Presenilin Deficiency Increases Susceptibility to Oxidative Damage in Fibroblasts
 Kun Zou1*
Kun Zou1*  Sadequl Islam1
Sadequl Islam1  Yang Sun1
Yang Sun1  Yuan Gao1
Yuan Gao1  Tomohisa Nakamura1 Hiroto Komano2 Taisuke Tomita3
Tomohisa Nakamura1 Hiroto Komano2 Taisuke Tomita3  Makoto Michikawa1*
Makoto Michikawa1*- 1Department of Biochemistry, Graduate School of Medical Sciences, Nagoya City University, Nagoya, Japan
- 2Advanced Prevention and Research Laboratory for Dementia, Faculty of Pharmaceutical Sciences, Hokkaido University, Sapporo, Japan
- 3Laboratory of Neuropathology and Neuroscience, Faculty of Pharmaceutical Sciences, University of Tokyo, Bunkyo City, Japan
Alzheimer’s disease (AD) is a genetic and sporadic neurodegenerative disease characterized by extracellular amyloid-β-protein (Aβ) aggregates as amyloid plaques and neuronal loss in the brain parenchyma of patients. Familial AD (FAD) is found to be genetically linked to missense mutations either in presenilin (PS) or amyloid precursor protein (APP). Most of PS mutations increase Aβ42/Aβ40 ratio, which is thought to result in early amyloid deposition in brain. However, PS deficiency in the fore brain of adult mouse leads to neuronal loss in an Aβ independent manner and the underlying mechanism is largely unknown. In this study, we found that reactive oxygen species (ROS) are increased in PS deficient fibroblasts and that H2O2 and ferrous sulfate treatment produced more ROS in PS deficient fibroblasts than in wild-type fibroblasts. PS deficient fibroblasts showed significantly decreased cellular ferritin levels compared with wild-type fibroblasts, suggesting reduced iron sequestrating capability in PS deficient cells. Blockade of γ-secretase activity by a γ-secretase inhibitor, DAPT, decreased ferritin levels, indicating that γ-secretase activity is important for maintaining its levels. Moreover, overexpression PS1 mutants in wild-type fibroblasts decreased ferritin light chain levels and enhanced intracellular ROS levels. Our results suggest that dysfunction of PS may reduce intracellular ferritin levels and is involved in AD pathogenesis through increasing susceptibility to oxidative damage.
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by extracellular amyloid plaques in brain parenchyma, intracellular neurofibrillary tangles and neuronal loss (McKhann et al., 1984; Knopman et al., 2021). The major components of amyloid plaques are amyloid β-proteins (Aβ) consisting of 40–43 amino acids, which are generated from amyloid precursor protein (APP) by β-secretase and γ-secretase (Shoji et al., 1992; De Strooper et al., 2012). The accumulation of Aβ occurs 15–20 years prior to AD symptoms and it is one of the earliest pathological events in AD brain (Scheltens et al., 2016). Aβ is hypothesized to be the causative molecule for AD because it has neurotoxic effects, which is called amyloid cascade hypothesis (Selkoe and Hardy, 2016).
AD is clinically divided into sporadic AD (SAD) caused by multiple risk factors, including lifestyle-related factors, and familial AD (FAD) caused by inherited mutations of specific genes. SAD accounts for more than 95% of all AD cases and it is hypothesized to be induced by reduced clearance of Aβ. Genetic causes for FAD include dominantly inherited mutations of the genes encoding amyloid precursor protein (APP), presenilin 1 (PSEN1, PS1) and PSEN2 (PS2). PS1 and PS2 provide the catalytic subunit to the γ secretase complex, which contains PS, nicastrin, Aph-1 and PEN-2 (De Strooper et al., 2012). Most of the mutations found in PSEN result in increased Aβ42 production or decreased Aβ40 production, leading to an increased Aβ42/Aβ40 ratio (Borchelt et al., 1996; De Strooper et al., 2012; Watanabe and Shen, 2017). The findings of amyloid inhibitory effect of Aβ40 against Aβ42 aggregation and the increased Aβ42/Aβ40 ratio in most of FAD patients with PS mutations provide molecular mechanisms for the pathogenesis of FAD. However, some of PSEN mutations lead to reduce Aβ42/Aβ40 ratio and PS conditional knockout mice in the forebrain show progressive neurodegeneration without Aβ deposition, suggesting that loss of PS function could contribute to neuronal damage independently of amyloid cascade (Saura et al., 2004; De Strooper et al., 2012; Watanabe and Shen, 2017).
In addition to amyloid accumulation, oxidative damage in the brain of AD patients also typifies AD pathology and is found to precede Aβ deposition (Nunomura et al., 2001; Ayton et al., 2013; Chen and Zhong, 2014). Redox-active iron is responsible for inducing oxidative damage and extensively deposits in amyloid plaques and neurofibrillary tangles of AD brain (Smith et al., 1997; Zecca et al., 2004; Belaidi and Bush, 2016). We have found that Aβ40 protects neurons against oxidative damage induced by iron, whereas Aβ42 enhanced neurotoxicity (Zou et al., 2002; Zou and Michikawa, 2008). These lines of evidence suggest that Aβ deposition could be a secondary event of oxidative damage in AD brain. Here, we studied whether loss of function of PS is involved in oxidative stress in cells and found that reactive oxygen species (ROS) generation increased in PS deficient fibroblasts compared with wild type fibroblasts. We also found that cellular levels of both ferritin light and heavy chains markedly decreased in PS deficient fibroblasts and that inhibition of γ-secretase activity suppressed cellular levels of ferritin light and heavy chains in the presence of ferrous sulfate. Our results suggest that dysfunction of PS or reduced γ-secretase activity may decrease iron sequestration and enhance oxidative stress.
Materials and Methods
Cell Culture and Western Blot Analysis
Wild-type and PS double-knockout (PS-DK) fibroblasts were provided by Dr. Bart De Strooper (Herreman et al., 1999). Cells were cultured in Dulbecco’s modified Eagle medium (DMEM; GIBCO) containing 10% fetal bovine serum (FBS). Wild-type and PS-DK fibroblasts were lysed in RIPA buffer [10 mM Tris/HCl (pH 7.5), 150 mM NaCl, 1% Non-idet P-40, 0.1% sodium dodecyl sulfate (SDS) and 0.2% sodium deoxycholate, containing a protease inhibitor cocktail (Roche)]. Equal amounts (20 μg) of protein from cell lysate were separated by SDS-PAGE in 12% gel and blotted onto polyvinylidene difluoride (PVDF) membranes (Immobilon). The ferritin proteins were detected by Western blotting using rabbit polyclonal anti-ferritin light chain and anti-ferritin heavy chain antibodies (abcam). The blots were visualized using Super Signal Chemiluminescence solution (Wako) according to the manufacturer’s instructions. Membranes were then stripped and re-probed with anti–α-tubulin (Sigma) or anti-β-actin (Proteintec) antibodies. Quantification of protein levels was performed using Image J software (National Institutes of Health).
