Corrigendum: Molecular Analysis of Bacterial Isolates From Necrotic Wheat Leaf Lesions Caused by Xanthomonas translucens, and Description of Three Putative Novel Species, Sphingomonas albertensis sp. nov., Pseudomonas triticumensis sp. nov. and Pseudomonas foliumensis sp. nov.
 James T. Tambong1*
James T. Tambong1* Renlin Xu1Suzanne Gerdis1Greg C. Daniels2
Renlin Xu1Suzanne Gerdis1Greg C. Daniels2 Denise Chabot1Keith Hubbard1
Denise Chabot1Keith Hubbard1 Michael W. Harding2
Michael W. Harding2- 1Ottawa Research and Development Centre, Agriculture and Agri-Food Canada, Ottawa, ON, Canada
- 2Crop Diversification Centre South, Alberta Agriculture and Forestry, Brooks, AB, Canada
Xanthomonas translucens is the etiological agent of the wheat bacterial leaf streak (BLS) disease. The isolation of this pathogen is usually based on the Wilbrink’s-boric acid–cephalexin semi-selective medium which eliminates 90% of other bacteria, some of which might be novel species. In our study, a general purpose nutrient agar was used to isolate 49 bacterial strains including X. translucens from necrotic wheat leaf tissues. Maximum likelihood cluster analysis of 16S rRNA sequences grouped the strains into 10 distinct genera. Pseudomonas (32.7%) and Pantoea (28.6%) were the dominant genera while Xanthomonas, Clavibacter and Curtobacterium had 8.2%, each. Erwinia and Sphingomonas had two strains, each. BLAST and phylogenetic analyses of multilocus sequence analysis (MLSA) of specific housekeeping genes taxonomically assigned all the strains to validly described bacterial species, except three strains (10L4B, 12L4D and 32L3A) of Pseudomonas and two (23L3C and 15L3B) of Sphingomonas. Strains 10L4B and12L4D had Pseudomonas caspiana as their closest known type strain while strain 32L3A was closest to Pseudomonas asturiensis. Sphingomonas sp. strains 23L3C and 15L3B were closest to S. faeni based on MLSA analysis. Our data on MLSA, whole genome-based cluster analysis, DNA-DNA hybridization and average nucleotide identity, matrix-assisted laser desorption/ionization-time-of-flight, chemotaxonomy and phenotype affirmed that these 5 strains constitute three novel lineages and are taxonomically described in this study. We propose the names, Sphingomonas albertensis sp. nov. (type strain 23L3CT = DOAB 1063T = CECT 30248T = LMG 32139T), Pseudomonas triticumensis sp. nov. (type strain 32L3AT = DOAB 1067T = CECT 30249T = LMG 32140T) and Pseudomonas foliumensis sp. nov. (type strain 10L4BT = DOAB 1069T = CECT 30250T = LMG 32142T). Comparative genomics of these novel species, relative to their closest type strains, revealed unique repertoires of core secretion systems and secondary metabolites/antibiotics. Also, the detection of CRISPR-Cas systems in the genomes of these novel species suggests an acquired mechanism for resistance against foreign mobile genetic elements. The results presented here revealed a cohabitation, within the BLS lesions, of diverse bacterial species, including novel lineages.
Introduction
Members of the Xanthomonas translucens group are gram-negative plant pathogenic bacteria that can cause serious diseases to important cereal crops as well as forage grasses (Hersemann et al., 2017; Curland et al., 2018; Sapkota et al., 2020). The group is subdivided into 10 different pathovars based on biochemical and molecular characteristics and host ranges (van den Mooter et al., 1987; Sapkota et al., 2020) and are taxonomically placed into the translucens and graminis subgroups (Vauterin et al., 1992; Langlois et al., 2017; Peng et al., 2019). Pathovars undulosa, translucens, hordei, cerealis, secalis and phleipratensis belong to the translucens subgroup and cause bacterial leaf streak (BLS) on wheat, barley, oat, triticale, and rye (Duveiller, 1994; Vauterin et al., 1995; Sapkota et al., 2020). The “graminis” subgroup comprises of four pathovars (arrhenatheri, graminis, poae, and phlei) that cause bacterial wilt diseases on forage grasses (Vauterin et al., 1995; Sapkota et al., 2020). Giblot-Ducray et al. (2009) published a new pathovar, pistaciae, that is pathogenic to pistachio and has not yet been classified to either subgroups.
Wheat BLS caused by X. translucens pv. undulosa is an important disease worldwide (Duveiller, 1990; Duveiller et al., 1997; Adhikari et al., 2012). In the last decade, the incidence of BLS in wheat has significantly increased in the Midwestern United States, the major wheat-growing regions (McMullen and Adhikari, 2011; Adhikari et al., 2012), and wheat-belt of western Canada1. The typical symptoms of BLS start with water-soaked streaks that subsequently become translucent necrotic leaf lesions (Smith et al., 1919; Duveiller et al., 1997; Sapkota et al., 2020), and under high disease pressure, the entire wheat leaf might be severely affected. Bacterial ooze can be seen on the leaf surface under humid and warm weather conditions. Traditional methods of isolation of the pathogen use a Wilbrink’s-boric acid-cephalexin (WBC) agar, a semi-selective medium, to eliminate other microbes (Duveiller, 1994). Duveiller (Duveiller, 1994) indicated the elimination of over 90% of “saprophytic” bacteria from washes of wheat and triticale lots using WBC, facilitating the identification of the pale yellow X. translucens pv. undulosa. It is clear that the main goal of the plant pathologist is to isolate the BLS causal agent for downstream studies such as accurate identification using biochemical, morphological and genomic evaluation, and enhancement of the biological collection as well as verification of Koch’s postulate. However, in so doing other bacterial species are eliminated, some of which might be novel genotypes.
Epi- and endo-phytic phyllosphere are habitats to millions of pathogenic and beneficial microorganisms that colonize leaves. Culture-dependent (Yashiro et al., 2011; Anguita-Maeso et al., 2019) and culture-independent methods (Grady et al., 2019; Singh et al., 2019; Regalado et al., 2020) have been used extensively to study the microbial diversity of healthy phyllosphere. Until now, however, the phylogenetic positions of cultivable bacteria associated with diseased leaf lesions, beside the causal pathogens, are not well studied.
The objectives of this study were to (i) isolate X. translucens pv. undulosa and other culturable bacteria associated with typical symptoms of BLS on wheat; (ii) characterize the diversity and taxonomic positions of the strains using 16S rRNA, multilocus sequence analysis (MLSA) and genome analysis; (iii) perform in silico analysis of orthologous genes, secretion systems and antimicrobial secondary metabolites of potential novel strains relative to their closest known/validly described bacterial species; and (iv) describe novel bacterial species. 16S rRNA sequence analysis is a reliable taxonomic marker for genus-level classification of bacteria (Gomila et al., 2015; Tambong, 2019) while MLSA, as single individual housekeeping genes or several housekeeping genes concatenated into pseudomolecules, has been routinely used for accurate and consistent phylogenetic resolution of strains at the species-level (Konstantinidis and Tiedje, 2007; Mulet et al., 2010; Liu et al., 2012; Tambong et al., 2014; Gomila et al., 2015; Tambong, 2019). Also the advent of genome sequencing using next generation technologies and recent advances in bioinformatics tools have revolutionized bacterial classification, providing highly reliable, reproducible and cumulative datasets and databases (Thompson et al., 2013; Parks et al., 2018; Paul et al., 2019). Genome-based parameters, such as comparative genomics, in silico DNA-DNA hybridization and average nucleotide identity; and matrix-assisted laser desorption/ionization-time-of-flight (MALDI-TOF) as well as biochemical characterization were used to validate the MLSA results and the hypothesis that three unique lineages discovered in this study constitute putative novel species within two bacterial genera, Pseudomonas and Sphingomonas.
Materials and Methods
Sample Collection and Isolation of Bacteria
Wheat flag leaves exhibiting symptoms of bacterial streak were collected from four southern Alberta fields of spring wheat (CWRS-AAC Viewfield). Four leaves per field were surface sterilized for 1 min in 2% sodium hypochlorite and rinsed three times with sterile water. The isolation of the bacteria was done as previously reported (Malette et al., 2020). Briefly, 0.25 cm2 section of symptomatic leaf tissue was aseptically excised, bisecting at least one bacterial lesion and immersed in a 100 μl droplet of sterile water and incubated at room temperature for 5 min. The droplet was examined under a dissecting microscope to confirm bacteria oozing into a suspension. Using a 10-μl loop, the suspensions were streaked onto nutrient agar (NA) and plates were incubated at 25°C and examined at 24 and 48 h for bacterial colonies. Up to four, morphologically-unique colonies were picked from each plate onto fresh NA plates using a sterile needle and incubated for 48 h. A total of 42 cultures were sent to the Ottawa Molecular Bacteriology Laboratory of Agriculture and Agri-Food Canada for identification. After repeated streaking (Tambong et al., 2017; Tchagang et al., 2018), 49 single colonies were obtained and preserved at −80°C in LB medium with 30% glycerol (v/v) for long-term storage. Gram-reaction of the strains was determined using the 3% KOH assay (Ryu, 1940).
DNA Extraction, PCR Amplification, and Gene Sequencing
Strains were grown overnight in nutrient broth and genomic DNA extracted using the Wizard® Genomic DNA Purification Kit (Promega Corp.) following the manufacturer’s protocol. 16S rRNA gene sequence was used for genus-level identification of the strains (Lane, 1991). Strain identification to species-level was done using housekeeping genes reported in previous taxonomic studies of Pantoea (leuS, gyrB, rpoB (Tambong et al., 2014; Tambong, 2019), Pseudomonas (rpoD, gyrB, rpoB (Mulet et al., 2010; Gomila et al., 2015; Tambong et al., 2017), Clavibacter (gyrB, ppkA and recA; Jacques et al., 2012), Sphingomonas (atpD, fusA and rpoB; Chen, 2012; Ogier et al., 2019), Xanthomonas (atpD and gyrB; Fischer-Le Saux et al., 2015; Ferreira et al., 2019), and Curtobacterium (atpD and rpoB; Gonçalves et al., 2019; Osdaghi et al., 2020). Erwinia strains were characterized to species-level based on leuS and rpoB genes and primers used for the genus Pantoea. All PCR amplifications were performed in a TProfessional thermal cycler (Biometra GmbH i.L., Göttingen, Germany) using Titanium Taq polymerase (Clontech, Inc., CA, United States) as previously described (Tambong et al., 2006). Sequencing was done with the ABI BigDye Terminator chemistry v3.1 (ThermoFisher Scientific, Canada) and run on an ABI 3500xl automated sequencer (ThermoFisher).
BLAST and Phylogenetic Analyses
The Basic Local Alignment Search Tool (BLAST; Altschul et al., 1990) program was used to query GenBank databases with the partial gene sequences obtained in this study for homology. The individual gene loci were separately aligned in SeqMan Pro ver 15.3.0 (DNASTAR) and the 5′ and 3′ ends of DNA sequences trimmed to the shortest sequence. Geneious 11.1.5 (Biomatters Ltd.) was used to concatenate corresponding gene sequences. The individual genes and concatenated pseudogene sequences were independently aligned using MUSCLE v3.8.1551 (Edgar, 2004); and modeltest-ng (Darriba et al., 2020) was implemented to identify the best-fit models of nucleotide substitution based on the Akaike’s Information Criterion (AIC). The best-fit model of each dataset is given in the phylogenetic tree captions. Maximum likelihood phylogenetic tree inference was performed using RAxML-NG (Kozlov et al., 2019) with 1,000 non-parametric bootstrap replicates to assess internal branch robustness (Felsenstein, 1985).
De novo Genome Sequencing and Analysis
Bacterial sample preparations for de novo genome sequencing of 7 representative strains were performed as described previously (Malette et al., 2020). Libraries were constructed using a Nextera DNA Flex Prep Kit (Illumina) following the manufacturer’s instructions. The draft genome sequences were determined by paired-end sequencing using Illumina NextSeq technology at the Molecular Technologies Laboratory, Ottawa Research and Development Centre, Ottawa, Canada. The quality of the paired-end reads, each 150 bp in length, were checked using FastQC version 0.11.3 (Andrews, 2010) and trimmed, if required. De novo assemblies were performed using ABySS version 1.5.2 (Simpson et al., 2009) or Unicyler v0.4.8 (Wick et al., 2017) as implemented in PATRIC (Wattam et al., 2014) and scaffolds < 300 bp in length were discarded.
