Tauopathy and Epilepsy Comorbidities and Underlying Mechanisms
 Kaylin Hwang
Kaylin Hwang Rahil N. Vaknalli
Rahil N. Vaknalli Kwaku Addo-Osafo
Kwaku Addo-Osafo Mariane Vicente
Mariane Vicente Keith Vossel*
Keith Vossel*- Mary S. Easton Center for Alzheimer’s Research and Care, Department of Neurology, David Geffen School of Medicine at UCLA, Los Angeles, CA, United States
Tau is a microtubule-associated protein known to bind and promote assembly of microtubules in neurons under physiological conditions. However, under pathological conditions, aggregation of hyperphosphorylated tau causes neuronal toxicity, neurodegeneration, and resulting tauopathies like Alzheimer’s disease (AD). Clinically, patients with tauopathies present with either dementia, movement disorders, or a combination of both. The deposition of hyperphosphorylated tau in the brain is also associated with epilepsy and network hyperexcitability in a variety of neurological diseases. Furthermore, pharmacological and genetic targeting of tau-based mechanisms can have anti-seizure effects. Suppressing tau phosphorylation decreases seizure activity in acquired epilepsy models while reducing or ablating tau attenuates network hyperexcitability in both Alzheimer’s and epilepsy models. However, it remains unclear whether tauopathy and epilepsy comorbidities are mediated by convergent mechanisms occurring upstream of epileptogenesis and tau aggregation, by feedforward mechanisms between the two, or simply by coincident processes. In this review, we investigate the relationship between tauopathies and seizure disorders, including temporal lobe epilepsy (TLE), post-traumatic epilepsy (PTE), autism spectrum disorder (ASD), Dravet syndrome, Nodding syndrome, Niemann-Pick type C disease (NPC), Lafora disease, focal cortical dysplasia, and tuberous sclerosis complex. We also explore potential mechanisms implicating the role of tau kinases and phosphatases as well as the mammalian target of rapamycin (mTOR) in the promotion of co-pathology. Understanding the role of these co-pathologies could lead to new insights and therapies targeting both epileptogenic mechanisms and cognitive decline.
Introduction
Tau is a microtubule-associated protein encoded in humans by the microtubule-associated protein tau gene, MAPT, on chromosome 17 (Wang and Mandelkow, 2016). In the brain, tau is most abundant in neurons, including neuronal axons, somatodendritic compartments, and nuclei, but it is also present in glia and, to a lesser degree, extracellularly (Morris et al., 2011; Wang and Mandelkow, 2016). The functions of tau in the brain are multifaceted, but its most well-characterized role is in microtubule binding and assembly. Tau is natively unfolded and highly soluble, thus exhibiting little tendency for aggregation. However, under pathological conditions, the hyperphosphorylation of tau reduces its affinity for tubulin and is thought to drive abnormal aggregations of phosphorylated tau (p-tau), such as neuropil threads or neurofibrillary tangles (NFTs), resulting in tauopathies (Wang and Mandelkow, 2016; Kovacs, 2017).
Endogenous tau is also implicated in neuronal activity (Wang and Mandelkow, 2016), though this role of tau is less well understood. Neuronal excitation, in turn, also regulates tau by promoting extracellular release and phosphorylation. Rapid and persisting increases in extracellular tau following in vivo (Yamada et al., 2014) or in vitro (Pooler et al., 2013) neuronal stimulation suggest that tau amplification is associated with pathological neuronal activation. Given that seizure and chronic epilepsy animal models result in prolonged tau phosphorylation (Liang et al., 2009; Liu et al., 2016; Alves et al., 2019), a growing body of research is examining the role of pathological tau in epilepsy and mechanisms underlying epilepsy and tauopathy comorbidities.
In Alzheimer’s disease (AD), which is the most common tauopathy, an estimated 60% of patients have seizures and subclinical epileptic activity (Vossel et al., 2016, 2017; Lam et al., 2020; Horvath et al., 2021). Seizures are more common in AD and dementia with Lewy bodies than in primary tauopathies, such as frontotemporal dementia and progressive supranuclear palsy (Beagle et al., 2017). However, the possibility of seizures and hyperexcitability in primary tauopathies should not be ruled out, as they occur more frequently in these diseases than in the general population (Beagle et al., 2017; Sanchez et al., 2018). Myoclonus, a sign of network hyperexcitability, is observed in a subset of patients with corticobasal degeneration (Armstrong et al., 2013), and epileptic activity is present in the FTDP-17 animal model of frontotemporal dementia with parkinsonism (Garcia-Cabrero et al., 2013).
Furthermore, tau pathology is repeatedly found in human epilepsy (Sanchez et al., 2018). In a post-mortem series of 138 refractory epilepsy cases of diverse causes, Braak staging of NFTs in the age group 40–65 years revealed increased Braak stages III/IV compared with data from an age-matched series of non-epilepsy cases (Thom et al., 2011). Abnormally high total tau and p-tau levels were also detected in cerebrospinal fluid of status epilepticus patients, with increased total tau correlating with greater risk of developing chronic epilepsy (Monti et al., 2015). Given that neurodegenerative conditions characterized by hyperphosphorylated tau aggregations exhibit increased rates of epilepsy, epilepsies are being re-conceptualized within a tauopathy context (Xi et al., 2011; Sanchez et al., 2018; Ali et al., 2019).
As such, the present review seeks to explore the following three main questions: (1) Does tau play a role in mediating network hyperexcitability and seizure activity across different epilepsy disorders? (2) Do comorbid tauopathies and epilepsies stem from independent or common mechanisms? (3) How do tauopathy and epilepsy comorbidities contribute to disease-related cognitive impairment? In light of evidence indicating tau-mediated epileptic activity and dysregulation of tau-related cell signaling pathways across seizure disorders, we propose a potential overarching mechanism (Figure 1) whereby endogenous tau helps enable network hyperexcitability, which triggers homeostatic responses aimed, in part, to disable tau activity by phosphorylation. Resulting tau hyperphosphorylation and aggregation may in turn further contribute to cognitive impairments seen in some seizure disorders.
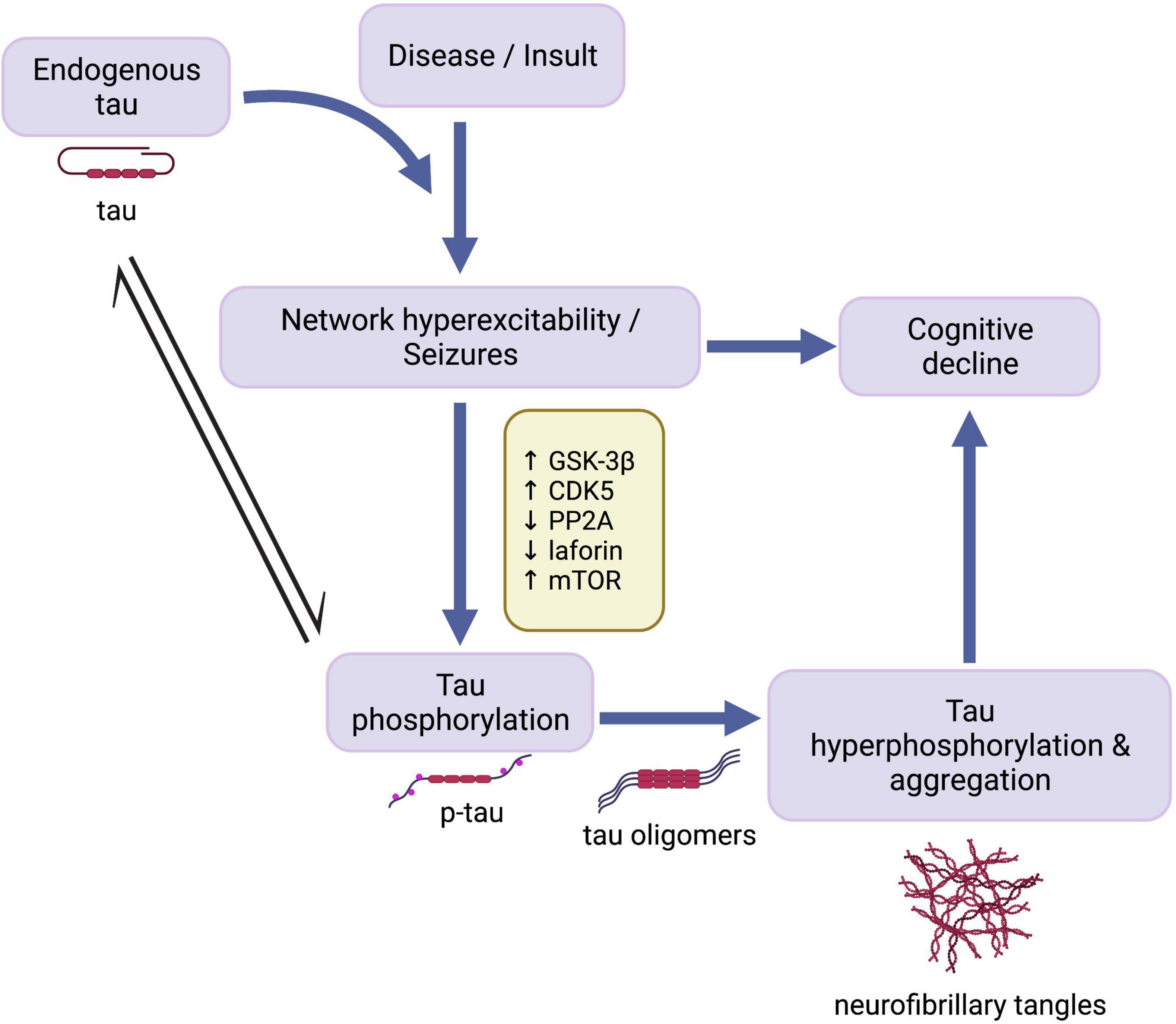
Figure 1. Cascade of events in development of seizures and tau pathology. Endogenous tau has an enabling function in the development of seizure activity following disease onset or traumatic insult. Network hyperexcitability in turn leads to cognitive decline and the activation of mechanisms involving mTOR and tau kinases and phosphatases, resulting in abnormal phosphorylation of tau. Overactivation of these cell signaling pathways increases susceptibility to pathological tau hyperphosphorylation and aggregation, which may also contribute to epilepsy-associated cognitive decline. GSK-3β, glycogen synthase kinase-3β; CDK5, cyclin-dependent kinase 5; PP2A, protein phosphatase 2A; mTOR, mammalian target of rapamycin; p-tau, abnormally phosphorylated tau. Created with BioRender.com.
