 Yong Nie
Yong Nie Heng Zhao
Heng Zhao Zimin Wang
Zimin Wang Zhengyu Zhou
Zhengyu Zhou Xiaoyong Liu
Xiaoyong Liu Bo Huang
Bo Huang- 1Anhui Provincial Key Laboratory for Microbial Pest Control, Anhui Agricultural University, Hefei, China
- 2School of Civil Engineering and Architecture, Anhui University of Technology, Ma’anshan, China
- 3School of Ecology and Nature Conservation, Institute of Microbiology, Beijing Forestry University, Beijing, China
- 4College of Life Sciences, Shandong Normal University, Jinan, China
- 5State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China
The genus Conidiobolus s.s. was newly delimited from Conidiobolus s.l. In order to gain insight into its mitochondrial genetic background, this study sequenced six mitochondrial genomes of the genus Conidiobolus s.s. These mitogenomes were all composed of circular DNA molecules, ranging from 29,253 to 48,417 bp in size and from 26.61 to 27.90% in GC content. The order and direction for 14 core protein-coding genes (PCGs) were identical, except for the atp8 gene lost in Conidiobolus chlamydosporus, Conidiobolus polyspermus, and Conidiobolus polytocus, and rearranged in the other Conidiobolus s.s. species. Besides, the atp8 gene split the cox1 gene in Conidiobolus taihushanensis. Phylogenomic analysis based on the 14 core PCGs confirmed that all Conidiobolus s.s. species formed a monophyly in the Entomophthoromycotina lineage. The number and length of introns were the main factors contributing to mitogenomic size, and deep variations and potential transfer were detected in introns. In addition, gene transfer occurred between the mitochondrial and nuclear genomes. This study promoted the understanding of the evolution and phylogeny of the Conidiobolus s.s. genus.
Introduction
As the largest group in the family Ancylistanceae (Entomophthorales), the Conidiobolus s.l. has a long list of 80 records in the Index Fungorum1. Most Conidiobolus s.l. species are saprobic and widely distributed in plant debris and soil, while some members are parasitic to insects (Humber, 2012; Gryganskyi et al., 2013; Nie et al., 2020b), and a few species were proposed as pathogens of mammals and humans (Vilela et al., 2010; Vilela and Mendoza, 2018). A recent multi-locus phylogenetic analysis divided the genus Conidiobolus s.l. into four lineages (Nie et al., 2020b), and consequently, four genera Capillidium, Conidiobolus s.s., Microconidiobolus, and Neoconidiobolus were delimited. The genus Conidiobolus s.s. holds 17 species only and is characterized by microspores arising from conidia (Nie et al., 2020a,b). Currently, four nuclear and four mitochondrial genomes of Conidiobolus s.l. are available in GenBank2 and JGI,3 involving only one mitogenome of the Conidiobolus s.s. Remarkably, Capillidium heterosporum was the first fungus whose nuclear genome harbors the gene families CYP5014 and CYP5136 encoding cytochrome P450s, and it contained the second greatest number of introns among basal fungi (Nie et al., 2019; Wang et al., 2020).
Mitochondria of eukaryotes were considered to be evolved from endosymbiotic Alphaproteobacteria (Lang et al., 1999). As an important organelle in the eukaryotic cell, mitochondria are in charge of energy production through oxidative phosphorylation and cellular respiration (Henze and Martin, 2003; Chan, 2006; Martin et al., 2015). Except for the anaerobic mitochondrion-free Neocallimastigomycota, fungal mitogenomes have a wide range of sizes (Aguileta et al., 2014; Sandor et al., 2018). As reported, the smallest is 12,100 bp in the basal fungus Rozella allomycis (James et al., 2013), and the largest is 287,403 bp of an ectomycorrhizal fungus Tuber calosporum (Li X. et al., 2020). Most fungi possess a circular mitochondrial genome (Sandor et al., 2018), and a few are known as linear such as Kluyveromyces marxianus (Inokuma et al., 2015) and Synchytrium endobioticum (van de Vossenberg et al., 2018). Fungal mitogenomes typically harbor indefinite numbers of transfer RNA genes (trn), two ribosomal RNA genes (rnl and rns), and 14 core protein-coding genes (PCGs) for NADH dehydrogenase subunits (nad1, nad2, nad3, nad4, nad4L, nad5, and nad6), cytochrome oxidase subunits (cox1, cox2, and cox3), apocytochrome b (cob), and ATP synthase subunits (atp6, atp8, and atp9) (Lang, 2018). A few of these genes are sometimes lost (James et al., 2013), while two other genes encoding the ribosomal protein S3 (rps3) and the RNA subunit of RNase P (rnpB) are present in certain fungal mitogenomes (Franco et al., 2017).
Aside from genomic size and gene organization, fungal mitogenome comparative analyses have widely paid attention to intron distribution and variation (Nadimi et al., 2016; Nie et al., 2019; Zhang et al., 2019; Li Q. et al., 2020). The number and length of introns vary and contribute to mitogenome size (Friedrich et al., 2012; Torriani et al., 2014). For instance, Clavaria fumosa mitogenome contains 93,365 bp of intronic sequence, accounting for 36.36% (Wang et al., 2020). Two groups (I and II) of mitochondrial introns were recognized based on secondary structure and splicing mechanism (Saldanha et al., 1993). Group I mitochondrial introns frequently exist in fungal mitogenomes, while group II introns dominate in plant mitogenomes (Lang et al., 2007). Group I introns are phylogenetically divided into six main subgroups (IA, IA3, IB, IC1, IC2, and ID), with open reading frames (ORFs) usually coding homing endonucleases (HEs: LAGLIDADG and GIY-YIG). Mitogenomic introns also vary in position among species and even isolates and, consequently, are divided into different position classes (Pcls) (Beaudet et al., 2013; Wang et al., 2018). Recently, a novel nomenclature of mitochondrial introns has been proposed to avoid confusion when comparing different fungal mitogenomes, and the nomenclature system was based on: (1) three-letter abbreviation of host scientific name; (2) host gene name; (3) one capital letter P (for group I introns), S (for group II introns), or U (for introns unclassified); and (4) intron insertion site in the host gene according to the cyclosporin-producing fungus Tolypocladium inflatum (Zhang and Zhang, 2019). In general, mitogenomic introns are highly similar and homologous in the same Pcl (Ferandon et al., 2010) and have an implication for inferring fungal evolution (Sandor et al., 2018). Besides the above characteristics, the codon usage, repetitive elements, and tRNA types have also been used to evaluate the evolutionary dynamics of the fungal mitogenomes (Yuan et al., 2017; Abboud et al., 2018; Li Q. et al., 2020, Li et al., 2021). Mitochondrial genomes are inherited by a single parent and have a higher number of copies, causing a faster evolution than nuclear (Ballard and Whitlock, 2004; Pantou et al., 2006; Cameron, 2014). Therefore, the core conserved PCGs were always used for phylogenetic analyses in both basal and higher fungi (Zhang et al., 2017; Nie et al., 2019; Cai et al., 2021; Li et al., 2021).
According to the NCBI database,4 only 29 complete mitogenomes of basal fungi were available. In this study, we reported and performed comparative analyses of six additional basal fungi including Conidiobolus chlamydosporus, Conidiobolus lichenicolus, Conidiobolus mycophagus, Conidiobolus polyspermus, Conidiobolus polytocus, and Conidiobolus taihushanensis. The aims of this study are: (1) to characterize the organization, codon usage, and repetitive element of Conidiobolus s.s. mitogenomes, (2) to reveal the diversity of intron and intronic ORFs among Conidiobolus s.s. mitogenomes, and (3) to investigate the taxonomic status of Conidiobolus s.s. in the subphylum Entomophthoromycotina based on the concatenated amino-acid sequences of 14 PCGs in mitogenomes. This work will promote the understanding of origin, evolution, and phylogeny of entomophthoroid fungi and related species.
Materials and Methods
Fungal Isolates and DNA Extraction
Six ex-types of Conidiobolus s.s. were obtained from the American Type Culture Collection, Manassas, United States (ATCC) and the China General Microbiological Culture Collection Center, Beijing, China (CGMCC) and duplicated in the Research Center for Entomogenous Fungi, Anhui Agricultural University, Hefei, Anhui, China (RCEF): ATCC 12242 = RCEF 6832 (C. chlamydosporus), ATCC 12244 = RCEF 6833 (C. polytocus), ATCC 14444 = RCEF 6834 (C. polyspermus), ATCC 16200 = RCEF 6835 (C. lichenicolus), ATCC 16201 = RCEF 6836 (C. mycophagus), and CGMCC 3.16016 = RCEF 6559 (C. taihushanensis). Fresh fungal biomass on potato dextrose agar (PDA: potato 200 g, dextrose 20 g, agar 20 g, H2O 1,000 ml) at 21°C for 7 days was collected. Genomic DNA was extracted using the CTAB method with liquid nitrogen grinding (Watanabe et al., 2010).
Mitogenome Sequencing, Assembly, and Annotation
Whole mitogenome sequencing of six Conidiobolus s.s. species was performed using an Illumina HiSeq X Ten sequencing platform (Nextomics Biosciences, Co., Ltd., Wuhan, China). The paired-end libraries with 300 bp inserts were constructed according to the manufacturer’s instruction (AIR Paired-End DNA Sequencing Kit; Bioo Scientific). Raw sequencing data were evaluated by FastQC 0.11.8 (Andrews, 2010) for quality, GC content, and length. Trimmomatic 0.39 (Bolger et al., 2014) was used to remove the adapter reads and filter low-quality sequences, resulting in a size of 15–18 Gb data per ex-type strain. The mitogenomes were assembled by Norgal 1.0 (Alnakeeb et al., 2017) from clean data without reference sequences and rechecked by NOVOPlasty 4.2 (Dierckxsens et al., 2017) with C. heterosporum (NC_040967) as reference sequence. The assembled mitogenomes were annotated automatically with the MFannot tool5 using the mitochondrial genetic code (also known as genetic code 4) and GeSeq (Tillich et al., 2017) to predict mitogenome organization (Zhang et al., 2017; Nie et al., 2019; Li Q. et al., 2020). Then, the boundaries of rRNAs were rechecked by aligning to other reported mitogenomes of entomophthoroid fungi including C. heterosporum (NC040967), Neoconidiobolus thromboides (MW795364), and Microconidiobolus nodosus (MW795365) (Nie et al., 2019, 2021; Cai et al., 2021). The transfer-RNA (tRNA) annotations were confirmed by tRNAscan-SE 1.21 (Schattner et al., 2005). Intronic and intergenic spacers were searched by ORF Finder6 and nucleotide 6-frame translation-protein BLAST (blastx, see text footnote 6). Exonerate v2.2 (Slater and Birney, 2005) was used to verify intron–exon borders of PCGs. Circular mitogenome maps were drawn with OGDraw v1.3.1 (Lohse et al., 2013; Greiner et al., 2019) after manual proofreading.
