Transcription factor Ap2b regulates the mouse autosomal recessive polycystic kidney disease genes, Pkhd1 and Cys1
 Maoqing Wu1
Maoqing Wu1  Naoe Harafuji
Naoe Harafuji Ljubica Caldovic
Ljubica Caldovic Lisa M. Guay-Woodford
Lisa M. Guay-Woodford- 1Center for Translational Research, Children’s National Hospital, Washington, DC, United States
- 2O’Neal Comprehensive Cancer Center, University of Alabama at Birmingham, Birmingham, AL, United States
- 3Center for Genetic Medicine Research, Children’s National Hospital, Washington, DC, United States
- 4Department of Genomics and Precision Medicine, School of Medical and Health Sciences, The George Washington University, Washington, DC, United States
Transcription factor Ap2b (TFAP2B), an AP-2 family transcription factor, binds to the palindromic consensus DNA sequence, 5′-GCCN3-5GGC-3’. Mice lacking functional Tfap2b gene die in the perinatal or neonatal period with cystic dilatation of the kidney distal tubules and collecting ducts, a phenotype resembling autosomal recessive polycystic kidney disease (ARPKD). Human ARPKD is caused by mutations in PKHD1, DZIP1L, and CYS1, which are conserved in mammals. In this study, we examined the potential role of TFAP2B as a common regulator of Pkhd1 and Cys1. We determined the transcription start site (TSS) of Cys1 using 5′ Rapid Amplification of cDNA Ends (5′RACE); the TSS of Pkhd1 has been previously established. Bioinformatic approaches identified cis-regulatory elements, including two TFAP2B consensus binding sites, in the upstream regulatory regions of both Pkhd1 and Cys1. Based on reporter gene assays performed in mouse renal collecting duct cells (mIMCD-3), TFAP2B activated the Pkhd1 and Cys1 promoters and electromobility shift assay (EMSA) confirmed TFAP2B binding to the in silico identified sites. These results suggest that Tfap2b participates in a renal epithelial cell gene regulatory network that includes Pkhd1 and Cys1. Disruption of this network impairs renal tubular differentiation, causing ductal dilatation that is the hallmark of recessive PKD.
1 Introduction
Autosomal recessive polycystic kidney disease (ARPKD), characterized by bilateral cystic kidneys and congenital hepatic fibrosis, is a rare inherited disorder with a prevalence of 1:26,500 live births (Guay-Woodford et al., 2014; Alzarka et al., 2017). In the majority of patients, ARPKD is caused by mutations in the gene PKHD1, that encodes the protein fibrocystin (FPC) (Onuchic et al., 2002; Ward et al., 2002), the function of which is unknown. A major obstacle to understanding normal FPC function and the pathogenic mechanisms that result from FPC deficiency is the lack of an orthologous animal model that fully recapitulates human PKHD1-associated renal disease (Outeda et al., 2017). Pkhd1 deficient mice develop fibrocystic liver disease, but typically express either no renal phenotype or mild renal cystic disease (O’Connor and Guay-Woodford, 2015). By contrast, the congenital polycystic kidney (cpk) mouse strain, with a genetic defect in the Cys1 gene, exhibits a phenotype that strikingly resembles human ARPKD (Hou et al., 2002; Nagao et al., 2012). Cys1 encodes the protein cystin. Similar to other cystoproteins, both cystin and FPC localize to the primary apical cilium of renal epithelial cells (Tao et al., 2009). Recently, we described the first patient with ARPKD associated with CYS1 mutations (Yang et al., 2021).
The AP2 family of transcription factors consists of five members designated TFAP2A, −B, −C, −D, and −E that influence promoter activity through binding to the consensus DNA sequence 5′-GCCN3-5GGC-3’ (Williams and Tjian, 1991a; Fornes et al., 2020). The AP2 family members are encoded by five genes that are found in both human and mouse genomes: TFAP2A/Tfap2a, TFAP2B/Tfap2b, TFAP2C/Tfap2c, TFAP2D/Tfap2d, and TFAP2E/Tfap2e (Hilger-Eversheim et al., 2000; Zhao et al., 2001; Cheng et al., 2002; Feng and Williams, 2003; Tummala et al., 2003; Zhao et al., 2003). All AP2 transcription factors share the same protein domain organization featuring a proline and glutamine rich transactivation domain at the N-terminus, followed by a basic DNA binding domain in the middle of the protein, and a helix-span-helix dimerization motif at the C-terminus (Williams and Tjian, 1991a; Williams and Tjian, 1991b; Moser et al., 1995; Eckert et al., 2005). AP2 family members can form both homo- and heterodimers that regulate gene expression (Eckert et al., 2005). All AP2 genes are expressed early in the developing mouse central nervous system, but later in embryonic development, each family member exhibits a tissue-specific expression pattern (Moser et al., 1997b; Eckert et al., 2005).
TFAP2B is expressed in developing and adult kidneys (Moser et al., 1995; Moser et al., 1997b). Single cell RNA-Seq analysis using mouse kidneys revealed that Tfap2b is expressed primarily in the cortical collecting duct cells (Park et al., 2018; Lamontagne et al., 2022). TFAP2B expression is associated with distal tubule cell fate determination (Miao et al., 2021). In humans, germline mutations of TFAP2B cause Char syndrome, an autosomal dominant disorder characterized by patent ductus arteriosis, digital abnormalities and facial dysmorphism (Raap et al., 2021). While TFAP2B can be detected in distal tubules of the adult kidney, there is no human renal phenotype, suggesting involvement of additional factors in human renal tubular differentiation. By contrast, in mice, Tfap2b gene disruption leads to a renal phenotype with cystic dilatation of distal tubules and collecting ducts, in a pattern that phenocopies recessive PKD (Moser et al., 1997a; Moser et al., 2003; Wang et al., 2018; Lamontagne et al., 2022). The observations linking Tfap2b with renal cystogenesis, together with its expression during kidney development, led us to speculate that TFAP2B may regulate the expression of mouse cystogenes, particularly those implicated in ARPKD. The paradigm of renal cystic disease linked to a transcription factor mutation with altered expression of multiple cystogenic genes has been previously established. Mutations in the gene encoding hepatocyte nuclear factor-1-beta (HNF1B) cause cystic kidney disease, and HNF1B has been shown to regulate expression of several cystogenic genes, including Pkhd1 and Cys1 (Shao et al., 2020).
In the current study, we evaluated whether TFAP2B regulates the expression of ARPKD-associated genes Pkhd1 and Cys1 in the inner medullary collecting duct cell line, mIMCD-3, a widely used experimental cell model. We identified putative TFAP2B binding sites in the upstream regulatory regions of both Pkhd1 and Cys1 using bioinformatic approaches. Binding sites identified in silico were then tested experimentally using reporter gene and electrophoretic mobility shift assays (EMSA) to show that Pkhd1 and Cys1 are indeed regulated by TFAP2B.
2 Materials and methods
2.1 Bioinformatic analysis of mouse Cys1 and Pkhd1 promoters
Grep command and regular expressions that describe TFAP2B consensus binding sites (Supplementary Figure S1) in the JASPAR database of transcription factor binding sites (Fornes et al., 2020) were used to query both sense and antisense strands of the Cys1 and Pkhd1 upstream regulatory regions, chr12:24681631-24684806 and chr1:20618038-20620038 of the GRCm38/mm10 mouse genome assembly, respectively.
