Lysosomes and α-synuclein form a dangerous duet leading to neuronal cell death
 Mathieu Bourdenx
Mathieu Bourdenx Erwan Bezard
Erwan Bezard Benjamin Dehay
Benjamin Dehay- 1Institut des Maladies Neurodégénératives, Université de Bordeaux, UMR 5293, Bordeaux, France
- 2CNRS, Institut des Maladies Neurodégénératives, UMR 5293, Bordeaux, France
Neurodegenerative diseases are (i) characterized by a selective neuronal vulnerability to degeneration in specific brain regions; and (ii) likely to be caused by disease-specific protein misfolding. Parkinson’s disease (PD) is characterized by the presence of intraneuronal proteinacious cytoplasmic inclusions, called Lewy Bodies (LB). α-Synuclein, an aggregation prone protein, has been identified as a major protein component of LB and the causative for autosomal dominant PD. Lysosomes are responsible for the clearance of long-lived proteins, such as α-synuclein, and for the removal of old or damaged organelles, such as mitochondria. Interestingly, PD-linked α-synuclein mutants and dopamine-modified wild-type α-synuclein block its own degradation, which result in insufficient clearance, leading to its aggregation and cell toxicity. Moreover, both lysosomes and lysosomal proteases have been found to be involved in the activation of certain cell death pathways. Interestingly, lysosomal alterations are observed in the brains of patients suffering from sporadic PD and also in toxic and genetic rodent models of PD-related neurodegeneration. All these events have unraveled a causal link between lysosomal impairment, α-synuclein accumulation, and neurotoxicity. In this review, we emphasize the pathophysiological mechanisms connecting α-synuclein and lysosomal dysfunction in neuronal cell death.
Introduction
Neurodegenerative diseases are (i) characterized by a selective neuronal vulnerability to degeneration in specific brain regions; and (ii) likely caused by disease-specific protein misfolding. Parkinson’s disease (PD), the second most common neurodegenerative disorder after Alzheimer’s disease, is notably characterized by the loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc). Loss of dopamine perturbates the cortico-basal ganglia-cortical control of voluntary movements. Current treatments have no proven protective or restorative effect and are only symptomatic. Moreover, their long-term use is associated with the onset of dramatic side effects i.e., fluctuated responses and L-Dopa induced dyskinesia. The important of understanding the mechanisms of neuronal death underlying neurodegenerative diseases is crucial for identifying targets for disease-modifying/curative strategies. In addition to dopaminergic neuronal cell loss, the main pathological hallmark of PD is the presence of intraneuronal proteinaceous cytoplasmic inclusions, named Lewy bodies (LB). α-Synuclein (α-syn), a major protein component of LB, has been identified as autosomal dominant cause of PD, which is found increased in expression in patients (Goedert et al., 2013; Lashuel et al., 2013). The presence of LB in PD suggests that defective protein handling contributes to the pathogenesis of the disease. Proteasomal and autophagic proteolysis are the two major pathways for degradation of cellular constituents in eukaryotic cells. Mounting evidence indicates that alterations in autophagy-lysosomal pathways (ALP) may be preferentially involved in PD. In this article, we review the close relationship between α-syn and the lysosome, two players involved in neuronal cell death in PD.
The Harmful α-Synuclein
α-Syn has a central role in the pathogenesis of PD and other synucleinopathies, as dementia with Lewy bodies (DLB) and Multiple System Atrophy (MSA; Spillantini and Goedert, 2000). In 1997, the first link between PD and α-syn was described with the identification of point mutations -A53T- in the SNCA gene in autosomal-dominant forms of PD (Polymeropoulos et al., 1997; Athanassiadou et al., 1999; Spira et al., 2001; Ki et al., 2007; Choi et al., 2008; Puschmann et al., 2009). To date, the list of missense mutations continues to grow with A30P, E46K, H50Q, G51D, A53E (all classified as PARK1 locus) (Krüger et al., 1998; Zarranz et al., 2004; Appel-Cresswell et al., 2013; Lesage et al., 2013; Proukakis et al., 2013; Pasanen et al., 2014). The subsequent identification of families with multiplication (duplication or triplication) of its allele (PARK4 locus) strengthen the link between α-syn and PD (Singleton et al., 2003; Chartier-Harlin et al., 2004), suggesting that increased expression levels of the normal α-syn can be causal for PD and others synucleinopathies. Furthermore, genome-wide association studies (GWAS) have linked single nucleotide polymorphisms (SNPs) in the SNCA gene with increased susceptibility of developing PD (Simón-Sánchez et al., 2009; Edwards et al., 2010; International Parkinson Disease Genomics et al., 2011). α-Syn is a 14 kDa neuronal protein consisting of 140 amino acids mainly localized to presynaptic terminals. While the exact physiological function of α-syn remains to be fully understood, several studies have implicated its capacity to interact directly with cellular membranes, such as vesicles (Auluck et al., 2010) or mitochondria-associated membrane, which is an endoplasmic reticulum subdomain involved in lipid and calcium homeostasis (Guardia-Laguarta et al., 2014). Nevertheless, substantial evidence suggests that α-syn function is related to vesicle dynamics, neurotransmission and synaptic plasticity, the mechanisms of which have been reviewed elsewhere (Bellani et al., 2010). In its native state, the previous paradigm was that α-syn behaves as an unfolded monomer. However, a recent report now hints at a more complex picture as the predominant physiological species of α-syn is a helically folded tetramer (Bartels et al., 2011). α-Syn is, however, intrinsically defined as an aggregation-prone protein. In PD brains, α-syn antibodies strongly react in LB (Spillantini et al., 1997) and Lewy neurites (Takeda et al., 1998). Biochemical analyses have shown that α-syn is a major protein component of LB and may be part of the β-sheet enriched fibrillar structure of these inclusions (Crowther et al., 2000). α-Syn can undergo several post-translational modification such as truncation, nitration, oxidation, sumoylation, ubiquitinylation and phosphorylation (Giasson et al., 2000; Fujiwara et al., 2002; Tofaris et al., 2003; Anderson et al., 2006; Dorval and Fraser, 2006; Krumova et al., 2011). Interestingly, post-translationally modified α-syn has been found in LB and some post-translational modifications, such as oxidation or nitration, have been shown to impact its aggregation process in favor to oligomeric species (Fujiwara et al., 2002; Norris et al., 2003; Yamin et al., 