 Tiancong Wang
Tiancong Wang Lin Luo1
Lin Luo1
 Hongbing Shao
Hongbing Shao Min Wang
Min Wang Cui Guo
Cui Guo- 1College of Marine Life Sciences, Frontiers Science Center for Deep Ocean Multispheres and Earth System, Institute of Evolution and Marine Biodiversity, Ocean University of China, Qingdao, China
- 2UMT-OUC Joint Centre for Marine Studies, Ocean University of China, Qingdao, China
- 3Haide College, Ocean University of China, Qingdao, China
- 4The Affiliated Hospital of Qingdao University, Qingdao, China
Cyanophages play a crucial role in the biogeochemical cycles of aquatic ecosystems by affecting the population dynamics and community structure of cyanobacteria. In this study, a novel cyanophage, Nanhaivirus ms29, that infects Synechococcus sp. MW02 was isolated from the ocean basin in the South China Sea. It was identified as a T4-like phage using transmission electron microscopy. Phylogenetic analysis demonstrated that this cyanophage is distinct from other known T4-like cyanophage, belonging to a novel genus named Nanhaivirus within the family Kyanoviridae, according to the most recent classification proposed by the International Committee on Taxonomy of Viruses (ICTV). The genome of this novel cyanophage is composed of 178,866 bp of double-stranded DNA with a G + C content of 42.5%. It contains 217 potential open reading frames (ORFs) and 6 tRNAs. As many as 30 auxiliary metabolic genes (AMGs) were identified in the genome, which related to photosynthesis, carbon metabolism, nutrient uptake and stress tolerance, possibly reflecting a genomic adaption to the oligotrophic environment. Read-mapping analysis showed that Nanhaivirus ms29 mainly distributed in temperate and tropical epipelagic waters. This study enriches of the virus gene database of cyanophages and provides valuable insights into the phylogeny of cyanophages and their interactions with their hosts.
1. Introduction
Marine unicellular cyanobacteria, including Prochlorococcus and Synechococcus, are among the oldest photosynthetic autotrophic organisms on Earth, and plays an important role in shaping the global biogeochemistry with their high abundance and key metabolic activities (Scanlan et al., 2009). Synechococcus, in particular, is widely distributed with the annual mean global abundances of ~7.0 × 1026 cells, contributing to around 16.7% of the ocean’s net primary production (Waterbury et al., 1986; Flombaum et al., 2013). It exhibit a remarkable genetic diversity and comprise at least 20 distinct phylogenetic lineages, as evidenced by molecular markers (Scanlan, 2003; Mazard et al., 2012). Based on the phylogenetic analysis, marine Synechococcus strains form a distinct branch known as cluster 5, which is divided into three subclusters: 5.1, 5.2, and 5.3. Among them, subgroup 5.1 is the most abundant and diverse subgroup in the marine environment, further classified into at least nine clades (Fuller et al., 2003; Xia et al., 2015). Certain strains of Synechococcus are known to exhibit niche preferences, such as temperature, light and salinity, and therefore show different geographical distributions (Pittera et al., 2014). However, the population dynamics of cyanobacteria can be influenced by infection of cyanophages, leading to changes in the structure and function of the marine microbial food web.
Cyanophages constitute a genetically diverse and highly abundant component of marine planktonic communities. They play a crucial role in regulating the abundance, diversity and function of cyanobacterial communities. It has been reported that these phages can cause mortality rates ranging from 0.005% to 30% of their cyanobacterial hosts on a daily basis (Waterbury and Valois, 1993). Moreover, during phage infection of their hosts, gene transfer and recombination events, i.e., horizontal transfer of auxiliary metabolic genes (AMGs), often occur, which can continuously influence their evolution in a long-term phage-host symbiotic environment (Sullivan et al., 2010; Chevallereau et al., 2022). These AMGs encode proteins involved in host photosynthesis, carbon metabolism, nucleotide biosynthesis, stress responses, and nutrient utilization (Ignacio-Espinoza and Sullivan, 2012; Fuchsman et al., 2021), and thus allow the cyanophage to drive coevolution with their hosts and achieve effective replication of progeny by providing complementary support during infection (Hurwitz et al., 2015).
Since publication of the first complete genome sequence of cyanophage P60 infected marine Synechococcus in 2002 (Chen and Jingrang, 2002), there has been an increasing number of cyanophage isolates, which largely enriched the genomic database and provided valuable insights into their biology and ecology. Nevertheless, compared with the number of phage sequences obtained by using metagenomic techniques, the available cyanophage isolates still represent a limited fraction, estimated to be only 1% to 5% of the total number of cyanophages in the ocean (Simmonds et al., 2017). Furthermore, most of the confirmed T4-like cyanophage isolates are from coastal estuary regions, while only about 20% originating from the open ocean areas (Jiang et al., 2020; Zhang et al., 2020). This highlights the possibility that our comprehension of cyanophages could be limited to particular geographical areas, indicating a potential for greater diversity in cyanophage genomes and behaviors yet to be uncovered across various marine habitats. In this study, a novel marine cyanophage, Nanhaivirus ms29, which infects Synechococcus MW02, was isolated from an oligotrophic open-ocean site in the South China Sea. Its morphological, physiological and genomic features were characterized. Comparative genomics, phylogenetic analysis, and metagenomic read-mapping were applied to investigate the evolution, taxonomy, and biogeography of this cyanophage, thereby expanding the database of the cyanophage distributed in the oligotrophic environment. This study contributes to our knowledge of phage-host interactions and their adaptation to the marine environment, shedding light on the ecological and evolutionary dynamics of cyanophages in the open ocean.
2. Materials and methods
2.1. Cyanophage isolation
Surface seawater (150 L) was collected from an oligotrophic ocean basin site MS-29 (12°0.0′ N, 116°0.0′ E) with a bottom depth of 4,079 m (Figure 1A). The collected seawater was filtered sequentially through 3-μm and 0.22-μm polycarbonate membranes (Isopore™, Millipore). The filtrate was concentrated by tangential flow filtration using a 50 kDa cartridge (Pellicon® XL Cassette, Biomax® 50 kDa; polyethersulfone, Millipore, United States) to increase the viral concentration by 300 times the initial concentration. The viral concentrate was stored at 4°C in the dark.