Closely-related type strain genomes were determined via the Type (Strain) Genome Server (TyGS)2 which implements the MASH algorithm (Ondov et al., 2016); and corresponded to the closest bacterial species determined by MLSA. The Genome BLAST Distance Phylogeny approach (GBDP) as previously described was used to compute precise distances and genome-based phylogenetic relationships were inferred under the algorithm ‘trimming’ as recommended previously (Meier-Kolthoff et al., 2013). The resulting intergenomic distances were used to infer, using FASTME 2.1.4 including SPR postprocessing (Lefort et al., 2015), balanced minimum evolution trees with branch support (100 pseudo-bootstrap values). Trees were rooted at the midpoint (Farris, 1972). Genome-based DNA-DNA hybridizations (gDDH)3 were calculated using the default parameters of the GGDC 2.1 (Meier-Kolthoff et al., 2013) and values compared at the recommended species-level cut-off threshold of 70%. Average nucleotide identity values (ANI), were computed with default parameters using the FASTANI tool (Jain et al., 2018) to assess the taxonomic position of strains relative to the closest taxa at a species delineation cut-off threshold of 96%. The FastANI tool fragments a given query genome into overlapping fragments of a specific size. The sized fragments are then mapped to the reference genome using Mashmap (Jain et al., 2018).
For gene content analysis, the assembled genome sequences were annotated using the NCBI Prokaryotic Genome Automatic Annotation Pipeline (PGAAP) and the output used as in put data. Identification and comparison of orthologs and orthologous gene network analysis were done using OrthoVenn2 (Xu et al., 2019). The detection of bacterial secretion systems and cas3-typing (CRISPR-associated proteins) were done using, respectively, the TXSScan and cas_finder modules of MacSyFinder (Abby et al., 2014). In silico searches for secondary metabolites and antibiotic profiling were done against antiSMASH database-bacterial version4 (Blin et al., 2019).
MALDI-TOF MS, Fatty Acid Analysis and Phenotypic Fingerprinting
Matrix-assisted laser desorption/ionization-time-of-flight MS was used to profile whole cells according to the ethanol/formic acid extraction protocol recommended by Bruker Daltonics5. Pseudomonas strains were cultured in tryptic soy agar medium and incubated at 28°C for 24 h while Sphingomonas strains were grown at 20°C for 72 h in trypticase soy agar. Strains were grown, processed and profiled in duplicates. Spectral measurements, mass range of 2,000-20,000 m/z, were recorded using the Microflex L20 instrument (Bruker Daltonics, Germany) and data analyzed using FlexAnalysis and BioTyper 3.1 Explorer (Bruker Daltonics). The spectra were obtained in linear, positive ion mode according to the manufacturer’s recommended settings (Bruker Daltonik, Bremen, Germany). Each final spectrum resulted from the sum of the spectra generated at random positions to a maximum of 240 shots per spectrum. Spectra were visualized and pairwise superimposed using mMass version 5.5.0 (Niedermeyer and Strohalm, 2012). Where required, log score values were generated from the main spectra and clustering and principal component (PCA) analyses done as previously described (Maier et al., 2006).
Whole cell fatty acids of the Pseudomonas strains were analyzed as previously reported (Tambong et al., 2017) while that of Sphingomonas sp. nov. strain 23L3C was analyzed as reported by Busse et al. (2003). The extraction and analysis of fatty acid methyl esters were done according to MIDI (Sherlock Microbial Identification System, version 6.2) recommended protocol (Sasser, 1990). The Agilent 7890B gas chromatograph was used to generate the profiles and automatically identified by the MIDI TSBA 6 database.
Standard biochemical and bacteriological characterizations were performed on the potential novel strains as previously described (Tambong et al., 2017) using GEN III MicroplateTM (Biolog) to analyze 71 carbon sources and 23 chemical sensitivity assays including pH, salt tolerance and antibiotics as recommended by manufacturer. In addition, API ZYM galleries (bioMérieux) were used to study the strains according to manufacturer’s protocol. Catalase assay was performed as previously reported (Tambong et al., 2017) and oxidase activity was tested using strips (Millipore-Sigma). Cell morphology was investigated using scanning (SEM) and transmission (TEM) electron microscopy as previously reported (Tambong et al., 2017). SEM images were captured using Hitachi SU7000 (Hitachi, Tokyo, Japan) field emission scanning electron microscopy, as recommended by manufacturer. Biolog and API ZYM assays were performed in duplicates with similar results.
Results
Genus-Level Identification Based on 16S rRNA
Thirty-nine (80%) out of the 49 strains were Gram-negative bacteria while 10 strains were Gram-positive suggesting that the former major group is more abundant in the infected wheat leaf that showed typical symptoms of the bacterial leaf streak caused by X. translucens (Figure 1). All of the Gram-negative strains belong to the phylum Proteobacteria while the 10 Gram positive strains are of the Actinobacteria phylum. The 49 strains were taxonomically affiliated to seven bacterial families. Families Erwiniaceae and Pseudomonadaceae encompassed 65% of the strains (Figure 1). The family Microbacteriaceae (9 out of 10 strains) was the more abundant of the two families within the Gram-positive major group.
FIGURE 1
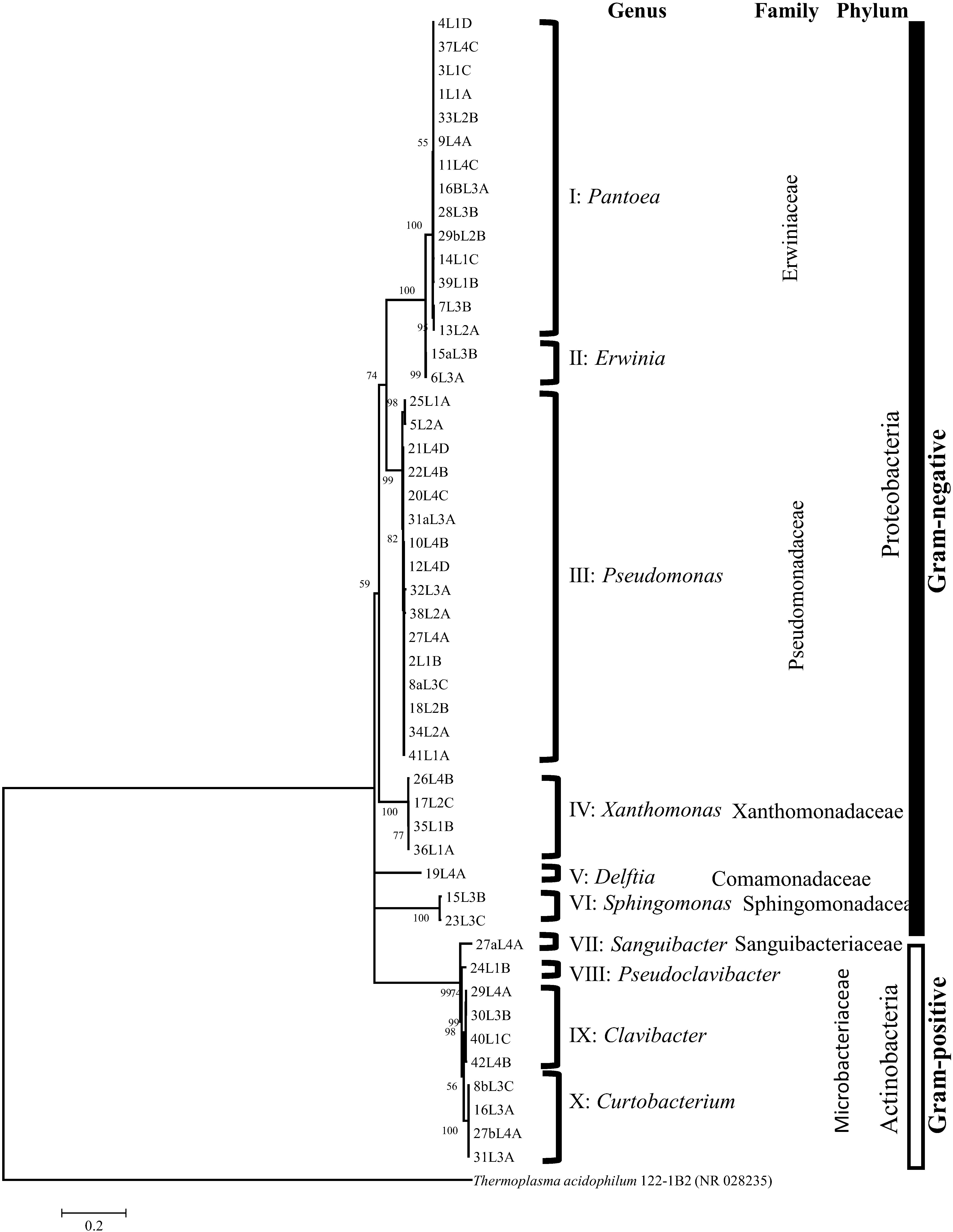
Figure 1. 16S rRNA tree based on maximum likelihood algorithm grouped the strains into 10 clusters corresponding to distinct and validly described bacterial genera. Based on Akaaike’s information criterion, the GTR + I + G4 substitution model was used. Bootstrap values >50% are shown at the branching points.
Forty-nine 16S rRNA sequences, l200 -1400 nt in length, were grouped phylogenetically by using the maximum likelihood algorithm into 10 unique clusters constituting different bacterial genera (Figure 1). All clusters had, at least, two strains with the exception of clusters V, VII and VIII with only one strain each (Figure 1). Comparative 16S rRNA sequence analysis using BLAST and publicly available GenBank databases taxonomically classified the strains of each of the 10 clusters into 10 valid bacterial genera. Fourteen of the 16 strains belonging to the family Erwiniaceae were classified as Pantoea (cluster I) while two strains were assigned to the genus Erwinia (Cluster II) (Figure 1 and Supplementary Table 1). Fourteen of the Pantoea strains had 16S rRNA sequence similarities in the range of 98.7 to 99.9% with Pantoea agglomerans DSM 3493T and the other two had a sequence homology of 98.8% with Pantoea allii BD 390T (Supplementary Table 1). Two of the Erwiniaceae strains (cluster II; Figure 1) exhibited 99.8% and 99.4% 16S rRNA sequence homologies to Erwinia persicina NBRC 102418T (Supplementary Table 1). Strains of cluster III (Figure 1) belonged to the genus Pseudomonas; and exhibited 16S rRNA sequence similarity values in the range of 98.9 to 99.9% (Supplementary Table 1) with type strains of species with validly published names, e.g., Pseudomonas caspiana FB102T (99.6% with strain 10L4B and 12L3D), Pseudomonas congelans DSM 14939T (99.8% with strain 27L4A), and Pseudomonas asturiensis LPPA 221T (99.5% with strain 32L3A) (Supplementary Table 1). Strains of cluster IV (Figure 1) were taxonomically assigned to the genus Xanthomonas (Family Xanthomonadaceae) exhibiting 16S rRNA similarities of 97.7% to 99.7% to Xanthomonas translucens pv. translucens DSM 18974T (Supplementary Table 1). The 2 strains in cluster VI (Figure 1 and Supplementary Table 1) were taxonomically classified as Sphinogomonas (Sphinogomonadaceae) with 98.5% and 98.7% 16S rRNA sequence similarities to Sphingomonas faeni MA-olkiT (Supplementary Table 1). 16S rRNA nucleotide sequences of strains that are highly similar to members of the genus Clavibacter (Microbacteriaceae) were in cluster IX (Figure 1) and had similarity values of 99.3 to 99.86% (30L3B, 40L1C and 42L4B) and 99.65% (29L4A) with Clavibacter tessellarius ATCC 33566T and Clavibacter michiganensis subsp. capsici PF008T, respectively (Supplementary Table 1). Four strains showing 16S rRNA nucleotide sequence homologies that are highly similar (98.6-99.4%; Supplementary Table 1) to Curtobacterium pusillum DSM 20527T were taxonomically placed in cluster X (Figure 1). Based on 16S rRNA sequence homologies (> 98%), only one strain was identified for clusters V, VII and VIII (Figure 1); and their respective strains were classified as Delftia (Comamonadaceae), Sanguibacter (Sanguibacteriaceae) and Pseudoclavibacter (Microbacteriaceae) (Supplementary Table 1). These 3 clusters, with only strain each, were not analyzed beyond 16S rRNA.
