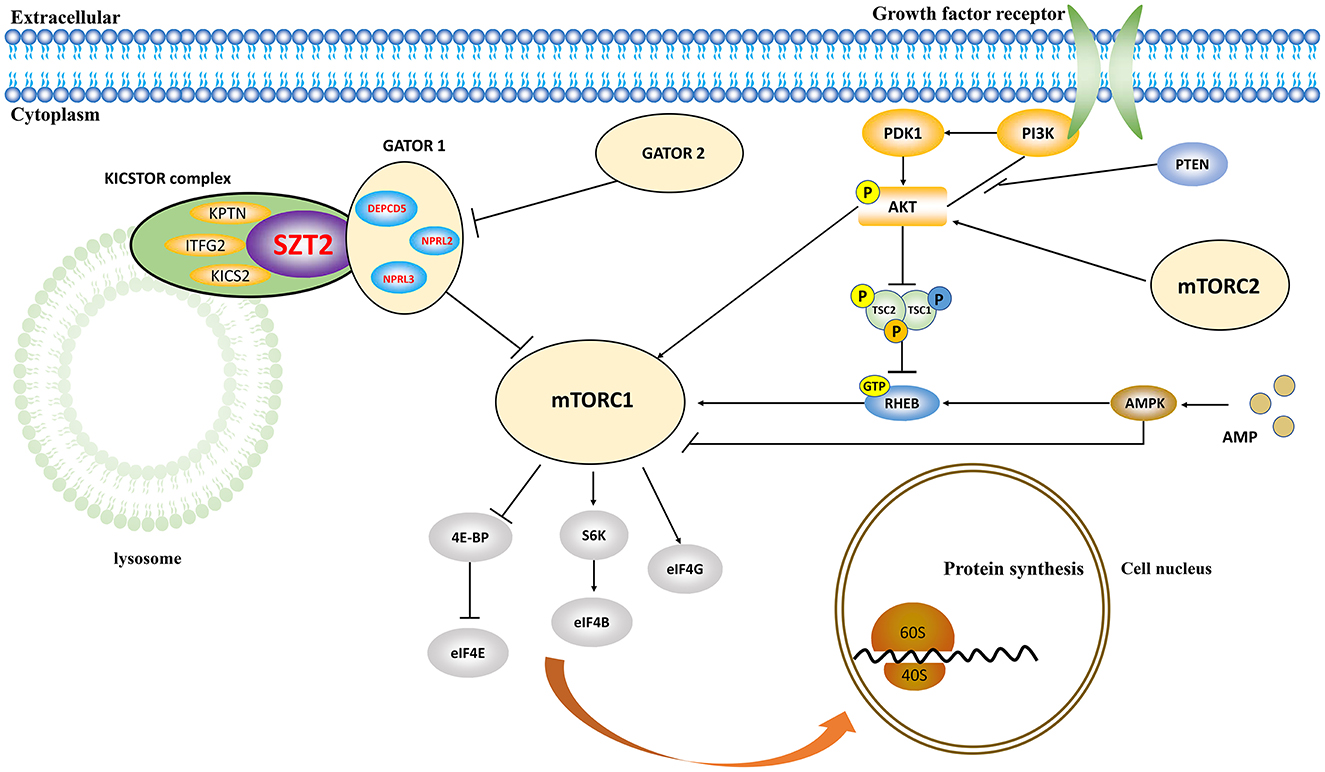
Figure 5. Schematic illustration of the role of SZT2 in the mTOR pathway. The SZT2 protein is the core subunit of the KICSTOR complex, negatively regulating mTORC1 signaling. It directly interacts with the GATOR1 complex, which was constituted by DEPDC5, NPRL2, and NPRL3.
The SZT2 protein directly interacts with the GATOR1 complex, which was constituted by DEPDC5, NPRL2, and NPRL3 (Figure 5). These genes were associated with heterogeneous epilepsy (Baldassari et al., 2019), ranging from mild partial epilepsy to severe epileptic encephalopathy (Baldassari et al., 2019; Iffland et al., 2019; Samanta, 2022). It was considered that SZT2 was associated with a similar phenotypic spectrum, like the three genes. Previously, the SZT2 variants have been reported to be associated with DEE and occasionally NDD without seizures (Supplementary Table S1). This study identified SZT2 variants in patients that presented mild partial epilepsy with favorable outcomes without NDD, expanding the phenotypic spectrum of SZT2.
In mice, heterozygous knockout of Szt2 caused minimal clonic seizures, whereas homozygous knockout led to maximal tonic hindlimb extension seizures and preweaning lethality with incomplete penetrance (Frankel et al., 2009). In this study, the patients with null mutations presented DEE; the patient with biallelic null variants presented further severer phenotype featured by frequent spasms/tonic seizures and brain structure abnormalities; patients with biallelic missense variants presented mild partial epilepsy. Similarly, the analysis of previously reported cases showed that patients with biallelic null mutations presented significantly higher percentages of refractory seizures and earlier onset age of seizure than those with other genotypes. These findings suggested a dose-dependent effect of SZT2, which is potentially one of the explanations for the phenotypic variation of SZT2 variants.