Comparative Analysis of Mitogenomes in Conidiobolus s.s.
The base compositions of the six Conidiobolus s.s. mitogenomes were tested by DNASTAR Lasergene v7.1.7 Strand asymmetries were calculated according to the following formulas: AT skew = [A - T]/[A + T] and GC skew = [G - C]/[G + C] (Li et al., 2021). The most frequently used codons and amino acids were predicted using the Sequence Manipulation Suite (Stothard, 2000). The pairwise genetic distances between each pair of the 13 core PCGs (excluding atp8) in the six Conidiobolus s.s. mitogenomes were detected using MEGAX based on the Kimura-2-parameter (K2P) substitution model (Kumar et al., 2018). The non-synonymous substitution rates (Ka) and synonymous substitution rates (Ks) were calculated under DnaSP v6.10.01 (Rozas et al., 2017). BLASTN searches were conducted to investigate the lateral gene transfer (LGT) between mitochondrial and nuclear genomes (Unpublished). Tandem repeats were analyzed using Tandem Repeat Finder with default parameters (Benson, 1999). The online analysis of SSRIT8 was used to identify simple sequence repeats (SSRs) by allowing the maximum motif-length group of decamer and the minimum repeat number of five. A gene synteny analysis was performed with Mauve (Darling et al., 2004) under default alignment parameters for the six Conidiobolus s.s. mitogenomes obtained herein and four mitogenomes of related species reported elsewhere (C. heterosporum, Conidiobolus sp., N. thromboides, and M. nodosus).
Intron Analysis
A novel nomenclature of mitochondrial introns was followed, and only intronic ORFs >300 bp were considered (Zhang et al., 2017, 2019). To reveal the intron evolution in the family Ancylistanceae, introns in core PCGs of six Conidiobolus s.s. and four related species mitogenomes were classified into different Pcls according to previously described methods (Ferandon et al., 2013). The Pcls of core PCGs in Ancylistanceae were named according to the insert position in the reference mitogenome of C. heterosporum (NC_040967) (Nie et al., 2019). Pcls had the same insertion site considered homologous, usually highly similar in sequence (Ferandon et al., 2010).
Phylogenomic Analysis
To detect the phylogenetic relationship of the six Conidiobolus s.s. in the basal fungi, all 14 conserved PCGs were used. A total of 38 fungal mitogenomes were downloaded from GenBank for comparison, and three plant and animal mitogenomes served as outgroups (Supplementary Table 1). The amino acid sequences of each locus were aligned with MAFFT v6.864 (Katoh and Standley, 2013) and concatenated with SequenceMatrix 1.7.8 (Vaidya et al., 2011). Phylogenetic trees were constructed by Maximum Likelihood (ML) and Bayesian Inference (BI) using the best model of amino acid assessed with Akaike Information Criterion (AIC) in Modeltest 3.7 (Posada and Crandall, 1998). Under the GTR + I + G model, the ML phylogram was constructed using RAxML 8.1.17 with 1,000 bootstrap replicates (Stamatakis, 2014). The BI analysis was processed with 500,000 generations and four chains (one cold and three hot) using MrBayes 3.2.2 (Ronquist and Huelsenbeck, 2003). Markov Chain Monte Carlo (MCMC) chains ran until the convergences met and the standard deviation fell below 0.01. The first 25% of the generations were discarded as “burn-in,” and a posterior probability was estimated for the remaining sampled generations.
Data Availability
The six mitogenome sequences of Conidiobolus s.s. were deposited in GenBank, and their accession numbers are listed in Table 1.
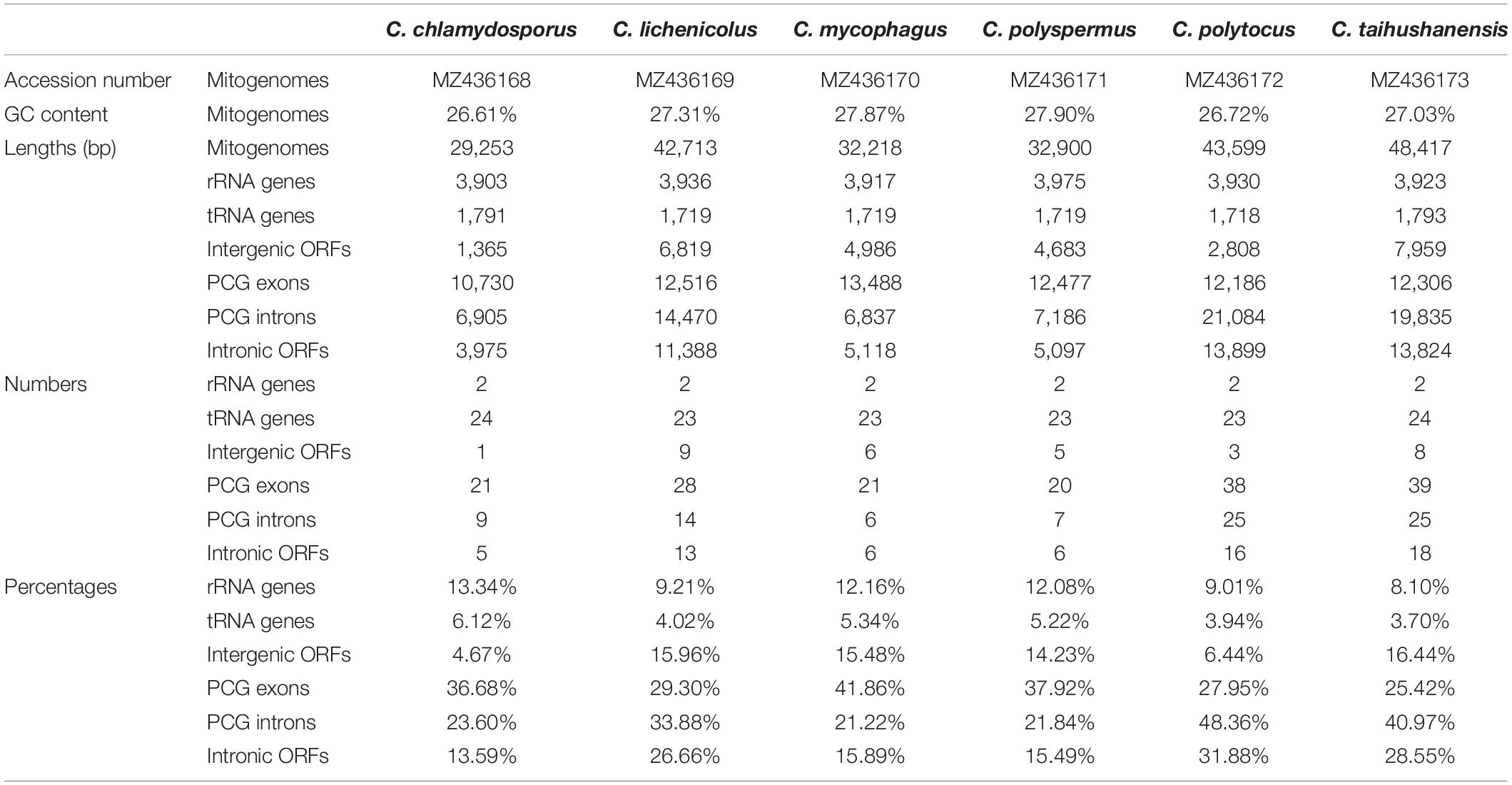
Table 1. Mitogenome information of six Conidiobolus s.s. species.
Results
Characterization of the Six Conidiobolus s.s. Mitogenomes
The complete circular mitogenomes of the six Conidiobolus s.s. were assembled (Figure 1 and Table 1). With a wide length range of 29,253–48,417 bp and a narrow GC content range of 26.61–27.90%, a single mitogenome contained 1–9 intergenic ORFs and a conserved set of 38–40 genes including two rRNA genes, 23–24 tRNA genes, and 13–14 PCGs. A total of 26 types of intergenic ORFs were found in all the six Conidiobolus s.s. mitogenomes: 24 were detected once, and two (orf324 and orf446) were examined four times (Supplementary Table 2). A total of 23 types of tRNA genes encoded 20 standard amino acids. The tRNA gene trnM(cat) had two copies in every mitogenome, while the tRNA gene trnT(tgt) had two copies in the mitogenome of C. taihushanensis. Two types of anticodons for each of the genes trnL and trnS were presented in mitogenome of every species, while the trnR gene with two types of anticodons was presented in C. chlamydosporus (Supplementary Table 3). Almost all 14 conserved PCGs existed in all species, except the atp8 gene not found in C. chlamydosporus, C. polyspermus, and C. polytocus (Figures 1, 2, 4). Exons of these PCGs had a length range of 10,730–13,488 bp and a GC content range of 25.42–41.86%. AT and GC skews were both positive in most conserved PCGs but negative in genes nad2 and nad6 (Figure 2). The genes nad5 and nad4L had the largest and smallest K2P genetic distances, respectively (Figure 3). The Ka values of nad6 and atp9 were the largest and smallest in the six Conidiobolus s.s. species, respectively. The cox2 gene had the largest synonymous substitutions rate (Ks), while nad3 had the smallest. Ka/Ks values were all <1.00. The highest Ka/Ks value was observed for nad6, while atp9 exhibited the lowest Ka/Ks value.