Fluorescence Microscopy
The levels of intracellular ROS were measured using dihydroethidium (DHE), which interacts with ROS to form ethidium and emits a red fluorescence in the cells, as described previously (Bindokas et al., 1996). A total of 2 × 104/ml wild-type and PS-DK fibroblasts were seeded and incubated at 37°C in 5% CO2 for 12 h and then treated with vehicle control or 1 mM H2O2. After 30 min, 5 μM DHE (Cayma Chemical) was added into the cells and the cells were incubated for another 30 min at 37°C in 5% CO2. Confocal microscopy images were captured using an Olympus FV3000 confocal microscope (Olympus). For quantification of fluorescence intensity, acquired images were imported to Image J software. Threshold intensity was determined using the automatically set thresholding function for all images. Pixel intensity for fluorescence images was expressed as fluorescence mean intensity.
Reactive Oxygen Species Activity Assay
Intracellular total ROS activity in live cells was analyzed using Amplite Fluorimetric ROS assay kit (AAT Bioquest). WT and PS-DK cells were seeded in a 96 well plate in triplicate and cultured with DMEM containing 10% FBS for 48h. Then the cells were incubated in serum free DMEM and 100 μl/well of Amplite™ ROS Red working solution at 37°C for 1 h. The cells were then treated with 30 and 100 μM FeSO4. After 16 h, ROS levels were measured using Amplite Fluorimetric ROS assay kit according to the manufacturer’s protocol. The fluorescence density was measured by plate reader (SPECTRA MAX GEMINI EM) at Ex/Em = 520/605 nm.
Infection of Presenilin Mutants
The expression of human PS mutants (PS1ΔE9, PS1G384A, PS1D257A, and PS1D385A) was performed as previously described (Islam et al., 2022). Briefly, the retroviral plasmids were transfected into platinum-E cells using FuGENE (Promega) for retroviral packaging. After 24 h, the conditioned medium was collected and used as viral stock. For highly efficient retroviral infection, fibroblasts were cultured with viral stock containing 5 μg/ml polybrene (Santa Cruz Biotechnology) and allowed to proceed for PS expression.
Statistical Analyses
Prism 7.0 software (GraphPad Software) was used for statistical analyses. All data are shown as the mean ± SEM of at least three independent experiments with P-values. P-value<0.05 was considered to represent a significant difference. Student’s t-test was used to determine whether the results were significantly different between two groups. Group differences were analyzed by one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison tests for multiple groups against the control group.
Results
Reactive Oxygen Species Levels in PS Double-Knockout Fibroblasts Increased at Basal Level and After H2O2 Treatment
To investigate whether PS function is involved in oxidative stress in cells, wild-type and PS1, 2 double knockout (PS-DK) fibroblasts were stained with DHE, which detects cellular ROS generation by oxidation to form ethidium with red fluorescence. The ratio of DHE-positive PS-DK fibroblasts was significantly higher than wild-type fibroblasts, 13% for wild-type and 86% for PS-DK cells, respectively (Figures 1A,B). This result suggests that the basal ROS levels in PS-DK fibroblasts is higher than wild-type fibroblasts. To examine whether PS-DK fibroblasts are more susceptible to oxidative stress, we treated wild-type and PS-DK fibroblasts with H2O2 and found that PS-DK fibroblasts generated markedly more ROS than wild-type fibroblasts (Figures 1C,D). This result suggests that PS-DK fibroblasts may have lower antioxidant capabilities than wild-type cells.
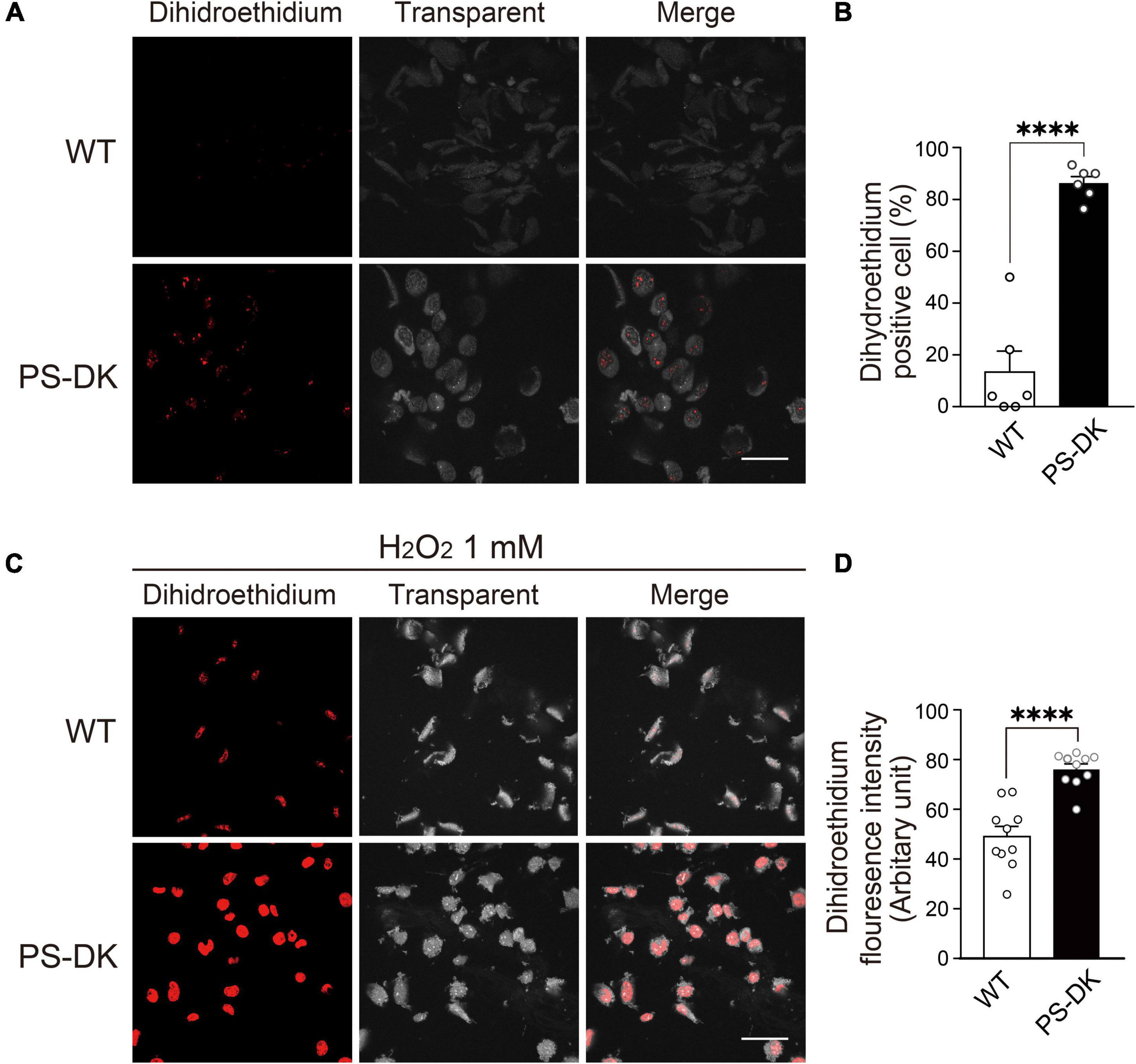
Figure 1. PS deficiency increased oxidative stress in PS-DK fibroblasts. (A) Confocal microscopy of DHE staining. Wild-type and PS-DK fibroblasts were incubated with DHE (5 μM) for 30 min in DMEM containing 10% FBS at 37°C in 5% CO2. The number of DHE-positive cells increased in PS-DK fibroblasts, indicating that presenilin deficiency enhanced ROS production. (B) Ratio of DHE-positive cells in wild-type and PS-DK fibroblasts. n = 6 images from 3 independent experiments, ****P< 0.0001, unpaired two-tailed Student’s t-test. (C) Confocal microscopy of DHE staining after H2O2 treatment. Wild-type and PS-DK fibroblasts were treated with 1 mM H2O2 for 30 min and then stained with DHE (5 μM). (D) Fluorescence intensity in wild-type and PS-DK fibroblasts were determined Image J software. n = 10 images from 3 independent experiments, ****P< 0.0001, unpaired two-tailed Student’s t-test. Scale bars, 20 μm. All quantitative data shown as mean±SEM.
