Early Origin and Evolution of the Angelman Syndrome Ubiquitin Ligase Gene Ube3a
 Masaaki Sato1,2*
Masaaki Sato1,2*- 1Graduate School of Science and Engineering and Brain and Body System Science Institute, Saitama University, Saitama, Japan
- 2RIKEN Brain Science Institute, Wako, Japan
The human Ube3a gene encodes an E3 ubiquitin ligase and exhibits brain-specific genomic imprinting. Genetic abnormalities that affect the maternal copy of this gene cause the neurodevelopmental disorder Angelman syndrome (AS), which is characterized by severe mental retardation, speech impairment, seizure, ataxia and some unique behavioral phenotypes. In this review article, I highlight the evolution of the Ube3a gene and its imprinting to provide evolutionary insights into AS. Recent comparative genomic studies have revealed that Ube3a is most phylogenetically similar to HECTD2 among the human HECT (homologous to the E6AP carboxyl terminus) family of E3 ubiquitin ligases, and its distant evolutionary origin can be traced to common ancestors of fungi and animals. Moreover, a gene more similar to Ube3a than HECTD2 is found in a range of eukaryotes from amoebozoans to basal metazoans, but is lost in later lineages. Unlike in mice and humans, Ube3a expression is biallelic in birds, monotremes, marsupials and insects. The imprinting domain that governs maternal expression of Ube3a was formed from non-imprinted elements following multiple chromosomal rearrangements after diversification of marsupials and placental mammals. Hence, the evolutionary origins of Ube3a date from long before the emergence of the nervous system, although its imprinted expression was acquired relatively recently. These observations suggest that exogenous expression and functional analyses of ancient Ube3a orthologs in mammalian neurons will facilitate the evolutionary understanding of AS.
Introduction
The evolution of the brain in mammals is characterized by dramatic increases in size and complexity, especially in the neocortex (Striedter, 2005). Previous advances in comparative genomics have revealed emerging principles of the genetic basis of brain evolution (Khaitovich et al., 2006; Vallender et al., 2008). Changes in protein-coding sequences and regulatory elements as well as emergence of new genes and loss of existing ones likely had profound phenotypic impacts on brain development and ultimately led to significant alterations in brain structure and function. Thus, understanding how genes that play essential roles in human brain development and cognition evolved is of great importance and interest.
The ubiquitin ligase gene Ube3a (also known as E6-associated protein; E6AP) provides an excellent model for studies of gene evolution because of its brain-specific imprinting and implication in the neurodevelopmental disorder Angelman syndrome (AS). Ube3a is a homologous to the E6AP carboxyl terminus (HECT) domain-containing E3 ubiquitin ligase that was initially discovered as the protein involved in human papillomavirus E6-mediated p53 degradation (Huibregtse et al., 1993). It is expressed monoallelically from the maternal allele in the brain in a parent-of-origin specific manner (Albrecht et al., 1997; Rougeulle et al., 1997; Vu and Hoffman, 1997). The imprinting of Ube3a and its neighboring genes is coordinated by a regulatory region known as the Prader-Willi syndrome (PWS)-AS imprinting center (IC), which is located upstream of the adjacent SNURF (SNRPN upstream reading frame)–SNRPN (small nuclear ribonucleoprotein-associated protein N) gene on the human 15q11-q13 chromosome region (Buiting et al., 1999; Ohta et al., 1999; Perk et al., 2002). Genetic abnormalities that affect the maternal copy of Ube3a are known to cause AS, which is characterized by a wide variety of symptoms such as severe mental retardation, speech impairment, seizure, ataxia and unique behavioral phenotypes such as frequent laughter (Angelman, 1965; Williams et al., 1995; Kishino et al., 1997; Matsuura et al., 1997; Clayton-Smith and Laan, 2003; Mabb et al., 2011; Buiting et al., 2016), whereas duplication or increased expression of this gene is linked to autism spectrum disorders (Bolton et al., 2001; Glessner et al., 2009; Smith et al., 2011; Urraca et al., 2013). Accordingly, Ube3a is essential for neural circuit maturation and experience-dependent plasticity in the mammalian cerebral cortex (Yashiro et al., 2009; Sato and Stryker, 2010). In this review article, I highlight the evolution of the Ube3a gene and its imprinting to gain evolutionary insights into AS.
Ancient Origin of the Ube3a Gene
Ube3a contains a single HECT domain at the C-terminal and no discernible functional domain at its N-terminal side. Recent analyses revealed that the Ube3a protein is phylogenetically closest to HECTD2 among 28 human HECT domain-containing ubiquitin ligases, and a group of proteins called small HERCs (HERC3-6) that possess a single N-terminal RCC1-like domain (RLD) and a C-terminal HECT domain are also similar to Ube3a (Marín, 2010; Grau-Bové et al., 2013; Scheffner and Kumar, 2014; Figure 1A). HECTD2 and small HERCs are expressed in the brain, although their functions are not well understood (Sánchez-Tena et al., 2016). HECTD2 has been associated with susceptibility to neurological diseases (Lloyd et al., 2009a,b). Two other HECT E3 ligases, Ube3b and Ube3c, were named after Ube3a (Gong et al., 2003), but carry a calmodulin-binding IQ domain in addition to a HECT domain and are categorized as a distinct class of HECT E3 ligases (Figure 1A). Notably, Ube3b has been strongly implicated in the human developmental disorder blepharophimosis-ptosis-intellectual-disability syndrome (Basel-Vanagaite et al., 2012).