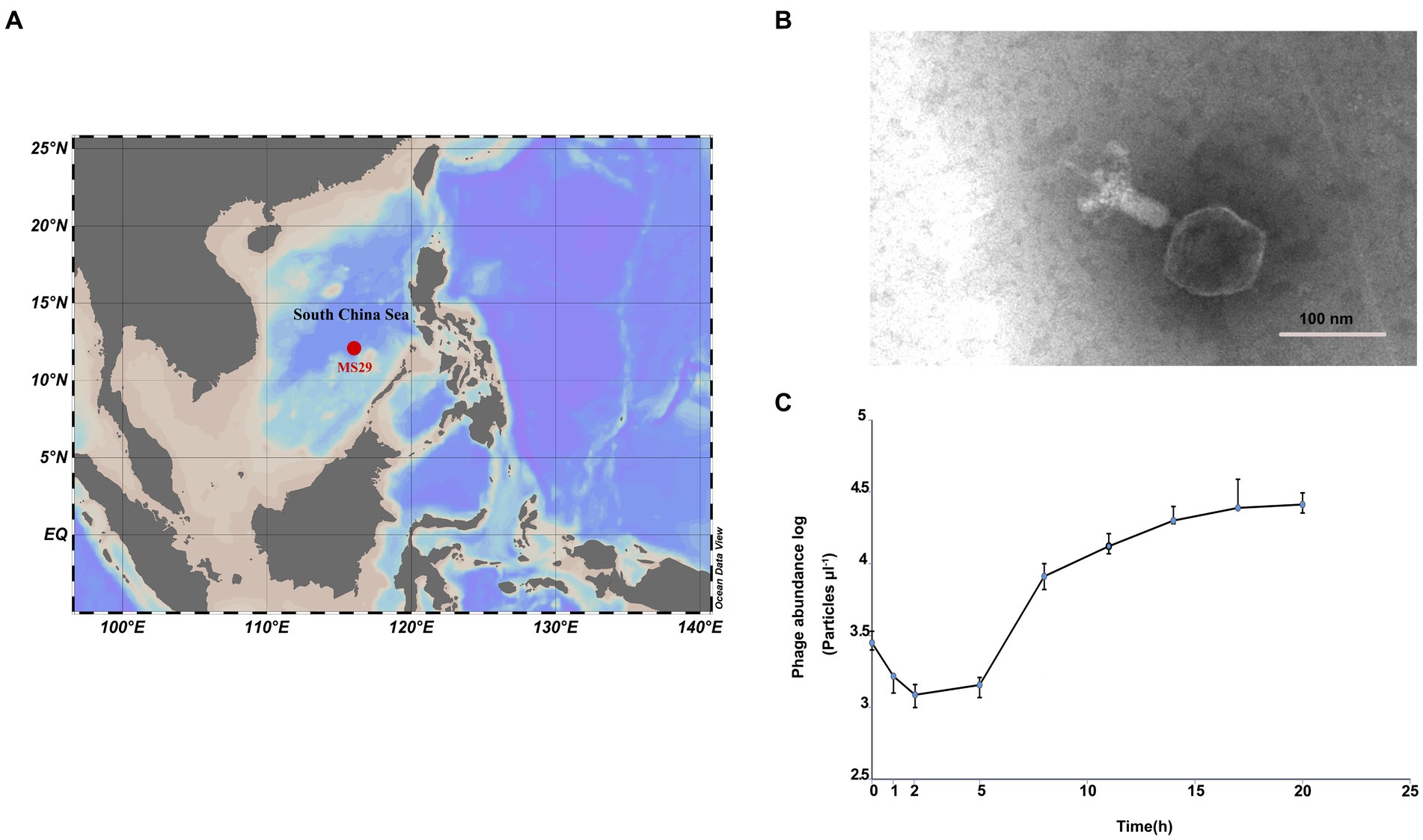
Figure 1. (A) The isolation site of Nanhaivirus ms29 in the South China Sea. (B) Transmission electron micrograph of Nanhaivirus ms29. (C) One-step growth curve of Nanhaivirus ms29.
The host of the cyanophages is Synechococcus sp. strain MW02 (NCBI accession number KP113680) belonging to clade IX, which belongs to clade IX in subcluster 5.1 and was originally isolated from the Hong Kong coastal waters impacted by various water masses (Xia et al., 2015). This clade appears to have a high tolerance to different environmental conditions and is capable of adapting to diverse ecological niches (Xia et al., 2015, 2021). The algal culture was grown in conical flasks with f/2 seawater medium under a constant illumination of approximately 25 μmol m−2 s−1 at 25°C in a 12-h/12-h light–dark cycle. Liquid infection was used to isolate the cyanophage, where aliquots of virus concentrate were added to the exponentially growing host culture in a ratio of 1:9. Phage-host suspensions were incubated for about 1 week until lysed host cells were observed based on the color and turbidity of the lysate. A separate control group was set up with pure algal culture.
2.2. Phage purification
The cyanophage obtained in this study was purified using a serial dilution method followed by sucrose density gradient centrifugation (Jiang et al., 2020). To begin with, the lysate was diluted at a series levels (10-fold dilution over 7 orders of magnitude) using sodium chloride-magnesium sulfate (SM) buffer (100 mM NaCl, 50 mM Tris, 10 mM MgSO4, and 0.01% gelatin, pH 7.5). The infectivity of each dilution was tested, and the most diluted sample that induced lysis was selected for next round of continuous dilution and infection assay using the same method. After three rounds of purification, a pure lysate containing a single phage strain was obtained theoretically (Wilhelm et al., 2006). The obtained cyanophage was concentrated using high recovery centrifugal filter units (Amicon® Ultra-15, MWCO 30 kDa, Millipore; Chénard and Suttle, 2008).
The concentrated phage suspensions were further purified by sucrose density gradient centrifugation. Four sucrose gradients of 20%, 30%, 40%, and 50% (w/v) were prepared, and the suspensions were centrifuged at 110,000 ×g for 2.5 h at 4°C in a Beckman-Coulter ultra-high-speed centrifuge. A visible band between the 40 and 50% gradient was carefully extracted using a syringe with a needle and diluted in TE buffer. The dilution was then centrifuged at 110,000 ×g at 4°C for 3 h to obtain the concentrated cyanophages. The purified phage precipitates were resuspended in SM buffer and stored at 4°C.
2.3. Morphological examination by transmission electron microscopy
The purified phage suspension (20 μL) was placed onto a 200-mesh copper grid. A drop of 1% (w/v) phosphotungstic acid (pH 7.2) was added onto the grid, covering the phage particles, and allowed to stain for 10 min (as described by Wilson et al., 1993). After staining, the excess stain was blotted off gently with filter paper. The stained phage particles on the grid were then observed using a transmission electron microscope (JEOL JEM-1200EX, Japan) operating at 100 kV.
2.4. One-step growth curve and host specificity test
To understand the growth kinetics of Nanhaivirus ms29 in its host, a one-step growth curve was conducted. Briefly, exponentially growing Synechococcus sp. strain MW02 were inoculated in medium. Cyanophage suspension was added to the host culture at a multiplicity of infection (MOI) of 0.1 to increase the chance of infection for the host cells during the first infection cycle. The samples were collected at various time points, including 0, 1, 2, 5, 8, 11, 14, 17, and 20 h after infection. The abundance of phage and host was measured using flow cytometry (McSharry, 2000).
The infectivity of Nanhaivirus ms29 was tested using nine Synechococcus strains, including Synechococcus WH7803, WH8102, MW02, MW03, LTWRed, LTWGreen, PSHK05, CCMP1333, and PCC7002. The cyanophage solution was added to each host Synechococcus culture in logarithmic growth phase at a volume ratio of 1:9, in triplicates. The cyanophage solution was replaced by the medium in the control group. The mixtures were incubated under the same conditions described above. Cell lysis was monitored and compared in the control and cyanophage solution groups every day for 2 weeks to examine the infectivity.
2.5. Phage DNA extraction, sequencing, and annotation
Phage genomic DNA was extracted using the TIANamp Virus DNA Kit (TIANGEN) and sequenced using Illumina Miseq 2 × 300 paired-end sequence method. Raw reads were cleaned up by trimming adaptor sequences and removing low-quality reads. Reads containing >40% low-quality bases (mass value ≤ 20), >10% N content, and overlap with the adapter for >15 bp with less than 3 mismatches, were removed. Clean reads were then assembled into a single contig using Velvet (Version 1.2.07). The GapCloser (v1.12) was applied to fill the gap in preliminary assembly results and the fragments shorter than 500 bp were eliminated. The complete genome of Nanhaivirus ms29 has been deposited in the GenBank database under the accession number OP807803. The prediction of the hypothetical open reading frames (ORFs) was performed with Prodigal (version 2.6.3; Hyatt et al., 2010). The prediction of the transfer RNA (tRNA) genes sequences was performed using the RAST online server1 (Brettin et al., 2015). After that, the translated amino acid sequences were scanned for homologs by BLASTp (version 2.9.0) in Non-Redundant (nr) protein database2 of the GenBank. The protein domains were predicted and analyzed by Pfam database3 (Hofle and Brettar, 1996), and Kyoto Encyclopedia of Genes and Genomes (KEGG) database.4 The genome information of all Kyanoviridae members was downloaded from the NCBI database, and the same steps as above were used to annotate the genome, the content of AMGs in Nanhaivirus ms29 and member genomes was determined.