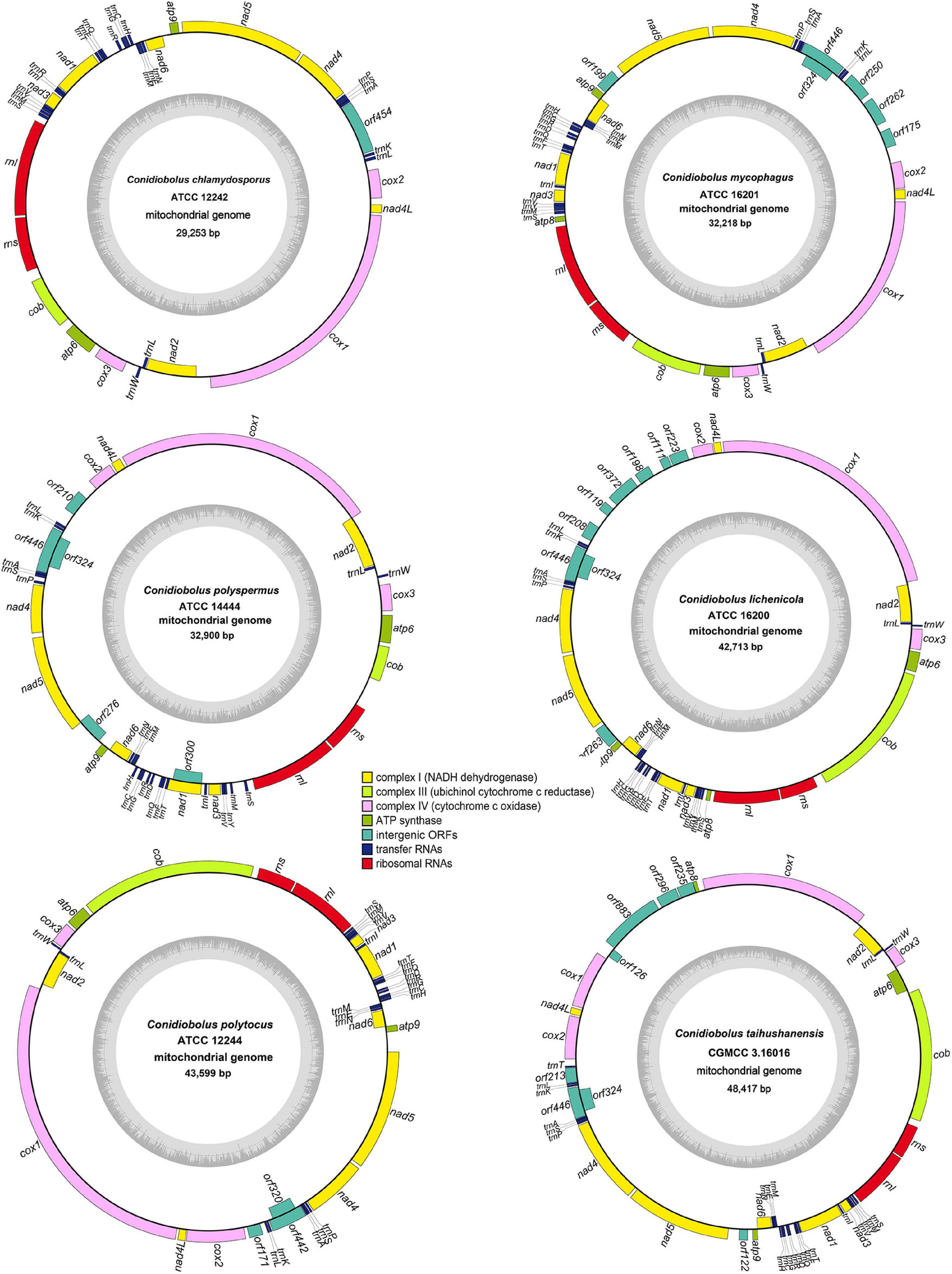
Figure 1. Circular complete mitogenome maps of six ex-types in Conidiobolus s.s. genes are represented by different colored blocks. Genes outside and inside the outermost black ring line are transcribed in a clockwise and counter-clockwise direction, respectively. Inner gray rings show GC contents.
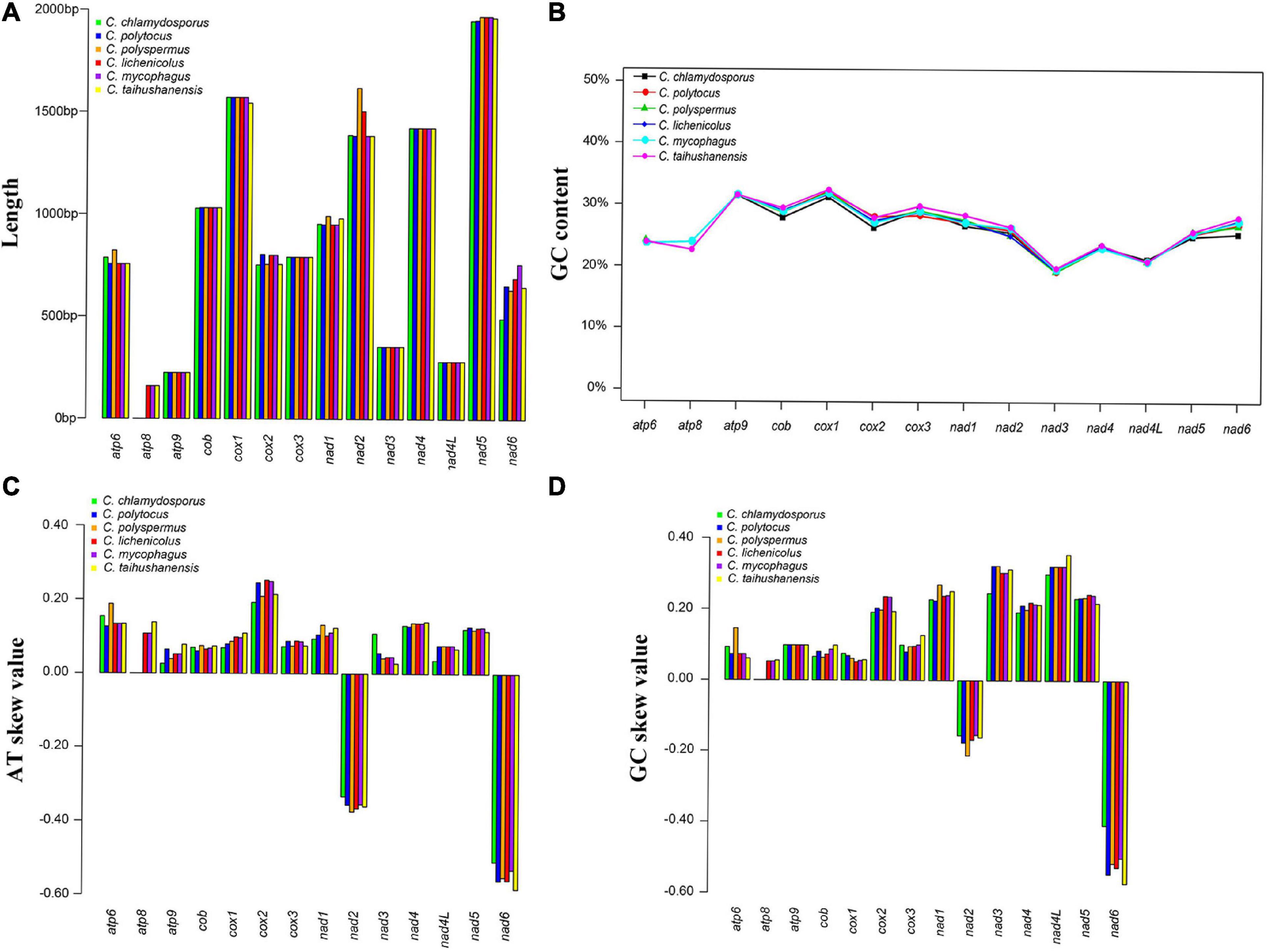
Figure 2. Variations of 14 protein-coding genes (PCGs) in the six Conidiobolus s.s. mitogenomes. (A) Length; (B) GC content; (C) AT skews; (D) GC skews.
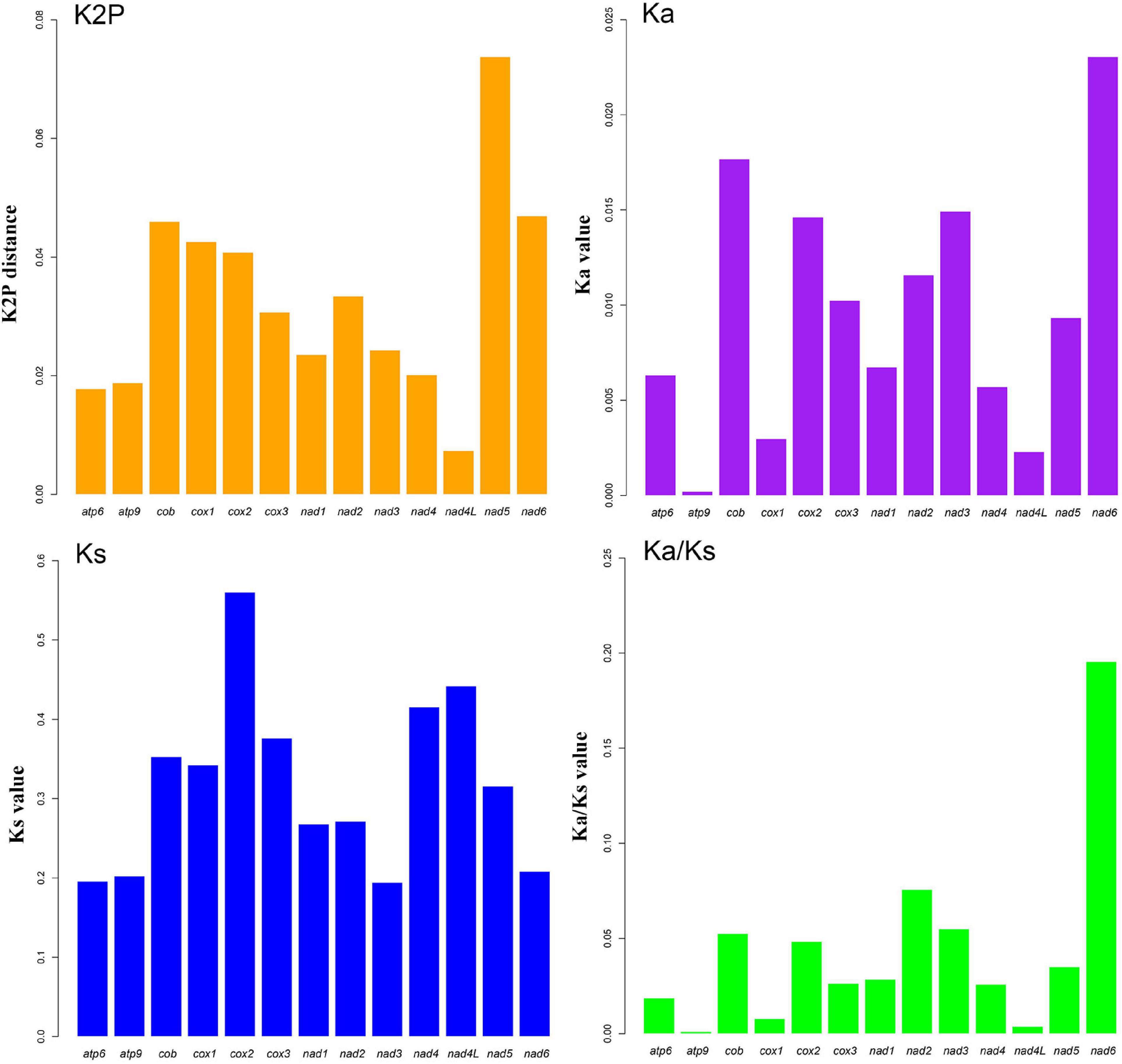
Figure 3. Genetic analysis of 13 protein-coding genes (PCGs) in six Conidiobolus s.s. mitogenomes. K2P, the Kimura-2-parameter distance; Ka, the mean number of non-synonymous substitutions per non-synonymous site; Ks, the mean number of synonymous substitutions per synonymous site. Gene atp8 is not included due to its loss in three species.