To our knowledge, the previously reported SZT2 mutations were all biallelic variants. This study reported a de novo monoallelic variant of SZT2 (c.6707-2A>G) in a patient with DEE, which was classified as “pathogenic” according to the ACMG criteria. Additionally, the clinical features of the patient coincided highly with DEE-18. This variant was therefore considered to be associated with the phenotype. It is unknown whether other factors, such as dominant negative effects or undetectable intron variants in trans by WES, are involved in the pathogenicity, which warrants further verification.
In the presented study, seizures were refractory in patients with null SZT2 mutations, whereas seizures-free was achieved in patients with biallelic missense variants by using VPA and LTG. Previously, VPA and LTG were also common effective drugs for patients with SZT2 mutations, including a seizure-free patient with biallelic null mutations (Figure 4D). It was considered that VPA and LTG may be used as basic therapeutic regimens for patients with SZT2 mutations, which will be potentially beneficial for patients if an early genetic diagnosis could be made.
This study has several limitations. The pathogenicity of the variants identified in this study was only supported by in silico analysis; the detailed functional alteration needs to be experimentally examined. Due to the limitation of WES, potentially disease-causing intron variants or copy number variants may not be detected, especially case 1 with only a heterozygous SZT2 variant. The genotype-phenotype correlation needs more cases to further elucidate in the future.
In summary, this study suggested that SZT2 variants were potentially associated with partial epilepsy with favorable outcomes without NDD, expanding the phenotypic spectrum of SZT2. The genotype-phenotype correlation and dose-dependent effect help in understanding the underlying mechanism of phenotypic variation.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the Foshan Maternity and Child Healthcare Hospital (FSFY-MEC-2022-069). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author contributions
SL, LJ, X-GY, and Y-JC contributed to the conception of the study, interpretation of clinical data, and drafting of the manuscript. HL, Y-YH, B-ZG, L-DG, X-YL, X-GL, and B-ML examined the patients and participated in the drafting of the manuscript. Y-JC and Z-GL provided a critical review and substantially revised the manuscript. All authors read and approved the manuscript before submitting it to the journal for publication.
Funding
This work was supported by the National Natural Science Foundation of China (82271505, 82171439, and 81971216), the Science and Technology Project of Guangzhou (201904010292 and 201904020028), Foshan Science and Technology Bureau (Grant No. 2020001003419), Multi-Center Clinical Research Fund Project of the Second Affiliated Hospital of Guangzhou Medical University (2020-LCYJ-DZX-03 and 2021-LCYJ-DZX-02), Key Projects of Hunan Provincial Health and Health Commission (20201910) and the 4310 Program of Clinical Medical Research of the University of South China (20224310NHYCG11). The funders had no role in study design, data collection and analysis, and the decision to publish or preparation of the manuscript.
Acknowledgments
The help of patients and clinicians participating in this work are greatly appreciated.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2023.1162408/full#supplementary-material
References
Baldassari, S., Picard, F., Verbeek, N. E., van Kempen, M., Brilstra, E. H., Lesca, G., et al. (2019). The landscape of epilepsy-related GATOR1 variants. Genet. Med. 21, 398–408. doi: 10.1038/s41436-018-0060-2
Basel-Vanagaite, L., Hershkovitz, T., Heyman, E., Raspall-Chaure, M., Kakar, N., Smirin-Yosef, P., et al. (2013). Biallelic SZT2 mutations cause infantile encephalopathy with epilepsy and dysmorphic corpus callosum. Am. J. Hum. Genet. 93, 524–529. doi: 10.1016/j.ajhg.2013.07.005
Capriotti, E., Fariselli, P., and Casadio, R. (2005). I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 33, W306-10. doi: 10.1093/nar/gki375
Deng, J., Fang, F., Wang, X. H., Dai, L. F., Tian, X. J., Chen, C. H., et al. (2019). Clinical and genetic characteristics of focal epilepsy in children caused by GATOR1 complex gene variation. Zhonghua Er Ke Za Zhi 57, 780–785. doi: 10.3760/cma.j.issn.0578-1310.2019.10.010
DePristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Maguire, J. R., Hartl, C., et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498. doi: 10.1038/ng.806