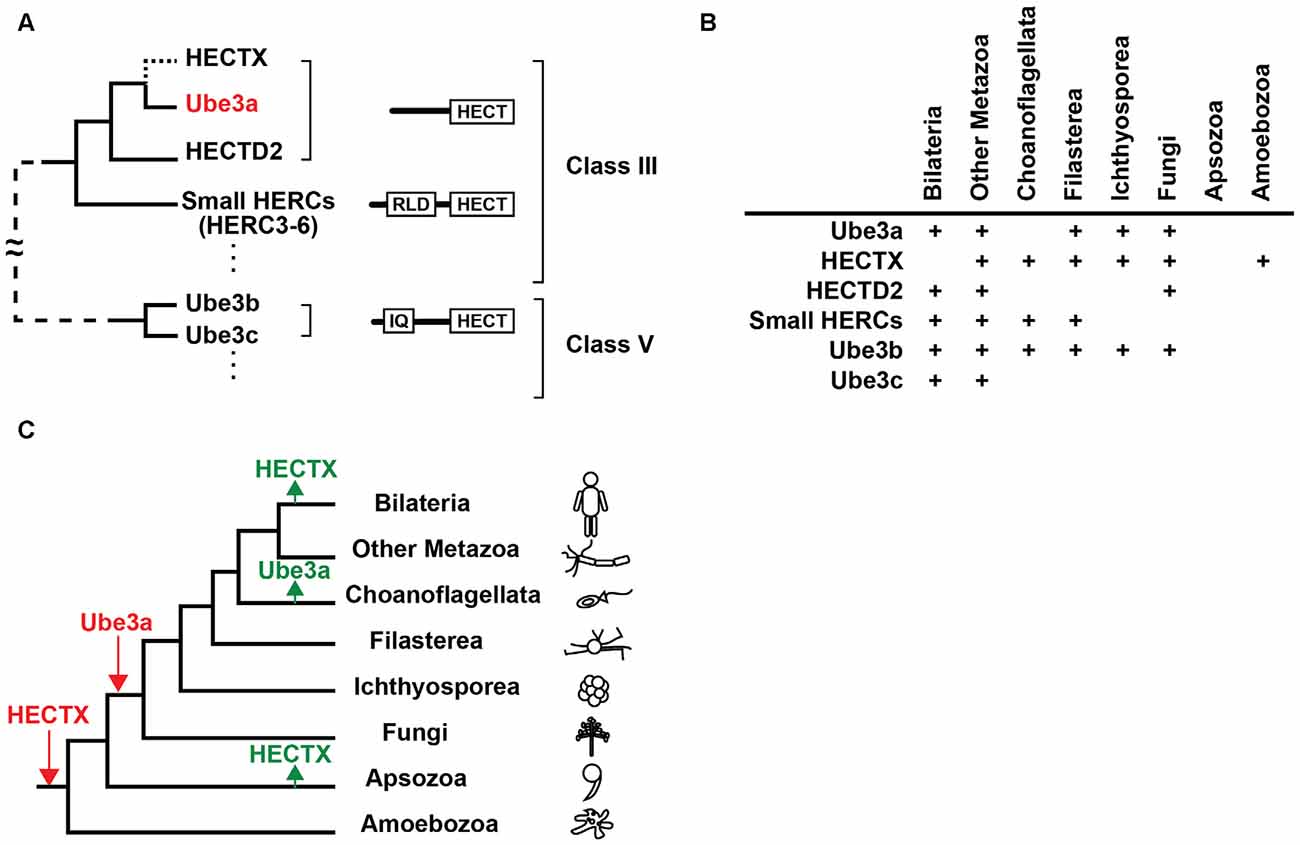
Figure 1. Evolutionary origin of Ube3a. (A) Phylogenetic relationships, domain structures, and classes of homologous to the E6AP carboxyl terminus (HECT) E3 ligases that are similar to Ube3a. The branch to HECTX that was lost in bilaterians is indicated by a dotted line. For a simplified diagram, branches that connect class III and class V E3 ligases are shown with a dashed line. IQ, IQ domain; RLD, RCC1-like domain. (B) Presence of orthologs of Ube3a, HECTX, HECTD2, small HERCs, Ube3b and Ube3c in eukaryotes. (C) Gains (red) and losses (green) of Ube3a and HECTX are overlaid on a eukaryotic tree of life. Figures were modified from Grau-Bové et al. (2013).
Orthologs of Ube3a are commonly found in bilaterians including vertebrates and Drosophila (Reiter et al., 2006; Wu et al., 2008; Hope et al., 2016) but not in Caenorhabditis elegans, implying that Ube3a was lost in some nematode lineages (Marín, 2010). The evolutionary origins of Ube3a can be traced to the genomes of basal metazoans, including the cnidarian Nematostella vectensis, the sponge Amphimedon queenslandica, and the placozoan Trichoplax adherens (Marín, 2010), and to fungi such as Mortierella verticillata (Grau-Bové et al., 2013; Figures 1B,C). However, no Ube3a orthologs are present in choanoflagellates, suggesting secondary loss in this lineage (Grau-Bové et al., 2013). Notably, the E3 ligase gene HECTX is more similar to Ube3a than HECTD2 and is found in the genomes of amoebozoans, fungi, choanoflagellates and early metazoans, but not in those of bilaterians (Marín, 2010; Grau-Bové et al., 2013; Figures 1A–C). These findings suggest that an ancient E3 ligase gene that was more similar to Ube3a than the extant HECTD2 was secondarily lost in the bilaterian lineage.
The evolution of Ube3a substrate specificity remains unclear. Several neuronal proteins have been identified to date as possible direct ubiquitination targets of Ube3a (Sell and Margolis, 2015; Sun et al., 2015). Although the precise modes of these protein interactions have not been characterized, specific substrate recognition by Ube3a is thought to be mediated by its non-catalytic N-terminal region (Cooper et al., 2004; Scheffner and Kumar, 2014). Whereas Ube3a orthologs share the conserved HECT domain at their C-termini, their N-terminal regions are more variable among lineages, suggesting that ancient Ube3a orthologs recognized differing sets of target proteins to those ubiquitinated by the present human Ube3a. New substrate specificity was likely acquired during evolution by changes in substrate binding regions and encounters of Ube3a with potential novel substrates, the latter of which were probably brought by changes in expression and subcellular localization, and the emergence of new proteins.