Species-Level Identification Based on BLAST and Phylogenetics Analyses of Housekeeping Genes
Table 1 summarizes the results of BLAST analysis of the nucleotide sequences of key taxonomic housekeeping genes used for species-level identification. Four strains exhibited 98.7% and 100.0% similarity levels with Xanthomonas translucens pv. translucens DSM 18974T and Xanthomonas translucens pv. undulosa LMG 892T. (Table 1). Four Curtobacterium strains were identified to be putative Curtobacterium flaccumfaciens based on homology of atpD and rpoB gene sequences (Table 1). Three of the 4 Clavibacter strains were identified as putative Clavibacter tessellarius with recA, gyrB and ppkA partial gene sequence homology values greater than 98% while strain 29L4A exhibited 97.7% (ppkA), 97.2% (gyrB) and 95.2% (recA) sequence similarity values with Clavibacter insidiosus LMG 3663T (Table 1). Nucleotide sequences of leuS, gyrB and rpoB (Tambong, 2019; Tambong et al., 2014) were used to delineate Pantoea strains into two species, P. agglomerans (type strain CIP 57.51T) and P. allii (type strain LMG 24248T) with homology values in the range of 98.0 to 99.0%. Fourteen strains exhibited leuS gene similarities of 98.9 to 100% with P. agglomerans CIP 57.51T (= DSM 3493T) while two strains had percent homologies of 98.4 and 100% with Pantoea allii LMG 24248T (Table 1). leuS and rpoB gene sequences were used to reliably and taxonomically associate the two Erwinia strains (6L3B and 15aL3B) with Erwinia persicina NBRC 102418T at percent similarity values of 99.6 and 100%, respectively (Table 1).
TABLE 1
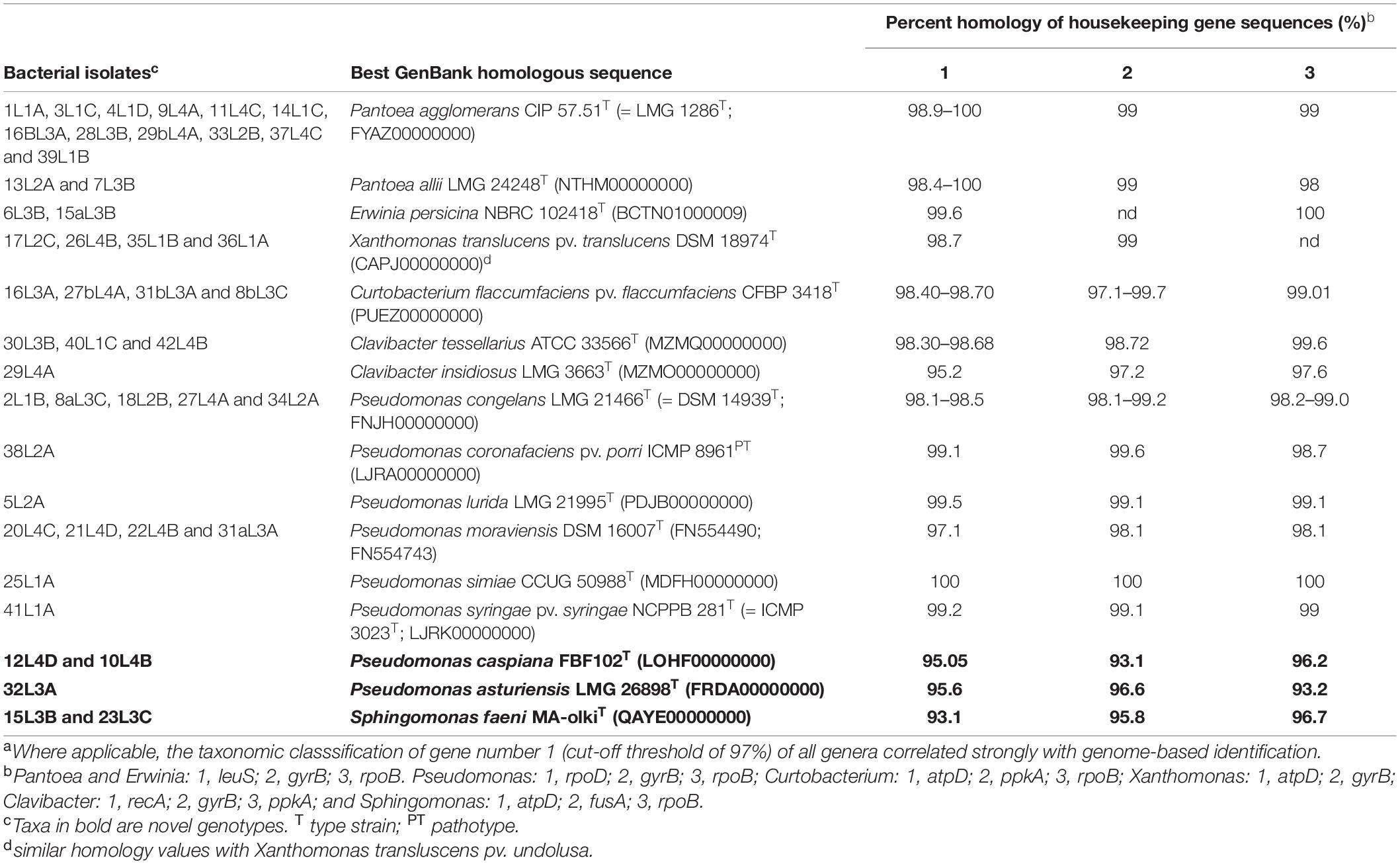
Table 1. Species-level identification of 46 bacterial isolates obtained in this study using housekeeping gene sequence homology to publicly available GenBank sequence entries.a
Species level identification of Pseudomonas strains was achieved by BLAST analysis of rpoD, gyrB, and rpoB as previously indicated (Tambong et al., 2017) at a cut-off threshold of 97%. The 16 Pseudomonas strains were classified to 8 closest validly described species based on the sequence similarity in the range of 95.0 to 100%, 94.0 to 99.0%, and 93.0 to 100%, for rpoD, rpoB and gyrB, respectively (Table 1). Based on this scheme, 13 Pseudomonas strains were accurately assigned to 6 validly described species (Table 1). For example, five strains (2L1B, 8aL3C, 18L2B, 27L4A, and 34L2A), with sequence homology values (98.1-98.5%) higher than the species cut-off threshold, were taxonomically assigned to Pseudomonas congelans (Table 1). Strain 32L3A, however, had the highest gene sequence homologies of 95.9% (rpoD), 93.2% (rpoB), and 96.6% (gyrB), with Pseudomonas asturiensis LPPA 221T (Table 1), with three genes showing percent homology values less than the species cut-off threshold of 97% (Gomila et al., 2015; Mulet et al., 2010). Also, two strains (10L4B and 12L4D) had low percent gyrB, rpoB and rpoD sequence homology values of 95.1, 93, and 93.0%, respectively, with Pseudomonas caspiana FBF102T, suggesting a potential novel genotype (Table 1). The two Sphingomonas strains recorded percent similarity values of 96.4, 96.4 and 92.6% for fusA, rpoB, and atpD genes, respectively, with strain MA-olkiT, the type strain of Sphingomonas faeni. These low homology levels (<97%) suggest that these two strains (15L3B and 23L3C) could be putative novel species within the genus Sphingomonas.
Multilocus sequence analysis phylogenetic analyses of individual and concatenated genes were used to further characterize the taxonomic positions of these five strains (10L4B,12L4D, 32L3A, 23L3C, and 15L3B). Single gene ML evolutionary trees showed these novel strains clustering uniquely from known validly described species and well supported by 100% bootstrap values (data not shown). The ML trees (Figures 2A-C) derived from concatenated partial gene sequences specific for the respective bacterial genera (Pseudomonas, rpoB-gyrB-rpoD, 8321 nt; and Sphingomonas, atpD-fusA-rpoB, 6997nt) clustered the strains similarly to those of single gene phylogenies. These analyses re-affirmed the uniqueness of these strains and suggest that they represented three putative novel species within Pseudomonas and Sphingomonas genera.
FIGURE 2
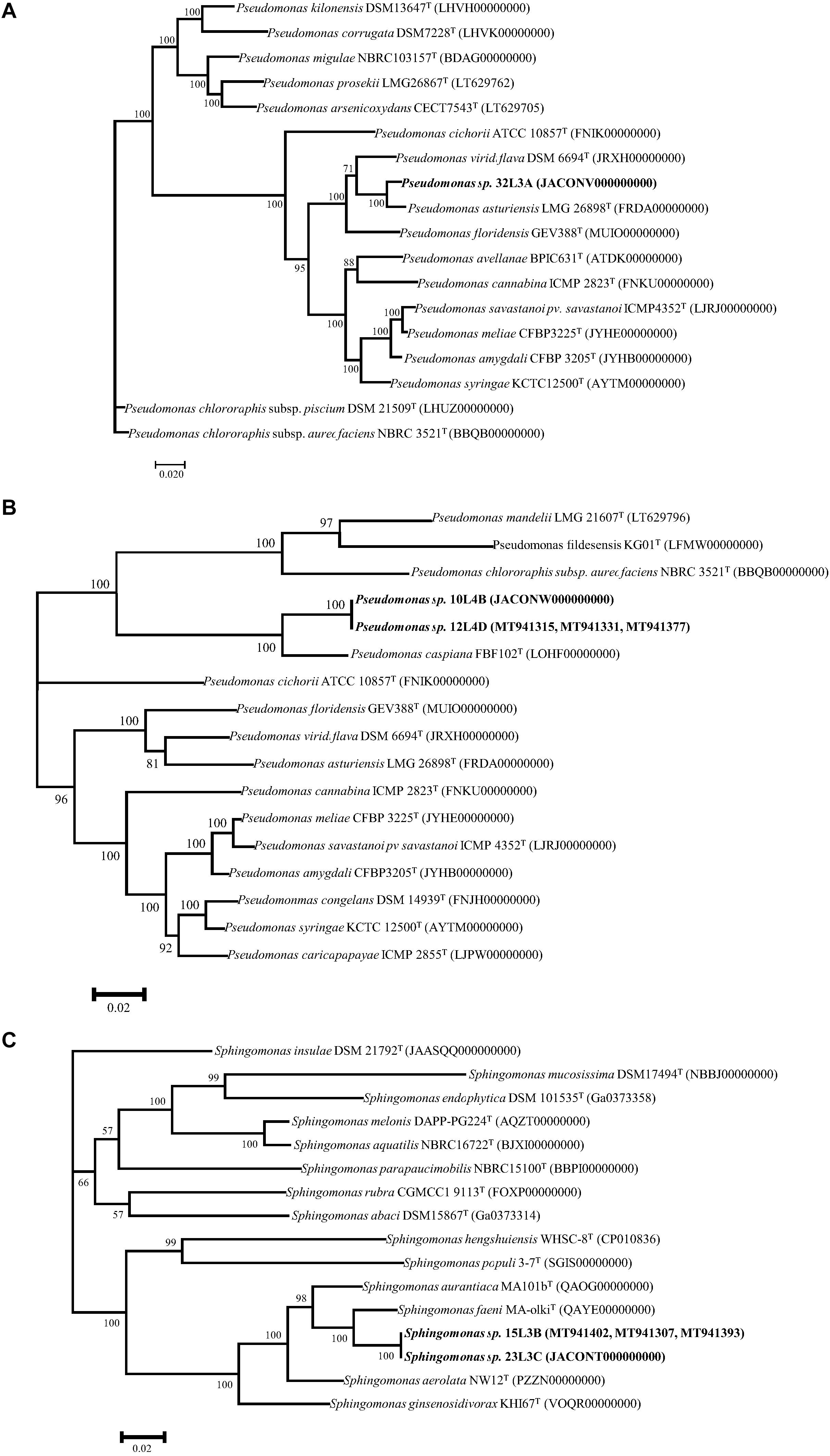
Figure 2. Maximum Likelihood phylogenetic trees of concatenated housekeeping gene sequences of the novel genotypes (in bold) within the genera Pseudomonas (A,B), and Sphingomonas (C) and corresponding closest type strains. rpoB-gyrB-rpoD (8321 nt) and atpD-fusA-rpoB (6997nt) were used for Pseudomonas, and Sphingomonas, respectively. Trees were infer using RAxML-ng (Kozlov et al., 2019) with TIM2 + I + G4 (A) and GTR + I + G4 (B,C) as the best substitution models (Darriba et al., 2020) based on Akaaike’s information criterions. Bootstrap values >50% are shown at the branching points. GenBank accession numbers of closest type strains are given in parentheses.
MALDI-TOF MS Data
Matrix-assisted laser desorption/ionization-time-of-flight MS is an emerging technology for quick and accurate identification of most bacterial strains by analyzing mass spectra of whole cell proteins. This technique was used to confirm that strains 32L3A, 10L4B, and 23L3C represented novel lineages relative to their corresponding closest known type strains. Pairwise comparative analysis of the spectra of the novel strains Pseudomonas sp. 32L3A or Sphingomonas sp. 23L3C and their corresponding closest known type strain revealed 8 or 7 peaks (m/z) indicated by asterisks that could be used to differentiate these strains from P. asturiensis or S. faeni, respectively (Supplementary Figure 1). For the relationship between strain 10L4B and its closest type strain (P. caspiana), we implemented cluster analysis to generate a dendrogram and pca (Figure 3). The novel Pseudomonas sp. strain 10L4B clustered distinctively at a distance level of about 250 with Pseudomonas caspiana CECT 9164T as the closest neighbor (Figure 3A). Principal component analysis of MALDI-TOF profiles of strain 10L4B confirmed its distinct taxonomic status relative to P. caspiana, P. cichorii, P. congelans and P. syringae (Figure 3B). These MALDI-TOF results are consistent with MLSA-based data, suggesting that these strains represent novel Pseudomonas and Sphingomonas species.