Role of Tau in Epileptic Activity
Across various animal models that exhibit epilepsy, reducing tau levels reduces network hyperexcitability, seizure severity, and latency to seizure stages (Roberson et al., 2007; Ittner et al., 2010; DeVos et al., 2013; Holth et al., 2013; Wang and Mandelkow, 2016; Ali et al., 2019). Endogenous levels of tau also positively correlate with chemically induced seizure susceptibility in wild-type mice (DeVos et al., 2013), suggesting a role of endogenous tau in mediating epileptic activity.
Though not a primary epilepsy, autism spectrum disorder (ASD) is a disease with links to both seizures and tau. Several studies reveal a strong relationship between epilepsy in individuals with autism and autism in those with epilepsy (Viscidi et al., 2013; Reilly et al., 2015; Sundelin et al., 2016), with the prevalence of epilepsy in ASD doubling in adolescence (26%) compared to childhood (12%) (Hara, 2007; Woolfenden et al., 2012; Sharma et al., 2021). Likely explanations supporting the conjugation of the two conditions include an imbalance in neuronal excitation/inhibition (Bourgeron, 2009; Nelson and Valakh, 2015; Specchio et al., 2022). In the Cntnap2–/– mouse model of autism with focal epilepsy, global tau knockdown prevents epileptic activity in addition to other autistic-like behaviors (Tai et al., 2020), indicating an epileptogenic role of tau in ASD.
Genetic ablation of tau, even by 50% by inactivation of a single Mapt allele, also reduces epileptic activity, high mortality rates, and cognitive deficits in the Scn1a mouse model of Dravet syndrome, a severe and intractable childhood epilepsy that is caused by mutations in the SCN1A gene and can lead to autism (Catterall et al., 2010; Li et al., 2011; Gheyara et al., 2014; Anwar et al., 2019; Tai et al., 2020). This effect also occurs in the Scn1a model following postnatal injection of tau-targeting antisense oligonucleotides (Shao et al., 2022), suggesting that antisense oligonucleotides may be a promising treatment avenue for children with Dravet syndrome. Shao et al. (2022) further found that selective genetic ablation of tau in hippocampal excitatory neurons but not in inhibitory neurons mediates the neuroprotective effects of tau reduction in the Scn1a model. The authors propose that the suppression of epileptic activity by tau reduction may therefore result from a lower hypersynchrony of excitatory neuronal activity rather than greater inhibitory regulation.
In some models, pathological tau, rather than endogenous tau, can contribute to epileptic activity. Temporal lobe epilepsy (TLE) is one of the most prevalent forms of focal epilepsy (Tellez-Zenteno and Hernandez-Ronquillo, 2012; Asadi-Pooya et al., 2017), and in the electrical amygdala kindling rodent model of TLE, tau-knockout mice do not differ from wild-type mice in seizure outcome following repeated kindling (Liu S. et al., 2017). However, kindling produces longer epileptic discharge durations and accelerated seizure progression in rTg4510 transgenic mice, which overexpress P301L tau in forebrain and develop increased p-tau and NFTs (Liu S. et al., 2017). These findings suggest that an increase in p-tau and tau aggregation promotes kindling-induced epileptogenesis.
Taken together, findings across different seizure disease models reveal a significant function of endogenous tau, as well as pathological tau, in the mediation of epileptic activity. Given tau’s role in modulating neuronal activity under normal physiological conditions (DeVos et al., 2013; Chang et al., 2021), endogenous tau likely contributes to network hyperexcitability across primary and secondary epilepsies. In humans, tau mRNA expression and protein levels in the brain can vary greatly (Trabzuni et al., 2012). And though exact reasons for individual differences in endogenous tau levels remain unknown, high levels may consequently predict a person’s susceptibility to epileptic activity. Elevated tau measurements in cerebrospinal fluid have in fact been shown to correlate with seizure type and duration in patients with epilepsy (Tumani et al., 2015). Higher levels of endogenous tau alone may not cause seizures, but it is possible that this may predispose an individual to seizure development upon pathogenesis.
Presence of P-Tau Pathology in Seizure Disorders and Links to Epileptic Activity
While levels of endogenous or total tau differ across examinations of patients with epilepsy, increased levels of p-tau in the brain are found across many seizure disorders (Thom et al., 2011). These include patients with TLE, Dravet syndrome, Nodding syndrome (Thom et al., 2011; Pollanen et al., 2018; Hotterbeekx et al., 2019), Niemann-Pick type C disease (NPC) (Auer et al., 1995; Love et al., 1995; Suzuki et al., 1995; Malnar et al., 2014), focal cortical dysplasia IIB (FCDIIb) (Sen et al., 2007; Iyer et al., 2014), and tuberous sclerosis complex (TSC) (Sarnat and Flores-Sarnat, 2015; Liu et al., 2020), as well as animal models of post-traumatic epilepsy (PTE) (Cho et al., 2020; Alyenbaawi et al., 2021), Lafora disease (Epm2a–/–) (Ganesh et al., 2002; Puri et al., 2009; Machado-Salas et al., 2012), and ASD (Gassowska-Dobrowolska et al., 2021). It should be noted that tau aggregation in the brain is associated with older age and is generally uncommon in healthy young adults (Braak et al., 2011; Crary et al., 2014). However, the presence of tau pathology in childhood or adolescent-onset epilepsies, such as Dravet syndrome, Nodding syndrome, Lafora disease, NPC, TSC, and ASD, and in relatively younger TLE patient cohorts (Puvenna et al., 2016; Smith et al., 2019; Gourmaud et al., 2020) suggest a causal link between seizure activity and p-tau accumulation.
In animal models of TLE, tau hyperphosphorylation is observed in relevant brain regions, including the amygdala, hippocampus, and cortex, following chemical and electrical amygdala kindling (Jones et al., 2012; Liu et al., 2016; Alves et al., 2019). And in humans with chronic epilepsy, elevated p-tau is present in post-mortem (Thom et al., 2011) as well as surgically resected tissue (Puvenna et al., 2016; Tai et al., 2016; Liu C. et al., 2017; Prada Jardim et al., 2018; Smith et al., 2019; Gourmaud et al., 2020). For instance, analysis of resected temporal lobe tissue by Tai et al. (2016) found pathological tau phosphorylation in the form of neuropil threads, NFTs, and pre-tangles in 31 of 33 TLE patients between 50 and 65 years of age. Interestingly, observations of subpial bands formed by cortical p-tau depositions have been made across separate studies (Puvenna et al., 2016; Tai et al., 2016; Smith et al., 2019), providing evidence for a novel pattern of tau pathology in TLE that may result from seizure-induced reorganization of temporal lobe networks (Tai et al., 2018).
Tau hyperphosphorylation is also consistently present in the initial and long-term secondary mechanisms initiated by traumatic brain injury (TBI) (Petraglia et al., 2014; Zheng et al., 2014; Rubenstein et al., 2015; Ali et al., 2019; Tan et al., 2020), a leading cause of morbidity and mortality worldwide (Hay et al., 2016). For instance, sustaining even a single TBI can result in progressive NFT formation that is more extensive and severe than what is expected with normal aging (Johnson et al., 2012; Zanier et al., 2018). Furthermore, it is estimated that over 50% of severe TBI cases will result in seizures or PTE (Kovacs et al., 2014), and animal models reveal increased p-tau levels in the brain associated with TBI-induced epileptic activity (Cho et al., 2020; Alyenbaawi et al., 2021). In a recent study, Alyenbaawi et al. (2021) presented a novel model of transgenic zebrafish expressing a fluorescent tau biosensor where TBIs by blast-like pressure waves induced progressive tauopathies. Tau aggregation positively correlated with TBI severity and the presence of seizure-like clonic shaking. Furthermore, tau aggregation following TBI administration was prevented by the anti-convulsant ezogabine and exacerbated by kainate treatment, demonstrating a role of seizure activity in mediating tauopathy development (Alyenbaawi et al., 2021).
A mechanism by which epileptogenesis gives rise to tau hyperphosphorylation may underlie the high incidence of tauopathy and epilepsy co-pathology that is found in diseases such as AD and dementia with Lewy bodies (Vossel et al., 2016, 2017; Beagle et al., 2017). Given that endogenous tau plays a role in regulating neuronal activity, disruption in the homeostatic balance of tau modifications may mediate seizure comorbidities observed in these diseases, and epileptic activity may in turn help drive tau hyperphosophorylation. In classical tauopathies where overt epilepsy infrequently occurs, tau hyperphosphorylation can arise from a variety of different causes (Kovacs, 2017). However, it is also possible that epileptic activity in these tauopathies is clinically underrecognized due to being non-motor or subclinical in nature, as suggested by the detection of subclinical epileptic activity in over 40% of AD patients during overnight electroencephalography and 1-h magnetoencephalogram recordings (Vossel et al., 2016). More studies involving extended periods of neurophysiological monitoring are therefore required to investigate the presence of epileptic activity and its potential contribution to tau hyperphosphorylation in primary tauopathies.
It should also be noted that tau pathology is not universally found in connection with epileptic activity. For instance, 31% of the post-mortem refractory epilepsy cases studied by Thom et al. (2011) were classified as Braak Stage 0, and analysis of surgically resected tissue from 56 TLE patients by Silva et al. (2021) found p-tau-positive neurons in only two samples. The absence of pathological tau deposition in these cases indicates that epileptogenesis does not always lead to tauopathies. However, the factors that determine the subsequent development of tau pathology in some cases of aberrant network excitability but not others remain unclear. It is possible that the formation of pathological tau deposits is linked to specific seizure disorders or that mechanisms mediating tau hyperphosphorylation are overactivated in cases of more severe epilepsy.
