 Wenli Yan
Wenli Yan Baolong Luo
Baolong Luo Xuyao Zhang
Xuyao Zhang Yongqing Ni
Yongqing Ni Fengwei Tian
Fengwei Tian- 1School of Food Science and Technology, Shihezi University, Shihezi, China
- 2School of Food Science and Technology, Jiangnan University, Wuxi, China
Breast milk acts as an intermediary for the transfer of functionally important commensal bacteria from mother to infant, especially for Bifidobacterium that can colonize the infant gut. However, the vast majority of rRNA amplicon-based studies reported the conspicuous intercohort and interindividual variation for the prevalence of Bifidobacterium in breast milk. In order to elucidate whether Bifidobacterium phylotypes persistently co-occured at the species or strain level in mother–breast milk–infant triads, we analyzed collectively the next-generation sequencing (NGS) datasets of bacterial 16S rRNA gene and the Bifidobacterium-specific groEL gene from maternal feces, breast milk, and infant feces in a small yet very homogeneous cohort of 25 healthy Uyghur mother–infant pairs (lactation for 7–720 days) in Kashgar, Xinjiang, China. Overall, 16S rRNA gene analysis showed that microbiome in the newborn gut was closer to that of breast milk in the first 4 months of lactation, and subsequently showed an obvious trend of adulthood at 6–12 months. Based on the BLAST accurate taxonomic result of the representative sequences of all ASVs (amplicon sequencing variants), only three sets of ASVs could be clearly assigned into Bifidobacterium species, whereas the remaining eight sets of ASVs corresponded to four indefinite Bifidobacterium species group. By contrast, the groEL gene dataset was partitioned into 376 ASVs, at least belonging to 13 well-known Bifidobacterium species or subspecies, of which 15 ASVs, annotated to seven well-known Bifidobacterium species or subspecies, showed triadic synchronism in most 23 mother–infant pairs tested. However, several other rare bifidobacterial phylotypes, which were frequently encountered in animals, were found to display no correspondence of the presence between the three ecosystems of mother–infant pairs. Our test results were obviously to support the hypothesis that breast milk acts as an intermediary for the transfer of probiotic commensal bacteria from mother to infant, especially for endosymbiotic Bifidobacterium that can colonize the infant gut. Some oxygen-insensitive exogenous Bifidobacterium phylotypes with a cosmopolitan lifestyle may be indirectly transferred to breast milk and the infant’s intestinal tract through environmental contamination. Thus, the groEL gene proved to be a very effective target for the depth resolution of Bifidobacterium community by high-throughput sequencing technologies.
Introduction
Breast milk is generally regarded as the best source of nutrients for healthy growth and development of infants (Bode, 2012; Ballard and Morrow, 2013). Over the past decade and a half, many studies have focused on the bacterial community of breast milk using both culture-dependent and culture-independent techniques. Culture-independent techniques, based on the amplicon analysis of 16S rRNA gene, revealed the presence of several hundred bacterial species from breast milk up to now, the most common genus of which includes Staphylococcus, Streptococcus, Flavobacterium, Propionibacterium, Burkholderia, Rothia, Corynebacterium, and Lactobacillus, commonly known as a core milk microbiota (Heikkila and Saris, 2003; Groenlund et al., 2007; Martin et al., 2007; Perez et al., 2007; Collado et al., 2009; Hunt et al., 2011; Fitzstevens et al., 2017).
The origins of the bacteria in breast milk are thought to include the maternal gut (via an entero-mammary pathway) and through bacterial exposure of the breast during nursing (skin microbiota and the oral cavity of the infant) (Mueller et al., 2015). Increasingly, accumulating evidence highlights that the maternal gut serves as the most relevant source of bacteria that are detectable in breast milk. With the consumption of breast milk, and the reduced oxygen/redox potential in the infant gut, the obligate anaerobic bacteria emerge in the infant gut, especially the Bifidobacterium species and Bacteroides species (Ferretti et al., 2018). In the skin microbiome of a healthy human, by contrast, a member of Bifidobacterium was not detected (Oh et al., 2016), and was reported occasionally in the oral cavity of infants and the vagina of their mothers (Sundin et al., 2017; Dzidic et al., 2018; Nyangahu and Jaspan, 2019). So, breast milk constitutes the main source of an array of potentially beneficial bacteria to the breastfed infant gut.
Previous studies showed that total bacteria concentration is lower in colostrum than in transitional and mature milk, with increasing levels of Bifidobacterium in breast milks over lactation time (Cabrera-Rubio et al., 2012; Khodayar-Pardo et al., 2014; Sakwinska et al., 2016). Indeed, in a previous study, no bifidobacteria were detected from colostrum, and Bifidobacterium strains were isolated only from breast milk samples obtained 7 days after birth or later. Again, despite the use of advanced next-generation sequencing (NGS) of 16S rRNA gene amplicon, presences of Bifidobacterium spp. were sporadically reported in a few colostrum samples or not at all. In contrast, the prevalence of bifidobacteria in transitional and mature milks was generally increased, but its relative abundance is only 0.1–1.3% or lower. More notably, Bifidobacterium populations were not also detected in a considerable proportion of transitional and mature milk samples, although an identical test method was applied to the same batch of samples (Hunt et al., 2011; Urbaniak et al., 2016; McGuire and McGuire, 2017; Murphy et al., 2017).
In general, bacterial loads of feces samples are five to seven orders of magnitude higher than in breast milk (Fernandez et al., 2013). From 1 to 6 months of age, members of the genus Bifidobacterium clearly dominate the infant gut microbiota, regardless of delivery mode, representing an average of 10–90% of the total infant gut microbiota (Turroni et al., 2012; Lim et al., 2016; Lundgren et al., 2018). However, a small proportion of infants have very low abundance or undetectable bifidobacteria as members of the fecal microbiota regardless of breast milk or formula feeding (Koenig et al., 2011; Yatsunenko et al., 2012; Subramanian et al., 2014).
While multiple studies have shown that specific Bifidobacterium strains in the maternal gut are transferred to the infant gut through breastfeeding (Makino et al., 2011, 2013; Milani et al., 2015; Duranti et al., 2017), the sources and ways of acquisition of these potential probiotic bacteria regarding establishing a sound intestinal microbiome for infants are poorly understood. As a whole, it is not clear how bifidobacterial community from the maternal gut or breast milk progressively transmits to the infant gut during the first 2 years of life, and whether there is the concordance of the presence of Bifidobacterium spp. between the three ecosystems, represented by maternal feces, breast milk, and neonatal feces.
Next-generation sequencing is a more sensitive and less biased analytical method than the culture-based method (Hunt et al., 2011; Jost et al., 2013; Ward et al., 2013). These methods generate tens of thousands of 16S rRNA gene sequences per DNA sample, but taxa present in very low abundance could still be missed (Meehan et al., 2018). In addition, 16S rRNA gene-based profiling of the human microbiota is strongly influenced by sample processing and the choice of PCR primers, leading to underrepresentation of bifidobacteria in 16S rRNA sequence dataset. Also, it is difficult to identify the members of Bifidobacterium at species level by 16S rRNA short variable region amplicon sequencing (Schloss and Westcott, 2011). All these reasons result in inconsistent detection of the proportional abundance of specific bacteria taxa of human microbiota, including Bifidobacterium spp. Particularly, breast milk samples are highly variable in bacterial load values. There are large individual differences over time between samples from the different mothers and, in some cases, even within individuals at different time points (Bode et al., 2014; Moossavi et al., 2019). Therefore, we are more interested in the co-occurrence and combination of gut symbiotic Bifidobacterium phylotypes in mother–breast milk–infant triads than in their absolute content.
More recently, the groEL gene proved to be a very effective target for the identification and quantification of Bifidobacterium spp. through high-throughput sequencing technologies or qPCR (Junick and Blaut, 2012; Hu et al., 2017). In this study, a comparative analysis of feces and breast milk microbiota in a small, yet very homogeneous cohort of 25 healthy mother–infant pairs in Kashgar, northwest China (n = 25, infants’ age from 7 days to 2 years), was presented, using high-throughput sequencing technologies of the 16S ribosomal RNA gene and groEL gene specific to the genus Bifidobacterium. Our research objective is to assess the association of bifidobacterial phylotypes between infant feces and their mothers’ breast milk and maternal feces in mother–breast milk–infant triads, with the emphasis on the number and changes of Bifidobacterium phylotypes in the infant feces and breast milk.
Materials and Methods
Sample Collection
We collected breast milk and feces samples from mothers and their infants between 7 and 720 days after birth, during clinic or home study visits and recruited mother–infant pairs meeting the following criteria: (i) the Uighur people native to Kashgar, Xingjiang, (ii) vaginal delivery at full-term (≥37 week gestation), (iii) exclusive breastfeeding during the first 5 months and the lactation continuing until sampling, and (iv) no antibiotic/probiotic exposure of either the mother or the infant during pregnancy, intrapartum, or postnatally. All the participants were healthy and did not require hospitalization. They were included for microbiota analysis with standard collection protocol (Sakwinska et al., 2016). Nevertheless, our samples were collected from all the mother–infant pairs for only one time, and a longitudinal study was not carried out.
Demographic and clinical data were recorded in a specific case report form. All participants responded to a general questionnaire including socioeconomic, lifestyle aspects, and body mass index (BMI) of the mother. The case report recorded the number of gestational weeks at delivery, delivery method, feeding patterns, the gender, height, weight, and head circumference (for the newborn) of the infant. All demographic data of our cohort is summarized in Table 1, and this cohort has been reported in a previous study, which was focused on Lactobacillus (Zhang et al., 2020).
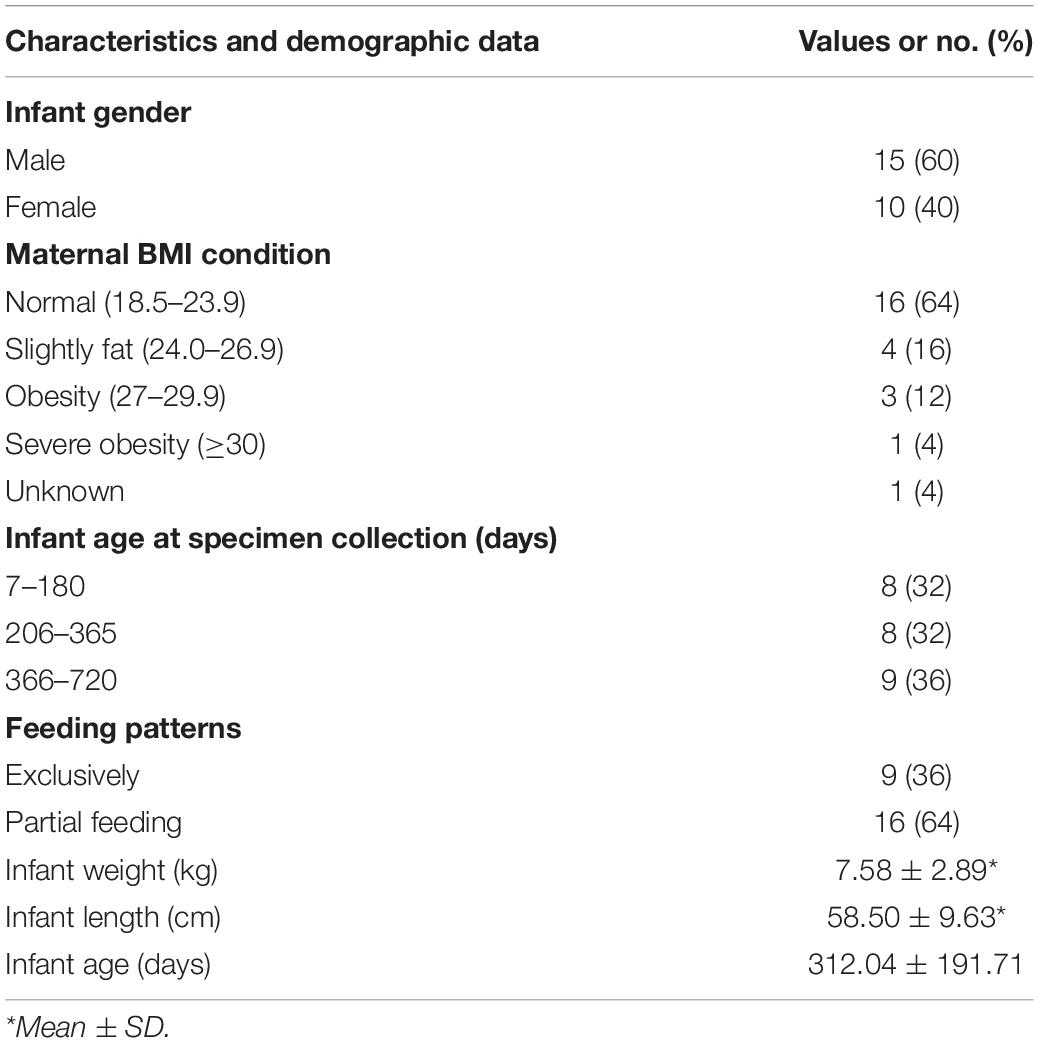
Table 1. Demographic characteristics of our cohort.
Standard sterile collection tubes were used to collect feces and breast milk (with the aid of a breast pump) samples. For breast milk, the first few drops (0.5–1 ml) were discarded, and the breast was thoroughly cleansed with chlorhexidine solution before manually collecting 3–5 ml of milk. Samples were immediately transported to the laboratory using portable refrigerators and ice packs. Each breast milk sample was divided into several 1-ml servings into sterile centrifuge tube and 500-mg feces sample were divided into a sterile centrifuge tube ready for DNA extraction and then were all frozen at −80°C in batches for processing and remained frozen until DNA extraction. All samples were collected and stored on the same day, and total bacterial DNA was extracted within 7 days of sampling as well as sequenced, to reduce the errors caused by the condition of storage, experiment, and sequencing.