Ube3a and the Evolution of the Nervous System
The early origin of Ube3a indicates that it predates the origins of nerve cells and synapses. The expression and function of the Ube3a protein in primitive organisms are currently unclear. Whether the nervous system evolved from single or multiple independent origins remains controversial (Miller, 2009; Ryan et al., 2013; Moroz et al., 2014; Liebeskind et al., 2016). At the base of the metazoan tree, sponges and placozoans lack nerve and muscle cells, but exhibit coordinated behaviors such as feeding and contraction (Ellwanger et al., 2007; Smith et al., 2015). On the other hand, cnidarians and ctenophores have diffuse nervous systems called nerve nets, which communicate by synapses (Anderson and Spencer, 1989; Tamm and Tamm, 1995; Marlow et al., 2009). Centralized nervous systems evolved in the bilaterian lineage (Arendt et al., 2016).
Searches for orthologs of specific postsynaptic density (PSD) proteins demonstrate that the genomes of nerve-less basal metazoans and unicellular choanoflagellates contain core sets of scaffold protein orthologs, and these are co-expressed in a distinct cell type of Amphimedon larvae (Sakarya et al., 2007; Alié and Manuel, 2010). Shank postsynaptic scaffold proteins have been implicated in autism spectrum disorders in humans (Durand et al., 2007; Berkel et al., 2010; Sato et al., 2012) and are also found in the choanoflagellate genome (Alié and Manuel, 2010), providing another remarkable example of the ancient origins of genes that are involved in human developmental disorders. More recent studies showed that human PSD proteins that are essential for basic cellular processes, such as amino acid biosynthesis and energy generation, are conserved between prokaryotes and eukaryotes, whereas the majority of structural and signaling molecules, including those involved in ubiquitination, are specific to eukaryotes (Emes and Grant, 2011). The ancient eukaryotic origin of Ube3a is thus consistent with the early origins of human postsynaptic proteins, many of which are linked to neurogenetic disorders (Bayés et al., 2011).
Assembly of the PWS-AS Imprinted Domain
Ube3a expression is imprinted in the brain but not in peripheral tissues in humans and mice (Albrecht et al., 1997; Rougeulle et al., 1997; Vu and Hoffman, 1997). Moreover, Ube3a expression is imprinted in neurons but not in glial cells of the brain (Yamasaki et al., 2003; Judson et al., 2014). Interestingly, imprinting of Ube3a is not fully established in the postnatal mouse brain and paternal Ube3a expression decreases as neurons mature (Sato and Stryker, 2010; Judson et al., 2014). Hence, imprinted expression of Ube3a is tissue- and cell type-specific and is developmentally regulated.
Genomic imprinting, or parent-of-origin specific epigenetic gene silencing, is widespread in placental mammals and also occurs in marsupials, suggesting evolution from common ancestors of marsupials and eutherians (Renfree et al., 2009). Genome-wide characterization of imprinted genes revealed parent-of-origin allelic effects in over 1300 loci in embryonic and adult mouse brains (Gregg et al., 2010). However, hypotheses regarding the origins and evolutionary advantages of genomic imprinting are few. Among these, the host defense hypothesis proposes that genomic imprinting evolved from the cellular mechanisms that mediate methylation and silencing of foreign DNA elements (Barlow, 1993). Alternatively, the kinship theory suggests fitness advantages of genomic imprinting. Specifically, paternally and maternally expressed genes have been shown to increase and decrease the transfer of maternal nutrients to the fetus during pregnancy, respectively, as observed for the paternally expressed IGF2 growth factor and the maternally expressed IGF2 receptor (IGF2R) growth repressor (Haig, 2004). Another hypothesis, the coadaptation theory, proposes that genomic imprinting coordinates placental and hypothalamic functions of the fetus and mother to optimize growth, postnatal suckling and maternal care, as demonstrated by the paternal Peg3 transcription factor that is expressed in these tissues (Li et al., 1999; Curley et al., 2004).
A comparative genomic study revealed an unexpected picture of the assembly of the PWS-AS imprinted domain during evolution (Rapkins et al., 2006). In human chromosome 15q and homologous mouse chromosome 7C regions, Ube3a is located downstream of SNURF–SNRPN, which forms a bicistronic transcript and is expressed from the paternal allele. Maternal expression of Ube3a and paternal expression of SNURF–SNRPN are controlled by the IC that lies upstream of SNURF–SNRPN (Figure 2A). This arrangement is conserved in eutherians including mice and humans, but is not present in marsupials such as the gray short-tailed opossum Monodelphis domestica and other animals of greater evolutionary age. In these animals, the gene CNGA3 is present downstream of Ube3a instead of SNURF–SNRPN (Figure 2A). Accordingly, expression of Ube3a is biallelic in the marsupial tammar wallaby, the montreme platypus, and in chickens and Drosophila (Colosi et al., 2006; Rapkins et al., 2006; Hope et al., 2016). The searches for the marsupial ortholog of SNRPN revealed that it resides beside the closely related SNRPB gene in the Monodelphis domestica genome (Figure 2B). Furthermore, the genomes of evolutionarily older animals including monotremes have SNRPB but no SNRPN orthologs. These findings suggest that SNRPN was formed by tandem duplication of the evolutionally older SNRPB gene in marsupials.
