 Magdalena Matloka
Magdalena Matloka Frédérique Rau
Frédérique Rau Denis Furling
Denis Furling- Sorbonne Université, INSERM, Association Institut de Myologie, Centre de Recherche en Myologie, Paris, France
Myotonic dystrophy type 1 (DM1 also known as Steinert disease) is a multisystemic disorder mainly characterized by myotonia, progressive muscle weakness and wasting, cognitive impairments, and cardiac defects. This autosomal dominant disease is caused by the expression of nuclear retained RNAs containing pathologic expanded CUG repeats that alter the function of RNA-binding proteins in a tissue-specific manner, leading ultimately to neuromuscular dysfunction and clinical symptoms. Although considerable knowledge has been gathered on myotonic dystrophy since its first description, the development of novel relevant disease models remains of high importance to investigate pathophysiologic mechanisms and to assess new therapeutic approaches. In addition to animal models, in vitro cell cultures provide a unique resource for both fundamental and translational research. This review discusses how cellular models broke ground to decipher molecular basis of DM1 and describes currently available cell models, ranging from exogenous expression of the CTG tracts to variable patients’ derived cells.
Introduction
Myotonic dystrophies (DM) are a group of dominant disorders that are among the most prevalent neuromuscular diseases in adults (1). The main characteristics of these multisystemic diseases are myotonia, progressive muscle weakness and wasting, cardiac-conduction defects, cognitive impairments together with other endocrine dysfunctions (1). Two DM forms have been identified so far: type 1 (DM1 also called Steinert Disease) and type 2 (DM2, previously known as PROMM), which is generally less severe than DM1. Both share a similar molecular mechanism in which deleterious expansion of microsatellite repeats in non-coding regions, (CTG)n in 3′UTR of dystrophia myotonica protein kinase (DMPK) gene in DM1 (2–4) and (CCTG)n in intron 1 of CNBP gene in DM2 (5) are transcribed into expanded C/CUG-RNA that are retained in the nucleus as discrete foci. These ribonuclear foci sequester muscleblind-like (MBNL) RNA-binding proteins, resulting in their functional loss and consequently, RNA metabolism alterations (6–11). Thus, misregulation of alternative splicing events within downstream effector genes were found in striated muscles of DM1 patients and associated with clinical symptoms, such as insulin resistance, myotonia, muscle weakness, and cardiac defects (12–18).
During the past 20 years, several animal models, including mouse, fly, zebrafish, and worm have been developed to investigate DM1 pathophysiologic mechanisms. They largely contributed to the current state of the art on myotonic dystrophies, which also benefited from research performed on cell cultures. At present, more than 100 years from the first descriptions of Steinert disease, there is still a need for cellular models to decipher disease-related molecular mechanisms and evaluate therapeutic approaches before in vivo validation. Because DM is a multisystemic disease affecting many tissues and cell types, various cell models are required to cover all DM-associated defects. Thanks to technological progresses, we have access to new cellular models allowing more comprehensive and adequate studies. Herein, we will discuss the use of in vitro cell models through the advances in myotonic dystrophy research and describe the available cellular models, from exogenous expression of CTG repeats to patient’s derived cells, which were developed for the study of DM1.
Cellular Models in DM1 Research History
In the early 1900s, Dr. Hans Steinert provided for the first time a detailed description of a neuromuscular disorder characterized by dystrophic progression with myotonia and degeneration of skeletal muscle (1). Since then, Steinert’s disease that was renamed as myotonic dystrophy type 1 or DM1 by the International Myotonic Dystrophy Consortium has been extensively investigated at both clinical and pathophysiologic level. Even before the discovery of the mutation responsible for DM1, primary cells derived from DM1 patients have been used to uncover differences in behavior or cytochemistry (19–22) to study metabolism (23–26) or to understand mechanisms leading to symptoms described in patients, like widely observed insulin resistance (27–30). However, besides learning about the clinical, physiological, and cellular manifestations of DM1, it was essential to define the molecular bases of the disease. The first breakthrough came in 1992, when the mutation responsible for DM1 was identified as an unstable CTG expansion within the 3′ non-coding region of the DMPK gene (2–4, 27, 31, 32). The next challenge was to understand how this expansion leads to molecular and cellular defects observed in DM1 cells. As it became striking that mutant DMPK mRNA was altered in DM1, the use of different cellular models provided initially confusing conclusions about its expression in the disease (33, 34). Nevertheless, the observation that the level of mutant DMPK mRNA decreased when the size of the repeats increased, led to the hypothesis that expanded repeats were rather impairing post-transcriptional processing of the mutant DM1 allele (35). The compelling evidence for this postulate came shortly after, when discrete ribonuclear foci were reported for the first time in DM1 fibroblasts (36). Additionally, experiments performed with patient-derived myoblasts and fibroblasts determined that mutant DMPK transcripts, while correctly spliced and polyadenylated, were not exported to the cytoplasm but retained in the nucleus (37, 38), causing approximately 50% reduction of the DMPK protein levels in DM1 myoblasts (39). These findings obtained from patient-derived cells gave rise to the idea of a RNA gain-of-function mechanism in DM1. This concept was proposed following the identification of a RNA-binding protein, CELF1 (also called CUG-BP) that could bind to single-stranded UG motifs and is aberrantly accumulated in the nucleus of cells derived from DM1 patients (40–43). Upregulation of CELF1 and its splicing regulatory activity have been associated with abnormal splicing of downstream targets, suggesting a trans-dominant effect of CUG repeats on RNA processing in DM1 (18, 44) which was further confirmed in cell models overexpressing exogenous CUG expanded tracts with increasing lengths (16). Finally, at the beginning of the 2000s, a second breakthrough has been reached with the identification of RNA-binding proteins that bind specifically to CUG repeats proportionally to the size of the expansion (45). These proteins belong to the MBNL family, which includes three paralogs (MBNL1, MBNL2, and MBNL3), and all of them are sequestered within the nuclear RNA foci in DM1 patient cells (46). Among their functions, MBNL proteins are splicing regulatory factors that control developmental switch between fetal and adult isoforms of many transcripts (47). Thus, titration of MBNL proteins by nuclear CUGexp-RNA results in alternative splicing misregulations of several pre-mRNAs in DM1, and some of them are associated to DM1 phenotypic features, establishing the deficiency of functional MBNLs as a central cause of the disease (12–18, 48–51).