DNA Extraction and High-Throughput Sequencing
For breast milk samples, TIANamp Blood DNA Kit (TIANGEN, Beijing, China) was utilized with some modifications, referring to Sakwinska et al. (2016) to extract bacteria DNA. One milliliter of breast milk was centrifuged at full speed (12,000 rpm) for 10 min at 4°C. The pellets were resuspended in 200 μl of Tris-EDTA buffer and treated with 10 μl of lysozyme (50 mg/ml) and 5 μl of DNase-free RNase (20 mg/ml) for 30 min at 37°C. Twenty-five milligrams of glass beads (10 μm) was added to the solution and treated with three bead-beating steps in a FastPrep instrument (MP Biomedicals, Irvine, CA, United States) at 5.5 movements per second for 1 min. After the instantaneous centrifugation, the supernatants were collected and treated with 20 μl of proteinase K for 20 min at 56°C. Two hundred microliters of GB buffer was added, and samples were incubated at 65°C for 10 min, and then 200 μl of ethanol was added. DNA was further purified using Spin Columns CB3 (TIANGEN) following the manufacturer’s instructions. The feces samples were processed with the TIANamp DNA Stool Kit (TIANGEN, Beijing, China) according to the manufacturer’s instructions. Extracted DNA was quantified using the nucleic acid quantifier (NanoDrop Technologies, Wilmington, DE, United States).
Sequencing Data Processing
For each sample, the V4–V5 region of the 16S rRNA gene and the groEL gene was amplified and sequenced according to the manufacturer’s instructions (Illumina, San Diego, CA, United States) by Shanghai Personal Biotechnology Co., Ltd., Shanghai, China1. Primer pairs for groEL sequences (Bif-groEL-F: 5-TCC GAT TAC GAY CGY GAG AAG CT-3/Bif-groEL-R: 5-CSG CYT CGG TSG TCA GGA ACA G-3) belonging to the target Bifidobacterium and available in GenBank release 234.0 (Benson et al., 2011) were designed by the Jiangnan University (Hu et al., 2017).
Raw sequences were processed by using a pipeline combining USEARCH v10.0 (Edgar, 2010) and QIIME (Caporaso et al., 2010). High-quality reads, as selected using the default values in USEARCH, were binned into ASVs (amplicon sequence variants) according to the denoising (error correcting) Illumina amplicon reads using Unoise3 (Edgar, 2017), through an open-reference strategy. Taxonomic identification of ASVs for the V4–V5 region sequences was assigned using the Naive Bayes classifier of the Ribosomal Database Project (RDP) against Greengenes database (August 2013 release). Meanwhile the taxonomy of ASVs for the groEL sequences was performed through comparison with the Chaperonin Sequence Database2 (Hill et al., 2004) and the National Center for Biotechnology Information (NCBI).
The diversity index was calculated by QIIME, and statistics and plot were performed using R software (version 4.0.2). Observed ASVs and Shannon index were analyzed as alpha rarefaction metrics. Weighted and unweighted UniFrac distances were computed as beta diversity, which was used for principal coordinate analyses (PCoA), and the function “adonis” of the vegan package of R software was utilized to test the significance of separation by permutational multivariate analysis of variance. p-values were corrected for multiple comparisons using the Benjamini–Hochberg method. p < 0.05 was considered statistically significant. The R function hclust() and package ggtree were utilized to cluster samples based on the Bray–Curtis similarity index using average linkage clustering and generate the dendrogram. Linear discriminant analysis (LDA) effect size (LEfSe) analysis was performed by R package dplyr and an open-reference strategy (Zhang et al., 2018). The bifidobacterial co-occurrence relationship of breast milk, infant feces, and maternal feces was determined based on the Spearman correlation coefficient and was visualized using the AnnotationDBi package.
Results
Microbial Community Structures in Breast Milk
The extracted bacterial DNA was phylogenetically characterized by the 16S rRNA gene (V4–V5 regions) Illumina sequencing. A total of 1,620,470 high-quality reads were obtained. Reads were binned into 1,936 ASVs. The bacterial community was distinct between breast milk, maternal feces, and infant feces in both composition and diversity. In the breast milk and feces samples, the most abundant phylum was, respectively, Proteobacteria (average relative abundance: 46.5%) and Firmicutes (mothers: 60.2%, infants: 58.3%; Figure 1A). The two most frequently present families in the breast milk were Enterobacteriaceae (25.5%) and Streptococcaceae (19.3%). In the infant feces samples, Streptococcaceae (26.1%), Lactobacillaceae (16.3%), and Enterobacteriaceae (11.5%), were the three most dominant families. In the maternal feces, Ruminococcaceae (17.6%) and Streptococcaceae (9.2%) constituted the predominant families. Pseudomonadaceae, as a family consisting of mostly aerobic bacteria, has an average relative abundance (rel. ab.) in breast milk that was obviously higher (2.2%) than those in the feces (infants: 0.2%, mothers: 0.04%), whereas the average rel. ab. of Ruminococcaceae, as one of the most important anaerobic bacterial families, was lower in breast milk (<0.1%) than in feces (infants: 3.8%; mothers: 17.6%). The results of the adonis analysis based on Euclidean matrix calculated from microbial relative abundance at the family level (Supplementary Table 1) showed that the correlation between the microbial community structure (the family level) of breast milk and maternal BMI, feeding patterns, infants’ age, gender, and weight status was not significant (R2 < 0.2, p > 0.05); the microbial community structure of the maternal feces was only significantly correlated with the weight status (R2 = 0.23, p = 0.001), while that of infants’ feces was only significantly correlated with age (day; R2 = 0.13, p < 0.01).
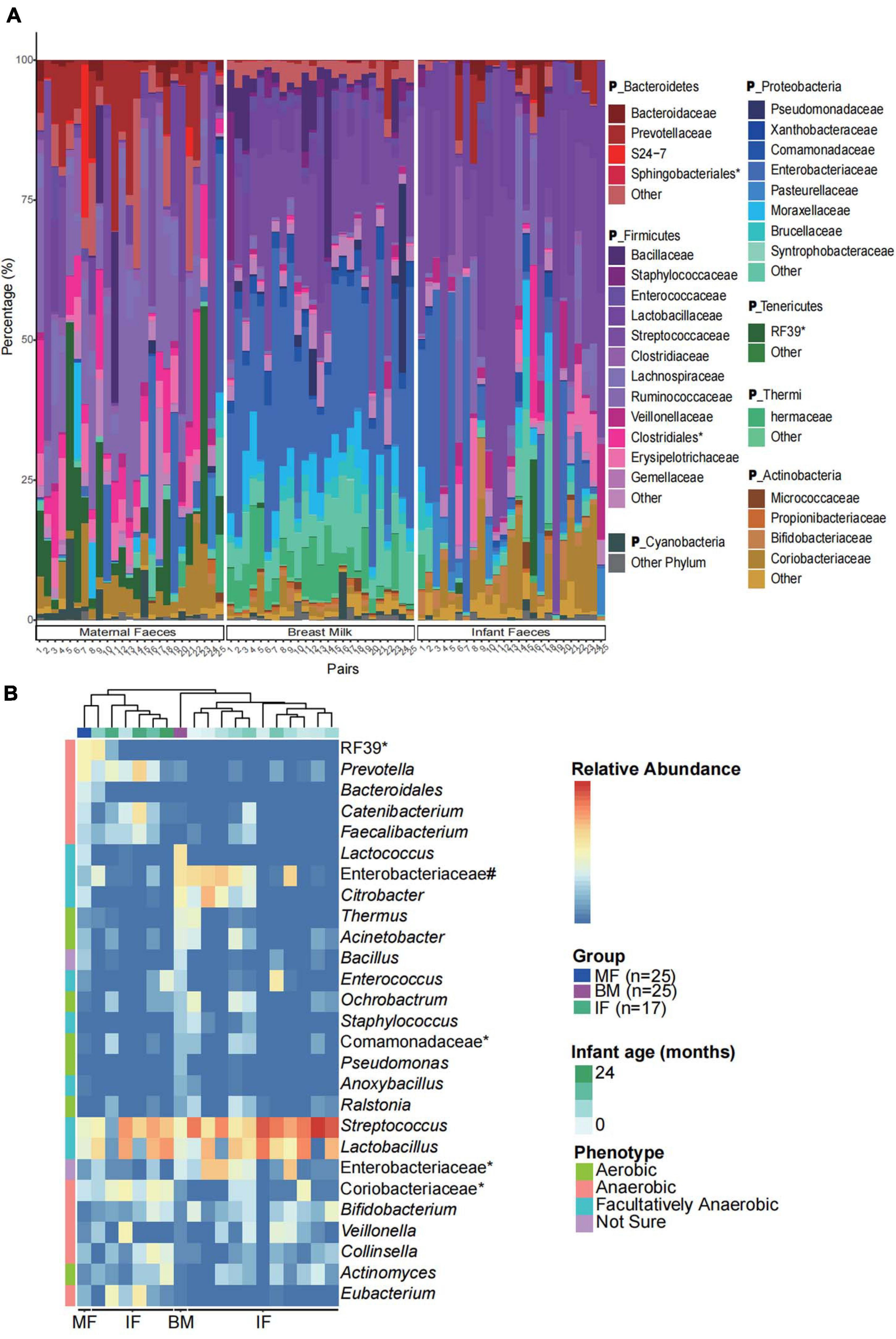
Figure 1. Microbial community characteristics of the infant feces, maternal feces, and breast milk samples. (A) Community structure of the dominant bacteria in breast milk and feces samples of all infants and their mothers at the family level. IF, infant feces; MF, maternal feces; BM, breast milk. *The unclassified bacteria at the family level. The P_ in taxonomy labels indicate that the level of taxonomy is phylum. (B) The relative abundance of the biomarkers (at the genus level) in feces samples of 17 infants aged 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 11,12,13, 15, 20, 21, and 24 months (only one infant feces sample per month) and in feces and breast milk samples of all mothers (n = 25). The groups are ordered by hierarchical clustering of Bray–Curtis dissimilarities. *The unclassified bacteria at the genus level. #Other genera of Enterobacteriaceae. The biologically interpretable phenotypes (oxygen tolerance) are predicted by algorithm BugBase.
We next used LEfSe to perform differentially abundant analysis at different taxonomic levels (from phylum to genus) between samples from three ecosystems (maternal feces, infant feces, and breast milk). Only the ASVs with relative abundance more than 0.01% were selected for the more accurate analysis. LEfSe analysis identified 109 differentially abundant genera and 198 differentially predominant families in three ecosystems (Supplementary Figure 1). From the cladogram that represents differentially abundant taxonomic level from phylum to genus level, it was evident that the breast milk harbored significantly more indicator taxa. In order to further observe the relationship of microbial composition of infants’ feces samples with that of samples from maternal feces and breast milk at different stages of lactation, we investigated the occurrence of biomarkers (LDA score > 3, p < 0.001) and dominant families in three ecosystems. Also, according to the results (Figure 1B), we found that feces microbiome of infants of younger age were more similar to breast milk microbiome (hierarchical clustering), which was represented by higher relative abundance of Enterobacteriaceae and Citrobacter, while the feces microbiome of infants with higher age were closer to those of mothers, mainly represented as higher relative abundance of Prevotella and Catenibacterium. It indicated that the temporal succession of the microbial community structure of the infants’ gut is actually a process by which the microbial composition similar to breast milk microbiome tends to be similar to the maternal gut microbiome.
Next, FEAST, a microbial sources tracking tool (Shenhav et al., 2019), was used to calculate the microbial source proportion of 25 infants’ feces, that from their mothers’ feces, and breast milk at ASV level. We found that the feces microbiome of the majority of infants (16/25) were more likely to be derived from their mothers’ breast milk than from their mother’s feces (Figure 2). The correlation between the ratio of source proportion of mother’s faces and breast milk and infants’ gender, weight status, and maternal obesity were not significant (p > 0.05; data not shown).