2.2 RT-PCR of Tfab2b mRNA isoforms from kidneys and mIMCD-3 cells
Kidney tissue from a 6-week-old female mouse was snap frozen, transferred to a gentleMACS M tube (Miltenyi Biotec, Cat. No. 130-093-236, RRID:SCR_020269) in Buffer RLT plus 2-Mercaptoethanol (as per RNeasy Mini Kit instructions, see below) and homogenized using a gentleMACS Dissociator (Miltenyi Biotec) using manufacturer’s program RNA-02. Homogenized samples were transferred to microcentrifuge tubes for total RNA extraction using the RNeasy Mini Kit (Qiagen, Cat. No. 74104) according to manufacturer’s instructions.
mIMCD-3 cells were purchased from American Type Culture Collection (ATCC, Cat. No. CRL-2123, RRID:CVCL_0429), and maintained in DMEM/F-12 medium (Thermo Fisher Scientific, Cat. No. 11330057) containing 10% heat-inactivated fetal bovine serum (Atlanta Biologicals, Cat. No. S11050H) and 1% penicillin/streptomycin (Thermo Fisher Scientific, Cat. No. 15140163). Cells were cultured at 37°C in 5% CO2/95% air. Total RNA was isolated from cells using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions.
RNA was treated with RQ1 RNase-Free DNase (Promega, Cat. No. M6101), and then repurified using the RNeasy Mini kit. For RT-PCR, RNA samples were reverse-transcribed using SuperScript III First-Strand Synthesis SuperMix (Thermo Fisher Scientific, Cat. No. 18080400) and oligo dT primers. PCR was performed using primer sets specific for each Tfap2b isoform (Supplementary Table S1), GoTaq Master Mix (Promega, Cat. No. M7123), .2 µM forward and reverse primer, and either 4 ng of cDNA in the first round of PCR or .4 µL of amplification product in nested PCR using the PCR conditions listed in Supplementary Table S2.
2.3 Plasmid construction
DNA fragments harboring presumptive upstream regulatory regions of the mouse Cys1 gene (Table 2) were generated by PCR using 300 nM primers listed in Supplementary Table S3, Expand Long Template Enzyme mix (Roche Cat. No. 1173264100), 350 µM dNTPs, 200 ng of genomic DNA from C57BL/6j mice as template, and amplification conditions listed in Supplementary Table S4. Amplification products were subcloned into the pGL3-basic reporter gene plasmid (Promega) using NheI and HindIII restriction enzymes for Cys1p-constructs. DNA fragments harboring presumptive upstream regulatory regions of the mouse Pkhd1 gene (Table 2) were generated by PCR using 300 nM primers listed in Supplementary Table S5, 1x Expand Long Template Enzyme mix, 350 µM dNTPs, 200 ng of genomic DNA from C57BL/6j mice as template, and amplification conditions listed in Supplementary Table S4. Amplification products for constructs Pkhd1p-1443 and Pkhd1p-1434 were cloned into pGL3-basic reporter gene plasmid (Promega) using restriction enzymes Acc65I and BglII, while BglII and HindIII were used for constructs Pkhd1p-981, Pkhd1p-950 and Pkhd1p-941.
To generate Tfap2b1 and Tfap2b2 expression vectors, the RNeasy Mini Kit (Qiagen) was used to isolate total RNA from 5 days post-confluent mIMCD-3 cells cultured as described above. Superscript First-strand Synthesis (Thermo Fisher Scientific, Cat. No. 11904-018) was used for cDNA generation following manufacturer’s instructions. Full-length Tfap2b1 and Tfap2b2 coding sequences were amplified from mIMCD-3 cDNA using primers 5′-GGG GTA CCC ACT CAC CTC CTA GAG ACC AGG-3’ and 5′-CCT TAA TTA AGC TCA TTT CCT GTG TTT CTC CTC CTT GTC-3′, and 5′-CGG AAT TCA CCA TGG CAT TAG TCC ACA CCT ATT CAT CCA TGG-3’ and 5′-CCT TAA TTA AGC TCA TTT CCT GTG TTT CTC CTC CTT GTC-3′, respectively, and Expand High Fidelity PCR System (Roche Cat. No. 11732641001), 200 µM each dNTP, 500 ng of cDNA, and the following amplification conditions: 2 min initial denaturation at 94°C, 35 cycles of 10 s denaturation at 94°C, 30 s annealing at 62°C, 50 s elongation at 72°C, and 5 min final elongation at 72°C. The Tfap2b1, Tfap2b2 amplification products were subcloned into pCMV-Myc-N (Clontech Laboratories Inc. Cat. No. 635689) expression plasmid using EcoRI and NotI restriction enzymes to yield the expression plasmids CMV-Tfap2b1 and CMV-Tfap2b2.
2.4 5′ Rapid amplification of cDNA ends (5′ RACE)
5′ RACE was performed using FirstChoice RLM-RACE (Ambion, Cat. No. 1700). Total RNA was extracted from 5 days post-confluent mIMCD-3 cells using RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions (and see above). RNA was treated with Calf Intestine Alkaline Phosphatase followed by Tobacco Acid Pyrophosphatase. A 45 nt RNA adapter oligonucleotide 5-GCU GAU GGC GAU GAA UGA ACA CUG CGU UUG CUG GCU UUG AUG AAA-3′ was ligated to the decapped mRNA using T4 RNA ligase, followed by reverse transcription using random hexamers. cDNA was generated and used as a template for nested PCR using first, the 5′ RACE outer adaptor primer 5′-GCT GAT GGC GAT GAA TGA ACA CTG-3′ and 3′ Cys1-specific outer primer 5′-GGG TGG GAG TCA TGC TGG GAG CAA G-3′, followed by PCR with the inner 5′ RACE adaptor primer 5′-CGC GGA TCC GAA CAC TGC GTT TGC TGG CTT TGA TG-3′ and 3′ Cys1-specific inner primer 5′-CCC AAG CTT GGA GAC TAG CAC TGT CGG AAA GGA GG-3′ to amplify the 5′ products containing potential Cys1 transcription start sites. PCR products were separated by agarose gel electrophoresis, isolated and purified from the gel, cloned into the pCR2.1-TOPO plasmid using the TOPO TA Cloning Kit, and used to transform One Shot TOP10 chemically competent E. coli cells (Thermo Fisher Scientific, Cat. No. K450001). Individual PCR products from single clones were then sequenced and mapped onto the mouse genomic DNA sequence.
2.5 Site-directed mutagenesis
QuikChange Site-Directed Mutagenesis (Stratagene, Cat. No. 200522) was used to generate altered promoter constructs. Design and use of mutagenic oligonucleotide primers was per manufacturer’s specifications and instructions. Mutations were confirmed in all constructs by sequence analysis.
2.6 Reporter gene assays
mIMCD-3 cells (ATCC) grown to 90%–95% confluence in 24-well plates were transfected using Lipofectamine 2000 Transfection Reagent (Thermo Fisher Scientific, Cat. No. 11668030). The transfection efficiency was normalized using the Renilla pRL plasmid (15 ng), and the total quantity of transfected DNA was kept constant using pcDNA3.1 (+) Mammalian Expression Vector (Thermo Fisher Scientific, Cat. No. V79020). Between 48 and 72 h after transfection, luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega, Cat. No. E1910) according to the manufacturer’s instructions. The luminometer was programmed to perform a 2-s premeasurement delay, followed by a 10-s measurement period for each reporter assay. After measurement of the firefly luciferase activity, Stop & Glo Reagent was added to each sample and enzymatic activity of Renilla luciferase was measured.
2.7 Electrophoretic mobility shift assay (EMSA)
Electrophoretic mobility shift assay (EMSA) was performed as previously described (Wu et al., 2013). Briefly, nuclear extracts, predicted to contain endogenous TFAP2B, were prepared from 5-day post-confluent mIMCD-3 cells using Nuclear and Cytoplasmic Extraction kits (Pierce, Cat. No. 78833). Biotin-labeled oligonucleotides were prepared as we described (Wu et al., 2013). Binding reactions were performed according to the instructions provided with the LightShift Chemiluminescent EMSA kit (Pierce, Cat. No. 20148). Samples were run on non-denaturing polyacrylamide gels. Biotin-labeled DNA oligonucleotides were detected with avidin-based Chemiluminescent Nucleic Acid Detection kit (Pierce, Cat. No.89880). Reduction in the mobility of protein-oligonucleotide complexes on the gels represented the binding of TFAP2B to the oligonucleotide.
