Electron Transfer Coupled to Conformational Dynamics in Cell Respiration
 Marco Reidelbach1
Marco Reidelbach1  Christoph Zimmer
Christoph Zimmer Brigitte Meunier
Brigitte Meunier Peter R. Rich
Peter R. Rich Vivek Sharma
Vivek Sharma- 1Department of Physics, University of Helsinki, Helsinki, Finland
- 2Department of Structural and Molecular Biology, University College London, London, United Kingdom
- 3Institute for Integrative Biology of the Cell (I2BC), Université Paris-Saclay, Gif-sur-Yvette, France
- 4HiLIFE Institute of Biotechnology, University of Helsinki, Helsinki, Finland
Cellular respiration is a fundamental process required for energy production in many organisms. The terminal electron transfer complex in mitochondrial and many bacterial respiratory chains is cytochrome c oxidase (CcO). This converts the energy released in the cytochrome c/oxygen redox reaction into a transmembrane proton electrochemical gradient that is used subsequently to power ATP synthesis. Despite detailed knowledge of electron and proton transfer paths, a central question remains as to whether the coupling between electron and proton transfer in mammalian mitochondrial forms of CcO is mechanistically equivalent to its bacterial counterparts. Here, we focus on the conserved span between H376 and G384 of transmembrane helix (TMH) X of subunit I. This conformationally-dynamic section has been suggested to link the redox activity with the putative H pathway of proton transfer in mammalian CcO. The two helix X mutants, Val380Met (V380M) and Gly384Asp (G384D), generated in the genetically-tractable yeast CcO, resulted in a respiratory-deficient phenotype caused by the inhibition of intra-protein electron transfer and CcO turnover. Molecular aspects of these variants were studied by long timescale atomistic molecular dynamics simulations performed on wild-type and mutant bovine and yeast CcOs. We identified redox- and mutation-state dependent conformational changes in this span of TMH X of bovine and yeast CcOs which strongly suggests that this dynamic module plays a key role in optimizing intra-protein electron transfers.
Introduction
All forms of cytochrome c oxidase (CcO) reduce molecular oxygen (O2) to water with four electrons provided by cytochrome c and four protons from the mitochondrial matrix or bacterial cytoplasmic aqueous phase. These charge transfers from the two opposite sides of the membrane result in membrane polarization, which is further enhanced with an energetically uphill transfer of four more protons across the membrane (Wikström, 1977) (Figure 1A). The resulting proton concentration difference and charge imbalance across the membrane (the protonmotive force) drives the synthesis of ATP. The reduction of O2 occurs at a highly conserved binuclear center (BNC) comprising heme a3 and CuB. The redox chemistry at the BNC drives the proton pump of CcO. Extensive structural (Iwata et al., 1995; Svensson-Ek et al., 2002; Tsukihara et al., 1995; Abramson et al., 2000), biochemical (Pfitzner et al., 2000; Fetter et al., 1995; Maréchal et al., 2012) and computational work (Sharma et al., 2013; Sharma et al., 2015; Kaila et al., 2008; Ghosh et al., 2009; Popović and Stuchebrukhov, 2004) on CcOs from varying sources has delineated a now widely-accepted mechanism of proton pumping in bacterial and yeast enzymes, based on a D channel role for redox-coupled proton movements (Rich, 2017; Wikström and Sharma, 2018). However, high-resolution structural data on CcO from bovine heart mitochondria (Yoshikawa and Shimada, 2015), has led to a coupling mechanism proposal which instead involves the transport of the additional pumped protons via an alternative H pathway. Mutations in this H pathway in bacterial and yeast enzymes have not affected catalytic turnover or proton pumping (Lee et al., 2000; Maréchal et al., 2020), prompting the suggestion that their H pathways may instead have a dielectric role that could aid internal electron transfer or mediate allosteric effects (Rich and Maréchal, 2013; Sharma et al., 2017; Malkamäki et al., 2019). This dichotomy of possible proton pumping mechanisms between bacterial/yeast and mammalian CcOs remains an unresolved issue, hindered in particular by technical difficulties in introducing equivalent point mutations into the mammalian mitochondrial DNA-encoded subunits of respiratory complexes [cf. (Mok et al., 2020)]. This leaves computational approaches, as applied here, as ideal methods to explore function, to predict effects of such point mutations on enzyme mechanism and to compare them where possible with experimentally-verifiable effects of these mutations in genetically-viable organisms such as yeast. Here, we performed microseconds long atomistic molecular dynamics (MD) simulations on a high-resolution (1.5 Å) structure of oxidized bovine CcO (Yano et al., 2016), focusing on the highly conserved H376-G384 span of transmembrane helix (TMH) X of subunit I. The H378 and H376 are ligands of hemes a and a3, respectively, and the V380-G384 segment is a conformationally-flexible region (Figure 1B) that has been proposed to gate H channel proton conductivity via redox- and ligand-induced movement of S382 (Yoshikawa and Shimada, 2015). The MD simulations were extended with additional computations in both bovine and yeast CcOs, together with functional studies in yeast CcO, of two point mutations, Val380Met (V380M) and Gly384Asp (G384D). These two mutations were initially identified in random mutagenesis trials (Ortwein et al., 1997; Meunier and Rich, 1998) and cause loss of CcO turnover activity, thus highlighting the importance of this region of helix X in the enzyme mechanism. The simulations predict redox- and mutation-induced conformational transitions in yeast and bovine CcOs, based on which we suggest that this dynamic and functionally critical module performs a similar function in both enzymes, perhaps controlling intra-protein electron transfer rather than having a role in proton transfers.
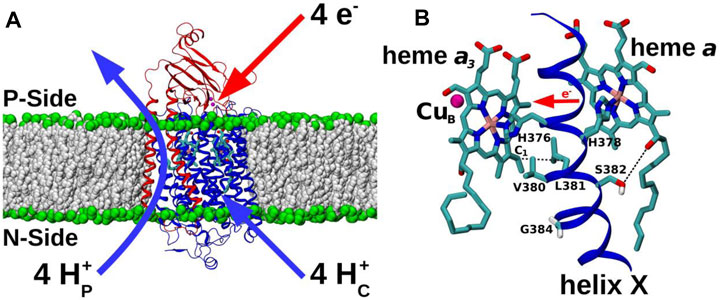
FIGURE 1. CcO and TMH X of catalytic subunit I. (A) Subunit I (blue) and II (red) of CcO from yeast Saccharomyces cerevisiae (PDB 6HU9) embedded in a POPC lipid bilayer (grey and green spheres). Redox-active centers are included, and the overall proton transfers (H+P – pumped protons; H+C – substrate protons) associated with a full four-electron catalytic cycle are indicated. P-side: positively-charged intermembrane space; N-side: negatively-charged mitochondrial matrix. (B) Redox-active cofactors of yeast CcO, heme a3 and heme a and their ligands H376 and H378, respectively, from TMH X of subunit I are shown. CuB (magenta sphere) and heme a3 form the BNC. Residues V380, L381, S382, and G384 are labelled. S382 sidechain oxygen – heme a hydroxyl oxygen and L381 Cβ – heme a3 vinyl C1 distances are indicated by dashed black lines. Forward direction of heme-heme electron transfer is marked with a red horizontal arrow.
Results
Catalytic Activity and Redox Properties of WT and Mutant Yeast CcO
The two conserved residues V380 and G384 are located in the vicinity of S382, the proposed H channel gate in bovine CcO. Although yeast mutation S382A itself had no significant effect on respiratory competence, catalytic turnover or proton coupling (Maréchal et al., 2020), the two mutations (V380M and G384D) resulted in a severely respiratory-deficient phenotype caused by an assembled but inactive CcO (Figure 2) [see also (Ortwein et al., 1997; Meunier and Rich, 1998)]. V380M and G384D are located on the helix X face opposite to S382 and extend towards the BNC (Figure 1B), and are quite separate from all three proposed proton channels (Supplementary Figure S1). Mutations of these two residues are thus not expected to kill enzymatic activity by directly influencing the proton transfer reactions. Instead, due to their proximity to hemes, inhibitory effects on redox reactions are possible. The inhibitory effects, together with the known conformational flexibility and very strong conservation of this span of TMH X (Supplementary Figure S2), certainly point to this domain having a crucial role to play.