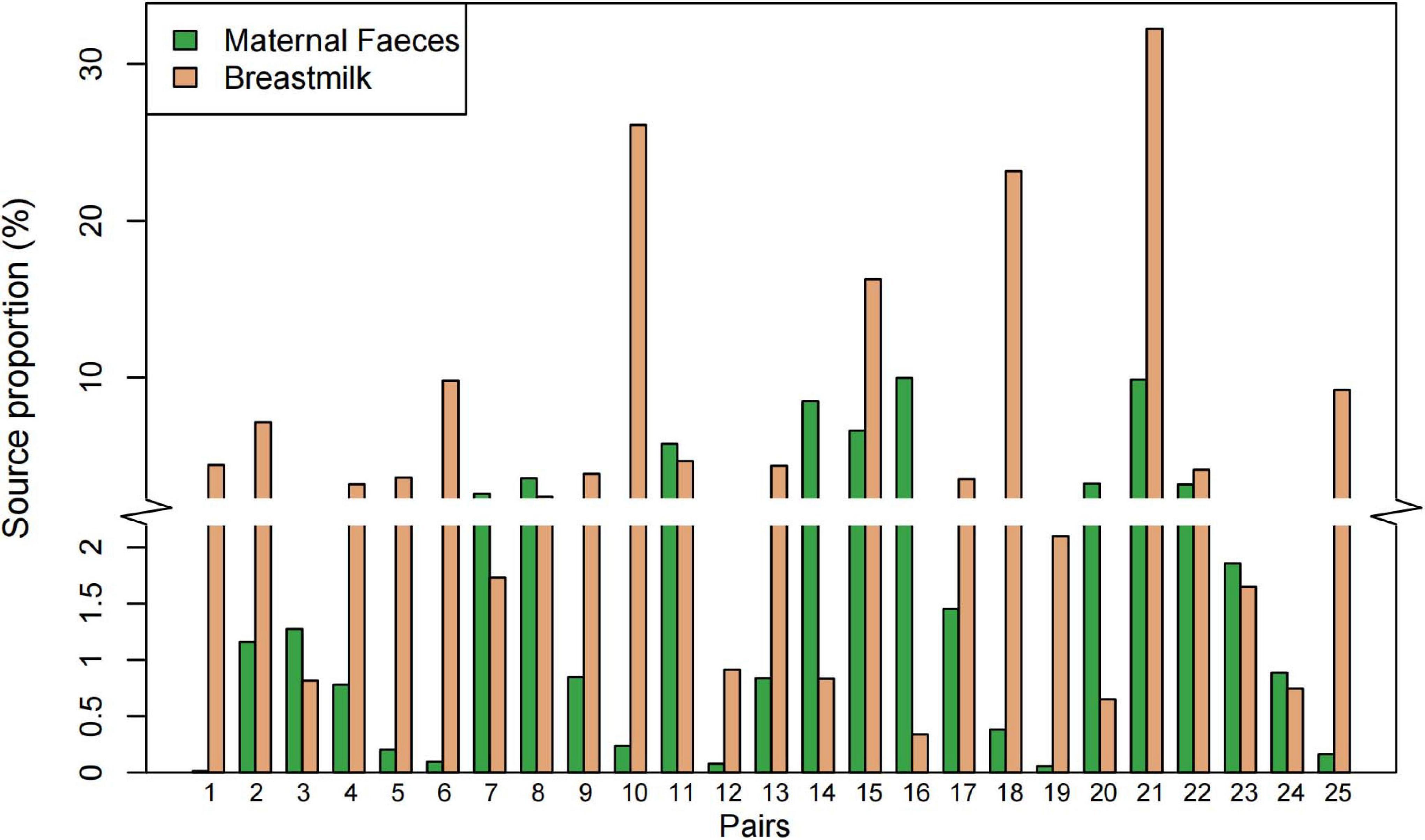
Figure 2. Proportion of the microbial sources at amplicon sequencing variants (ASV) level in infant feces using the FEAST model. The y-axis truncation value is 2.1–2.9.
Intra- and Intergroup Diversity Analysis
A constrained principal coordinate analyses (CPCoA) based on the Bray–Curtis distance showed that the microbiota of breast milk, maternal feces, and infant feces, as expected, clustered separately, and the adonis test confirmed that the reported separation was significant (Figure 3A). The average microbiota profile obtained for breast milk was significantly more diverse (observed ASV index = 204 ± 109; Figure 3B) than both maternal and infant feces (176 ± 81 and 148 ± 118, respectively); interestingly, according to the unweighted UniFrac metric, the variability among breast milk samples was the lowest across all samples (0.30 ± 0.05; Kruskal–Wallis test p < 0.0001; Figure 3C). Meanwhile, the same result is presented in Figure 3D, which considers both phylogeny and relative abundance (α-diversity and β-diversity determined by Rao’s diversity decomposition at the ASVs level) (Rao, 1984). In other words, the breast milk ecosystem of the 25 enrolled mothers was more complex and less heterogeneous among samples (in terms of bacterial species) than the fecal ecosystem, suggesting that the bacteria from other ecosystems, except the maternal gut, also could be transferred to breast milk.
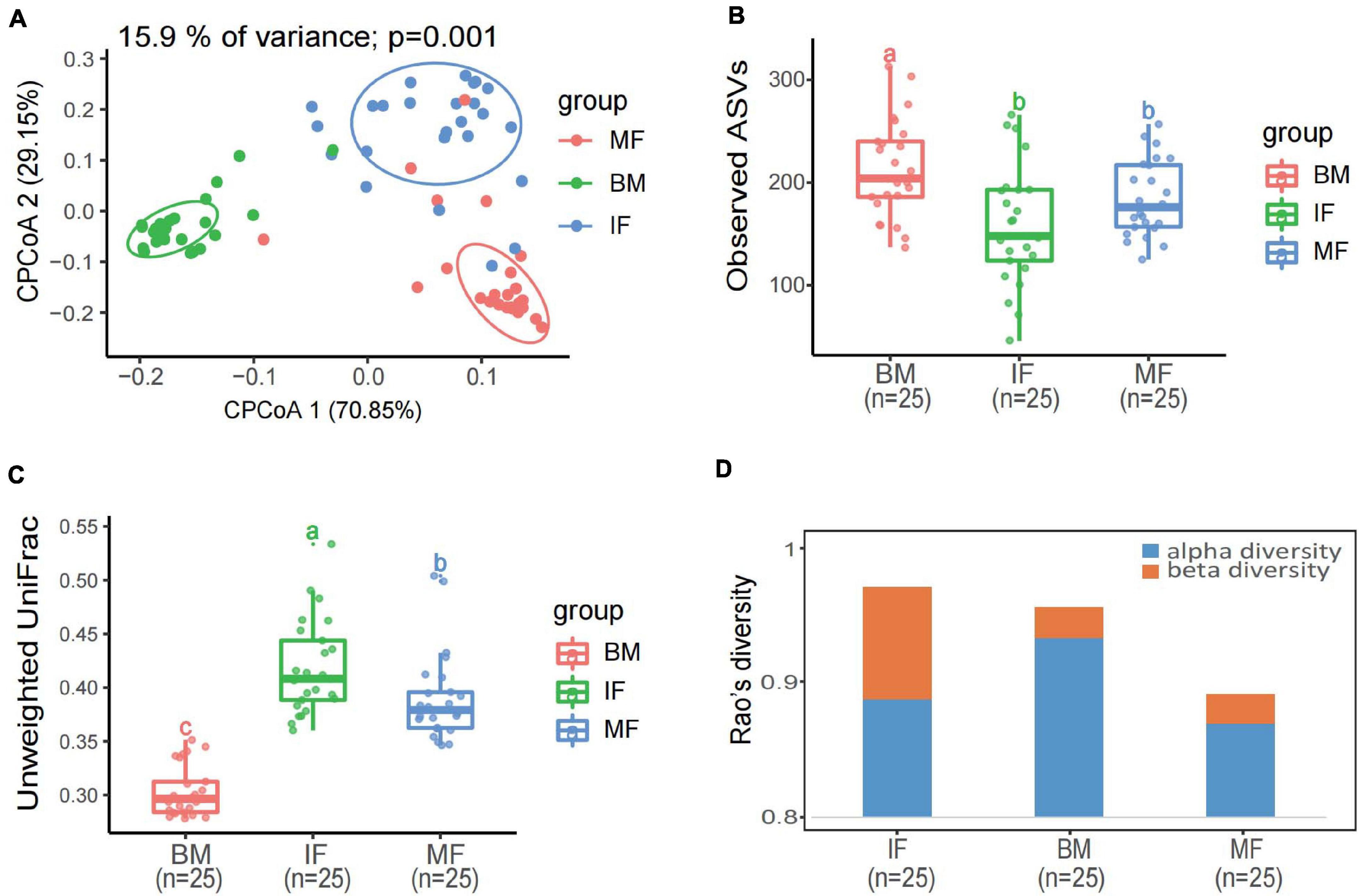
Figure 3. Diversity analysis of bacterial communities in infant feces, maternal feces, and breast milk samples. (A) Constrained principal coordinate analysis plot of Bray–Curtis distances between samples including three groups (15.9% of variance, p = 0.001; n = 75). Ellipses represent a 95% confidence interval around the cluster centroid. IF, infant feces; MF, maternal feces; BM, breast milk. (B) Box plot of the observed ASV index and (C) intragroup unweighted UniFrac distances calculated for maternal feces group (red), breast milk group (green), and infant’s feces group (blue) samples. Statistical analysis was performed using the permutational multivariate analysis of variance. (D) The α-diversity and β-diversity of three groups determined by Rao’s diversity decomposition at the ASVs level, considering both phylogeny and relative abundance.
Bacterial Groups With Statistical Differences
Bifidobaterium Profile Identified by 16S rRNA Gene Sequencing
It may be clear that seeding of early life microbiota with maternal microbes leaves a lasting imprint on the biology of infants. Considering the importance of Bifidobacterium as a health-promoting commensal of the gut microbiome in populations, it is necessary for us to delve deeply into the subdata of bifidobacteria in the whole dataset of mother and child pairs, rather than simply categorizing them into the “other” group in each sample as some reports have done. In the present study, in order to obtain more accurate taxonomic results at species level, the representative sequences of all 11 ASVs (which were identified as members of the Bifidobaterium genus) were extracted to homology search using NCBI BLAST3. Due to the limited resolving power of the 16S rRNA gene in the identification of different bacteria species, only three ASVs were clearly assigned into the Bifidobacterium species level: Bifidobacterium adolescentis, B. pseudolongum, and B. bifidum, respectively. The representative sequences of the remaining eight ASVs corresponded to four species groups (Query Coverage and Percent Identity were both 100%) of Bifidobacterium (each contains multiple closely related species recognized), namely, B. breve/longum (three ASVs), B. pseudocatenulatum/kashiwanohense (three ASVs), B. adolescentis/faecale (one ASV), and B. angulatum/merycium (one ASV). Overall, the average relative abundance of Bifidobacterium species groups varied depending on the types of samples, and their prevalence differed significantly among sample sets of three ecosystems. As shown in Table 2, the most predominant phylotype was the one belonging to the B. breve/longum group, the prevalence of which was 100% in infant feces. As expected, the average rel. ab of the B. adolescentis in maternal feces was higher than that in breast milk and infant feces, but its detection rate was only 72% in maternal feces, with seven maternal feces being negative. The B. pseudocatenulatum/kashiwanohense group was detected in approximately 80% of infant feces. The detection rate of B. bifidum in breast milk and infant feces was higher than in maternal feces. B. pseudolongum was detected in only four breast milk and undetected at any feces samples. Because the sequences that were annotated to match Bifidobacterium accounted for a very low proportion in the 16S rRNA gene datasets of all three types of samples, a limited number of ASVs were found to share among the three ecosystems.
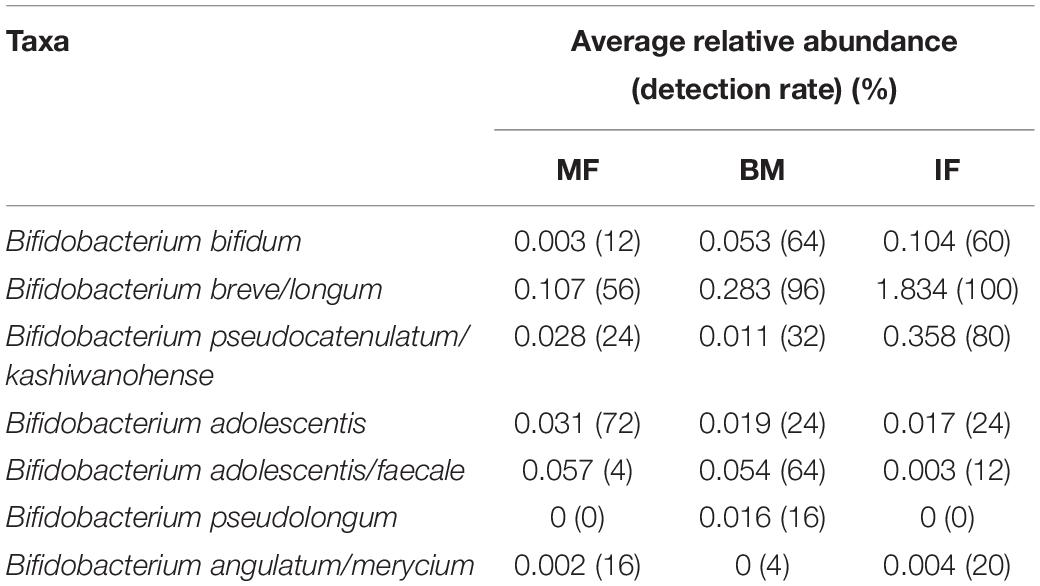
Table 2. The relative abundance and detection rate of bifidobacteria group obtained by the National Center for Biotechnology Information (NCBI) comparison in the three ecosystems.
Bifidobacterium Profile Identified by groEL Gene Sequencing
In groEL gene Illumina sequencing dataset, two samples (a maternal feces sample and an infant feces sample from a different mother–infant pair) with extremely low reads (<800) were removed. A total of 1,702,445 high-quality reads were obtained, and then were binned into ASVs (n = 376) according to the denoising (error correcting). As expected, the large majority of the recovered reads (90%) matched the DNA of the members of the genus Bifidobacterium. In order to accurately assess the association between maternal breast milk and fecal bifidobacterial community, our analysis was performed at the species level as much as possible. Consequently, multiple ASVs that hit the same nearest neighbor were identified as belonging to a specific species and/or subspecies regardless of the small sequence divergence. Taxonomic annotation showed that 376 ASVs were assigned to at least 13 members of Bifidobacterium that contain five well known subspecies, including B. adolescentis (counts of ASVs: 49), B. angulatum (34), B. animalis subsp. animalis (4), B. animalis ssp. lactis (4), B. bifidum (36), B. breve (16), B. kashiwanohense (85), B. longum ssp. infantis (58), B. longum ssp. longum (36), B. pseudocatenulatum (22), B. pseudolongum (8), B. pseudolongum ssp. globosum (6), and B. ruminantium (18). A neighbor-joining phylogenetic tree containing the representative sequences of all ASVs and closely related bifidobacterial taxon was constructed (Supplementary Figure 2).