3 Results
3.1 Identification of the Cys1 transcription start site (TSS)
The proximal regulatory regions of Pkhd1 and Cys1 extend upstream from the transcription start sites (TSS). The TSS of Pkhd1 has been identified (Hiesberger et al., 2004), therefore we used 5′ rapid amplification of cDNA ends (5′ RACE) to identify the Cys1 TSS. A Cys1 gene-specific reverse primer annealing within the first exon and an adaptor forward primer were used to amplify the 5′ ends of Cys1 cDNAs prepared from mouse inner medullary collecting duct cell line (mIMCD-3) total RNA (Figure 1A). We cloned and sequenced five distinctly-sized 5′ RACE products (Figures 1B–D). Only the 5′ RACE product representing the longest transcript fully conforms to the consensus TSS (PyPyA (T/A)PyPy) (Haberle and Stark, 2018). The four other products failed to match the consensus sequence, and that was also true of a TSS identified in NCBI (Figure 1D). Based on our findings, Cys1 may have more than one TSS, and we note that DNA upstream of Cys1 includes a GC-rich region, a feature common among genes with multiple TSSs (Shao et al., 2020). Throughout we refer to the TSS defined by the longest transcript in assigning Cys1 DNA sequence position designations.
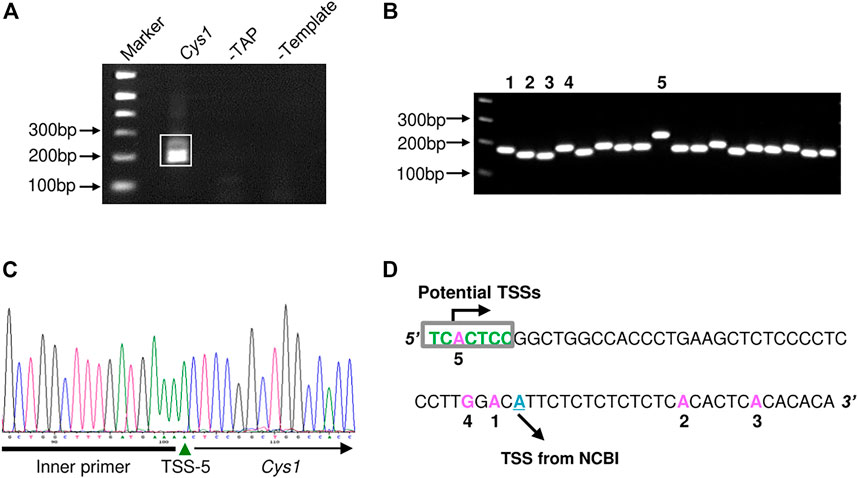
FIGURE 1. Determination of the Cys1 transcription start site (TSS) using 5′ RACE. (A) Agarose gel electrophoresis of Cys1 5′ RACE PCR products. The boxed PCR reaction products (lane Cys1) were cloned and sequenced. Remaining lanes show the results of negative control PCR reactions: middle lane, template mRNA was not decapped by TAP treatment (-TAP, a negative control for RACE); right lane, non-reverse transcribed mRNA subjected directly to PCR (-Template, a negative control for PCR amplification). (B) Individual colony-based PCR reaction products generated using cloned Cys1 amplicon plasmid DNA as template. Clones 1–5 were analyzed by DNA sequencing. (C) Clone 5 DNA sequencing data. Inner primer indicates sequence corresponding to the 5′ RACE adaptor primer from the FirstChoice RLM-RACE kit. (D) Locations of TSS from the five clones are shown in pink; initiator consensus sequence [PyPyAN (T/A)PyPy] is shown in green (Py denotes pyrimidine, C or T). Only the candidate TSS from clone 5 fully conforms to the initiator consensus sequence. The putative TSS from NCBI is depicted in cyan.
3.2 Identification of TFAP2B binding sites in mouse Cys1 and Pkhd1 upstream regulatory regions
Genomic DNA sequences spanning 3,175 and 2,000 bp upstream of the Cys1 and Pkhd1 TSSs, respectively, were analyzed using bioinformatics approaches to identify candidate TFAP2B binding sites (Supplementary Figure S1) (Fornes et al., 2020). Both Cys1 and Pkhd1 regulatory regions were each found to harbor two TFAP2B binding sites, at −175 and −35 for Cys1, and −1443 and −950 for Pkhd1 (Table 1). Inspection of the UCSC Genome Browser conservation tracks (Felsenstein and Churchill, 1996; Siepel et al., 2005; Pollard et al., 2010), ENCODE candidate cis-regulatory elements (cCRE) (Leung et al., 2015) and vertebrate conserved elements (Chiaromonte et al., 2002) revealed predicted promoter regions and regulatory elements upstream of Cys1 and Pkhd1 TSSs (Figure 2A; Figure 3A). Three of the four candidate TFAP2B binding sites are located within the predicted regulatory elements of Cys1 and Pkhd1 (Figure 2A, Figure 3A). For Cys1, both TFAP2B binding sites are located within the predicted promoter region (Figure 2A, black rectangles). In the case of Pkhd1, one TFAP2B binding site is located within a predicted enhancer, while the second is between predicted enhancers (Figure 3A, black rectangles). These data are consistent with the reported renal cystic phenotype of mice deficient in TFAP2B (Moser et al., 1997a; Moser et al., 2003; Wang et al., 2018).
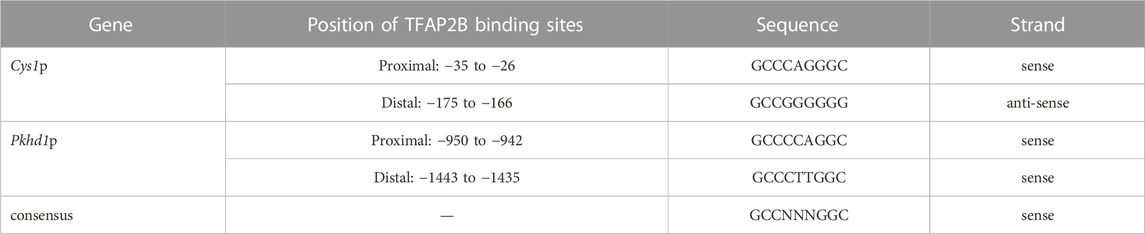
TABLE 1. Predicted TFAP2B binding sites in the Cys1 and Pkhd1 promoters.
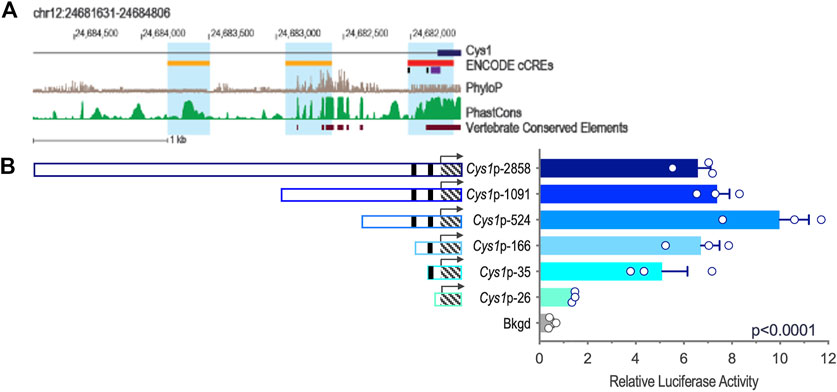
FIGURE 2. Cys1 upstream regulatory region and reporter gene analysis of Cys1 promoter constructs. (A) Map of the Cys1 upstream regulatory region showing UCSC tracks for predicted regulatory elements (cCRE and Vertebrate Conserved Elements) and sequence conservation (phyloP and PhastCOns). Black rectangles indicate TFAP2B binding sites. (B) Cys1 promoter constructs and corresponding relative luciferase activities in transfected mIMCD-3 cells. Black rectangles indicate TFAP2B binding sites. Hatched boxes indicate Cys1 5′-UTR sequences. Arrows indicate TSS. The Cys1 TSS genomic coordinate is Chr12:24,681,806 from the GRCm38/mm10 mouse genome assembly. The error bars indicate S.E.M. One-way ANOVA was used to determine the significance of differences among mean luciferase activities associated with each construct.