2003). In the past few years, substantial progress has been made not only at elucidating how α-syn undergoes spontaneous self-aggregation, but also in its highly heterogenous aggregation process that turns its monomers into multiple oligomeric forms, then protofibrils, fibrils and aggregates. The identification of pathological species of α-syn involved in the perturbation of cellular function is an expanding area of research. Recent studies support the concept of soluble oligomers as the prominent toxic α-syn species in in vitro and in vivo settings, although the precise size and type of the toxic oligomeric species remains to be determined (Auluck et al., 2010; Winner et al., 2011; Cremades et al., 2012). Recent evidence piles up for prion-like propagation mechanisms in synucleinopathies, including PD. Indeed, α-syn might behave as a prion, responsible for initiating and spreading the pathological process in PD. Supporting this concept, α-syn can be transmitted to neighboring neurons and neuronal precursor cells (Puschmann et al., 2009; Hansen et al., 2011). In vivo studies have added a further piece to the puzzle with the observation that intracerebral inoculation of synthetic recombinant α-syn fibrils (Pffs) can mimic α-syn pathology in mice (Luk et al., 2012). More recently, through an innovative strategy based on the purification of aggregated α-syn from the SNpc of PD patients, intranigral or intrastriatal inoculations of PD-derived LB extracts resulted in progressive nigrostriatal neurodegeneration in both mice and monkeys (Recasens et al., 2014), which were found to originate at striatal dopaminergic terminals, Overall, these results demonstrated that human α-syn species contained in PD-derived LB are pathogenic and have the capacity to initiate a PD-like pathological process, not only in rodents but also in non-human primates (Recasens et al., 2014). Taken together, α-syn has multiple ways to cause cellular perturbations and lead to cell death. The presence of undegraded proteinaceous inclusions led the research community to wonder how is handled α-syn degradation? It is now understood that this involves both the ubiquitin-proteasome system (UPS) and the ALP. α-Syn is, however, predominantly degraded inside lysosomes, through chaperone-mediated autophagy (CMA) or endocytosis (Webb et al., 2003; Cuervo et al., 2004; Martinez-Vicente and Vila, 2013). The signals responsible for targeting α-syn (although it contains a KFERQ-like sequence, i.e., a motif recognized by heat shock cognate70 (hsc70) allowing direct lysosomal import) to a given degradation pathway are not yet fully understood, but may heavily depend on its folding state. Aggregated proteins will be preferentially routed for degradation to the lysosome through macroautophagy, whereas soluble forms would be both targeted to the proteasome or to the CMA. Overall, defective α-syn protein degradation can be recognized as an important pathogenic factor.
Lysosome: White Knight or Two-Face
Lysosomes are dynamic acidic organelles that contain hydrolytic enzyme capable of degrading intracellular components, which were discovered by Christian de Duve more than 50 years ago (De Duve et al., 1955; Luzio et al., 2007). Acidic pH (around 4.6) is maintained in the lumen by proton-pumping vacuolar ATPases. Around 200 proteins have been reported as lysosomal membrane proteins such as proton pumps, secretory, plasma membrane, signaling or transport proteins (Schröder et al., 2007). The most abundant proteins are the lysosomal-associated membrane protein (LAMP)-1 and LAMP-2 as well as the lysosomal integral membrane protein (LIMP)-2 and CD63 (Saftig et al., 2010). Interestingly, lysosomes have a high intravesicular Ca2+ concentration (around 500-600 μM). Defective lysosomal Ca2+ uptake has been associated with human diseases, such as Niemann-Pick type C (Lloyd-Evans and Platt, 2011). Several lysosomal storage disorders are caused by lysosomal membrane dysfunctions (Ruivo et al., 2009). These defects are mostly due to non-enzymatic transport defects, highlighting the importance of transport and channel proteins in lysosome physiology (Ruivo et al., 2009). As mitochondrial outer membrane permeabilization (MOMP) is a major checkpoint of apoptosis pathway, lysosomal membrane permeabilization (LMP) has also been shown to induce cell death (Boya and Kroemer, 2008). Following LMP, cell death can occur through several pathway including canonical MOMP/caspase pathway but also MOMP- and caspase-independent pathways (Boya and Kroemer, 2008). The two main effects of LMP are the release of lysosomal proteases, such as cathepsins B or D (CSTB/CTSD), and cytosolic acidification. CTSD or CTSB could then directly or indirectly promote cytochrome C release from mitochondria (Boya and Kroemer, 2008). Currently, the principal inducer of LMP remains to be reactive oxygen species (ROS), although Bcl-2-associated X protein (Bax) has also shown to initiate this process (Kågedal et al., 2005).
Several pathways converge to the lysosome: phagocytosis, endocytosis, and autophagy through three different means respectively named microautophagy, CMA and macroautophagy. Autophagy (which comes from the Greek: “self-eating”) is an evolutionary conserved mechanism that allows cells to degrade their own components and recycle important molecules (Wong and Cuervo, 2010; Cuervo, 2011; Boya et al., 2013). Briefly, CMA involves selective recognition by a chaperone and import through LAMP-2a, while microautophagy and macroautophagy involve direct sequestration of a portion of the cytosol (including proteins and organelles). While microautophagy requires the direct invagination of lysosomal membrane, macroautphagy involves the formation of a vesicle named autophagosome that will then fused with lysosomes to allow degradation of the sequestered material. In regards to protein aggregation, macroautophagy has been suggested to be the mammalian counterpart of the cytosolic-to-vacuole (Cvt)-pathway in yeast responsible for cargo-selective degradation (Yamamoto and Simonsen, 2011). Selective degradation of protein aggregates, named aggrephagy, has been characterized based on the observation of autophagosomes specifically containing aggregates (Filimonenko et al., 2010). Moreover, a phosphatidylinositol 3-phosphate-binding protein, Alfy, has been shown to specifically recognize and promote degradation of huntingtin aggregates (Filimonenko et al., 2010). For several decades, lysosomes have been only considered as terminal degradative compartments. However, recent studies suggest that lysosomes are involved in a vast number of cellular functions including lysosome-to-nucleus signaling, secretion, energy metabolism and cell death pathways (Rodriguez et al., 1997; Settembre et al., 2012, 2013).