The bifidobacterial community profiles presented in all samples among three ecosystems are shown in Figure 4, which showed noticeable differences between sample groups in the composition, relative abundance, and diversity. In infant feces samples, the bifidobacterial community structure (composition and abundance) were more similar to that in breast milk samples (Figure 4A). However, the beta diversity analysis exhibited diametrically opposed results. As was shown in the result of hierarchical clustering and PCoA based on the unweighted UniFrac distance (ignored the abundance of all phylotypes in each sample), the bifidobacterial flora in the infant feces is more similar to that in the maternal feces even if there were significant differences between the three ecosystems (p < 0.001) (Figures 4B,C). The result of PCoA based on weighted UniFrac distance indicated that Bifidobacterium microbiome of maternal feces samples was more distinctive compared with that of breast milk and infant feces (Figure 4D).
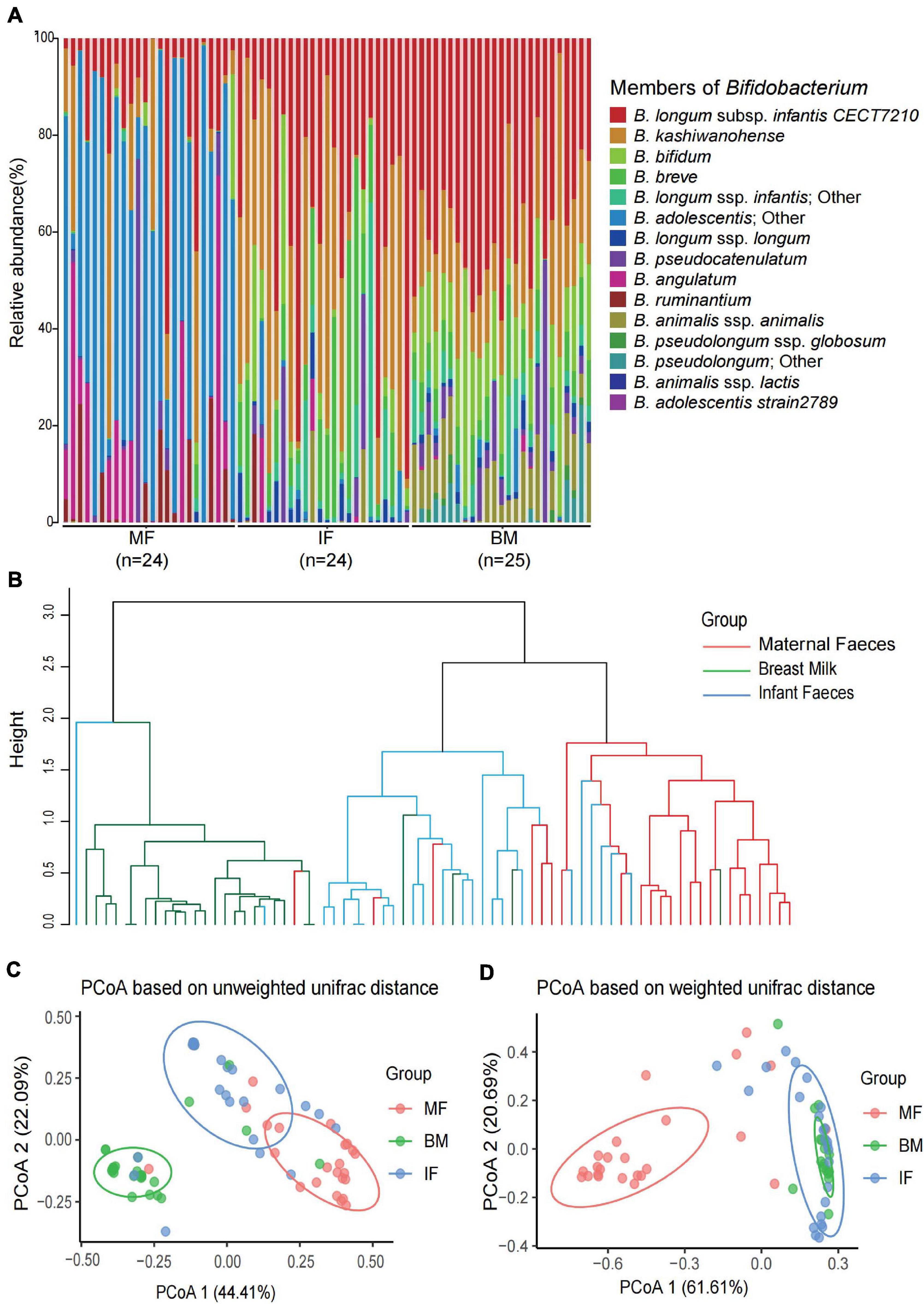
Figure 4. The composition and diversity of Bifidobacterium phylotypes in each infant feces, maternal feces, and breast milk samples. (A) The composition of Bifidobacterium phylotypes in each infant feces, maternal feces, and breast milk samples. (B) The hierarchical clustering based on unweighted UniFrac distance among the three ecosystems samples. Principal coordinate analysis based on unweighted UniFrac distances (C) and weighted UniFrac distances (D) among the three ecosystems. Ellipses represent a 95% confidence interval around the cluster centroid. In both PCoAs, first and second principal components (PCoA1 and PCoA2) were plotted. The significant difference of each two groups in panels (C,D) was evaluated by permutational multivariate analysis of variance. All p-values of each two groups are less than 0.001.
Analysis of proportion abundances and detection rates (presence/absence), based on the whole groEL gene amplicons (Figure 5), shows that the dominant bifidobacteria taxon in breast milk and infant fecal samples were B. longum ssp. infantis (the average rel. ab. Of 43.5 and 39.0% in breast milk and infant feces, respectively, of the groEL gene amplicons of the genus Bifidobacterium) and B. kashiwanohense (14.8% in breast milk and 29.1% in infant feces), followed by B. breve and B. longum ssp. longum. On the contrary, the fecal samples from the mothers were dominated by B. adolescentis and B. angulatum, accounting for 52.0 and 12.6%, respectively, of the groEL gene amplicons of the genus Bifidobacterium. In contrast to maternal feces samples, the average rel. ab. of these two bifidobacterial species were very low in breast milk (1.2 and 0.2%) and infant feces (1.7 and 1.3%). Intriguingly, B. bifidum, considered as one of the infant-type bifidobacteria, was more abundant in the feces samples of mothers than that of infants. Another special bifidobacteria was B. ruminantium with the highest detection rates (100%) and lower relative abundance (0.06%) in maternal feces. However, its mean relative abundance in breast milk (<0.001%) and infant feces (0.8%) was significantly decreased, and no B. ruminantium ASV was detected in quite a few samples, especially in breast milk samples (detection rates in breast milk and infant feces: 30.4 and 70.0%).
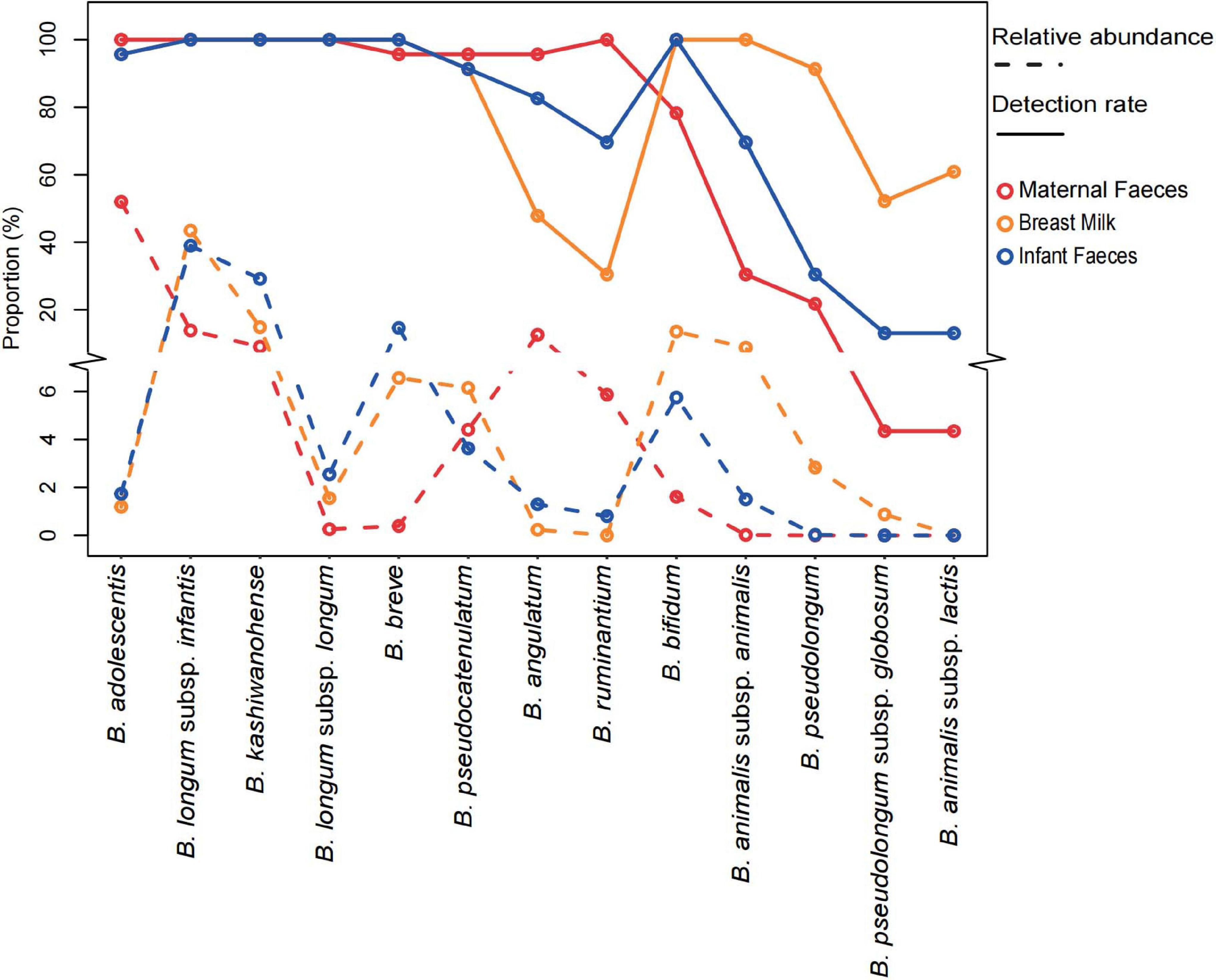
Figure 5. The relative abundance and detection rate of 13 Bifidobacterium species or subspecies in the three systems of 23 mother–infant pairs. The y-axis truncation value is 6.1–8.9.
The Co-occurrence of Bifidobacterium Phylotype in Mother–Breast Milk–Infant Triads Based on groEL Gene
To get a better view of the bifidobacterial co-occurrence and association between mother–infant pairs, the detection rates (presence/absence) and average rel. ab. of each Bifidobacterium phylotype and its corresponding ASVs in breast milk and feces samples were analyzed deeply in mother–breast milk–infant triads (23 pairs with three ecosystem data simultaneously). Of 13 Bifidobacterium phylotypes (species or subspecies), 7 phylotypes, namely, B. adolescentis, B. kashiwanohense, B. longum ssp. infantis, B. bifidum, B. breve, B. longum ssp. longum, and B. pseudocatenulatum, were identified to be universally distributed in more than 90% samples of three ecosystems, with the exception of B. bifidum whose prevalence in the maternal feces was 78%, implying that they are shared nearly by all 23 mother–infant pairs analyzed (Table 3). ASV with an average rel. ab. greater than 1% in any one ecosystem was defined as dominant ASV, and 50 dominant ASVs were obtained. Among these 50 dominant ASVs, the co-occurrence of different ASVs belonging to the same Bifidobacterium phylotype was not exactly the same in infant–milk–mother triads (Figure 6). In these dominant ASVs, 14 ASVs were annotated as B. kashiwanohense, and they could be clustered into three clusters by a phylogenetic tree. Among them, the cluster composed of ASV_26, ASV_43, ASV_71, and ASV_82 had lower detection rates in breast milk samples than in feces samples; on the contrary, the detection rate of ASV_21 in breast milk was higher. It is worth noting that there were nine dominant ASVs annotated as B. adolescentis, but only one ASV has a high co-occurrence rate in infant–milk–mother triads (21/23). Moreover, out of a total of 49 ASVs, assigned to B. adolescentis, 18 ASVs were detected only in maternal feces samples in more than half of mother–infant pairs (more than 11 pairs) (Supplementary Table 2). In contrast, of the eight dominant ASVs annotated as B. longum ssp. infantis, six ASVs were co-occurrence in more than half of the infant–milk–mother triads of mother–infant pairs. Moreover, B. ruminantium and B. angulatum presented in almost all maternal feces samples, but were absent in more than half of the breast milk samples. In 11 mother–infant pairs, B. ruminantium was concurrently detected in the feces samples of infants and their mothers, but was not detected in the corresponding breast milk samples. Interestingly, we found that among the dominant ASVs annotated as B. ruminantium, if they can be detected in the infant feces sample, they can also be detected in feces samples of their mothers. In addition, the detection frequency of B. animalis and B. pseudolongum in breast milk samples (100 and 91.3%) was significantly higher than that in the feces samples of mothers (30.4 and 21.7%) and infants (30.4 and 69.6%). In terms of the same mother–infant pair, the dominant ASV (ASV_11), annotated as B. animalis ssp. Animalis, was concurrently detected in infants’ feces samples and their mothers’ breast milk samples of 16 mother–infant dyads, but present only in the feces samples of the corresponding five mothers.