Cell Models Expressing Exogenous CTG Repeats
Several years after the identification of the mutation, the expression of exogenously expressed CTG tracts in cellular models was widely used as a tool to confirm the direct role of the repeats in the pathologic mechanisms of DM1. The repeats, usually inserted in the 3′UTR of a truncated DMPK gene commonly under the control of a CMV promoter, are transiently or stably expressed in well-characterized human or murine cell lines, such as HeLa, HEK, or C2 cells. Even if they are lacking the entire genomic context of the CTG expansion and its own specific promoter regulation, they still recapitulate several DM1-associated features like the formation of ribonuclear foci that colocalizes with MBNL proteins and the splicing defect (16, 52, 53). Thus, they provide fast and reproducible tools for informative screening readout. Up to date, several constructs containing expanded CTG repeats have been described. A construct expressing interrupted 960 CTG repeats has been used in a wide range of studies including molecular mechanism investigations and validation of therapeutic approaches (54). To ensure the stability of the expansion, the CTG tracts are interrupted with TCGA sequences every 20 repeats (16). Several similar constructs with different lengths of CTG repeats and/or promoters were developed (16, 52, 55, 56), however, the potential impact of these interruptions is not well defined yet. Additionally, constructs expressing short but pure repeats were also developed. Stable muscle cell lines expressing 200 CTG showed nuclear aggregates of mutated RNA that may cause disruptions in myogenic differentiation according to the 3′UTR-DMPK environments of the CTG repeats (57, 58). Overexpression of large pure repeats is more challenging due to their instability and technical issues associated with the cloning of long tracts of CTG repeats that substantially restricts their length (59, 60). However, some works partially overcame this constraint by expressing plasmids reaching 800 and 914 uninterrupted CTG repeats in the 3′UTR context of the DMPK gene or in inducible construct expressing GFP, respectively (10, 61–63).
In vitro cell models expressing exogenous CTG repeats have been widely used for small molecules screenings, therapeutic approaches, or molecular investigations (16, 54–56). However, these models may encounter some limitations associated with the level of CTG overexpression that is not under the control of the endogenous DMPK promoter and the absence of the complete DMPK genomic context that could limit their use in specific tissue or molecular mechanism studies.
DM1 Patients’ Derived Cell Models
Cells obtained directly from patients are of great utility in modeling human genetic disorders if they reproduce molecular hallmarks of the disease. Regarding DM1 patients’ derived cells, they express the whole range of mutation lengths observed in affected individuals within their natural genomic context and reproduce other canonical features of the disease (Figure 1) such as CUGexp-RNA foci that colocalize with the MBNL family members (45, 46, 64–70), alternative splicing misregulations (48, 65, 67, 70–73), and alterations of metabolic pathways (74, 75). However, considering variable parameters, like for instance samples from patients with different forms of the disease (from congenital to adult), culture conditions, or replicative senescence of primary cells, one should be aware of experimental variability between them. The generation of patient’s cell lines or stem cells with their vast reprogramming abilities represent additional tools for deciphering molecular DM1 pathogenesis but also for translational research including drugs screening (72, 76) and therapeutics development (65, 70, 77–79).
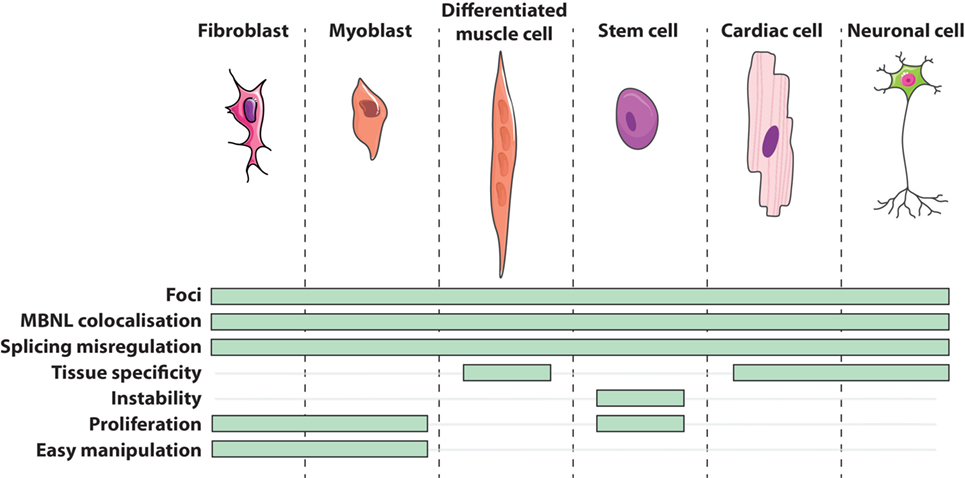
Figure 1. Features of DM1 cells in vitro.
Human Primary Cells
Currently in the DM1 field, primary cell cultures mainly consist of dermal fibroblasts and skeletal muscle cells, also called myoblasts. Isolated directly from patient’s tissue by either an enzymatic digestion of the extracellular matrix or an explant technique (small sliced pieces of biopsy from which cells grow out), primary cells potentially maintain physiological characteristics of their origin tissue environment. Primary fibroblasts are often used because of their relative accessibility from patients and their ease of manipulation in culture. On the other hand, primary myoblasts have the advantage to initiate a myogenic differentiation that results in their fusion into multinucleated cells or myotubes (Figure 1). Concomitantly, the expression of the DMPK gene is upregulated during the myogenic differentiation process (39) and differentiated DM1 muscle cells conserve some features found in DM1 muscles such as alternative splicing misregulation of muscle-specific transcripts (49, 78–88). Given the relative difficulty to have access to muscle biopsies of dystrophic patients, an alternative source of muscle-like cells was designed. Thus, primary fibroblasts were transduced with a viral construct expressing the key myogenic factor MYOD1 to force the expression of the myogenic program. This trans-differentiation leads to the formation of differentiated muscle-like cells expressing muscle-specific transcripts presenting similar splicing abnormalities that those found in DM1 primary myoblasts and patients’ muscles (18, 89, 90).
However, working with primary cells has also some constraints. Asides from the limited accessibility and availability of biopsies from patients, all somatic cells enter into replicative senescence after a define number of divisions that is inversely correlated with the age of the donor (91, 92). This phenomenon is even more pronounced in DM cells as their proliferative capacity is reduced when compared with age-matched control due to a premature entry into replicative senescence (81). Another difficulty, except the lengths of the repeats itself, is that primary cells could reflect the variability of the individual they are isolated from. Indeed, the age of the donor (fetal vs. adult), the tissue origin (distal vs. proximal muscle), and impairment, and the severity of the patient symptoms could influence cells behavior when grown in culture. In addition, various optimizations of cell cultures and medias or manipulation conditions may potentially lead to discrepancies between results.
Immortalized Human Fibroblast and Myoblasts
To circumvent the limitation of replicative senescence and keep the cells in a proliferative state, immortalized cell lines from DM1 primary fibroblasts, trans-differentiated fibroblasts, and myoblasts have been established (Figure 2) (71, 76, 93–95). The immortalization process of human fibroblasts requires the stable re-expression of the human telomerase (hTERT) to prevent the excessive shortening of telomeres that triggers the entry in replicative senescence. Additional inhibition of the dominant p16 pathway by overexpressing CDK4 (the natural ligand of p16) in association, or not, with CCND1 is needed for the immortalization of human myoblasts (96–98). As a result, the immortalized DM1 cell lines display potentially unlimited number of divisions while keeping most of the tissue- and disease-specific characteristics. Furthermore, clonal selection leads to homogeneous cell cultures, which allow to provide more consistent and reproducible results. Immortalized cells due to their unlimited lifespan are of special interest when considerably large amounts of cellular material is needed, e.g., for high-throughput screenings (54, 76, 99).