FIGURE 3
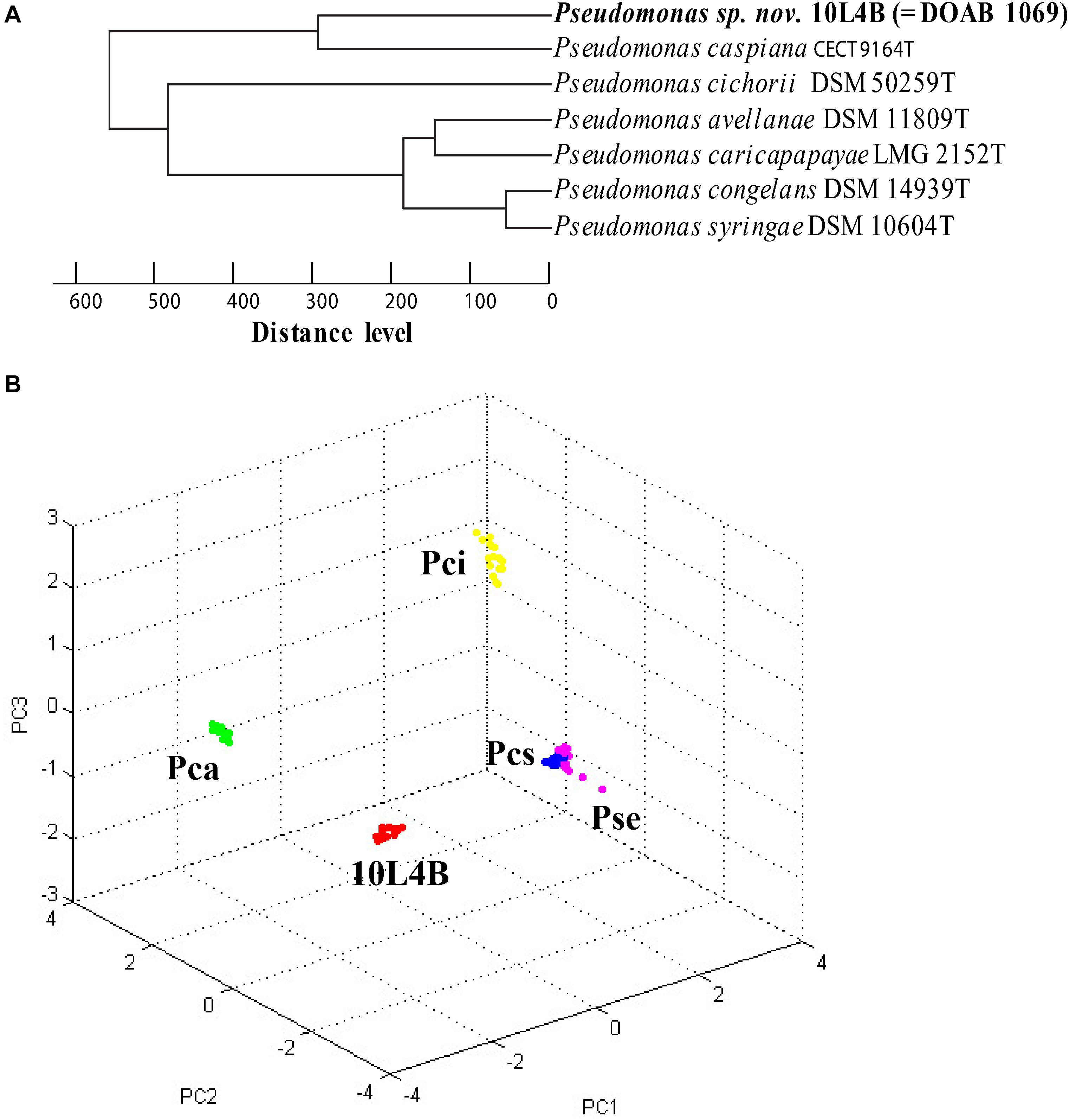
Figure 3. Cluster analysis of MALDI-TOF MS data of Pseudomonas sp. nov. 10L4B (red) compared with validly described type strains of the genus Pseudomonas (P. caspiana CECT 9164T, P. cichorii DSM 50259T, P. avellanae DSM 11809T, P. caricapapayae LMG 2152T, P. congelans DSM 14939T and P. syringae DSM 106604T) (A) dendrogram, and (B) principal component analysis, Pca, P. caspiana CECT 9164T (green); Pci, P. cichorii DSM 50259T(yellow); Pcs, P. congelans DSM 14939T(blue); and Pse, P. syringae DSM 106604T (pink). Each dot represents a spectrum with 24 spectra acquired per strain.
Genome-Based DNA-DNA Homology and Phylogenomics
To further validate strain identification, we sequenced and analyzed the genomes of 9 representative strains: 6 identified based on MLSA data to the species-level and the 3 putative novel lineages. Supplementary Table 2 shows the basic statistics of the whole genome sequences obtained in this study. All the genome sequences were of good quality. Genome size ranged from 4.1 Mb (Sphingomonas sp. 23L3C) to 6.0 Mb (Pseudomonas sp. 10L4B) with a completeness of 94.7 to 100% (Supplementary Table 2). Protein-coding genes of 3,821 (Sphingomonas) to 5,710 (Pseudomonas) (Supplementary Table 2) are consistent with the corresponding genus. Genome-based DDH (gDDH; Meier-Kolthoff et al., 2013) and average nucleotide identity (ANI; Jain et al., 2018) analyses, at the respective species cut-off threshold of 70 and 96%, validated the species-level taxonomic positions of the 6 bacterial strains identified using housekeeping genes (Table 2). For example, the genome sequence of strain 7L3B had the highest gDDH and ANI values (> threshold values of 70 and 96%, respectively) of 91.5 and 99.0%, respectively, with the type strain of Pantoea allii LMG 24248T (NTMH00000000) while strain 1L1A was confirmed to be a putative Pantoea agglomerans exhibiting gDDH and ANI values of 88.6 and 98.7%, respectively, with the type strain, DSM 3493T (FYAZ00000000) (Table 2). Also, the draft genome sequence of strain 17L2C had gDDH and ANI values greater than the respective threshold values with the type strain, Xanthomonas translucens pv. translucens DSM 18974T (78.0 and 97.7%) and showed 94.9 and 99.7% with a reference, Xanthomonas translucens pv. undulosa Xtu 4699. This confirms that this strain belongs to pathovar undulosa as indicated by the pathogenic reaction on wheat and barley (data not shown). Similarly, the gDDH and ANI values of the 3 putative novel bacterial genotypes were lower than the respective species cut-off threshold levels (Table 2). For example, strain 32L3A had the highest gDDH and ANI values of 58.8 and 80.9%, respectively, with the type strain of Pseudomonas asturiensis LMG 26898T (FRDA00000000) (Table 2). Also, strain 23L3C had only 34.4% (gDDH) and 88.8% (ANI) genome homology values with Sphingomonas faeni MA-olkiT (QAYE00000000 (Table 2). The gDDH and ANI results were corroborated by whole genome-based phylogenetic analysis inferred using GBDP-derived distances of strains 10L4B, 32L3A, and 23L3C and their corresponding closest validly described bacterial type strains (Supplementary Figure 2). For example, Sphingomonas sp. strain 23L3C grouped uniquely but close to Sphingomonas faeni MA-olkiT with a branch support of 100% pseudo-bootstrap values (Supplementary Figure 2). These whole genome sequence data strongly corroborated the other results and supported the hypothesis that these strains constitute putative novel genotypes within the genera Pseudomonas and Sphingomonas.
TABLE 2
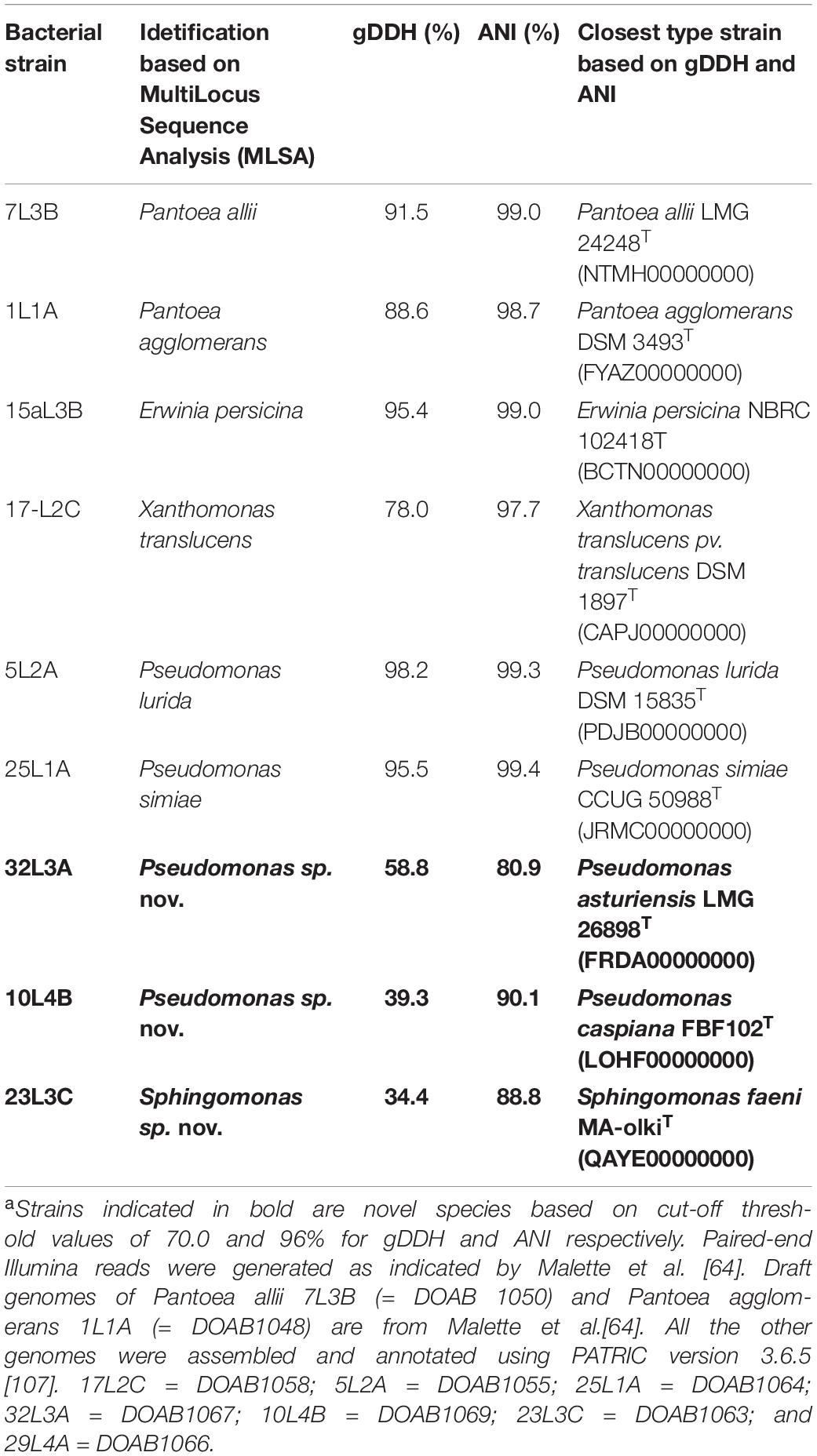
Table 2. In silico DNA-DNA hybridization (gDDH) and average nucleotide identity (ANI) values of de novo whole genome sequences of 6 representative strains identified to species level and the 3 putative novel bacterial strainsa.
Orthologous Gene Analysis
To provide a better insight of the genomic make-up of the proposed novel species, we analyzed the orthologous genes of the novel lineages relative to their closest type strains of species with validly published names. The gene content comparisons of the potential novel species and their closest described species indicated distinct genetic make-up.