2.6. Phylogenetic analysis
To classify Nanhaivirus ms29, 57 isolated reference genome sequences of the Kyanoviridae were downloaded from the NCBI Virus database, and 56 complete genome sequences were selected for analysis. A proteome clustering tree based on genome-wide sequence similarities computed by tBLASTx was generated with the genomes of Nanhaivirus ms29 and the other 56 selected cyanophages using ViPTree online server (Nishimura et al., 2017). The conserved portal protein sequences were used to construct a single gene phylogenetic tree (Zhong et al., 2002; Weinheimer and Aylward, 2022). These specific amino acid sequences were aligned with MEGA (version 7.0.18) and trimmed with TrimAl (version 1.4.15). The maximum-likelihood (ML) phylogenetic tree was constructed using IQ-tree (multicore version 2.0.3), and the bootstrap values were based on 1,000 replicates (Nguyen et al., 2015). The phylogenetic tree was visualized with iTOL.5
2.7. Global distribution and relative abundance of Nanhaivirus ms29
To determine the relative abundances of Nanhaivirus ms29, RPKM (Reads Per Kilobase per Million mapped reads) was calculated using CoverM (v0.6.0).6 The Nanhaivirus ms29 contigs were mapped to quality-controlled reads from the 154 Global Ocean Viromes (GOV 2.0) dataset using the minimap2 (v2.17) program (Li et al., 2010, 2021). To compare the abundance distribution of this novel cyanophage with other T4-like cyanophages in global oceans, 45 cyanophage sequences from different cyanophage genera in Kyanoviridae were selected as references. The relative abundance of Nanhaivirus ms29 was compared among five marine viral ecological zones (VEZs) defined by the Global Ocean Viromes (GOV 2.0) dataset: Arctic (ARC), Antarctic (ANT), temperate and tropical epipelagic (EPI), temperate and tropical mesopelagic (MES), and bathypelagic (BATHY; Gregory et al., 2019). To map the global distribution of Synechococcus phage Nanhaivirus ms29, the relative abundance was log10(x + 1) transformed and visualized using a heatmap.
3. Results and discussion
3.1. Morphology and biological characterization of Nanhaivirus ms29
Transmission electron microscopy examination showed that Nanhaivirus ms29 exhibited a T4-like cyanophage with an icosahedral head (approximately 89.945 nm in diameter) and a long straight contractile tail (length 124.166 nm; Figure 1B). The one-step growth curve showed that Nanhaivirus ms29 had an adsorption period of 1–2 h, followed by a latent period of 2–5 h (Figure 1C). The first peak release time occurred approximately 17 h after inoculation. The infection cycle of this cyanophage is similar to that of the cyanophage Ormenosvirus syn9 (Doron et al., 2016), which is its closest relative on the protein taxonomic tree. Host specificity test showed that Nanhaivirus ms29 could not infect the other 9 strains of Synechococcus.
3.2. Phylogenetic analyses and taxonomy—proposal of a new cyanophage genus
In the new virus classification system proposed by ICTV in October 2020, T4-like cyanophages were grouped under the family Kyanoviridae. So far, this family has been classified into 45 genera and 56 species. However, it appears that this proposal has not gained widespread adoption and many studies still uses the previous classification method. Here, we used genome sequences of Nanhaivirus ms29 and other 56 representative Kyanoviridae isolates to perform phylogenetic analysis. Newly identified cyanophages were usually classified using phylogenetic trees constructed from viral marker genes such as portal proteins, major capsid proteins, and large subunits of terminases (Dion et al., 2020). Here, we used the portal-protein-encoding homolog, gene 20 (g20), to construct a single-gene phylogenetic tree of 57 phages (Figure 2A). This approach has been previously used to classify T4-like phages (Zhong et al., 2002). Based on the results of the phylogenetic tree, the 57 cyanophages strains were classified into three clusters: Cluster I, Cluster II and Cluster III. The Nanhaivirus ms29 is clustered separately in Cluster III, forming a distinct branch. To further explore the phylogenetic relationship between Nanhaivirus ms29 and the other T4-like cyanophages and evaluate their taxonomic position, we constructed a genome-wide proteomic tree (Figure 2B). The result showed that Ormenosvirus syn9, Shandvirus sh35 and Shandvirus sb64 were most closely related to Nanhaivirus ms29 in the proteome tree. Shandvirus sh35 and Shandvirus sb64 infect Synechococcus sp. WH8102 and were isolated from the Yellow and Bohai estuaries in China (You et al., 2019), while Ormenosvirus syn9 was isolated from Woods Hole Harbor in the United States and also hosted by Synechococcus sp. WH8102 (Weigele et al., 2007). Nanhaivirus ms29 is the first cyanophage isolated from the open ocean to be included in this little clade and to infect Synechococcus MW02. According to the nucleotide homology heat map of 57 cyanophages, Shandvirus sh35 had the highest nucleotide identity with Nanhaivirus ms29, but the observed similarity was only 54.7% (Figure 2C). Therefore, according to the latest genus classification criteria proposed by ICTV (nucleotide homology with existing members < 70%), we proposed that the newly discovered Synechococcus phage represents a new cyanophage genus in Kyanoviridae and named it Nanhaivirus. The new genus Nanhaivirus has been submitted to ICTV.
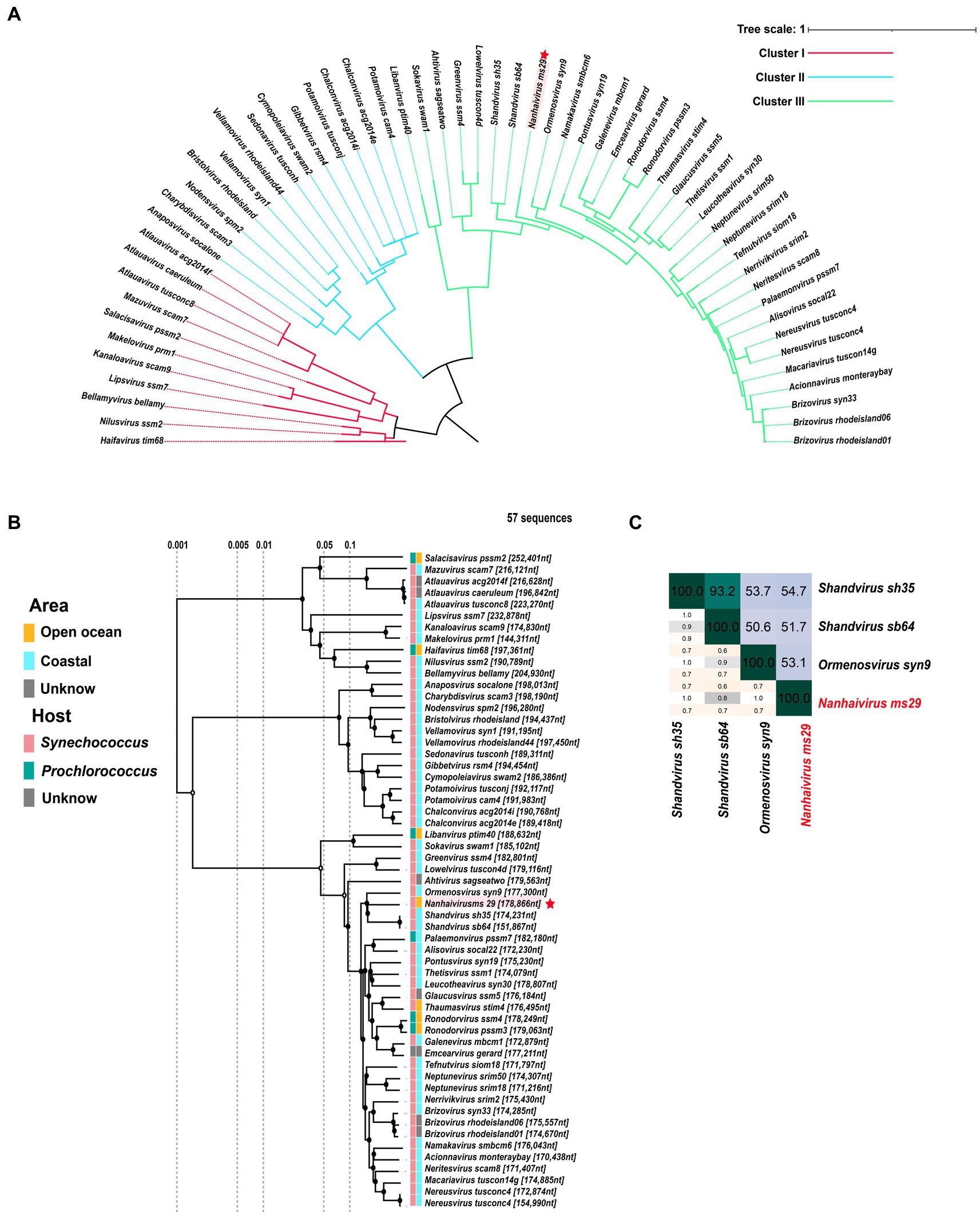
Figure 2. (A) The maximum-likelihood phylogenomic tree based on g20 gene that encodes the portal vertex protein. (B) The proteome cluster tree of Nanhaivirus ms29 with members in Kyanoviridae. (C) Heatmap of average nucleotide identity (ANI) identity between Nanhaivirus ms29 and its close relatives.