Dysregulation of Cell Signaling Activity Upstream of Tau Phosphorylation
Kinases
Given that the balance of tau phosphorylation states is regulated by enzymatic activity, investigations into the impairment of tau kinases and phosphatases in seizure disorders reveal links between epileptic activity and tau hyperphosphorylation. Investigations into novel pharmacological interventions targeting tau hyperphosphorylation in epilepsy have therefore concentrated on inhibiting and enhancing related phosphorylation and dephosphorylation mechanisms, respectively (Zheng et al., 2014; Ali et al., 2019).
One relevant kinase responsible for tau phosphorylation is glycogen synthase kinase-3β (GSK-3β) (Toral-Rios et al., 2020). Upregulation of GSK-3β is found in surgically resected tissue samples from patients with intractable epilepsy (Xi et al., 2009; Liu X. et al., 2017), and GSK-3β overactivation co-occurs with increased p-tau levels in mesial TLE patients (Liu C. et al., 2017). Inhibition of GSK-3β may have dual benefits given that GSK-3β inhibition reduces tau hyperphosphorylation and NFT formation in tau-overexpressing transgenic mice (Noble et al., 2005; Engel et al., 2006; Leroy et al., 2010) and produces anticonvulsant effects against pentylenetetrazol-induced seizures in zebrafish larvae (Aourz et al., 2019). However, the observation of sustained increases in p-tau levels following kainic acid administration being accompanied by only transient increases in GSK-3β activity (Liang et al., 2009) and the lack of effect on hippocampal p-tau by GSK-3β inhibitor pretreatment in the intra-amygdala kainic acid-induced status epilepticus mouse model (Alves et al., 2019) indicate that GSK-3β is not the only kinase responsible for tau phosphorylation following epileptic activity.
Another protein kinase highly implicated in tau phosphorylation is cyclin-dependent kinase 5 (CDK5). Dysregulation of CDK5 signaling can contribute to neurodegeneration, excitotoxicity, and tau hyperphosphorylation (Cruz et al., 2003). As is seen with GSK-3β, CDK5 overactivation is present in resected tissue from refractory epilepsy patients (Xi et al., 2009; Liu X. et al., 2017), and dysplastic cortical neurons in FCD patients express CDK5 aggregations (Sisodiya et al., 2002). Furthermore, progressive activation of CDK5 co-occurs with increasing tau phosphorylation in rodent seizure models (Chen et al., 2000; Liang et al., 2009), indicating significant mediation of seizure-associated tau hyperphosphorylation by CDK5. For example, in the genetic mouse model of NPC, increased activation of CDK5 and its activator, p25, coincides spatially and temporally with tau pathology, and CDK5 inhibition by roscovitine and olomoucine prevents cytoskeletal protein phosphorylation (Bu et al., 2002; Zhang et al., 2004, 2008).
Both GSK-3β and CDK5 play a role in neuronal excitability through involvement in GABAergic and glutamatergic neurotransmission, and inhibiting their activity can affect network activity through various mechanisms (Sen et al., 2008; Jaworski, 2020; Toral-Rios et al., 2020; Banerjee et al., 2021). Therefore, inhibiting these kinases should be approached with caution. For instance, genetic ablation of the CDK5 activator, p35, increases susceptibility to spontaneous seizures in mice (Chae et al., 1997). Tau levels were not measured in this study, but it is possible that the absence of activated CDK5 in this genetic model results in higher levels of dephosphorylated tau that contribute to neuronal hyperexcitability. These considerations highlight the complexity of kinase regulation in the setting of normal activity and hyperexcitable states. Targeting tau rather than upstream kinases may therefore be a more viable intervention option for seizure disorders (Figure 1).
Phosphatases
In addition to kinase activity, tau hyperphosphorylation associated with seizure activity may also be due to a lack of tau dephosphorylation by tau phosphatases. Following kainic acid administration in mice, biphasic changes in p-tau levels occur, where decreased phosphorylation is first observed within the first 6-h period followed by a gradual 3–5-fold increase until a 48-h endpoint. This progression is accompanied by a corresponding increase and then decrease in the activation of protein phosphatase 2A (PP2A) (Liang et al., 2009), which is estimated to account for 70% of human brain tau dephosphorylation (Wang and Mandelkow, 2016). It is possible that phosphatase activity is initially triggered to offset elevated tau phosphorylation caused by upregulated kinase activity following an epileptic event. For instance, GSK-3β upregulation causes PP2A activation (Wang et al., 2015). However, long-lasting phosphatase downregulation ultimately occurs, as evidenced by decreased PP2A activity paired with increased p-tau levels observed in epileptogenic brain regions following post-kainic acid status epilepticus, amygdala kindling, and fluid percussion injury in rats (Liu et al., 2016).
Similarly, abnormal p-tau in the form of NFTs are also observed in the Epm2a–/– mouse model (Puri et al., 2009), which replicates many of the features of Lafora disease caused by EPM2A mutations, including laforin deficiency, neuronal degeneration, spontaneous epileptic activity, and the development of Lafora bodies (Ganesh et al., 2002). Laforin is another tau phosphatase (Puri et al., 2009), though further research is required to investigate connections between hyperexcitability states and laforin downregulation in other seizure disorders. At least in the Epm2a–/– model, pathological tau levels are also associated with increased GSK-3β activation (Puri et al., 2009), suggesting that tau hyperphosphorylation is not mediated by the absence of laforin alone in Lafora disease.
Interestingly, the lack of phosphatase activity may also contribute to epileptic activity. Sodium selenate is a specific agonist for PP2A expressing the regulatory B subunit, an essential subunit for tau dephosphorylation by PP2A (Jones et al., 2012; Liu et al., 2016), and shows promise as a potential antiepileptic treatment option. Sodium selenate treatment attenuates seizure activity and tau hyperphosphorylation and accumulation following administration of pentylenetetrazol or kainic acid as well as in the TLE model of amygdala kindling and the fluid percussion injury model of PTE (Jones et al., 2012; Liu et al., 2016). The antiepileptic effects of sodium selenate persist following drug washout in animal TBI models (Liu et al., 2016), highlighting a potential disease-modifying effect of PP2A upregulation by sodium selenate during epileptogenesis when applied in early PTE disease stage.
The mechanisms through which tau dephosphorylation by phosphatase function alleviates epileptic activity remain unclear. Dephosphorylated tau at sufficient levels may be favorable in chronic epileptic states, or phosphatases may participate in independent signaling pathways that abate neuronal hyperexcitability. Regardless, as was proposed with tau kinases, long-term phosphatase inactivation may serve as a homeostatic response aimed at maintaining higher levels of phosphorylated tau and preventing endogenous tau from enabling network hyperexcitability (Figure 1). Taken together, the discussed findings indicate that seizures give rise to disruptions in the intricate balance of tau kinase and phosphatase activity and that the combined effects of kinase upregulation and phosphatase downregulation contribute to progressive tau hyperphosphorylation and accumulation in seizure disorders.
Mammalian Target of Rapamycin Pathways
The mammalian target of rapamycin (mTOR) is a highly conserved protein kinase that is implicated in a wide array of cellular and metabolic functions, including cell survival, growth, proliferation, migration, and differentiation (Wong, 2010; Mueed et al., 2018). Activation of mTOR is also a proposed driver of tau pathology given the involvement of tau-related kinases both upstream and downstream of mTOR signaling (Figure 2) and the contribution of mTOR-mediated autophagy dysfunction to tau hyperphosphorylation (Tramutola et al., 2017; Mueed et al., 2018). For instance, the downstream targets of mTOR activation include signaling cascades involving 4EBP1, S6K1, and CDK5, all of which result in tau phosphorylation (Mueed et al., 2018).
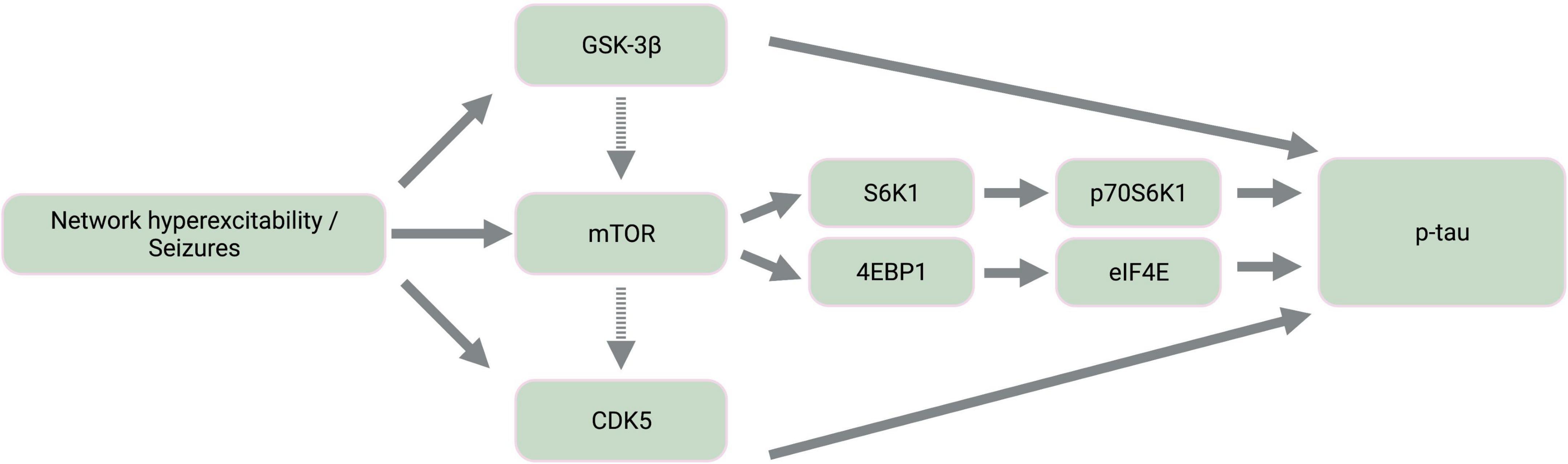
Figure 2. Simplified diagram of activated kinase signaling cascades in epilepsy. Epileptic activity leads to the activation of tau kinases, GSK-3β and CDK5, as well as mTOR. Dashed-line arrows indicate indirect activation of mTOR by GSK-3β through the mTOR complex 1 and of CDK5 by mTOR through amyloid-β aggregation and calpain activation. The downstream targets of mTOR activation involve the activation of additional tau kinases, p70S6K1 and eIF4E. GSK-3β, glycogen synthase kinase-3β; mTOR, mammalian target of rapamycin; CDK5, cyclin-dependent kinase 5; S6K1, ribosomal protein S6 kinase 1; 4EBP1, 4E binding protein 1; p70S6K1, phosphorylated S6K1; eIF4E, eukaryotic translation initiation factor 4E; p-tau, abnormally phosphorylated tau. Created with BioRender.com.