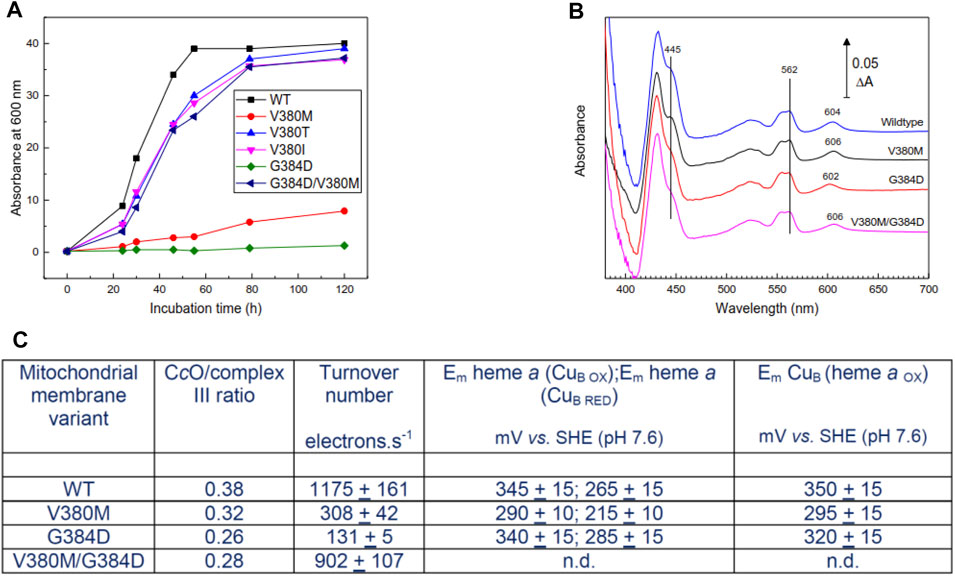
FIGURE 2. Properties of mutant yeast cells and mitochondrial membranes. (A) Respiratory growth rates of intact yeast cells in ethanol—containing medium. (B) Dithionite-reduced minus oxidized spectra of mitochondrial membranes derived from WT and mutant cell lines. Spectra were normalized to the same complex III concentrations assessed from ΔA at 562–575 nm. The 602–606 nm band arises primarily from heme a; hemes a and a3 contribute roughly equally to the 445 nm band. (C) Summary of ratio of CcO/complex III, CcO turnover numbers and redox properties of metal centers of CcO in mitochondrial membranes. Specific experimental details for all panels can be found in Methods. n.d. – not determined, but expected to resemble WT.
Cell respiratory growth profiles (Figure 2A) confirmed the respiratory-deficiency phenotype of the V380M and G384D point mutations. The revertants M380T and M380I (V380T and V380I, respectively) restored respiratory competence as, surprisingly, did the double mutant V380M/G384D [Figure 2A, see also (Namslauer et al., 2011)]. The inhibition of growth of V380M and G384D cells was not due to impaired expression of CcO, which was expressed above WT levels (3.2 and 2.6, respectively, compared to 2.4 nmol/gm wet weight in WT). CcO contents and turnover numbers in mitochondrial membranes further confirmed that respiratory deficiency arose primarily from impairment of CcO turnover in the point mutants and its recovery in the double mutant (Figures 2B,C). Reduced minus oxidized difference spectra revealed a 2 nm blueshift in G384D and a 2 nm redshift of the heme a α-band in V380M which surprisingly persisted in the double mutant (Figure 2B). In contrast, the α-band of heme a3 was not noticeably shifted, based on the reduced+CO minus reduced difference spectra which arise solely from heme a3 (Supplementary Figure S3). The redox behavior of WT and mutant yeast CcO was studied in the cyanide-inhibited mitochondrial membranes (see methods and Supplementary Figure S4). A rough estimate of the redox properties of heme a in cyanide-inhibited membranes (Figure 2C) indicated a lowering of its redox potential in V380M but little or no change in G384D. In the cyanide-inhibited state heme a3 is fixed in its oxidized state, but CuB still changes redox state and distorts the redox curve of heme a through electrostatic interaction; hence, the detailed shape of the heme a reduction versus Eh plot also provides information of the midpoint potential of CuB (Moody and Rich, 1990). This analysis suggested a lowering of Em(CuB) in V380M but a much smaller possible decrease in G384D. In WT yeast cells, heme a is typically around 5–10% reduced during steady state turnover. Heme a remained fully reduced during steady state aerobic turnover in whole G384D cells, showing definitively that the inhibited step was electron transfer into the BNC. This was less clear in V380M cells, where heme a remained 40–50% reduced during aerobic turnover. Overall, these spectroscopic and redox data indicate structural perturbation of heme a in both mutants. The reason for inhibition in G384D is most likely a low Em of heme a3 which prevents electron transfer from heme a. The cause of inhibition of V380M is less clear, but most likely involves electron transfers both into and from heme a. Hence, both mutations V380M and G384D in the conserved segment V380-G384 perturb internal electron transfer reactions of CcO through redox and/or structural changes. The presence of both mutations together must alleviate the effects of the single mutations in order to restore turnover and respiratory competence.
Molecular Dynamics Simulations of WT and Mutant Forms of Yeast and Bovine CcOs
To obtain molecular insights into the loss of oxidoreductase activity in the helix X mutants, we performed fully atomistic classical MD simulations of WT and mutant yeast and bovine CcOs in both oxidized and fully reduced states. Redox-dependent changes in distances between the S382 sidechain and heme a farnesyl hydroxyl, and between L381 (Cβ) and the C1 atom of the vinyl group of heme a3 (Figure 1B) were identified in high-resolution structural data of bovine CcO and their functional importance was emphasized (Shimada et al., 2017). In agreement with these structural data, in both bovine and yeast WT CcO simulations we observed a shorter S382 – farnesyl hydroxyl (heme a) distance (3–4 Å) in the fully oxidized state in comparison to the fully reduced state (greater than 4.5 Å) (Figure 3A). In further agreement are the coupled dynamics of the L381 (Cβ) – heme a3 vinyl (C1) distance which lengthens when the S382 – farnesyl hydroxyl distance shortens. This coupling was found to be more strongly-linked in bovine in comparison to yeast CcO simulations (Figure 3B). Overall, however, these simulated structural changes closely resemble the experimentally-observed conformational states and suggest that the in-silico model predictions are accurate.
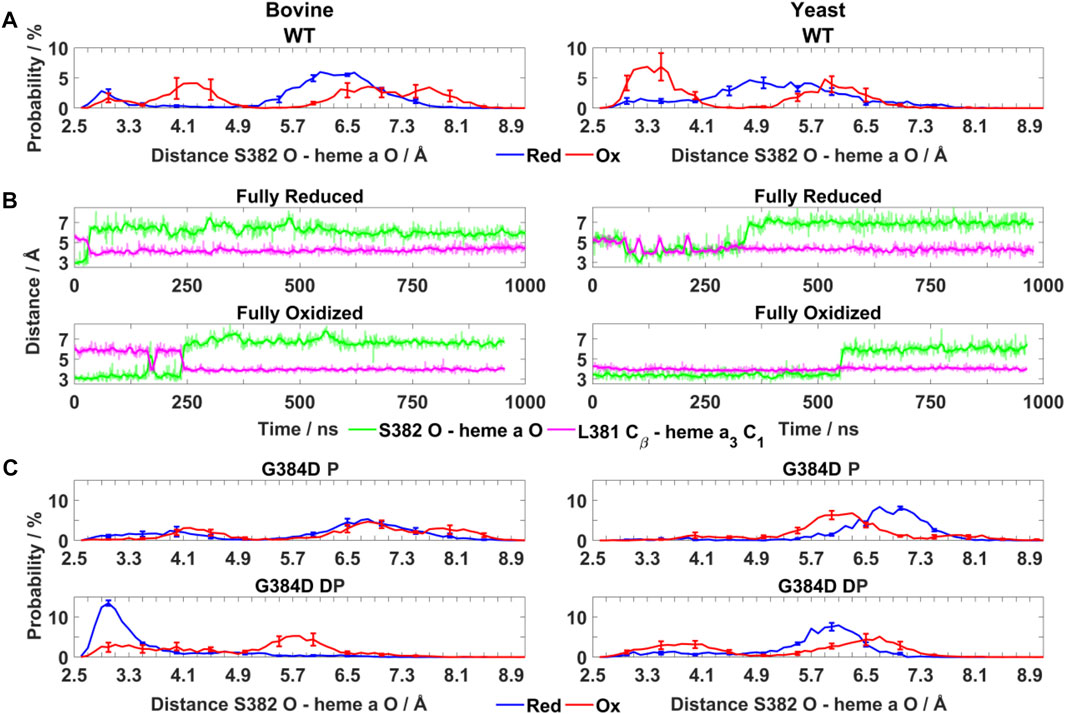
FIGURE 3. Conformational dynamics of TMH X residues in WT and mutant bovine and yeast CcOs. (A) Probability distribution (%) of S382 hydroxyl (O atom) – heme a farnesyl hydroxyl (O atom) distance in bovine and yeast WT CcO in the fully reduced and oxidized states. (B) Typical time-series of the S382 hydroxyl – heme a farnesyl hydroxyl and L381 Cβ – heme a3 C1 distance in bovine (left) and yeast (right) WT CcO in the fully reduced and oxidized states. (C) Probability distribution (%) of S382 hydroxyl – heme a farnesyl hydroxyl distance in G384D bovine (left) and yeast (right) CcOs with D384 (P - protonated/DP - deprotonated) in their fully reduced and oxidized states. The distributions are averaged over all simulation replicas and error bars correspond to standard deviations.