Presenilin Deficiency Leads to Increased Susceptibility to Iron-Induced Oxidative Damage and Decreased Cellular Ferritin Levels
Because H2O2 generates ROS through Fenton reaction and free iron ion is involved in this reaction, Fe2++H2O2→Fe3++HO⋅+OH–, we examined whether iron treatment can also induce more ROS in PS-DK fibroblasts than in wild-type fibroblasts. The cellular ROS level in PS-DK fibroblasts treated with 30 μM ferrous sulfate showed a tendency toward increase compared with wild-type fibroblasts. The treatment with 100 μM ferrous sulfate significantly increased the cellular ROS level in PS-DK fibroblasts compared with wild-type fibroblasts (Figure 2A). This result suggests that PS-DK fibroblasts have lower antioxidant molecules against iron-induced free radical generation. Antioxidant molecules include direct ROS scavengers and indirect metal binding proteins that quench metal ions to inhibit secondary generation of free radicals. Ferritin is a major intracellular iron binding and storage protein that provides protective effects to cells against free iron-induced oxidative stress (Philpott et al., 2017). We then examined the cellular levels of ferritin in wild-type and PS-DK fibroblasts. Interestingly, total ferritin, ferritin light chain and ferritin heavy chain markedly decreased in PS-DK fibroblasts compared with wild-type fibroblasts (Figures 2B–D). These results suggest that the decrease in ferritin levels may result in increased free iron and increased susceptibility to iron-induced oxidative damage.
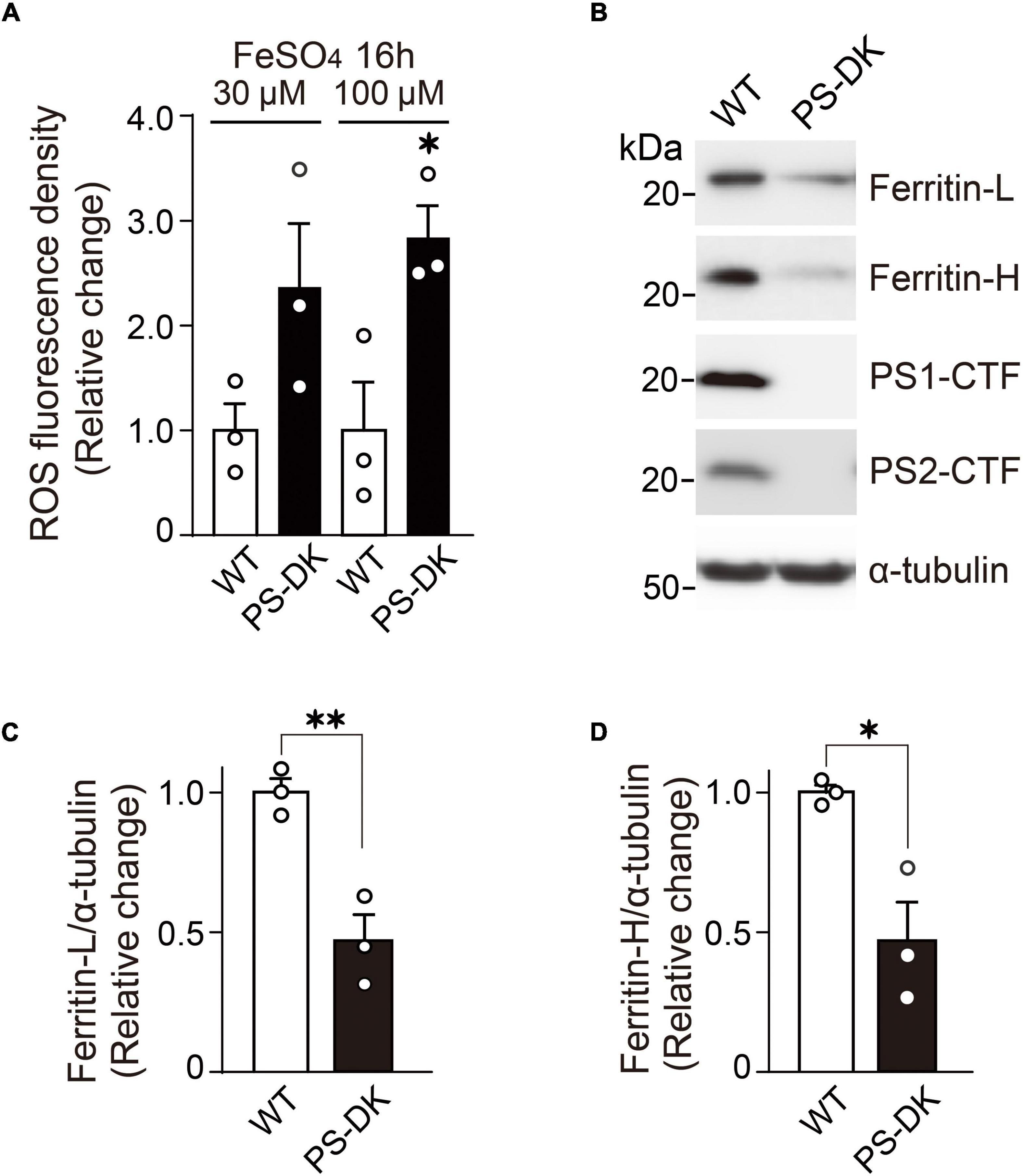
Figure 2. PS-DK fibroblasts generated more ROS by iron-treatment and showed lower intracellular ferritin levels than wild-type fibroblasts. (A) ROS fluorescence density were measured 16 h after treatment with ferrous sulfate. PS-DK fibroblasts showed higher ROS levels after iron-treatment. n = 3, from 3 independent experiments, *P< 0.05, unpaired two-tailed Student’s t-test. (B) Western blot analysis of intracellular ferritin light and heavy chains, PS1 C-terminal fragment (CTF) and PS2 CTF. (C,D) Quantification of intracellular ferritin light and heavy chain levels. Their levels were normalized to α-tubulin protein levels. n = 3, from 3 independent experiments, *P< 0.05, **P< 0.01, unpaired two-tailed Student’s t-test. All quantitative data shown as mean±SEM. Ferritin-L, ferritin light chain. Ferritin-H, ferritin heavy chain.