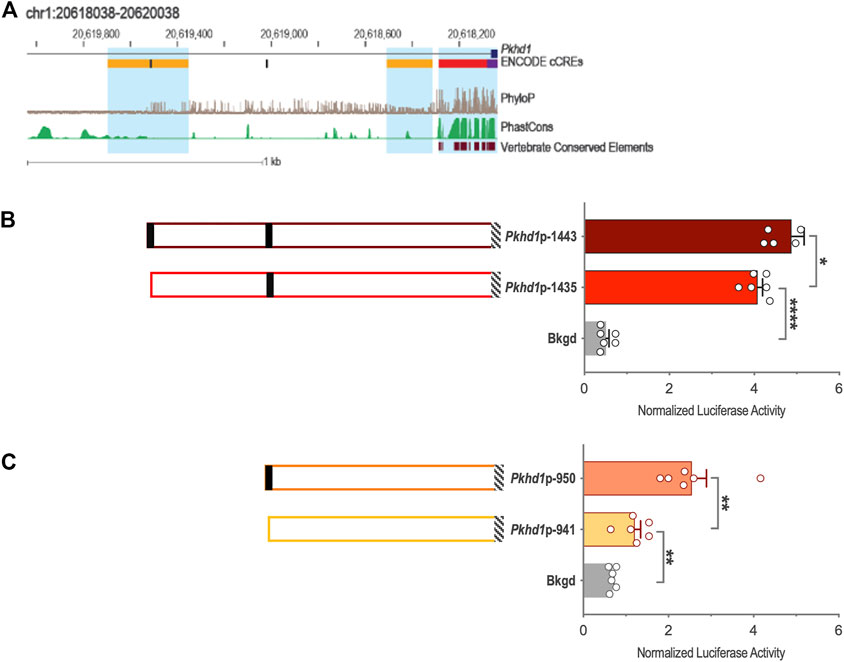
FIGURE 3. Pkhd1 upstream regulatory region and reporter gene analysis of the Pkhd1 promoter constructs. (A) Map of the Pkhd1 upstream regulatory region showing UCSC tracks for predicted regulatory elements (cCRE and Vertebrate Conserved Elements) and sequence conservation (phyloP and PhastCOns). Black rectangles indicate TFAP2B binding sites. (B and C) Pkhd1 promoter constructs and corresponding normalized luciferase activities in transfected mIMCD-3 cells. Black rectangles indicate TFAP2B binding sites. Hatched boxes indicate Pkhd1 promoter and 5′-UTR sequences. The error bar indicates S.E.M.; * indicates p < .05; ** indicates p < .01.
3.3 Expression of Tfap2b derived mRNAs in mouse kidney and collecting duct cells
Query of the NCBI database revealed that mouse Tfap2b mRNA is alternatively spliced to produce three transcripts corresponding to three protein isoforms. The Tfap2b transcripts NM_009334.3 and NM_001025305.2 that encode TFAP2B1 (NP_033360.2) and TFAP2B2 (NP_001020476.1) isoforms are 6132 and 6167 nt in length, respectively, while the TFAP2B3 (NP_001273269.1) isoform is encoded by 3813 nt transcript NM_001286340.1. We focused our studies on the TFAP2B1 and TFAP2B2 isoforms because their transcripts correspond in size to Tfap2b mRNA reported in developing mice (Moser et al., 1995). We used isoform-specific RT-PCR to confirm the expression of Tfap2b derived transcripts in mouse kidney and in the mIMCD-3 cell line (Supplementary Figure S2). Single round of RT-PCR amplification using kidney tissue was sufficient to detect Tfap2b1 and Tfap2b2 transcripts while nested PCR was required to detect Tfap2b3 transcript (Supplementary Figure S2). In mIMCD-3 cells, Tfap2b1 and Tfap2b2 transcripts were detected using nested PCR, but no Tfap2b3 transcript was detectable (Supplementary Figure S2). Although we did not perform a quantitative RT-PCR analysis, our results suggest that expression levels of Tfap2b1 and Tfap2b2 isoforms exceed levels of Tfap2b3 in mouse kidneys, and that in mIMCD-3 cells, only Tfap2b1 and Tfap2b2 isoforms are expressed at levels detectable by PCR.
3.4 Deletion analysis of Cys1 and Pkhd1 upstream regulatory regions
We tested whether the predicted Cys1 and Pkhd1 upstream regulatory elements, including TFAP2B binding sites, influenced gene expression. Deletion fragments of Cys1 and Pkhd1 upstream DNA (Table 2) were linked to a luciferase reporter gene and assayed in mIMCD-3 cells expressing Tfap2b1 and Tfap2b2 mRNAs. Six Cys1 promoter reporter constructs were tested, spanning genomic regions −2858, −1091, −524, −166, −35, and −26 to +175 bp (Table 2; Figure 2B). Reporter activity directed by Cys1p-35 with one TFAP2B binding site was notably increased relative to the Cys1p-26 construct lacking a TFAP2B binding site (Figure 2B), suggesting that TFAP2B indeed regulates Cys1 gene expression by binding to its promoter. Further evidence of a potential role for TFAP2B in Cys1 regulation was demonstrated by a notable difference in luciferase activities between Cys1p-166, which harbors one TFAP2B binding site, and Cys1p-524 with two TFAP2B binding sites (Figure 2B), suggesting that both TFAP2B binding sites play roles in activating Cys1 gene expression. Luciferase activities derived from Cys1p-524 were the highest, while luciferase activities derived from Cys1p-1091 and Cys1p-2858 constructs were lower (Figure 2B). These findings suggest the presence of negative regulatory elements between Cys1p-524 and Cys1p-2858 of the Cys1 upstream regulatory region.
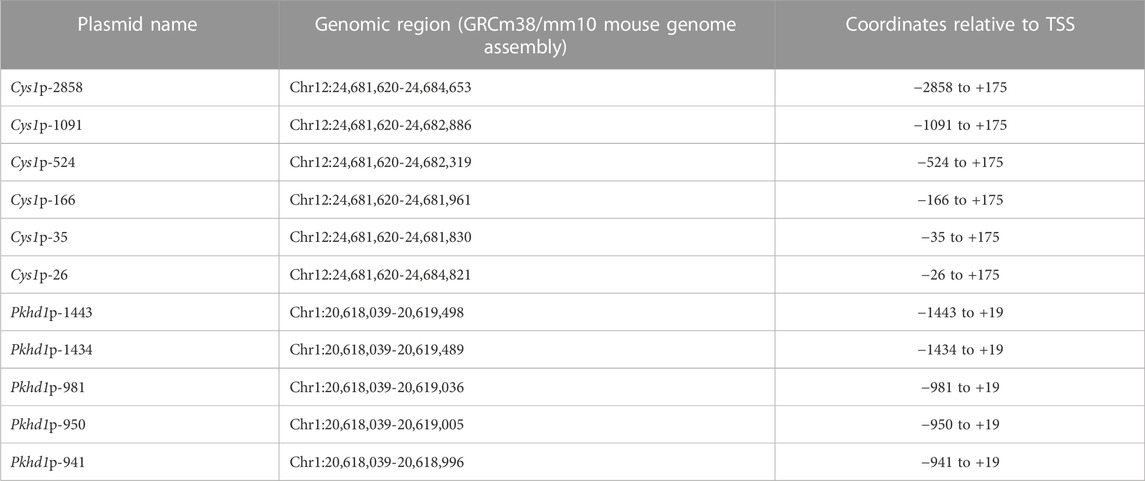
TABLE 2. Cys1 and Pkhd1 genomic DNA regions cloned into pGL3.