Next, the behaviors of bovine and yeast CcOs were simulated with the G384D mutation in both anionic and neutral forms (DP and P forms, respectively, see Supplementary Table S1). While neutral D384 (P) didn’t perturb much the geometry of TMH X in bovine or yeast CcO simulations, its anionic D384 (DP) form caused a large structural change resulting in significant loss of α-helicity in the 380-384 region, primarily in their fully oxidized states (Figure 4A). In contrast, in the fully reduced states, α-helicity was maintained and was similar to WT. Due to the electrostatic repulsion from reduced hemes, anionic D384 (DP) was displaced away from hemes by ca. 1–2 Å (D384 Cα – heme a3 Fe distance) from its original position, and formed hydrogen bonds to water molecules which diffused into the protein interior (ca. 7.4 ± 1.6 and 8.0 ± 1.8 water molecules with D384 locus) for ca. 95.6 ± 1.1% and 83.4 ± 1.1% of bovine and yeast CcO simulation times, respectively. On the other hand, in the fully oxidized state, anionic D384 (DP) was stabilized with strong hydrogen bonds to backbones of L381 and S382 (70.3 ± 7.3%) in bovine CcO and the sidechain of unique S388 (61.2 ± 3.4%) in yeast CcO (Figure 4B), resulting in a dominant loss of helicity. Overall, with neutral D384 (P), the TMH X helicity was preserved as in WT enzyme for both redox states in both enzymes. However, with anionic D384 (DP), a loss of helicity was observed in the oxidized state for both yeast and bovine CcOs. This suggests that it is most likely the anionic nature of aspartate in oxidized state of enzyme that perturbs the WT conformation of TMH X and is in part the reason for the loss of catalytic activity of yeast CcO.
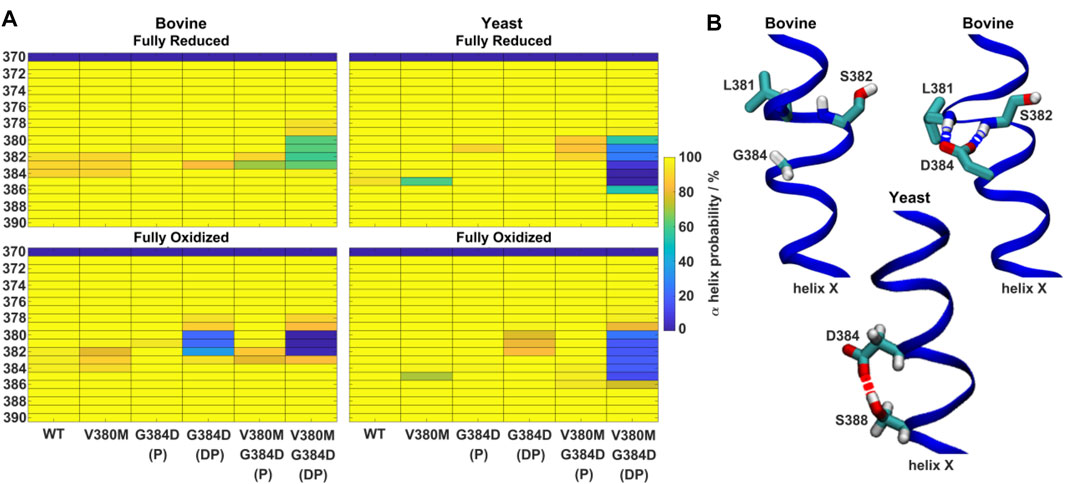
FIGURE 4. α-helicity of TMH X and hydrogen bonding rearrangements. (A) α-helix probability (%) of TMH X in WT, V380M, G384D (P/DP) and double mutant V380M/G384D (P/DP) bovine (left) and yeast (right) CcOs in their fully reduced (top) and oxidized (bottom) states. P (protonated) and DP (deprotonated) denote the protonation state of D384. (B) Conformation of TMH X in WT and the G384D (DP) mutant of bovine CcO (top) and the G384D (DP) mutant of yeast CcO (bottom). L381, S382, G384/D384, and S388 are labelled. Hydrogen bonds connecting D384 with the backbones of L381 and S382 or the sidechain of S388 are indicated by dashed blue and red lines, respectively.
As noted above, the S382 – farnesyl hydroxyl (heme a) distance displays redox-state dependency (Figure 3A), we therefore next studied how this distance behaved in the G384D mutant. The bovine G384D (DP) simulation data showed stabilization of a shorter S382 – farnesyl hydroxyl distance in the fully reduced state of enzyme, in stark contrast to WT simulations (Figures 3A,C). A similar effect was less clear in simulations of the lower-resolution yeast structure. However, in both yeast and bovine CcO simulations, the S382 – farnesyl hydroxyl distance, which was found to be 3–4 Å in the WT fully oxidized state (Figure 3A), was found to partly lengthen in the G384D (DP) case (Figure 3C). This was also observed when bovine and yeast CcO simulations were performed with neutral G384D (P) (Figure 3C), suggesting that the G384D mutant disrupts the functionally important redox-state dependency of the S382 – farnesyl hydroxyl (heme a) distance.
Overall, the above data highlight that the loss of α-helicity, redox-state dependent hydrogen bonding re-arrangements as well as perturbation of redox-state dependent conformational dynamics of the 380-384 region of helix X may all contribute to the observed loss of activity in the yeast G384D CcO mutant. Given that both bovine and yeast CcO share high structural and sequence similarity in the TMH X region (Supplementary Figure S2), and given the resemblance of the conformational transitions of S382 – farnesyl hydroxyl distance and TMH X helical propensity in WT and mutant enzymes, it is expected that a similar activity loss would be found in an equivalent mutant of bovine CcO, which currently remains inaccessible to genetic manipulation.
In addition to these conformational effects in the G384D mutant, our density functional theory (DFT) calculations on small and large model systems (see Computational Methods) revealed a lower electron affinity of heme a3 in the anionic G384D (DP) mutant compared to the WT enzyme (by ca. −11.5 to −9.1 kcal/mol, Table 1). In contrast, when modeled with neutral G384D (P), WT like electron affinity was observed. The effect of lowered electron affinity was also seen when DFT calculations were performed on WT and anionic G384D (DP) MD simulation snapshots (n = 5, see methods). We suggest that in addition to the conformational effects discussed above for G384D mutant, lowered electron affinity of heme a3 also contributes to the observed enzymatic inactivity, and is in agreement with our experimental data that show that electron transfer from heme a to heme a3/CuB is blocked.

TABLE 1. Electron affinity of heme a3 in WT and mutants.