Impairment of ALP, which is essential to maintain proper protein and organelle quantity and quality within cells, is increasingly regarded as a major pathogenic event in neurodegenerative diseases, including PD. The presence of LB in brains of PD patients made the first connection with ALP and led to the hypothesis that defective protein handling system might contribute to the pathogenesis of the disease. Several studies from independent groups reported ALP impairment associated with lysosomal depletion in brain tissue from idiopathic PD patients (Chu et al., 2009; Alvarez-Erviti et al., 2010; Dehay et al., 2010). More precisely, accumulation of undegraded microtubule-associated protein light chain 3 (LC3)-positive vesicles, decreased cytosolic hsc70, LAMP-1 and LAMP-2a have been reported (Chu et al., 2009; Alvarez-Erviti et al., 2010; Dehay et al., 2010). Genetic studies further strengthen the connection between PD and ALP dysfunction, which have indicated that lysosomal impairments may play a primary pathogenic role in the disease process. Interestingly, both CMA and proteasome can degrade the two proteins associated with autosomal dominant inheritance of PD, i.e., α-syn (PARK1/PARK4 locus) and Leucine-rich repeat kinase 2 (LRRK2–PARK8 locus) (Webb et al., 2003; Cuervo et al., 2004; Orenstein et al., 2013). However, PD-linked α-syn mutants (as well as post-translationally dopamine-modified wild-type α-syn) and mutant forms of LRRK2 block CMA activity, resulting in insufficient clearance and subsequent accumulation and aggregation of α-syn (Cuervo et al., 2004; Martinez-Vicente et al., 2008; Mak et al., 2010; Orenstein et al., 2013). Notably, two other genes encoding for lysosomal proteins have been linked to PD: the lysosomal type 5 P-type ATPase (ATP13A2—PARK9 locus) (Ramirez et al., 2006) and the enzyme glucocerebrosidase (GBA; Aharon-Peretz et al., 2004; Di Fonzo et al., 2007; Sidransky et al., 2009). While the former has been characterized in rare families with prominent parkinsonism (Ramirez et al., 2006; Di Fonzo et al., 2007), the latter has been identified as risk factor in multicenter genetic analysis of patients (Sidransky et al., 2009). Recently, genetic analysis suggested that lysosomal dysfunction may play an important role in the etiology of DLB (Bras et al., 2014). Relevant to PD, these two proteins have been reported to be components of LB (Goker-Alpan et al., 2010; Dehay et al., 2012). Defects in one of these two proteins may result in insufficient clearance of α-syn through lysosomes, hence leading to the accumulation of this protein in both cytosol and lysosome lumen (Dehay et al., 2012). Such vicious pathogenic loop has been reported between GBA and α-syn (Mazzulli et al., 2011). One can thus imagine a similar scenario in which toxic species of α-syn “damage” lysosomes, hence leading to an impairment of α-syn clearance that subsequently favor α-syn-aggregation. Such aggregates then cause, in return, other damages, while concomitantly accumulating in lysosomes/autolysosomes to form LB (Dehay et al., 2012).
In addition to the aforementioned genes, hereditary parkinsonism has been identified in families carrying mutations for ALP-related pathways. For instance, mutations in parkin (PARK2 locus), in the phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1) (PARK6 locus) or in DJ-1 (PARK7 locus), which are all involved in mitophagy, lead to autosomal recessive forms of PD (Corti et al., 2011). While PINK1 and parkin belong to the same pathway, DJ-1 has been shown to be involved in an independent parallel pathway, which can rescue a loss of function of PINK1 (Hao et al., 2010; Thomas et al., 2011). A defective degradation of dysfunctional mitochondria leads to maintaining those in the neuron and hence promotes the mitochondrial dysfunctions that have been characterized in PD patients (i.e., decrease in complex I activity and accumulation of large-scale mitochondrial DNA mutations) (Schapira et al., 1989; Bender et al., 2006). Mutations in the PD-associated gene UCH-L1 (PARK5) abnormally interact with LAMP-2A, also causing an increase amount of α-syn (Kabuta et al., 2008). From a genetic point of view, all genes that have been positively associated with PD (Corti et al., 2011) are also connected to ALP, which shed light on the lysosome as an important player in PD-induced cell death.
Neuronal Cell Death: The Third Partner
Lysosomal function impairment and α-syn aggregation can both induce cell death either on their own or through a dramatic additive effect. Of importance, α-syn seems to induce cell toxicity through its different pathological α-syn species, which include post-translationally modified, mutant, oligomeric and aggregated forms. These can (i) disrupt its typical function in neurotransmission release (Abeliovich et al., 2000; Jenco et al., 1998); (ii) impair mitochondrial dynamics, structure and function (Martin et al., 2006; Nakamura et al., 2011; Stefanovic et al., 2014); and (iii) disrupt ER-Golgi vesicle trafficking (Cooper et al., 2006; Gitler et al., 2008) and mitochondria-associated ER membrane (Mercado et al., 2013; Guardia-Laguarta et al., 2014), which results in ER stress. Further supporting the α-syn species toxicity, CMA inhibition by either PD-linked α-syn mutants or dopamine-modified wild-type α-syn results in an accumulation of α-syn, but also of undegraded CMA-substrates, involved for instance in the regulation of neuronal survival through the degradation of the neuronal survival factor myocyte enhancer factor 2D (MEF2D; Yang et al., 2009).
Regarding the lysosome, LMP is one mechanism for the induction of certain cell death pathways. As mentioned above, disruption of lysosomal membrane provokes cell death through release of CTSs and other hydrolases from the lysosomal lumen to the cytosol. These lysosomal proteases can remain active at cytosolic pH and induce cellular damages by degradation of vital proteins or activation of caspases. In relation to PD pathophysiology, mechanistic studies using the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of PD have reported a lysosomal dysfunction, characterized by lysosomal depletion and autophagosome accumulation. Such lysosomal deficiency was secondary to abnormal LMP induced by Complex I inhibition-mediated ROS production (Dehay et al., 2010; Vila et al., 2011). Recent studies reported that the pro-apoptotic Bax protein, which mediates MOMP, is activated in PD patients (Bové et al., 2014). In experimental PD mouse model, Bax translocates to the lysosome and mediates LMP before MOMP (Bové et al., 2014). Interestingly, pharmacological inhibition of Bax-mediated LMP and MOMP results in an overall attenuation of MPTP-mediated cell death, even if the treatment is administered once pathogenic neuronal changes are already in motion (Bové et al., 2014), suggesting that the phenomenon at work is reversible.