Ferritin Light and Heavy Chains in Response to Iron Treatment Decreased in PS Double-Knockout Fibroblasts
Ferritin sequesters iron in a non-toxic form and its expression level is regulated by labile iron for protecting cells from damage triggered by excess iron (Picard et al., 1998; Torti and Torti, 2002). We examined whether the regulation of cellular ferritin levels by iron treatment is impaired in PS-DK fibroblasts. Wild-type and PS-DK fibroblasts were treated with 1, 3, and 10 μM ferrous sulfate, respectively. Both wild-type and PS-DK cells showed iron dose-dependent increases in ferritin light chain and ferritin heavy chain levels (Figures 3A,C). However, the cellular levels of ferritin light chain were significantly lower in PS-DK fibroblasts than wild-type fibroblasts at all concentration of iron treatment. Ferritin light chain levels in PS-DK fibroblasts were 57, 42, and 46% compared with wild fibroblasts at 1, 3, and 10 μM ferrous sulfate treatment, respectively (Figure 3B). Ferritin heavy chain levels in PS-DK fibroblasts were 17, 25, and 40% compared with wild fibroblasts at 1, 3, and 10 μM ferrous sulfate treatment, respectively (Figure 3D). These results suggest that PS deficiency does not only reduce the basal level of cellular ferritin, but also reduce the levels of ferritin in response to iron over load. Thus, the increase of ROS levels in PS-DK fibroblasts may result from decreased cellular ferritin levels and increased labile iron ions.
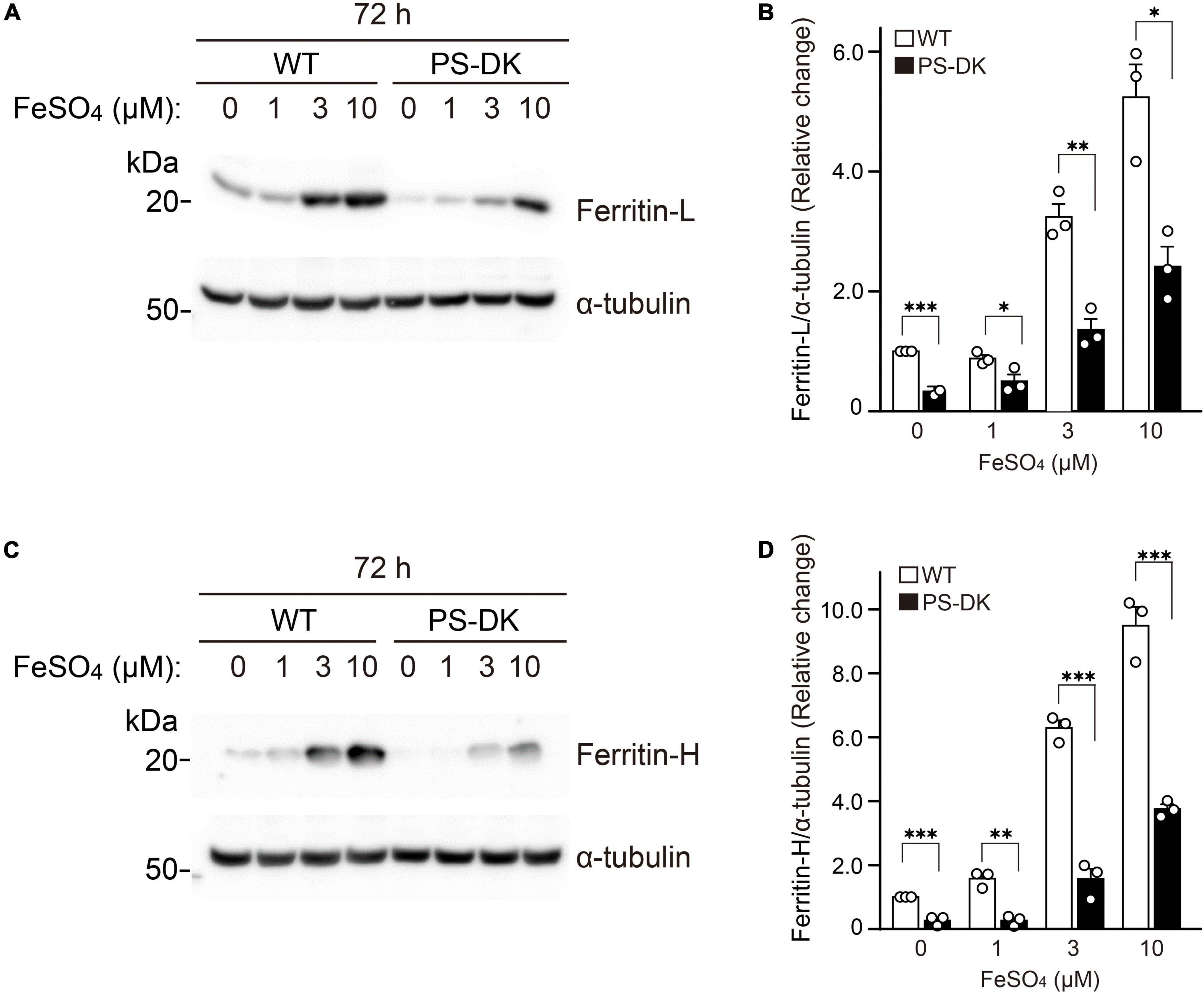
Figure 3. PS deficiency reduced ferritin levels in response to iron treatment. (A) Western blot analysis of ferritin light chain in wild-type and PS-DK fibroblasts treated with ferrous sulfate. (B) Quantification of intracellular ferritin light chain levels. Their levels were normalized to α-tubulin protein levels. n = 3, from 3 independent experiments, *P< 0.05, **P< 0.01, ***P< 0.001, one-way ANOVA followed by Tukey’s multiple-comparison tests. (C) Western blot analysis of ferritin heavy chain in wild-type and PS-DK fibroblasts treated with ferrous sulfate. (D) Quantification of intracellular ferritin heavy chain levels. Their levels were normalized to α-tubulin protein levels. n = 3, from 3 independent experiments, **P< 0.01, ***P< 0.001, one-way ANOVA followed by Tukey’s multiple-comparison tests. All quantitative data shown as mean ± SEM. Ferritin-L, ferritin light chain. Ferritin-H, ferritin heavy chain.