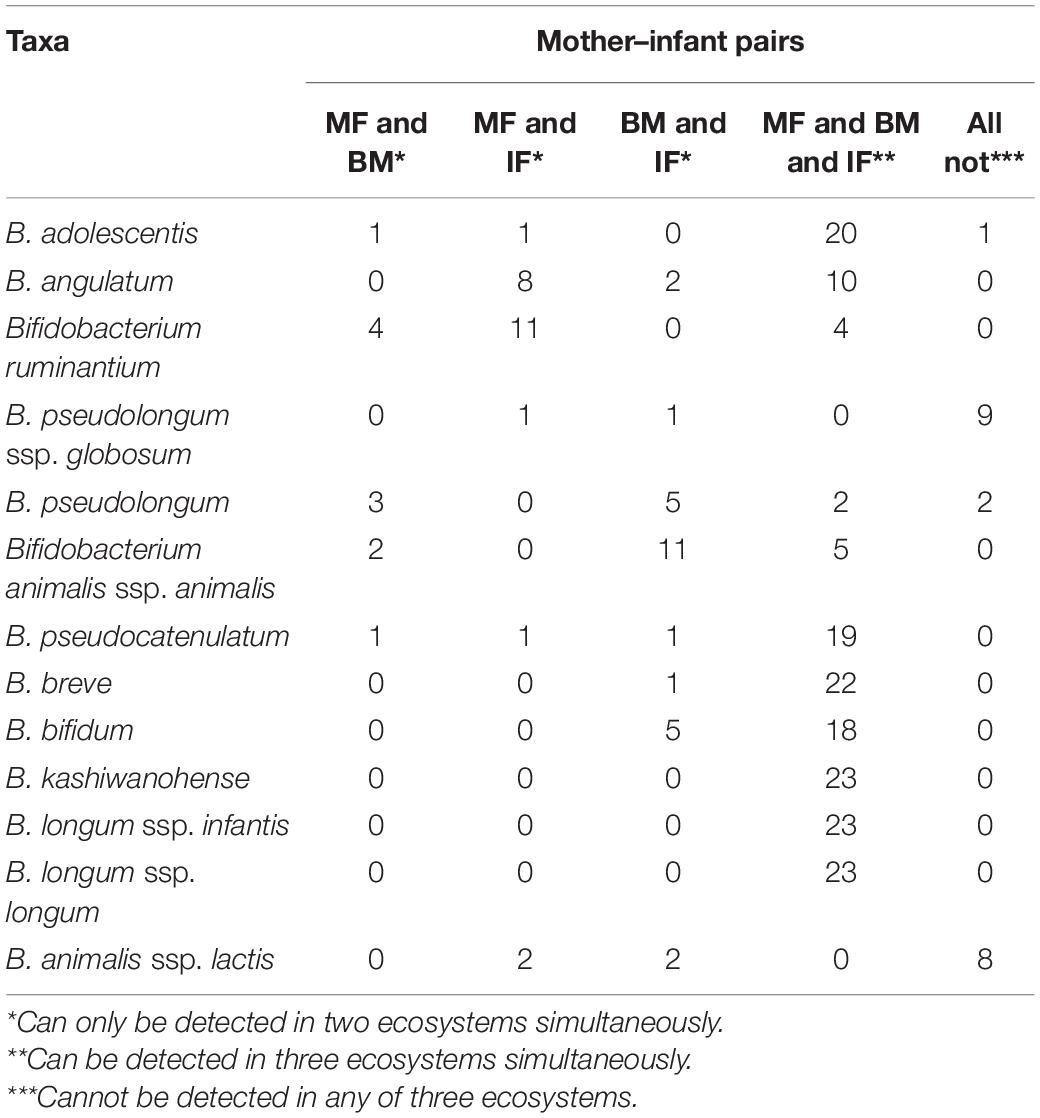
Table 3. The occurrence frequency of 13 Bifidobacterium species or subspecies in samples from 23 mother–infant pairs.
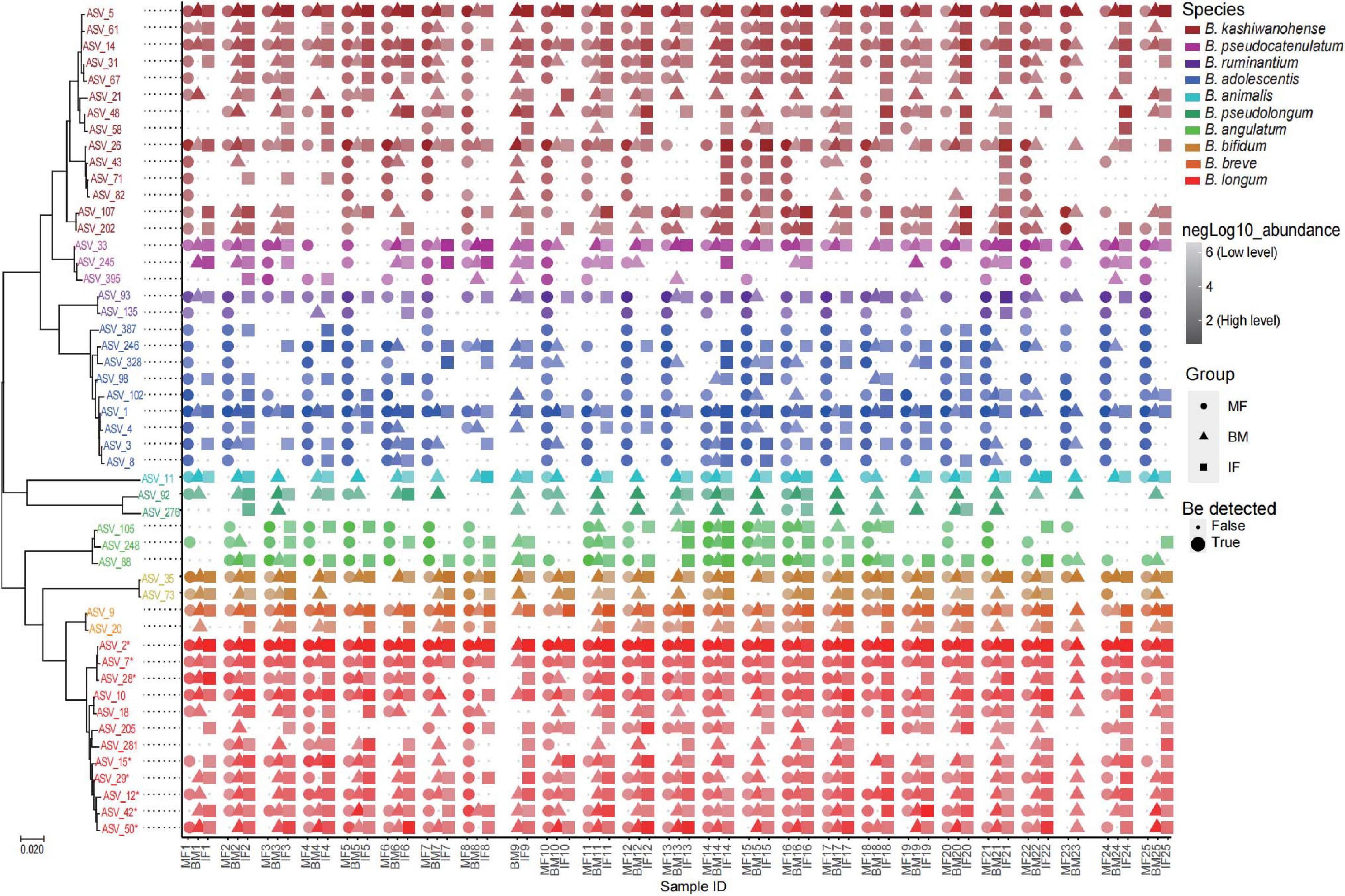
Figure 6. Phylogenetic tree of the 50 dominant ASVs and their co-occurrence in samples from 25 mother–breast milk–infant triads. The color of the text in the tree represents different bifidobacteria species. The color gradient of symbols represents the level of logarithm of relative abundance (the darker the color, the higher the relative abundance), the shapes represent different ecosystems, and the size is the logical value of whether the ASV can be detected in the sample. *B. longum subsp. infantis.
To investigate the concordance of microbial co-occurrence between the three ecosystems, we next assessed the correlations between ASVs, which were classified into eight common Bifidobacterium species (or subspecies) and rare groups. Approximately 87–122 ASVs (nodes) and 219–524 connections (edges) were retained in the co-occurrence networks across all ecosystems (Figure 7). The components and topographies of the networks of the breast milk group were significantly different from those of the feces sample groups, and the most remarkable difference was the connections of adult- to infant-type bifidobacteria. The topographies of the network of the breast milk group (Figure 7A) were relatively simple with a connection index of 0.059, compared with 0.062 and 0.071, respectively, for the infant feces (Figure 7B) and maternal feces groups (Figure 7C). The connections of adult-type bifidobacteria (B. adolescentis and B. angulatum) showed low frequencies of co-occurrence across breast milk samples with other bifidobateria, especially the so-called infant-type bifidobacterial species. For example, the ASVs annotated to B. adolescentis in the network of breast milk samples emerged as a separate cluster and had no co-occurrence with ASVs of other bifidobacteria (Figure 7A). In the network of infant fecal Bifidobacterium microbiota, a cluster with tight positive correlation was formed by typical infant-type bifidobacteria, B. breve, and B. longum ssp. infantis, along with B. longum ssp. longum, while adult-type bifidobacteria (B. adolescentis and B. angulatum) formed another cluster with B. ruminantium and a small number of ASVs annotated to B. kashiwanohense. It is noted especially that these two major clusters showed a significant negative correlation by multiple connections (Figure 7B). In the bifidobacterial co-occurrence network of maternal feces, one obvious characteristic is that there is significant negative correlation between one B. adolescentis ASV with the highest average relative abundance, and multiple ASVs belong to the B. kashiwanohense and B. longum ssp. longum. In addition, the vast majority of adult-type bifidobacterial ASVs (B. adolescentis and B. angulatum) had very few connections with other ASVs, although some B. angulatum ASVs were positively correlated with one B. longum ssp. infantis ASV (Figure 7C).
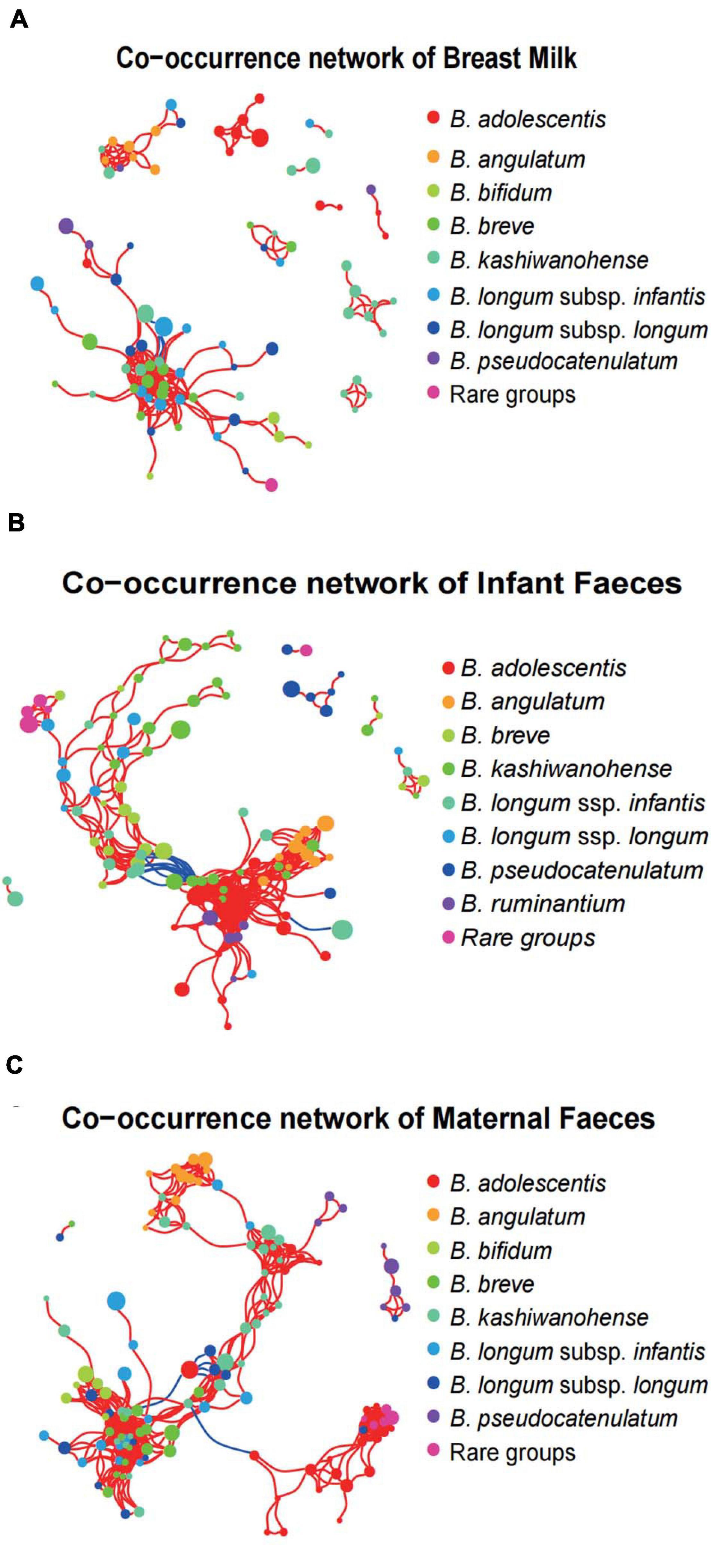
Figure 7. Bifidobacterial co-occurrence networks in the three systems. Co-occurrence networks of Bifidobacterium phylotypes in breast milk (A), infant feces (B), and maternal feces (C). Each node in the network indicates an ASV, and the color represents bifidobacterial phylotypes. Each co-occurring pair among bacterial populations has an absolute Spearman rank correlation above 0.7 [red line: positive correlation (R > 0.70); blue line: negative (blue) correlation (R < –0.70)] with an FDR-corrected significance level under 0.01.