One of the meeting points between α-syn and lysosome involves ROS. Recent reports suggest that α-syn oligomers can induce both MOMP and in particular LMP (Freeman et al., 2013; Stefanovic et al., 2014). α-Syn aggregation underlies a bidirectional relationship with ROS production. Specific α-syn oligomers increase ROS production, whereas oxidized α-syn inhibits fibril formation in favor to toxic species (Norris et al., 2003; Cremades et al., 2012). Hence, α-syn-mediated ROS production can lead to LMP, as previously characterized in PD, and subsequently to cell death. All these studies suggest that oxidative stress impact both lysosomes and α-syn aggregation. In the past few years, another piece has been added to the puzzle, suggesting that α-syn might potentially spread in a prion-like manner, from cell to cell and region to region. Although mechanisms of α-syn release are not yet elucidated, α-syn may be released by exocytosis in a calcium-dependent manner (Lee et al., 2005; Emmanouilidou et al., 2010), a phenomenon exacerbated after lysosomal inhibition (Alvarez-Erviti et al., 2011), subsequently enhancing disease progression and the lysosomal contribution to the pathology. Non-genetic factors, however, cannot be excluded as important risks to PD. This includes ageing for instance which remains the most compelling risk factor for PD. Ageing is also associated with mitochondrial and lysosomal impairments as well as ROS production (Dufour and Larsson, 2004; Mattson and Magnus, 2006), linking the several key events that occur in neuronal cell death in PD.
Of interest, pharmacological or genetic enhancement of autophagy has been shown to be beneficial in PD models. For example, in the MPTP-treated mouse model, pharmacological activation of ALP with the mammalian target of rapamycin (mTOR) inhibitor, rapamycin, attenuates neurodegeneration and lysosomal dysfunction (Dehay et al., 2010; Malagelada et al., 2010). Consistent with this approach, viral-mediated overexpression of ALP components, such as transcription factor EB (TFEB), LAMP2a or Beclin-1, provided neuroprotection in viral-mediated α-syn-overexpressing rodent models of PD (Spencer et al., 2009; Decressac et al., 2013; Xilouri et al., 2013). With regards to the development of therapeutic approaches, we must keep in mind that a balance needs to be maintained between boosting and inhibiting processes of autophagy. Indeed, autophagy has been shown to have both survival promoting and death promoting roles (Eskelinen, 2005). Hence, enhancement of lysosomal biogenesis or specific activation of late steps of the autophagy machinery might provide more successful approach compared to a broad activation of the whole autophagy machinery, potentially leading to a deleterious effect and eventually cell death. Increasing the ability of neurons under attack to degrade protein aggregates remains a promising strategy for PD.
Concluding Remarks
Seventeen years after its association with PD, α-syn is now considered as a central player in PD pathogenesis, linking genetic and idiopathic forms of parkinsonism. Two key elements strongly associate α-syn aggregation and lysosomal dysfunction: (i) aggregated or post-translationally modified forms of α-syn can directly or indirectly inhibit lysosomal function; and (ii) the occurrence of a lysosomal depletion in brains from PD patients as well as in several experimental models of PD. Consistent with these evidences, LB formation might be the result of the combination of both α-syn aggregation and lysosomal failure, as key components of autophagy and α-syn have been localized in LB. Altogether, this suggests that α-syn aggregation and lysosomal impairment, enhanced with ageing, could play a deleterious duet leading to dopaminergic cell death (Figure 1).
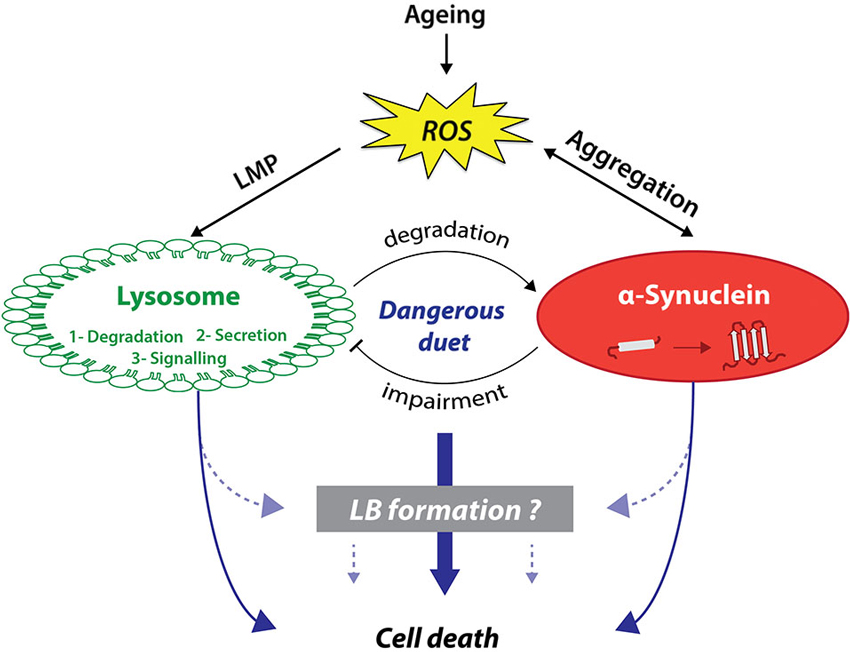
Figure 1. Lysosomes and α-synuclein are involved in a vicious pathogenic loop eventually leading to cell death and LB formation. On the one hand, lysosomes have been shown to be involved in cell death activation through canonical or non-canonical pathways. On the other hand, α-synuclein (α-syn) can also trigger cell death through several distinct pathways including membranes permeabilization. Lysosomes and α-syn display a bidirectional relationship. While lysosomes predominantly degrade α-syn, α-syn aggregation can lead to lysosomal dysfunction in return. α-Syn-mediated lysosome impairment can lead to alterations of one of lysosomal main function: protein degradation, lysosome-to-nucleus signaling, and secretion. This pathogenic loop can be worsened with age and in particular ROS production, which can induce both LMP and α-syn aggregation. Interestingly, α-syn aggregation, especially specific oligomeric species, can increase ROS production. We previously suggested that this loop might be the template for the formation of LB, which remains currently unknown.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Daniel Ko for valuable comments on the manuscript. The University of Bordeaux and the Centre National de la Recherche Scientifique provided infrastructural support. This work was supported by Marie Curie Reintegration Grant FP7-PEOPLE-2009-ERG256303 from the European Commission (to Benjamin Dehay); by a grant from the Fondation pour la Recherche Médicale (to Benjamin Dehay); by Agence Nationale de la Recherche Grants ANR-08-MNP-018, ANR-07-MNP-Trafinlid and ANR-12-BSV4-0001-01 (to Erwan Bezard) and Mathieu Bourdenx is a recipient of an MESR fellowship.