Inhibition of γ-Secretase Activity Reduced the Increase of Ferritin in Response to Iron Treatment
PS is a multifunctional molecule and its major function is serving as a catalytic component of γ-secretase complex (De Strooper et al., 2012). To examine whether γ-secretase activity is involved in regulating ferritin levels in response to iron treatment, wild-type fibroblasts were treated with a γ-secretase inhibitor, DAPT (5 μM), and ferrous sulfate (0, 1, 2, 5, and 10 μM). In the presence of DAPT, ferritin light and heavy chain levels induced by iron treatment was significantly lower than that without DAPT treatment (Figures 4A,C). DAPT treatment suppressed ferritin light chain levels to 37% at 5 μM ferrous sulfate treatment, and to 56% at 10 μM ferrous sulfate treatment compared to those without DAPT treatment (Figure 4B). Ferritin heavy chain levels were also decreased by DAPT to 82% at 5 μM ferrous sulfate treatment and to 53% at 10 μM ferrous sulfate treatment (Figure 4D). These results suggest that γ-secretase activity can regulate ferritin levels in response to iron treatment and that impairment of γ-secretase activity may increase susceptibility to iron-induced oxidative damage.
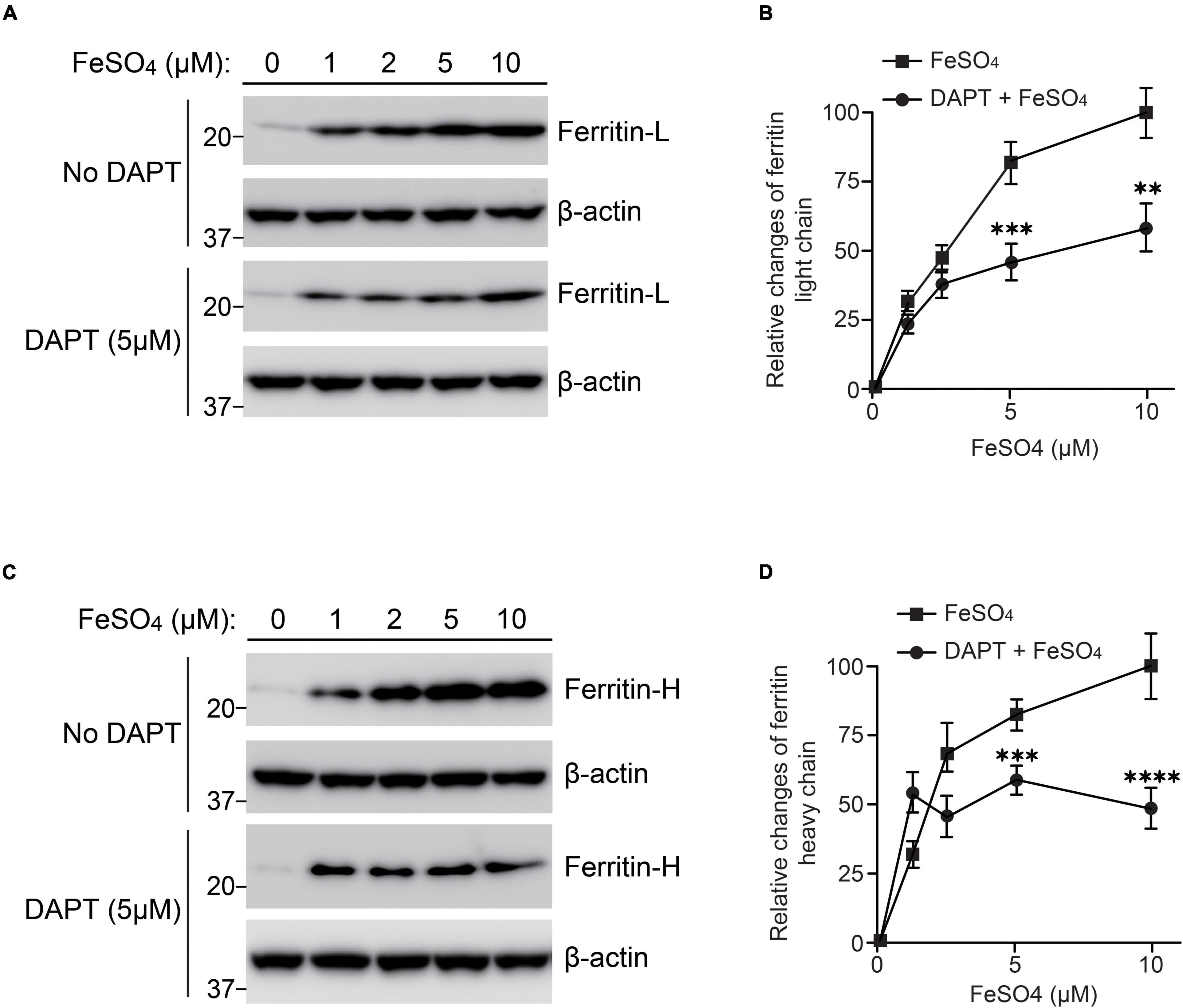
Figure 4. Inhibition of γ-secretase activity reduced ferritin levels in response to iron treatment in wild-type fibroblasts. Wild-type fibroblasts were treated with or without 5 μM DAPT in DMEM containing 10% FBS after seeding. After 1 h, ferrous sulfate was added and the cells were incubated at 37°C in 5% CO2 for 72 h. (A) Western blot analysis of ferritin light chain in wild-type fibroblasts treated with or without ferrous sulfate or DAPT. (B) Quantification of intracellular ferritin light chain levels. Ferritin light chain levels at 10 μM ferrous sulfate treatment (without DAPT) were normalized to α-tubulin protein levels and adjusted to 100%. The relative change of ferritin light chain at each concentration was calculated. DAPT treatment significantly inhibited intracellular ferritin light chain levels. n = 3, from 3 independent experiments, **P< 0.01, ***P< 0.001, one-way ANOVA followed by Tukey’s multiple-comparison tests. (C) Western blot analysis of ferritin heavy chain in wild-type fibroblasts treated with or without ferrous sulfate or DAPT. (D) Quantification of intracellular ferritin heavy chain levels. Ferritin heavy chain levels in wild-type fibroblasts with 10 μM ferrous sulfate treatment (without DAPT) were normalized to α-tubulin protein levels and adjusted to 100%. The relative change of ferritin light chain at each concentration was calculated. DAPT treatment significantly inhibited intracellular ferritin heavy chain levels. n = 3, from 3 independent experiments, ***P< 0.001, ****P< 0.0001, one-way ANOVA followed by Tukey’s multiple-comparison tests. ■, ferrous sulfate treatment. 🌑, DAPT and ferrous sulfate treatment. All quantitative data shown as mean±SEM. Ferritin-L, ferritin light chain. Ferritin-H, ferritin heavy chain.