3.3. Genomic features
The genome of Nanhaivirus ms29 is a linear double-stranded DNA of 178,866 bp with a GC content of 42.5%. The GC content of most Synechococcus phages genomes typically ranges from 35.4% to 51.7% (Jiang et al., 2020). It has been shown that organisms such as bacteria, phages, and plasmids, which are dependent on parasitism for survival, tend to have low G + C content in their genomes (Rocha and Danchin, 2002), probably due to differences in the cost of relevant metabolites in cells and the limited availability of G and C relative to A and T/U (Rocha and Danchin, 2002). The Nanhaivirus ms29 contains 217 ORFs and six different tRNA types that encode for 5 amino acids (Ser, Arg, Val, Pro, Gly). After comparison in the NCBI non-redundant (NR) protein database, Pfam protein database, and KEGG protein database, 67 out of 217 ORFs (30.87%) matched with homologs of other known functional proteins, while the remaining 150 ORFs (69.13%) were predicted to be hypothetical proteins due to the lack of relevant genetic information (Figure 3). The predicted functions of these 67 ORFs included structuring (18 ORFs), DNA replication and regulation (16 ORFs), auxiliary metabolic genes (AMGs, 30 ORFs), and packaging proteins (3 ORFs).
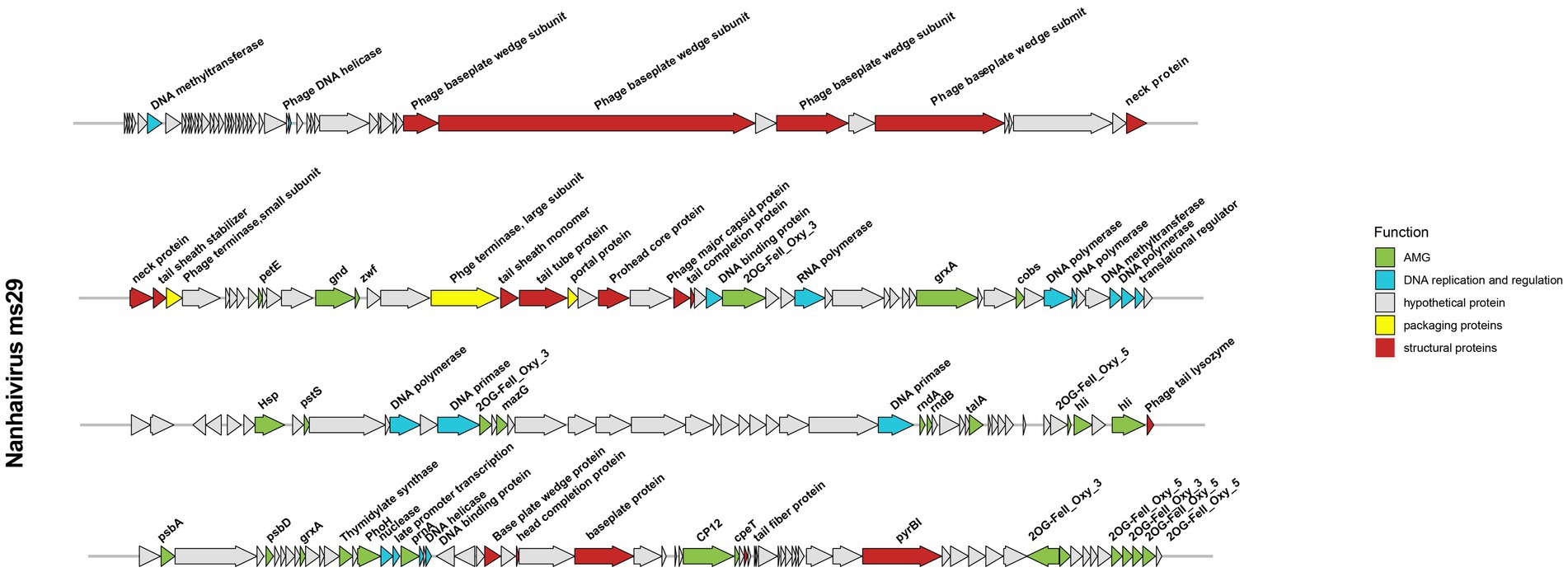
Figure 3. Genome map of Nanhaivirus ms29 and functional annotation of the predicted proteins.
The structural proteins of Nanhaivirus ms29 include the neck protein (ORF 50, 51), prohead core protein (ORF 72), baseplate protein (ORF 40, 41, 43, 45, 176, 180, 188), tail tube protein (ORF 69, 150), tail fiber protein (ORF 201), phage tail sheath protein (ORF 52, 68), head completion protein (ORF 178), phage tail completion protein (ORF 75), and the major capsid proteins (ORF 74). The large and small terminase subunits (ORF 67 and ORF 53) plus the portal protein (ORF 70) belong to the packaging module of Nanhaivirus ms29 (Fujisawa and Morita, 1997). The DNA replication and regulation module consist of 16 ORFs, including four DNA polymerase-associated proteins (ORF 93, 94, 98, 112), DNA methyltransferase (ORF 6, 112), DNA primase (ORF 114, 131), DNA helicase (ORF 29, 170), DNA binding protein (ORF 77, 172), transcriptional regulator (ORF 99, 168) and RNA polymerase (ORF 81), nuclease (ORF 167). These genes, which are directly involved in DNA replication and transcription, are ubiquitous in most living organisms (Stubbe, 2000).
3.4. Large numbers of AMGs to enhance the fitness of Nanhaivirus ms29 in the oligotrophic environment
Autonomous replication of viruses inside host cells leads to the expression of viral genes, including AMGs, which are associated with a variety of physiological activities in virus-infected hosts, including photosynthesis (Mann et al., 2003), carbon metabolism (Thompson et al., 2011), and nucleotide metabolism (Dwivedi et al., 2013). Recently, Class II AMGs, which do not exist in the KEGG database or lack explicit annotations of metabolic pathways, were also suggested to be important in cyanophage function (Hurwitz and U’Ren, 2016). In the genome of Nanhaivirus ms29, 30 AMGs were identified, including 26 Class I AMGs and 4 Class II AMGs (Figure 4). AMGs involved in photosynthesis (i.e., petE, hli, psbA, psbD, cpeT) may play an important role in maintaining the photosynthetic activity of infected host cells, providing energy for phage replication and increasing phage fitness (Lindell et al., 2005). Genes related to carbon metabolism included CP12, gnd, zwf, and talA. Previous studies have demonstrated that phage-encoded CP12 and talA can redirect the energy flow of ATP and NADPH produced during photosynthesis away from the Calvin cycle toward the biosynthesis of dNTPs for phage replication during host infection (Thompson et al., 2011). AMGs related to phosphate regulation (pstS, phoH) may involve in phosphate uptake and starvation. Nucleotide metabolism (MazG, nrdA, nrdB, thyX, pyrBI, cobS) are genes related to genome replication and transcription in cyanophages. For example, the phage-encoded MazG was proposed to regulate the cellular level of ppGpp and therefore affect transcription and translation in the host and extend the period of cell survival under the stress of phage infection (Clokie and Mann, 2006). In addition, Class II AMGs (ORF 78, 115, 146, 208, 213, 214, 215, 216, 2OG-Fe(II)_Oxy, ORF 88, 160 grxA, ORF 107 hsp., ORF 169 prnA) were also identified in genome of Nanhaivirus ms29. For example, the 2OG-Fe(II) oxygenase superfamily proteins have the highest copy number among all AMGs in the genome of Nanhaivirus ms29, which may involve in performing multiple functions (van Staalduinen and Jia, 2014).