Discussion
There is evidence that breast milk acts as an intermediary for the transfer of functionally important bacteria from mother to infant (Lyons et al., 2020). In particular, the interest in the gut symbiotic members of Lactobacillus and Bifidobacterium that can colonize the infant gut has grown significantly, mainly because their presence has been associated with a healthy microbiota (Arboleya et al., 2016; Ojo-Okunola et al., 2019). Breast milk is an important source of Bifidobacterium for the newborn gut (Oh et al., 2016). Due to the fact that total bacterial loads of breast milk are five to seven orders of magnitude lower than in feces samples (Fernandez et al., 2013), the amount of bifidobacteria in breast milk samples is very low compared with feces samples, especially the feces samples of infants. As a result, most studies based on culture techniques have reported very inconsistent results regarding the number and combination of Bifidobacterium phylotypes in breast milks within a specific population or across cohorts (Martin et al., 2009; Jost et al., 2013; Soto et al., 2014; Milani et al., 2015; Damaceno et al., 2017). However, most studies on vertical transmission of Bifidobacterium strains between the mother infant pair confirmed their co-occurrence among maternal intestine, breast milk, and the corresponding infant’s intestine. Nevertheless, such potential vertical transmission and the co-occurrence of Bifidobacterium is still only partially understood.
In the current study, the microbial composition of the three ecosystems was remarkably different: the abundance of aerobic bacteria (such as Pseudomonadaceae) in breast milk and that of anaerobic bacteria (such as Ruminococcaceae) in the maternal gut was significantly higher than another two ecosystems. This observation could be explained by the different conditions of the resident communities in the three ecosystems for pH, oxygen levels, and nutrients availability as reported also in earlier studies (Chen et al., 2018). However, within the cohort as a whole, Bifidobacterium spp. were detected at low relative abundances (mean relative abundances of 0.23, 0.44, and 2.32%) in the samples of the three ecosystems (maternal feces, breast milk, and neonatal feces). Based on very accurate and painstaking analyses of 16S rRNA gene dataset, our results showed that except for one maternal feces sample where no reads assigned to Bifidobacterium was observed, the presence of 16S rRNA reads assigned to Bifidobacterium could be identified in all breast milk and other feces samples, but its relative abundance was as low as 0.004% in a few breast milk and 0.02% in a few infant feces samples (data not shown). Previous studies showed that total bacteria concentration is lower in colostrum than in transitional and mature milk, with increasing levels of Bifidobacterium in breast milk over lactation time (Cabrera-Rubio et al., 2012; Khodayar-Pardo et al., 2014). In fact, the presence of Bifidobacterium was sporadically reported in a few colostrum samples or not at all (Hunt et al., 2011; Boix-Amoros et al., 2016; Drago et al., 2017; Chen et al., 2018). In our other study, in only about 5.4% of colostrum samples were retrieved 16S rRNA ASVs corresponding to Bifidobacterium, with <0.05% mean relative abundance (data not shown).
According to the published literature, most studies using high-throughput sequencing of 16S rRNA gene amplicon have reported conspicuous intercohort variation regarding the prevalence and relative abundance of Bifidobacterium in microbiome of breast milks and the infant gut, when compared with the stability of the adult gut microbiome (Murphy et al., 2017; Lackey et al., 2019; Moossavi et al., 2019). In the present study, it is curious that the relative abundance of Bifidobaterium (2.32%) in the microbiome of the infant gut was much lower than that in similar studies by other research groups, who reported that bifidobacteria achieved large proportions of the gut microbiota in the first few months after birth, ranging from 10 to 90% (Turroni et al., 2012; Lim et al., 2016; Duranti et al., 2017; Lundgren et al., 2018). The high abundance of bifidobacteria in the gut of breastfeeding infants was explained by the fact that host-derived glycans, like gut mucins and breast milk oligosaccharides in breast milk as specific growth substrates fertilize bifidobacterial growth, especially B. longum subsp. infantis, B. bifidum, and B. breve containing specific gene clusters encoding enzymes that are capable of hydrolyzing HMOs (Aakko et al., 2017; Turroni et al., 2017; Lawson et al., 2020). Thus, further research is needed to determine the reason for such a low relative abundance of Bifidobacterium in the intestinal tract of infants across our cohort, especially the association with breast milk oligosaccharides and diet and lifestyle, and environmental factors. In fact, some previous studies similarly reported that a proportion of infants targeting other populations have very low abundance or undetectable bifidobacteria as members of the fecal microbiota regardless of breast milk or formula feeding (Tannock et al., 2013, 2016). Duranti et al. (2017) reported very large interindividual variations for the relative abundance of Bifidobacterium in infant gut microbiota, with an abundance as low as 0.05%. In adults, bifidobacteria have been reported to commonly make up 1–10% of the gut microbiota (Arboleya et al., 2016). In our cohort, Bifidobacterium spp. were detected at low proportions (0.23%) in maternal feces samples. In fact, it was surprising that the complete absence of bifidobacteria in the Hadza gut microbiota is reported (Schnorr et al., 2014).
Given that determining the bacterial species with partial 16S rRNA sequences has to be taken with care, the representative sequences of all 11 Bifidobaterium ASVs in the 16S rRNA gene dataset were NCBI BLAST homology searched in order to obtain more accurate taxonomic results. Our results showed that they were annotated to seven species or species groups, of which the most predominant phylotype was the B. breve/longum group in breast milk and infant gut, with the prevalence being 100%. Although the average rel. ab of the B. adolescentis in maternal gut was higher than that in breast milk and infant gut, its prevalence was only 72% in the maternal gut, with seven maternal gut samples being negative. By contrast, the relative abundance of Bifidobaterium in the breast milk microbiome is similar to what has been reported in other studies. For example, in a study that targeted HBM samples collected between 6 and 10 weeks postpartum from lactating South African women, the average relative abundances of the genus Bifidobacterium was about 1.0%, yet with 28% of breast milk samples being free of bifidobacteria (Ojo-Okunola et al., 2019). In another study (Moossavi et al., 2019), NGS data based on 16S rRNA gene sequencing of V4 hypervariable region revealed that only 39% of breast milk samples contained Bifidobacterium, with mean relative abundance of 0.25 ± 0.98%. A recent study (Kumar et al., 2016) investigated the influence of geographical location on breast mature milk microbiota of healthy mothers, and the genus Bifidobacterium was found only in samples from South African women and not from Finland, Spain, and Beijing, China. Furthermore, in a study of 145 American women at approximately 6 weeks postpartum, mean relative abundances of the genus Bifidobacterium was about 0.65%. However, the relative abundances of Bifidobacterium were significantly different between breast milk microbiome types (Lundgren et al., 2018). In addition, a previous study in Taiwan and mainland China similarly reported that B. longum was the predominant bifidobacterial species with mean relative abundance of 0.3%, but its prevalence was only 62.4% (Li et al., 2017). Some studies have not even reported the presence and relative abundance of Bifidobacterium in breast milk during the first months postpartum (Browne et al., 2019), while other studies reported that Bifidobacterium spp. were detected at low proportions in human breast milk samples, but their prevalence was not described (Boix-Amoros et al., 2016; Chen et al., 2018; Meehan et al., 2018).
As described in all studies mentioned above, most studies on breast milk microbiome using the NGS of specific 16S variable gene regions reported only their results at the taxonomic level of genus. Particularly, the pitfalls inherent to 16S subregions, such as V3–V5, including the limited discriminating power among sequences belonging to the phylum Actinobacteria and the underrepresentation of rare OTUs with low abundance, may lead to skewed estimates of bifidobacterial community (Ellegaard and Engel, 2016; Johnson et al., 2019). Consequently, the choice of PCR primers can lead to underrepresentation of bifidobacterial community in 16S rRNA sequencing dataset, concealing the number and diversity of species- and strain-level sequence variants (Sim et al., 2012; Alcon-Giner et al., 2017).
In addition to the above reasons, another possible explanation for the underrepresentation of Bifidobacterium, which is in the downstream data analysis methods, some rare bacterial genus with very low abundance, including Bifidobacterium, which might be categorized into the “other” group in each sample, cannot be ruled out (Meehan et al., 2018). Particularly at the ASV level, taxa exhibiting such a low relative abundance (reads less than 10) are often dismissed in data analysis of high throughput for microbial community and diversity analysis, thereby leading to inconsistent results regarding the occurrence of bifidobacteria in mother–infant pairs (Chen et al., 2018). All of the above situations may be the reasons why there are always portions of the same set of samples from each of multiple independent cohort studies showing Bifidobacterium negative, despite using the same method.
A very interesting phenomenon was that the publications that investigated the bacteria vertical transfer from mother to infant have not reported the vertical transfer of B. longum subsp. infantis to date. The most likely explanation of this finding is that, in contrast to B. longum subsp. longum, which can be detected in the intestinal tracts of both adults and infants, the B. longum subsp. infantis, as a typical infant-type bifidobacterial phylotype is difficult to capture because it is a very low abundance taxon in the adult gut, even using metagenomic methods (Ward et al., 2013; Zhang et al., 2015; Vatanen et al., 2019). For example, only 10% of Finnish infants harbored Bifidobacterium longum subsp. infantis, whereas Russian infants commonly maintained a probiotic Bifidobacterium bifidum strain in infancy (Vatanen et al., 2019). More recently, the groEL gene proved to be a very effective target for the identification and quantification of Bifidobacterium spp. through high-throughput sequencing technologies or qPCR (Junick and Blaut, 2012; Hu et al., 2017). In our study, according to the denoising, taxonomic annotation showed that 376 Bifidobaterium ASVs in the groEL gene dataset were assigned to at least 13 well-known Bifidobacterium species or subspecies. Of them, seven phylotypes, including B. longum subsp. infantis, were identified to be universally distributed in all 23 mother–infant pairs analyzed. More remarkably, the so-called infant-type Bifidobacterium phylotype, such as B. bifidum, B. breve, and B. longum subsp. Infantis, was found to be present in the gut of all mothers, while B. adolescentis, a typical adult-type bifidobacterial phylotype, was almost detected in all infant feces and breast milk samples. Therefore, in terms of the occurrence and ecological distribution of human-associated bifidobacterial species, there should not be a very strict infant vs. adult subdivision. Our test results were obviously to support the hypothesis that breast milk as a seeding mediator inoculated the infant gut microbiome, regardless of the infant-type Bifidobacterium phylotype and adult-type bifidobacterial phylotype (Turroni et al., 2017). Of interest, Bifidobaterium ASV data obtained by the groEL gene sequences revealed relatively high intrasubject variability in B. longum, including B. longum ssp. infantis (58 ASVs) and B. longum ssp. longum (36 ASVs), compared with other common Bifidobacterium species, B. bifidum (36 ASVs), or B. breve (16 ASVs), which was consistent with the results reported in a recent study on strain-specific functional adaptation in the human gut microbiome during early life (Vatanen et al., 2019). Notably, comparison of the groEL gene-based ASVs identified in the data sets indicated the presence of identical ASVs in different sample pairs, implying that these identical sequences correspond to very closely related strains that presented in non-corresponding mother–infant dyads. Different ASVs of B. kashiwanohense showed different co-occurrence relationships in breast milk and feces samples, which may be caused by the bias of different strains to different ecosystems (Vatanen et al., 2019). Despite this genetic similarity, it can be determined that there may be great phenotypic variation between strains of the same species (homotypic strains) (Van Rossum et al., 2020). Further studies are needed to prove whether the phenotypic variation of strains in the mother-to-infant transmission process will be changed due to the ecosystem.
Moreover, there were very low relative proportion abundances of other six bifidobacterial taxa, whose prevalence (presence/absence) displayed both interindividual and sample set variations. According to the definition of microbial ecology, these Bifidobacterium phylotypes should belong to the rare taxa. However, thus far, little is known about the occurrence and the ecological relevance of rare Bifidobacterium phylotypes in human body environments. For example, in our cohort, rare Bifidobacterium phylotypes presented in the majority of maternal and infant gut, such as B. ruminantium and B. angulatum, with the highest detection rate (100%) and lower relative abundance (0.02%) in maternal gut, were not detected in quite a few breast milk samples. On the contrary, rare phylotypes like B. animalis and B. pseudolongum, showing almost 100% the detection frequency in breast milk samples, were absent in quite a few guts across all mother–infant pairs, especially in the mother gut. Among them, B. animalis ssp. animalis was concurrently detected in infants’ gut and their mothers’ breast milk of 16 mother–infant dyads, but presented only in the gut of the corresponding five mothers. In fact, B. animalis subsp. lactis/animalis and B. pseudolongum, which are frequently found in various animals, are scarcely encountered in the human intestinal tract, suggesting that they are not shared between mothers and their respective infant (Delcenserie et al., 2011; Lugli et al., 2019). Possibly, the probiotic endosymbiotic Bifidobacterium species are transmitted from mother to infant by direct vertical transmission during early life in most cases. However, another possibility that some oxygen-insensitive Bifidobacterium phylotypes with a cosmopolitan lifestyle (B. animalis and B. adolescentis) are indirectly transferred to breast milk and the infant’s intestinal tract through environmental contamination cannot be ruled out (Bondue and Delcenserie, 2015). Coincidentally, a study from an Argentine population described B. animalis subsp. lactis strains that originated from 16 breast milk samples (Zacarias et al., 2011). Besides, analogous to our result, a recent study also reported the presence of B. pseudolongum in the breast milk (Chen et al., 2018). Furthermore, our investigations are under way to clarify the importance and generality of the latter transmission route.