The two potential novel Pseudomonas strains and their corresponding closest validly described species (P. caspiana and P. asturiensis) were analyzed together. The four whole genome sequences had a total of 18,463 protein-coding sequences. The genes were grouped into 5,523 clusters consisting of 2,061 orthologous clusters with at least two species and 3,462 single-copy gene clusters. The four Pseudomonas genome sequences shared 3,493 orthologous protein clusters (Figure 4A) that are involved in biological processes, molecular function and cellular component. The novel strain 10L4B shared 682 orthologous protein family clusters with its closest known type strain, P. capsiana FBF102T while strain 32L3A shared 608 protein family clusters with P. asturiensis LMG 26894T (Figure 4A). Strain 32L3A, however, shared only 12 protein cluster families with P. caspiana FBF102T while strains 10L4B and P. asturiensis 26894T uniquely shared only 36 protein family clusters. Of the 3,493 protein family clusters shared by the 4 Pseudomonas strains, cluster 1 had the highest protein count of 18 and is annotated as P:antibiotic biosynthetic process; IEA:UniProtKB-KW (GO:0017000; Swiss-protein hit # P0C064). The gene network of cluster 1 shows the similarity levels of Pseudomonas sp. nov. strain 32L3A and its closest validly described species, P. asturiensis and had 6 and 7 predicted protein sequences, respectively (Figure 4B). For the other pair, Pseudomonas sp. nov. 10L4B and its closest known bacterial relative, P. caspiana FBF102T exhibited significantly lower numbers, 2 and 3 respectively, of this protein (Figure 4B). Protein similarity/homology was confirmed by phylogenetic analysis which revealed 5 distinct groupings, three of which are unique to Pseudomonas sp. 32L3A and its closest known bacterial species, P. asturiensis LMG 26894T (Figure 4C).
FIGURE 4
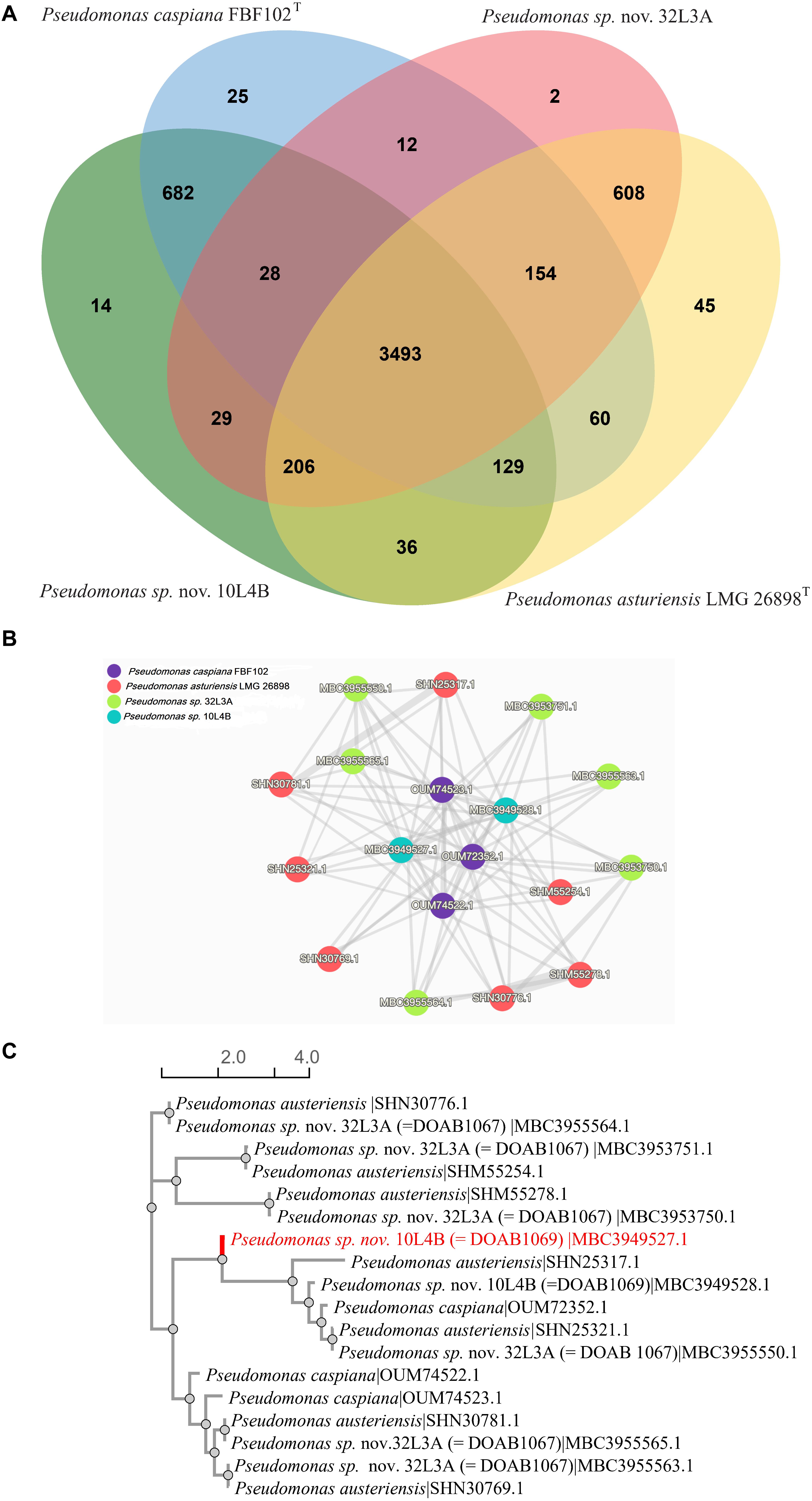
Figure 4. Venn diagram of the distribution of shared gene families (orthologous clusters) among the Pseudomonas strains (A); similarity networks (B) and phylogenetic relationship (C) of the 18 protein sequences in cluster 1. Analysis was done using OrthoVenn 2 software (Xu et al., 2019).
The two novel species, Pseudomonas sp. nov. 32L3A and Pseudomonas sp. nov. 10L4B, had 2 and 14 unique protein family clusters, respectively that were not detected in the whole genome sequences of their respective closest type strains. The two clusters (clusters 5,279 and 5,280) of strain 32L3A had 4 proteins with no significant match to entries of the Swiss-protein databases. BLASTp analysis of the two proteins of cluster 5,279 indicates high homology to integrase, an enzyme required for integration of the phage into the host genome by site-specific recombination (Van Duyne, 2005). Cluster 5280 proteins seem to be a calcium-binding protein with 87% homology to that of Pseudomonas corrugata strain C8A5 isolated from the rhizosphere of painted nettles (Plectranthus scutellarioides).
Seven of the 14 unique protein family clusters in the novel strain 10L4B were annotated to be involved in biological processes such as ion transport (GO:0006811), nitrogen and organic acid metabolic processes. The 7 other clusters consist of 27 proteins with four protein sequences in cluster 3,534 showing low homology (42%) to BapA prefix-like domain-containing protein (WP_039297424) of Cedecea neteri that has been implicated in biofilm formation and host colonization (Latasa et al., 2005). The two proteins of cluster 5,286 or 5,287 were 98 or 100% similar to the demethoxyubiquinone hydroxylase family protein (WP_095100952) or SMI1/KNR4 family protein (WP_095101549), respectively in Pseudomonas sp. Irchel 3A5, an unclassified Pseudomonas strain. Protein family cluster 5,290 contains two proteins that are unique to the novel strain 10L4B. The two proteins in cluster 5,292 had about 77% homology to the suppressor of fused domain protein of Pseudomonas sp. Irchel 3A5 and Pseudomonas savastanoi. All the other clusters were annotated as hypothetical proteins and exhibited high sequence homologies (>95%) with Pseudomonas sp. Irchel 3A5.
Pairwise orthologous gene analysis for the novel Sphingomonas sp. strain 23L3C revealed sharing 2,778 protein family clusters with S. faeni MA-olkiT, its taxonomically closest known type species; and, also, each had 40 or 71 unique protein families, respectively (Figure 5A). Cluster 1 protein family, shared by both strains, had 8 protein sequences with only one identified in the genome sequence of strain 23L3C. The protein similarity network (Figure 5B) and phylogenetic tree (Figure 5C) of cluster 1 suggest that the only protein identified in strain 23L3C is distinct. Protein sequences in cluster 1 matched the Swiss-protein # P20384, a putative transposon Tn552 DNA-invertase bin3 that facilitates horizontal transfer and recombination events related to multidrug resistance (Shore et al., 2010; Abriouel et al., 2015). This disproportionately high number (7) of protein sequences suggests that the Sphingomonas faeni MA-okiT could be prone to acquiring antimicrobial resistance genes from the environment. Cluster 3 of the protein shared between the two strains is a family identified as a multidrug efflux pump subunit AcrB. This cluster matched entry number P31224 in the Swiss-Protein database; and seems to constitute part of the AcrA-AcrB-AcrZ-TolC complex with broad spectrum and uses the proton motive force to export substrates including antimicrobial compounds (Sennhauser et al., 2007; Hobbs et al., 2012).
FIGURE 5
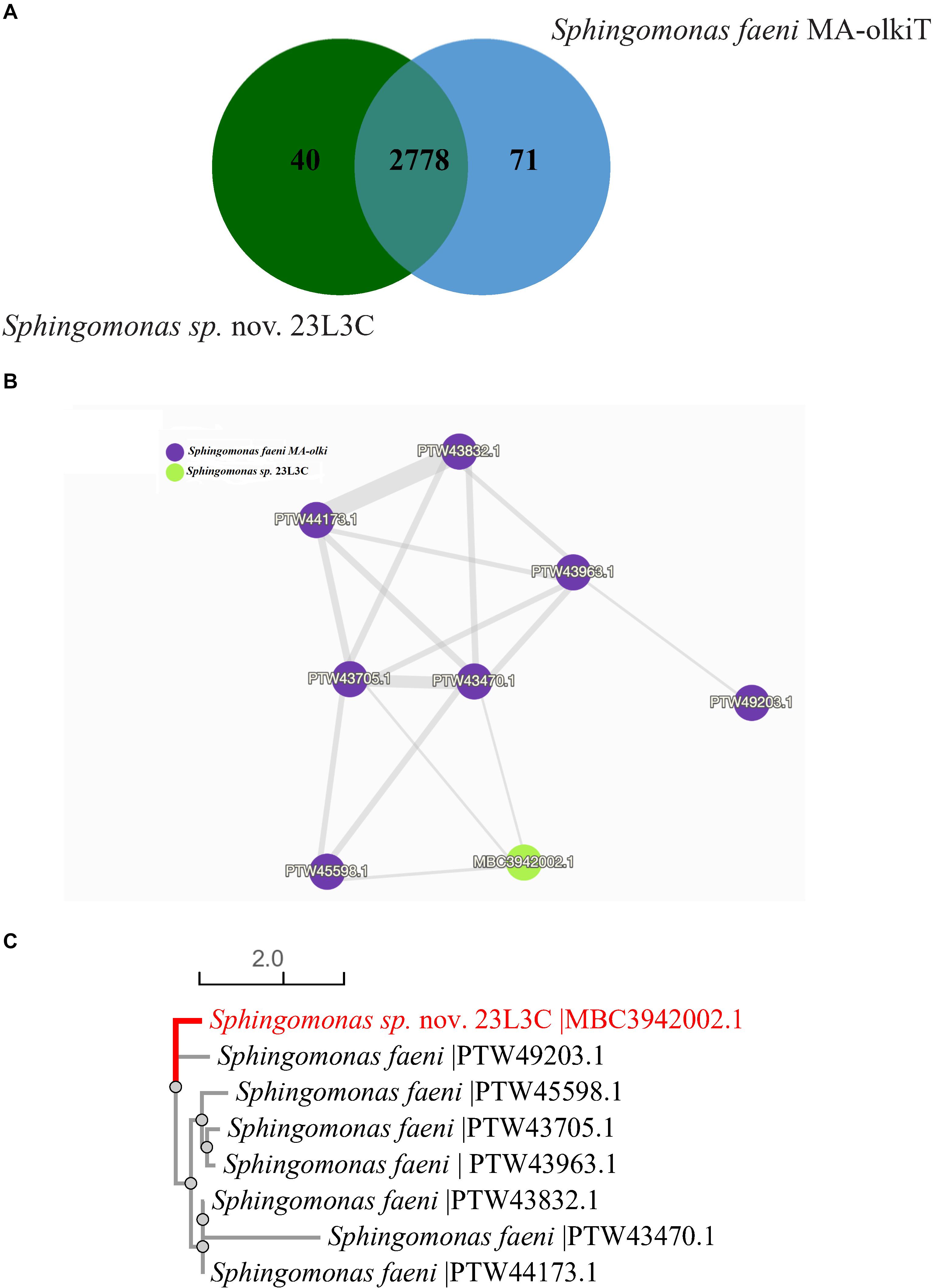
Figure 5. Distribution of shared gene families (orthologous clusters) between Sphingomonas sp. nov. 23L3C (= DOAB 1063) and Sphingomonas faeni MA-olkiT (A); similarity networks (B) and phylogenetic relationship (C) of the 8 protein sequences in cluster 1. Data generated using the OrthoVenn 2 software (Xu et al., 2019).