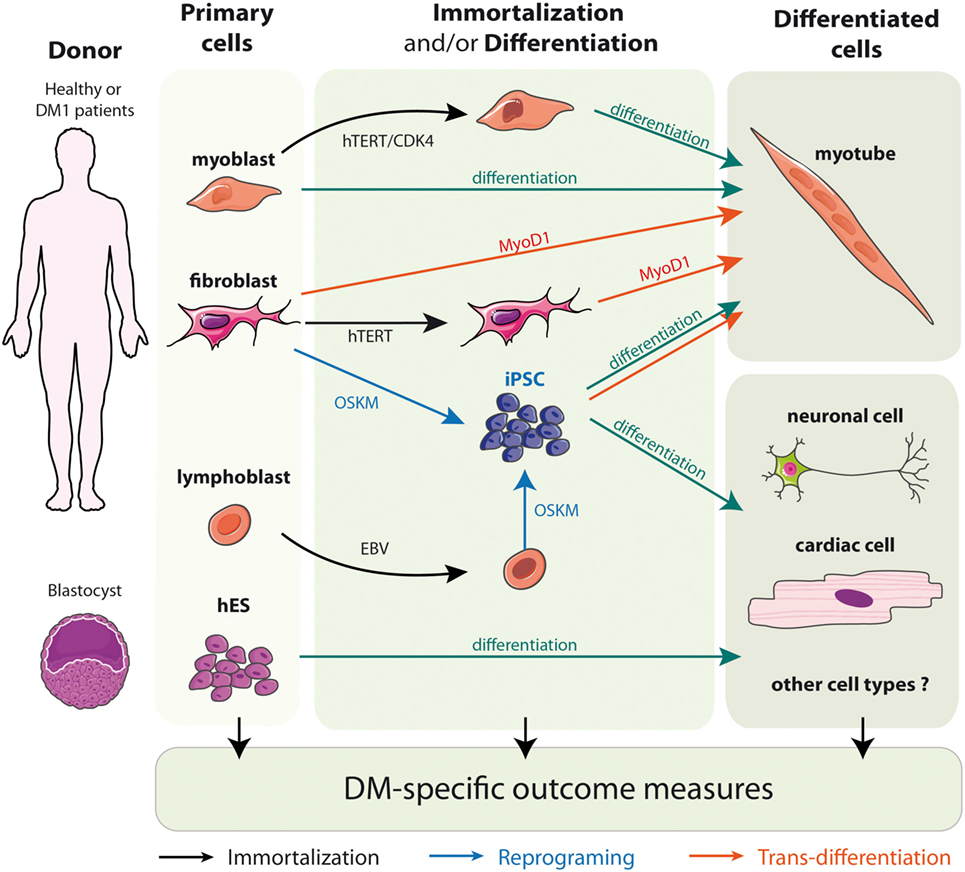
Figure 2. Models of DM1 patients’ derived cells.
Although the immortalized cell lines present a high value, it is not fully determined yet whether the immortalization process that requires viral transduction for genomic integration and stable expression of hTERT and CDK4 transgenes has any consequences on cellular behavior. Further validations and the use of different cell lines might also be needed to determine whether increasing number of divisions may alter disease- or tissues-characteristics of immortalized cells.
Human Pluripotent Stem Cells (hPSCs)
Cultures of some primary cells types like human neuronal cells constitute a major challenge due to limited biopsies availability and delay of tissue harvesting. The difficulty to obtain material reflecting early stages of the disease process also represents considerable limitations for disease investigations (100). Fortunately, the ability to generate defined cell types from hPSCs offers a unique opportunity to study disease mechanism in a cell-specific manner. hPSCs comprising both embryonic and induced pluripotent stem cells (iPSCs), carry the potential to differentiate them into a wide spectrum of cell types in vitro, including the recapitulation of early human embryo development states (101–103). Therefore, they provide an attractive prospect for modeling cell-type-specific disorders.
Embryonic stem cells (hESCs) are isolated from the inner mass of the blastocyst, and can be distinguished by their remarkable long-term proliferative potential along with the ability to differentiate into practically any cell type (Figure 2) (102). Although hESCs allow the generation of diverse cell types, up to date most of the work in the DM1 field has been performed in neural stem cells. Lately, they proved to be additionally useful in identification of new pathways misregulated in the context of DM1 mutation-like disruptions in mTor signaling or defective neuritogenesis (64, 67). However, hESC research raises several ethical issues and has been a subject of controversy over the past 15 years. To obviate the concerns of hESCs, a more recent method of generating patient-specific cells has arisen. It is built on the discovery that somatic cell nuclei can be “reprogrammed” to an embryonic-like state. The whole process begins for obtaining somatic cells, e.g., fibroblasts, from the affected individuals, which are then subjected to delivery of reprogramming factor cocktails (Figure 2). Such modified cells are referred to as iPSCs (101). Most importantly, this approach can be applied to human somatic cells, offering a unique opportunity to derive patient-specific cell lines from readily available material (100, 101). Several hiPSc derived from primary fibroblasts of DM patients have already been described (65, 73, 104–107). Interestingly, the ability to reprogram DM1 immortalized lymphoblastic cells carrying 200 CTG repeats into hiPSC also opens the possibility to obtain, in the near future, hiPSC directly from patient’s blood samples rather than skin biopsies (106).
Intriguingly, regarding the pluripotent state of the cells, CTG repeats are highly unstable during both reprogramming and subsequent passages, with a more rapid expansion when the initial CTG tract is longer (108). Contradictory to what is observed in vivo, several reports have shown CTG repeats instability during culture of undifferentiated DM1-hPSCs but not in differentiated cells, which might be related to some epigenetic differences in these cells (73, 108, 109). In DM1 maternally derived hESC lines, hypermethylation occurs upstream of CTG repeats when repeats number exceeded 300, however, the hypermethylation observed during reprogramming of patients fibroblasts into hiPSCs is not associated with the expansion of CTG repeats (110, 111).
In the past years, a substantial progress in the culture and differentiation technologies associated with hiPSC has been done. Comparing to tissue harvesting, hiPSCs have the advantage of a nearly endless supply. They might be expanded to large quantities and stored for a future expansion or manipulation. Nevertheless, the equivalence of iPS-derived cells to mature in vivo cells might vary because they often do not capture the entire mature phenotype. Also, other issues such as homogenous culture of differentiated cells, chromosomal rearrangement during reprogramming and relatively high cost of hPSC maintenance, constitute additional challenges. However, this technology offers a unique opportunity to investigate specific human disease cell types such as neuronal cells or cardiomyocytes for which there are none other or highly limited biological resources.
DM1 Cell Models as a Tool for the Development of Therapeutic Approaches
In vitro studies using DM1 cell models contribute also to the development of therapeutic approaches for myotonic dystrophies. Comprehensive studies have been performed in various DM1 cell models to determine and support translational potential of new strategies. Thus, different approaches aim to degrade mutant DMPK mRNAs have been tested in DM1 derived cells including gapmer antisense oligonucleotides (ASOs) directed against the CUGexp repeats (55, 112) or the DMPK transcript it-self (113) as well as shRNA (114), which have showed significant efficacy in decreasing the level of CUGexp-transcripts. In another hand, CUGexp-steric blocking approaches by using fully modified ASOs or viral-derived antisense RNA proved also to be effective in reversal disease molecular features when tested in DM1 cellular models (77, 78, 112, 115, 116). As a matter of fact, DM1 cells are not only used for therapeutic compounds validation. Indeed this tool is also utilized in screening assays allowing the identification of molecules that either interfere with the abnormal MBNL1:CUGexp interaction such as pentamidine (117, 118) or lomofungin (119), reduce the expression of mutant DMPK mRNAs like actinomycin D, modulate splicing changes (72) or affect the behavior of nuclear foci (120–122). Besides, reliable DM1 cell models are essential in the perspective of the recent progress made in genome engineering. TALEN and CRISPR-Cas approaches are being successfully applied in different disease cellular models giving rise to a wide range of possibilities for future therapeutic interventions (65, 70, 95, 123–125). DM1 cell models constitute, therefore, an inescapable source and a flexible platform for thorough studies and validation of disease therapeutics.