It is already known that there is a mutualistic cross-feeding or resource-sharing phenomenon in the bifidobacteria community of breastfed infant gut (Turroni et al., 2017). In the present study, we employed network-based analyses to elucidate the association and co-occurrence between Bifidobacterium phylotypes present in each of the three ecosystems of mother–breast milk–infant triads at the species or strain level (ASV). Strikingly, as shown in the network of infant gut, the major cluster where typical infant-type bifidobacterial ASVs intertwined with each other showed a significant negative correlation with a major cluster structured by adult-type bifidobacteria ASVs through multiple connections, indicating the presence of co-exclusion association. This result seems not to be in accordance with the reports by Turroni and cooperator (Turroni et al., 2016), who found that a strain of B. adolescentis exhibited mutalisitic cross-feeding behaviors when cocultured with B. bifidum, B. breve, and B. longum subsp. infantis strain, respectively. However, in the network of breast milk, the ASVs of different bifidobacterial phylotypes, including adult types, emerged as a separate cluster and displayed weak correlation with each other, suggesting that such ecological relationships might represent the random associations of bifidobacterial species in the poorly competitive ecosystem of breast milk. At present, our investigations are under way to clarify the association between maternal breast milk and infant fecal bifidobacterial profiles at the different stages of lactation, to pay close attention to the combination and longitudinal changes of Bifidobacterium phylotypes (species or strain level) between different ethnic groups during early life.
Conclusion
By analyzing bacterial 16S rRNA gene datasets from the maternal stool, breast milk, and infant stool in a small yet very homogeneous cohort of 25 healthy Uyghur mother–infant pairs in Kashgar, Xinjiang, China, only three sets of ASVs could be clearly assigned into B. adolescentis, B. pseudolongum, and B. bifidum, respectively, whereas the remaining eight sets of ASVs corresponded to four Bifidobacterium species groups containing more than two closely related species. More remarkably, the groEL gene proved to be a very effective mark gene for the depth resolution of Bifidobacterium community by high-throughput sequencing technology, allowing all Bifidobaterium ASVs to be assigned to at least 13 well-known Bifidobacterium species or subspecies. Among them, seven well-known Bifidobacterium phylotypes showed synchronism in 23 mother–infant pairs. However, several other rare bifidobacterial phylotypes, which were frequently encountered in animals, were found to display no correspondence of the presence between the three ecosystems of mother–infant pairs. Consequently, our test results were obviously to support the hypothesis that breast milk acts as an intermediary for the transfer of functionally important commensal bacteria from mother to infant, especially for endosymbiotic Bifidobacterium that can colonize the infant gut. In contrast, some oxygen-insensitive exogenous Bifidobacterium phylotypes with a cosmopolitan lifestyle may be indirectly transferred to breast milk and the infant’s intestinal tract through environmental contamination. Furthermore, the so-called infant-type Bifidobacterium phylotype, was found to be present in the gut of all mothers, while adult-type bifidobacterial phylotypes were almost detected in all infant gut and breast milk samples, indicating that there should not be a very strict infant vs. adult subdivision in terms of the occurrence and ecological distribution of human-associated bifidobacterial species.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: www.ncbi.nlm.nih.gov/, BioProject ID PRJNA659245 and PRJNA702483.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of the First Affiliated Hospital, Shihezi University School of Medicine. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
YN and FT conceptualized the study and acquired the funding. WY and XZ conducted the experimental investigation. WY was in charge of the bioinformatics, statistics, figures, and wrote the original draft. YN wrote, reviewed, and edited the manuscript. BL collected the samples. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the joint key funds of the National Natural Science Foundation of China and the Autonomous Region Government of Xinjiang, China (grant no. U1903205).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.669442/full#supplementary-material
Supplementary Figure 1 | Differentially abundant bacterial taxa between maternal feces (MF), breast milk (BM) and infant feces (IF) samples. (A) LEfSe comparison of microbiota in feces and breast milk samples. The genera listed in green describe breast milk, genera listed in blue describe infant feces, and genera listed in red describe maternal feces samples. Significant bacterial genera were determined by Kruskal–Wallis test (P < 0.05) with LDA score greater than 3. (B) Cladogram representation of differentially abundant bacterial taxonomic group detected using LEfSe. Different colors indicate the group in which clade was most abundant.
Supplementary Figure 2 | Phylogenetic tree constructed based on the bifidobacterial ASV sequences. Outer circle color represents the phylogenetic strains.
Footnotes
References
Aakko, J., Kumar, H., Rautava, S., Wise, A., Autran, C., Bode, L., et al. (2017). Human milk oligosaccharide categories define the microbiota composition in human colostrum. Benef. Microbes 8, 563–567. doi: 10.3920/bm2016.0185
Alcon-Giner, C., Caim, S., Mitra, S., Ketskemety, J., Wegmann, U., Wain, J., et al. (2017). Optimisation of 16S rRNA gut microbiota profiling of extremely low birth weight infants. BMC Genomics 18:841. doi: 10.1186/s12864-017-4229-x
Arboleya, S., Watkins, C., Stanton, C., and Ross, R. P. (2016). Gut bifidobacteria populations in human health and aging. Front. Microbiol. 7:1204. doi: 10.3389/fmicb.2016.01204
Ballard, O., and Morrow, A. L. (2013). Human milk composition nutrients and bioactive factors. Pediatr. Clin. N. Am. 60, 49–74. doi: 10.1016/j.pcl.2012.10.002
Benson, D. A., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J., and Sayers, E. W. (2011). GenBank. Nucleic Acids Res. 39, D32–D37. doi: 10.1093/nar/gkq1079
Bode, L. (2012). Human milk oligosaccharides: every baby needs a sugar mama. Glycobiology 22, 1147–1162. doi: 10.1093/glycob/cws074
Bode, L., Mcguire, M., Rodriguez, J. M., Geddes, D. T., Hassiotou, F., Hartmann, P. E., et al. (2014). It’s alive: microbes and cells in human milk and their potential benefits to mother and infant. Adv. Nutr. 5, 381–396. doi: 10.3945/an.114.006643
Boix-Amoros, A., Collado, M. C., and Mira, A. (2016). Relationship between milk microbiota, bacterial load, macronutrients, and human cells during lactation. Front. Microbiol. 7:492. doi: 10.5389/fmicb.7016.00492
Bondue, P., and Delcenserie, V. (2015). Genome of bifidobacteria and carbohydrate metabolism. Korean J. Food Sci. An. 35, 1–9. doi: 10.5851/kosfa.2015.35.1.1
Browne, P. D., Aparicio, M., Alba, C., Hechler, C., Beijers, R., Miguel Rodriguez, J., et al. (2019). Human milk microbiome and maternal postnatal psychosocial distress. Front. Microbiol. 10:2333. doi: 10.3389/fmicb.2019.02333
Cabrera-Rubio, R., Carmen Collado, M., Laitinen, K., Salminen, S., Isolauri, E., and Mira, A. (2012). The human milk microbiome changes over lactation and is shaped by maternal weight and mode of delivery. Am. J. Clin. Nutr. 96, 544–551. doi: 10.3945/ajcn.112.037382
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chen, P.-W., Lin, Y.-L., and Huang, M.-S. (2018). Profiles of commensal and opportunistic bacteria in human milk from healthy donors in Taiwan. J. Food Drug Anal. 26, 1235–1244. doi: 10.1016/j.jfda.2018.03.004
Collado, M. C., Delgado, S., Maldonado, A., and Rodriguez, J. M. (2009). Assessment of the bacterial diversity of breast milk of healthy women by quantitative real-time PCR. Lett. Appl. Microbiol. 48, 523–528. doi: 10.1111/j.1472-765X.2009.02567.x
Damaceno, Q. S., Souza, J. P., Nicoli, J. R., Paula, R. L., Assis, G. B., Figueiredo, H. C., et al. (2017). Evaluation of potential probiotics isolated from human milk and colostrum. Probiotics Antimicro. 9, 371–379. doi: 10.1007/s12602-017-9270-9271
Delcenserie, V., Gavini, F., China, B., and Daube, G. (2011). Bifidobacterium pseudolongum are efficient indicators of animal fecal contamination in raw milk cheese industry. BMC Microbiol. 11:178. doi: 10.1186/1471-2180-11-178
Drago, L., Toscano, M., De Grandi, R., Grossi, E., Padovani, E. M., and Peroni, D. G. (2017). Microbiota network and mathematic microbe mutualism in colostrum and mature milk collected in two different geographic areas: Italy versus Burundi. ISME J. 11, 875–884. doi: 10.1038/ismej.2016.183
Duranti, S., Lugli, G. A., Mancabelli, L., Armanini, F., Turroni, F., James, K., et al. (2017). Maternal inheritance of bifidobacterial communities and bifidophages in infants through vertical transmission. Microbiome 5:66. doi: 10.1186/s40168-017-0282-286
Dzidic, M., Abrahamsson, T. R., Artacho, A., Collado, M. C., Mira, A., and Jenmalm, M. C. (2018). Oral microbiota maturation during the first 7 years of life in relation to allergy development. Allergy 73, 2000–2011. doi: 10.1111/all.13449
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edgar, R. C. (2017). UNBIAS: an attempt to correct abundance bias in 16S sequencing, with limited success. bioRxiv [preprint] doi: 10.1101/124149
Ellegaard, K. M., and Engel, P. (2016). Beyond 16S rRNA community profiling: intra-species diversity in the gut microbiota. Front. Microbiol. 7:1475. doi: 10.3389/fmicb.2016.01475
Fernandez, L., Langa, S., Martin, V., Maldonado, A., Jimenez, E., Martin, R., et al. (2013). The human milk microbiota: origin and potential roles in health and disease. Pharmacol. Res. 69, 1–10. doi: 10.1016/j.phrs.2012.09.001
Ferretti, P., Pasolli, E., Tett, A., Asnicar, F., Gorfer, V., Fedi, S., et al. (2018). Mother-to-infant microbial transmission from different body sites shapes the developing infant gut microbiome. Cell Host Microbe 24, 133–145. doi: 10.1016/j.chom.2018.06.005
Fitzstevens, J. L., Smith, K. C., Hagadorn, J. I., Caimano, M. J., Matson, A. P., and Brownell, E. A. (2017). Systematic review of the human milk microbiota. Nutr. Clin. Pract. 32, 354–364. doi: 10.1177/0884533616670150
Groenlund, M. M., Gueimonde, M., Laitinen, K., Kociubinski, G., Groenroos, T., Salminen, S., et al. (2007). Maternal breast-milk and intestinal bifidobacteria guide the compositional development of the Bifidobacterium microbiota in infants at risk of allergic disease. Clin. Exp. Allergy 37, 1764–1772. doi: 10.1111/j.1365-2222.2007.02849.x
Heikkila, M. P., and Saris, P. E. J. (2003). Inhibition of Staphylococcus aureus by the commensal bacteria of human milk. J. Appl. Microbiol. 95, 471–478. doi: 10.1046/j.1365-2672.2003.02002.x
Hill, J. E., Penny, S. L., Crowell, K. G., Goh, S. H., and Hemmingsen, S. M. (2004). cpnDB: a chaperonin sequence database. Genome Res. 14, 1669–1675. doi: 10.1101/gr.2649204
Hu, L. J., Lu, W. W., Wang, L. L., Pan, M. L., Zhang, H., Zhao, J. X., et al. (2017). Assessment of bifidobacterium species using groEL gene on the basis of illumina MiSeq high-throughput sequencing. Genes 8:336. doi: 10.3390/genes8110336
Hunt, K. M., Foster, J. A., Forney, L. J., Schuette, U. M. E., Beck, D. L., Abdo, Z., et al. (2011). Characterization of the diversity and temporal stability of bacterial communities in human milk. PLoS One 6:e21313. doi: 10.1371/journal.pone.0021313
Johnson, J. S., Spakowicz, D. J., Hong, B. Y., Petersen, L. M., Demkowicz, P., Chen, L., et al. (2019). Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 10:5029. doi: 10.1038/s41467-019-13036-13031
Jost, T., Lacroix, C., Braegger, C., and Chassard, C. (2013). Assessment of bacterial diversity in breast milk using culture-dependent and culture-independent approaches. Br. J. Nutr. 110, 1253–1262. doi: 10.1017/s0007114513000597