Forty-one protein family clusters were identified only in the novel Sphingomonas sp. strain 23L3C and consisted of 89 coding sequences, ranging from 2 to 6 per cluster. Seventeen (42.5%) of the 41 family clusters could not be matched to entries in the curated Swiss-protein database. Cluster 2 showed the highest number of proteins (6) and matched entry P25184 in the Swiss-protein database with a gene ontology of P:siderophore transport, a receptor for specific transmembrane uptake of the ferric pseudobactin 358. Cluster 4 had four proteins that matched Swiss-protein number D3Z7P3, a glutaminase isoform that has been implicated in the catabolism of glutamine and plays a role in acid-base homeostasis (Cassago et al., 2012; Li et al., 2016).
A multidrug export proteins (EmrA) grouped in cluster 8 corresponded to P27303 protein of the Swiss-protein database, a part of the EMrAB-TolC tripartite efflux system that confers antibiotics resistance to compounds such as nalidixic acid and 2,4-dinitrophenol (Lomovskaya and Lewis, 1992; Borges-Walmsley et al., 2003).
In silico Detection of Secondary Metabolites, and Secretion Systems
In silico secondary metabolite detection using AntiSMASH tool found at least three gene clusters within the genome sequence of the strains (Table 3). The types of metabolites detected included NRPS, NAGGN, acryl polyene, bacteriocin, siderophores, T3PKS, lasso peptide and terpene (Table 3). Metabolites of the aryl polylene, siderophores and bacteriocin types were more prominent in strains of Pseudomonas while the T3PKS type metabolites were detected only in Sphingomonas strains (Table 3). All the Pseudomonas strains have a cluster that is similar to pyoverdine with the exception of strain 10L4B (Table 3). A cluster involved in the biosynthesis of cichofactin A/cichofactin B was detected in Pseudomonas sp. nov. 32L3A (100% similarity) but not in its closest known species, P. asturiensis LMG 26898T. The taiwachelin biosynthetic cluster was detected in Pseudomonas sp. nov. 10L4B but absent in P. caspiana FBF102T (Table 3). The novel genotype Sphingomonas sp. strain 23L3C had an emulsan-like cluster of the lantidin type which was not detected in the whole genome sequence of its corresponding closest known type strain, S. faeni Ma-olkiT (Table 3).
TABLE 3
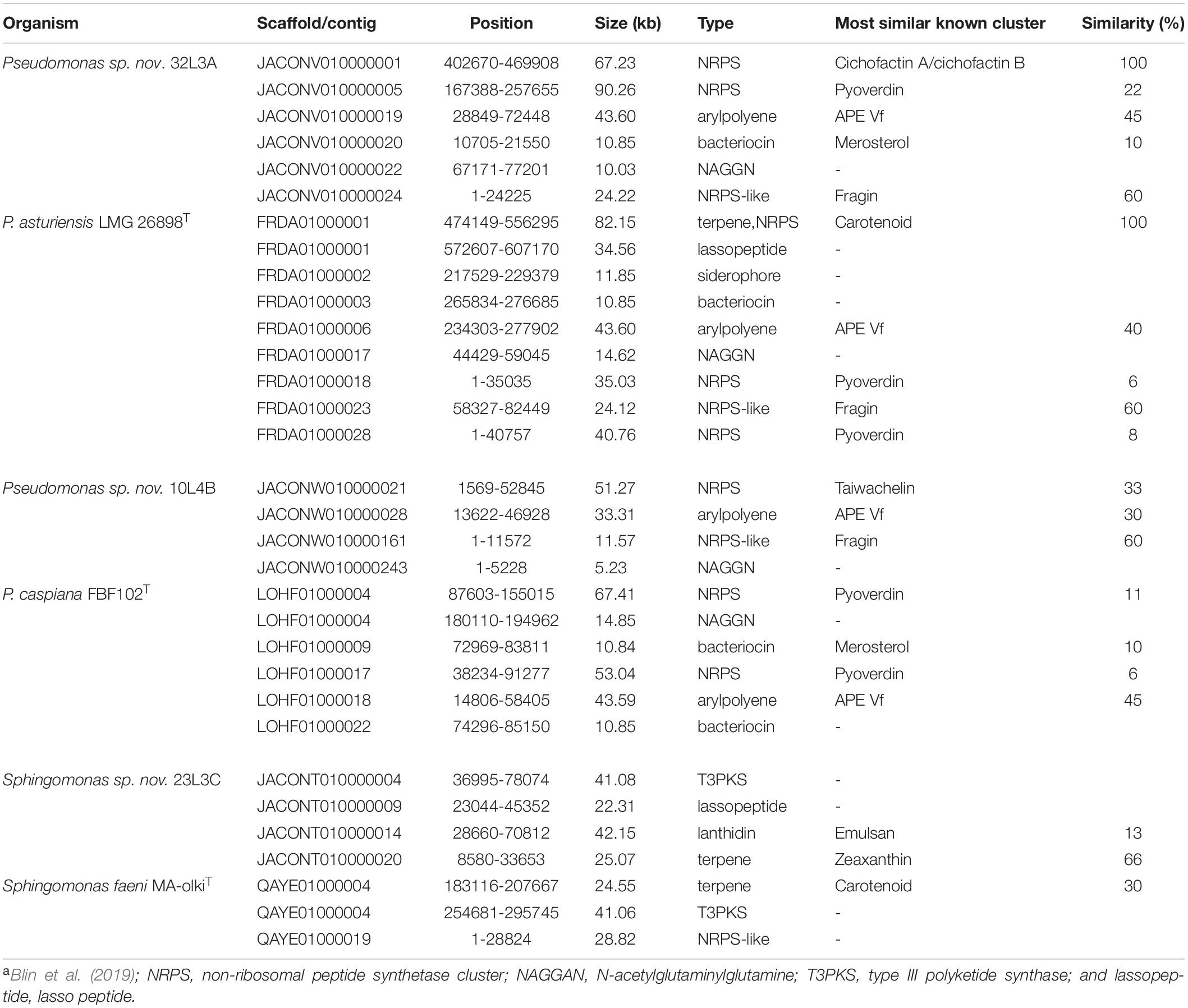
Table 3. In silico profiling of bacteriocin and antibiotic peptides within the genomes of the novel Pseudomonas and Sphingomonas species and their corresponding closest known type strains using the bacterial antiSMASH databasea.
Bacterial secretion systems are essential in the virulence, pathogenicity and global cell function. We detected non-flagellar and flagellar secretion systems, showing differences between the analyzed strains (Figure 6). The presence of the T1SS, T2SS and the flagellar systems were comparable across the strains while the T3SS and T6SSii contents differed between Pseudomonas and Sphingomonas strains (Figure 6). Sphingomonas strains (23L3C and Ma-okiT) had very few genes annotated as T3SS and T6SSii, respectively (Figure 6). The novel strains 32L3A and 10L4B as well as the type strain of P. caspiana FBF102T had very few protein coding sequences that were annotated as T4SS (Figure 6). Also, fewer counts of components of the tight adherence (Tad) pilus were detected in the novel genotype 10L4B and its corresponding closest known type strain, P. caspiana compared to the other strains. Sphingomonas strains had fewer components of the Type IV pilus (T4P) compared to the other bacterial strains studied (Figure 6) while those of the new Pseudomonas strains and their corresponding closest type strains were similar. Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR-Cas), a defense mechanism against foreign genetic elements in prokaryotes, were detected by using MacSyFinder tool. Five or six putative and mandatory CRISPR-Cas3 type I systems were detected, with variable sequence lengths of 445-829 bp or 194-810 bp, for the Pseudomonas or Sphingomonas strains, respectively.
FIGURE 6
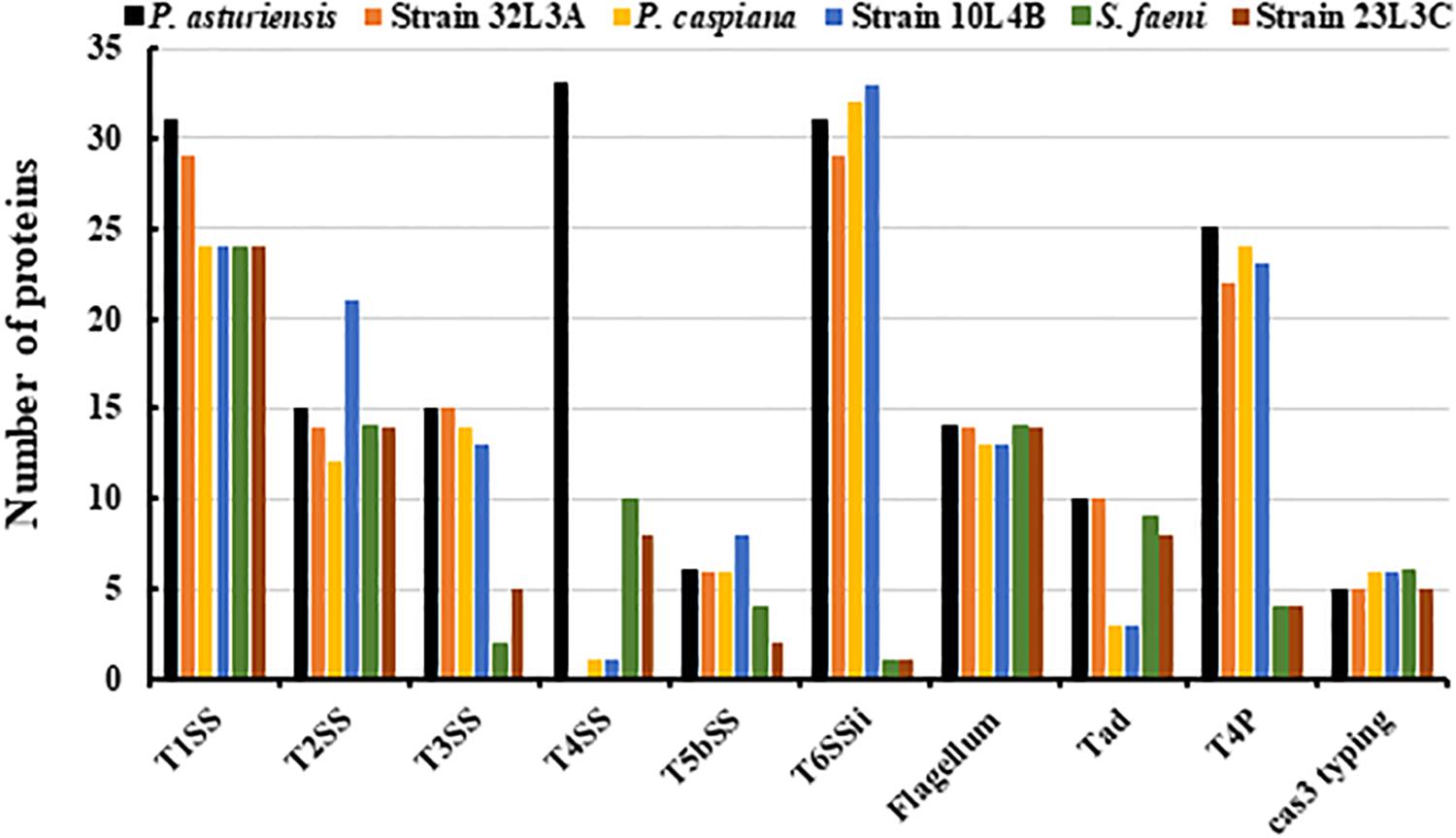
Figure 6. In silico detection of bacterial secretion systems and cas-3 sequences/genes within the genome sequences of the novel bacterial strains (Pseudomonas sp. nov. 10L4B (= DOAB1069); Pseudomonas sp. nov. 32L3A (= DOAB 1067) and Sphingomonas sp. nov. 23L3C (= DOAB 1063) and their respective corresponding closest validly described type strains Pseudomonas asturiensis, Pseudomonas caspiana and Sphingomonas faeni. Secretion systems- and Cas3-typing-related proteins were detected using, respectively, the TXSSCan and Cas_finder modules of MacSyFinder [1]. T1SS, T2SS, T3SS, T4SS, T5SS, and T6SS represent type I, type II, type III, type IV, type V and type VI secretion systems, respectively. Tad, Tad pili; and T4P, Type IV pili. Cas3 represents CRISPR-Cas, Clustered Regularly Interspaced Short Palindromic Repeats.