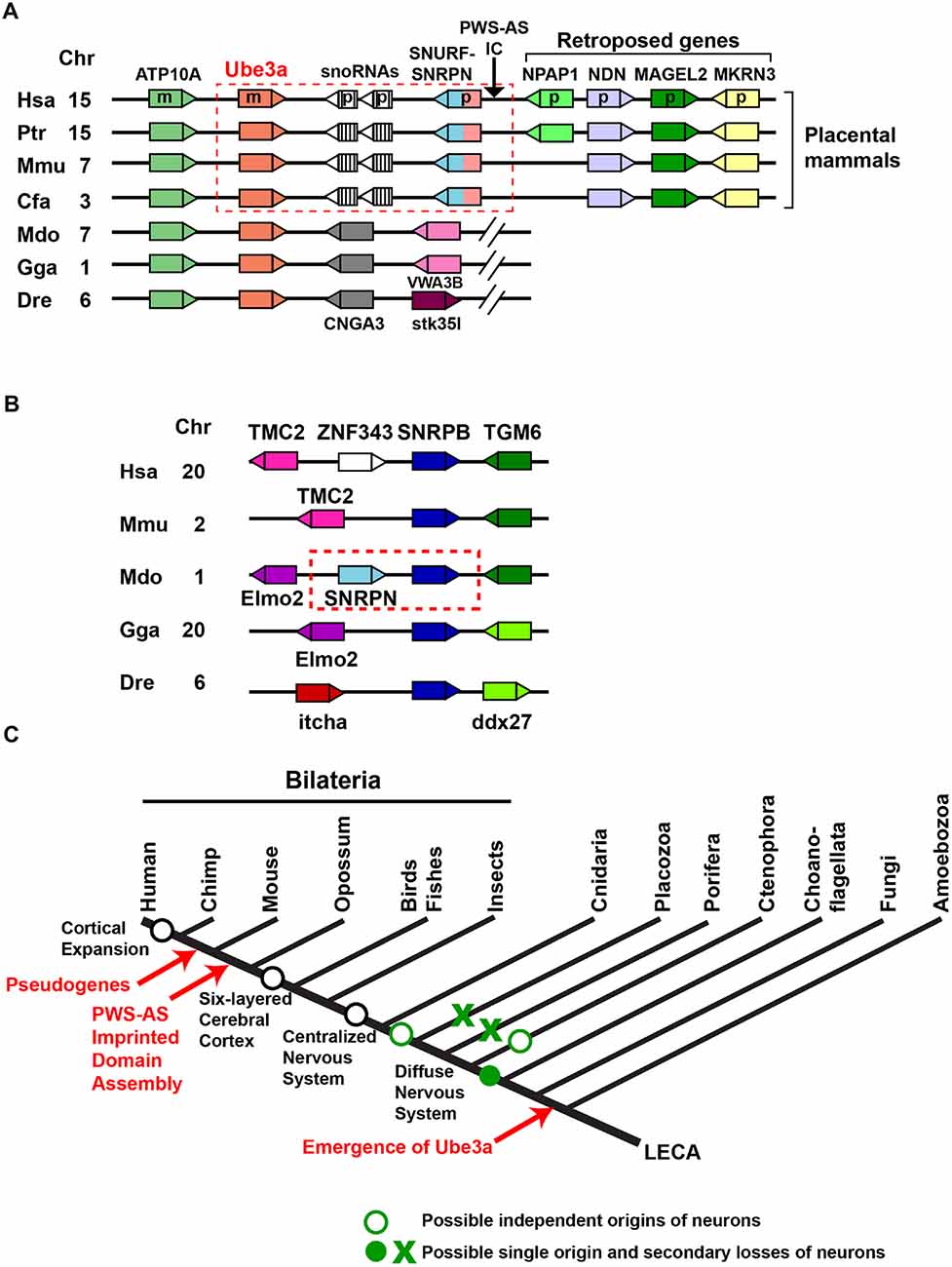
Figure 2. Assembly of the imprinted Ube3a locus in the genomes of placental mammals. (A) A schematic representation of arrangements of Ube3a in vertebrate genomes. Genes are represented as boxes, and triangles indicate their transcriptional directions. The same colors indicate the same genes across species. The conserved arrangement of Ube3a and SNRPN upstream reading frame (SNURF)–small nuclear ribonucleoprotein-associated protein N (SNRPN) in placental mammals is enclosed by a red dashed box. The arrow indicates the chromosomal position of the Prader-Willi syndrome-Angelman syndrome imprinting center (PWS-AS IC) located upstream of the SNURF–SNRPN gene in placental mammals. Hsa, Homo sapiens; Ptr, Pan troglodytes; Mmu, Mus musculus; Cfa, Canis lupus familiaris; Mdo, Monodelphis domestica; Gga, Gallus gallus; Dre, Danio rerio; Chr, chromosomal location in each species; m, maternally expressed gene in humans; p, paternally expressed gene in humans. (B) A schematic representation of the arrangement of SNRPB in vertebrate genomes. The tandem duplication of SNRPN from SNRPB in the opossum genome is enclosed by a red dashed box. (C) Early origin and evolution of Ube3a in relation to the evolution of the nervous system. LECA, last eukaryotic common ancestor.
The PWS-AS imprinted domain encompasses a genomic region of about 2 Mb in humans and comprises a smaller AS subdomain that includes two maternally expressed genes (Ube3a and ATP10A) and a larger PWS subdomain that contains six paternally expressed genes (MKRN3, MAGEL2, NDN, NPAP1, SNURF and SNRPN) and two major clusters of the paternally expressed small nucleolar RNAs (snoRNAs) SNORD115 (HBII-52) and SNORD116 (HBII-85). Numbers of snoRNA genes in this region are highly variable across eutherian lineages (Zhang et al., 2014). In addition, the paternally expressed NPAP1 is primate specific and lacks an ortholog in other eutherians (Neumann et al., 2014), suggesting that this imprinted domain is a highly dynamic genomic region. Currently, it is held that the PWS-AS imprinted domain formed from non-imprinted components by genome rearrangement in an eutherian ancestor after divergence from marsupials. The following ordered or concurrent steps have been proposed (Hore et al., 2007; Renfree et al., 2009): (1) fission of Ube3a-CNGA3 border; (2) translocation of SNRPN next to Ube3a; (3) generation of SNURF and IC; (4) insertion and expansion of snoRNA repeats; (5) insertion of the three retroposed genes MKRN3, MAGEL2, and NDN, followed by the integration of NPAP1 in the primate lineage. A few key questions remain unsolved regarding the assembly of the PWS-AS imprinted domain. In particular, it is unclear why Ube3a was fused to SNRPN and became a part of the PWS-AS imprinting domain, and no marsupial progenitors of SNURF and IC have yet been identified (Renfree et al., 2009).