In contrast to the blocked electron transfer from heme a to heme a3 observed in the G384D mutant, the reduction of heme a was most likely slowed in the V380M mutant due to the perturbation in its environment and the drop in its midpoint potential (Figure 2). In the V380M mutant simulations, we found a partial loss of helicity in TMH X (Figure 4A) and an increased sidechain fluctuation along the helix (Figure 5). These effects were seen in both bovine and yeast CcOs. In addition, we observed two distinct populations of the methionine sidechain in both yeast and bovine CcO MD runs (Figures 6A,B), while no such effect was observed with V380 in WT and G384D mutant simulations. The M380 conformation vicinal to heme a3 (5–5.5 Å) was found to induce a weak effect of lowered electron affinity of heme a3 (−2.50 to −2.33 kcal/mol in small and large DFT cluster models, Table 1) relative to the WT valine. Overall, these data point to electron transfer into heme a and onwards being likely hindered in the V380M mutant by perturbation in electron affinities, primarily of heme a and possibly CuB, resulting from the structural changes induced in helix X residues.
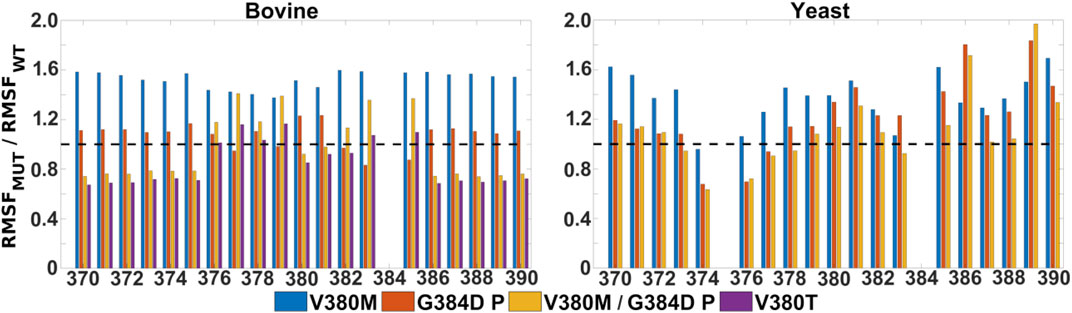
FIGURE 5. Mobility of TMH X residues in WT, single and double mutant bovine and yeast CcO simulations. Ratio of RMSF (root mean square fluctuations) of the sidechains of helix X from mutant and WT simulations. Bovine (left) and yeast (right) CcO simulations in their fully oxidized states are displayed with data from V380M (blue), protonated G384D (P, red), protonated V380M/G384D (P, yellow) and V380T (purple). The dashed black line (y = 1) indicates a WT-like RMSF.
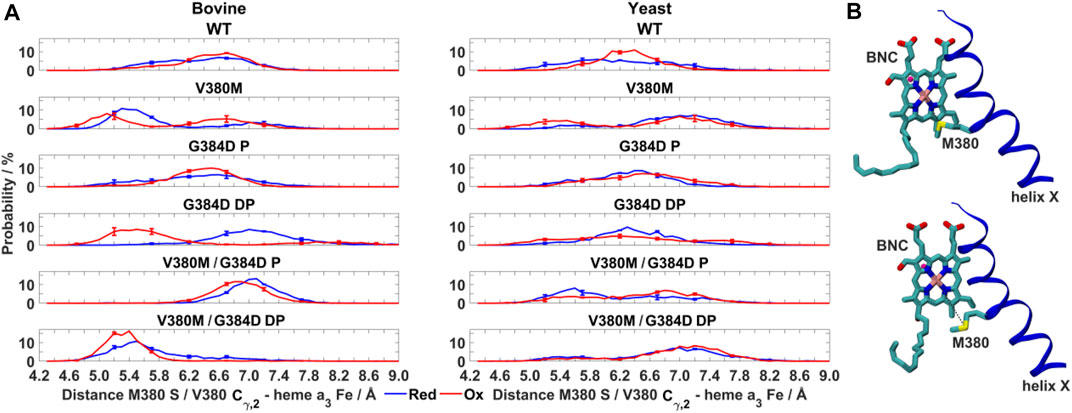
FIGURE 6. Dynamics of V380M mutant in bovine and yeast CcO. (A) Probability distribution (%) of V380/M380– heme a3 Fe distance in WT, V380M, and V380M/G384D (P/DP) bovine (left) and yeast (right) CcOs in their fully reduced and oxidized states. (B) Conformation of the BNC and TMH X in bovine V380M CcO simulations. Residue M380 is labelled. The M380 S – heme a3 Fe distance (short, top and long, bottom) is indicated by dashed black lines. The distributions are averaged over all simulation replicas and error bars correspond to standard deviations.
It is remarkable that the yeast CcO double mutant carrying both V380M and G384D mutations is fully respiratory competent with close to WT CcO turnover activity (Figure 2). As stated above, the structural and redox changes caused by the single mutations must be ameliorated when both mutations are present together. In our simulations of fully oxidized bovine CcO, the double mutant with anionic G384D (DP) showed a loss of α-helicity similar to that found in the single anionic G384D (DP) mutant (Figure 4A). Similar to bovine CcO, the yeast CcO double mutant with anionic G384D (DP) also showed α-helicity loss in helix X in both redox states. In contrast, in both bovine and yeast CcOs with neutral G384D (P), α-helicity of helix X remained similar to WT in both single and double mutants and in both redox states (Figure 4A). Likewise, sidechain fluctuations along the TMH X with neutral G384D (P) in both single and double mutants are rather comparable to WT, and also to the respiratory competent yeast mutant V380T when simulated in oxidized bovine CcO (Figure 5). This raises the possibility that the restored activity of the double mutant might be because the neutral state of D384 is preferred, hence preventing significant loss of helicity of helix X and, hence, minimizing redox potential changes. Indeed, MD simulation data reveal that there are fewer water molecules in the vicinity of anionic D384 (DP) in the double mutant (3.9 ± 2.9) compared to the single mutant G384D (7.3 ± 1.5), further suggesting that lower level of hydration may stabilize the neutral state of G384D (P) in the double mutant case. When we analyzed the dynamics of M380, which showed two distinct populations (vicinal and distal to heme a3 configurations, Figure 6), we found the vicinal conformation was entirely lost in the neutral D384 (P) double mutant, and the distal configuration retained higher occupancy in bovine simulations. Thus, we propose that the latter conformation, as well as the neutral state of D384 (P), would render the electron affinities of the hemes close to WT values, hence sustaining normal oxidoreductase activities. This is also supported by our DFT calculations which show a WT-like electron affinity of heme a3 in the double mutant (Table 1), suggesting that D384 may indeed attain a neutral state thus rescuing the respiratory activity.
Discussion
To date the highest resolution structures of Bos taurus CcO (1.5 Å) were obtained by X-ray crystallography. These structures have provided remarkable insights into its protein interior (Yano et al., 2016). However, due to current limitations on mammalian mitochondrial genome editing, mutation-based experimental tests of predicted functions using single point mutations have not been available to confirm whether a unified underlying mechanism operates in between CcOs from different domains of life. Yeast mitochondrial CcO, a near evolutionary relative of mammalian CcOs, was genetically altered to study effects of two point mutations (V380M and G384D) in the conserved H376-G384 span of TMH X, combined with molecular dynamics simulations of WT and mutant enzymes. From WT CcO simulations, we confirmed the known concerted redox-coupled dynamics of the S382 sidechain - heme a farnesyl hydroxyl distance with that of the L381 (Cβ) - C1 of vinyl group of heme a3 distance. This provided confidence into the modeling and simulation protocol applied in this study. The yeast G384D mutation caused a blockage of electron transfer from heme a to the BNC most likely due to the observed MD-based conformational and hydrogen-bonding changes in the TMH X segment of both bovine and yeast CcO, together with the negative charge of aspartate, causing a lowered midpoint potential of heme a3, a notion supported by DFT calculations. Simulations of the V380M mutant of both bovine and yeast CcOs showed a higher level of flexibility in helix X residues as well as stabilization of a unique conformation of methionine closer to heme a3. These lead to redox potential changes of heme a, and also possibly of the binuclear center, hence inducing the decrease in enzymatic activity. The restored activity of double mutant V380M/G384D is indeed remarkable and puzzling at the same time. Our combined experimental and simulation data provide indirect support for the neutral state of aspartic acid in the V380M/G384D double mutant, in contrast to its more favored anionic character in the single mutant G384D. However, further direct investigations of protonation state, for example by FTIR spectroscopy, would be needed to confirm this proposal. Overall, comparisons of our MD simulations of mutant yeast and bovine CcOs in two different redox states reveal several similarities (as detailed in Results section), but also some differences, which in part are likely to arise from the lower resolution of the yeast CcO structure. Nevertheless, given high sequence similarity and the similarity of simulated behaviors of this TMH X span, we envisage that it fulfills the same functions in both bovine and yeast CcOs, and that introduction of the same mutations into bovine CcO would result in the same loss of function.