References
Abeliovich, A., Schmitz, Y., Farinas, I., Choi-Lundberg, D., Ho, W. H., Castillo, P. E., et al. (2000). Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25, 239–252. doi: 10.1016/s0896-6273(00)80886-7
Aharon-Peretz, J., Rosenbaum, H., and Gershoni-Baruch, R. (2004). Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N. Engl. J. Med. 351, 1972–1977. doi: 10.1056/nejmoa033277
Alvarez-Erviti, L., Rodriguez-Oroz, M. C., Cooper, J. M., Caballero, C., Ferrer, I., Obeso, J. A., et al. (2010). Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 67, 1464–1472. doi: 10.1001/archneurol.2010.198
Alvarez-Erviti, L., Seow, Y., Schapira, A. H., Gardiner, C., Sargent, I. L., Wood, M. J., et al. (2011). Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 42, 360–367. doi: 10.1016/j.nbd.2011.01.029
Anderson, J. P., Walker, D. E., Goldstein, J. M., De Laat, R., Banducci, K., Caccavello, R. J., et al. (2006). Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 281, 29739–29752. doi: 10.1074/jbc.m600933200
Appel-Cresswell, S., Vilarino-Guell, C., Encarnacion, M., Sherman, H., Yu, I., Shah, B., et al. (2013). Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 28, 811–813. doi: 10.1002/mds.25421
Athanassiadou, A., Voutsinas, G., Psiouri, L., Leroy, E., Polymeropoulos, M. H., Ilias, A., et al. (1999). Genetic analysis of families with Parkinson disease that carry the Ala53Thr mutation in the gene encoding alpha-synuclein. Am. J. Hum. Genet. 65, 555–558. doi: 10.1086/302486
Auluck, P. K., Caraveo, G., and Lindquist, S. (2010). alpha-Synuclein: membrane interactions and toxicity in Parkinson’s disease. Annu. Rev. Cell Dev. Biol. 26, 211–233. doi: 10.1146/annurev.cellbio.042308.113313
Bartels, T., Choi, J. G., and Selkoe, D. J. (2011). alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477, 107–110. doi: 10.1038/nature10324
Bellani, S., Sousa, V. L., Ronzitti, G., Valtorta, F., Meldolesi, J., and Chieregatti, E. (2010). The regulation of synaptic function by alpha-synuclein. Commun. Integr. Biol. 3, 106–109. doi: 10.4161/cib.3.2.10964
Bender, A., Krishnan, K. J., Morris, C. M., Taylor, G. A., Reeve, A. K., Perry, R. H., et al. (2006). High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 38, 515–517. doi: 10.1038/ng1769
Bové, J., Martinez-Vicente, M., Dehay, B., Perier, C., Recasens, A., Bombrun, A., et al. (2014). BAX channel activity mediates lysosomal disruption linked to Parkinson disease. Autophagy 10, 889–900. doi: 10.4161/auto.28286
Boya, P., and Kroemer, G. (2008). Lysosomal membrane permeabilization in cell death. Oncogene 27, 6434–6451. doi: 10.1038/onc.2008.310
Boya, P., Reggiori, F., and Codogno, P. (2013). Emerging regulation and functions of autophagy. Nat. Cell Biol. 15, 713–720. doi: 10.1038/ncb2788
Bras, J., Guerreiro, R., Darwent, L., Parkkinen, L., Ansorge, O., Escott-Price, V., et al. (2014). Genetic analysis implicates APOE, SNCA and suggests lysosomal dysfunction in the etiology of dementia with Lewy bodies. Hum. Mol. Genet. doi: 10.1093/hmg/ddu334. [Epub ahead of print].
Chartier-Harlin, M. C., Kachergus, J., Roumier, C., Mouroux, V., Douay, X., Lincoln, S., et al. (2004). Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364, 1167–1169. doi: 10.1016/s0140-6736(04)17103-1
Choi, J. M., Woo, M. S., Ma, H. I., Kang, S. Y., Sung, Y. H., Yong, S. W., et al. (2008). Analysis of PARK genes in a Korean cohort of early-onset Parkinson disease. Neurogenetics 9, 263–269. doi: 10.1007/s10048-008-0138-0
Chu, Y., Dodiya, H., Aebischer, P., Olanow, C. W., and Kordower, J. H. (2009). Alterations in lysosomal and proteasomal markers in Parkinson’s disease: relationship to alpha-synuclein inclusions. Neurobiol. Dis. 35, 385–398. doi: 10.1016/j.nbd.2009.05.023
Cooper, A. A., Gitler, A. D., Cashikar, A., Haynes, C. M., Hill, K. J., Bhullar, B., et al. (2006). Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 313, 324–328. doi: 10.1126/science.1129462
Corti, O., Lesage, S., and Brice, A. (2011). What genetics tells us about the causes and mechanisms of Parkinson’s disease. Physiol. Rev. 91, 1161–1218. doi: 10.1152/physrev.00022.2010
Cremades, N., Cohen, S. I., Deas, E., Abramov, A. Y., Chen, A. Y., Orte, A., et al. (2012). Direct observation of the interconversion of normal and toxic forms of alpha-synuclein. Cell 149, 1048–1059. doi: 10.1016/j.cell.2012.03.037
Crowther, R. A., Daniel, S. E., and Goedert, M. (2000). Characterisation of isolated alpha-synuclein filaments from substantia nigra of Parkinson’s disease brain. Neurosci. Lett. 292, 128–130. doi: 10.1016/s0304-3940(00)01440-3
Cuervo, A. M. (2011). Cell biology. Autophagy’s top chef. Science 332, 1392–1393. doi: 10.1126/science.1208607
Cuervo, A. M., Stefanis, L., Fredenburg, R., Lansbury, P. T., and Sulzer, D. (2004). Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295. doi: 10.1126/science.1101738
Decressac, M., Mattsson, B., Weikop, P., Lundblad, M., Jakobsson, J., and Bjorklund, A. (2013). TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc. Natl. Acad. Sci. U S A 110, E1817–E1826. doi: 10.1073/pnas.1305623110
De Duve, C., Pressman, B. C., Gianetto, R., Wattiaux, R., and Appelmans, F. (1955). Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J. 60, 604–617.