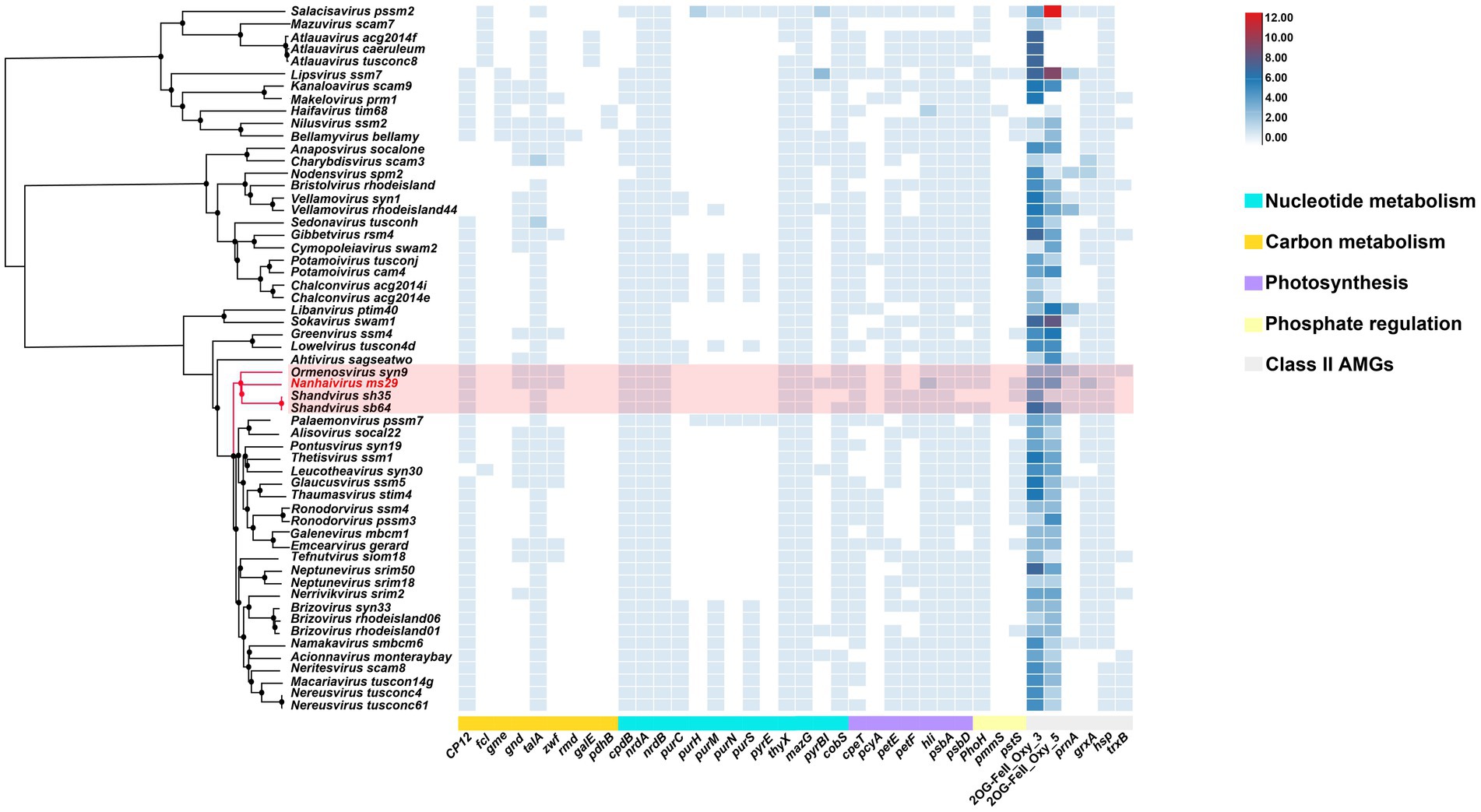
Figure 4. AMGs copy number heat map in 57 viral genomes. The genome-wide phylogenetic tree of the 57 viruses is shown next to the viral name. CP12, Calvin cycle Inhibitor; fcl, GDP-L-fucose synthase; gme, GDP-D-mannose 3′, 5′-epimerase; gnd, 6-phosphogluconate dehydrogenase; talA, Transaldolase; zwf, Glucose-6-phosphate 1-dehydrogenase; rmd, GDP-4-dehydro-6-deoxy-D-mannose reductase; galE, UDP-glucose 4-epimerase; pdhB, Pyruvate dehydrogenase; cpdB, 2′,3′-cyclic-nucleotide 2′-phosphodiesterase; nrdA, Ribonucleoside-diphosphate reductase alpha chain; nrdB, Ribonucleoside-diphosphate reductase beta chain; purC, Phosphoribosylaminoimidazole-succinocarboxamide synthase; purH, Phosphoribosylaminoimidazolecarboxamide formyltransferase; purM, Phosphoribosylformylglycinamidine cyclo-ligase; purN, Phosphoribosylaminoimidazolecarboxamide formyltransferase; purS, Phosphoribosylformylglycinamidine synthase subunit; purE, 5-(carboxyamino) imidazole ribonucleotide mutase; thyX, Thymidylate synthase; MazG, Nucleoside triphosphate diphosphatase; pyrBI, Carbamoyltransferase; cpeT, Photosynthesis-antenna proteins; pcyA, Phycocyanobilin:ferredoxin oxidoreductase; petF, Ferredoxin; petE, Plastocyanin; hli, Highlight inducing protein; psbA, Photosystem II reaction center D1 protein; psbD, Photosystem II reaction center D2 protein; phoH, Phosphohistidine phosphatase; pmmS, 2-phosphinomethylmalate synthase; pstS, Phosphate transport system substrate-binding protein; prnA, Tryptophan 7-halogenase; cobS, Cobaltochelatase; grxA, Glutaredoxin; hsp., Heat shock protein; trxB, Thioredoxin; 2OG-FeII_Oxy_3 and 2OG-FeII_Oxy_5 are different subfamilies within the 2OG-Fe(II) oxygenase superfamily.
To compare the distribution of different AMGs present in the genomes of representative T4-like cyanophages, a quantitative heatmap was constructed and the cyanophages were clustered based on their proteome (Figure 4). Among the 57 phages compared, the genome of Nanhaivirus ms29 contains a higher number of AMGs, exceeds approximately 85% of the phages analyzed. It has been suggested that the AMG repertoire of virus is correlated with its isolated environment (coastal vs. open ocean; Crummett et al., 2016). The presence of relatively large number of AMGs in the genome of Nanhaivirus ms29, which was isolated from open ocean, may indicate its coping strategies and adaptive evolution toward oligotrophic environments.