Pseudogenes of Ube3a
The two processed pseudogenes Ube3ap1 and Ube3ap2 have been identified in the human genome, although there is no evidence of their expression (Kishino and Wagstaff, 1998). These pseudogenes are also found in chimpanzees but not in mice and macaques, indicating that they formed in a common ancestor of chimpanzees and humans. Ube3ap1 and Ube3ap2 are located on chromosome 2 and 21, respectively, in the human and chimpanzee genomes.
Conclusion and Perspective
Ube3a is an ancient gene that emerged prior to nervous systems, and its imprinted expression was acquired much later (Figure 2C). These findings accord with the current view that genes involved in human neurogenetic disorders are not necessarily evolutionarily new. To deepen the understanding of the evolution of Ube3a, comparison with the evolution of genes involved in brain disorders other than neurodevelopmental disorders, such as neurological disorders, is instructive. For example, presenilins (encoded by PSEN1 and PSEN2 in humans) form the catalytic center of γ-secretase that processes amyloid precursor protein (APP) to produce amyloid-β (Aβ) peptide, and mutations in PSEN1, PSEN2 and APP are found in early-onset familial Alzheimer’s disease (Bertram et al., 2010). Presenilin orthologs are widespread among eukaryotes, including amoebozoans, metazoans and plants, suggesting that their ancestral gene was already present in the last common eukaryotic ancestor (Gazave et al., 2009). On the other hand, orthologs of the APP gene family (amyloid precursor-like protein 1 (APLP1), amyloid precursor-like protein 2 (APLP2), and APP) have been identified only in multicellular metazoans, including Nematostella vectensis, and the amyloidogenic Aβ motif and γ-secretase cleavage sites are conserved only across APP orthologs from jawed vertebrates (Tharp and Sarkar, 2013; Moore et al., 2014). Although phylogenetic studies of the proposed Ube3a substrates are yet to be conducted and searches for additional candidates of AS-relevant substrates should be continued, the evolutions of Ube3a and presenilin suggest that the ancient emergence of disease-related enzymes and more recent appearance of their relevant substrates could be a common evolutionary scheme of the key signal transduction components across different brain disorders.
Recent studies suggest that diverse symptoms of AS are mediated by distinct circuits, cell types, substrates and downstream pathways that act at different developmental stages (Mandel-Brehm et al., 2015; Silva-Santos et al., 2015; Judson et al., 2016). From an evolutionary point of view, it can be suggested that the key events in the evolutionary history of Ube3a led to the current etiology of AS. These events likely include: (1) expression of the Ube3a protein in nerve cells and its localization at functionally important subcellular compartments such as synapses; (2) colocalization and interaction with substrates that play essential roles in neuronal development and function; and (3) acquisition of genomic imprinting, leading to increased vulnerability of Ube3a to genetic damage. Thus, further studies of the expression and localization of Ube3a orthologs in primitive extant organisms, and exogenous expression and functional analyses of these orthologs in mammalian neurons, will broaden the evolutionary perspective of AS, as described for a few other synaptic proteins (Burkhardt et al., 2014; Yang et al., 2015).
Author Contributions
MS wrote the manuscript.
Funding
This work was supported in part by a grant from the Angelman Syndrome Foundation.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer DT and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
Acknowledgments
I thank Toru Takumi and Junichi Nakai for support and Takeshi Kawashima for helpful discussion.
References
Albrecht, U., Sutcliffe, J. S., Cattanach, B. M., Beechey, C. V., Armstrong, D., Eichele, G., et al. (1997). Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nat. Genet. 17, 75–78. doi: 10.1038/ng0997-75
Alié, A., and Manuel, M. (2010). The backbone of the post-synaptic density originated in a unicellular ancestor of choanoflagellates and metazoans. BMC Evol. Biol. 10:34. doi: 10.1186/1471-2148-10-34
Anderson, P. A., and Spencer, A. N. (1989). The importance of cnidarian synapses for neurobiology. J. Neurobiol. 20, 435–457. doi: 10.1002/neu.480200513
Angelman, H. (1965). ‘Puppet’ children: a report on three cases. Dev. Med. Child Neurol. 7, 681–688. doi: 10.1111/j.1469-8749.1965.tb07844.x
Arendt, D., Tosches, M. A., and Marlow, H. (2016). From nerve net to nerve ring, nerve cord and brain—evolution of the nervous system. Nat. Rev. Neurosci. 17, 61–72. doi: 10.1038/nrn.2015.15
Barlow, D. P. (1993). Methylation and imprinting: from host defense to gene regulation? Science 260, 309–310. doi: 10.1126/science.8469984