Conclusion
Through the years, cultured cells showed to be an essential model for both fundamental and translational research on myotonic dystrophy. Despite the fact that cells do not reflect the complexity of a whole organ or body, each cellular model, from the patients’ derived cells to more artificial models overexpressing CTG expanded tracts, is suitable for different investigations. They were and are used in many studies addressing various questions related to myotonic dystrophy diseases like mutation lengths, instability, polymorphisms or tissue-specific mechanisms, molecular alterations, and effect of therapeutic approaches. Understanding all of those features paved the way to decipher molecular basics of DM1 and DM2, as both forms share common features, i.e., abnormal expansion of repeated sequences, formation of RNA-positive foci and trans-dominant effect on alternative splicing. Even though DM1 cells served as archetype for DM research, it is noteworthy that some cellular models have been also established for myotonic dystrophy type 2. Further investigations in those cells may emphasize the differences between both DM forms and promote better understanding of their pathological mechanisms. Besides, recent advances in cell-availability and -engineering have given rise to unprecedented experimental opportunities to study disease mechanisms and therapeutic strategies. Late genome engineering tools, with the particular use of emerging development of TALENs and the CRISPR–Cas9 systems, facilitate the next generation of therapeutic interventions and hold a great promise for permanent genetic corrections (65, 70, 95, 123–125). Furthermore, it opens the door for the development of isogenic cell lines providing a genetically matched “control cells.” Alternate possibilities brought by genome editing tools combined with hiPSC technologies promise the generation of novel tissue-specific cell lines opening new horizons for the development of more refined wide-ranging myotonic dystrophy cell models, which will push forward future disease investigations.
Author Contributions
MM, AK, FR and DF wrote the review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by Sorbonne Université, Inserm, CNRS and Association Institut de Myologie.
References
2. Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3’ end of a transcript encoding a protein kinase family member. Cell (1992) 69(2):385.
3. Fu YH, Pizzuti A, Fenwick RG Jr, King J, Rajnarayan S, Dunne PW, et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science (1992) 255(5049):1256–8. doi:10.1126/science.1546326
4. Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3’ untranslated region of the gene. Science (1992) 255(5049):1253–5. doi:10.1126/science.1546325
5. Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science (2001) 293(5531):864–7. doi:10.1126/science.1062125
6. Batra R, Charizanis K, Manchanda M, Mohan A, Li M, Finn DJ, et al. Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol Cell (2014) 56(2):311–22. doi:10.1016/j.molcel.2014.08.027
7. Du H, Cline MS, Osborne RJ, Tuttle DL, Clark TA, Donohue JP, et al. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat Struct Mol Biol (2010) 17(2):187–93. doi:10.1038/nsmb.1720
8. Ebralidze A, Wang Y, Petkova V, Ebralidse K, Junghans RP. RNA leaching of transcription factors disrupts transcription in myotonic dystrophy. Science (2004) 303(5656):383–7. doi:10.1126/science.1088679
9. Fernandez-Costa JM, Llamusi MB, Garcia-Lopez A, Artero R. Alternative splicing regulation by muscleblind proteins: from development to disease. Biol Rev Camb Philos Soc (2011) 86(4):947–58. doi:10.1111/j.1469-185X.2011.00180.x
10. Huichalaf C, Sakai K, Jin B, Jones K, Wang GL, Schoser B, et al. Expansion of CUG RNA repeats causes stress and inhibition of translation in myotonic dystrophy 1 (DM1) cells. FASEB J (2010) 24(10):3706–19. doi:10.1096/fj.09-151159
11. Rau F, Freyermuth F, Fugier C, Villemin J-P, Fischer M-C, Jost B, et al. Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat Struct Mol Biol (2011) 18(7):840–5. doi:10.1038/nsmb.2067
12. Charlet BN, Savkur RS, Singh G, Philips AV, Grice EA, Cooper TA. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol Cell (2002) 10(1):45–53. doi:10.1016/S1097-2765(02)00572-5
13. Freyermuth F, Rau F, Kokunai Y, Linke T, Sellier C, Nakamori M, et al. Splicing misregulation of SCN5A contributes to cardiac-conduction delay and heart arrhythmia in myotonic dystrophy. Nat Commun (2016) 7:11067. doi:10.1038/ncomms11067
14. Fugier C, Klein AF, Hammer C, Vassilopoulos S, Ivarsson Y, Toussaint A, et al. Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat Med (2011) 17(6):720–5. doi:10.1038/nm.2374
15. Mankodi A, Takahashi MP, Jiang H, Beck CL, Bowers WJ, Moxley RT, et al. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell (2002) 10(1):35–44. doi:10.1016/S1097-2765(02)00563-4
16. Philips AV, Timchenko LT, Cooper TA. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science (1998) 280(5364):737–41. doi:10.1126/science.280.5364.737
17. Rau F, Laine J, Ramanoudjame L, Ferry A, Arandel L, Delalande O, et al. Abnormal splicing switch of DMD’s penultimate exon compromises muscle fibre maintenance in myotonic dystrophy. Nat Commun (2015) 6:7205. doi:10.1038/ncomms8205
18. Savkur RS, Philips AV, Cooper TA. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet (2001) 29(1):40–7. doi:10.1038/ng704
19. Hartwig GB, Miller SE, Frost AP, Roses AD. Myotonic muscular dystrophy: morphology, histochemistry, and growth characteristics of cultured skin fibroblasts. Muscle Nerve (1982) 5(2):125–30. doi:10.1002/mus.880050208
20. Lo Curto F, Castello A, Magrini U, Nappi G. Cytochemistry of cultured fibroblasts in myotonic muscular dystrophy. J Genet Hum (1975) 23(Suppl):173–8.