Furthermore, mTOR hyperactivation accompanies epileptic activity across different seizure models including animal models of TLE, PTE, FCDII, Dravet syndrome, ASD, and TSC (Meikle et al., 2007; Zeng et al., 2008; Sha et al., 2012; Guo et al., 2013; Gheyara et al., 2014; Butler et al., 2015; Marsan and Baulac, 2018; Tai et al., 2020; Shao et al., 2022). mTOR therefore likely contributes to tau and seizure co-pathology, warranting further pharmaceutical consideration of mTOR inhibition by rapamycin. Rapamycin treatment inhibits both tau hyperphosphorylation (Liu et al., 2013; Ozcelik et al., 2013; Tramutola et al., 2017) and the development of status epilepticus and chronic epilepsy in models of pharmacological seizure induction (Zeng et al., 2008; Huang et al., 2010), TLE (Drion et al., 2016), and PTE (Guo et al., 2013; Butler et al., 2015). Given that active GSK-3β also activates mTOR (Mueed et al., 2018), tau hyperphosphorylation resulting from seizure-associated GSK-3β upregulation may be further exacerbated by GSK-3β-mediated mTOR activation (Caccamo et al., 2013; Figure 2).
While the dysregulation of mTOR signaling pathways may manifest differentially across seizure disorders, the many connections between tau and mTOR highlight the significance of maintaining an optimal ratio of dephosphorylated and phosphorylated tau through balanced kinase/phosphatase regulation. Endogenous tau enables mTOR activation through a disinhibition mechanism whereby tau inhibits phosphatase and tensin homolog deleted chromosome 10 (PTEN), which normally inhibits mTOR (Tai et al., 2020; Figure 3). In genetic mouse models of ASD and Dravet syndrome, tau ablation prevents epilepsy and normalizes mTOR overactivation (Gheyara et al., 2014; Tai et al., 2020; Shao et al., 2022), suggesting that reducing tau may be beneficial in these diseases via PTEN disinhibition. mTOR also functions as a negative regulator of autophagy (Tramutola et al., 2017). Therefore, mTOR hyperactivity could prevent clearance of both normal and pathological tau (Chesser et al., 2013). Furthermore, reduced autophagy resulting from mTOR overactivation is implicated in elevated endogenous tau levels in the TSC2 mouse model of TSC (Caccamo et al., 2013). In seizure disorders such as TSC and NPC that are characterized by autophagy dysregulation (Pacheco et al., 2009; Caccamo et al., 2013), increase in normal tau levels may in turn contribute to both seizure activity and PTEN inhibition, creating a feedback loop of mTOR overactivation that results in further hyperphosphorylation of tau (Figure 3).
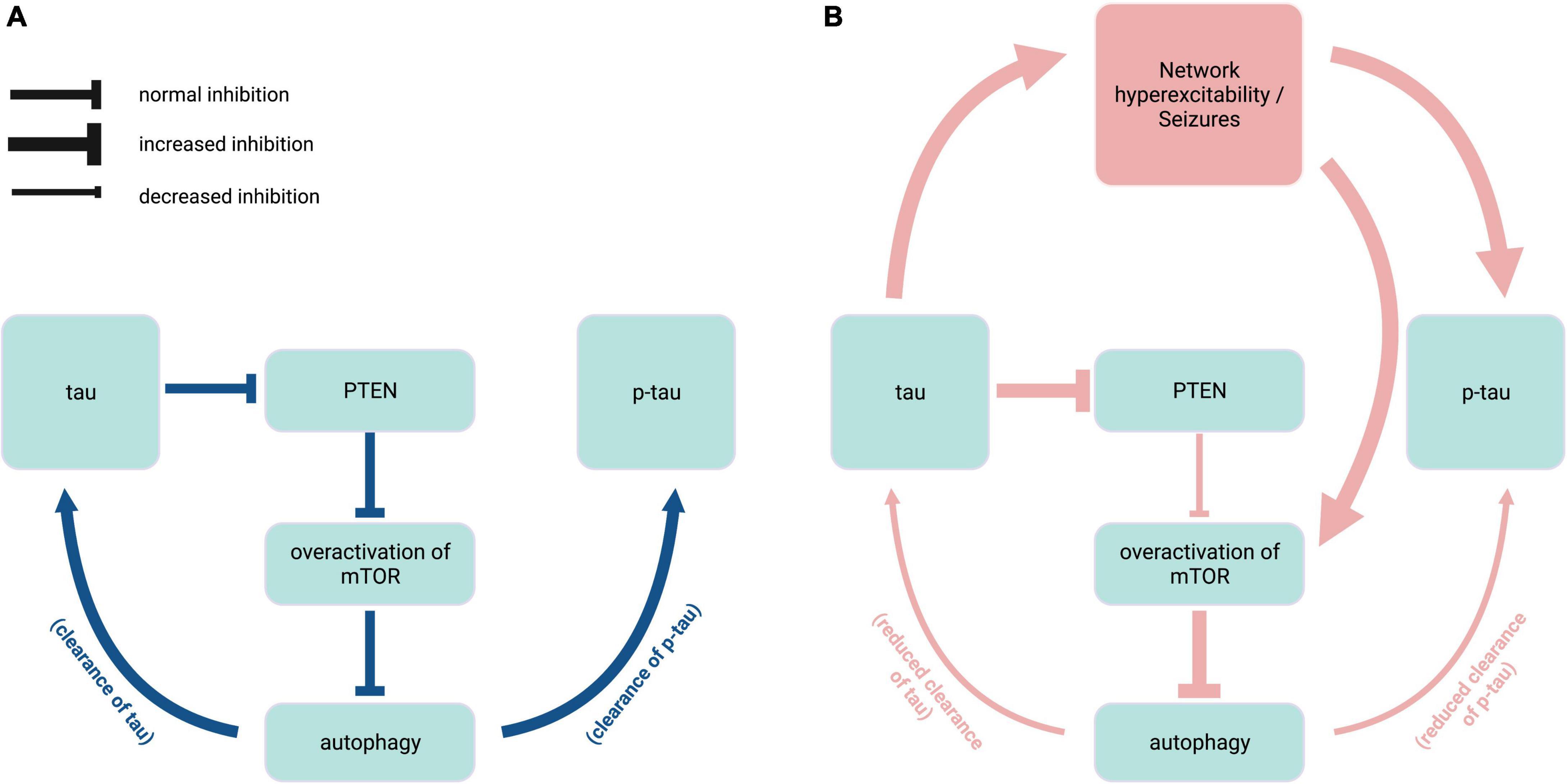
Figure 3. Interplay of endogenous tau, mTOR, and autophagy in epilepsy. (A) Under normal physiological conditions (blue), endogenous tau positively regulates mTOR activity via PTEN inhibition, and mTOR in turn negatively regulates autophagy mechanisms that contribute to the clearance of tau and p-tau. (B) In hyperexcitability states found in pathophysiological conditions such as tuberous sclerosis complex and Niemann-Pick type C disease (red), overactivation of mTOR due to increased PTEN inhibition causes excess inhibition of autophagy, resulting in reduced clearance of tau species. Elevated levels of normal tau in turn exacerbate epileptic activity and mTOR disinhibition. PTEN, phosphatase and tensin homolog deleted chromosome 10; mTOR, mammalian target of rapamycin; p-tau, phosphorylated tau. Created with BioRender.com.
Tau-Associated Cognitive Decline in Epilepsy Disorders
Cognitive impairment is a common comorbidity of seizure disorders. Though cognitive deficits can independently occur in seizure disorders as a direct result of disease etiology, such as trauma, epileptic activity contributes to, and exacerbates cognitive decline (Holmes, 2015). However, there are also investigations into a potential role of tau pathology in epilepsy-associated cognitive decline. While individuals with dementia have higher rates of epilepsy, seizures are experienced more frequently in tauopathy-associated dementias like AD than in other dementias (Sanchez et al., 2018). Cognitive decline is also accelerated in patients who have both AD and seizures compared to those with only AD (Vossel et al., 2017), suggesting that pathological tau and seizures can synergistically worsen cognitive outcomes.
Correlations between tau pathology and cognition are in fact observed in epilepsy. Post-mortem analysis of 138 refractory epilepsy cases revealed that 77% of patients with Braak staging III or higher exhibited progressive cognitive decline (Thom et al., 2011), and increased total and p-tau levels measured in surgically resected tissue from TLE patients are inversely correlated with cognitive scores (Kandratavicius et al., 2013; Tai et al., 2016; Gourmaud et al., 2020). In younger TLE patients, an association between post-operative naming decline and subtle tau hyperphosphorylation localized to only the subiculum and dentate gyrus suggest that tau-associated pathological changes in relevant brain regions over time may underlie progressive cognitive impairment seen in TLE (Prada Jardim et al., 2018). Furthermore, the neuroprotective effects of tau ablation against not only seizures but also cognitive deficits in animal models of ASD and Dravet syndrome (Gheyara et al., 2014; Tai et al., 2020) provide evidence for a role of tau in mediating cognitive impairment in these diseases as well. Therefore, tau pathology present in seizure disorders may exacerbate cognitive decline resulting from epileptic states, with seizure-driven tau hyperphosphorylation further compounding this effect with disease progression.