In addition, we observed that that the clade composed of Nanhaivirus ms29 and three other cyanophages exhibit a similar AMG composition, with only minor differences for some low-frequency AMGs among the clades, such as gnd, zwf, pyrBI, and pstS. To acquire phosphorus, cyanobacteria typically use an ABC-type phosphate transporter, consisting of a high affinity phosphate-binding protein (pstS) and two transmembrane proteins (pstC and pstA; Kamennaya et al., 2020). Studies have shown that the pstS gene in cyanophages can be effectively expressed during host infection and improve the phosphate uptake rate of the host after infection (Zhao et al., 2022), which may contribute to adaptation of the cyanophage to the oligotrophic environment. Coincide with our observation, pstS has been suggested to be one of the genes contributing to the difference in AMG content between coastal and open ocean phage isolates (Fuchsman et al., 2023). The enzymes produced by the gnd and zwf genes in cyanophages are involved in the pentose phosphate pathway, an important metabolic pathway providing reducing capacity and the carbon skeleton required for biosynthesis (Thompson et al., 2011). The enzymes produced by these two genes can convert 6-phosphogluconate to NADPH and ketones. This process could be important for the growth and replication of oligotrophic cyanophages, as NADPH is a crucial cofactor needed for energy production and biosynthesis (Doron et al., 2016). PyrBI that encodes catalytic subunit of aspartate carbamoyltransferase catalyzes the first step in the pyrimidine biosynthetic pathway. However, the role of pyrBI in cyanophages is still not clear. In brief, variation in content of AMGs among closely related cyanophages reflect the different horizontal evolutionary paths resulting from selection under diverse environmental pressures during vertical evolution of homologous cyanophages. Therefore, further research on AMGs among cyanophages of different habitats will deepen our understanding of cyanophage-host interactions in the environment.
3.5. Ecological distribution of Nanhaivirus ms29
The biogeographical distribution of Nanhaivirus ms29 was examined in 154 viral metagenomes from five VEZs (Vertical Ecological Zones) of the Global Ocean Viromes (GOV2.0) dataset, which included the Arctic (ARC), Antarctic (ANT), temperate and tropical epipelagic (EPI), temperate and tropical mesopelagic (MES), and bathypelagic (BATHY) regions (Gregory et al., 2019). We selected 45 cyanophage sequences from different cyanophage genera under Kyanoviridae as references and compared their absolute abundance in GOV2.0 (Figure 5A). In T4-like phages, the abundance of Nanhaivirus ms29 is relatively low, with the highest abundance observed in the EPI and MES regions. Despite its low abundance, Nanhaivirus-ms29 was widely distributed across the Indian, Pacific, Atlantic, Arctic, and Antarctic Oceans, with a higher relative abundance in the Indian Ocean than other regions (Figure 5B). Notably, Nanhaivirus ms29 was highly abundant in the nearshore waters of the Arabian Sea and the Red Sea compared to other sea areas. Furthermore, Nanhaivirus ms29 was also detected in the arctic pole.
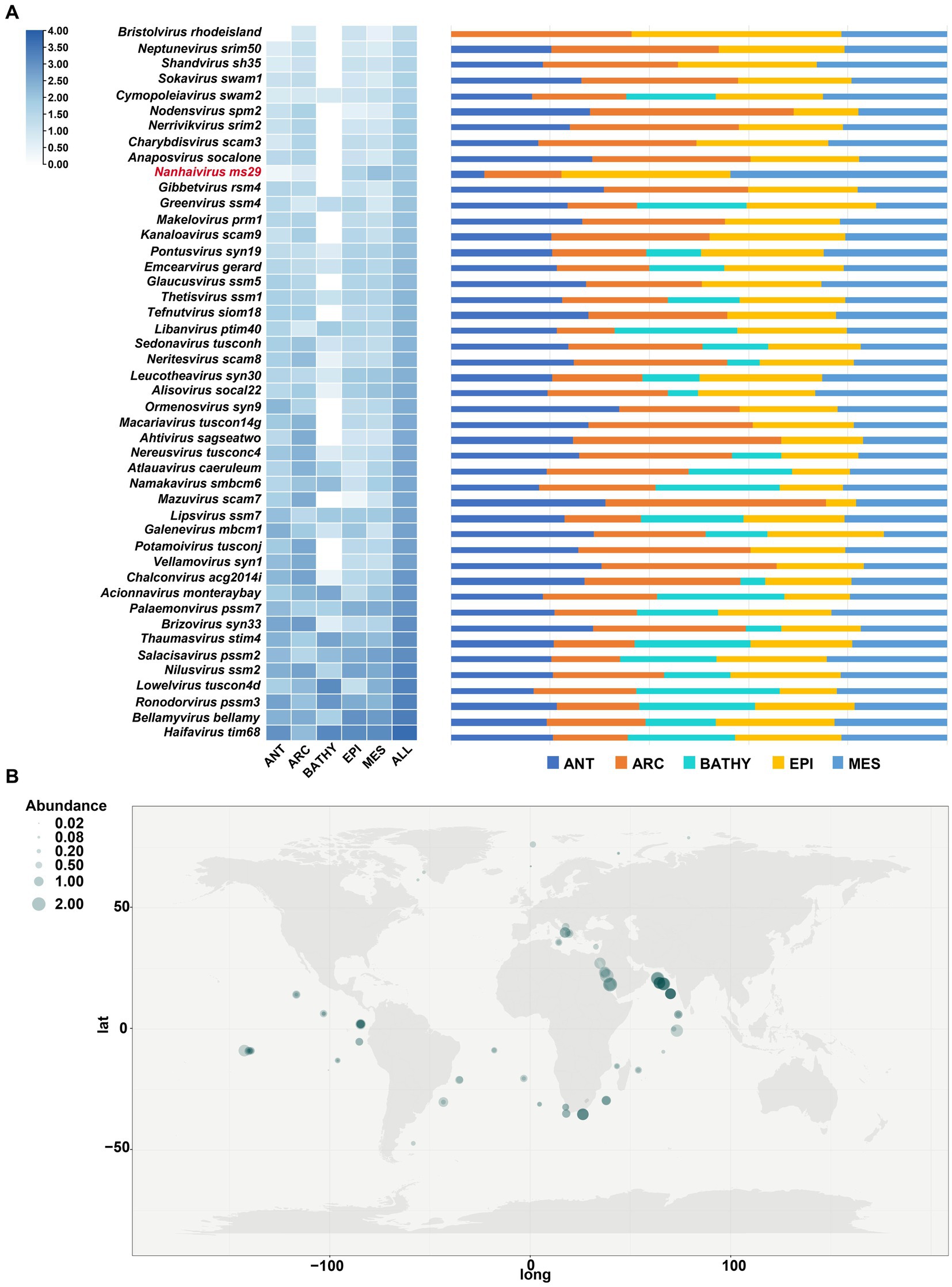
Figure 5. Relative abundance of Nanhaivirus ms29 in marine environments. (A) The relative abundance of Nanhaivirus ms29 and other members in Kyanoviridae family calculated in 154 viromes from the Global Ocean Viromes (GOV 2.0) data set. The abundance was expressed as RPKM (Reads Per Kilobase per Million mapped reads) and calculated using CoverM. The values were normalized by the number of databases in each viral ecological zone (VEZ) and transformed by log10 (X + 1). The five marine VEZs are Arctic (ARC), Antarctic (ANT), temperate and tropical epipelagic (EPI), temperate and tropical mesopelagic (MES), and bathypelagic (BATHY). (B) Global geographical distribution of Nanhaivirus ms29. The size of the circles represents the relative abundance of Nanhaivirus ms29.
4. Conclusion
Isolating and culturing novel cyanophage infecting major bacterial populations, such as Synechococcus, is fundamental for understanding cyanophage taxonomy and the relationship between cyanophage and their hosts. In this study, we isolated a novel Synechococcus phage Nanhaivirus ms29 from the oligotrophic ocean basin in the South China Sea. The morphological, growth, genomic, phylogenetic and biogeographical characteristics of Nanhaivirus ms29 were described. It has a genome of 178,866 bp with G + C content of 42.5%, which contains 217 potential ORFs and 6 tRNAs. We identified as many as 30 auxiliary metabolic genes (AMGs) that could potentially promote important metabolic processes such as photosynthesis, carbon metabolism, nitrogen metabolism, and phosphorus metabolism, which may play an important role in enhancing cyanophage adaptation and promoting host metabolism in the oligotrophic environment. Based on phylogenetic analysis and comparative genomics, we proposed a novel genus, Nanhaivirus, in the family of Kyanoviridae.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Author contributions
TW: investigation, methodology, formal analysis, and writing—original draft. LL, YX, CW, and HS: investigation, software, validation, and visualization. MW: conceptualization and supervision. CG: conceptualization, supervision, funding acquisition, and writing—review and editing. All authors contributed to the article and approved the submitted version.