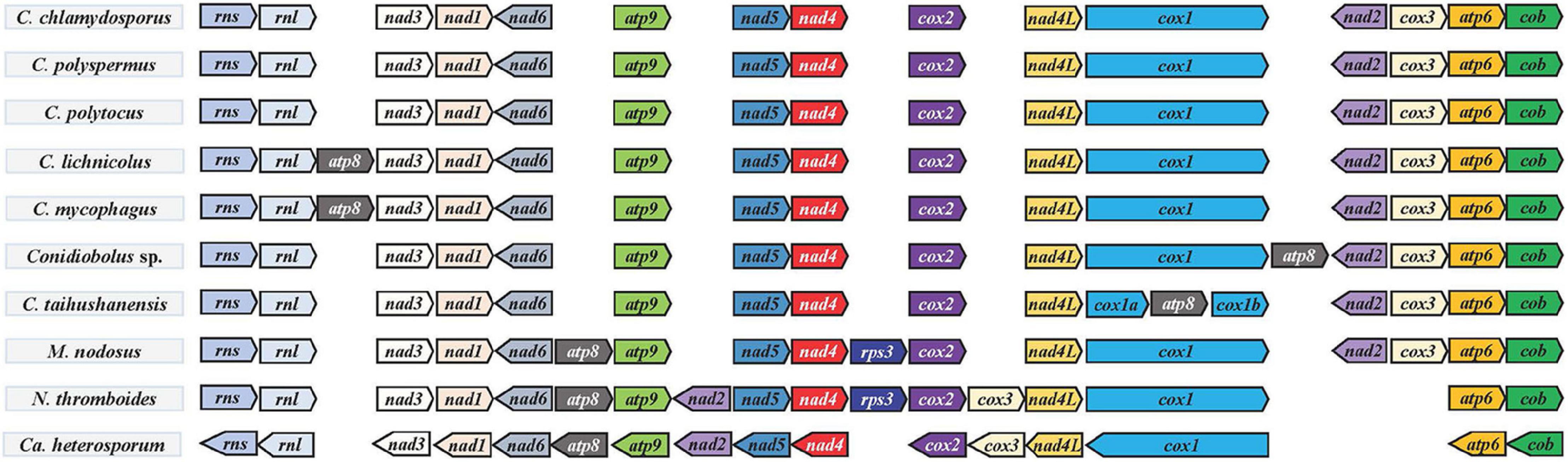
Figure 4. Gene order in Conidiobolus s.s. mitogenomes and four related species. All species were adjusted to start with the gene rns. Genes on the forward and reverse strands were indicated by right and left arrows, respectively.
Gene Order in Conidiobolus s.l.
The order of PCGs and rRNA genes was compared among Conidiobolus s.l. mitogenomes including seven Conidiobolus species and three allied species C. heterosporum (NC040967), M. nodosus (MW795365), and N. thromboides (MW795364) (Figure 4). The result showed that most genes were arranged in the same order on the same strand, whereas genes nad2 and nad6 were on the other strand. Furthermore, all genes of C. heterosporum were on the other strand. Other changes involved the gene loss of rps3 in almost all Conidiobolus s.l. except M. nodosus and N. thromboides, the gene atp8 lost in three species (C. chlamydosporus, C. polyspermus, and C. polytocus) and rearranged in the other four Conidiobolus species, the gene rearrangement of nad2 and cox3 in N. thromboides and C. heterosporum, and the gene split of cox1 by atp8 in C. taihushanensis.
A similar synteny was revealed for the group of Conidiobolus spp. and M. nodosus, and another similar synteny was revealed for the other group of N. thromboides and C. heterosporum. Between these two groups was an obvious rearrangement (Figure 5).
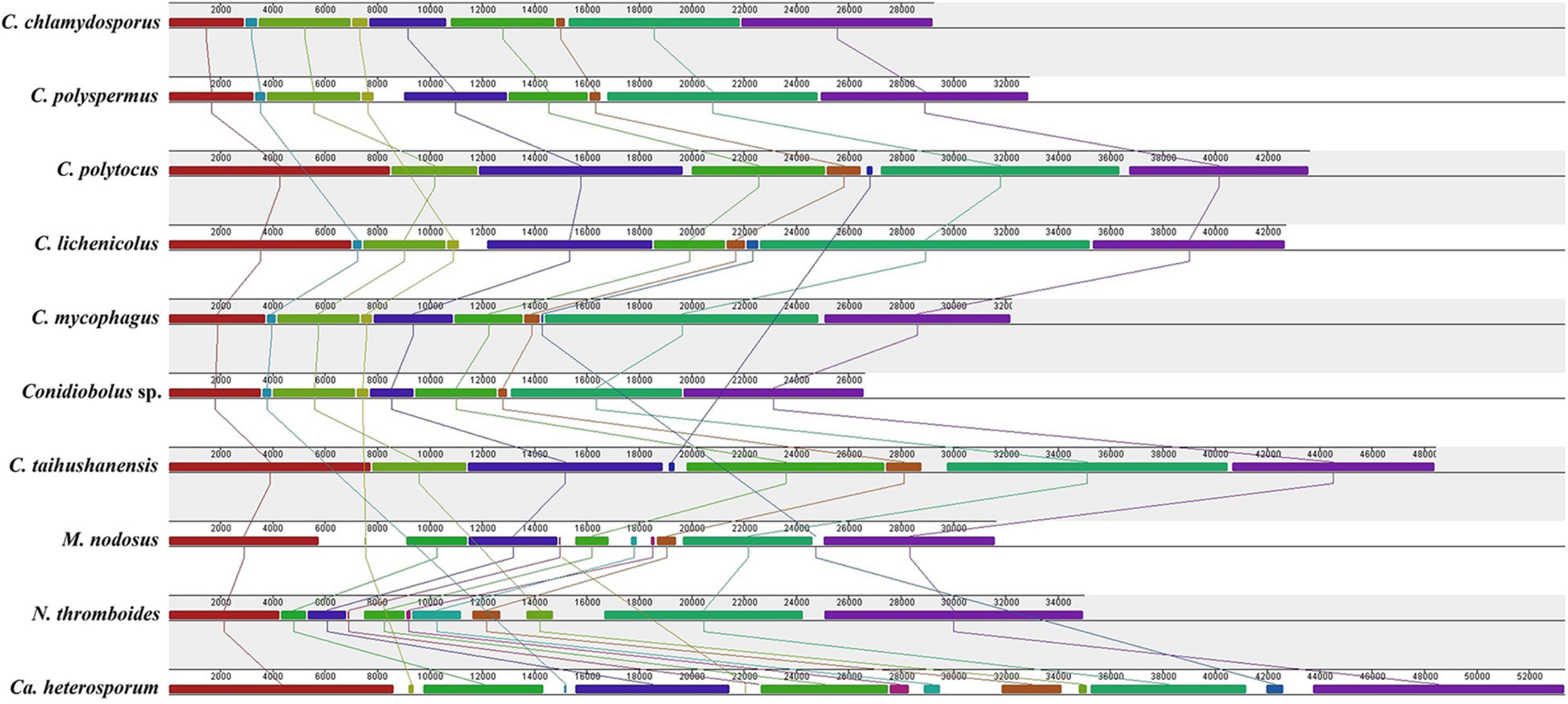
Figure 5. Synteny comparison of mitogenomes among six Conidiobolus s.s. and four related species.
Introns and Intronic Open Reading Frames in Conidiobolus s.l.
Seven of the 14 conserved PCGs of the six Conidiobolus s.s. mitogenomes assembled herein had no introns and intronic ORFs. The other seven PCGs contained introns and intronic ORFs (Supplementary Table 4). They totally possessed 86 introns, but varied remarkably among species in number, ranging from 6 in C. mycophagus to 25 in C. polytocus and C. taihushanensis, as well as in length percentage, ranging from 17.00% in C. mycophagus to 42.60% in C. polytocus (Tables 1, 2). These introns were annotated to three subgroups IA, IB, and ID of the group I type, except three unknown each in the cob, nad1, and nad5 genes of C. polytocus and one unknown in the cox1 gene of C. taihushanensis (Supplementary Table 4).