Dehay, B., Bové, J., Rodriguez-Muela, N., Perier, C., Recasens, A., Boya, P., et al. (2010). Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 30, 12535–12544. doi: 10.1523/jneurosci.1920-10.2010
Dehay, B., Ramirez, A., Martinez-Vicente, M., Perier, C., Canron, M. H., Doudnikoff, E., et al. (2012). Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc. Natl. Acad. Sci. U S A 109, 9611–9616. doi: 10.1073/pnas.1112368109
Di Fonzo, A., Chien, H. F., Socal, M., Giraudo, S., Tassorelli, C., Iliceto, G., et al. (2007). ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology 68, 1557–1562. doi: 10.1212/01.wnl.0000260963.08711.08
Dorval, V., and Fraser, P. E. (2006). Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and alpha-synuclein. J. Biol. Chem. 281, 9919–9924. doi: 10.1074/jbc.m510127200
Dufour, E., and Larsson, N. G. (2004). Understanding aging: revealing order out of chaos. Biochim. Biophys. Acta 1658, 122–132. doi: 10.1016/j.bbabio.2004.04.020
Edwards, T. L., Scott, W. K., Almonte, C., Burt, A., Powell, E. H., Beecham, G. W., et al. (2010). Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann. Hum. Genet. 74, 97–109. doi: 10.1111/j.1469-1809.2009.00560.x
Emmanouilidou, E., Melachroinou, K., Roumeliotis, T., Garbis, S. D., Ntzouni, M., Margaritis, L. H., et al. (2010). Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 30, 6838–6851. doi: 10.1523/JNEUROSCI.5699-09.2010
Eskelinen, E. L. (2005). Doctor Jekyll and Mister Hyde: autophagy can promote both cell survival and cell death. Cell Death Differ. 12(Suppl. 2), 1468–1472. doi: 10.1038/sj.cdd.4401721
Filimonenko, M., Isakson, P., Finley, K. D., Anderson, M., Jeong, H., Melia, T. J., et al. (2010). The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol. Cell 38, 265–279. doi: 10.1016/j.molcel.2010.04.007
Freeman, D., Cedillos, R., Choyke, S., Lukic, Z., Mcguire, K., Marvin, S., et al. (2013). Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS One 8:e62143. doi: 10.1371/journal.pone.0062143
Fujiwara, H., Hasegawa, M., Dohmae, N., Kawashima, A., Masliah, E., Goldberg, M. S., et al. (2002). alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 4, 160–164. doi: 10.1038/ncb748
Giasson, B. I., Duda, J. E., Murray, I. V., Chen, Q., Souza, J. M., Hurtig, H. I., et al. (2000). Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 290, 985–989. doi: 10.1126/science.290.5493.985
Gitler, A. D., Bevis, B. J., Shorter, J., Strathearn, K. E., Hamamichi, S., Su, L. J., et al. (2008). The Parkinson’s disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. U S A 105, 145–150. doi: 10.1073/pnas.0710685105
Goedert, M., Spillantini, M. G., Del Tredici, K., and Braak, H. (2013). 100 years of Lewy pathology. Nat. Rev. Neurol. 9, 13–24. doi: 10.1038/nrneurol.2012.242
Goker-Alpan, O., Stubblefield, B. K., Giasson, B. I., and Sidransky, E. (2010). Glucocerebrosidase is present in alpha-synuclein inclusions in Lewy body disorders. Acta Neuropathol. 120, 641–649. doi: 10.1007/s00401-010-0741-7
Guardia-Laguarta, C., Area-Gomez, E., Rüb, C., Liu, Y., Magrane, J., Becker, D., et al. (2014). alpha-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 34, 249–259. doi: 10.1523/jneurosci.2507-13.2014
Hansen, C., Angot, E., Bergstrom, A. L., Steiner, J. A., Pieri, L., Paul, G., et al. (2011). alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Invest. 121, 715–725. doi: 10.1172/JCI43366
Hao, L. Y., Giasson, B. I., and Bonini, N. M. (2010). DJ-1 is critical for mitochondrial function and rescues PINK1 loss of function. Proc. Natl. Acad. Sci. U S A 107, 9747–9752. doi: 10.1073/pnas.0911175107
International Parkinson Disease Genomics, C., Nalls, M. A., Plagnol, V., Hernandez, D. G., Sharma, M., Sheerin, U. M., et al. (2011). Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet 377, 641–649. doi: 10.1016/S0140-6736(10)62345-8
Jenco, J. M., Rawlingson, A., Daniels, B., and Morris, A. J. (1998). Regulation of phospholipase D2: selective inhibition of mammalian phospholipase D isoenzymes by alpha- and beta-synucleins. Biochemistry 37, 4901–4909. doi: 10.1021/bi972776r
Kabuta, T., Setsuie, R., Mitsui, T., Kinugawa, A., Sakurai, M., Aoki, S., et al. (2008). Aberrant molecular properties shared by familial Parkinson’s disease-associated mutant UCH-L1 and carbonyl-modified UCH-L1. Hum. Mol. Genet. 17, 1482–1496. doi: 10.1093/hmg/ddn037
Kågedal, K., Johansson, A. C., Johansson, U., Heimlich, G., Roberg, K., Wang, N. S., et al. (2005). Lysosomal membrane permeabilization during apoptosis–involvement of Bax? Int. J. Exp. Pathol. 86, 309–321. doi: 10.1111/j.0959-9673.2005.00442.x
Ki, C. S., Stavrou, E. F., Davanos, N., Lee, W. Y., Chung, E. J., Kim, J. Y., et al. (2007). The Ala53Thr mutation in the alpha-synuclein gene in a Korean family with Parkinson disease. Clin. Genet. 71, 471–473. doi: 10.1111/j.1399-0004.2007.00781.x
Krüger, R., Kuhn, W., Müller, T., Woitalla, D., Graeber, M., Kosel, S., et al. (1998). Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 18, 106–108.