Chemotaxonomic and Phenotypic Characterization
Whole cell fatty acid contents (Supplementary Table 3) of the two Pseudomonas novel strains were consistent with those of sensu stricto pseudomonads (Oyaizu and Komagata, 1983; Tambong et al., 2017). The major cellular fatty acid peaks (Supplementary Table 3) of the Pseudomonas strains were C16:0, C16:1 ω6c/C16:1 ω7c (summed feature 3), C18:1 ω7c/C18:1 ω6c (summed feature 8), C17:0 cyclo; C12:0 2-OH, C12:0 3-OH and C10:0 3-OH. Predominant fatty acid peaks in Sphingomonas sp. nov. 23L3C were summed feature 8 (C18:1 ω7c and/or C18:1 ω6c), summed feature 3 (C16:1 ω7c and/or C16:1 ω6c), C14:0 2-OH and C16:0 (Supplementary Table 3). The absence of any 3-OH and the presence of C14:0 2-OH fatty acids in the whole cell fatty acid content of strain 32L3A (Supplementary Table 3) is in agreement with members of the genus Sphingomonas, sensu stricto (Kampfer et al., 1997; Denner et al., 2001).
Based on GENIII microplate assay (Biolog), 52, 43, or 35 carbon sources out of the 77 substrates tested were readily utilized by strain 10L4B, 32L2A or 23L3C respectively (Supplementary Table 4) and phenotypic patterns differed from those of their closest type strains. The strains were observed to be rod-shaped of variable sizes with Sphingomonas sp. nov. strain 23L3C being the smallest (Figure 7). Multiple polar flagella were observed on the cells of Pseudomonas sp. nov. strain 32L3A while strain 10L4B had one visible polar flagellum; and cells of Sphingomonas sp. nov. strain 23L3C did not show the presence of flagella (Figure 7).
FIGURE 7
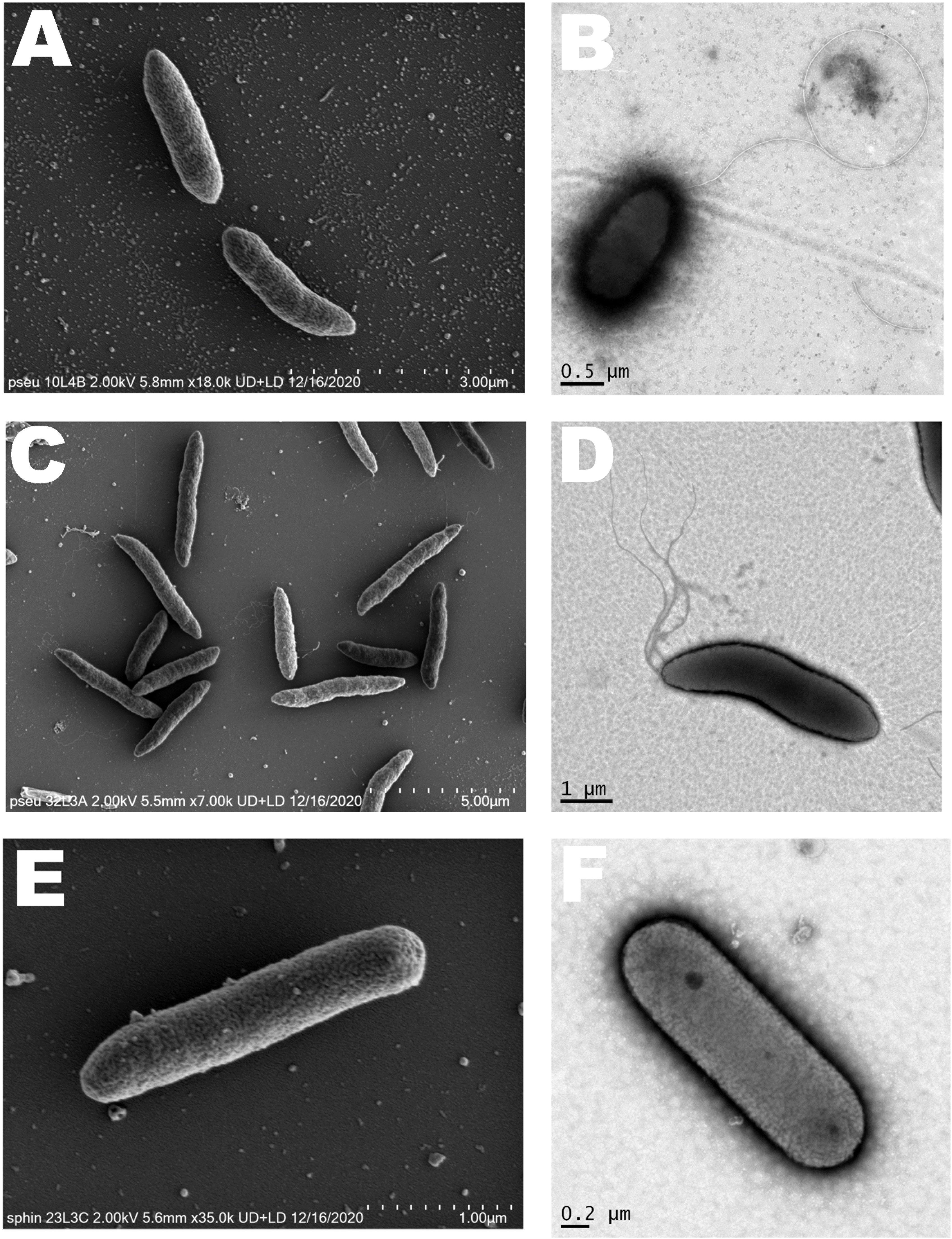
Figure 7. Cell morphology of the novel bacterial species. Pseudomonas sp. nov. strain 10L3B (A,B); Pseudomonas sp. nov. strain 32L3A (C,D) and Sphingomonas sp. nov. strain 23L3C (E,F) based on scanning (left) and transmission (right) electron microscopy. Strain 10L4B has a visible polar flagellum, strain 32L3A shows multiple flagella while strain 23L3C has no visible flagellum.
Also, both novel Pseudomonas strains tolerated pH 6.0 (neutral pH) while Sphingomonas strain 23L3C preferred pH 5.0 (acidic condition) (Supplementary Table 4). Pseudomonas strains grew well at 4% salt tolerance level while the Sphingomonas strain did not seem to tolerate high salt concentrations (Supplementary Table 4). Both novel Pseudomonas strains were resistant to troleandomycin, rifamycin SV, vancomycin, lincomycin but sensitive to nalidixic acid and minocycline. Strain 23L3C is resistant to vancomycin and nalidixic acid, but sensitive to rifamycin SV, minocycline and lincomycin (Supplementary Table 4).
Using API ZYM assays, the Pseudomonas strains were positive for esterase (C4), esterase lipase (C 8), leucine arylamidase, valine arylamidase, cystine arylamidase, acid phosphatase, naphthol-AS-BI-phosphohydrolase, and β-glucosidase, but negative for α-glucosidase, α-galactosidases, β-galactosidases and β-galactosidases. Strain 23L3C exhibited the same enzymatic activities that were positive for the Pseudomonas strains but, also, uniquely exhibited high activity of β-galactosidases.
Discussion
This study reports the analysis of cultivable bacterial strains isolated from a unique and unexploited niche, the wheat necrotic lesions caused by Xanthomonas translucens (the causal agent of bacterial leaf streak disease), and the identification of three novel and previously undescribed bacterial species.
Until now, research on the characteristic BLS lesions only focused on the isolation of the target pathogen for downstream studies such as accurate identification, verification of Koch’s postulate and population dynamics. To achieve this isolation objective, the Wilbrink’s-boric acid–cephalexin semi-selective medium is generally employed since it eliminates 90% of saprophytic and epiphytic bacteria (Duveiller, 1994). It is clear why plant pathologists use this semi-selective medium, but in so doing a vast amount of bacterial biodiversity is not being documented. Several other semi-selective media are routinely used for the isolation of agricultural important bacterial pathogens e.g., Clavibacter michiganensis on tomato seeds (Ftayeh et al., 2011) and Pseudomonas syringae on kiwifruit (Miyoshi and Tachibana, 1994) and thus, eliminating other bacteria, some of which might be novel taxa of unknown ecological significance.
We used a general purpose medium, nutrient agar, and isolated forty-nine bacterial strains, consisting of 10 known and validly described genera (Figure 1). The 10 bacterial genera were reliably identified to the genus-level based on 16S rRNA sequence analysis, which revealed that Pseudomonas (32.7%) and Pantoea (28.6%) were the dominant genera. Consistent with previous reports (Mulet et al., 2010; Gomila et al., 2015; Tambong et al., 2017), 16S rRNA gene sequences showed low resolution at the intrageneric level. Housekeeping genes, such as leuS, atpD, rpoD, gyrB, fusA, rpoB, ppkA, and recA, in different combinations, have been used routinely to refine interspecfic phylogenetic and taxonomic positions of species of Pseudomonas, Pantoea, Clavibacter, Sphingomonas and Curtobacterium (Konstantinidis and Tiedje, 2007; Mulet et al., 2010; Tambong et al., 2017). rpoD, rpoB and gyrB are key housekeeping genes for species-level identification of Pseudomonas with rpoD reported to be the most discriminatory (Mulet et al., 2010; Gomila et al., 2015; Tambong et al., 2017) and is routinely used as the key gene for identification of species of Pseudomonas. leuS, rpoB and gyrB have been reported to provide accurate species delineation within the genus Pantoea (Deletoile et al., 2009; Tambong et al., 2014; Tambong, 2019). leuS gene fragment used in this study is a reliable marker for identification and differentiation of members of the genus Pantoea and is highly congruent to genome-based approach (Tambong et al., 2014; Rahimi-Khameneh et al., 2019; Tambong, 2019).
BLAST and phylogenetic (single and concatenated genes) analyses of these housekeeping genes were employed in our study and resulted in accurate species-level identification of the majority (80%) of the strains. Strains of Pantoea were identified to belong to either Pantoea agglomerans or Pantoea allii. Pantoea agglomerans is a versatile bacterium isolated from a variety of sources including wheat (Lindh et al., 1991; Kobayashi et al., 2018; Cherif-Silini et al., 2019). It was, however, surprising to isolate from wheat two strains of Pantoea allii, a known potent pathogen of onion (Brady et al., 2011; Rahimi-Khameneh et al., 2019). The strains of P. allii isolated in this study were found to be pathogenic to onion but not wheat (data not shown). Taxonomic assignment of the 4 Xanthomonas strains to X. translucens was done using atpD and gyrB, housekeeping genes that have been routinely used for differentiation of species within this genus (Parkinson et al., 2009; Fischer-Le Saux et al., 2015; Ferreira et al., 2019). Based on the rpoD, rpoB and gyrB (Mulet et al., 2010; Gomila et al., 2015), the Pseudomonas strains were accurately classified to six validly described species with the exception of five strains (32L3A, 12L4D, 10L4B, 23l3C, and 15L3B). Strain 32L3A had Pseudomonas asturiensis LMG 26898T as its closest validly described type strain. Strains 10L4B and 12L4D were found to be identical based on high homology values (>99.0%) of their 16S rRNA, rpoD, rpoB, and gyrB nucleotide sequences and had Pseudomonas caspiana as the closest type strain. Also, the 2 strains (23L3C and 15L3B) of Sphingomonas were similar but taxonomically differed from their closest type strain, Sphingomonas faeni MA-olkiT. As such the taxonomical positions of these three strains of Pseudomonas and two of Sphingomonas were further analyzed to determine whether they constitute novel genotypes. We used MALDI-TOF and genome-based analyses to verify the uniqueness of these bacterial strains.
Matrix-assisted laser desorption/ionization-time-of-flight MS analysis of the whole cell protein content based on spectra allowed for a straightforward separation of the novel strains from their respective closest validly described type strains. MALDI-TOF MS is an innovative tool for rapid and cost-effective bacterial identification (Bizzini and Greub, 2010; Camoez et al., 2016; Sauget et al., 2017) that has recently be integrated in clinical microbial identification workflows (Bizzini and Greub, 2010; Sauget et al., 2017). This phenotypic analysis is becoming key in rapid identification of novel bacterial species/genotypes (Moore and Rossello-Mora, 2011; Spitaels et al., 2014; Waleron et al., 2019; Lassalle et al., 2020). Even though MALDI-TOF analysis is a valuable screening tool, it might show a low discriminative power in delineating closely related bacterial species, e.g., P. congelans and P. syringae. In such cases, the corresponding results should be interpreted with caution and other methods used to validate the identification.
Rapid advances in high-throughput sequencing technologies and the development of bioinformatics tools have enabled the analysis of whole genome sequences for accurate identification, classification and taxonomy/systematics of bacterial strains (Gomila et al., 2015; Tambong, 2019). dDDH and ANI analyses based on whole genome sequence data corroborated the hypothesis that strains 32L3A, 10L4B, and 23L3C constitute novel species within the genera Pseudomonas and Sphingomonas since the computed values were lower than the cut-off threshold levels for species delineation. Genome-based techniques are reliable alternatives to wet lab DNA-DNA hybridization (wDDH) in bacterial species and strain identification (Thompson et al., 2013; Gomila et al., 2015; Parks et al., 2018; Tambong, 2019). Genome-based techniques eliminate the inherent shortcomings of wDDH such as irreproducibility between laboratories and high error (Sneath, 1989; Stackebrandt, 2003).