Conclusion
As presented in this review, a mounting body of literature has elucidated connections between tau pathology and epilepsy disorders of diverse etiologies. The antiseizure effect of tau ablation that can be reproduced in a variety of seizure models indicates a significant mediating role of endogenous tau in epileptogenesis. Given findings of upregulated kinase and downregulated phosphatase activity across different seizure disorders, we propose that epileptic activity can trigger homeostatic responses whereby enzymatic pathways disable endogenous tau by increased phosphorylation to stabilize aberrant network hyperexcitability. Subsequent hyperphosphorylation and accumulation of tau results from overactivation of such mechanisms, especially with recurring epileptic activity, and continuous epileptic states. Furthermore, growing evidence indicates a potential contribution of tau hyperphosphorylation to progressive cognitive decline in seizure disorders. Though the exact degrees to which tau involvement in seizures and cognitive decline are mediated by convergent or divergent mechanisms in distinct diseases remains unclear, the overlapping of tau-related cell signaling pathways and prevalence of tau hyperphosphorylation found throughout different types of epilepsies (Table 1) warrant continuing efforts into understanding epilepsies from a tauopathy perspective. Greater focus on tau in epileptic pathophysiology may yield advances in diagnostic and prognostic tools and novel therapeutic approaches targeting tau and tau-associated pathways.
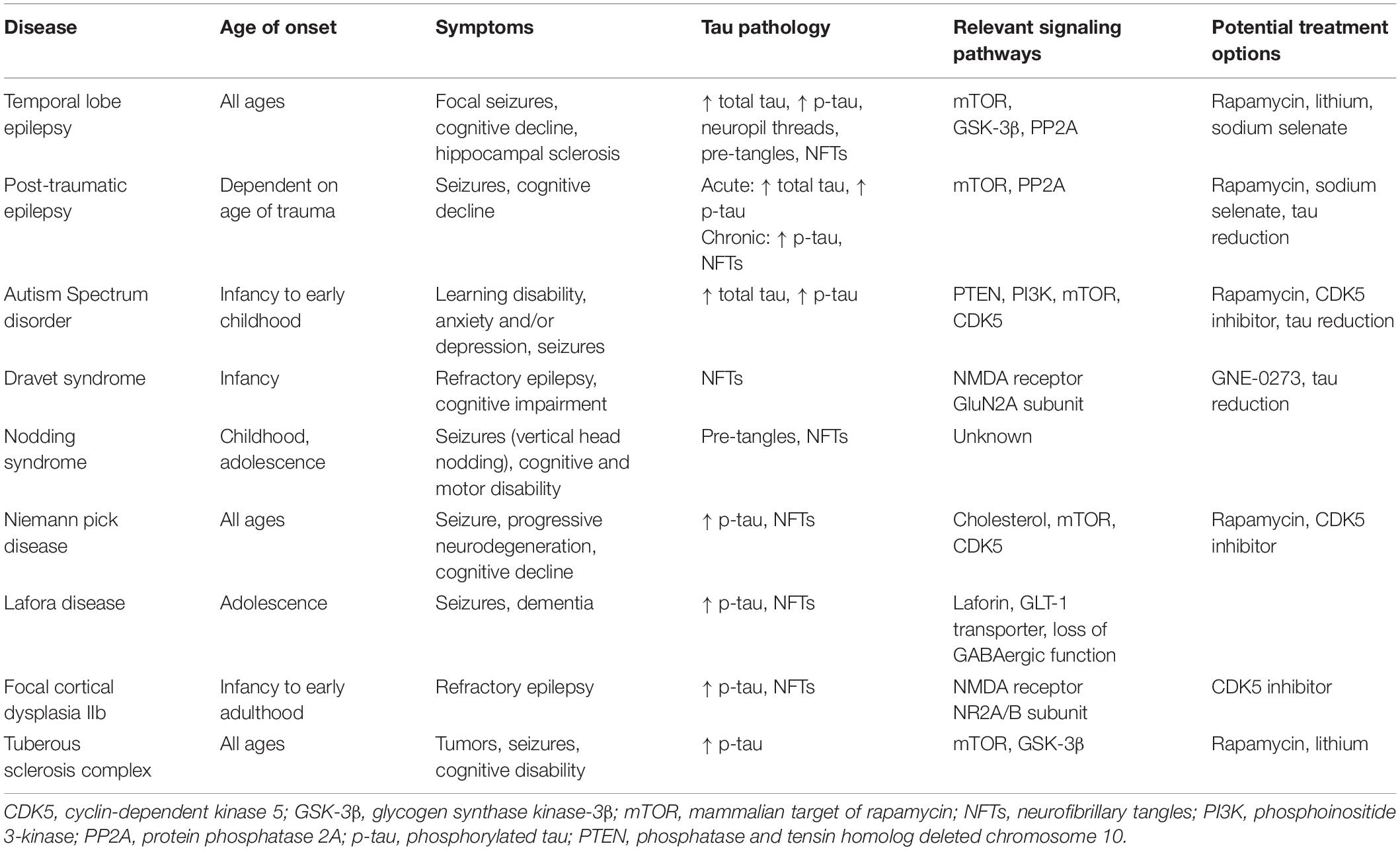
Table 1. Characterization of seizure disorders and their links to tau pathology and tau-associated mechanisms.
Author Contributions
KV and KH developed the manuscript concept. KH prepared the figure and table. All authors contributed to the manuscript and approved the final submitted version.
Funding
This work was supported by the NIH grants R01 AG058820, R01 AG075955, and R56 AG074473, and funding from the Fineberg Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ali, I., Silva, J. C., Liu, S., Shultz, S. R., Kwan, P., Jones, N. C., et al. (2019). Targeting neurodegeneration to prevent post-traumatic epilepsy. Neurobiol. Dis. 123, 100–109. doi: 10.1016/j.nbd.2018.08.006
Alves, M., Kenny, A., de Leo, G., Beamer, E. H., and Engel, T. (2019). Tau phosphorylation in a mouse model of temporal lobe epilepsy. Front. Aging Neurosci. 11:308. doi: 10.3389/fnagi.2019.00308
Alyenbaawi, H., Kanyo, R., Locskai, L. F., Kamali-Jamil, R., DuVal, M. G., Bai, Q., et al. (2021). Seizures are a druggable mechanistic link between TBI and subsequent tauopathy. eLife 10:e058744. doi: 10.7554/eLife.58744
Anwar, A., Saleem, S., Patel, U. K., Arumaithurai, K., and Malik, P. (2019). Dravet syndrome: an overview. Cureus 11:e5006. doi: 10.7759/cureus.5006
Aourz, N., Serruys, A. K., Chabwine, J. N., Balegamire, P. B., Afrikanova, T., Edrada-Ebel, R., et al. (2019). Identification of GSK-3 as a potential therapeutic entry point for epilepsy. ACS Chem. Neurosci. 10, 1992–2003. doi: 10.1021/acschemneuro.8b00281
Armstrong, M. J., Litvan, I., Lang, A. E., Bak, T. H., Bhatia, K. P., Borroni, B., et al. (2013). Criteria for the diagnosis of corticobasal degeneration. Neurology 80, 496–503. doi: 10.1212/WNL.0b013e31827f0fd1
Asadi-Pooya, A. A., Stewart, G. R., Abrams, D. J., and Sharan, A. (2017). Prevalence and incidence of drug-resistant mesial temporal lobe epilepsy in the United States. World Neurosurg. 99, 662–666. doi: 10.1016/j.wneu.2016.12.074
Auer, I. A., Schmidt, M. L., Lee, V. M., Curry, B., Suzuki, K., Shin, R. W., et al. (1995). Paired helical filament tau (PHFtau) in Niemann-Pick type C disease is similar to PHFtau in Alzheimer’s disease. Acta Neuropathol. 90, 547–551. doi: 10.1007/BF00318566
Banerjee, J., Srivastava, A., Sharma, D., Dey, S., Tripathi, M., Sharma, M. C., et al. (2021). Differential regulation of excitatory synaptic transmission in the hippocampus and anterior temporal lobe by cyclin dependent kinase 5 (Cdk5) in mesial temporal lobe epilepsy with hippocampal sclerosis (MTLE-HS). Neurosci. Lett. 761:136096. doi: 10.1016/j.neulet.2021.136096
Beagle, A. J., Darwish, S. M., Ranasinghe, K. G., La, A. L., Karageorgiou, E., and Vossel, K. A. (2017). Relative incidence of seizures and myoclonus in Alzheimer’s disease, dementia with lewy bodies, and frontotemporal dementia. J. Alzheimers Dis. 60, 211–223. doi: 10.3233/JAD-170031
Bourgeron, T. (2009). A synaptic trek to autism. Curr. Opin. Neurobiol. 19, 231–234. doi: 10.1016/j.conb.2009.06.003
Braak, H., Thal, D. R., Ghebremedhin, E., and Del Tredici, K. (2011). Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 70, 960–969. doi: 10.1097/NEN.0b013e318232a379
Bu, B., Li, J., Davies, P., and Vincent, I. (2002). Deregulation of cdk5, hyperphosphorylation, and cytoskeletal pathology in the Niemann-Pick type C murine model. J. Neurosci. 22, 6515–6525.