Basel-Vanagaite, L., Dallapiccola, B., Ramirez-Solis, R., Segref, A., Thiele, H., Edwards, A., et al. (2012). Deficiency for the ubiquitin ligase UBE3B in a blepharophimosis-ptosis-intellectual-disability syndrome. Am. J. Hum. Genet. 91, 998–1010. doi: 10.1016/j.ajhg.2012.10.011
Bayés, à., van de Lagemaat, L. N., Collins, M. O., Croning, M. D. R., Whittle, I. R., Choudhary, J. S., et al. (2011). Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat. Neurosci. 14, 19–21. doi: 10.1038/nn.2719
Berkel, S., Marshall, C. R., Weiss, B., Howe, J., Roeth, R., Moog, U., et al. (2010). Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat. Genet. 42, 489–491. doi: 10.1038/ng.589
Bertram, L., Lill, C. M., and Tanzi, R. E. (2010). The genetics of Alzheimer disease: back to the future. Neuron 68, 270–281. doi: 10.1016/j.neuron.2010.10.013
Bolton, P. F., Dennis, N. R., Browne, C. E., Thomas, N. S., Veltman, M. W., Thompson, R. J., et al. (2001). The phenotypic manifestations of interstitial duplications of proximal 15q with special reference to the autistic spectrum disorders. Am. J. Med. Genet. 105, 675–685. doi: 10.1002/ajmg.1551
Buiting, K., Lich, C., Cottrell, S., Barnicoat, A., and Horsthemke, B. (1999). A 5-kb imprinting center deletion in a family with Angelman syndrome reduces the shortest region of deletion overlap to 880 bp. Hum. Genet. 105, 665–666. doi: 10.1007/s004390051160
Buiting, K., Williams, C., and Horsthemke, B. (2016). Angelman syndrome—insights into a rare neurogenetic disorder. Nat. Rev. Neurol. 12, 584–593. doi: 10.1038/nrneurol.2016.133
Burkhardt, P., Grønborg, M., McDonald, K., Sulur, T., Wang, Q., and King, N. (2014). Evolutionary insights into premetazoan functions of the neuronal protein homer. Mol. Biol. Evol. 31, 2342–2355. doi: 10.1093/molbev/msu178
Clayton-Smith, J., and Laan, L. (2003). Angelman syndrome: a review of the clinical and genetic aspects. J. Med. Genet. 40, 87–95. doi: 10.1136/jmg.40.2.87
Colosi, D. C., Martin, D., Moré, K., and Lalande, M. (2006). Genomic organization and allelic expression of UBE3A in chicken. Gene 383, 93–98. doi: 10.1016/j.gene.2006.07.019
Cooper, E. M., Hudson, A. W., Amos, J., Wagstaff, J., and Howley, P. M. (2004). Biochemical analysis of Angelman syndrome-associated mutations in the E3 ubiquitin ligase E6-associated protein. J. Biol. Chem. 279, 41208–41217. doi: 10.1074/jbc.m401302200
Curley, J. P., Barton, S., Surani, A., and Keverne, E. B. (2004). Coadaptation in mother and infant regulated by a paternally expressed imprinted gene. Proc. Biol. Sci. 271, 1303–1309. doi: 10.1098/rspb.2004.2725
Durand, C. M., Betancur, C., Boeckers, T. M., Bockmann, J., Chaste, P., Fauchereau, F., et al. (2007). Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 39, 25–27. doi: 10.1038/ng1933
Ellwanger, K., Eich, A., and Nickel, M. (2007). GABA and glutamate specifically induce contractions in the sponge Tethya wilhelma. J. Comp. Physiol. A Neuroethol. Sens. Neural Behav. Physiol. 193, 1–11. doi: 10.1007/s00359-006-0165-y
Emes, R. D., and Grant, S. G. N. (2011). The human postsynaptic density shares conserved elements with proteomes of unicellular eukaryotes and prokaryotes. Front. Neurosci. 5:44. doi: 10.3389/fnins.2011.00044
Gazave, E., Lapébie, P., Richards, G. S., Brunet, F., Ereskovsky, A. V., Degnan, B. M., et al. (2009). Origin and evolution of the Notch signalling pathway: an overview from eukaryotic genomes. BMC Evol. Biol. 9:249. doi: 10.1186/1471-2148-9-249
Glessner, J. T., Wang, K., Cai, G., Korvatska, O., Kim, C. E., Wood, S., et al. (2009). Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459, 569–573. doi: 10.1038/nature07953
Gong, T. W. L., Huang, L., Warner, S. J., and Lomax, M. I. (2003). Characterization of the human UBE3B gene: structure, expression, evolution and alternative splicing. Genomics 82, 143–152. doi: 10.1016/s0888-7543(03)00111-3
Grau-Bové, X., Sebé-Pedrós, A., and Ruiz-Trillo, I. (2013). A genomic survey of HECT ubiquitin ligases in eukaryotes reveals independent expansions of the HECT system in several lineages. Genome Biol. Evol. 5, 833–847. doi: 10.1093/gbe/evt052
Gregg, C., Zhang, J., Weissbourd, B., Luo, S., Schroth, G. P., Haig, D., et al. (2010). High-resolution analysis of parent-of-origin allelic expression in the mouse brain. Science 329, 643–648. doi: 10.1126/science.1190830
Haig, D. (2004). Genomic imprinting and kinship: how good is the evidence? Annu. Rev. Genet. 38, 553–585. doi: 10.1146/annurev.genet.37.110801.142741
Hope, K. A., LeDoux, M. S., and Reiter, L. T. (2016). The Drosophila melanogaster homolog of UBE3A is not imprinted in neurons. Epigenetics 11, 637–642. doi: 10.1080/15592294.2016.1214783
Hore, T. A., Rapkins, R. W., and Graves, J. A. M. (2007). Construction and evolution of imprinted loci in mammals. Trends Genet. 23, 440–448. doi: 10.1016/j.tig.2007.07.003
Huibregtse, J. M., Scheffner, M., and Howley, P. M. (1993). Cloning and expression of the cDNA for E6-AP, a protein that mediates the interaction of the human papillomavirus E6 oncoprotein with p53. Mol. Cell. Biol. 13, 775–784. doi: 10.1128/mcb.13.2.775