Krumova, P., Meulmeester, E., Garrido, M., Tirard, M., Hsiao, H. H., Bossis, G., et al. (2011). Sumoylation inhibits alpha-synuclein aggregation and toxicity. J. Cell Biol. 194, 49–60. doi: 10.1083/jcb.201010117
Lashuel, H. A., Overk, C. R., Oueslati, A., and Masliah, E. (2013). The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 14, 38–48. doi: 10.1038/nrn3406
Lee, H. J., Patel, S., and Lee, S. J. (2005). Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J. Neurosci. 25, 6016–6024. doi: 10.1523/jneurosci.0692-05.2005
Lesage, S., Anheim, M., Letournel, F., Bousset, L., Honore, A., Rozas, N., et al. (2013). G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 73, 459–471. doi: 10.1002/ana.23894
Lloyd-Evans, E., and Platt, F. M. (2011). Lysosomal Ca(2+) homeostasis: role in pathogenesis of lysosomal storage diseases. Cell Calcium 50, 200–205. doi: 10.1016/j.ceca.2011.03.010
Luk, K. C., Kehm, V., Carroll, J., Zhang, B., O’brien, P., Trojanowski, J. Q., et al. (2012). Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953. doi: 10.1126/science.1227157
Luzio, J. P., Pryor, P. R., and Bright, N. A. (2007). Lysosomes: fusion and function. Nat. Rev. Mol. Cell Biol. 8, 622–632. doi: 10.1038/nrm2217
Mak, S. K., Mccormack, A. L., Manning-Bog, A. B., Cuervo, A. M., and Di Monte, D. A. (2010). Lysosomal degradation of alpha-synuclein in vivo. J. Biol. Chem. 285, 13621–13629. doi: 10.1074/jbc.m109.074617
Malagelada, C., Jin, Z. H., Jackson-Lewis, V., Przedborski, S., and Greene, L. A. (2010). Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J. Neurosci. 30, 1166–1175. doi: 10.1523/jneurosci.3944-09.2010
Martin, L. J., Pan, Y., Price, A. C., Sterling, W., Copeland, N. G., Jenkins, N. A., et al. (2006). Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. 26, 41–50. doi: 10.1523/jneurosci.4308-05.2006
Martinez-Vicente, M., Talloczy, Z., Kaushik, S., Massey, A. C., Mazzulli, J., Mosharov, E. V., et al. (2008). Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J. Clin. Invest. 118, 777–788. doi: 10.1172/JCI32806
Martinez-Vicente, M., and Vila, M. (2013). Alpha-synuclein and protein degradation pathways in Parkinson’s disease: a pathological feed-back loop. Exp. Neurol. 247, 308–313. doi: 10.1016/j.expneurol.2013.03.005
Mattson, M. P., and Magnus, T. (2006). Ageing and neuronal vulnerability. Nat. Rev. Neurosci. 7, 278–294. doi: 10.1038/nrn1886
Mazzulli, J. R., Xu, Y. H., Sun, Y., Knight, A. L., Mclean, P. J., Caldwell, G. A., et al. (2011). Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146, 37–52. doi: 10.1016/j.cell.2011.06.001
Mercado, G., Valdés, P., and Hetz, C. (2013). An ERcentric view of Parkinson’s disease. Trends Mol. Med. 19, 165–175. doi: 10.1016/j.molmed.2012.12.005
Nakamura, K., Nemani, V. M., Azarbal, F., Skibinski, G., Levy, J. M., Egami, K., et al. (2011). Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J. Biol. Chem. 286, 20710–20726. doi: 10.1074/jbc.p110.213538
Norris, E. H., Giasson, B. I., Ischiropoulos, H., and Lee, V. M. (2003). Effects of oxidative and nitrative challenges on alpha-synuclein fibrillogenesis involve distinct mechanisms of protein modifications. J. Biol. Chem. 278, 27230–27240. doi: 10.1074/jbc.m212436200
Orenstein, S. J., Kuo, S. H., Tasset, I., Arias, E., Koga, H., Fernandez-Carasa, I., et al. (2013). Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 16, 394–406. doi: 10.1038/nn.3350
Pasanen, P., Myllykangas, L., Siitonen, M., Raunio, A., Kaakkola, S., Lyytinen, J., et al. (2014). A novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 35, 2180.e1–2180.e5. doi: 10.1016/j.neurobiolaging.2014.03.024
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Proukakis, C., Dudzik, C. G., Brier, T., Mackay, D. S., Cooper, J. M., Millhauser, G. L., et al. (2013). A novel alpha-synuclein missense mutation in Parkinson disease. Neurology 80, 1062–1064. doi: 10.1212/wnl.0b013e31828727ba
Puschmann, A., Ross, O. A., Vilarino-Guell, C., Lincoln, S. J., Kachergus, J. M., Cobb, S. A., et al. (2009). A Swedish family with de novo alpha-synuclein A53T mutation: evidence for early cortical dysfunction. Parkinsonism Relat. Disord. 15, 627–632. doi: 10.1016/j.parkreldis.2009.06.007
Ramirez, A., Heimbach, A., Gründemann, J., Stiller, B., Hampshire, D., Cid, L. P., et al. (2006). Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 38, 1184–1191. doi: 10.1038/ng1884
Recasens, A., Dehay, B., Bové, J., Carballo-Carbajal, I., Dovero, S., Perez-Villalba, A., et al. (2014). Lewy body extracts from Parkinson disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann. Neurol. 75, 351–362. doi: 10.1002/ana.24066
Rodriguez, A., Webster, P., Ortego, J., and Andrews, N. W. (1997). Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J. Cell Biol. 137, 93–104. doi: 10.1083/jcb.137.1.93
Ruivo, R., Anne, C., Sagné, C., and Gasnier, B. (2009). Molecular and cellular basis of lysosomal transmembrane protein dysfunction. Biochim. Biophys. Acta 1793, 636–649. doi: 10.1016/j.bbamcr.2008.12.008
Saftig, P., Schröder, B., and Blanz, J. (2010). Lysosomal membrane proteins: life between acid and neutral conditions. Biochem. Soc. Trans. 38, 1420–1423. doi: 10.1042/bst0381420
Schapira, A. H., Cooper, J. M., Dexter, D., Jenner, P., Clark, J. B., and Marsden, C. D. (1989). Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1, 1269.