Butler, C. R., Boychuk, J. A., and Smith, B. N. (2015). Effects of rapamycin treatment on neurogenesis and synaptic reorganization in the dentate gyrus after controlled cortical impact injury in mice. Front. Syst. Neurosci. 9:163. doi: 10.3389/fnsys.2015.00163
Caccamo, A., Magri, A., Medina, D. X., Wisely, E. V., Lopez-Aranda, M. F., Silva, A. J., et al. (2013). mTOR regulates tau phosphorylation and degradation: implications for Alzheimer’s disease and other tauopathies. Aging Cell 12, 370–380. doi: 10.1111/acel.12057
Catterall, W. A., Kalume, F., and Oakley, J. C. (2010). NaV1.1 channels and epilepsy. J. Physiol. 588(Pt 11), 1849–1859. doi: 10.1113/jphysiol.2010.187484
Chae, T., Kwon, Y. T., Bronson, R., Dikkes, P., Li, E., and Tsai, L. H. (1997). Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron 18, 29–42. doi: 10.1016/s0896-6273(01)80044-1
Chang, C. W., Evans, M. D., Yu, X., Yu, G. Q., and Mucke, L. (2021). Tau reduction affects excitatory and inhibitory neurons differently, reduces excitation/inhibition ratios, and counteracts network hypersynchrony. Cell Rep. 37:109855. doi: 10.1016/j.celrep.2021.109855
Chen, J., Zhang, Y., Kelz, M. B., Steffen, C., Ang, E. S., Zeng, L., et al. (2000). Induction of cyclin-dependent kinase 5 in the hippocampus by chronic electroconvulsive seizures: role of [Delta]FosB. J. Neurosci. 20, 8965–8971. doi: 10.1523/JNEUROSCI.20-24-08965.2000
Chesser, A. S., Pritchard, S. M., and Johnson, G. V. (2013). Tau clearance mechanisms and their possible role in the pathogenesis of Alzheimer disease. Front. Neurol. 4:122. doi: 10.3389/fneur.2013.00122
Cho, S. J., Park, E., Telliyan, T., Baker, A., and Reid, A. Y. (2020). Zebrafish model of posttraumatic epilepsy. Epilepsia 61, 1774–1785. doi: 10.1111/epi.16589
Crary, J. F., Trojanowski, J. Q., Schneider, J. A., Abisambra, J. F., Abner, E. L., Alafuzoff, I., et al. (2014). Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 128, 755–766. doi: 10.1007/s00401-014-1349-0
Cruz, J. C., Tseng, H. C., Goldman, J. A., Shih, H., and Tsai, L. H. (2003). Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron 40, 471–483. doi: 10.1016/s0896-6273(03)00627-5
DeVos, S. L., Goncharoff, D. K., Chen, G., Kebodeaux, C. S., Yamada, K., Stewart, F. R., et al. (2013). Antisense reduction of tau in adult mice protects against seizures. J. Neurosci. 33, 12887–12897. doi: 10.1523/JNEUROSCI.2107-13.2013
Drion, C. M., Borm, L. E., Kooijman, L., Aronica, E., Wadman, W. J., Hartog, A. F., et al. (2016). Effects of rapamycin and curcumin treatment on the development of epilepsy after electrically induced status epilepticus in rats. Epilepsia 57, 688–697. doi: 10.1111/epi.13345
Engel, T., Goni-Oliver, P., Lucas, J. J., Avila, J., and Hernandez, F. (2006). Chronic lithium administration to FTDP-17 tau and GSK-3beta overexpressing mice prevents tau hyperphosphorylation and neurofibrillary tangle formation, but pre-formed neurofibrillary tangles do not revert. J. Neurochem. 99, 1445–1455. doi: 10.1111/j.1471-4159.2006.04139.x
Ganesh, S., Delgado-Escueta, A. V., Sakamoto, T., Avila, M. R., Machado-Salas, J., Hoshii, Y., et al. (2002). Targeted disruption of the Epm2a gene causes formation of Lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum. Mol. Genet. 11, 1251–1262. doi: 10.1093/hmg/11.11.1251
Garcia-Cabrero, A. M., Guerrero-Lopez, R., Giraldez, B. G., Llorens-Martin, M., Avila, J., Serratosa, J. M., et al. (2013). Hyperexcitability and epileptic seizures in a model of frontotemporal dementia. Neurobiol. Dis. 58, 200–208. doi: 10.1016/j.nbd.2013.06.005
Gassowska-Dobrowolska, M., Kolasa-Wolosiuk, A., Cieslik, M., Dominiak, A., Friedland, K., and Adamczyk, A. (2021). Alterations in tau protein level and phosphorylation state in the brain of the autistic-like rats induced by prenatal exposure to valproic acid. Int. J. Mol. Sci. 22:3209. doi: 10.3390/ijms22063209
Gheyara, A. L., Ponnusamy, R., Djukic, B., Craft, R. J., Ho, K., Guo, W., et al. (2014). Tau reduction prevents disease in a mouse model of Dravet syndrome. Ann. Neurol. 76, 443–456. doi: 10.1002/ana.24230
Gourmaud, S., Shou, H., Irwin, D. J., Sansalone, K., Jacobs, L. M., Lucas, T. H., et al. (2020). Alzheimer-like amyloid and tau alterations associated with cognitive deficit in temporal lobe epilepsy. Brain 143, 191–209. doi: 10.1093/brain/awz381
Guo, D., Zeng, L., Brody, D. L., and Wong, M. (2013). Rapamycin attenuates the development of posttraumatic epilepsy in a mouse model of traumatic brain injury. PLoS One 8:e64078. doi: 10.1371/journal.pone.0064078
Hara, H. (2007). Autism and epilepsy: a retrospective follow-up study. Brain Dev. 29, 486–490. doi: 10.1016/j.braindev.2006.12.012
Hay, J., Johnson, V. E., Smith, D. H., and Stewart, W. (2016). Chronic traumatic encephalopathy: the neuropathological legacy of traumatic brain injury. Annu. Rev. Pathol. 11, 21–45. doi: 10.1146/annurev-pathol-012615-044116
Holmes, G. L. (2015). Cognitive impairment in epilepsy: the role of network abnormalities. Epileptic Disord. 17, 101–116. doi: 10.1684/epd.2015.0739
Holth, J. K., Bomben, V. C., Reed, J. G., Inoue, T., Younkin, L., Younkin, S. G., et al. (2013). Tau loss attenuates neuronal network hyperexcitability in mouse and Drosophila genetic models of epilepsy. J. Neurosci. 33, 1651–1659. doi: 10.1523/JNEUROSCI.3191-12.2013
Horvath, A. A., Papp, A., Zsuffa, J., Szucs, A., Luckl, J., Radai, F., et al. (2021). Subclinical epileptiform activity accelerates the progression of Alzheimer’s disease: a long-term EEG study. Clin. Neurophysiol. 132, 1982–1989. doi: 10.1016/j.clinph.2021.03.050
Hotterbeekx, A., Lammens, M., Idro, R., Akun, P. R., Lukande, R., Akena, G., et al. (2019). Neuroinflammation and not tauopathy is a predominant pathological signature of nodding syndrome. J. Neuropathol. Exp. Neurol. 78, 1049–1058. doi: 10.1093/jnen/nlz090
Huang, X., Zhang, H., Yang, J., Wu, J., McMahon, J., Lin, Y., et al. (2010). Pharmacological inhibition of the mammalian target of rapamycin pathway suppresses acquired epilepsy. Neurobiol. Dis. 40, 193–199. doi: 10.1016/j.nbd.2010.05.024
Ittner, L. M., Ke, Y. D., Delerue, F., Bi, M., Gladbach, A., van Eersel, J., et al. (2010). Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 142, 387–397. doi: 10.1016/j.cell.2010.06.036
Iyer, A., Prabowo, A., Anink, J., Spliet, W. G., van Rijen, P. C., and Aronica, E. (2014). Cell injury and premature neurodegeneration in focal malformations of cortical development. Brain Pathol. 24, 1–17. doi: 10.1111/bpa.12060
Jaworski, T. (2020). Control of neuronal excitability by GSK-3beta: epilepsy and beyond. Biochim. Biophys. Acta Mol. Cell Res. 1867:118745. doi: 10.1016/j.bbamcr.2020.118745
Johnson, V. E., Stewart, W., and Smith, D. H. (2012). Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol. 22, 142–149. doi: 10.1111/j.1750-3639.2011.00513.x
Jones, N. C., Nguyen, T., Corcoran, N. M., Velakoulis, D., Chen, T., Grundy, R., et al. (2012). Targeting hyperphosphorylated tau with sodium selenate suppresses seizures in rodent models. Neurobiol. Dis. 45, 897–901. doi: 10.1016/j.nbd.2011.12.005
Kandratavicius, L., Monteiro, M. R., Hallak, J. E., Carlotti, C. G. Jr., Assirati, J. A. Jr., and Leite, J. P. (2013). Microtubule-associated proteins in mesial temporal lobe epilepsy with and without psychiatric comorbidities and their relation with granular cell layer dispersion. Biomed. Res. Int. 2013:960126. doi: 10.1155/2013/960126
Kovacs, G. G. (2017). Tauopathies. Handb. Clin. Neurol. 145, 355–368. doi: 10.1016/B978-0-12-802395-2.00025-0
Kovacs, S. K., Leonessa, F., and Ling, G. S. (2014). Blast TBI models, neuropathology, and implications for seizure risk. Front. Neurol. 5:47. doi: 10.3389/fneur.2014.00047
Lam, A. D., Sarkis, R. A., Pellerin, K. R., Jing, J., Dworetzky, B. A., Hoch, D. B., et al. (2020). Association of epileptiform abnormalities and seizures in Alzheimer disease. Neurology 95, e2259–e2270. doi: 10.1212/WNL.0000000000010612
Leroy, K., Ando, K., Heraud, C., Yilmaz, Z., Authelet, M., Boeynaems, J. M., et al. (2010). Lithium treatment arrests the development of neurofibrillary tangles in mutant tau transgenic mice with advanced neurofibrillary pathology. J. Alzheimers Dis. 19, 705–719. doi: 10.3233/JAD-2010-1276
Li, B. M., Liu, X. R., Yi, Y. H., Deng, Y. H., Su, T., Zou, X., et al. (2011). Autism in Dravet syndrome: prevalence, features, and relationship to the clinical characteristics of epilepsy and mental retardation. Epilepsy Behav. 21, 291–295. doi: 10.1016/j.yebeh.2011.04.060
Liang, Z., Liu, F., Iqbal, K., Grundke-Iqbal, I., and Gong, C. X. (2009). Dysregulation of tau phosphorylation in mouse brain during excitotoxic damage. J. Alzheimers Dis. 17, 531–539. doi: 10.3233/JAD-2009-1069
Liu, A. J., Staffaroni, A. M., Rojas-Martinez, J. C., Olney, N. T., Alquezar-Burillo, C., Ljubenkov, P. A., et al. (2020). Association of cognitive and behavioral features between adults with tuberous sclerosis and frontotemporal dementia. JAMA Neurol. 77, 358–366. doi: 10.1001/jamaneurol.2019.4284
Liu, C., Russin, J., Heck, C., Kawata, K., Adiga, R., Yen, W., et al. (2017). Dysregulation of PINCH signaling in mesial temporal epilepsy. J. Clin. Neurosci. 36, 43–52. doi: 10.1016/j.jocn.2016.10.012
Liu, S., Shen, Y., Shultz, S. R., Nguyen, A., Hovens, C., Adlard, P. A., et al. (2017). Accelerated kindling epileptogenesis in Tg4510 tau transgenic mice, but not in tau knockout mice. Epilepsia 58, e136–e141. doi: 10.1111/epi.13847
Liu, S. J., Zheng, P., Wright, D. K., Dezsi, G., Braine, E., Nguyen, T., et al. (2016). Sodium selenate retards epileptogenesis in acquired epilepsy models reversing changes in protein phosphatase 2A and hyperphosphorylated tau. Brain 139(Pt 7), 1919–1938. doi: 10.1093/brain/aww116
Liu, X., Ou, S., Yin, M., Xu, T., Wang, T., Liu, Y., et al. (2017). N-methyl-D-aspartate receptors mediate epilepsy-induced axonal impairment and tau phosphorylation via activating glycogen synthase kinase-3beta and cyclin-dependent kinase 5. Discov. Med. 23, 221–234.