Overexpression of Mutant PS1 Decreased Ferritin Light Chain Levels and Increased Reactive Oxygen Species Levels in Wild-Type Fibroblasts
To examine whether PS mutants found in FAD (PS1ΔE9 and PS1G384A) or PS mutants without γ-secretase activity (PS1D257A and PS1D385A) can reduce ferritin level and increase ROS level, we overexpressed PS1ΔE9, PS1G384A, PS1D257A, or PS1D385A in wild-type fibroblasts. PS1ΔE9, PS1D257A, and PS1D385A significantly decreased ferritin light chain levels, whereas did not affect ferritin heavy chain levels. PS1G384A did not change ferritin light or heavy chain levels (Figures 5A–D). ROS levels in fibroblasts overexpressing PS1D257A or PS1D385A are markedly more than control cells (Figure 5E). These results suggest that PS mutants with less γ-secretase activity have dominant negative effects on reducing ferritin light chain levels and increasing ROS levels.
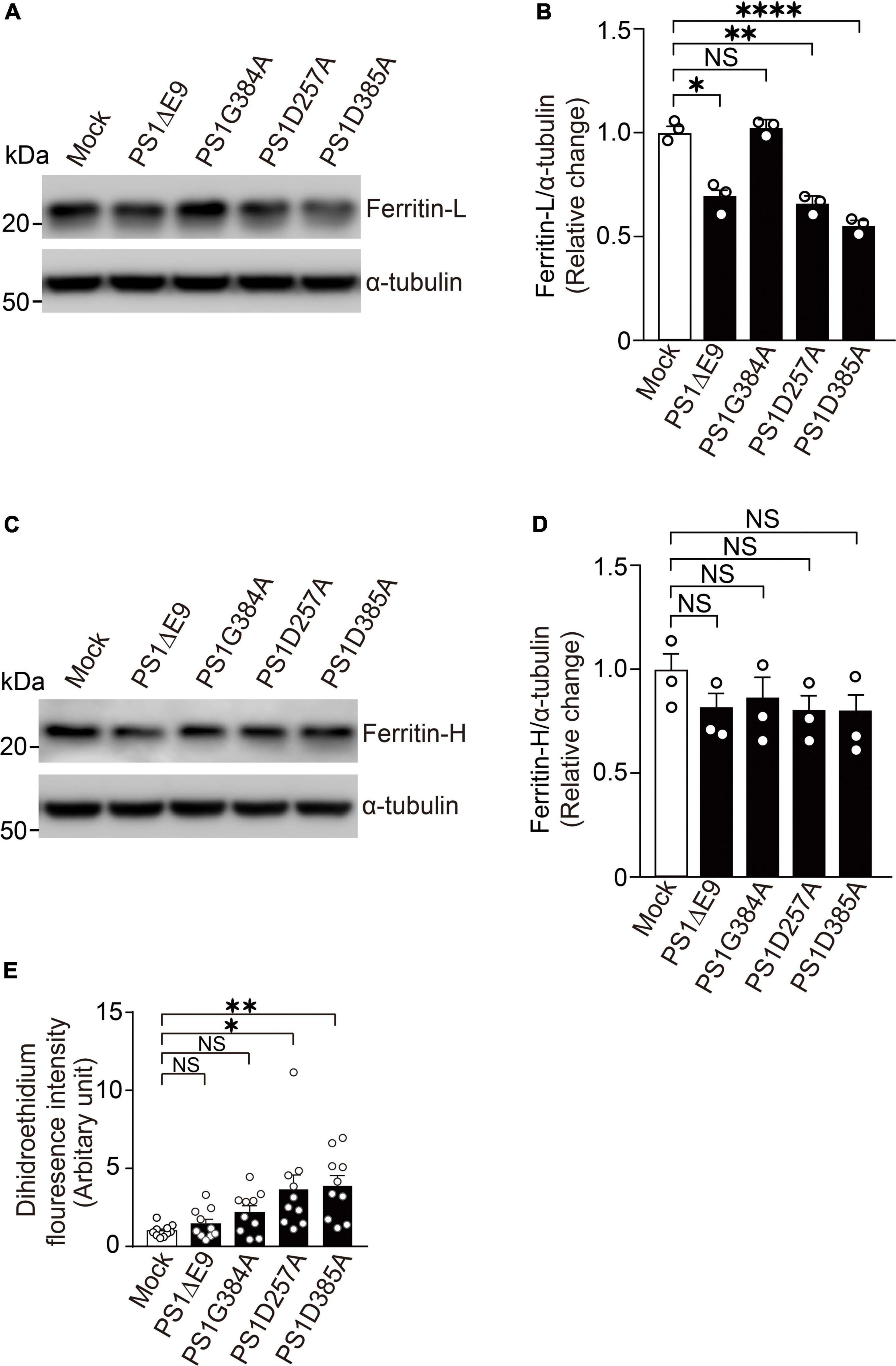
Figure 5. Overexpression of PS1 mutants decreased ferritin light chain levels and increased ROS levels in wild-type fibroblasts. (A) Western blot analysis of intracellular ferritin light chain in wild-type fibroblasts overexpressing PS1ΔE9, PS1G384A, PS1D257A, or PS1D385A. (B) Quantification of intracellular ferritin light chain levels. Their levels were normalized to α-tubulin protein levels. n = 3, from 3 independent experiments, *P< 0.05, **P< 0.01, ****P< 0.0001, one-way ANOVA followed by Tukey’s multiple-comparison tests. (C) Western blot analysis of intracellular ferritin heavy chain in wild-type fibroblasts overexpressing PS1ΔE9, PS1G384A, PS1D257A, or PS1D385A. (D) Quantification of intracellular ferritin heavy chain levels. Their levels were normalized to α-tubulin protein levels. n = 3, from 3 independent experiments, NS, not significant, one-way ANOVA followed by Tukey’s multiple-comparison tests. (E) Fluorescence intensity in wild-type fibroblasts infected with retrovirus bearing human PS1 mutations (PS1ΔE9, PS1G384A, PS1D257A, and PS1D385A) were determined by Image J software. n = 10 images from 3 independent experiments, *P< 0.05, **P< 0.01. NS, not significant, by one-way ANOVA followed by Tukey’s multiple-comparison tests.