Frankel, W. N., Yang, Y., Mahaffey, C. L., Beyer, B. J., and O'Brien, T. P. (2009). Szt2, a novel gene for seizure threshold in mice. Genes Brain Behav. 8, 568–576. doi: 10.1111/j.1601-183X.2009.00509.x
Iffland, P. H. II, Carson, V., Bordey, A., and Crino, P. B. (2019). GATORopathies: the role of amino acid regulatory gene mutations in epilepsy and cortical malformations. Epilepsia 60, 2163–2173. doi: 10.1111/epi.16370
Jiang, T., Gao, J., Jiang, L., Xu, L., Zhao, C., Su, X., et al. (2021). Application of trio-whole exome sequencing in genetic diagnosis and therapy in Chinese children with epilepsy. Front. Mol. Neurosci. 14, 699574. doi: 10.3389/fnmol.2021.699574
Li, J., Lin, S. M., Qiao, J. D., Liu, X. R., Wang, J., Jiang, M., et al. (2021). CELSR3 variants are associated with febrile seizures and epilepsy with antecedent febrile seizures. CNS Neurosci. Ther. 28, 382–389. doi: 10.1111/cns.13781
Liu, J., Tong, L., Song, S., Niu, Y., Li, J., Wu, X., et al. (2018). Novel and de novo mutations in pediatric refractory epilepsy. Mol. Brain 11, 48. doi: 10.1186/s13041-018-0392-5
Liu, L., Chen, Z.-R., Xu, H.-Q., Liu, D.-T., Mao, Y., Liu, H.-K., et al. (2020). DEPDC5 variants associated malformations of cortical development and focal epilepsy with febrile seizure plus/febrile seizures: the role of molecular sub-regional effect. Front. Neurosci. 14, 821. doi: 10.3389/fnins.2020.00821
Marsan, E., and Baulac, S. (2018). Review: mechanistic target of rapamycin (mTOR) pathway, focal cortical dysplasia and epilepsy. Neuropathol. Appl. Neurobiol. 44, 6–17. doi: 10.1111/nan.12463
Meng, X. F., Yu, J. T., Song, J. H., Chi, S., and Tan, L. (2013). Role of the mTOR signaling pathway in epilepsy. J. Neurol. Sci. 332, 4–15. doi: 10.1016/j.jns.2013.05.029
Møller, R. S., Weckhuysen, S., Chipaux, M., Marsan, E., Taly, V., Bebin, E. M., et al. (2016). Germline and somatic mutations in the MTOR gene in focal cortical dysplasia and epilepsy. Neurol. Genet 2, e118. doi: 10.1212/NXG.0000000000000118
Moloney, P. B., Cavalleri, G. L., and Delanty, N. (2021). Epilepsy in the mTORopathies: opportunities for precision medicine. Brain Commun. 3, fcab222. doi: 10.1093/braincomms/fcab222
Peng, M., Yin, N., and Li, M. O. (2017). SZT2 dictates GATOR control of mTORC1 signalling. Nature 543, 433–437. doi: 10.1038/nature21378
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 17, 405–424. doi: 10.1038/gim.2015.30
Samanta, D. (2022). DEPDC5-related epilepsy: a comprehensive review. Epilepsy Behav. 130, 108678. doi: 10.1016/j.yebeh.2022.108678
Toutzaris, D., Lewerenz, J., Albrecht, P., Jensen, L. T., Letz, J., Geerts, A., et al. (2010). A novel giant peroxisomal superoxide dismutase motif-containing protein. Free Radic. Biol. Med. 48, 811–820. doi: 10.1016/j.freeradbiomed.2009.12.023
Wang, J., Lin, Z. J., Liu, L., Xu, H. Q., Shi, Y. W., Yi., et al. (2017). Epilepsy-associated genes. Seizure 44, 11–20. doi: 10.1016/j.seizure.2016.11.030
Wang, J., Qiao, J. D., Liu, X. R., Liu, D. T., Chen, Y. H., Wu, Y., et al. (2021). UNC13B variants associated with partial epilepsy with favourable outcome. Brain 144, 3050–3060. doi: 10.1093/brain/awab164
Wang, J.-Y., Zhou, P., Wang, J., Tang, B., Su, T., Liu, X.-R., et al. (2018). ARHGEF9 mutations in epileptic encephalopathy/intellectual disability: toward understanding the mechanism underlying phenotypic variation. Neurogenetics 19, 9–16. doi: 10.1007/s10048-017-0528-2
Wolfson, R. L., Chantranupong, L., Wyant, G. A., Gu, X., Orozco, J. M., Shen, K., et al. (2017). KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature 543, 438–442. doi: 10.1038/nature21423
Keywords: SZT2 gene, partial epilepsy, developmental and epileptic encephalopathy, phenotypic variation, genotype-phenotype correlation
Citation: Luo S, Ye X-G, Jin L, Li H, He Y-Y, Guan B-Z, Gao L-D, Liang X-Y, Wang P-Y, Lu X-G, Yan H-J, Li B-M, Chen Y-J and Liu Z-G (2023) SZT2 variants associated with partial epilepsy or epileptic encephalopathy and the genotype-phenotype correlation. Front. Mol. Neurosci. 16:1162408. doi: 10.3389/fnmol.2023.1162408
Received: 09 February 2023; Accepted: 10 April 2023;
Published: 05 May 2023.
Edited by:
Qian Chen, Massachusetts Institute of Technology, United StatesReviewed by:
Zhonghua Lu, Chinese Academy of Sciences (CAS), ChinaZhenhua Liu, Central South University, China
Copyright © 2023 Luo, Ye, Jin, Li, He, Guan, Gao, Liang, Wang, Lu, Yan, Li, Chen and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong-Jun Chen, chenyj-usc@foxmail.com; Zhi-Gang Liu, zhigangliu88@163.com
†These authors have contributed equally to this work and share first authorship