Our integrated approach combining yeast biochemistry and molecular simulations reveals that perturbation of the conformation of conserved helix X segment (residues 380–384) can have a profound effect on internal electron transfer reactions of CcO. This is in line with the observations from structural analysis, where this segment undergoes conformational changes depending on the redox state of the enzyme (Yoshikawa and Shimada, 2015). However, since electron transfer from heme a into the BNC is obligatorily coupled to proton transfer into the proton trap/loading site (Rich, 2017; Wikström and Sharma, 2018), turnover inhibition could also arise from interference in this linked proton transfer. We note that these helix X mutations are 10–15 Å from the three putative proton channels, but are much closer to the redox-active hemes a and a3 (Figure 1 and Supplementary Figure S1), which suggests a more direct effect of mutations on redox transitions than proton transfers in the three channels. Moreover, the distance of helix X residues from E242 of the D channel is 13–15 Å and from the proton trap is ca. 13 Å. Hence, assuming the widely held model of proton translocation via these elements (Rich, 2017; Wikström and Sharma, 2018), it is unlikely to have any major influence on proton movements via this route. The mutations are however closer to the S382 residue (ca. 6 Å), which resides on the opposite side of TMH X and is a key element of the H channel proton pump proposal (Yoshikawa and Shimada, 2015). However, H+/e− coupling data from yeast CcO show no effect upon mutation of S382 to alanine (Maréchal et al., 2020), which argues against a role in H channel proton transfers. It is well-known that heme-heme electron transfer rate is extremely fast (ns tunneling) in CcO due to the proximity of the two heme edges (Verkhovsky et al., 2001). This fast electron transfer is unlikely to be sufficiently slowed by the point mutations discussed above to cause overall inhibition. Instead, we suggest that it is the overall slowing of the net electron transfer rate caused by changes in the redox potentials of cofactors that results in the mutant-induced turnover inhibition. Thus, we propose that the natural conformational dynamics of this critical segment may well control and facilitate intra-protein electron transfer (between heme a and the BNC), rather than having primary role in facilitating and gating a proton pumping channel.
Experimental Methods
Yeast Mutant Constructs and Mitochondrial Membrane Preparation
Yeast extract was purchased from Ohly GmbH, Germany. All other reagents were purchased from Sigma Aldrich. Yeast Saccharomyces cerevisiae strains were constructed from a modified strain W303-1B (Alpha ade2 HIS3 leu2 trp1 ura3) that expressed wild type CcO with a 6-his tag sequence attached to Cox13 for ease of CcO purification. The respiratory growth defective mutants V380M and G384D were obtained by random mutagenesis as described in (Meunier et al., 1993) [see also (Ortwein et al., 1997; Meunier and Rich, 1998)]. The respiratory growth competent mutants or revertants V380T/I and V380M/G384D were derived from V380M and G384D by direct selection on respiratory medium. Subsequent confirmatory sequencing revealed that V380M and its revertants also had a silent mutation A308T. Protocols for growth of the yeast cells in galactose-containing medium and preparation of mitochondrial membrane fragments were as detailed in (Meunier et al., 2012). Mitochondrial membranes were either assayed immediately after preparation or were stored at −80°C in 50 mM potassium phosphate, 2 mM potassium EDTA at pH 7.4.
CcO Content and Steady State Redox Poise in Whole Cells
For quantitation of level of expression of CcO, cells were resuspended to 80 mg wet weight cells/mL in 440 mM sucrose (to prevent sedimentation) and 50 mM potassium phosphate at pH 7.2. They were left for 1 min before scanning a baseline. 10 µM myxothiazol and 0.01% w/v hydrogen peroxide were then added to ensure that CcO was fully oxidized and a new spectrum was recorded. Dithionite was then added to fully reduce components and a further spectrum was taken after stabilisation. CcO was quantitated at 604–619 nm (WT), 606–621 nm (V380M and V380M/G384D) and 602–617 nm (G384D) using an extinction coefficient of 26 mM−1 cm−1 (Rich and Moody, 1997).
In order to determine the steady state redox poise of heme a and cytochrome c, cells were diluted to 20 mg/ml into aerobic 50 mM potassium phosphate (pH 7.2) and 440 mM sucrose. A baseline spectrum was recorded rapidly before anaerobiosis occurred. After anaerobiosis, causing CcO and cytochrome c to become fully reduced, a second scan was taken. Finally, 10 µM antimycin A (or 10 µM myxothiazol) and 0.01% w/v (∼3 mM) H2O2 were added and the sample was rescanned. These rapidly blocked the respiratory chain and released oxygen into the medium, causing CcO and cytochrome c to become fully oxidized. From these spectra, the total CcO concentration and the aerobic steady state redox poise of heme a and cytochrome c could be deduced.
Turnover Numbers in Mitochondrial Membranes
CcO concentrations were measured from sodium dithionite-reduced minus oxidized difference spectra of mitochondrial membranes resuspended in 50 mM potassium phosphate and 2 mM EDTA pH 7.4. CcO was quantitated at 604–619 nm (WT), 602–617 nm (G384D) or 606–621 nm (V380M and V380M/G384D) with an extinction coefficient, Δε, of 26 mM−1 cm−1 (based on Δε at 606–621 nm of bovine CcO (Rich and Moody, 1997). Steady-state oxygen consumption rates were measured in a stirred reaction vessel of a Clark-type O2 electrode at 25°C. Assays were carried out using mitochondrial membranes containing 2–10 nM CcO in 10 mM potassium phosphate at pH 6.6, 50 mM KCl, 0.05% (w/v) UDM, 50 µM horse heart cytochrome c and 40 µM TMPD (Dodia et al., 2014). A baseline was measured in the absence of cyt c and the reaction was initiated by addition of 2 mM sodium ascorbate. Turnover numbers (TN) are expressed in terms of the number of electrons transferred from cyt c per second per CcO (e.s−1).
Redox Potential Determinations
Redox behaviour of the 602–6 nm bands were determined in cyanide-inhibited mitochondrial membrane fragments. Since cyanide binds to heme a3 and substantially lowers its midpoint potential, these titrations represent the redox behaviour of heme a only. However, CuB remains still redox-active and interacts electrostatically with heme a, splitting the heme a redox plot into high potential (CuB oxidized) and low potential (CuB reduced) waves (Moody and Rich, 1990). Mitochondrial membranes were suspended in a buffer of 50 mM potassium phosphate and 2 mM EDTA at pH 7.4 and 23°C. 2 µM horse heart cytochrome c and 40 µM potassium ferricyanide were then added to fully oxidize the CcO and a baseline spectrum from 500 to 650 nm was recorded. 5mM potassium cyanide was then added. This rapidly bound to heme a3, after which time the cytochrome c and heme a began to reduce slowly with a very slow leak of endogenous reductant that forms in all types of mitochondrial membrane preparations (probably arising from slow lipid and protein oxidations), allowing a series of spectra to be recorded as these components became reduced. Finally, full reduction of cytochrome c and heme a was induced by addition of 4 mM sodium ascorbate. At each fractional reduction step the ambient potential, Eh, was calculated from the fractional reduction of cytochrome c at 550–542 nm using a midpoint value of +255 mV vs. SHE. Fractional reduction of the 602–6 nm band was determined from the size of the 602–6 nm peak relative to the weighted average of reference points either side of the peak at λmax ± 16 nm. A correction was made for the small contribution of cytochrome c at these wavelength triplets by subtraction of the appropriate fraction of its 550–542 nm absorbance change (0.014, 0.013 or 0.0115 for the 602, 604 and 606 nm triplets, respectively). Data were fitted to a model in which heme a interacts anticooperatively with CuB, resulting in high potential (CuB oxidized) and low potential (CuB reduced) components of heme a (Moody and Rich, 1990). Heme a3 redox potential determinations require accurate titration and deconvolution of the 445 nm band contributions of hemes a and a3 in the unligated enzyme. In contrast to bovine mitochondria, this cannot be achieved with sufficient accuracy in these yeast mitochondrial membranes due to their much lower CcO/complex III ratios.