Liu, Y., Su, Y., Wang, J., Sun, S., Wang, T., Qiao, X., et al. (2013). Rapamycin decreases tau phosphorylation at Ser214 through regulation of cAMP-dependent kinase. Neurochem. Int. 62, 458–467. doi: 10.1016/j.neuint.2013.01.014
Love, S., Bridges, L. R., and Case, C. P. (1995). Neurofibrillary tangles in niemann-pick disease type C. Brain 118(Pt 1), 119–129. doi: 10.1093/brain/118.1.119
Machado-Salas, J., Avila-Costa, M. R., Guevara, P., Guevara, J., Duron, R. M., Bai, D., et al. (2012). Ontogeny of Lafora bodies and neurocytoskeleton changes in Laforin-deficient mice. Exp. Neurol. 236, 131–140. doi: 10.1016/j.expneurol.2012.04.008
Malnar, M., Hecimovic, S., Mattsson, N., and Zetterberg, H. (2014). Bidirectional links between Alzheimer’s disease and Niemann-Pick type C disease. Neurobiol. Dis. 72, 37–47. doi: 10.1016/j.nbd.2014.05.033
Marsan, E., and Baulac, S. (2018). Review: mechanistic target of rapamycin (mTOR) pathway, focal cortical dysplasia and epilepsy. Neuropathol. Appl. Neurobiol. 44, 6–17. doi: 10.1111/nan.12463
Meikle, L., Talos, D. M., Onda, H., Pollizzi, K., Rotenberg, A., Sahin, M., et al. (2007). A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J. Neurosci. 27, 5546–5558. doi: 10.1523/JNEUROSCI.5540-06.2007
Monti, G., Tondelli, M., Giovannini, G., Bedin, R., Nichelli, P. F., Trenti, T., et al. (2015). Cerebrospinal fluid tau proteins in status epilepticus. Epilepsy Behav. 49, 150–154. doi: 10.1016/j.yebeh.2015.04.030
Morris, M., Maeda, S., Vossel, K., and Mucke, L. (2011). The many faces of tau. Neuron 70, 410–426. doi: 10.1016/j.neuron.2011.04.009
Mueed, Z., Tandon, P., Maurya, S. K., Deval, R., Kamal, M. A., and Poddar, N. K. (2018). Tau and mTOR: the hotspots for multifarious diseases in Alzheimer’s development. Front. Neurosci. 12:1017. doi: 10.3389/fnins.2018.01017
Nelson, S. B., and Valakh, V. (2015). Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron 87, 684–698. doi: 10.1016/j.neuron.2015.07.033
Noble, W., Planel, E., Zehr, C., Olm, V., Meyerson, J., Suleman, F., et al. (2005). Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc. Natl. Acad. Sci. U.S.A. 102, 6990–6995. doi: 10.1073/pnas.0500466102
Ozcelik, S., Fraser, G., Castets, P., Schaeffer, V., Skachokova, Z., Breu, K., et al. (2013). Rapamycin attenuates the progression of tau pathology in P301S tau transgenic mice. PLoS One 8:e62459. doi: 10.1371/journal.pone.0062459
Pacheco, C. D., Elrick, M. J., and Lieberman, A. P. (2009). Tau normal function influences Niemann-Pick type C disease pathogenesis in mice and modulates autophagy in NPC1-deficient cells. Autophagy 5, 548–550. doi: 10.4161/auto.5.4.8364
Petraglia, A. L., Plog, B. A., Dayawansa, S., Dashnaw, M. L., Czerniecka, K., Walker, C. T., et al. (2014). The pathophysiology underlying repetitive mild traumatic brain injury in a novel mouse model of chronic traumatic encephalopathy. Surg. Neurol. Int. 5:184. doi: 10.4103/2152-7806.147566
Pollanen, M. S., Onzivua, S., Robertson, J., McKeever, P. M., Olawa, F., Kitara, D. L., et al. (2018). Nodding syndrome in Uganda is a tauopathy. Acta Neuropathol. 136, 691–697. doi: 10.1007/s00401-018-1909-9
Pooler, A. M., Phillips, E. C., Lau, D. H., Noble, W., and Hanger, D. P. (2013). Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 14, 389–394. doi: 10.1038/embor.2013.15
Prada Jardim, A., Liu, J., Baber, J., Michalak, Z., Reeves, C., Ellis, M., et al. (2018). Characterising subtypes of hippocampal sclerosis and reorganization: correlation with pre and postoperative memory deficit. Brain Pathol. 28, 143–154. doi: 10.1111/bpa.12514
Puri, R., Suzuki, T., Yamakawa, K., and Ganesh, S. (2009). Hyperphosphorylation and aggregation of Tau in laforin-deficient mice, an animal model for Lafora disease. J. Biol. Chem. 284, 22657–22663. doi: 10.1074/jbc.M109.009688
Puvenna, V., Engeler, M., Banjara, M., Brennan, C., Schreiber, P., Dadas, A., et al. (2016). Is phosphorylated tau unique to chronic traumatic encephalopathy? Phosphorylated tau in epileptic brain and chronic traumatic encephalopathy. Brain Res. 1630, 225–240. doi: 10.1016/j.brainres.2015.11.007
Reilly, C., Atkinson, P., Das, K. B., Chin, R. F., Aylett, S. E., Burch, V., et al. (2015). Features of autism spectrum disorder (ASD) in childhood epilepsy: a population-based study. Epilepsy Behav. 42, 86–92. doi: 10.1016/j.yebeh.2014.11.014
Roberson, E. D., Scearce-Levie, K., Palop, J. J., Yan, F., Cheng, I. H., Wu, T., et al. (2007). Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 316, 750–754. doi: 10.1126/science.1141736
Rubenstein, R., Chang, B., Davies, P., Wagner, A. K., Robertson, C. S., and Wang, K. K. (2015). A novel, ultrasensitive assay for tau: potential for assessing traumatic brain injury in tissues and biofluids. J. Neurotrauma 32, 342–352. doi: 10.1089/neu.2014.3548
Sanchez, M. P., Garcia-Cabrero, A. M., Sanchez-Elexpuru, G., Burgos, D. F., and Serratosa, J. M. (2018). Tau-induced pathology in epilepsy and dementia: notions from patients and animal models. Int. J. Mol. Sci. 19:1092. doi: 10.3390/ijms19041092
Sarnat, H. B., and Flores-Sarnat, L. (2015). Infantile tauopathies: hemimegalencephaly; tuberous sclerosis complex; focal cortical dysplasia 2; ganglioglioma. Brain Dev. 37, 553–562. doi: 10.1016/j.braindev.2014.08.010
Sen, A., Thom, M., Martinian, L., Harding, B., Cross, J. H., Nikolic, M., et al. (2007). Pathological tau tangles localize to focal cortical dysplasia in older patients. Epilepsia 48, 1447–1454. doi: 10.1111/j.1528-1167.2007.01107.x
Sen, A., Thom, M., Nikolic, M., and Sisodiya, S. M. (2008). The potential role of cyclin-dependent kinase 5 in focal cortical dysplasia. Dev. Neurosci. 30, 96–104. doi: 10.1159/000109855
Sha, L. Z., Xing, X. L., Zhang, D., Yao, Y., Dou, W. C., Jin, L. R., et al. (2012). Mapping the spatio-temporal pattern of the mammalian target of rapamycin (mTOR) activation in temporal lobe epilepsy. PLoS One 7:e39152. doi: 10.1371/journal.pone.0039152
Shao, E., Chang, C. W., Li, Z., Yu, X., Ho, K., Zhang, M., et al. (2022). TAU ablation in excitatory neurons and postnatal TAU knockdown reduce epilepsy, SUDEP, and autism behaviors in a Dravet syndrome model. Sci. Transl. Med. 14:eabm5527. doi: 10.1126/scitranslmed.abm5527
Sharma, V., Saini, A. G., Malhi, P., and Singhi, P. (2021). Epilepsy and EEG abnormalities in children with autism spectrum disorders. Indian J. Pediatr. [Epub ahead of print]. doi: 10.1007/s12098-021-03928-w
Silva, J. C., Vivash, L., Malpas, C. B., Hao, Y., McLean, C., Chen, Z., et al. (2021). Low prevalence of amyloid and tau pathology in drug-resistant temporal lobe epilepsy. Epilepsia 62, 3058–3067. doi: 10.1111/epi.17086
Sisodiya, S. M., Thom, M., Lin, W. R., Bajaj, N. P., Cross, J. H., and Harding, B. N. (2002). Abnormal expression of cdk5 in focal cortical dysplasia in humans. Neurosci. Lett. 328, 217–220. doi: 10.1016/s0304-3940(02)00520-7
Smith, K. M., Blessing, M. M., Parisi, J. E., Britton, J. W., Mandrekar, J., and Cascino, G. D. (2019). Tau deposition in young adults with drug-resistant focal epilepsy. Epilepsia 60, 2398–2403. doi: 10.1111/epi.16375