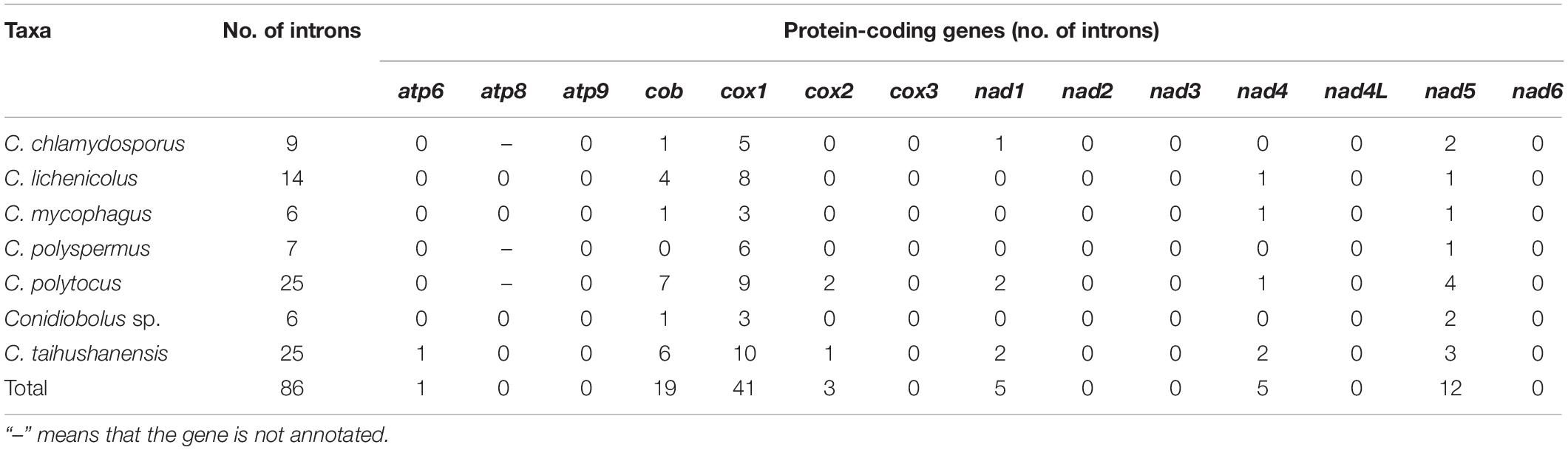
Table 2. Introns in Conidiobolus s.s. mitogenomes.
In total, 64 intronic ORFs were detected in the six Conidiobolus mitogenomes (Table 1 and Supplementary Table 4). These intronic ORFs were mainly encoded for LAGLIDADG and GIY-YIG endonucleases, and most were specific. Only two intronic ORFs (orf327 and orf290) were shared by three species, and seven (orf217, orf291, orf295, orf298, orf328, orf331, and orf333) were shared by two species.
Altogether, 77 Pcls were detected in nine conserved PCGs in all the 10 Conidiobolus s.l. species, with 31 in the cox1 gene and 19 in the cob gene. Only one Pcl was detected in each of the atp9 and cox3 genes (Supplementary Figure 1). Pcls P9, P12, P19, and P30 were widely distributed in the cox1 gene in more than five species. Pcl P6 was the most widely distributed in the cob gene.
Codon Usage and Repeat Elements in Conidiobolus s.l.
Most conserved PCGs in the Conidiobolus s.s. mitogenomes started with ATG (Supplementary Table 5), except atp6 in C. polyspermus (GTG), cox2 in C. lichenicolus, C. mycophagus, and C. polytocus (GTG), nad2 in C. polyspermus (TTG), and nad6 in C. mycophagus (TTG). The TAA was the most commonly used stop codon, and AAA, TAG, and TAA were also used. The most frequently used codons were ATA (isoleucine; Ile), AAT (asparagine; Asn), TTA (leucine; Leu), TCA (serine; Ser), and GGT (glycine; Gly) (Supplementary Figure 2).
Variation of repeat sequence regions among Conidiobolus s.l. mitogenomes ranged from 1 to 30 in number and 45 to 1,904 bp in length (Supplementary Table 6). A total of 111 types of tandem repeat motifs varied from 17 to 55 in number, 35 to 293 bp in length, and 80.42 to 100% in nucleotide similarity (Supplementary Table 7). The longest tandem repeat with 200 bp was found in C. chlamydosporus, which was located in the protein-coding regions of orf454. In all, there were 83.78% tandem repeat sequences that were repeated two to six times in the mitogenome of six Conidiobolus s.s., and the highest copy number was 24.7 in the C. chlamydosporus mitogenome. A total of 50 SSRs were identified in six Conidiobolus s.s. mitogenomes. The dimer (AT or TA) and trimer (GAA, TAT, ATT, and AAT) motifs were the most commonly detected types, while four (AATA, seven repeats) and seven (AAAAAAC, five repeats) motifs were only detected in C. chlamydosporus mitogenome (Supplementary Table 8).
Gene transfer analyses showed that 62–150 fragments were identified with a total percent identity from 81.29 to 100% in the six Conidiobolus s.s. mitogenomes (Supplementary Table 9). Correspondingly, the total alignment sequences ranged from 21,708 to 39,339 bp. The most fragment was detected in the C. taihushanensis mitogenome. The largest aligned fragment with 2,536 bp was located between cox1 and nad4L genes in the C. chlamydosporus mitogenome. The similarity of this large alignment was 99.65% compared with its nuclear genome, with five mismatches and four gaps.
Phylogenomic Analyses for Conidiobolus s.l.
The combined dataset was composed of 5,941 characters and 47 taxa. The best model applied in the ML and BI analyses was GTR + I + G. The final average standard deviation of split frequencies was 0.000748. The ML and BI phylogenetic trees shared a similar topology, and the ML tree was presented with ML bootstrap/BI posterior probability values of robust clades at relative branches (Figure 6). The seven Conidiobolus s.s. species formed a monophyletic group (100/1.00). Three related Conidiobolus s.l. species C. heterosporum, M. nodosus, and N. thromboides were basal to the seven Conidiobolus s.s. species within the Entomophthoromycotina lineage.
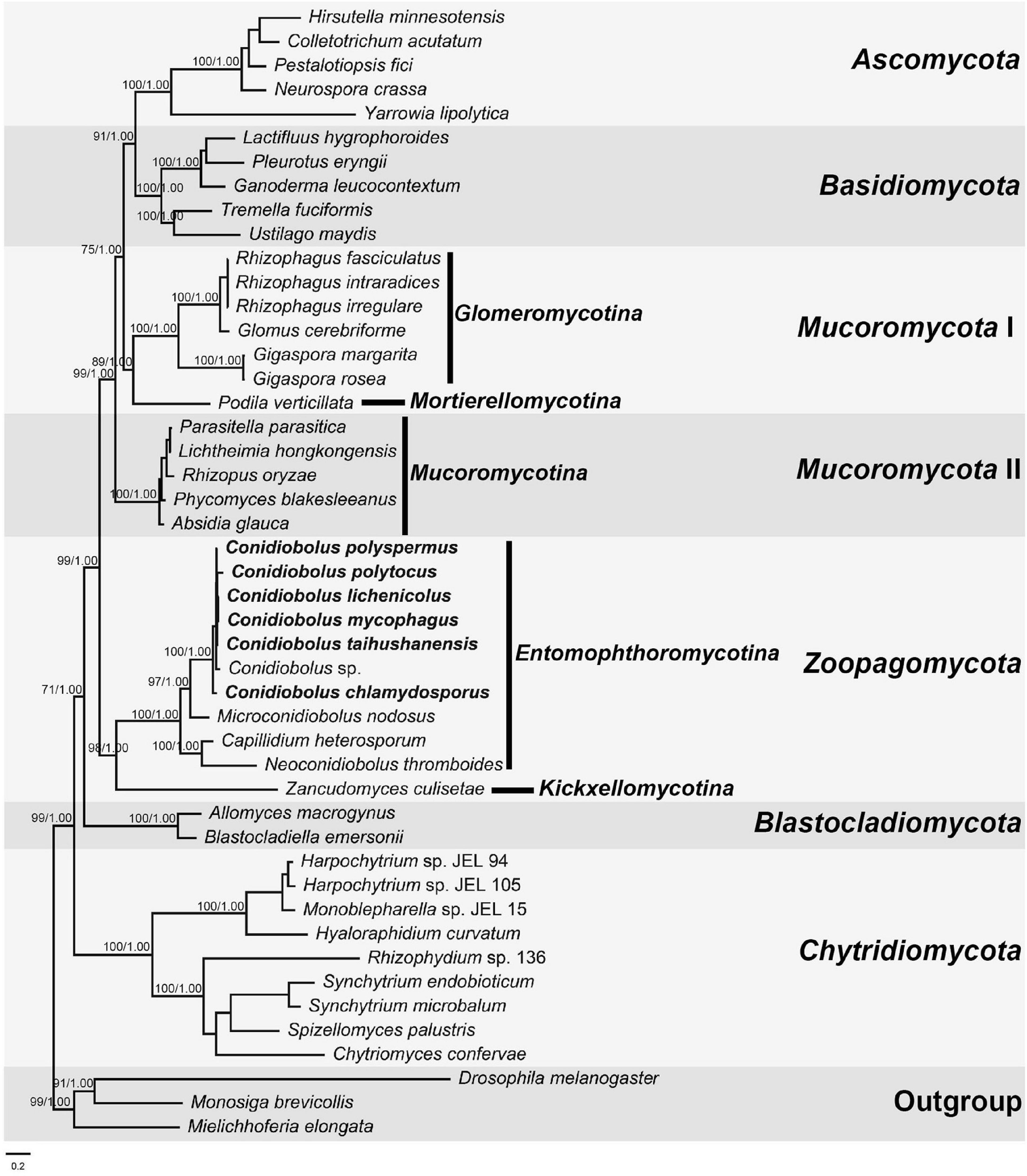
Figure 6. The Maximum Likelihood (ML) phylogenomic tree of fungi based on 14 protein-coding genes (PCGs): cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, atp6, atp8, and atp9. Support values for ML and Bayesian analysis (BI) greater than 70% and 0.95, respectively, are given on relative clade. Drosophila melanogaster, Mielichhoferia elongata, and Monosiga brevicollis serve as outgroups. The lower left bar represents 0.2 substitutes per site.
Discussion
Fungal mitogenomes differ greatly in gene content, gene arrangement, repeat elements, and intronic content among species and even closely related ones (Sandor et al., 2018; Li Q. et al., 2020, Li et al., 2021). These differences contain important information about the origin and evolution of species (Sankoff et al., 1992). In this article, we focused on these characteristics to evaluate the evolution of Conidiobolus s.s.
The seven Conidiobolus s.s. mitogenomes (26,612–48,417 bp) were moderate in size compared with those of other basal fungi (12,100–225,604 bp; Nie et al., 2019). The size of mitogenomes was mainly determined by its introns and intergenic regions (Table 1) as denoted elsewhere (Sandor et al., 2018; Liu W. et al., 2020). A much wider range was observed in a single genus of higher fungi. Taking Schizosaccharomyces as an example, its mitogenome sizes ranged from approximately 19,000–80,000 bp (Bullerwell et al., 2003). The wide size range of fungal mitogenomes might imply a variable function in oxidative phosphorylation and cellular respiration.