Computational Methods
Molecular Dynamics Simulations
We performed molecular dynamics (MD) simulations of small model systems (∼100,000 atoms), representing the core subunits (SU) I and II of bovine and yeast CcO using a high resolution (1.5 Å) bovine [5B1A (Yano et al., 2016)] and a lower resolution (3.35 Å) yeast [S. cerevisiae; 6HU9 (Hartley et al., 2019)] structure.
To represent the CcO variants generated by site-directed mutagenesis, V380 and G384 of SU I were altered to M/T and D, respectively (Supplementary Table S1). The CcO model system was embedded in a bilayer of POPC lipids (85 lipids in the lower and upper leaflet) and solvated in TIP3P water with 0.15 M NaCl using CHARMM-GUI and associated tools (Jo et al., 2008). The redox-active sites were modeled in their fully oxidized state with H2O and OH− as ligands of heme a3 and CuB, respectively, and the catalytic tyrosine (Y244) in its anionic form or, in their fully reduced state, without the oxygenous ligands and Y244 in its neutral form (Johansson et al., 2008; Sharma et al., 2013). Residues E242, K319, and D364 of SU I were patched neutral while standard protonation states were assumed for all other residues except the mutated residue D384 which was modeled in both its neutral and anionic form. N- and C-terminals of the protein were treated by the CHARMM NTER and CTER patches, while the CHARMM force field was used for the protein (CHARMM22/36) (MacKerell et al., 1998; MacKerell et al., 2004; Best et al., 2012), membrane (CHARMM27/36) (Feller et al., 1997; Klauda et al., 2010), water (CHARMM36) (Jorgensen et al., 1983), and ions (CHARMM36) (Beglov and Roux, 1994).
All model systems were subjected to two consecutive energy minimizations using Gromacs 2019.3 (Abraham et al., 2015), with and without frozen protein, of 50,000 steps and a maximal force <1,000 kJ mol−1 nm−1 using the steepest decent algorithm and a subsequent equilibration of 0.2 ns using the Berendsen barostat (Berendsen et al., 1984), Nosé-Hoover thermostat (Nosé, 1984; Hoover, 1985), constraints on all bonds including hydrogen atoms [LINCS (Hess, 2008)], and a timestep of 2 fs. For the final production runs (see Supplementary Table S1 for individual simulation lengths) the Parrinello-Rahman barostat (Parrinello and Rahman, 1981) and Nosé-Hoover thermostat were applied. Analysis and visualization of all simulation trajectories was performed with VMD software (Humphrey et al., 1996) (Supplementary Figure S5).
To test the reversibility of the conformational changes of helix X, we performed additional WT simulations using the final protein conformations of the G384D and V380M/G384D mutant simulations with deprotonated D384 in the fully oxidized state of bovine CcO as starting point. These conformations were chosen because they showed the largest deviation of helix X from an α-helical structure. In all simulations an α-helical structure of helix X was restored when the mutations were reversed, thus further supporting our modeling and simulation protocol.
For the double mutant simulations, both V380 and G384 were mutated during the system setup. To complement these simulations, we performed six additional bovine double mutant simulations with anionic D384 in the fully oxidized state (Supplementary Table S1). Three of these simulations were started from simulations in which G384 was mutated to anionic D384 in the fully oxidized state. In two of these simulations anionic D384 formed (as before) stable hydrogen bonds to the backbones of L381 and S382, while no such hydrogen bonds were formed in the third simulation. The remaining three simulations were started from simulations in which V380 was mutated to M380 in the fully oxidized state (Supplementary Table S1). In two of these simulations anionic D384 formed (as before) stable hydrogen bonds to the backbones of L381 and S382, while the hydrogen bonds formed in the third simulation were rather unstable. Overall, similar results were obtained despite starting MD simulations from different initial conditions. This further strengthens data obtained from simulations and our results and conclusions.
pKa calculations were also performed on the bovine and yeast CcO MD simulation snapshots using Propka (Olsson et al., 2011; Søndergaard et al., 2011) and Delphi (Wang et al., 2015; Wang et al., 2016) software to estimate the pKa of D384 in single and double mutants and in different redox and protonation states. These calculations, however, did not support the neutral state of D384 in the double (V380M/G384D) mutant (see Results section).
Density Functional Theory Calculations
We determined the energy difference between the oxidized and reduced state of heme a3 to investigate the role of the charge change of heme a3 in the different variants of CcO without the coupled proton transfer. The calculations were performed in two model systems (“small” and “large”), both constructed from the high-resolution bovine CcO structure (PDB 5B1A) and contained heme a3 in its reduced or oxidized state, as well as residues H376, V380 (or M380), and G384 (or D384) of SU I. The “large” model systems contain in addition residues F344, F348, L381, S382, and M383 of SU I. For M380 and D384 two states were considered, i.e., “vicinal”/“distal” (short/long) conformation of M380 according to MD simulations and protonated (P)/deprotonated (DP) form of D384, in the “small” model systems, while only the “vicinal” conformation of M380 and the deprotonated form of D384 was considered in the “large” model systems. To reduce the size of the model systems and to guarantee a charge close to neutral, the propionate groups of heme a3, the farnesyl chain of heme a3, and the backbones of all protein residues were replaced by fixed capping atoms (H). For “small” model systems with deprotonated D384, nine to twelve additional water molecules were included, according to the MD simulations. The remaining coordinates were obtained as before. In addition, we also performed DFT calculations on small model systems (n = 5) scooped out of the simulation snapshots obtained from WT and G384D (DP) bovine MD runs.
All model systems were subjected to a geometry optimization using TURBMOLE 7.3 (Ahlrichs et al., 1989) with the DFT functional BP86 (Perdew, 1986; Becke, 1988), a convergence criterion of 10−6 Hartree, and a Becke-Johnson damped dispersion correction (Grimme et al., 2010; Grimme et al., 2011). The Fe atom of heme a3 was treated by the def2-TZVP basis set, while all remaining atoms were treated by the def2-SVP basis set (Weigend and Ahlrichs, 2005). To speed up the geometry optimizations the multipole accelerated R-I approximation (Sierka et al., 2003) was used. Single-point energy calculations were performed for all optimized structures using the DFT functional B3-LYP (Lee et al., 1988; Becke, 1993) with the same convergence criterion and dispersion corrections as before. All atoms were treated by the def2-TZVP basis set. To account for the removed protein environment the conductor-like screening model (Klamt and Schüürmann, 1993) was used with a dielectric constant (ε) of four. Table 1 lists the data obtained from DFT calculations.
Data Availability Statement
All data are included in the article. The simulation snapshots are available upon request from the corresponding author VS, vivek.sharma@helsinki.fi.
Author Contributions
PR and VS designed research. MR performed all MD simulations and DFT calculations. BM generated the mutant yeast strains. CZ and PR performed functional assays. MR, PR, and VS analyzed data and wrote the text.