Funding
Data and samples were collected onboard of R/V “Tan Kah Kee” implementing the open research cruise NORC2021-06 supported by NSFC Shiptime Sharing Project (project number: 42049906). This study was supported by National Natural Science Foundation of China (nos. 42176149, 41906126, 41976117, and 42120104006).
Acknowledgments
We thank the support of the high-performance servers of Center for High Performance Computing and System Simulation, Pilot National Laboratory for Marine Science and Technology (Qingdao), the Marine Big Data Center of Institute for Advanced Ocean Study of Ocean University of China, the IEMB-1, a high performance computing cluster operated by the Institute of Evolution and Marine Biodiversity, and the high-performance servers of Frontiers Science Center for Deep Ocean Multispheres and Earth System.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
References
Brettin, T., Davis, J. J., Disz, T., Edwards, R. A., Gerdes, S., Olsen, G. J., et al. (2015). RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 5:8365. doi: 10.1038/srep08365
Chen, F., and Jingrang, L. (2002). Genomic sequence and evolution of marine Cyanophage P60: a new insight on lytic and lysogenic phages. Appl. Environ. Microbiol. 68, 2589–2594. doi: 10.1128/AEM.68.5.2589-2594.2002
Chénard, C., and Suttle, C. A. (2008). Phylogenetic diversity of sequences of Cyanophage photosynthetic gene PsbA in marine and freshwaters. Appl. Environ. Microbiol. 74, 5317–5324. doi: 10.1128/AEM.02480-07
Chevallereau, A., Pons, B. J., van Houte, S., and Westra, E. R. (2022). Interactions between bacterial and phage communities in natural environments. Nat. Rev. Microbiol. 20, 49–62. doi: 10.1038/s41579-021-00602-y
Clokie, M. R. J., and Mann, N. H. (2006). Marine Cyanophages and light. Environ. Microbiol. 8, 2074–2082. doi: 10.1111/j.1462-2920.2006.01171.x
Crummett, L. T., Puxty, R. J., Weihe, C., Marston, M. F., and Martiny, J. B. H. (2016). The genomic content and context of auxiliary metabolic genes in marine cyanomyoviruses. Virology 499, 219–229. doi: 10.1016/j.virol.2016.09.016
Dion, M. B., Oechslin, F., and Moineau, S. (2020). Phage diversity, genomics and phylogeny. Nat. Rev. Microbiol. 18, 125–138. doi: 10.1038/s41579-019-0311-5
Doron, S., Fedida, A., Hernández-Prieto, M. A., Sabehi, G., Karunker, I., Stazic, D., et al. (2016). Transcriptome dynamics of a broad host-range Cyanophage and its hosts. ISME J. 10, 1437–1455. doi: 10.1038/ismej.2015.210
Dwivedi, B., Xue, B., Lundin, D., Edwards, R. A., and Breitbart, M. (2013). A Bioinformatic analysis of ribonucleotide reductase genes in phage genomes and metagenomes. BMC Evol. Biol. 13:33. doi: 10.1186/1471-2148-13-33
Flombaum, P., Gallegos, J. L., Gordillo, R. A., Rincón, J., Zabala, L. L., Jiao, N., et al. (2013). Present and future global distributions of the marine Cyanobacteria Prochlorococcus and Synechococcus. Proc. Natl. Acad. Sci. U. S. A. 110, 9824–9829. doi: 10.1073/pnas.1307701110
Fuchsman, C. A., Carlson, M. C. G., Prieto, D. G., Hays, M. D., and Rocap, G. (2021). Cyanophage host-derived genes reflect contrasting selective pressures with depth in the Oxic and anoxic water column of the eastern tropical North Pacific. Environ. Microbiol. 23, 2782–2800. doi: 10.1111/1462-2920.15219
Fuchsman, C. A., Prieto, D. G., Hays, M. D., and Cram, J. A. (2023). Associations between Picocyanobacterial ecotypes and Cyanophage host genes across ocean basins and depth. PeerJ 11:e14924. doi: 10.7717/peerj.14924
Fujisawa, H., and Morita, M. (1997). Phage DNA Packaging. Genes Cells 2, 537–545. doi: 10.1046/j.1365-2443.1997.1450343.x
Fuller, N. J., Marie, D., Partensky, F., Vaulot, D., Post, A. F., and Scanlan, D. J. (2003). Clade-specific 16S ribosomal DNA oligonucleotides reveal the predominance of a single marine Synechococcus clade throughout a stratified water column in the Red Sea. Appl. Environ. Microbiol. 69, 2430–2443. doi: 10.1128/AEM.69.5.2430-2443.2003
Gregory, A. C., Zayed, A. A., Conceição-Neto, N., Temperton, B., Bolduc, B., Alberti, A., et al. (2019). Marine DNA viral macro- and microdiversity from pole to pole. Cells 177, 1109–1123.e14. doi: 10.1016/j.cell.2019.03.040
Hofle, M. G., and Brettar, I. (1996). Genotyping of heterotrophic Bacteria from the Central Baltic Sea by use of low-molecular-weight RNA profiles. Appl. Environ. Microbiol. 62, 1383–1390. doi: 10.1128/aem.62.4.1383-1390.1996
Hurwitz, B. L., Brum, J. R., and Sullivan, M. B. (2015). Depth-stratified functional and taxonomic niche specialization in the ‘core’ and ‘flexible’ Pacific Ocean Virome. ISME J. 9, 472–484. doi: 10.1038/ismej.2014.143
Hurwitz, B. L., and U’Ren, J. M. (2016). Viral metabolic reprogramming in marine ecosystems. Curr. Opin. Microbiol. 31, 161–168. doi: 10.1016/j.mib.2016.04.002
Hyatt, D., Chen, G.-L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Ignacio-Espinoza, J. C., and Sullivan, M. B. (2012). Phylogenomics of T4 Cyanophages: lateral gene transfer in the ‘core’ and origins of host genes. Environ. Microbiol. 14, 2113–2126. doi: 10.1111/j.1462-2920.2012.02704.x
Jiang, T., Guo, C., Wang, M., Wang, M., Zhang, X., Liu, Y., et al. (2020). Genome analysis of two novel Synechococcus phages that lack common auxiliary metabolic genes: possible reasons and ecological insights by comparative analysis of Cyanomyoviruses. Viruses 12:E800. doi: 10.3390/v12080800
Kamennaya, N. A., Geraki, K., Scanlan, D. J., and Zubkov, M. V. (2020). Accumulation of ambient phosphate into the periplasm of marine bacteria is proton motive force dependent. Nature communications 11:2642. doi: 10.1038/s41467-020-16428-wIF:16.6
Li, Z., Pan, D., Wei, G., Pi, W., Zhang, C., Wang, J.-H., et al. (2021). Deep Sea sediments associated with cold seeps are a subsurface reservoir of viral diversity. ISME J. 15, 2366–2378. doi: 10.1038/s41396-021-00932-y
Li, B., Ruotti, V., Stewart, R. M., Thomson, J. A., and Dewey, C. N. (2010). RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 26, 493–500. doi: 10.1093/bioinformatics/btp692
Lindell, D., Jaffe, J. D., Johnson, Z. I., Church, G. M., and Chisholm, S. W. (2005). Photosynthesis genes in marine viruses yield proteins during host infection. Nature 438, 86–89. doi: 10.1038/nature04111
Mann, N. H., Cook, A., Millard, A., Bailey, S., and Clokie, M. (2003). Marine ecosystems: bacterial photosynthesis genes in a virus. Nature 424:741. doi: 10.1038/424741a