Discussion
Neurodegeneration in the brain of patients with AD has long been considered to be caused by the aggregation of toxic form of Aβ, Aβ42. This notion is strongly supported by the findings that most of genetic mutations in APP, PSEN1, and PSEN2 found in FAD showed increased Aβ42 generation or Aβ42/Aβ40 ratio (Scheltens et al., 2016; Sun et al., 2017). However, oxidative damage is found to precede Aβ deposition and iron extensively deposits in amyloid plaques and neurofibrillary tangles in the brain of patient with SAD (Nunomura et al., 2001; Ayton et al., 2013). In support of this oxidative damage pathogenesis theory for AD, we have found that Aβ40, the major form all secreted Aβ, has neuroprotective effects against iron-induced oxidative damage (Zou et al., 2002; Zou and Michikawa, 2008). The findings that iron overload can accelerate Aβ production also support this notion (Becerril-Ortega et al., 2014; Shen et al., 2019). Here we demonstrated that PS deficiency leads to increased ROS generation in cells at basal level and after treatment with H2O2 or labile iron. In addition, cellular ferritin light and heavy chain levels significantly decreased in PS-DK fibroblasts compared with wild-type fibroblasts, suggesting that the lack of iron sequestration by ferritin increases ROS generation in PS-DK cells.
Multitude of omics studies have characterized expression profiles of thousands of molecular changes and uncovered neuronal gene subnetworks as the most dysregulated in human AD brain and AD mouse models. Interestingly, ferritin heavy and light chain genes, FTH1 and FTL, have been found to be dysregulated in human AD brain and AD mouse models, indicating an important role of ferritin in AD pathophysiology (Bai et al., 2020; Wang et al., 2021). Ferritin plays a key role in maintaining iron homeostasis by capturing and buffering the intracellular labile iron pool (Torti and Torti, 2002). The translation of ferritin is controlled by RNA-binding proteins, iron regulatory protein-1 (IRP1) and IRP2 that interact with iron responsive element (IRE) (Gray and Hentze, 1994; Pantopoulos, 2004; Zhou and Tan, 2017). Microglia highly express FTL with increased Iba1 expression, a marker for activated microglia and represented as iron-accumulating microglia, whereas FTL expressed to a lower extent in neurons in human brain (Kenkhuis et al., 2021). Microglia are in a constant cross-talk with neurons and are capable of tight regulation of neuronal homeostasis. Activated microglia reacts against disease and injury, protect from chronic neuroinflammation and oxidative damage, a hallmark feature of neurodegenerative diseases (Ndayisaba et al., 2019). Thus, PS may be involved in maintaining neuronal homeostasis by regulating iron metabolism in both microglia and neurons.
We found that suppression of γ-secretase activity significantly inhibited the elevation ferritin light and heavy chains in response to iron treatment (Figure 4), suggesting that γ-secretase activity may be involved in IRP- and IRE-mediated pathway in iron metabolism. Among the PSEN1 mutations examined in this study, PS1ΔE9 causes a reduction in Aβ40 production, whereas PS1G384A mutant significantly increases Aβ42 production, suggesting that PS1ΔE9 results in a decreased γ-secretase activity (Bentahir et al., 2006). PS1D257A and PS1D385A also greatly reduce γ-secretase activity (Wolfe et al., 1999). Thus, the decreased ferritin light chain levels and increase ROS levels in PS1D257A and PS1D385A overexpressing cells may result from loss of γ-secretase activity (Figure 5). It is possible that γ-secretase inhibitors using in clinical trials may reduce ferritin levels or increase ROS levels. This should be studied in human samples from clinical trials in future. There are more than 100 substrates of γ-secretase were identified (De Strooper et al., 2012), that which substrate is responsible for regulating cellular ferritin levels needs to be further studied. Although most of mutations of PSEN found in FAD result in impaired γ-secretase activity, dysregulated Aβ42/40 ratio and APP cleavage, our findings suggest FAD PSEN mutations may also lead to increased oxidative damage by affecting cellular ferritin levels and provide a novel molecular mechanism underlying the neuronal degeneration in FAD induced by PS mutations.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
KZ, SI, YS, YG, TN, HK, and TT: data curation. KZ, SI, and YS: data analysis. KZ: conceptualization and writing the manuscript. KZ and MM: supervision and funding. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Grant-in-Aid for Scientific Research C 19K07846 and 22K07352 (to KZ), Grant-in-Aid for Scientific Research B 16H05559 (to MM), and Challenging Exploratory Research 15K15712 (to MM) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan. This work was also supported by AMED under grant number JP20dk0207050h001 and JP20de010702 (to MM), and by Grants-in-aid from “the 24th General Assembly of the Japanese Association of Medical Sciences” (to KZ), the Daiko Foundation (to KZ), the Hirose International Scholarship Foundation (to KZ), and the Hori Sciences and Arts Foundation (to KZ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Bart De Stropper for providing wild-type and PS double knockout (PS-DK) mouse embryonic fibroblasts.
References
Ayton, S., Lei, P., and Bush, A. I. (2013). Metallostasis in alzheimer’s disease. Free Radic. Biol. Med. 62, 76–89. doi: 10.1016/j.freeradbiomed.2012.10.558
Bai, B., Wang, X., Li, Y., Chen, P.-C., Yu, K., Dey, K. K., et al. (2020). Deep multilayer brain proteomics identifies molecular networks in alzheimer’s disease progression. Neuron 105, 975.e–991.e.
Becerril-Ortega, J., Bordji, K., Freret, T., Rush, T., and Buisson, A. (2014). Iron overload accelerates neuronal amyloid-beta production and cognitive impairment in transgenic mice model of Alzheimer’s disease. Neurobiol. Aging 35, 2288–2301. doi: 10.1016/j.neurobiolaging.2014.04.019
Belaidi, A. A., and Bush, A. I. (2016). Iron neurochemistry in alzheimer’s disease and parkinson’s disease: targets for therapeutics. J. Neurochem. 139(Suppl. 1), 179–197. doi: 10.1111/jnc.13425
Bentahir, M., Nyabi, O., Verhamme, J., Tolia, A., Horre, K., Wiltfang, J., et al. (2006). Presenilin clinical mutations can affect gamma-secretase activity by different mechanisms. J. Neurochem. 96, 732–742. doi: 10.1111/j.1471-4159.2005.03578.x
Bindokas, V. P., Jordan, J., Lee, C. C., and Miller, R. J. (1996). Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J. Neurosci. 16, 1324–1336. doi: 10.1523/JNEUROSCI.16-04-01324.1996
Borchelt, D. R., Thinakaran, G., Eckman, C. B., Lee, M. K., Davenport, F., Ratovitsky, T., et al. (1996). Familial alzheimer’s disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron 17, 1005–1013. doi: 10.1016/s0896-6273(00)80230-5
Chen, Z., and Zhong, C. (2014). Oxidative stress in alzheimer’s disease. Neurosci. Bull. 30, 271–281.