Funding
PR was funded by UK Biotechnology and Biological Sciences Research Council grants BB/K001094/1 and BB/L020165/1. VS is supported by research funding from the Academy of Finland, the Magnus Ehrnrooth Foundation, the University of Helsinki and the Sigrid Jusélius Foundation (project grant and Senior Researcher grant).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Center for Scientific Computing, Finland is acknowledged for computational resources, including pilot grand challenge resources for project complexIty2 (mahti supercomputer). We acknowledge PRACE for awarding us access to Marconi100 at CINECA, Italy.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2021.711436/full#supplementary-material
References
Abraham, M. J., Murtola, T., Schulz, R., Páll, S., Smith, J. C., Hess, B., et al. (2015). GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 1-2, 19–25. doi:10.1016/j.softx.2015.06.001
Abramson, J., Riistama, S., Larsson, G., Jasaitis, A., Svensson-Ek, M., Laakkonen, L., et al. (2000). The Structure of the Ubiquinol Oxidase from Escherichia coli and its Ubiquinone Binding Site. Nat. Struct. Biol. 7, 910–917. doi:10.1038/82824
Ahlrichs, R., Bär, M., Häser, M., Horn, H., and Kölmel, C. (1989). Electronic Structure Calculations on Workstation Computers: The Program System Turbomole. Chem. Phys. Lett. 162, 165–169. doi:10.1016/0009-2614(89)85118-8
Becke, A. D. (1988). Density-functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A. 38, 3098–3100. doi:10.1103/physreva.38.3098
Becke, A. D. (1993). Density‐functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 98, 5648–5652. doi:10.1063/1.464913
Beglov, D., and Roux, B. (1994). Finite Representation of an Infinite Bulk System: Solvent Boundary Potential for Computer Simulations. J. Chem. Phys. 100, 9050–9063. doi:10.1063/1.466711
Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., DiNola, A., and Haak, J. R. (1984). Molecular Dynamics with Coupling to an External bath. J. Chem. Phys. 81, 3684–3690. doi:10.1063/1.448118
Best, R. B., Zhu, X., Shim, J., Lopes, P. E. M., Mittal, J., Feig, M., et al. (2012). Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J. Chem. Theor. Comput. 8, 3257–3273. doi:10.1021/ct300400x
Dodia, R., Meunier, B., Kay, C. W. M., and Rich, P. R. (2014). Comparisons of Subunit 5A and 5B Isoenzymes of Yeast Cytochrome C Oxidase. Biochem. J. 464, 335–342. doi:10.1042/bj20140732
Feller, S. E., Yin, D., Pastor, R. W., and MacKerell, A. D. (1997). Molecular Dynamics Simulation of Unsaturated Lipid Bilayers at Low Hydration: Parameterization and Comparison with Diffraction Studies. Biophysical J. 73, 2269–2279. doi:10.1016/s0006-3495(97)78259-6
Fetter, J. R., Qian, J., Shapleigh, J., Thomas, J. W., Garcia-Horsman, A., Schmidt, E., et al. (1995). Possible Proton Relay Pathways in Cytochrome C Oxidase. Proc. Natl. Acad. Sci. 92, 1604–1608. doi:10.1073/pnas.92.5.1604
Ghosh, N., Prat-Resina, X., Gunner, M. R., and Cui, Q. (2009). Microscopic pKa Analysis of Glu286 in Cytochrome C Oxidase (Rhodobacter Sphaeroides): Toward a Calibrated Molecular Model. Biochemistry 48, 2468–2485. doi:10.1021/bi8021284
Grimme, S., Antony, J., Ehrlich, S., and Krieg, H. (2010). A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 132, 154104. doi:10.1063/1.3382344
Grimme, S., Ehrlich, S., and Goerigk, L. (2011). Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 32, 1456–1465. doi:10.1002/jcc.21759
Hartley, A. M., Lukoyanova, N., Zhang, Y., Cabrera-Orefice, A., Arnold, S., Meunier, B., et al. (2019). Structure of Yeast Cytochrome C Oxidase in a Supercomplex with Cytochrome Bc1. Nat. Struct. Mol. Biol. 26, 78–83. doi:10.1038/s41594-018-0172-z
Hess, B. (2008). P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theor. Comput. 4, 116–122. doi:10.1021/ct700200b
Hoover, W. G. (1985). Canonical Dynamics: Equilibrium Phase-Space Distributions. Phys. Rev. A. 31, 1695–1697. doi:10.1103/physreva.31.1695
Humphrey, W., Dalke, A., and Schulten, K. (1996). VMD: Visual Molecular Dynamics. J. Mol. graphics 14, 33–38. doi:10.1016/0263-7855(96)00018-5
Iwata, S., Ostermeier, C., Ludwig, B., and Michel, H. (1995). Structure at 2.8 Å Resolution of Cytochrome C Oxidase from Paracoccus Denitrificans. Nature 376, 660–669. doi:10.1038/376660a0
Jo, S., Kim, T., Iyer, V. G., and Im, W. (2008). CHARMM-GUI: A Web-Based Graphical User Interface for CHARMM. J. Comput. Chem. 29, 1859–1865. doi:10.1002/jcc.20945
Johansson, M. P., Kaila, V. R. I., and Laakkonen, L. (2008). Charge Parameterization of the Metal Centers in Cytochromecoxidase. J. Comput. Chem. 29, 753–767. doi:10.1002/jcc.20835
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., and Klein, M. L. (1983). Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 79, 926–935. doi:10.1063/1.445869
Kaila, V. R. I., Verkhovsky, M. I., Hummer, G., and Wikström, M. (2008). Glutamic Acid 242 Is a Valve in the Proton Pump of Cytochrome C Oxidase. Proc. Natl. Acad. Sci. 105, 6255–6259. doi:10.1073/pnas.0800770105
Klamt, A., and Schüürmann, G. (1993). COSMO: a New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and its Gradient. J. Chem. Soc. Perkin Trans. 2 2, 799–805. doi:10.1039/p29930000799
Klauda, J. B., Venable, R. M., Freites, J. A., O’Connor, J. W., Tobias, D. J., Mondragon-Ramirez, C., et al. (2010). Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 114, 7830–7843. doi:10.1021/jp101759q
Lee, C., Yang, W., and Parr, R. G. (1988). Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 37, 785–789. doi:10.1103/physrevb.37.785
Lee, H.-m., Das, T. K., Rousseau, D. L., Mills, D., Ferguson-Miller, S., and Gennis, R. B. (2000). Mutations in the Putative H-Channel in the CytochromecOxidase fromRhodobactersphaeroidesShow that This Channel Is Not Important for Proton Conduction but Reveal Modulation of the Properties of Hemea†. Biochemistry 39, 2989–2996. doi:10.1021/bi9924821
MacKerell, A. D., Feig, M., and Brooks, C. L. (2004). Extending the Treatment of Backbone Energetics in Protein Force fields: Limitations of Gas-phase Quantum Mechanics in Reproducing Protein Conformational Distributions in Molecular Dynamics Simulations. J. Comput. Chem. 25, 1400–1415. doi:10.1002/jcc.20065
MacKerell, A. D., Bashford, D., Bellott, M., Dunbrack, R. L., Evanseck, J. D., Field, M. J., et al. (1998). All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins†. J. Phys. Chem. B 102, 3586–3616. doi:10.1021/jp973084f
Malkamäki, A., Meunier, B., Reidelbach, M., Rich, P. R., and Sharma, V. (2019). The H Channel Is Not a Proton Transfer Path in Yeast Cytochrome C Oxidase. Biochim. Biophys. Acta (BBA) - Bioenerg. 1860, 717–723. doi:10.1016/j.bbabio.2019.07.012
Maréchal, A., Meunier, B., Lee, D., Orengo, C., and Rich, P. R. (2012). Yeast Cytochrome C Oxidase: A Model System to Study Mitochondrial Forms of the Haem-Copper Oxidase Superfamily. Biochim. Biophys. Acta (BBA) - Bioenerg. 1817, 620–628. doi:10.1016/j.bbabio.2011.08.011
Maréchal, A., Xu, J.-Y., Genko, N., Hartley, A. M., Haraux, F., Meunier, B., et al. (2020). A Common Coupling Mechanism for A-type Heme-Copper Oxidases from Bacteria to Mitochondria. Proc. Natl. Acad. Sci. USA 117, 9349–9355. doi:10.1073/pnas.2001572117