To functionally assess TFAP2B regulation of Pkhd1 gene expression, we generated four reporter gene constructs containing genomic DNA regions −1443, −1435, −950, and −941 to +19 (Table 2; Figures 3B, C) relative to the Pkhd1 TSS. Using reporter gene assays as described above, we compared luciferase activity in mIMCD-3 cells after transfection of constructs lacking one or both TFAP2B binding sites (Figures 3B, C). Removal of the distal TFAP2B binding site resulted in lower luciferase activity, as indicated by comparison of Pkhd1p-1443 vs. Pkhd1p-1435 transfected cells (Figure 3B). Similarly, removal of the proximal TFAP2B binding site led to reduced luciferase activity as shown by comparison of Pkhd1p-950 to Pkhd1p-941 transfected cells (Figure 3C). These data are consistent with the presence of functional TFAP2B binding sites within the Pkhd1 upstream regulatory region and that TFAP2B can regulate Pkhd1 transcription.
Taken together, our results support the role of TFAP2B in the activation of both Pkhd1 and Cys1 transcription. These findings in turn support the hypothesis that Pkhd1 and Cys1 expression are downregulated in Tfap2b−/− cystic mice, in a manner analogous to HNF1B deficiency leading to reduced cystogenic gene expression and a cystic phenotype (Shao et al., 2020). Further studies are required to test this hypothesis in vivo, including a careful analysis of the potential for Cys1 transgene overexpression to confer partial or complete rescue of the renal cystic phenotype in Tfap2b−/− mice, as was previously demonstrated for Cys1 transgene rescue of renal cystic disease in Cys1cpk/cpk (cpk) mice (Yang et al., 2021).
3.5 Cys1 and Pkhd1 promoters are both regulated by TFAP2B1 and TFAP2B2 isoforms
To characterize TFAP2B effects on Cys1 and Pkhd1 promoter reporter activity in the presence of overexpressed TFAP2B1 or TFAP2B2 isoforms, we cloned TFAP2B1 and TFAP2B2 coding sequences and generated expression vectors CMV-Tfap2b1 and CMV-Tfap2b2, which were co-transfected into mIMCD-3 cells together with either Cys1p-166 or Pkhd1p-981 luciferase reporter constructs. Both TFAP2B1 and TFAP2B2 increased Cys1p-166-linked luciferase activity, and TFAP2B2 was more effective than TFAP2B1 (Figure 4A). Similarly, both TFAP2B1 and TFAP2B2 enhanced luciferase activity from Pkhd1p-981. In this case, TFAP2B1 was the more active isoform (Figure 4B). We suggest that the observed differences in the magnitudes of activation of Cys1 and Pkhd1 promoters by TFAP2B1 and TFAP2B2 isoforms may reflect differences in the abundance of differentially active homo- and heterodimers that they may form. However, overexpression of either TFAP2B1 or TFAP2B2 in mIMCD-3 cells did not substantially impact the expression levels of endogenous Pkhd1 and Cys1 (Supplementary Figure S3).
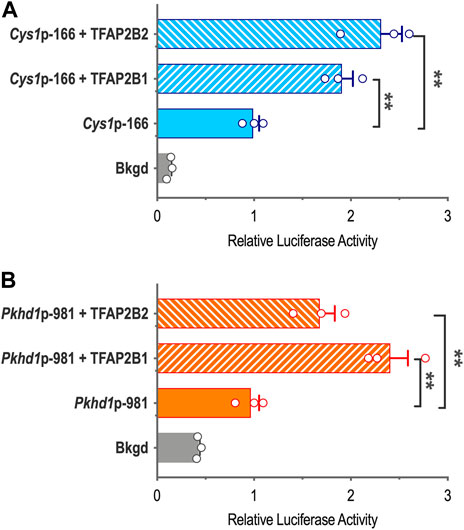
FIGURE 4. Cys1 and Pkhd1 promoter activation by TFAP2B1 and TFAP2B2. (A) The Cys1p-166 luciferase reporter construct (containing one TFAP2B binding site, see Figure 3) was transfected into mIMCD-3 cells with either TFAP2B2 or TFAP2B1 expression vectors. Relative luciferase activity (normalized to Cys1p-166 transfected alone) is shown. (B) A Pkhd1p-981 luciferase reporter construct (containing one TFAP2B binding site, similar to Pkhd1p-950 in Figure 4C) analyzed as described above. For both Cys1 and Pkhd1 constructs, TFAP2B1 and TFAP2B2 co-transfection significantly (**p < .01) increased luciferase activity compared to transfection of the Cys1 or Pkhd1 luciferase reporter alone. The error bar indicates S.E.M.
We further validated a role for TFAP2B in the regulation of Cys1 and Pkhd1 by assessing activity of Cys1p-35 and Pkhd1p-1443 luciferase reporter constructs in which the TFAP2B binding sites were mutated, such that conserved GCC nucleotides within the TFAP2B binding site were changed to ATT (Figures 5A, B). Mutation of the proximal TFAP2B binding site in Cys1p-35 resulted in lower luciferase activity (Figure 5A). Likewise, mutation of the distal TFAP2B binding site in the Pkhd1p-1443 Pkhd1 upstream regulatory region showed a tendency to reduce luciferase activity, though this effect was not statistically significant (p = .14, n = 6) (Figure 5B). These findings support a role for TFAP2B in regulating Cys1 and Pkhd1 transcription.
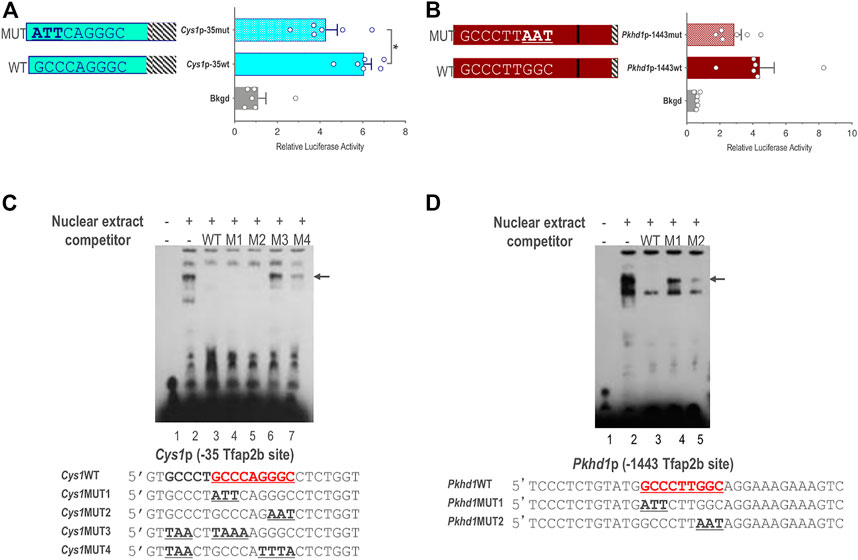
FIGURE 5. Endogenous TFAP2B binding to sites in Cys1 and Pkhd1 promoters. (A) Luciferase activity in mIMCD-3 cells transfected with Cys1p-35 reporter constructs bearing either WT or mutant versions of the proximal Cys1 TFAP2B consensus binding site. (B) Luciferase assays in cells transfected with Pkhd1p-1443 reporter constructs with either WT or mutant forms of the distal Pkhd1 TFAP2B binding site. (C and D) EMSA analysis comparing competitive binding of nuclear extract to WT vs. mutant forms of the Cys1p-35 (C) and Pkhd1p-1443 (D) TFAP2B site. WT TFAP2B binding site and mutated bp are shown in bold and underlined typeface. Arrows indicate shifted bands of interest. The error bar indicates S.E.M.; * indicates p < .05.