Schröder, B., Wrocklage, C., Pan, C., Jäger, R., Kösters, B., Schäfer, H., et al. (2007). Integral and associated lysosomal membrane proteins. Traffic 8, 1676–1686. doi: 10.1111/j.1600-0854.2007.00643.x
Settembre, C., Fraldi, A., Medina, D. L., and Ballabio, A. (2013). Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 14, 283–296. doi: 10.1038/nrm3565
Settembre, C., Zoncu, R., Medina, D. L., Vetrini, F., Erdin, S., Erdin, S., et al. (2012). A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 31, 1095–1108. doi: 10.1038/emboj.2012.32
Sidransky, E., Nalls, M. A., Aasly, J. O., Aharon-Peretz, J., Annesi, G., Barbosa, E. R., et al. (2009). Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 361, 1651–1661. doi: 10.1056/NEJMoa0901281
Simón-Sánchez, J., Schulte, C., Bras, J. M., Sharma, M., Gibbs, J. R., Berg, D., et al. (2009). Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 41, 1308–1312. doi: 10.1038/ng.487
Singleton, A. B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., et al. (2003). alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302, 841. doi: 10.1126/science.1090278
Spencer, B., Potkar, R., Trejo, M., Rockenstein, E., Patrick, C., Gindi, R., et al. (2009). Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J. Neurosci. 29, 13578–13588. doi: 10.1523/jneurosci.4390-09.2009
Spillantini, M. G., and Goedert, M. (2000). The alpha-synucleinopathies: Parkinson’s disease, dementia with Lewy bodies and multiple system atrophy. Ann. N Y Acad. Sci. 920, 16–27. doi: 10.1111/j.1749-6632.2000.tb06900.x
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Spira, P. J., Sharpe, D. M., Halliday, G., Cavanagh, J., and Nicholson, G. A. (2001). Clinical and pathological features of a Parkinsonian syndrome in a family with an Ala53Thr alpha-synuclein mutation. Ann. Neurol. 49, 313–319. doi: 10.1002/ana.67.abs
Stefanovic, A. N., Stöckl, M. T., Claessens, M. M., and Subramaniam, V. (2014). alpha-Synuclein oligomers distinctively permeabilize complex model membranes. FEBS J. 281, 2838–2850. doi: 10.1111/febs.12824
Takeda, A., Mallory, M., Sundsmo, M., Honer, W., Hansen, L., and Masliah, E. (1998). Abnormal accumulation of NACP/alpha-synuclein in neurodegenerative disorders. Am. J. Pathol. 152, 367–372.
Thomas, K. J., Mccoy, M. K., Blackinton, J., Beilina, A., Van Der Brug, M., Sandebring, A., et al. (2011). DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum. Mol. Genet. 20, 40–50. doi: 10.1093/hmg/ddq430
Tofaris, G. K., Razzaq, A., Ghetti, B., Lilley, K. S., and Spillantini, M. G. (2003). Ubiquitination of alpha-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. J. Biol. Chem. 278, 44405–44411. doi: 10.1074/jbc.m308041200
Vila, M., Bové, J., Dehay, B., Rodriguez-Muela, N., and Boya, P. (2011). Lysosomal membrane permeabilization in Parkinson disease. Autophagy 7, 98–100. doi: 10.4161/auto.7.1.13933
Webb, J. L., Ravikumar, B., Atkins, J., Skepper, J. N., and Rubinsztein, D. C. (2003). Alpha-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 278, 25009–25013. doi: 10.1074/jbc.m300227200
Winner, B., Jappelli, R., Maji, S. K., Desplats, P. A., Boyer, L., Aigner, S., et al. (2011). In vivo demonstration that alpha-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. U S A 108, 4194–4199. doi: 10.1073/pnas.1100976108
Wong, E., and Cuervo, A. M. (2010). Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 13, 805–811. doi: 10.1038/nn.2575
Xilouri, M., Brekk, O. R., Landeck, N., Pitychoutis, P. M., Papasilekas, T., Papadopoulou-Daifoti, Z., et al. (2013). Boosting chaperone-mediated autophagy in vivo mitigates alpha-synuclein-induced neurodegeneration. Brain 136, 2130–2146. doi: 10.1093/brain/awt131
Yamamoto, A., and Simonsen, A. (2011). The elimination of accumulated and aggregated proteins: a role for aggrephagy in neurodegeneration. Neurobiol. Dis. 43, 17–28. doi: 10.1016/j.nbd.2010.08.015
Yamin, G., Uversky, V. N., and Fink, A. L. (2003). Nitration inhibits fibrillation of human alpha-synuclein in vitro by formation of soluble oligomers. FEBS Lett. 542, 147–152. doi: 10.1016/s0014-5793(03)00367-3
Yang, Q., She, H., Gearing, M., Colla, E., Lee, M., Shacka, J. J., et al. (2009). Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science 323, 124–127. doi: 10.1126/science.1166088
Keywords: α-synuclein, lysosome, Parkinson’s disease, cell death
Citation: Bourdenx M, Bezard E and Dehay B (2014) Lysosomes and α-synuclein form a dangerous duet leading to neuronal cell death. Front. Neuroanat. 8:83. doi: 10.3389/fnana.2014.00083
Received: 30 June 2014; Accepted: 28 July 2014;
Published online: 14 August 2014.
Edited by:
Javier Blesa, Columbia University, USACopyright © 2014 Bourdenx, Bezard and Dehay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Benjamin Dehay, Institut des Maladies Neurodégénératives, Université de Bordeaux, 146 rue Léo Saignat, UMR 5293, 33076 Bordeaux cedex, France e-mail: benjamin.dehay@u-bordeaux.fr