All the 14 conserved PCGs were all detected in the Conidiobolus s.s. mitogenomes, except that the atp8 gene was absent in the mitogenomes of C. chlamydosporus, C. polyspermus, and C. polytocus. This kind of gene loss is not uncommon. The atp9 gene was lost from the Trichoderma gamsii mitogenome (Kwak, 2021). The nad3 gene was not annotated in the Colletotrichum lindemuthianum mitogenome (De-Queiroz et al., 2018), neither was other NADH genes in some yeast mitogenomes (Friedrich et al., 2012). Most scholars considered that some mitochondrial genes have been transferred to the nuclear genome during the evolution of eukaryotes (Adams and Palmer, 2003). The arrangement of mitochondrial genes in fungal mitogenomes reflected their phylogenetic relationship to certain degrees (Zhang et al., 2018; Wang et al., 2020). As expected, the gene orders were identical in the monophyletic Conidiobolus s.s., which was confirmed by the phylogenomic analyses (Figure 6). The gene direction in C. heterosporum mitogenome was almost opposite to others, implying its distant phylogenetic relationship to other species. As a complement to gene order analysis, the mitogenomic synteny analysis showed more detail about gene length and rearrangement (Figure 6). Basal fungal groups frequently exhibited intra-genus gene rearrangements (Li et al., 2019, 2021; Zhang et al., 2020), similar to the result herein. It is worth to note that the cox1 gene in C. taihushanensis mitogenome was split by the atp8 gene. This phenomenon often appeared in basal fungi, especially in the mitogenomes of Glomeromycetes (Pelin et al., 2012; Nadimi et al., 2016; Arcila-Galvis et al., 2021). It may be a long size and abundant introns of cox1 gene that caused a trans-splicing of cox1 genes located on different RNA molecules and transcribed separately (James et al., 2013; Nie et al., 2019). It is also worth to note that the rps3 gene was exhibited in M. nodosus and N. thromboides mitogenomes. The rps3 gene encodes a key protein component of the ribosome for protein translation, and it is often fused with an rnl intron that harbors an ORF homologous to HEs or a partial sequence of cox1 gene (Bullerwell et al., 2000; Sethuraman et al., 2009; Franco et al., 2017; Zhang et al., 2019).
As shown in Figure 2, the lengths of atp6, cox2, nad1, nad2, and nad6 varied among the six Conidiobolus s.s. mitogenomes species. The GC contents of all PCGs varied across the six mitogenomes, which indicated that the conserved PCGs of Conidiobolus s.s. mitogenomes mutate frequently. The nad4L gene bore the smallest pairwise genetic distance, indicating that it was highly conserved. The Ka/Ks values of the 13 PCGs were all <1, indicating that these genes were subjected to purifying selection (Figure 3).
A total of 41 introns were detected in the cox1 gene that occupied mostly half of the total introns in the Conidiobolus s.s. mitogenomes (Tables 1, 2). Usually, the cox1 gene contains many introns in fungal mitogenomes causing its large variation, and it was not suitable as DNA barcode to identify fungal species (Freel et al., 2015; Nadimi et al., 2016; Sandor et al., 2018). Pcls analysis of cox1 gene and other core PCGs was often used to evaluate the dynamics of introns in basidiomycetes mitogenomes. For example, two large-scale intron loss events were detected in the evolution of Boletales species based on the Pcls analysis of the cox1 gene (Li et al., 2021). The Pcls analysis of several core PCGs that contained introns was conducted in 27 Agaricales species. The result showed that few Pcls were widely distributed indicating that these introns may be inherited from the common ancestors, while the same Pcls were detected in distant species from different lineages providing the evidence of intron loss/gain in the evolution of Agaricales species by intron transfer (Huang et al., 2021). This study revealed that Pcl of Ancylistanceae introns varied greatly. Some introns (e.g., P19 and P30) were widely distributed in the cox1 gene and P6 in the cob gene. These introns may be inherited from the ancestors of Ancylistanceae. This phenomenon indicated the potential transfer of introns in the evolution of Ancylistanceae. However, the intronic comparative analysis was conducted within only 10 Ancylistanceae mitogenomes, so the intron loss/gain events were absent of strong evidences.
Repetitive elements in six Conidiobolus s.s. mitogenomes were obtained by BLAST analysis against themselves, resulting in a length variation of 0.2–5.8%. Strangely, C. chlamydosporus mitogenome was detected in most tandem repeats (50), while it possessed the smallest mitogenome size among six Conidiobolus s.s. mitogenomes. With BLAST to their nuclear genomes, 62–150 aligned fragments were examined between nuclear and mitochondrial genomes of the six Conidiobolus s.s. species. This result indicated that the natural gene fragment transfer events may have occurred between mitogenomes and nuclear genomes during the evolution of six Conidiobolus s.s. species.
Traditionally, the ITS, LSU rDNA sequences, and morphological characteristics were widely used in species identification and evolution (Zheng et al., 2007; Zhao et al., 2021; Zong et al., 2021). In addition, single- or multi-gene loci, SSRs, and single-nucleotide polymorphisms (SNPs) have been widely used to study the evolution and polymorphism of fungi during the last two decades (Tsykun et al., 2017). Phylogenetic analyses based on multi-gene loci and even whole genomic data developed rapidly as the cost of sequencing decreased (Spatafora et al., 2016; Nie et al., 2019). Especially, SNPs called from whole genome were extensively used to study polymorphisms in fungi (Duan et al., 2018; Ju et al., 2020), and SNP-based genome-wide association studies (GWAS) were widely used to explore the control mechanism for wheat leaf rust (Liu F. et al., 2020).
The genus Conidiobolus s.l. was divided into four clades representing four genera: Capillidium, Microconidiobolus, Neoconidiobolus, and Conidiobolus s.s. based on the multiple genetic locus phylogeny (Nie et al., 2020b). Phylogenomic analysis based on the 14 PCGs of mitogenomes revealed that six Conidiobolus s.s. species were grouped together with three related species: C. heterosporum, M. nodosus, and N. thromboides in the Entomophthoromycotina lineage. This phylogram was consistent with our previous study and the phylogenomic analysis based on the 192 orthologous proteins of nuclear genomes (Spatafora et al., 2016; Nie et al., 2019). Also, this phylogram revealed that four lineages among 10 Conidiobolus s.l. species demonstrated that 14 mitochondrial PCGs are suitable to clarify phylogenetic relationships among this fungal groups.
In our previous study, the complete mitogenome of C. heterosporum (synonym with Conidiobolus heterosporus) has been reported, and the comparative analysis with other 22 mitogenomes of basal fungi revealed a high variation in mitogenomes sizes, gene order, GC content, genetic code, etc. (Nie et al., 2019). It was worth to note that the horizontal transfer event of C. heterosporum introns has failed to detect strong evidence due to rarely available mitogenomes of basal fungi. In this study, we have sequenced, assembled, and annotated six additional mitogenomes of basal fungi in the genus Conidiobolus s.s., and the comparative analysis has also been provided to give a comprehensive evolution of mitogenomes in six Conidiobolus s.s. species. The result confirmed that the number and length of intron were the main factors contributing to size variations of Conidiobolus s.s. mitogenomes and confirmed our previous taxonomical treatment of Conidiobolus s.l. Meanwhile, the comparison within six Conidiobolus s.s. mitogenomes showed high intronic variations and gene rearrangement. However, the evidence of intron loss/gain events needs further comparative analysis by using more mitogenomes in the family Ancylistanceae, even in the order Entomophthorales. Fortunately, more and more reported mitogenomes of basal fungi will help us to promote the understanding of its evolution and phylogeny in the future. In addition, mitogenomic sequences from a sufficient number of individuals within a certain species can play essential potential in population heredity, pan-genome, and species definition (Duan et al., 2018; McCarthy and Fitzpatrick, 2019; Nie et al., 2019).
Conclusion
We have sequenced, assembled, and annotated six Conidiobolus s.s. mitogenomes. The result confirmed that the number and length of introns were the main factors contributing to size variations of Conidiobolus s.s. mitogenomes. Phylomitogenomics backed up our previous taxonomical treatment of Conidiobolus s.l., i.e., dividing into four genera. Comparative analysis within Conidiobolus s.s. mitogenomes showed a high gene synteny along with deep intronic variations and gene rearrangement. Potential intron loss/gain and transfer events occurred during the evolution of the family Ancylistanceae.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
BH and XL conceived and designed the experiments. YN, HZ, ZW, and ZZ analyzed the data. YN, XL, and BH wrote the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Nos. 31900008, 30770008, and 31970009).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.765733/full#supplementary-material
Supplementary Figure 1 | Position class (Pcl) analysis of nine conserved protein-coding genes (PCGs) within 10 Conidiobolus s.l. species. The phylogenetic tree is constructed by maximum-likelihood (ML) method based on 14 concatenated mitochondrial core proteins.
Supplementary Figure 2 | Codon usage analyses of Conidiobolus s.l. mitogenomes. (A) C. chlamydosporus, (B) C. lichenicolus, (C) C. mycophagus, (D) C. polyspermus, (E) C. polytocus, (F) C. taihushanensis, (G) Capillidium heterosporum, (H) Conidiobolus sp., (I) Microconidiobolus nodosus, and (J) Neoconidiobolus thromboides.
Footnotes
- ^ http://www.indexfungorum.org/, accessed on September 1, 2021.
- ^ https://www.ncbi.nlm.nih.gov/genome/
- ^ https://genome.jgi.doe.gov
- ^ https://www.ncbi.nlm.nih.gov, accessed on May 1, 2021.
- ^ http://megasun.bch.umontreal.ca/cgi-bin/mfannot/mfannotInterface.pl
- ^ http://www.ncbi.nlm.nih.gov
- ^ http://www.dnastar.com/
- ^ http://archive.gramene.org/db/markers/ssrtool
References
Abboud, T. G., Zubaer, A., Wai, A., and Hausner, G. (2018). The complete mitochondrial genome of the dutch elm disease fungus ophiostoma novo-ulmi subsp. novo-ulmi. Can. J. Microbiol. 64, 339–348. doi: 10.1139/cjm-2017-0605
Adams, K. L., and Palmer, J. D. (2003). Evolution of mitochondrial gene content: gene loss and transfer to the nucleus. Mol. Phylogenet. Evol. 29, 380–395. doi: 10.1016/s1055-7903(03)00194-5
Aguileta, G., de Vienne, D. M., Ross, O. N., Hood, M. E., Giraud, T., Petit, E., et al. (2014). High variability of mitochondrial gene order among fungi. Genome Biol. Evol. 6, 451–465. doi: 10.1093/gbe/evu028
Alnakeeb, K., Petersen, T. N., and Sicheritzpontén, T. (2017). Norgal: extraction and de novo assembly of mitochondrial DNA from whole-genome sequencing data. BMC Bioinformatics 18:510. doi: 10.1186/s12859-017-1927-y
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Cambridge: Babraham Bioinformatics.