3.6 Endogenous TFAP2B binds to the predicted Cys1 and Pkhd1 regulatory sequences
We used electrophoretic mobility shift assay (EMSA) to determine whether TFAP2B binds directly to the predicted sequences in the Pkhd1 and Cys1 promoters. mIMCD-3 cell nuclear extracts predicted to contain TFAP2B were incubated with biotin-labeled oligonucleotide probes with sequences either representing the proximal TFAP2B binding site from the Cys1 (Figure 5C) or the Pkhd1 promoter (Figure 5D). Binding of TFAP2B resulted in a mobility shift (retardation) of the oligonucleotide on the non-denaturing PAGE (Figure 5C, lane 2), indicated binding of endogenous TFAP2B to the oligonucleotides. Unlabeled oligonucleotides representing the wild-type (WT) TFAP2B binding sites (no avidin-based detection) showed competition with the labeled oligonucleotide for TFAP2B binding resulting in the elimination of the higher molecular weight band on the gel (Figure 5C, lane 3). These observations, together with the results from the reporter gene assays, confirm the presence of functional TFAP2B binding sites (Figure 5A). However, two oligonucleotides with GCC mutated to ATT/AAT on either side of the binding site also showed competition with the labeled WT oligonucleotide (Figure 5C, lanes 4-5).
We further examined the proximal TFAP2B binding region within the Cys1 promoter by mutating another GCC motif five nucleotides upstream of the TFAP2B binding site (Figure 5C, oligo Cys1WT). Mutating this GCC to TAA, in tandem with mutation of the predicted TFAP2B binding site (Figure 5C, oligos Cys1MUT3 and Cys1MUT4), abrogated the ability of unlabeled mutant oligonucleotides to compete with labeled WT oligonucleotide probe (Figure 5C, lanes 6 and 7). These observations suggest that DNA sequences flanking the TFAP2B binding site may assist the attachment of TFAP2B to its binding site in the Cys1 promoter. We note that the DNA sequences immediately upstream of the TFAP2B binding site in the Cys1WT, Cys1MUT1 and Cys1MUT2 oligos were derived from the mouse genome and differed from the vector-derived DNA sequences immediately upstream of the TFAP2B binding site in the Cys1p-35mut construct and these differences are responsible for the unanticipated binding of Cys1MUT1and Cys1MUT2.
In the case of Pkhd1, an oligonucleotide probe representing the distal upstream TFAP2B binding site was observed to undergo a mobility shift when incubated with mIMCD-3 cell nuclear extract (Figure 5D, lane 2), confirming TFAP2B binding to this site in vitro. Unlabeled WT oligonucleotide competed for TFAP2B binding (Figure 5D, lane 3) while two oligonucleotides with mutated TFAP2B binding sites did not (Figure 5D, lanes 4 and 5), confirming TFAP2B binding to the predicted sequences in the Pkhd1 promoter. DNA sequences flanking the TFAP2B binding site in the Pkhd1 promoter did not facilitate in vitro binding of TFAP2B to its binding site.
4 Discussion
While PKHD1 mutations are the major cause of ARPKD in human, Pkhd1 mutant mice do not recapitulate the ARPKD renal phenotype for yet undefined reasons. The cpk mouse, by contrast, is the best-characterized animal model which similarly resembles to that of human ARPKD. The renal cystic phenotype in Tfap2b−/− mice (Moser et al., 1997a; Moser et al., 2003; Wang et al., 2018; Lamontagne et al., 2022) and our results indicate that TFAP2B may activate both the Pkhd1 and Cys1 promoters. Identification of TFAP2B as a common activating transcription factor for Cys1 and Pkhd1 further support a functional link between these two cystogenes in collecting duct cells.
While patients with ARPKD due to PKHD1 defects and Pkhd1 mutant mice both have the liver ductal plate malformation phenotype, TFAP2B/Tfap2b apparently is not expressed in adult human and mouse livers or during embryonic development1, 2, 3. This could be either because TFAP2B/Tfap2b does not function in the liver or more likely, because cholangiocytes represent a small fraction (∼<5%) of the total liver epithelial cell mass. This issue remains unresolved as the scRNA-seq data in ENCODE database and published single-cell RNA-seq data (Aizarani et al., 2019) reveal absent or very low expression of TFAP2A and TFAP2B in adult human cholangiocytes; publicly available mouse scRNA-seq data (Tabula Muris database4) does not have an entry for cholangiocytes.
DNA binding sites of human TFAP2A, −B, −C, and −E transcription factors identified either through analysis of ChIP-seq data or SELEX experiments (Castro-Mondragon et al., 2022) are similar to TFAP2B binding sites we evaluated in the current report (Supplementary Figure S1). Furthermore, we have demonstrated similarity between DNA binding sites of human and mouse TFAP2B orthologues. Given the similarity in the DNA binding sites of the AP2 transcription factor family, the specificity in target gene activation appears to result from differential expression of TFAP2 family member(s) in specific cell types. For example, single cell RNA-seq experiments demonstrate Tfap2b expression in cortical collecting duct cells and Tfap2a expression in medullary collecting duct cells of adult mouse kidneys (Humphreys labor5) (Park et al., 2018; Ransick et al., 2019). Evaluation of conditional knockout mice clarify the roles of Tfap2a and Tfap2b in the developing and mature mouse kidney. Mice lacking functional Tfap2a in nephron progenitor cells have phenotypically normal kidneys while Tfap2a inactivation in adult medullary collecting ducts results in cystic dilatation (Lamontagne et al., 2022), suggesting that Tfap2a functions in the maintenance of medullary collecting duct cell terminal differentiation. In contrast, heterozygosity of Tfap2b in nephron progenitor cells causes progressive distal convoluted tubule abnormalities with associated renal fibrosis and cysts; whereas complete loss abolishes the development of distal convoluted tubules, and causes renal cysts, fibrosis, and early postnatal death (Marneros, 2020; Lamontagne et al., 2022). Therefore, Tfap2a and Tfap2b have non-redundant, distinct, spatiotemporal functions in discrete segments of the distal nephron.
We note that in both human PKHD1-related disease and the Cys1cpk/cpk mouse model of ARPKD, cystic disease is initiated in the fetal proximal tubule and transitions to collecting duct dilation as the predominant lesion in the perinatal period (Nakanishi et al., 2000). Based on these phenotypic observations and our data indicating that TFAP2B regulates Pkhd1 and Cys1, we propose that Tfap2b, with Pkhd1 and Cys1, are components of a transcriptional network that modulates renal tubular cell functional differentiation, which when disrupted leads to an ARPKD-like phenotype. However, there is complexity in the renal phenotype with differences between human and mice depending upon which gene is defective. Genetic defects in TFAP2B/Tfap2b genes result in ARPKD-like phenotype in mice (Moser et al., 1997a; Moser et al., 2003; Wang et al., 2018; Lamontagne et al., 2022) while humans with Char syndrome have no renal phenotype6. Genetic defects in PKHD1/Pkhd1 genes result in ARPKD in humans, while mice have either no or mild renal phenotype (O’Connor and Guay-Woodford, 2016). Genetic defects in CYS1/Cys1 genes result in ARPKD in both humans and mice (Yang et al., 2021). This phenotype variation suggests differential molecular functions of these genes in human and mouse renal epithelia. These differences reflect either susceptibility to cyst development in human kidneys or protective mechanisms in mouse kidneys and can be exploited for identifying therapeutic targets for ARPKD.
Our reporter gene assays are a simplified experimental system designed to test specific protein-DNA interactions. Expression of endogenous Pkhd1 and Cys1 in mIMCD-3 cells was not affected by overexpression of either TFAP2B1or TFAP2B2. Several factors could account for this result. First, mIMCD-3 cells originate from the inner medullary collecting duct cells, which do not express Tfap2b (Lamontagne et al., 2022) and may lack other transcription factors, such as KCTD1 (Marneros, 2020) and GAS41/YEATS4 (Ding et al., 2006) needed to activate expression of TFAP2B-regulated genes. Second, in addition to direct binding of upstream regulatory regions, TFAP2B could regulate Pkhd1 and Cys1 gene expression by modulating chromatin structure through interactions with YEATS4, which is a component of histone acetyltransferase machinery (Cai et al., 2003; Doyon et al., 2004).