Mazard, S., Ostrowski, M., Partensky, F., and Scanlan, D. J. (2012). Multi-locus sequence analysis, taxonomic resolution and biogeography of marine Synechococc Dang us. Environ. Microbiol. 14, 372–386. doi: 10.1111/j.1462-2920.2011.02514.x
McSharry, J. J. (2000). Analysis of virus-infected cells by flow cytometry. Methods 21, 249–257. doi: 10.1006/meth.2000.1005
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nishimura, Y., Yoshida, T., Kuronishi, M., Uehara, H., Ogata, H., and Goto, S. (2017). ViPTree: the viral proteomic tree server. Bioinformatics 33, 2379–2380. doi: 10.1093/bioinformatics/btx157
Pittera, J., Humily, F., Thorel, M., Grulois, D., Garczarek, L., and Six, C. (2014). Connecting thermal physiology and latitudinal niche partitioning in marine Synechococcus. ISME J. 8, 1221–1236. doi: 10.1038/ismej.2013.228
Rocha, E. P. C., and Danchin, A. (2002). Base composition Bias might result from competition for metabolic resources. Trends Genet. 18, 291–294. doi: 10.1016/S0168-9525(02)02690-2
Scanlan, D. J. (2003). Physiological diversity and niche adaptation in marine Synechococcus. Adv. Microb. Physiol. 47, 1–64. doi: 10.1016/s0065-2911(03)47001-x
Scanlan, D. J., Ostrowski, M., Mazard, S., Dufresne, A., Garczarek, L., Hess, W. R., et al. (2009). Ecological genomics of marine Picocyanobacteria. Microbiol. Mol. Biol. Rev. 73, 249–299. doi: 10.1128/MMBR.00035-08
Simmonds, P., Adams, M. J., Benkő, M., Mya Breitbart, J., Brister, R., Carstens, E. B., et al. (2017). Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 15, 161–168. doi: 10.1038/nrmicro.2016.177
Stubbe, J. (2000). Ribonucleotide reductases: the link between an RNA and a DNA world? Curr. Opin. Struct. Biol. 10, 731–736. doi: 10.1016/s0959-440x(00)00153-6
Sullivan, M. B., Huang, K. H., Ignacio-Espinoza, J. C., Berlin, A. M., Kelly, L., Weigele, P. R., et al. (2010). Genomic analysis of oceanic cyanobacterial Myoviruses compared with T4-like Myoviruses from diverse hosts and environments. Environ. Microbiol. 12, 3035–3056. doi: 10.1111/j.1462-2920.2010.02280.x
Thompson, L. R., Zeng, Q., Kelly, L., Huang, K. H., Singer, A. U., Stubbe, J., et al. (2011). Phage auxiliary metabolic genes and the redirection of cyanobacterial host carbon metabolism. Proc. Natl. Acad. Sci. U. S. A. 108, E757–E764. doi: 10.1073/pnas.1102164108
van Staalduinen, L. M., and Jia, Z. (2014). Post-translational hydroxylation by 2OG/Fe(II)-dependent Oxygenases as a novel regulatory mechanism in Bacteria. Front. Microbiol. 5:798. doi: 10.3389/fmicb.2014.00798
Waterbury, J. B., and Valois, F. W. (1993). Resistance to co-occurring phages enables marine Synechococcus communities to coexist with Cyanophages abundant in seawater. Appl. Environ. Microbiol. 59, 3393–3399. doi: 10.1128/aem.59.10.3393-3399.1993
Waterbury, J., Watson, S., Valois, F., and Franks, D. (1986). Biological and ecological characterization of the marine unicellular cyanobacterium Synechococcus. Can.Bull.Fish.Aquat 214, 71–120.
Weigele, P. R., Pope, W. H., Pedulla, M. L., Houtz, J. M., Smith, A. L., Conway, J. F., et al. (2007). Genomic and structural analysis of Syn9, a Cyanophage infecting marine Prochlorococcus and Synechococcus. Environ. Microbiol. 9, 1675–1695. doi: 10.1111/j.1462-2920.2007.01285.x
Weinheimer, A. R., and Aylward, F. O. (2022). Infection strategy and biogeography distinguish cosmopolitan groups of marine jumbo bacteriophages. ISME J. 16, 1657–1667. doi: 10.1038/s41396-022-01214-x
Wilhelm, S. W., Carberry, M. J., Eldridge, M. L., Poorvin, L., Saxton, M. A., and Doblin, M. A. (2006). Marine and freshwater Cyanophages in a Laurentian great Lake: evidence from infectivity assays and molecular analyses of G20 genes. Appl. Environ. Microbiol. 72, 4957–4963. doi: 10.1128/AEM.00349-06
Wilson, W. H., Joint, I. R., Carr, N. G., and Mann, N. H. (1993). Isolation and molecular characterization of five marine cyanophages propagated on Synechococcus sp. Strain WH7803. Applied and environmental microbiology 59(11), 3736–3743. doi: 10.1128/aem.59.11.3736-3743.1993
Xia, X., Vidyarathna, N. K., Palenik, B., Lee, P., and Liu, H. (2015). Comparison of the seasonal variations of Synechococcus assemblage structures in estuarine waters and coastal waters of Hong Kong. Appl. Environ. Microbiol. 81, 7644–7655. doi: 10.1128/AEM.01895-15
Xia, X., Zheng, Q., Leung, S. K., Wang, Y., Lee, P. Y., Jing, H., et al. (2021). Distinct metabolic strategies of the dominant heterotrophic bacterial groups associated with marine Synechococcus. Sci. Total Environ. 798:149208. doi: 10.1016/j.scitotenv.2021.149208
You, S., Wang, M., Jiang, Y., Jiang, T., Liu, Y., Liu, X., et al. (2019). The genome sequence of a novel Cyanophage S-B64 from the Yellow Sea, China. Curr. Microbiol. 76, 681–686. doi: 10.1007/s00284-019-01680-1
Zhang, D., You, F., He, Y., Te, S. H., and Gin, K. Y.-H. (2020). Isolation and characterization of the first freshwater Cyanophage infecting Pseudanabaena. J. Virol. 94, e00682–e00620. doi: 10.1128/JVI.00682-20
Zhao, F., Lin, X., Cai, K., Jiang, Y., Ni, T., Chen, Y., et al. (2022). Biochemical and structural characterization of the cyanophage-encoded phosphate-binding protein: implications for enhanced phosphate uptake of infected cyanobacteria. Environmental Microbiology, 24, 3037–3050. doi: 10.1111/1462-2920.16043
Zhong, Y., Chen, F., Wilhelm, S. W., Poorvin, L., and Hodson, R. E. (2002). Phylogenetic diversity of marine Cyanophage isolates and natural virus communities as revealed by sequences of viral capsid assembly protein gene G20. Appl. Environ. Microbiol. 68, 1576–1584. doi: 10.1128/AEM.68.4.1576-1584.2002
Keywords: cyanophage, Synechococcus, Kyanoviridae, novel genus, auxiliary metabolic genes, genomic and phylogenetic analysis, biogeographical distribution
Citation: Wang T, Luo L, Xiong Y, Wang C, Shao H, Wang M and Guo C (2023) Characterization and genomic analysis of an oceanic cyanophage infecting marine Synechococcus reveal a novel genus. Front. Microbiol. 14:1231279. doi: 10.3389/fmicb.2023.1231279
Edited by:
Min Jin, Third Institute of Oceanography, State Oceanic Administration, ChinaReviewed by:
Shengwei Hou, Southern University of Science and Technology, ChinaTianliang He, Anhui Agricultural University, China
Copyright © 2023 Wang, Luo, Xiong, Wang, Shao, Wang and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cui Guo, guocui@ouc.edu.cn