Arcila-Galvis, J. E., Arango, R. E., Torres-Bonilla, J. M., and Arias, T. (2021). The mitochondrial genome of a plant fungal pathogen Pseudocercospora fijiensis (Mycosphaerellaceae), comparative analysis and diversification times of the Sigatoka disease complex using fossil calibrated phylogenies. Life 11:215. doi: 10.3390/life11030215
Ballard, J. W. O., and Whitlock, M. C. (2004). The incomplete natural history of mitochondria. Mol. Ecol. 13, 729–744. doi: 10.1046/j.1365-294X.2003.02063.x
Beaudet, D., Nadimi, M., Iffis, B., and Hijri, M. (2013). Rapid mitochondrial genome evolution through invasion of mobile elements in two closely related species of arbuscular mycorrhizal fungi. PLoS One 8:12. doi: 10.1371/journal.pone.0060768
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580.
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120.
Bullerwell, C. E., Burger, G., and Lang, B. F. (2000). A novel motif for identifying rps3 homologs in fungal mitochondrial genomes. Trends Biochem. Sci. 25, 363–365. doi: 10.1016/S0968-0004
Bullerwell, C. E., Leigh, J., Forget, L., and Lang, B. F. (2003). A comparison of three fission yeast mitochondrial genomes. Nucleic Acids Res. 31, 759–768. doi: 10.1093/nar/gkg134
Cai, Y., Nie, Y., Wang, Z. M., and Huang, B. (2021). The complete mitochondrial genome of Microconidiobolus nodosus (Entomophthorales: Ancylistaceae). Mitochondrial DNA B Resour. 6, 1743–1744. doi: 10.1080/23802359.2021.1930219
Cameron, S. L. (2014). Insect mitochondrial genomics: implications for evolution and phylogeny. Annu. Rev. Entomol. 59, 95–117. doi: 10.1146/annurev-ento-011613-162007
Chan, D. C. (2006). Mitochondria: dynamic organelles in disease, aging, and development. Cell 125, 1241–1252. doi: 10.1016/j.cell.2006.06.010
Darling, A. C., Mau, B., FBlattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403.
De-Queiroz, C. B., Santana, M. F., Pereira-Vidigal, P. M., and De-Queiroz, M. V. (2018). Comparative analysis of the mitochondrial genome of the fungus Colletotrichum lindemuthianum, the causal agent of anthracnose in common beans. Appl. Microbiol. Biotechnol. 102, 2763–2778. doi: 10.1007/s00253-018-8812-0
Dierckxsens, N., Mardulyn, P., and Smits, G. (2017). NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45, 1–9. doi: 10.1093/nar/gkw955
Duan, S. F., Han, P. J., Wang, Q. M., Liu, W. Q., Shi, J. Y., Li, K., et al. (2018). The origin and adaptive evolution of domesticated populations of yeast from Far East Asia. Nat. Commun. 9:2690. doi: 10.1038/s41467-018-05106-7
Ferandon, C., Moukha, S., Callac, P., Benedetto, J. P., Castroviejo, M., and Barroso, G. (2010). The Agaricus bisporus cox1 gene: the longest mitochondrial gene and the largest reservoir of mitochondrial group I introns. PLoS One 5:e14048. doi: 10.1371/journal.pone.0014048
Ferandon, C., Xu, J., and Barroso, G. (2013). The 135 kbp mitochondrial genome of Agaricus bisporus is the largest known eukaryotic reservoir of group I introns and plasmid-related sequences. Fungal Genet. Biol. 55, 85–91. doi: 10.1016/j.fgb.2013.01.009
Franco, M. E. E., Lo’pez, S. M. Y., Medina, R., Lucentini, C. G., Troncozo, M. I., Pastorino, G. N., et al. (2017). The mitochondrial genome of the plant pathogenic fungus Stemphylium lycopersici uncovers a dynamic structure due to repetitive and mobile elements. PLoS One 12:e0185545. doi: 10.1371/journal.pone.0185545
Freel, K. C., Friedrich, A., and Schacherer, J. (2015). Mitochondrial genome evolution in yeasts: an all-encompassing view. FEMS Yeast Res. 15:9. doi: 10.1093/femsyr/fov023
Friedrich, A., Jung, P. P., Hou, J., Neuveglise, C., and Schacherer, J. (2012). Comparative mitochondrial genomics within and among yeast species of the Lachancea genus. PLoS One 7:6. doi: 10.1371/journal.pone.0047834
Greiner, S., Lehwark, P., and Bock, R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64. doi: 10.1101/545509
Gryganskyi, A. P., Humber, R. A., Smith, M. E., Hodge, K., Huang, B., Voigt, K., et al. (2013). Phylogenetic lineages in entomophthoromycota. Persoonia 30, 94–105. doi: 10.3767/003158513X666330
Henze, K., and Martin, W. (2003). Evolutionary biology: essence of mitochondria. Nature 426, 127–128. doi: 10.1038/426127a
Huang, W., Feng, H., Tu, W., Xiong, C., Jin, X., Li, P., et al. (2021). Comparative mitogenomic analysis reveals dynamics of intron within and between Tricholoma species and phylogeny of basidiomycota. Front. Genet. 12:534871. doi: 10.3389/fgene.2021.534871
Humber, R. A. (2012). Entomophthoromycota: a new phylum and reclassification for entomophthoroid fungi. Mycotaxon 120, 477–492. doi: 10.5248/120.477
Inokuma, K., Ishii, J., Hara, K. Y., Mochizuki, M., Hasunuma, T., and Kondo, A. (2015). Complete genome sequence of Kluyveromyces marxianus NBRC1777, a nonconventional thermotolerant yeast. Genome Announc. 3:e00389-15. doi: 10.1128/genomeA.00389-15
James, T. Y., Pelin, A., Bonen, L., Ahrendt, S., Sain, D., Corradi, N., et al. (2013). Shared signatures of parasitism and phylogenomics unite the Cryptomycota and Microsporidia. Curr. Biol. 23, 1548–1553. doi: 10.1016/j.cub.2013.06.057
Ju, X., Zhang, M. Z., Zhao, H., Liu, Z., Jia, B. S., Timothy, Y., et al. (2020). Genomic SNPs reveal population structure of Rhizopus arrhizus. Mycosystema 39, 2285–2303. doi: 10.13346/j.mycosystema.200105
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kwak, Y. (2021). An update on Trichoderma Mitogenomes: complete de novo mitochondrial genome of the fungal biocontrol agent Trichoderma harzianum (Hypocreales, Sordariomycetes), an ex-neotype strain CBS 226.95, and tracing the evolutionary divergences of Mitogenomes in Trichoderma. Microorganisms 9:1564. doi: 10.3390/microorganisms9081564
Lang, B. F. (2018). “Mitochondrial genomes in Fungi,” in Molecular Life Sciences, eds R. D. Wells, J. S. Bond, J. Klinman, and B. S. S. Masters (New York: Springer), 722–728.
Lang, B. F., Gray, M. W., and Burger, G. (1999). Mitochondrial genome evolution and the origin of eukaryotes. Annu. Rev. Genet. 33, 351–397. doi: 10.1146/annurev.genet.33.1.351
Lang, B. F., Laforest, M. J., and Burger, G. (2007). Mitochondrial introns: a critical view. Trends Genet. 23, 119–125. doi: 10.1016/j.tig.2007.01.006
Li, Q., Wang, Q., Jin, X., Chen, Z., Xiong, C., Li, P., et al. (2019). Characterization and comparative analysis of six complete mitochondrial genomes from ectomycorrhizal fungi of the Lactarius genus and phylogenetic analysis of the Agaricomycetes. Int. J. Biol. Macromol. 121, 249–260. doi: 10.1016/j.ijbiomac.2018.10.029
Li, Q., Wu, P., Li, L. J., Feng, H. Y., Tu, W. Y., Bao, Z. J., et al. (2021). The first eleven mitochondrial genomes from the ectomycorrhizal fungal genus (Boletus) reveal intron loss and gene rearrangement. Int. J. Biol. Macromol. 172, 560–572. doi: 10.1016/j.ijbiomac.2021.01.087
Li, Q., Yang, L., Xiang, D., Wan, Y., Wu, Q., Huang, W., et al. (2020). The complete mitochondrial genomes of two model ectomycorrhizal fungi (Laccaria): features, intron dynamics and phylogenetic implications. Int. J. Biol. Macromol. 145, 974–984. doi: 10.1016/j.ijbiomac.2019.09.188
Li, X., Li, L., Bao, Z., Tu, W., He, X., Zhang, B., et al. (2020). The 287,403 bp mitochondrial genome of ectomycorrhizal fungus tuber calosporum reveals intron expansion, tRNA loss, and gene rearrangement. Front. Microbiol. 11:591453. doi: 10.3389/fmicb.2020.591453
Liu, F., Jiang, Y., Zhao, Y. S., Schulthess, A. W., and Reif, J. C. (2020). Haplotype-based genome-wide association increases the predictability of leaf rust (Puccinia triticina) resistance in wheat. J. Exp. Bot. 71, 6958–6968. doi: 10.1093/jxb/eraa387
Liu, W., Cai, Y. L., Zhang, Q. Q., Chen, L. F., Shu, F., Ma, X. L., et al. (2020). The mitochondrial genome of Morchella importuna (272.2 kb) is the largest among fungi and contains numerous introns, mitochondrial non-conserved open reading frames and repetitive sequences. Int. J. Biol. Macromol. 143, 373–381. doi: 10.1016/j.ijbiomac.2019.12.056
Lohse, M., Drechsel, O., Kahlau, S., and Bock, R. (2013). OrganellarGenomeDRAW–a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41, W575–W581. doi: 10.1093/nar/gkt289
Martin, W. F., Garg, S., and Zimorski, V. (2015). Endosymbiotic theories for eukaryote origin. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 370:18. doi: 10.1098/rstb.2014.0330
McCarthy, C. G., and Fitzpatrick, D. A. (2019). Pan-genome analyses of model fungal species. Microb. Genomics 5:e000243. doi: 10.1099/mgen.0.000243
Nadimi, M., Daubois, L., and Hijri, M. (2016). Mitochondrial comparative genomics and phylogenetic signal assessment of mtDNA among arbuscular mycorrhizal fungi. Mol. Phylogenet. Evol. 98, 74–83. doi: 10.1016/j.ympev.2016.01.009
Nie, Y., Cai, Y., Gao, Y., Yu, D. S., Wang, Z. M., Liu, X. Y., et al. (2020a). Three new species of Conidiobolus sensu stricto from plant debris in eastern China. MycoKeys 73, 133–149. doi: 10.3897/mycokeys.73.56905
Nie, Y., Wang, L., Cai, Y., Tao, W., Zhang, Y.-J., and Huang, B. (2019). Mitochondrial genome of the entomophthoroid fungus Conidiobolus heterosporus provides insights into evolution of basal fungi. Appl. Microbiol. Biotechnol. 103, 1379–1391. doi: 10.1007/s00253-018-9549-5
Nie, Y., Wang, Z. M., Zhao, H., Liu, X. Y., and Huang, B. (2021). Complete mitochondrial genome of Neoconidiobolus thromboides (Entomophthorales: Ancylistaceae). Mitochondrial DNA B Resour. 6, 1840–1841. doi: 10.1080/23802359.2021.1934167
Nie, Y., Yu, D. S., Wang, C. F., Liu, X. Y., and Huang, B. (2020b). A taxonomic revision of the genus Conidiobolus (Ancylistaceae. Entomophthorales): four clades including three new genera. MycoKeys 66, 55–81. doi: 10.3897/mycokeys.66.46575
Pantou, M. P., Kouvelis, V. N., and Typas, M. A. (2006). The complete mitochondrial genome of the vascular wilt fungus Verticillium dahliae: a novel gene order for Verticillium and a diagnostic tool for species identification. Curr. Genet. 50, 125–136. doi: 10.1007/s00294-006-0079-9
Pelin, A., Pombert, J. F., Salvioli, A., Bonen, L., Bonfante, P., and Corradi, N. (2012). The mitochondrial genome of the arbuscular mycorrhizal fungus Gigaspora margarita reveals two unsuspected trans-splicing events of group I introns. New Phytol. 194, 836–845.