Our in vitro findings set the stage for further studies to decipher the in vivo role for TFAP2B in the regulation of Cys1 and Pkhd1 expression. Such studies should include an analysis of the extent to which an overexpressed Tfap2b, Pkhd1 or Cys1 transgene can rescue the Tfap2b−/− cystic kidney phenotype, as we have recently demonstrated for the cpk mouse renal phenotype (Yang et al., 2021). Furthermore, from a clinical perspective, we speculate that among the approximately 20% of patients with ARPKD who lack mutations in PKHD1, DZIP1 or CYS1 coding sequences, there may be patients for whom disease is due to sequence variants in the TFAP2B binding sites of PKHD1 or CYS1. Alternatively, these patients may carry novel mutations directly affecting TFAP2B-regulated genes that are expressed in renal collecting duct epithelia.
Characterization of TFAP2B regulation of Pkhd1, Cys1 and potentially other cystogenic genes will expand our understanding of transcriptional networks that modulate renal tubular cell functional differentiation and provide new mechanistic insights into how disruption of these regulatory networks contributes to the ductal dilatation that is the hallmark of recessive PKD.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
MW and NH designed and performed the experiments, evaluated data, prepared figures, and wrote initial drafts of the manuscript. AO evaluated data and contributed to the preparation of an early version of this report. LC performed bioinformatic analysis of Pkhd1 and Cys1 upstream regulatory regions, evaluated data, revised figures, and prepared a revised version of the report. LG-W conceived of the study, designed the experiments, evaluated the data, edited and revised the manuscript.
Funding
This work was supported by NIH grants (R01DK121530), PKD Foundation, and The Moran Family Foundation to LG-W.
Acknowledgments
The authors thank Su Bu for technical assistance, Adam Richman for manuscript editing, current and former members of the LG-W laboratory and Dr. Zsuzsa Bebok (University of Alabama at Birmingham) for review and critical evaluation of the reported studies.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2022.946344/full#supplementary-material
Footnotes
1https://www.gtexportal.org/home/gene/TFAP2B
2https://www.ncbi.nlm.nih.gov/gene/702
3https://www.ncbi.nlm.nih.gov/gene/21419
4https://tabula-muris.ds.czbiohub.org
5http://humphreyslab.com/SingleCell/search.php
6https://monarchinitiative.org/gene/HGNC:11743#phenotype
References
Aizarani, N., Saviano, A., SagarMailly, L., Durand, S., and Herman, J. S. (2019). A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 572 (7768), 199–204. doi:10.1038/s41586-019-1373-2
Alzarka, B., Morizono, H., Bollman, J. W., Kim, D., and Guay-Woodford, L. M. (2017). Design and implementation of the hepatorenal fibrocystic disease core center clinical database: A centralized resource for characterizing autosomal recessive polycystic kidney disease and other hepatorenal fibrocystic diseases. Front. Pediatr. 5, 80. doi:10.3389/fped.2017.00080
Cai, Y., Jin, J., Tomomori-Sato, C., Sato, S., Sorokina, I., Parmely, T. J., et al. (2003). Identification of new subunits of the multiprotein mammalian TRRAP/TIP60-containing histone acetyltransferase complex. J. Biol. Chem. 278 (44), 42733–42736. doi:10.1074/jbc.C300389200
Castro-Mondragon, J. A., Riudavets-Puig, R., Rauluseviciute, I., Lemma, R. B., Turchi, L., Blanc-Mathieu, R., et al. (2022). Jaspar 2022: The 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 50 (D1), D165–d173. doi:10.1093/nar/gkab1113
Cheng, C., Ying, K., Xu, M., Zhao, W., Zhou, Z., Huang, Y., et al. (2002). Cloning and characterization of a novel human transcription factor AP-2 beta like gene (TFAP2BL1). Int. J. Biochem. Cell. Biol. 34 (1), 78–86. doi:10.1016/s1357-2725(01)00098-x
Chiaromonte, F., Yap, V. B., and Miller, W. (2002). Scoring pairwise genomic sequence alignments. Pac Symp. Biocomput 2002, 115–126. doi:10.1142/9789812799623_0012
Ding, X., Fan, C., Zhou, J., Zhong, Y., Liu, R., Ren, K., et al. (2006). GAS41 interacts with transcription factor AP-2beta and stimulates AP-2beta-mediated transactivation. Nucleic Acids Res. 34 (9), 2570–2578. doi:10.1093/nar/gkl319
Doyon, Y., Selleck, W., Lane, W. S., Tan, S., and Côté, J. (2004). Structural and functional conservation of the NuA4 histone acetyltransferase complex from yeast to humans. Mol. Cell. Biol. 24 (5), 1884–1896. doi:10.1128/mcb.24.5.1884-1896.2004
Eckert, D., Buhl, S., Weber, S., Jager, R., and Schorle, H. (2005). The AP-2 family of transcription factors. Genome Biol. 6 (13), 246. doi:10.1186/gb-2005-6-13-246
Felsenstein, J., and Churchill, G. A. (1996). A Hidden Markov Model approach to variation among sites in rate of evolution. Mol. Biol. Evol. 13 (1), 93–104. doi:10.1093/oxfordjournals.molbev.a025575
Feng, W., and Williams, T. (2003). Cloning and characterization of the mouse AP-2 epsilon gene: A novel family member expressed in the developing olfactory bulb. Mol. Cell. Neurosci. 24 (2), 460–475. doi:10.1016/s1044-7431(03)00209-4
Fornes, O., Castro-Mondragon, J. A., Khan, A., van der Lee, R., Zhang, X., Richmond, P. A., et al. (2020). Jaspar 2020: Update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 48 (D1), D87–D92. doi:10.1093/nar/gkz1001
Guay-Woodford, L. M., Bissler, J. J., Braun, M. C., Bockenhauer, D., Cadnapaphornchai, M. A., Dell, K. M., et al. (2014). Consensus expert recommendations for the diagnosis and management of autosomal recessive polycystic kidney disease: Report of an international conference. J. Pediatr. 165 (3), 611–617. doi:10.1016/j.jpeds.2014.06.015
Haberle, V., and Stark, A. (2018). Eukaryotic core promoters and the functional basis of transcription initiation. Nat. Rev. Mol. Cell. Biol. 19 (10), 621–637. doi:10.1038/s41580-018-0028-8
Hiesberger, T., Bai, Y., Shao, X., McNally, B. T., Sinclair, A. M., Tian, X., et al. (2004). Mutation of hepatocyte nuclear factor-1beta inhibits Pkhd1 gene expression and produces renal cysts in mice. J. Clin. Invest. 113 (6), 814–825. doi:10.1172/JCI20083
Hilger-Eversheim, K., Moser, M., Schorle, H., and Buettner, R. (2000). Regulatory roles of AP-2 transcription factors in vertebrate development, apoptosis and cell-cycle control. Gene 260 (1-2), 1–12. doi:10.1016/s0378-1119(00)00454-6
Hou, X., Mrug, M., Yoder, B. K., Lefkowitz, E. J., Kremmidiotis, G., D'Eustachio, P., et al. (2002). Cystin, a novel cilia-associated protein, is disrupted in the cpk mouse model of polycystic kidney disease. J. Clin. Invest. 109 (4), 533–540. doi:10.1172/JCI14099
Lamontagne, J. O., Zhang, H., Zeid, A. M., Strittmatter, K., Rocha, A. D., Williams, T., et al. (2022). Transcription factors AP-2α and AP-2β regulate distinct segments of the distal nephron in the mammalian kidney. Nat. Commun. 13 (1), 2226. doi:10.1038/s41467-022-29644-3
Leung, D., Jung, I., Rajagopal, N., Schmitt, A., Selvaraj, S., Lee, A. Y., et al. (2015). Integrative analysis of haplotype-resolved epigenomes across human tissues. Nature 518 (7539), 350–354. doi:10.1038/nature14217
Marneros, A. G. (2020). AP-2β/KCTD1 control distal nephron differentiation and protect against renal fibrosis. Dev. Cell. 54 (3), 348–366. e345. doi:10.1016/j.devcel.2020.05.026
Miao, Z., Balzer, M. S., Ma, Z., Liu, H., Wu, J., Shrestha, R., et al. (2021). Single cell regulatory landscape of the mouse kidney highlights cellular differentiation programs and disease targets. Nat. Commun. 12 (1), 2277. doi:10.1038/s41467-021-22266-1
Moser, M., Dahmen, S., Kluge, R., Grone, H., Dahmen, J., Kunz, D., et al. (2003). Terminal renal failure in mice lacking transcription factor AP-2 beta. Lab. Invest. 83 (4), 571–578. doi:10.1097/01.lab.0000064703.92382.50
Moser, M., Imhof, A., Pscherer, A., Bauer, R., Amselgruber, W., Sinowatz, F., et al. (1995). Cloning and characterization of a second AP-2 transcription factor: AP-2 beta. Development 121 (9), 2779–2788. doi:10.1242/dev.121.9.2779
Moser, M., Pscherer, A., Roth, C., Becker, J., Mucher, G., Zerres, K., et al. (1997a). Enhanced apoptotic cell death of renal epithelial cells in mice lacking transcription factor AP-2beta. Genes. Dev. 11 (15), 1938–1948. doi:10.1101/gad.11.15.1938
Moser, M., Ruschoff, J., and Buettner, R. (1997b). Comparative analysis of AP-2 alpha and AP-2 beta gene expression during murine embryogenesis. Dev. Dyn. 208 (1), 115–124. doi:10.1002/(SICI)1097-0177(199701)208:1<115:AID-AJA11>3.0.CO;2-5
Nagao, S., Kugita, M., Yoshihara, D., and Yamaguchi, T. (2012). Animal models for human polycystic kidney disease. Exp. Anim. 61 (5), 477–488. doi:10.1538/expanim.61.477
Nakanishi, K., Sweeney, W. E., Zerres, K., Guay-Woodford, L. M., and Avner, E. D. (2000). Proximal tubular cysts in fetal human autosomal recessive polycystic kidney disease. J. Am. Soc. Nephrol. 11 (4), 760–763. doi:10.1681/ASN.V114760
O’Connor, A., and Guay-Woodford, W. L. (2015). “The polycystic kidney diseases and other hepato-renal fibrocystic diseases: Clinical phenotypes, molecular pathobiology, and variation between mouse and man,” in Kidney development, repair, and regeneration (Amsterdam, Netherlands: Elsevier), 241–264.