Meunier, B., Lemarre, P., and Colson, A.-M. (1993). Genetic Screening in Saccharomyces cerevisiae for Large Numbers of Mitochondrial point Mutations Which Affect Structure and Function of Catalytic Subunits of Cytochrome-C Oxidase. Eur. J. Biochem. 213, 129–135. doi:10.1111/j.1432-1033.1993.tb17742.x
Meunier, B., Maréchal, A., and Rich, P. R. (2012). Construction of Histidine-Tagged Yeast Mitochondrial Cytochrome C Oxidase for Facile Purification of Mutant Forms. Biochem. J. 444, 199–204. doi:10.1042/bj20120116
Meunier, B., and Rich, P. R. (1998). Second-site Reversion Analysis Is Not a Reliable Method to Determine Distances in Membrane Proteins: an Assessment Using Mutations in Yeast Cytochrome C Oxidase Subunits I and II 1 1Edited by R. Huber. J. Mol. Biol. 283, 727–730. doi:10.1006/jmbi.1998.2132
Mok, B. Y., de Moraes, M. H., Zeng, J., Bosch, D. E., Kotrys, A. V., Raguram, A., et al. (2020). A Bacterial Cytidine Deaminase Toxin Enables CRISPR-free Mitochondrial Base Editing. Nature 583, 631–637. doi:10.1038/s41586-020-2477-4
Moody, A. J., and Rich, P. R. (1990). The Effect of pH on Redox Titrations of Haem a in Cyanide-Liganded Cytochrome-C Oxidase: Experimental and Modelling Studies. Biochim. Biophys. Acta (BBA) - Bioenerg. 1015, 205–215. doi:10.1016/0005-2728(90)90022-v
Namslauer, I., Dietz, M. S., and Brzezinski, P. (2011). Functional Effects of Mutations in Cytochrome C Oxidase Related to Prostate Cancer. Biochim. Biophys. Acta (BBA) - Bioenerg. 1807, 1336–1341. doi:10.1016/j.bbabio.2011.02.005
Nosé, S. (1984). A Unified Formulation of the Constant Temperature Molecular Dynamics Methods. J. Chem. Phys. 81, 511–519. doi:10.1063/1.447334
Olsson, M. H. M., Søndergaard, C. R., Rostkowski, M., and Jensen, J. H. (2011). PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theor. Comput. 7, 525–537. doi:10.1021/ct100578z
Ortwein, C., Link, T. A., Meunier, B., Colson-Corbisier, A.-M., Rich, P. R., and Brandt, U. (1997). Structural and Functional Analysis of Deficient Mutants in Subunit I of Cytochrome C Oxidase from Saccharomyces cerevisiae. Biochim. Biophys. Acta (BBA) - Bioenerg. 1321, 79–92. doi:10.1016/s0005-2728(97)00035-2
Parrinello, M., and Rahman, A. (1981). Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 52, 7182–7190. doi:10.1063/1.328693
Perdew, J. P. (1986). Density-functional Approximation for the Correlation Energy of the Inhomogeneous Electron Gas. Phys. Rev. B 33, 8822–8824. doi:10.1103/physrevb.33.8822
Pfitzner, U., Hoffmeier, K., Harrenga, A., Kannt, A., Michel, H., Bamberg, E., et al. (2000). Tracing the D-Pathway in Reconstituted Site-Directed Mutants of CytochromecOxidase fromParacoccus Denitrificans†. Biochemistry 39, 6756–6762. doi:10.1021/bi992235x
Popović, D. M., and Stuchebrukhov, A. A. (2004). Electrostatic Study of the Proton Pumping Mechanism in Bovine Heart Cytochrome C Oxidase. J. Am. Chem. Soc. 126, 1858–1871. doi:10.1021/ja038267w
Rich, P. R., and Maréchal, A. (2013). Functions of the Hydrophilic Channels in Protonmotive Cytochrome C Oxidase. J. R. Soc. Interf. 10, 20130183. doi:10.1098/rsif.2013.0183
Rich, P. R. (2017). Mitochondrial Cytochrome C Oxidase: Catalysis, Coupling and Controversies. Biochem. Soc. Trans. 45, 813–829. doi:10.1042/bst20160139
Rich, P. R., and Moody, A. J. (1997). “Cytochrome C Oxidase,” in Bioenergetics (Springer), 418–456. doi:10.1007/978-3-0348-8994-0_10
Sharma, V., Enkavi, G., Vattulainen, I., Róg, T., and Wikström, M. (2015). Proton-coupled Electron Transfer and the Role of Water Molecules in Proton Pumping by Cytochrome C Oxidase. Proc. Natl. Acad. Sci. USA 112, 2040–2045. doi:10.1073/pnas.1409543112
Sharma, V., Jambrina, P. G., Kaukonen, M., Rosta, E., and Rich, P. R. (2017). Insights into Functions of the H Channel of Cytochrome C Oxidase from Atomistic Molecular Dynamics Simulations. Proc. Natl. Acad. Sci. USA 114, E10339–E10348. doi:10.1073/pnas.1708628114
Sharma, V., Karlin, K. D., and Wikström, M. (2013). Computational Study of the Activated OH State in the Catalytic Mechanism of Cytochrome C Oxidase. Proc. Natl. Acad. Sci. 110, 16844–16849. doi:10.1073/pnas.1220379110
Shimada, A., Kubo, M., Baba, S., Yamashita, K., Hirata, K., Ueno, G., et al. (2017). A Nanosecond Time-Resolved XFEL Analysis of Structural Changes Associated with CO Release from Cytochrome C Oxidase. Sci. Adv. 3. doi:10.1126/sciadv.1603042
Sierka, M., Hogekamp, A., and Ahlrichs, R. (2003). Fast Evaluation of the Coulomb Potential for Electron Densities Using Multipole Accelerated Resolution of Identity Approximation. J. Chem. Phys. 118, 9136–9148. doi:10.1063/1.1567253
Søndergaard, C. R., Olsson, M. H., Rostkowski, M., and Jensen, J. H. (2011). Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theor. Comput 7, 2284–2295. doi:10.1021/ct200133y
Svensson-Ek, M., Abramson, J., Larsson, G., Törnroth, S., Brzezinski, P., and Iwata, S. (2002). The X-ray Crystal Structures of Wild-type and EQ(I-286) Mutant Cytochrome C Oxidases from Rhodobacter Sphaeroides. J. Mol. Biol. 321, 329–339. doi:10.1016/s0022-2836(02)00619-8
Tsukihara, T., Aoyama, H., Yamashita, E., Tomizaki, T., Yamaguchi, H., Shinzawa-Itoh, K., et al. (1995). Structures of Metal Sites of Oxidized Bovine Heart Cytochrome C Oxidase at 2.8 A. Science 269, 1069–1074. doi:10.1126/science.7652554
Verkhovsky, M. I., Jasaitis, A., and Wikström, M. (2001). Ultrafast Haem-Haem Electron Transfer in Cytochrome C Oxidase. Biochim. Biophys. Acta (BBA) - Bioenerg. 1506, 143–146. doi:10.1016/s0005-2728(01)00220-1
Wang, L., Li, L., and Alexov, E. (2015). pK a Predictions for Proteins, RNAs, and DNAs with the Gaussian Dielectric Function Using DelPhi pK a. Proteins 83, 2186–2197. doi:10.1002/prot.24935
Wang, L., Zhang, M., and Alexov, E. (2016). DelPhiPKa Web Server: Predicting pKaof Proteins, RNAs and DNAs. Bioinformatics 32, 614–615. doi:10.1093/bioinformatics/btv607
Weigend, F., and Ahlrichs, R. (2005). Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305. doi:10.1039/b508541a
Wikström, M. K. F. (1977). Proton Pump Coupled to Cytochrome C Oxidase in Mitochondria. Nature 266, 271–273. doi:10.1038/266271a0
Wikström, M., and Sharma, V. (2018). Proton Pumping by Cytochrome C Oxidase - A 40 Year Anniversary. Biochim. Biophys. Acta (BBA) - Bioenerg. 1859, 692–698. doi:10.1016/j.bbabio.2018.03.009
Yano, N., Muramoto, K., Shimada, A., Takemura, S., Baba, J., Fujisawa, H., et al. (2016). The Mg2+-Containing Water Cluster of Mammalian Cytochrome C Oxidase Collects Four Pumping Proton Equivalents in Each Catalytic Cycle. J. Biol. Chem. 291, 23882–23894. doi:10.1074/jbc.m115.711770
Keywords: proton pumping, molecular dynamics simulations, density functional theory, yeast bioenergetics, mitochondrial respiration
Citation: Reidelbach M, Zimmer C, Meunier B, Rich PR and Sharma V (2021) Electron Transfer Coupled to Conformational Dynamics in Cell Respiration. Front. Mol. Biosci. 8:711436. doi: 10.3389/fmolb.2021.711436
Received: 18 May 2021; Accepted: 26 July 2021;
Published: 06 August 2021.
Edited by:
Giulia Palermo, University of California, Riverside, United StatesReviewed by:
Jinan Wang, University of Kansas, United StatesMiłosz Wieczór, Institute for Research in Biomedicine, Spain
Copyright © 2021 Reidelbach, Zimmer, Meunier, Rich and Sharma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vivek Sharma, vivek.sharma@helsinki.fi; Peter R. Rich, prr@ucl.ac.uk