21. Swift MR, Finegold MJ. Myotonic muscular dystrophy: abnormalities in fibroblast culture. Science (1969) 165(3890):294–5. doi:10.1126/science.165.3890.294
22. Wertz RL, Hartwig GB, Frost AP, Brophy JJ, Atwater SK, Roses AD. Patients with myotonic dystrophy, a possible segmental progeroid syndrome, and duchenne muscular dystrophy have fibroblasts with normal limits for in vitro lifespan and growth characteristics. J Cell Physiol (1981) 107(2):255–60. doi:10.1002/jcp.1041070212
23. Ohno K, Nakano C, Eda I, Takeshita K. Fibroblasts from patients with myotonic muscular dystrophy: cholesterol requirement for proliferation and sensitivity to polyene antibiotics. Brain Dev (1984) 6(6):566–70. doi:10.1016/S0387-7604(84)80102-3
24. Ohno K, Nakano C, Ishii S, Takeshita K. Cytotoxicity of lectins toward skin fibroblasts from patients with Duchenne muscular dystrophy and myotonic dystrophy. Jinrui Idengaku Zasshi (1986) 31(2):85–91. doi:10.1007/BF01871402
25. Sakai T, Antoku Y. Decreased plasmalogen ratios in cultured skin fibroblasts from myotonic dystrophy. Muscle Nerve (1987) 10(7):626–8. doi:10.1002/mus.880100707
26. Thomas NS, Harper PS. Myotonic dystrophy: studies on the lipid composition and metabolism of erythrocytes and skin fibroblasts. Clin Chim Acta (1978) 83(1–2):13–23. doi:10.1016/0009-8981(78)90201-2
27. Hudson AJ, Huff MW, Wright CG, Silver MM, Lo TC, Banerjee D. The role of insulin resistance in the pathogenesis of myotonic muscular dystrophy. Brain (1987) 110(Pt 2):469–88. doi:10.1093/brain/110.2.469
28. Kakehi T, Kuzuya H, Kosaki A, Yamada K, Yoshimasa Y, Okamoto M, et al. Binding activity and autophosphorylation of the insulin receptor from patients with myotonic dystrophy. J Lab Clin Med (1990) 115(6):688–95.
29. Lam L, Hudson AJ, Strickland KP, Tevaarwerk GJ. Insulin binding to myotonic dystrophy fibroblasts. J Neurol Sci (1983) 58(2):289–95. doi:10.1016/0022-510X(83)90223-X
30. Nakano C, Ohno K. Insulin binding and internalization in cultured fibroblasts from myotonic muscular dystrophy. J Neurol Sci (1987) 80(2–3):229–35. doi:10.1016/0022-510X(87)90157-2
31. Buxton J, Shelbourne P, Davies J, Jones C, Perryman MB, Ashizawa T, et al. Characterization of a YAC and cosmid contig containing markers tightly linked to the myotonic dystrophy locus on chromosome 19. Genomics (1992) 13(3):526–31. doi:10.1016/0888-7543(92)90120-H
32. Shutler G, Korneluk RG, Tsilfidis C, Mahadevan M, Bailly J, Smeets H, et al. Physical mapping and cloning of the proximal segment of the myotonic dystrophy gene region. Genomics (1992) 13(3):518–25. doi:10.1016/0888-7543(92)90119-D
33. Carango P, Noble JE, Marks HG, Funanage VL. Absence of myotonic dystrophy protein kinase (DMPK) mRNA as a result of a triplet repeat expansion in myotonic dystrophy. Genomics (1993) 18(2):340–8. doi:10.1006/geno.1993.1474
34. Sabouri LA, Mahadevan MS, Narang M, Lee DS, Surh LC, Korneluk RG. Effect of the myotonic dystrophy (DM) mutation on mRNA levels of the DM gene. Nat Genet (1993) 4(3):233–8. doi:10.1038/ng0793-233
35. Krahe R, Ashizawa T, Abbruzzese C, Roeder E, Carango P, Giacanelli M, et al. Effect of myotonic dystrophy trinucleotide repeat expansion on DMPK transcription and processing. Genomics (1995) 28(1):1–14. doi:10.1006/geno.1995.1099
36. Taneja KL, McCurrach M, Schalling M, Housman D, Singer RH. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J Cell Biol (1995) 128(6):995–1002. doi:10.1083/jcb.128.6.995
37. Davis BM, McCurrach ME, Taneja KL, Singer RH, Housman DE. Expansion of a CUG trinucleotide repeat in the 3’ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc Natl Acad Sci U S A (1997) 94(14):7388–93. doi:10.1073/pnas.94.14.7388
38. Hamshere MG, Newman EE, Alwazzan M, Athwal BS, Brook JD. Transcriptional abnormality in myotonic dystrophy affects DMPK but not neighboring genes. Proc Natl Acad Sci U S A (1997) 94(14):7394–9. doi:10.1073/pnas.94.14.7394
39. Furling D, Lemieux D, Taneja K, Puymirat J. Decreased levels of myotonic dystrophy protein kinase (DMPK) and delayed differentiation in human myotonic dystrophy myoblasts. Neuromuscul Disord (2001) 11(8):728–35. doi:10.1016/S0960-8966(01)00226-7
40. Roberts R, Timchenko NA, Miller JW, Reddy S, Caskey CT, Swanson MS, et al. Altered phosphorylation and intracellular distribution of a (CUG)n triplet repeat RNA-binding protein in patients with myotonic dystrophy and in myotonin protein kinase knockout mice. Proc Natl Acad Sci U S A (1997) 94(24):13221–6. doi:10.1073/pnas.94.24.13221
41. Timchenko LT, Miller JW, Timchenko NA, DeVore DR, Datar KV, Lin L, et al. Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucleic Acids Res (1996) 24(22):4407–14. doi:10.1093/nar/24.22.4407
42. Timchenko LT, Timchenko NA, Caskey CT, Roberts R. Novel proteins with binding specificity for DNA CTG repeats and RNA CUG repeats: implications for myotonic dystrophy. Hum Mol Genet (1996) 5(1):115–21. doi:10.1093/hmg/5.1.115
43. Timchenko NA, Cai ZJ, Welm AL, Reddy S, Ashizawa T, Timchenko LT. RNA CUG repeats sequester CUGBP1 and alter protein levels and activity of CUGBP1. J Biol Chem (2001) 276(11):7820–6. doi:10.1074/jbc.M005960200
44. Kuyumcu-Martinez NM, Wang GS, Cooper TA. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol Cell (2007) 28(1):68–78. doi:10.1016/j.molcel.2007.07.027
45. Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, et al. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J (2000) 19(17):4439–48. doi:10.1093/emboj/19.17.4439
46. Fardaei M, Rogers MT, Thorpe HM, Larkin K, Hamshere MG, Harper PS, et al. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum Mol Genet (2002) 11(7):805–14. doi:10.1093/hmg/11.7.805
47. Konieczny P, Stepniak-Konieczna E, Sobczak K. MBNL proteins and their target RNAs, interaction and splicing regulation. Nucleic Acids Res (2014) 42(17):10873–87. doi:10.1093/nar/gku767
48. Buj-Bello A, Furling D, Tronchère H, Laporte J, Lerouge T, Butler-Browne GS, et al. Muscle-specific alternative splicing of myotubularin-related 1 gene is impaired in DM1 muscle cells. Hum Mol Genet (2002) 11(19):2297–307. doi:10.1093/hmg/11.19.2297