Junick, J., and Blaut, M. (2012). Quantification of human fecal bifidobacterium species by use of quantitative real-time PCR analysis targeting the groEL gene. Appl. Environ. Microbiol. 78, 2613–2622. doi: 10.1128/aem.07749-7711
Khodayar-Pardo, P., Mira-Pascual, L., Collado, M. C., and Martinez-Costa, C. (2014). Impact of lactation stage, gestational age and mode of delivery on breast milk microbiota. J. Perinatol. 34, 599–605. doi: 10.1038/jp.2014.47
Koenig, J. E., Spor, A., Scalfone, N., Fricker, A. D., Stombaugh, J., Knight, R., et al. (2011). Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl. Acad. Sci. U S A. 108, 4578–4585. doi: 10.1073/pnas.1000081107
Kumar, H., Du Toit, E., Kulkarni, A., Aakko, J., Linderborg, K. M., Zhang, Y., et al. (2016). Distinct patterns in human milk microbiota and fatty acid profiles across specific geographic locations. Front. Microbiol. 7:1619. doi: 10.3389/fmicb.2016.01619
Lackey, K. A., Williams, J. E., Meehan, C. L., Zachek, J. A., Benda, E. D., Price, W. J., et al. (2019). What’s normal? microbiomes in human milk and infant feces are related to each other but vary geographically: the inspire study. Front. Nutr. 6:45. doi: 10.3389/fnut.2019.00045
Lawson, M. A. E., O’neill, I. J., Kujawska, M., Javavdi, S. G., Wijeyesekera, A., Flegg, Z., et al. (2020). Breast milk-derived human milk oligosaccharides promote Bifidobacterium interactions within a single ecosystem. ISME J. 14, 635–648. doi: 10.1038/s41396-019-0553-552
Li, S.-W., Watanabe, K., Hsu, C.-C., Chao, S.-H., Yang, Z.-H., Lin, Y.-J., et al. (2017). Bacterial composition and diversity in breast milk samples from mothers living in Taiwan and Mainland China. Front. Microbiol. 8:965. doi: 10.3389/fmicb.2017.00965
Lim, E. S., Wang, D., and Holtz, L. R. (2016). The bacterial microbiome and virome milestones of infant development. Trends Microbiol. 24, 801–810. doi: 10.1016/j.tim.2016.06.001
Lugli, G. A., Duranti, S., Albert, K., Mancabelli, L., Napoli, S., Viappiani, A., et al. (2019). Unveiling genomic diversity among members of the species Bifidobacterium pseudolongum, a widely distributed gut commensal of the animal kingdom. Appl. Environ. Microbiol. 85:e03065. doi: 10.1128/aem.03065-3018
Lundgren, S. N., Madan, J. C., Emond, J. A., Morrison, H. G., Christensen, B. C., Karagas, M. R., et al. (2018). Maternal diet during pregnancy is related with the infant stool microbiome in a delivery mode-dependent manner. Microbiome 6:109. doi: 10.1186/s40168-018-0490-498
Lyons, K. E., Ryan, C. A., Dempsey, E. M., Ross, R. P., and Stanton, C. (2020). Breast milk, a source of beneficial microbes and associated benefits for infant health. Nutrients 12:1039. doi: 10.3390/nu12041039
Makino, H., Kushiro, A., Ishikawa, E., Kubota, H., Gawad, A., Sakai, T., et al. (2013). Mother-to-infant transmission of intestinal bifidobacterial strains has an impact on the early development of vaginally delivered infant’s microbiota. PLoS One 8:e78831. doi: 10.1371/journal.pone.0078331
Makino, H., Kushiro, A., Ishikawa, E., Muylaert, D., Kubota, H., Sakai, T., et al. (2011). Transmission of intestinal Bifidobacterium longum subsp longum strains from mother to infant, determined by multilocus sequencing typing and amplified fragment length polymorphism. Appl. Environ. Microbiol. 77, 6788–6793. doi: 10.1128/aem.05346-5311
Martin, R., Heilig, G. H. J., Zoetendal, E. G., Smidt, H., and Rodriguez, J. M. (2007). Diversity of the Lactobacillus group in breast milk and vagina of healthy women and potential role in the colonization of the infant gut. J. Appl. Microbiol. 103, 2638–2644. doi: 10.1111/j.1365-2672.2007.03497.x
Martin, R., Jimenez, E., Heilig, H., Fernandez, L., Marin, M. L., Zoetendal, E. G., et al. (2009). Isolation of bifidobacteria from breast milk and assessment of the bifidobacterial population by PCR-Denaturing gradient gel electrophoresis and quantitative real-time PCR. Appl. Environ. Microbiol. 75, 965–969. doi: 10.1128/aem.02063-2068
McGuire, M. K., and McGuire, M. A. (2017). Got bacteria? The astounding, yet not-so-surprising, microbiome of human milk. Curr. Opin. Biotech. 44, 63–68. doi: 10.1016/j.copbio.2016.11.013
Meehan, C. L., Lackey, K. A., Hagen, E. H., Williams, J. E., Roulette, J., Helfrecht, C., et al. (2018). Social networks, cooperative breeding, and the human milk microbiome. Am. J. Hum. Biol. 30:e23131. doi: 10.1002/ajhb.23131
Milani, C., Mancabelli, L., Lugli, G. A., Duranti, S., Turroni, F., Ferrario, C., et al. (2015). Exploring vertical transmission of bifidobacteria from mother to child. Appl. Environ. Microbiol. 81, 7078–7087. doi: 10.1128/aem.02037-2015
Moossavi, S., Sepehri, S., Robertson, B., Bode, L., Goruk, S., Field, C. J., et al. (2019). Composition and variation of the human milk microbiota are influenced by maternal and early-life factors. Cell Host Microbe 25, 324–335. doi: 10.1016/j.chom.2019.01.011
Mueller, N. T., Bakacs, E., Combellick, J., Grigoryan, Z., and Dominguez-Bello, M. G. (2015). The infant microbiome development: mom matters. Trends Mol. Med. 21, 109–117. doi: 10.1016/j.molmed.2014.12.002
Murphy, K., Curley, D., O’callaghan, T. F., O’shea, C.-A., Dempsey, E. M., O’toole, P. W., et al. (2017). The composition of human milk and infant faecal microbiota over the first three months of life: a pilot study. Sci. Rep. 7:40597. doi: 10.1038/srep40597
Nyangahu, D. D., and Jaspan, H. B. (2019). Influence of maternal microbiota during pregnancy on infant immunity. Clin. Exp. Immunol. 198, 47–56. doi: 10.1111/cei.13331
Oh, J., Byrd, A. L., Park, M., Program, N. C. S., Kong, H. H., et al. (2016). Temporal stability of the human skin microbiome. Cell 165, 854–866. doi: 10.1016/j.cell.2016.04.008
Ojo-Okunola, A., Claassen-Weitz, S., Mwaikono, K. S., Gardner-Lubbe, S., Stein, D. J., Zar, H. J., et al. (2019). Influence of socio-economic and psychosocial profiles on the human breast milk bacteriome of South African women. Nutrients 11:1390. doi: 10.3390/nu11061390
Perez, P. F., Dore, J., Leclerc, M., Levenez, F., Benyacoub, J., Serrant, P., et al. (2007). Bacterial imprinting of the neonatal immune system: lessons from maternal cells? Pediatrics 119, e724–e732. doi: 10.1542/peds.2006-1649
Rao, C. R. (1984). Convexity Properties of Entropy Functions and Analysis of Diversity. Michigan, MICH: Institute of Mathematical Statistics.
Sakwinska, O., Moine, D., Delley, M., Combremont, S., Rezzonico, E., Descombes, P., et al. (2016). Microbiota in breast milk of Chinese lactating mothers. PLoS One 11:e0160856. doi: 10.1371/journal.pone.0160856
Schloss, P. D., and Westcott, S. L. (2011). Assessing and improving methods used in operational taxonomic unit-based approaches for 16S rRNA gene sequence analysis. Appl. Environ. Microbiol. 77, 3219–3226. doi: 10.1128/aem.02810-2810
Schnorr, S. L., Candela, M., Rampelli, S., Centanni, M., Consolandi, C., Basaglia, G., et al. (2014). Gut microbiome of the Hadza hunter-gatherers. Nat. Commun. 5:3654. doi: 10.1038/ncomms4654
Shenhav, L., Thompson, M., Joseph, T. A., Briscoe, L., Furman, O., Bogumil, D., et al. (2019). FEAST: fast expectation-maximization for microbial source tracking. Nat. Methods 16, 627–632. doi: 10.1038/s41592-019-0431-x
Sim, K., Cox, M. J., Wopereis, H., Martin, R., Knol, J., Li, M.-S., et al. (2012). Improved detection of bifidobacteria with optimised 16S rRNA-gene based pyrosequencing. PLoS One 7:e32543. doi: 10.1371/journal.pone.0032543
Soto, A., Martin, V., Jimenez, E., Mader, I., Rodriguez, J. M., and Fernandez, L. (2014). Lactobacilli and bifidobacteria in human breast milk: influence of antibiotherapy and other host and clinical factors. J. Pediatr. Gastr. Nutr. 59, 78–88. doi: 10.1097/mpg.0000000000000347
Subramanian, S., Huq, S., Yatsunenko, T., Haque, R., Mahfuz, M., Alam, M. A., et al. (2014). Persistent gut microbiota immaturity in malnourished Bangladeshi children. Nature 510, 417–421. doi: 10.1038/nature13421
Sundin, O. H., Mendoza-Ladd, A., Zeng, M., Diaz-Arevalo, D., Morales, E., Fagan, B. M., et al. (2017). The human jejunum has an endogenous microbiota that differs from those in the oral cavity and colon. BMC Microbiol. 17:160. doi: 10.1186/s12866-017-1059-1056
Tannock, G. W., Lawley, B., Munro, K., Pathmanathan, S. G., Zhou, S. J., Makrides, M., et al. (2013). Comparison of the compositions of the stool microbiotas of infants fed goat milk formula, cow milk-based formula, or breast milk. Appl. Environ. Microbiol. 79, 3040–3048. doi: 10.1128/aem.03910-3912
Tannock, G. W., Lee, P. S., Wong, K. H., and Lawley, B. (2016). Why don’t all infants have bifidobacteria in their stool? Front. Microbiol. 7:834. doi: 10.3389/fmicb.2016.00834
Turroni, F., Berry, D., and Ventura, M. (2017). Editorial: bifidobacteria and their role in the human gut microbiota. Front. Microbiol. 7:2148. doi: 10.3389/fmicb.2016.02148
Turroni, F., Milani, C., Duranti, S., Mancabelli, L., Mangifesta, M., Viappiani, A., et al. (2016). Deciphering bifidobacterial-mediated metabolic interactions and their impact on gut microbiota by a multi-omics approach. ISME J. 10, 1656–1668. doi: 10.1038/ismej.2015.236
Turroni, F., Peano, C., Pass, D. A., Foroni, E., Severgnini, M., Claesson, M. J., et al. (2012). Diversity of bifidobacteria within the infant gut microbiota. PLoS One 7:e36957. doi: 10.1371/journal.pone.0036957
Urbaniak, C., Angelini, M., Gloor, G. B., and Reid, G. (2016). Human milk microbiota profiles in relation to birthing method, gestation and infant gender. Microbiome 4:1. doi: 10.1186/s40168-015-0145-y
Van Rossum, T., Ferretti, P., Maistrenko, O. M., and Bork, P. (2020). Diversity within species: interpreting strains in microbiomes. Nat. Rev. Microbiol. 18, 491–506. doi: 10.1038/s41579-020-0368-361
Vatanen, T., Plichta, D. R., Somani, J., Munch, P. C., Arthur, T. D., Hall, A. B., et al. (2019). Genomic variation and strain-specific functional adaptation in the human gut microbiome during early life. Nat. Microbiol. 4, 470–479. doi: 10.1038/s41564-018-0321-325
Ward, T. L., Hosid, S., Ioshikhes, I., and Altosaar, I. (2013). Human milk metagenome: a functional capacity analysis. BMC Microbiol. 13:116. doi: 10.1186/1471-2180-13-116
Yatsunenko, T., Rey, F. E., Manary, M. J., Trehan, I., Dominguez-Bello, M. G., Contreras, M., et al. (2012). Human gut microbiome viewed across age and geography. Nature 486, 222–227. doi: 10.1038/nature11053
Zacarias, M. F., Binetti, A., Laco, M., Reinheimer, J., and Vinderola, G. (2011). Preliminary technological and potential probiotic characterisation of bifidobacteria isolated from breast milk for use in dairy products. Int. Dairy. J. 21, 548–555. doi: 10.1016/j.idairyj.2011.03.007
Zhang, J., Zhang, N., Liu, Y.-X., Zhang, X., Hu, B., Qin, Y., et al. (2018). Root microbiota shift in rice correlates with resident time in the field and developmental stage. Sci. China Life. Sci. 61, 613–621. doi: 10.1007/s11427-018-9284-9284
Zhang, M., Hang, X., Tan, J., and Yang, H. (2015). The host genotype and environment affect strain types of Bifidobacterium longum subsp. longum inhabiting the intestinal tracts of twins. Appl. Environ. Microbiol. 81, 4774–4781. doi: 10.1128/AEM.00249-215
Keywords: human breast milk, mother–infant dyads, microbiome, Bifidobacterium, association
Citation: Yan W, Luo B, Zhang X, Ni Y and Tian F (2021) Association and Occurrence of Bifidobacterial Phylotypes Between Breast Milk and Fecal Microbiomes in Mother–Infant Dyads During the First 2 Years of Life. Front. Microbiol. 12:669442. doi: 10.3389/fmicb.2021.669442
Received: 18 February 2021; Accepted: 30 April 2021;
Published: 07 June 2021.
Edited by:
Abelardo Margolles, Consejo Superior de Investigaciones Científicas, SpainReviewed by:
Stefano Raimondi, University of Modena and Reggio Emilia, ItalyAnders P. Hakansson, Lund University, Sweden
Copyright © 2021 Yan, Luo, Zhang, Ni and Tian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongqing Ni, niyqlzu@sina.com; Fengwei Tian, fwtian@jiangnan.edu.cn