Judson, M. C., Sosa-Pagan, J. O., Del Cid, W. A., Han, J. E., and Philpot, B. D. (2014). Allelic specificity of Ube3a expression in the mouse brain during postnatal development. J. Comp. Neurol. 522, 1874–1896. doi: 10.1002/cne.23507
Judson, M. C., Wallace, M. L., Sidorov, M. S., Burette, A. C., Gu, B., van Woerden, G. M., et al. (2016). GABAergic neuron-specific loss of Ube3a causes Angelman syndrome-like EEG abnormalities and enhances seizure susceptibility. Neuron 90, 56–69. doi: 10.1016/j.neuron.2016.02.040
Khaitovich, P., Enard, W., Lachmann, M., and Pääbo, S. (2006). Evolution of primate gene expression. Nat. Rev. Genet. 7, 693–702. doi: 10.1038/nrg1940
Kishino, T., Lalande, M., and Wagstaff, J. (1997). UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 15, 70–73. doi: 10.1038/ng0197-70
Kishino, T., and Wagstaff, J. (1998). Genomic organization of the UBE3A/E6-AP gene and related pseudogenes. Genomics 47, 101–107. doi: 10.1006/geno.1997.5093
Li, L., Keverne, E. B., Aparicio, S. A., Ishino, F., Barton, S. C., and Surani, M. A. (1999). Regulation of maternal behavior and offspring growth by paternally expressed Peg3. Science 284, 330–333. doi: 10.1126/science.284.5412.330
Liebeskind, B. J., Hillis, D. M., Zakon, H. H., and Hofmann, H. A. (2016). Complex homology and the evolution of nervous systems. Trends Ecol. Evol. 31, 127–135. doi: 10.1016/j.tree.2015.12.005
Lloyd, S. E., Maytham, E. G., Pota, H., Grizenkova, J., Molou, E., Uphill, J., et al. (2009a). HECTD2 is associated with susceptibility to mouse and human prion disease. PLoS Genet. 5:e1000383. doi: 10.1371/journal.pgen.1000383
Lloyd, S. E., Rossor, M., Fox, N., Mead, S., and Collinge, J. (2009b). HECTD2, a candidate susceptibility gene for Alzheimer’s disease on 10q. BMC Med. Genet. 10:90. doi: 10.1186/1471-2350-10-90
Mabb, A. M., Judson, M. C., Zylka, M. J., and Philpot, B. D. (2011). Angelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 34, 293–303. doi: 10.1016/j.tins.2011.04.001
Mandel-Brehm, C., Salogiannis, J., Dhamne, S. C., Rotenberg, A., and Greenberg, M. E. (2015). Seizure-like activity in a juvenile Angelman syndrome mouse model is attenuated by reducing Arc expression. Proc. Natl. Acad. Sci. U S A 112, 5129–5134. doi: 10.1073/pnas.1504809112
Marín, I. (2010). Animal HECT ubiquitin ligases: evolution and functional implications. BMC Evol. Biol. 10:56. doi: 10.1186/1471-2148-10-56
Marlow, H. Q., Srivastava, M., Matus, D. Q., Rokhsar, D., and Martindale, M. Q. (2009). Anatomy and development of the nervous system of Nematostella vectensis, an anthozoan cnidarian. Dev. Neurobiol. 69, 235–254. doi: 10.1002/dneu.20698
Matsuura, T., Sutcliffe, J. S., Fang, P., Galjaard, R. J., Jiang, Y. H., Benton, C. S., et al. (1997). De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat. Genet. 15, 74–77. doi: 10.1038/ng0197-74
Miller, G. (2009). On the origin of the nervous system. Science 325, 24–26. doi: 10.1126/science.325_24
Moore, D. B., Gillentine, M. A., Botezatu, N. M., Wilson, K. A., Benson, A. E., and Langeland, J. A. (2014). Asynchronous evolutionary origins of Aβ and BACE1. Mol. Biol. Evol. 31, 696–702. doi: 10.1093/molbev/mst262
Moroz, L. L., Kocot, K. M., Citarella, M. R., Dosung, S., Norekian, T. P., Povolotskaya, I. S., et al. (2014). The ctenophore genome and the evolutionary origins of neural systems. Nature 510, 109–114. doi: 10.1038/nature13400
Neumann, L. C., Feiner, N., Meyer, A., Buiting, K., and Horsthemke, B. (2014). The imprinted NPAP1 gene in the Prader-Willi syndrome region belongs to a POM121-related family of retrogenes. Genome Biol. Evol. 6, 344–351. doi: 10.1093/gbe/evu019
Ohta, T., Gray, T. A., Rogan, P. K., Buiting, K., Gabriel, J. M., Saitoh, S., et al. (1999). Imprinting-mutation mechanisms in Prader-Willi syndrome. Am. J. Hum. Genet. 64, 397–413. doi: 10.1086/302233
Perk, J., Makedonski, K., Lande, L., Cedar, H., Razin, A., and Shemer, R. (2002). The imprinting mechanism of the Prader-Willi/Angelman regional control center. EMBO J. 21, 5807–5814. doi: 10.1093/emboj/cdf570
Rapkins, R. W., Hore, T., Smithwick, M., Ager, E., Pask, A. J., Renfree, M. B., et al. (2006). Recent assembly of an imprinted domain from non-imprinted components. PLoS Genet. 2:e182. doi: 10.1371/journal.pgen.0020182
Reiter, L. T., Seagroves, T. N., Bowers, M., and Bier, E. (2006). Expression of the Rho-GEF Pbl/ECT2 is regulated by the UBE3A E3 ubiquitin ligase. Hum. Mol. Genet. 15, 2825–2835. doi: 10.1093/hmg/ddl225
Renfree, M. B., Hore, T. A., Shaw, G., Graves, J. A. M., and Pask, A. J. (2009). Evolution of genomic imprinting: insights from marsupials and monotremes. Annu. Rev. Genomics Hum. Genet. 10, 241–262. doi: 10.1146/annurev-genom-082908-150026
Rougeulle, C., Glatt, H., and Lalande, M. (1997). The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain. Nat. Genet. 17, 14–15. doi: 10.1038/ng0497-411d
Ryan, J. F., Pang, K., Schnitzler, C. E., Nguyen, A. D., Moreland, R. T., Simmons, D. K., et al. (2013). The genome of the ctenophore Mnemiopsis leidyi and its implications for cell type evolution. Science 342:1242592. doi: 10.1126/science.1242592