Posada, D., and Crandall, K. A. (1998). MODELTEST: testing the model of DNA substitution. Bioinformatics 14, 817–818. doi: 10.1093/bioinformatics/14.9.817
Ronquist, F., and Huelsenbeck, J. P. (2003). MrBayes 3: bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Rozas, J., Ferrer-Mata, A., Sanchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Saldanha, R., Mohr, G., Belfort, M., and Lambowitz, A. M. (1993). Group I and group II introns. FASEB J. 7, 15–24.
Sandor, S., Zhang, Y. J., and Xu, J. P. (2018). Fungal mitochondrial genomes and genetic polymorphisms. Appl. Microbiol. Biotechnol. 102, 9433–9448. doi: 10.1007/s00253-018-9350-5
Sankoff, D., Leduc, G., Antoine, N., Paquin, B., Lang, B. F., and Cedergren, R. (1992). Gene order comparisons for phylogenetic inference: evolution of the mitochondrial genome. Proc. Natl. Acad. Sci. U. S. A. 89, 6575–6579. doi: 10.1073/pnas.89.14.6575
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the 862 detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689. doi: 10.1093/nar/gki366
Sethuraman, J., Majer, A., Iranpour, M., and Hausner, G. (2009). Molecular evolution of the mtDNA encoded rps3 gene among filamentous ascomycetes fungi with an emphasis on the ophiostomatoid fungi. J. Mol. Evol. 69, 372–385. doi: 10.1007/s00239-009-9291-9
Slater, G. S. C., and Birney, E. (2005). Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6:31. doi: 10.1186/1471-2105-6-31
Spatafora, J. W., Chang, Y., Benny, G. L., Lazarus, K., Smith, M. E., Berbee, M. L., et al. (2016). A phylum-level phylogenetic classification of zygomycete fungi based on genome scale data. Mycologia 108, 1028–1046. doi: 10.3852/16-042
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stothard, P. (2000). The sequence manipulation suite: javaScript programs for analyzing and formatting protein and DNA sequences. BioTechniques 28, 1102–1104. doi: 10.2144/00286ir01
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Torriani, S. F. F., Penselin, D., Knogge, W., Felder, M., Taudien, S., Platzer, M., et al. (2014). Comparative analysis of mitochondrial genomes from closely related Rhynchosporium species reveals extensive intron invasion. Fungal Genet. Biol. 62, 34–42. doi: 10.1016/j.fgb.2013.11.001
Tsykun, T., Rellstab, C., Dutech, C., Sipos, G., and Prospero, S. (2017). Comparative assessment of SSR and SNP markers for inferring the population genetic structure of the common fungus Armillaria cepistip. Heredity 119, 371–380. doi: 10.1038/hdy.2017.48
Vaidya, G., Lohman, D. J., and Meier, R. (2011). SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 27, 171–180. doi: 10.1111/j.1096-0031.2010.00329.x
van de Vossenberg, B. T. L. H., Brankovics, B., Nguyen, H. D. T., van Gent, M. P. E., Smith, D., Dadej, K., et al. (2018). The linear mitochondrial genome of the quarantine chytrid Synchytrium endobioticum; insights into the evolution and recent history of an obligate biotrophic plant pathogen. BMC Evol. Biol. 18:136. doi: 10.1186/s12862-018-1246-6
Vilela, R., and Mendoza, L. (2018). Human pathogenic entomophthorales. Clin. Microbiol. Rev. 31, 1–40. doi: 10.1128/CMR.00014-18
Vilela, R., Silva, S. M. S., Correa, F. R., Dominguez, E., and Mendoza, L. (2010). Morphologic and phylogenetic characterization of Conidiobolus lamprauges recovered from infected sheep. J. Clin. Microbiol. 48, 427–432. doi: 10.1128/JCM.01589-09
Wang, L., Zhang, S., Li, J. H., and Zhang, Y. J. (2018). Mitochondrial genome, comparative analysis and evolutionary insights into the entomopathogenic fungus Hirsutella thompsonii. Environ. Microbiol. 20, 3393–3405. doi: 10.1111/1462-2920.14379
Wang, X., Wang, Y. J., Yao, W., Shen, J. W., Chen, M. Y., Gao, M., et al. (2020). The 256 kb mitochondrial genome of Clavaria fumosa is the largest among phylum Basidiomycota and is rich in introns and intronic ORFs. IMA Fungus 11:26. doi: 10.1186/s43008-020-00047-7
Watanabe, M., Lee, K., Goto, K., Kumagai, S., Sugita-Konishi, Y., and Hara-Kudo, Y. (2010). Rapid and effective DNA extraction method with bead grinding for a large amount of fungal DNA. J. Food Prot. 73, 1077–1084. doi: 10.4315/0362-028X-73.6.1077
Yuan, X. L., Mao, X. X., Liu, X. M., Cheng, S., Zhang, P., and Zhang, Z. F. (2017). The complete mitochondrial genome of Engyodontium album and comparative analyses with Ascomycota mitogenomes. Genet. Mol. Biol. 40, 844–854. doi: 10.1590/1678-4685-GMB-2016-0308
Zhang, S., and Zhang, Y. J. (2019). Proposal of a new nomenclature for introns in protein-coding genes in fungal mitogenomes. IMA Fungus 10:15. doi: 10.1186/s43008-019-0015-5
Zhang, S., Zhang, Y. J., and Li, Z. (2019). Complete mitogenome of the entomopathogenic fungus Sporothrix insectorum RCEF 264 and comparative mitogenomics in ophiostomatales. Appl. Microbiol. Biotechnol. 13, 5797–5809. doi: 10.1007/s00253-019-09855-3
Zhang, Y., Sun, J., Rouse, G. W., Wiklund, H., Pleijel, H. F., Watanabe, H. K., et al. (2018). Phylogeny, evolution and mitochondrial gene order rearrangement in scaleworms (Aphroditiformia, Annelida). Mol. Phylogenet. Evol. 125, 220–231. doi: 10.1016/j.ympev.2018.04.002
Zhang, Y., Yang, G. Z., Fang, M. L., Deng, C., Zhang, K. Q., Yu, Z. F., et al. (2020). Comparative analyses of mitochondrial genomes provide evolutionary insights into nematode-trapping Fungi. Front. Microbiol. 11:617. doi: 10.3389/fmicb.2020.00617
Zhang, Y. J., Yang, X. Q., Zhang, S., Humber, R. A., and Xu, J. (2017). Genomic analyses reveal low mitochondrial and high nuclear diversity in the cyclosporinproducing fungus Tolypocladium inflatum. Appl. Microbiol. Biotechnol. 101, 8517–8531. doi: 10.1007/s00253-017-8574-0
Zhao, H., Zhu, J., Zong, T. K., Liu, X. L., Ren, L. Y., Lin, Q., et al. (2021). Two new species in the family Cunninghamellaceae from China. Mycobiology 49, 142–150. doi: 10.1080/12298093.2021.1904555
Zheng, R. Y., Chen, G. Q., Huang, H., and Liu, X. Y. (2007). A monograph of Rhizopus. Sydowia 59, 273–372.
Keywords: basal fungi, Entomophthorales, mitogenome, intron, gene order
Citation: Nie Y, Zhao H, Wang Z, Zhou Z, Liu X and Huang B (2021) The Gene Rearrangement, Loss, Transfer, and Deep Intronic Variation in Mitochondrial Genomes of Conidiobolus. Front. Microbiol. 12:765733. doi: 10.3389/fmicb.2021.765733
Received: 27 August 2021; Accepted: 01 October 2021;
Published: 11 November 2021.
Edited by:
David W. Ussery, University of Arkansas for Medical Sciences, United StatesReviewed by:
Chuan Xu, Shanghai Jiao Tong University, ChinaYanchun Shao, Huazhong Agricultural University, China
Copyright © 2021 Nie, Zhao, Wang, Zhou, Liu and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bo Huang, bhuang@ahau.edu.cn; Xiaoyong Liu, liuxiaoyong@im.ac.cn
†These authors have contributed equally to this work and share first authorship