O’Connor, A. K., and Guay-Woodford, L. M. (2016). “Chapter 20 - polycystic kidney diseases and other hepatorenal fibrocystic diseases: Clinical phenotypes, molecular pathobiology, and variation between mouse and man,” in Kidney development, disease, repair and regeneration. Editor M. H. Little (San Diego: Academic Press), 241–264.
Onuchic, L. F., Furu, L., Nagasawa, Y., Hou, X., Eggermann, T., Ren, Z., et al. (2002). PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am. J. Hum. Genet. 70 (5), 1305–1317. doi:10.1086/340448
Outeda, P., Menezes, L., Hartung, E. A., Bridges, S., Zhou, F., Zhu, X., et al. (2017). A novel model of autosomal recessive polycystic kidney questions the role of the fibrocystin C-terminus in disease mechanism. Kidney Int. 92 (5), 1130–1144. doi:10.1016/j.kint.2017.04.027
Park, J., Shrestha, R., Qiu, C., Kondo, A., Huang, S., Werth, M., et al. (2018). Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360 (6390), 758–763. doi:10.1126/science.aar2131
Pollard, K. S., Hubisz, M. J., Rosenbloom, K. R., and Siepel, A. (2010). Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 20 (1), 110–121. doi:10.1101/gr.097857.109
Raap, M., Gierendt, L., Kreipe, H. H., and Christgen, M. (2021). Transcription factor AP-2beta in development, differentiation and tumorigenesis. Int. J. Cancer 149 (6), 1221–1227. doi:10.1002/ijc.33558
Ransick, A., Lindström, N. O., Liu, J., Zhu, Q., Guo, J. J., Alvarado, G. F., et al. (2019). Single-cell profiling reveals sex, lineage, and regional diversity in the mouse kidney. Dev. Cell. 51 (3), 399–413. e397. doi:10.1016/j.devcel.2019.10.005
Shao, A., Chan, S. C., and Igarashi, P. (2020). Role of transcription factor hepatocyte nuclear factor-1β in polycystic kidney disease. Cell. Signal 71, 109568. doi:10.1016/j.cellsig.2020.109568
Siepel, A., Bejerano, G., Pedersen, J. S., Hinrichs, A. S., Hou, M., Rosenbloom, K., et al. (2005). Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15 (8), 1034–1050. doi:10.1101/gr.3715005
Tao, B., Bu, S., Yang, Z., Siroky, B., Kappes, J. C., Kispert, A., et al. (2009). Cystin localizes to primary cilia via membrane microdomains and a targeting motif. J. Am. Soc. Nephrol. 20 (12), 2570–2580. doi:10.1681/ASN.2009020188
Tummala, R., Romano, R. A., Fuchs, E., and Sinha, S. (2003). Molecular cloning and characterization of AP-2 epsilon, a fifth member of the AP-2 family. Gene 321, 93–102. doi:10.1016/s0378-1119(03)00840-0
Wang, J., Ji, W., Zhu, D., Wang, W., Chen, Y., Zhang, Z., et al. (2018). Tfap2b mutation in mice results in patent ductus arteriosus and renal malformation. J. Surg. Res. 227, 178–185. doi:10.1016/j.jss.2018.02.038
Ward, C. J., Hogan, M. C., Rossetti, S., Walker, D., Sneddon, T., Wang, X., et al. (2002). The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat. Genet. 30 (3), 259–269. doi:10.1038/ng833
Williams, T., and Tjian, R. (1991a). Analysis of the DNA-binding and activation properties of the human transcription factor AP-2. Genes. Dev. 5 (4), 670–682. doi:10.1101/gad.5.4.670
Williams, T., and Tjian, R. (1991b). Characterization of a dimerization motif in AP-2 and its function in heterologous DNA-binding proteins. Science 251 (4997), 1067–1071. doi:10.1126/science.1998122
Wu, M., Yang, C., Tao, B., Bu, S., and Guay-Woodford, L. M. (2013). The ciliary protein cystin forms a regulatory complex with necdin to modulate Myc expression. PLoS One 8 (12), e83062. doi:10.1371/journal.pone.0083062
Yang, C., Harafuji, N., O'Connor, A. K., Kesterson, R. A., Watts, J. A., Majmundar, A. J., et al. (2021). Cystin genetic variants cause autosomal recessive polycystic kidney disease associated with altered Myc expression. Sci. Rep. 11 (1), 18274. doi:10.1038/s41598-021-97046-4
Zhao, F., Lufkin, T., and Gelb, B. D. (2003). Expression of Tfap2d, the gene encoding the transcription factor Ap-2 delta, during mouse embryogenesis. Gene Expr. Patterns 3 (2), 213–217. doi:10.1016/s1567-133x(02)00067-4
Keywords: autosomal recessive polycystic kidney disease (ARPKD), Cys1, Pkhd1, Tfap2b, TFAP2B
Citation: Wu M, Harafuji N, O’Connor AK, Caldovic L and Guay-Woodford LM (2023) Transcription factor Ap2b regulates the mouse autosomal recessive polycystic kidney disease genes, Pkhd1 and Cys1. Front. Mol. Biosci. 9:946344. doi: 10.3389/fmolb.2022.946344
Received: 17 May 2022; Accepted: 29 December 2022;
Published: 12 January 2023.
Edited by:
Miguel Angel Garcia-Gonzalez, Complejo Hospitalario Universitario de Santiago, SpainReviewed by:
Adrian Cordido, Yale University, United StatesRachel Gallagher, Yale University, United States
Copyright © 2023 Wu, Harafuji, O’Connor, Caldovic and Guay-Woodford. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lisa M. Guay-Woodford, LGuaywoo@childrensnational.org