Specchio, N., Di Micco, V., Trivisano, M., Ferretti, A., and Curatolo, P. (2022). The epilepsy-autism spectrum disorder phenotype in the era of molecular genetics and precision therapy. Epilepsia 63, 6–21. doi: 10.1111/epi.17115
Sundelin, H. E., Larsson, H., Lichtenstein, P., Almqvist, C., Hultman, C. M., Tomson, T., et al. (2016). Autism and epilepsy: a population-based nationwide cohort study. Neurology 87, 192–197. doi: 10.1212/WNL.0000000000002836
Suzuki, K., Parker, C. C., Pentchev, P. G., Katz, D., Ghetti, B., D’Agostino, A. N., et al. (1995). Neurofibrillary tangles in Niemann-Pick disease type C. Acta Neuropathol. 89, 227–238. doi: 10.1007/BF00309338
Tai, C., Chang, C. W., Yu, G. Q., Lopez, I., Yu, X., Wang, X., et al. (2020). Tau reduction prevents key features of autism in mouse models. Neuron 106, 421.e1–437.e1. doi: 10.1016/j.neuron.2020.01.038
Tai, X. Y., Bernhardt, B., Thom, M., Thompson, P., Baxendale, S., Koepp, M., et al. (2018). Review: neurodegenerative processes in temporal lobe epilepsy with hippocampal sclerosis: clinical, pathological and neuroimaging evidence. Neuropathol. Appl. Neurobiol. 44, 70–90. doi: 10.1111/nan.12458
Tai, X. Y., Koepp, M., Duncan, J. S., Fox, N., Thompson, P., Baxendale, S., et al. (2016). Hyperphosphorylated tau in patients with refractory epilepsy correlates with cognitive decline: a study of temporal lobe resections. Brain 139(Pt 9), 2441–2455. doi: 10.1093/brain/aww187
Tan, X. L., Zheng, P., Wright, D. K., Sun, M., Brady, R. D., Liu, S., et al. (2020). The genetic ablation of tau improves long-term, but not short-term, functional outcomes after experimental traumatic brain injury in mice. Brain Inj 34, 131–139. doi: 10.1080/02699052.2019.1667539
Tellez-Zenteno, J. F., and Hernandez-Ronquillo, L. (2012). A review of the epidemiology of temporal lobe epilepsy. Epilepsy Res. Treat. 2012:630853. doi: 10.1155/2012/630853
Thom, M., Liu, J. Y., Thompson, P., Phadke, R., Narkiewicz, M., Martinian, L., et al. (2011). Neurofibrillary tangle pathology and Braak staging in chronic epilepsy in relation to traumatic brain injury and hippocampal sclerosis: a post-mortem study. Brain 134(Pt 10), 2969–2981. doi: 10.1093/brain/awr209
Toral-Rios, D., Pichardo-Rojas, P. S., Alonso-Vanegas, M., and Campos-Pena, V. (2020). GSK3beta and Tau protein in Alzheimer’s disease and epilepsy. Front. Cell Neurosci. 14:19. doi: 10.3389/fncel.2020.00019
Trabzuni, D., Wray, S., Vandrovcova, J., Ramasamy, A., Walker, R., Smith, C., et al. (2012). MAPT expression and splicing is differentially regulated by brain region: relation to genotype and implication for tauopathies. Hum. Mol. Genet. 21, 4094–4103. doi: 10.1093/hmg/dds238
Tramutola, A., Lanzillotta, C., and Di Domenico, F. (2017). Targeting mTOR to reduce Alzheimer-related cognitive decline: from current hits to future therapies. Expert Rev. Neurother. 17, 33–45. doi: 10.1080/14737175.2017.1244482
Tumani, H., Jobs, C., Brettschneider, J., Hoppner, A. C., Kerling, F., and Fauser, S. (2015). Effect of epileptic seizures on the cerebrospinal fluid–A systematic retrospective analysis. Epilepsy Res. 114, 23–31. doi: 10.1016/j.eplepsyres.2015.04.004
Viscidi, E. W., Triche, E. W., Pescosolido, M. F., McLean, R. L., Joseph, R. M., Spence, S. J., et al. (2013). Clinical characteristics of children with autism spectrum disorder and co-occurring epilepsy. PLoS One 8:e67797. doi: 10.1371/journal.pone.0067797
Vossel, K. A., Ranasinghe, K. G., Beagle, A. J., Mizuiri, D., Honma, S. M., Dowling, A. F., et al. (2016). Incidence and impact of subclinical epileptiform activity in Alzheimer’s disease. Ann. Neurol. 80, 858–870. doi: 10.1002/ana.24794
Vossel, K. A., Tartaglia, M. C., Nygaard, H. B., Zeman, A. Z., and Miller, B. L. (2017). Epileptic activity in Alzheimer’s disease: causes and clinical relevance. Lancet Neurol. 16, 311–322. doi: 10.1016/S1474-4422(17)30044-3
Wang, Y., and Mandelkow, E. (2016). Tau in physiology and pathology. Nat. Rev. Neurosci. 17, 5–21. doi: 10.1038/nrn.2015.1
Wang, Y., Yang, R., Gu, J., Yin, X., Jin, N., Xie, S., et al. (2015). Cross talk between PI3K-AKT-GSK-3beta and PP2A pathways determines tau hyperphosphorylation. Neurobiol. Aging 36, 188–200. doi: 10.1016/j.neurobiolaging.2014.07.035
Wong, M. (2010). Mammalian target of rapamycin (mTOR) inhibition as a potential antiepileptogenic therapy: from tuberous sclerosis to common acquired epilepsies. Epilepsia 51, 27–36. doi: 10.1111/j.1528-1167.2009.02341.x
Woolfenden, S., Sarkozy, V., Ridley, G., Coory, M., and Williams, K. (2012). A systematic review of two outcomes in autism spectrum disorder - epilepsy and mortality. Dev. Med. Child Neurol. 54, 306–312. doi: 10.1111/j.1469-8749.2012.04223.x
Xi, Z. Q., Wang, X. F., Shu, X. F., Chen, G. J., Xiao, F., Sun, J. J., et al. (2011). Is intractable epilepsy a tauopathy? Med. Hypotheses. 76, 897–900. doi: 10.1016/j.mehy.2011.03.003
Xi, Z. Q., Xiao, F., Yuan, J., Wang, X. F., Wang, L., Quan, F. Y., et al. (2009). Gene expression analysis on anterior temporal neocortex of patients with intractable epilepsy. Synapse 63, 1017–1028. doi: 10.1002/syn.20681
Yamada, K., Holth, J. K., Liao, F., Stewart, F. R., Mahan, T. E., Jiang, H., et al. (2014). Neuronal activity regulates extracellular tau in vivo. J. Exp. Med. 211, 387–393. doi: 10.1084/jem.20131685
Zanier, E. R., Bertani, I., Sammali, E., Pischiutta, F., Chiaravalloti, M. A., Vegliante, G., et al. (2018). Induction of a transmissible tau pathology by traumatic brain injury. Brain 141, 2685–2699. doi: 10.1093/brain/awy193
Zeng, L. H., Xu, L., Gutmann, D. H., and Wong, M. (2008). Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann. Neurol. 63, 444–453. doi: 10.1002/ana.21331
Zhang, M., Hallows, J. L., Wang, X., Bu, B., Wang, W., and Vincent, I. (2008). Mitogen-activated protein kinase activity may not be necessary for the neuropathology of Niemann-Pick type C mice. J. Neurochem. 107, 814–822. doi: 10.1111/j.1471-4159.2008.05657.x
Zhang, M., Li, J., Chakrabarty, P., Bu, B., and Vincent, I. (2004). Cyclin-dependent kinase inhibitors attenuate protein hyperphosphorylation, cytoskeletal lesion formation, and motor defects in Niemann-Pick Type C mice. Am. J. Pathol. 165, 843–853. doi: 10.1016/S0002-9440(10)63347-0
Keywords: tau, epilepsy, mTOR, hyperexcitability, hyperphosphorylation of tau, cognitive decline
Citation: Hwang K, Vaknalli RN, Addo-Osafo K, Vicente M and Vossel K (2022) Tauopathy and Epilepsy Comorbidities and Underlying Mechanisms. Front. Aging Neurosci. 14:903973. doi: 10.3389/fnagi.2022.903973
Received: 24 March 2022; Accepted: 22 June 2022;
Published: 18 July 2022.
Edited by:
Salvatore Spina, University of California, San Francisco, United StatesReviewed by:
Ettore Salsano, IRCCS Carlo Besta Neurological Institute Foundation, ItalyVictoria Campos-Peña, Manuel Velasco Suárez Instituto Nacional de Neurología y Neurocirugía, Mexico
Copyright © 2022 Hwang, Vaknalli, Addo-Osafo, Vicente and Vossel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kaylin Hwang, khhwang@g.ucla.edu; Keith Vossel, KVossel@mednet.ucla.edu