49. Nakamori M, Kimura T, Fujimura H, Takahashi MP, Sakoda S. Altered mRNA splicing of dystrophin in type 1 myotonic dystrophy. Muscle Nerve (2007) 36(2):251–7. doi:10.1002/mus.20809
50. Sergeant N, Sablonniere B, Schraen-Maschke S, Ghestem A, Maurage CA, Wattez A, et al. Dysregulation of human brain microtubule-associated tau mRNA maturation in myotonic dystrophy type 1. Hum Mol Genet (2001) 10(19):2143–55. doi:10.1093/hmg/10.19.2143
51. Tang ZZ, Yarotskyy V, Wei L, Sobczak K, Nakamori M, Eichinger K, et al. Muscle weakness in myotonic dystrophy associated with misregulated splicing and altered gating of CaV1.1 calcium channel. Hum Mol Genet (2012) 21(6):1312–24. doi:10.1093/hmg/ddr568
52. Ho TH, Charlet BN, Poulos MG, Singh G, Swanson MS, Cooper TA. Muscleblind proteins regulate alternative splicing. EMBO J (2004) 23(15):3103–12. doi:10.1038/sj.emboj.7600300
53. Warf MB, Berglund JA. MBNL binds similar RNA structures in the CUG repeats of myotonic dystrophy and its pre-mRNA substrate cardiac troponin T. RNA (2007) 13(12):2238–51. doi:10.1261/rna.610607
54. Konieczny P, Selma-Soriano E, Rapisarda AS, Fernandez-Costa JM, Perez-Alonso M, Artero R. Myotonic dystrophy: candidate small molecule therapeutics. Drug Discov Today (2017) 22(11):1740–8. doi:10.1016/j.drudis.2017.07.011
55. Lee JE, Bennett CF, Cooper TA. RNase H-mediated degradation of toxic RNA in myotonic dystrophy type 1. Proc Natl Acad Sci U S A (2012) 109(11):4221–6. doi:10.1073/pnas.1117019109
56. Rolfsmeier ML, Lahue RS. Stabilizing effects of interruptions on trinucleotide repeat expansions in Saccharomyces cerevisiae. Mol Cell Biol (2000) 20(1):173–80. doi:10.1128/MCB.20.1.173-180.2000
57. Amack JD, Mahadevan MS. The myotonic dystrophy expanded CUG repeat tract is necessary but not sufficient to disrupt C2C12 myoblast differentiation. Hum Mol Genet (2001) 10(18):1879–87. doi:10.1093/hmg/10.18.1879
58. Amack JD, Paguio AP, Mahadevan MS. Cis and trans effects of the myotonic dystrophy (DM) mutation in a cell culture model. Hum Mol Genet (1999) 8(11):1975–84. doi:10.1093/hmg/8.11.1975
59. Kang S, Jaworski A, Ohshima K, Wells RD. Expansion and deletion of CTG repeats from human disease genes are determined by the direction of replication in E. coli. Nat Genet (1995) 10(2):213–8. doi:10.1038/ng0695-213
60. Kim S-H, Cai L, Pytlos MJ, Edwards SF, Sinden RR. Generation of long tracts of disease-associated DNA repeats. Biotechniques (2005) 38(2):247–53. doi:10.2144/05382RR01
61. Nakamori M, Pearson CE, Thornton CA. Bidirectional transcription stimulates expansion and contraction of expanded (CTG)*(CAG) repeats. Hum Mol Genet (2011) 20(3):580–8. doi:10.1093/hmg/ddq501
62. Nakamori M, Taylor K, Mochizuki H, Sobczak K, Takahashi MP. Oral administration of erythromycin decreases RNA toxicity in myotonic dystrophy. Ann Clin Transl Neurol (2016) 3(1):42–54. doi:10.1002/acn3.271
63. Sobczak K, Wheeler TM, Wang W, Thornton CA. RNA interference targeting CUG repeats in a mouse model of myotonic dystrophy. Mol Ther (2013) 21(2):380–7. doi:10.1038/mt.2012.222
64. Denis JA, Gauthier M, Rachdi L, Aubert S, Giraud-Triboult K, Poydenot P, et al. mTOR-dependent proliferation defect in human ES-derived neural stem cells affected by myotonic dystrophy type 1. J Cell Sci (2013) 126(8):1763–72. doi:10.1242/jcs.116285
65. Gao Y, Guo X, Santostefano K, Wang Y, Reid T, Zeng D, et al. Genome therapy of myotonic dystrophy type 1 iPS cells for development of autologous stem cell therapy. Mol Ther (2016) 24(8):1378–87. doi:10.1038/mt.2016.97
66. Holt I, Mittal S, Furling D, Butler-Browne GS, Brook JD, Morris GE. Defective mRNA in myotonic dystrophy accumulates at the periphery of nuclear splicing speckles. Genes Cells (2007) 12(9):1035–48. doi:10.1111/j.1365-2443.2007.01112.x
67. Marteyn A, Maury Y, Gauthier MM, Lecuyer C, Vernet R, Denis JA, et al. Mutant human embryonic stem cells reveal neurite and synapse formation defects in type 1 myotonic dystrophy. Cell Stem Cell (2011) 8(4):434–44. doi:10.1016/j.stem.2011.02.004
68. Xia G, Ashizawa T. Dynamic changes of nuclear RNA foci in proliferating DM1 cells. Histochem Cell Biol (2015) 6:1–8. doi:10.1007/s00418-015-1315-5
69. Xia G, Santostefano KE, Goodwin M, Liu J, Subramony SH, Swanson MS, et al. Generation of neural cells from DM1 induced pluripotent stem cells as cellular model for the study of central nervous system neuropathogenesis. Cell Reprogram (2013) 15(2):166–77. doi:10.1089/cell.2012.0086
70. Xia G, Gao Y, Jin S, Subramony S, Terada N, Ranum LPW, et al. Genome modification leads to phenotype reversal in human myotonic dystrophy type 1 iPS-cell derived neural stem cells. Stem Cells (2015) 33(6):1829–38. doi:10.1002/stem.1970
71. Arandel L, Polay Espinoza M, Matloka M, Bazinet A, De Dea Diniz D, Naouar N, et al. Immortalized human myotonic dystrophy muscle cell lines to assess therapeutic compounds. Dis Model Mech (2017) 10(4):487–97. doi:10.1242/dmm.027367
72. Laustriat D, Gide J, Barrault L, Chautard E, Benoit C, Auboeuf D, et al. In vitro and in vivo modulation of alternative splicing by the biguanide metformin. Mol Ther Nucleic Acids (2015) 4(11):e262. doi:10.1038/mtna.2015.35
73. Ueki J, Nakamori M, Nakamura M, Nishikawa M, Yoshida Y, Tanaka A, et al. Myotonic dystrophy type 1 patient-derived iPSCs for the investigation of CTG repeat instability. Sci Rep (2017) 7:42522. doi:10.1038/srep42522
74. Botta A, Malena A, Tibaldi E, Rocchi L, Loro E, Pena E, et al. MBNL142 and MBNL143 gene isoforms, overexpressed in DM1-patient muscle, encode for nuclear proteins interacting with Src family kinases. Cell Death Dis (2013) 4:e770. doi:10.1038/cddis.2013.291
75. Renna LV, Bosè F, Iachettini S, Fossati B, Saraceno L, Milani V, et al. Receptor and post-receptor abnormalities contribute to insulin resistance in myotonic dystrophy type 1 and type 2 skeletal muscle. PLoS One (2017) 12(9):e0184987. doi:10.1371/journal.pone.0184987