De Strooper, B., Iwatsubo, T., and Wolfe, M. S. (2012). Presenilins and gamma-secretase: structure, function, and role in alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a006304. doi: 10.1101/cshperspect.a006304
Gray, N. K., and Hentze, M. W. (1994). Iron regulatory protein prevents binding of the 43S translation pre-initiation complex to ferritin and eALAS mRNAs. Embo J. 13, 3882–3891. doi: 10.1002/j.1460-2075.1994.tb06699.x
Herreman, A., Hartmann, D., Annaert, W., Saftig, P., Craessaerts, K., Serneels, L., et al. (1999). Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proc. Natl. Acad. Sci. U.S.A 96, 11872–11877. doi: 10.1073/pnas.96.21.11872
Islam, S., Sun, Y., Gao, Y., Nakamura, T., Noorani, A. A., Li, T., et al. (2022). Presenilin Is essential for ApoE secretion, a novel role of presenilin involved in alzheimer’s disease pathogenesis. J. Neurosci. 42, 1574–1586. doi: 10.1523/JNEUROSCI.2039-21.2021
Kenkhuis, B., Somarakis, A., de Haan, L., Dzyubachyk, O., Ijsselsteijn, M. E., de Miranda, N. F. C. C., et al. (2021). Iron loading is a prominent feature of activated microglia in Alzheimer’s disease patients. Acta Neuropathol. Commun. 9:27. doi: 10.1186/s40478-021-01126-5
Knopman, D. S., Amieva, H., Petersen, R. C., Chetelat, G., Holtzman, D. M., Hyman, B. T., et al. (2021). Alzheimer disease. Nat. Rev. Dis. Primers 7:33.
McKhann, G., Drachman, D., Folstein, M., Katzman, R., Price, D., and Stadlan, E. M. (1984). Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA work group under the auspices of department of health and human services task force on alzheimer’s disease. Neurology 34, 939–944. doi: 10.1212/wnl.34.7.939
Ndayisaba, A., Kaindlstorfer, C., and Wenning, G. K. (2019). Iron in neurodegeneration – cause or consequence? Front. Neurosci. 13:180 doi: 10.3389/fnins.2019.00180
Nunomura, A., Perry, G., Aliev, G., Hirai, K., Takeda, A., Balraj, E. K., et al. (2001). Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 60, 759–767. doi: 10.1093/jnen/60.8.759
Pantopoulos, K. (2004). Iron metabolism and the IRE/IRP regulatory system: an update. Ann. N Y Acad. Sci. 1012, 1–13. doi: 10.1196/annals.1306.001
Philpott, C. C., Ryu, M. S., Frey, A., and Patel, S. (2017). Cytosolic iron chaperones: proteins delivering iron cofactors in the cytosol of mammalian cells. J. Biol. Chem. 292, 12764–12771. doi: 10.1074/jbc.R117.791962
Picard, V., Epsztejn, S., Santambrogio, P., Cabantchik, Z. I., and Beaumont, C. (1998). Role of ferritin in the control of the labile iron pool in murine erythroleukemia cells*. J. Biol. Chem. 273, 15382–15386. doi: 10.1074/jbc.273.25.15382
Saura, C. A., Choi, S. Y., Beglopoulos, V., Malkani, S., Zhang, D., Shankaranarayana Rao, B. S., et al. (2004). Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron 42, 23–36. doi: 10.1016/s0896-6273(04)00182-5
Scheltens, P., Blennow, K., Breteler, M. M., de Strooper, B., Frisoni, G. B., Salloway, S., et al. (2016). Alzheimer’s disease. Lancet 388, 505–517.
Selkoe, D. J., and Hardy, J. (2016). The amyloid hypothesis of alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608. doi: 10.15252/emmm.201606210
Shen, X., Liu, J., Fujita, Y., Liu, S., Maeda, T., Kikuchi, K., et al. (2019). Iron treatment inhibits Abeta42 deposition in vivo and reduces Abeta42/Abeta40 ratio. Biochem. Biophys. Res. Commun. 512, 653–658. doi: 10.1016/j.bbrc.2019.01.119
Shoji, M., Golde, T. E., Ghiso, J., Cheung, T. T., Estus, S., Shaffer, L. M., et al. (1992). Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science 258, 126–129. doi: 10.1126/science.1439760
Smith, M. A., Harris, P. L., Sayre, L. M., and Perry, G. (1997). Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc. Natl. Acad. Sci. U.S.A 94, 9866–9868. doi: 10.1073/pnas.94.18.9866
Sun, L., Zhou, R., Yang, G., and Shi, Y. (2017). Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Aβ42 and Aβ40 peptides by γ-secretase. Proc. Natl. Acad. Sci. U.S.A 114, E476–E485. doi: 10.1073/pnas.1618657114
Torti, F. M., and Torti, S. V. (2002). Regulation of ferritin genes and protein. Blood 99, 3505–3516.
Wang, M., Li, A., Sekiya, M., Beckmann, N. D., Quan, X., Schrode, N., et al. (2021). Transformative network modeling of multi-omics data reveals detailed circuits, key regulators, and potential therapeutics for alzheimer’s disease. Neuron 109, 257.e–272.e. doi: 10.1016/j.neuron.2020.11.002
Watanabe, H., and Shen, J. (2017). Dominant negative mechanism of Presenilin-1 mutations in FAD. Proc. Natl. Acad. Sci. U.S.A 114, 12635–12637. doi: 10.1073/pnas.1717180114
Wolfe, M. S., Xia, W., Ostaszewski, B. L., Diehl, T. S., Kimberly, W. T., and Selkoe, D. J. (1999). Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature 398, 513–517. doi: 10.1038/19077
Zecca, L., Youdim, M. B., Riederer, P., Connor, J. R., and Crichton, R. R. (2004). Iron, brain ageing and neurodegenerative disorders. Nat. Rev. Neurosci. 5, 863–873.
Zhou, Z. D., and Tan, E. K. (2017). Iron regulatory protein (IRP)-iron responsive element (IRE) signaling pathway in human neurodegenerative diseases. Mol. Neurodegener. 12:75. doi: 10.1186/s13024-017-0218-4
Zou, K., Gong, J. S., Yanagisawa, K., and Michikawa, M. (2002). A novel function of monomeric amyloid beta-protein serving as an antioxidant molecule against metal-induced oxidative damage. J. Neurosci. 22, 4833–4841. doi: 10.1523/JNEUROSCI.22-12-04833.2002
Keywords: presenilin, oxidative, ferritin, iron, Alzheimer’s disease
Citation: Zou K, Islam S, Sun Y, Gao Y, Nakamura T, Komano H, Tomita T and Michikawa M (2022) Presenilin Deficiency Increases Susceptibility to Oxidative Damage in Fibroblasts. Front. Aging Neurosci. 14:902525. doi: 10.3389/fnagi.2022.902525
Received: 23 March 2022; Accepted: 27 May 2022;
Published: 16 June 2022.
Edited by:
Allison B. Reiss, New York University, United StatesReviewed by:
Satoru Funamoto, Doshisha University, JapanTimothy Y. Huang, Sanford Burnham Prebys Medical Discovery Institute, United States
Copyright © 2022 Zou, Islam, Sun, Gao, Nakamura, Komano, Tomita and Michikawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kun Zou, kunzou@med.nagoya-cu.ac.jp; Makoto Michikawa, michi@med.nagoya-cu.ac.jp