Sakarya, O., Armstrong, K. A., Adamska, M., Adamski, M., Wang, I. F., Tidor, B., et al. (2007). A post-synaptic scaffold at the origin of the animal kingdom. PLoS One 2:e506. doi: 10.1371/journal.pone.0000506
Sánchez-Tena, S., Cubillos-Rojas, M., Schneider, T., and Rosa, J. L. (2016). Functional and pathological relevance of HERC family proteins: a decade later. Cell. Mol. Life Sci. 73, 1955–1968. doi: 10.1007/s00018-016-2139-8
Sato, D., Lionel, A. C., Leblond, C. S., Prasad, A., Pinto, D., Walker, S., et al. (2012). SHANK1 deletions in males with Autism spectrum disorder. Am. J. Hum. Genet. 90, 879–887. doi: 10.1016/j.ajhg.2012.03.017
Sato, M., and Stryker, M. P. (2010). Genomic imprinting of experience-dependent cortical plasticity by the ubiquitin ligase gene Ube3a. Proc. Natl. Acad. Sci. U S A 107, 5611–5616. doi: 10.1073/pnas.1001281107
Scheffner, M., and Kumar, S. (2014). Mammalian HECT ubiquitin-protein ligases: biological and pathophysiological aspects. Biochim. Biophys. Acta 1843, 61–74. doi: 10.1016/j.bbamcr.2013.03.024
Sell, G. L., and Margolis, S. S. (2015). From UBE3A to Angelman syndrome: a substrate perspective. Front. Neurosci. 9:322. doi: 10.3389/fnins.2015.00322
Silva-Santos, S., van Woerden, G. M., Bruinsma, C. F., Mientjes, E., Jolfaei, M. A., Distel, B., et al. (2015). Ube3a reinstatement identifies distinct developmental windows in a murine Angelman syndrome model. J Clin. Invest. 125, 2069–2076. doi: 10.1172/jci80554
Smith, C. L., Pivovarova, N., and Reese, T. S. (2015). Coordinated feeding behavior in trichoplax, an animal without synapses. PLoS One 10:e0136098. doi: 10.1371/journal.pone.0136098
Smith, S. E. P., Zhou, Y. D., Zhang, G., Jin, Z., Stoppel, D. C., and Anderson, M. P. (2011). Increased gene dosage of Ube3a results in autism traits and decreased glutamate synaptic transmission in mice. Sci. Transl. Med. 3:103ra97. doi: 10.1126/scitranslmed.3002627
Sun, J., Zhu, G., Liu, Y., Standley, S., Ji, A., Tunuguntla, R., et al. (2015). UBE3A regulates synaptic plasticity and learning and memory by controlling SK2 channel endocytosis. Cell Rep. 12, 449–461. doi: 10.1016/j.celrep.2015.06.023
Tamm, S., and Tamm, S. L. (1995). A giant nerve net with multi-effector synapses underlying epithelial adhesive strips in the mouth of Beroë (Ctenophora). J. Neurocytol. 24, 711–723. doi: 10.1007/bf01179820
Tharp, W. G., and Sarkar, I. N. (2013). Origins of amyloid-β. BMC Genomics 14:290. doi: 10.1186/1471-2164-14-290
Urraca, N., Cleary, J., Brewer, V., Pivnick, E. K., McVicar, K., Thibert, R. L., et al. (2013). The interstitial duplication 15q11.2–q13 syndrome includes autism, mild facial anomalies and a characteristic EEG signature. Autism Res. 6, 268–279. doi: 10.1002/aur.1284
Vallender, E. J., Mekel-Bobrov, N., and Lahn, B. T. (2008). Genetic basis of human brain evolution. Trends Neurosci. 31, 637–644. doi: 10.1016/j.tins.2008.08.010
Vu, T. H., and Hoffman, A. R. (1997). Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nat. Genet. 17, 12–13. doi: 10.1038/ng0997-12
Williams, C. A., Angelman, H., Clayton-Smith, J., Driscoll, D. J., Hendrickson, J. E., Knoll, J. H., et al. (1995). Angelman syndrome: consensus for diagnostic criteria. Angelman syndrome foundation. Am. J. Med. Genet. 56, 237–238. doi: 10.1002/ajmg.1320560224
Wu, Y., Bolduc, F. V., Bell, K., Tully, T., Fang, Y., Sehgal, A., et al. (2008). A Drosophila model for Angelman syndrome. Proc. Natl. Acad. Sci. U S A 105, 12399–12404. doi: 10.1073/pnas.0805291105
Yamasaki, K., Joh, K., Ohta, T., Masuzaki, H., Ishimaru, T., Mukai, T., et al. (2003). Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. Hum. Mol. Genet. 12, 837–847. doi: 10.1093/hmg/ddg106
Yang, X., Pei, J., Kaeser-Woo, Y. J., Bacaj, T., Grishin, N. V., and Südhof, T. C. (2015). Evolutionary conservation of complexins: from choanoflagellates to mice. EMBO Rep. 16, 1308–1317. doi: 10.15252/embr.201540305
Yashiro, K., Riday, T. T., Condon, K. H., Roberts, A. C., Bernardo, D. R., Prakash, R., et al. (2009). Ube3a is required for experience-dependent maturation of the neocortex. Nat. Neurosci. 12, 777–783. doi: 10.1038/nn.2327
Keywords: brain evolution, genomic imprinting, developmental disorder, autism, intellectual disability, HECT domain, synapse
Citation: Sato M (2017) Early Origin and Evolution of the Angelman Syndrome Ubiquitin Ligase Gene Ube3a. Front. Cell. Neurosci. 11:62. doi: 10.3389/fncel.2017.00062
Received: 13 December 2016; Accepted: 22 February 2017;
Published: 07 March 2017.
Edited by:
Andrew Harkin, Trinity College, Dublin, IrelandReviewed by:
Daniela Tropea, Trinity College, Dublin, IrelandRobert Weissert, University of Regensburg, Germany
Edgar Richard Kramer, Medical Research Council Harwell (MRC), UK
Copyright © 2017 Sato. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Masaaki Sato, masasato@mail.saitama-u.ac.jp; msato@brain.riken.jp