76. O’Leary DA, Vargas L, Sharif O, Garcia ME, Sigal YJ, Chow S-K, et al. HTS-compatible patient-derived cell-based assay to identify small molecule modulators of aberrant splicing in myotonic dystrophy type 1. Curr Chem Genomics (2010) 4:9–18. doi:10.2174/1875397301004010009
77. Francois V, Klein AF, Beley C, Jollet A, Lemercier C, Garcia L, et al. Selective silencing of mutated mRNAs in DM1 by using modified hU7-snRNAs. Nat Struct Mol Biol (2011) 18(1):85–7. doi:10.1038/nsmb.1958
78. Furling D, Doucet G, Langlois MA, Timchenko L, Belanger E, Cossette L, et al. Viral vector producing antisense RNA restores myotonic dystrophy myoblast functions. Gene Ther (2003) 10(9):795–802. doi:10.1038/sj.gt.3301955
79. Langlois MA, Lee NS, Rossi JJ, Puymirat J. Hammerhead ribozyme-mediated destruction of nuclear foci in myotonic dystrophy myoblasts. Mol Ther (2003) 7(5 Pt 1):670–80. doi:10.1016/S1525-0016(03)00068-6
80. Beaulieu D, Thebault P, Pelletier R, Chapdelaine P, Tarnopolsky M, Furling D, et al. Abnormal prostaglandin E2 production blocks myogenic differentiation in myotonic dystrophy. Neurobiol Dis (2012) 45(1):122–9. doi:10.1016/j.nbd.2011.06.014
81. Bigot A, Klein AF, Gasnier E, Jacquemin V, Ravassard P, Butler-Browne G, et al. Large CTG repeats trigger p16-dependent premature senescence in myotonic dystrophy type 1 muscle precursor cells. Am J Pathol (2009) 174(4):1435–42. doi:10.2353/ajpath.2009.080560
82. Cardani R, Mancinelli E, Giagnacovo M, Sansone V, Meola G. Ribonuclear inclusions as biomarker of myotonic dystrophy type 2, even in improperly frozen or defrozen skeletal muscle biopsies. Eur J Histochem (2009) 53(2):107–11. doi:10.4081/ejh.2009.107
83. Furling D, Coiffier L, Mouly V, Barbet JP, St Guily JL, Taneja K, et al. Defective satellite cells in congenital myotonic dystrophy. Hum Mol Genet (2001) 10(19):2079–87. doi:10.1093/hmg/10.19.2079
84. Furling D, Lam LT, Agbulut O, Butler-Browne GS, Morris GE. Changes in myotonic dystrophy protein kinase levels and muscle development in congenital myotonic dystrophy. Am J Pathol (2003) 162(3):1001–9. doi:10.1016/S0002-9440(10)63894-1
85. Loro E, Rinaldi F, Malena A, Masiero E, Novelli G, Angelini C, et al. Normal myogenesis and increased apoptosis in myotonic dystrophy type-1 muscle cells. Cell Death Differ (2010) 17(8):1315–24. doi:10.1038/cdd.2010.33
86. Nakamori M, Sobczak K, Puwanant A, Welle S, Eichinger K, Pandya S, et al. Splicing biomarkers of disease severity in myotonic dystrophy. Ann Neurol (2013) 74(6):862–72. doi:10.1002/ana.23992
87. Perdoni F, Malatesta M, Cardani R, Giagnacovo M, Mancinelli E, Meola G, et al. RNA/MBNL1-containing foci in myoblast nuclei from patients affected by myotonic dystrophy type 2: an immunocytochemical study. Eur J Histochem (2009) 53(3):e18. doi:10.4081/ejh.2009.e18
88. Nakamori M, Kimura T, Kubota T, Matsumura T, Sumi H, Fujimura H, et al. Aberrantly spliced alpha-dystrobrevin alters alpha-syntrophin binding in myotonic dystrophy type 1. Neurology (2008) 70(9):677–85. doi:10.1212/01.wnl.0000302174.08951.cf
89. Kuyumcu-Martinez NM, Cooper TA. Misregulation of alternative splicing causes pathogenesis in myotonic dystrophy. Prog Mol Subcell Biol (2006) 44:133–59. doi:10.1007/978-3-540-34449-0_7
90. Ravel-Chapuis A, Bélanger G, Yadava RS, Mahadevan MS, DesGroseillers L, Côté J, et al. The RNA-binding protein Staufen1 is increased in DM1 skeletal muscle and promotes alternative pre-mRNA splicing. J Cell Biol (2012) 196(6):699–712. doi:10.1083/jcb.201108113
91. Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res (1965) 37:614–36. doi:10.1016/0014-4827(65)90211-9
92. Renault V, Piron-Hamelin G, Forestier C, DiDonna S, Decary S, Hentati F, et al. Skeletal muscle regeneration and the mitotic clock. Exp Gerontol (2000) 35(6–7):711–9. doi:10.1016/S0531-5565(00)00151-0
93. Larsen J, Pettersson OJ, Jakobsen M, Thomsen R, Pedersen CB, Hertz JM, et al. Myoblasts generated by lentiviral mediated MyoD transduction of myotonic dystrophy type 1 (DM1) fibroblasts can be used for assays of therapeutic molecules. BMC Res Notes (2011) 4:490. doi:10.1186/1756-0500-4-490
94. Pantic B, Borgia D, Giunco S, Malena A, Kiyono T, Salvatori S, et al. Reliable and versatile immortal muscle cell models from healthy and myotonic dystrophy type 1 primary human myoblasts. Exp Cell Res (2016) 342(1):39–51. doi:10.1016/j.yexcr.2016.02.013
95. Provenzano C, Cappella M, Valaperta R, Cardani R, Meola G, Martelli F, et al. CRISPR/Cas9-mediated deletion of CTG expansions recovers normal phenotype in myogenic cells derived from myotonic dystrophy 1 patients. Mol Ther Nucleic Acids (2017) 9:337–48. doi:10.1016/j.omtn.2017.10.006
96. Mamchaoui K, Trollet C, Bigot A, Negroni E, Chaouch S, Wolff A, et al. Immortalized pathological human myoblasts: towards a universal tool for the study of neuromuscular disorders. Skelet Muscle (2011) 1:34. doi:10.1186/2044-5040-1-34
97. Shiomi K, Kiyono T, Okamura K, Uezumi M, Goto Y, Yasumoto S, et al. CDK4 and cyclin D1 allow human myogenic cells to recapture growth property without compromising differentiation potential. Gene Ther (2011) 18(9):857–66. doi:10.1038/gt.2011.44
98. Zhu C-H, Mouly V, Cooper RN, Mamchaoui K, Bigot A, Shay JW, et al. Cellular senescence in human myoblasts is overcome by human telomerase reverse transcriptase and cyclin-dependent kinase 4: consequences in aging muscle and therapeutic strategies for muscular dystrophies. Aging Cell (2007) 6(4):515–23. doi:10.1111/j.1474-9726.2007.00306.x
99. Ketley A, Chen CZ, Li X, Arya S, Robinson TE, Granados-Riveron J, et al. High-content screening identifies small molecules that remove nuclear foci, affect MBNL distribution and CELF1 protein levels via a PKC-independent pathway in myotonic dystrophy cell lines. Hum Mol Genet (2014) 23(6):1551–62. doi:10.1093/hmg/ddt542
100. Han SSW, Williams LA, Eggan KC. Constructing and deconstructing stem cell models of neurological disease. Neuron (2011) 70(4):626–44. doi:10.1016/j.neuron.2011.05.003
101. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell (2006) 126(4):663–76. doi:10.1016/j.cell.2006.07.024
102. Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, et al. Embryonic stem cell lines derived from human blastocysts. Science (1998) 282(5391):1145–7. doi:10.1126/science.282.5391.1145
103. Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science (2007) 318(5858):1917–20. doi:10.1126/science.1151526
104. Dumevska B, Main H, McKernan R, Goel D, Schmidt U. Derivation of DM1 affected human embryonic stem cell line Genea067. Stem Cell Res (2016) 16(2):437–9. doi:10.1016/j.scr.2016.02.014
105. Kalra S, Montanaro F, Denning C. Can human pluripotent stem cell-derived cardiomyocytes advance understanding of muscular dystrophies? J Neuromuscul Dis (2016) 3(3):309–32. doi:10.3233/JND-150133
106. Martineau L, Racine V, Benichou SA, Puymirat J. Lymphoblastoids cell lines-derived iPSC line from a 26-year-old myotonic dystrophy type 1 patient carrying (CTG)200 expansion in the DMPK gene: CHUQi001-A. Stem Cell Res (2018) 26:103–6. doi:10.1016/j.scr.2017.12.010
107. Mateizel I, De Temmerman N, Ullmann U, Cauffman G, Sermon K, Van de Velde H, et al. Derivation of human embryonic stem cell lines from embryos obtained after IVF and after PGD for monogenic disorders. Hum Reprod (2006) 21(2):503–11. doi:10.1093/humrep/dei345
108. Du J, Campau E, Soragni E, Jespersen C, Gottesfeld JM. Length-dependent CTG·CAG triplet-repeat expansion in myotonic dystrophy patient-derived induced pluripotent stem cells. Hum Mol Genet (2013) 22(25):5276–87. doi:10.1093/hmg/ddt386
109. De Temmerman N, Seneca S, Van Steirteghem A, Haentjens P, Van der Elst J, Liebaers I, et al. CTG repeat instability in a human embryonic stem cell line carrying the myotonic dystrophy type 1 mutation. Mol Hum Reprod (2008) 14(7):405–12. doi:10.1093/molehr/gan034
110. Barbé L, Lanni S, López-Castel A, Franck S, Spits C, Keymolen K, et al. CpG methylation, a parent-of-origin effect for maternal-biased transmission of congenital myotonic dystrophy. Am J Hum Genet (2017) 100(3):488–505. doi:10.1016/j.ajhg.2017.01.033
111. Yanovsky-Dagan S, Mor-Shaked H, Eiges R. Modeling diseases of noncoding unstable repeat expansions using mutant pluripotent stem cells. World J Stem Cells (2015) 7(5):823–38. doi:10.4252/wjsc.v7.i5.823
112. González-Barriga A, Mulders SA, van de Giessen J, Hooijer JD, Bijl S, van Kessel ID, et al. Design and analysis of effects of triplet repeat oligonucleotides in cell models for myotonic dystrophy. Mol Ther Nucleic Acids (2013) 2:e81. doi:10.1038/mtna.2013.9
113. Jauvin D, Chrétien J, Pandey SK, Martineau L, Revillod L, Bassez G, et al. Targeting DMPK with antisense oligonucleotide improves muscle strength in myotonic dystrophy type 1 mice. Mol Ther Nucleic Acids (2017) 7:465–74. doi:10.1016/j.omtn.2017.05.007
114. Langlois M-A, Boniface C, Wang G, Alluin J, Salvaterra PM, Puymirat J, et al. Cytoplasmic and nuclear retained DMPK mRNAs are targets for RNA interference in myotonic dystrophy cells. J Biol Chem (2005) 280(17):16949–54. doi:10.1074/jbc.M501591200
115. Mulders SA, van den Broek WJ, Wheeler TM, Croes HJ, van Kuik-Romeijn P, de Kimpe SJ, et al. Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc Natl Acad Sci U S A (2009) 106(33):13915–20. doi:10.1073/pnas.0905780106
116. Wojtkowiak-Szlachcic A, Taylor K, Stepniak-Konieczna E, Sznajder LJ, Mykowska A, Sroka J, et al. Short antisense-locked nucleic acids (all-LNAs) correct alternative splicing abnormalities in myotonic dystrophy. Nucleic Acids Res (2015) 43(6):3318–31. doi:10.1093/nar/gkv163
117. Coonrod LA, Nakamori M, Wang W, Carrell S, Hilton CL, Bodner MJ, et al. Reducing levels of toxic RNA with small molecules. ACS Chem Biol (2013) 8(11):2528–37. doi:10.1021/cb400431f
118. Warf MB, Nakamori M, Matthys CM, Thornton CA, Berglund JA. Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc Natl Acad Sci U S A (2009) 106(44):18551–6. doi:10.1073/pnas.0903234106
119. Hoskins JW, Ofori LO, Chen CZ, Kumar A, Sobczak K, Nakamori M, et al. Lomofungin and dilomofungin: inhibitors of MBNL1-CUG RNA binding with distinct cellular effects. Nucleic Acids Res (2014) 42(10):6591–602. doi:10.1093/nar/gku275
120. Siboni RB, Bodner MJ, Khalifa MM, Docter AG, Choi JY, Nakamori M, et al. Biological efficacy and toxicity of diamidines in myotonic dystrophy type 1 models. J Med Chem (2015) 58(15):5770–80. doi:10.1021/acs.jmedchem.5b00356
121. Pushechnikov A, Lee MM, Childs-Disney JL, Sobczak K, French JM, Thornton CA, et al. Rational design of ligands targeting triplet repeating transcripts that cause RNA dominant disease: application to myotonic muscular dystrophy type 1 and spinocerebellar ataxia type 3. J Am Chem Soc (2009) 131(28):9767–79. doi:10.1021/ja9020149
122. Childs-Disney JL, Parkesh R, Nakamori M, Thornton CA, Disney MD. Rational design of bioactive, modularly assembled aminoglycosides targeting the RNA that causes myotonic dystrophy type 1. ACS Chem Biol (2012) 7(12):1984–93. doi:10.1021/cb3001606
123. van Agtmaal EL, André LM, Willemse M, Cumming SA, van Kessel IDG, van den Broek WJAA, et al. CRISPR/Cas9-induced (CTG CAG)n repeat instability in the myotonic dystrophy type 1 locus: implications for therapeutic genome editing. Mol Ther (2017) 25(1):24–43. doi:10.1016/j.ymthe.2016.10.014
124. Batra R, Nelles DA, Pirie E, Blue SM, Marina RJ, Wang H, et al. Elimination of toxic microsatellite repeat expansion RNA by RNA-targeting Cas9. Cell (2017) 170(5):899–912.e10. doi:10.1016/j.cell.2017.07.010
Keywords: dm1, cells, cultured, CTG repeats, models, biological, pathophysiology, human cells
Citation: Matloka M, Klein AF, Rau F and Furling D (2018) Cells of Matter—In Vitro Models for Myotonic Dystrophy. Front. Neurol. 9:361. doi: 10.3389/fneur.2018.00361
Received: 16 March 2018; Accepted: 03 May 2018;
Published: 23 May 2018
Edited by:
Benedikt Schoser, Klinikum der Universität München, GermanyCopyright: © 2018 Matloka, Klein, Rau and Furling. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Denis Furling, denis.furling@sorbonne-universite.fr