The availability of whole genome sequences allowed us to investigate the gene content of the novel species relative to their respective closest type strains. Analysis of several clusters of orthologous proteins showed similarities and differences. In the Pseudomonas, for example, all the strains had protein sequences in cluster 1 but the number of proteins was significantly different (Figure 4). The novel strain 32L3A and its corresponding type strain, Pseudomonas asturiensis, had 13 of the 18 protein sequences in cluster 1 while the novel strain 10L4B and its corresponding type strain (Pseudomonas caspiana) only had 3 and 2 proteins, respectively. This protein cluster is involved in the pathway for gramicidin S biosynthesis, an antimicrobial compound effective against some gram-positive, gram-negative bacteria and fungi (Gause and Brazhnikova, 1944; Kondejewski et al., 1996). The presence of this protein family cluster in all the Pseudomonas strains suggests a potential role in ecological fitness of these bacteria, especially in nutrient-restrictive niches. Still to be investigated is whether the high number of genes of this protein cluster family in Pseudomonas sp. nov. strain 32L3A and its corresponding closest known type strain could translate to higher competitiveness/fitness in their unique niches.
In silico analysis of secondary metabolites using AntiSMASH detected the presence of a pyoverdine cluster in all the genome sequences of Pseudomonas strains with the exception of strain 10L4B (Table 3). The lack of pyoverdine in the whole genome sequence of strain 10L4B could explain why this novel strain, when grown on King’s B medium, is non-fluorescent under ultra-voilet. Pyoverdin is a powerful iron-chelating and -transporting siderophore, and represents a ready marker for identification of some Pseudomonas (Meyer, 2000; Bodilis et al., 2009; Cezard et al., 2015). It is a yellow-green fluorescent pigment present in fluorescent Pseudomonas species (Meyer, 2000; Cezard et al., 2015).
Comparative orthologous gene analysis of the whole genome sequences of Sphingomonas sp. 23L3C and S. faeni Ma-okiT identified 89 unique protein coding sequences in the former strain only. While about 42.5% of the 41 protein family clusters could not be matched to entries in the curated Swiss-protein database, three of the annotated clusters provided insight to how strain 23L3C could be competitive in the environment. Cluster 2 was annotated as a receptor for specific transmembrane uptake of ferric pseudobactin 358. BLAST analysis suggests that this could be similar to pupB gene, an inducible ferric-pseudobactin receptor of Pseudomonas putida WCS358 (Koster et al., 1993). This PupB receptor is reported to facilitate iron transport using two (pseudobactin BN7 and pseudobactin BN8) distinct heterologous siderophores (Koster et al., 1993). The presence of the ferric-pseudobactin receptor suggests that strain 23L3C could be competitive in iron-limiting environments. Also, protein family cluster 8 matched a multidrug export proteins, EmrA (P27303), a part of EMrAB-TolC tripartite efflux system that confers antibiotics resistance to compounds such as nalidixic acid and 2,4-dinitrophenol (Lomovskaya and Lewis, 1992; Borges-Walmsley et al., 2003). This could explain why, in antibiotic sensitivity studies carry out in our study, the growth of strain 23L3C was not inhibited by nalidixic acid.
Sixteen bacterial species were isolated from necrotic tissues of wheat caused by Xanthomonas translucens including three previously undescribed taxa (Table 1). With the exception of the undescribed taxa, Pseudomonas lurida, Pseudomonas moraviensis, and Pseudomonas simiae, all the other isolated species have been reported to cause diseases on field crops including wheat with distinct and characteristic symptoms. However, the only characteristic symptoms observed during sample collection were those that are specific to Xanthomonas translucens, the causal agent of the bacterial leaf streak disease of wheat. Preliminary pathogenicity results using some of the other known plant pathogenic bacterial species did not induce compatible reaction on wheat seedlings (data not shown). Also, the presence of some of these other bacterial species on plant leaves is not surprising. For example, Pseudomonas lurida (Behrendt et al., 2007) and P. congelans (Behrendt et al., 2003) were initially isolated from phyllosphere of healthy grasses. These arguments suggest that these other bacterial species are either saprophytes, epiphytes, or endophytes. Further work is required to elucidate the ecological role(s) of these other species.
As a taxonomic conclusion, this study isolated 49 bacterial strains from the BLS lesions on wheat using a general purpose medium, nutrient agar, instead of the frequently used Wilbrink’s-boric acid-cephalexin semi-selective medium. All the strains were identified to the species-level with the exception of these five strains: 32L3A, 10L4B, 12L4D, 15L3B and 23L3C. Polyphasic classification of these strains determined that the first three strains are members of the genus Pseudomonas while the latter strains belonged to Sphingomonas sensu stricto (Takeuchi et al., 2001). Analyses of the 16S rDNA, multilocus gene sequences, genome-based DNA–DNA hybridization and average nucleotide identity values, MALDI-TOF analysis and electron microscopy as well as biochemical/physiological traits separated these strains from their closest validly described Pseudomonas or Sphingomonas species. Based on the data from these genotypic and phenotypic analyses presented here, it is concluded that these isolates represent three novel species. We propose the names Pseudomonas triticumensis sp. nov. for strain 32L3AT; Pseudomonas foliumensis sp. nov. for strains 10L4BT and 12L4D, and Sphingomonas albertensis sp. nov. for strains 23L3CT and 15L3B.
Description of Pseudomonas triticumensis sp. nov.
Pseudomonas triticumensis (tri.ti.cum.en’sis N.L. fem. adj., triticumensis from or originating from wheat, the plant from which strain 32L3AT (= DOAB 1067T = CECT 30249T = LMG 32140T) was isolated.
Cells are aerobic, Gram-reaction-negative, non-spore-forming rods (0.6–1.0 μm wide and 2.0 – 4.5 μm long), motile with one, or multiple polar flagella. After 48h on KB, colonies are white-yellowish and circular (average 2-4 mm, in diameter), convex with regular margins and produce fluorescent pigments. Growth in different NaCl concentrations optimal at 4%, and grows at 4°C with optimal growth at 28-30°C and no growth at 40°C. The major cellular fatty acid peaks of the bacteria were C16:0, C16:1 ω6c/C16:1 ω7c (summed feature 3), C18:1 ω7c/C18:1 ω6c (summed feature 8), C17:0 cyclo; C12:0 2-OH, C12:0 3-OH and C10:0 3-OH. Based on Biolog GENIII microplate assays, strain 32L3A utilized 37 carbon sources including d-melibiose, d-arabitol, α-d-glucose, stachyose, myo- inositol, d-mannitol, d-sorbitol, d-cellobiose, gentiobiose, L-serine but not L-lactic acid, citric acid, α-keto-glutaric acid, d-malic acid, l-malic acid, α-keto-butyric acid, acetoacetic acid, propionic acid, acetic acid, d-glucuronic acid, glucuronamide, mucic acid, quinic acid, d-saccharic acid. Using API ZYM assays, these bacteria are positive for alkaline phosphatase, esterase (C4), esterase lipase (C8), leucine arylamidase, valine arylamidase cystine arylamidase, acid phosphatase, Naphthol-AS-BI-phosphohydrolase, and β-glucosidase, but negative for α-glucosidase, α-galactosidases, β-galactosidases and β-glucuronidases. The most abundant fatty acids are C16:0, C16:1ω7c and/or C16:1ω6c (summed feature 3) and C18:1ω7c and/or C18:1ω7c (summed feature 8). These bacteria are resistant to troleandomycin, rifamycin SV, vancomycin, lincomycin but sensitive to nalidixic acid and minocycline. The type strain is 32L3AT (= DOAB 1067T = CECT 30249T = LMG 32140T), isolated from necrotic wheat leaf tissues naturally infected by Xanthomonas translucens from Alberta, Canada. The DNA G + C content of type strain 32L3AT is 59.3mol%.
Description of Pseudomonas foliumensis sp. nov.
Pseudomonas foliumensis (fo.lium’ensis N.L. fem. adj., foliumensis originating from leaf, the wheat leaf from which strains 12L4D and 10L4BT (= DOAB 1069T = CECT 30250T = LMG 32142T) were isolated.
Cells are aerobic, Gram-reaction-negative, non-spore-forming rods (0.6 - 1.0 μm wide and 1.5 – 2.5 μm long), motile with one or two polar flagella. After 48h on KB, colonies are white-yellowish and circular (average 4 mm, in diameter), convex with regular margins and do not produce fluorescent pigments. Growth in NaCl concentrations optimal at 4%, and grows at 4°C with optimal growth at 28–30°C and no growth at 40°C. Strong growth at pH 6 but very weak growth at pH 5. The major cellular fatty acid peaks of the Pseudomonas strains were C16:0, C16:1 ω6c/C16:1 ω7c (summed feature 3), C18:1 ω7c/C18:1 ω6c (summed feature 8), C17:0 cyclo; C12:0 2-OH, C12:0 3-OH and C10:0 3-OH. Based on Biolog GENIII microplate assays, these bacteria readily utilize 47 carbon sources e.g., α-d-glucose, d-mannose, gelatin, pectin, d-fructose, d-galactose, 3-methyl glucose, d-fucose, l-rhamnose, d-raffinose, D-fucose, α-d-Lactose, d-melibiose, β-methyl-d-glucoside, d-salicin, dextrin but not gentiobiose, sucrose, l-aspartic acid, l-glutamic acid, l-histidine, d-glucuronic acid, glucuronamide, mucic acid, quinic acid, citric acid, α-keto-glutaric acid, d-malic acid and l-malic acid. Using API ZYM assays, these bacteria are positive for alkaline phosphatase, esterase (C4), esterase lipase (C8), leucine arylamidase, valine arylamidase cystine arylamidase, acid phosphatase, naphthol-AS-BI-phosphohydrolase, and β-glucosidase, but negative for α-glucosidase, α-galactosidases, β-galactosidases and β-glucuronidases. The most abundant fatty acids are C16:0, C16:1ω7c and/or C16:1ω6c (summed feature 3) and C18:1ω7c and/or C18:1ω7c (summed feature 8). These bacteria are resistant to troleandomycin, rifamycin SV, vancomycin, lincomycin but sensitive to nalidixic acid and minocycline. The type strain is 10L4BT (= DOAB 1069T = CECT 30250T = LMG 32142T), isolated from necrotic wheat leaf tissues naturally infected by Xanthomonas translucens from Alberta, Canada. The DNA G + C content of the type strain 10L4BT is 57.2%.
Description of Sphingomonas albertensis sp. nov.
Sphingomonas albertensis (al.bert.en’sis N.L. fem. adj., albertensis from or originating from Alberta, the Canadian province where strains 15L3B and 23L3CT (= DOAB 1063T = CECT 30248T = LMG 32139T) were isolated. Gram-negative as determined by KOH tests. Growth is observed on LB, NA, NA yeast extract and TSA at a temperature range of 5–28°C but not at 37°C. Colonies are circular, slightly convex, opaque and orange-pigmented. Cells of these bacteria are small rods, 0⋅6–0⋅7 μm × 1⋅7–2⋅6 μm. Catalase- and oxidase-positive. Flagella or endospores not observed. Aerobic. Grew at pH 5.0 but not pH 6.0. Predominant fatty acid peaks in Sphingomonas sp. nov. 23L3C were summed feature 8 (C18:1 ω7c and/or C18:1 ω6c), summed feature 3 (C16:1 ω7c and/or C16:1 ω6c), C14:0 2-OH and C16:0. These bacteria were able to utilize 29 carbon source e.g., d-maltose, d-melibiose, β-methyl-d-glucoside, d-salicin, n-acetyl-β-d-mannosamine, n-acetyl-d-galactosamine, α-d-glucose, d-mannose, l-rhamnose, inosine, d-serine, d-arabitol, d-fructose-6-PO4, glycyl-l-proline, l-galactonic acid lactone, glucuronamide, d-malic acid, l-malic acid; but did not assimilate gentiobiose, sucrose, d-turanose, n-acetyl-d-glucosamine, n-acetyl neuraminic acid, 3-methyl glucose, d-fucose, mucic acid, quinic acid, d-saccharic acid, and citric acid. Based on API ZYM (bioMérieux), these bacteria showed high activity for alkaline phosphatase, esterase (C4), esterase lipase (C 8), leucine arylamidase, valine arylamidase, cystine arylamidase, acid phosphatase, naphthol-AS-BI-phosphohydrolase, β-galactosidases and β-glucosidase, but negative for α-glucosidase, α-galactosidases and β-glucuronidase. These bacteria are resistant to vancomycin and nalidixic acid, but sensitive to rifamycin SV, minocycline and lincomycin.
The type strain, 23L3CT (= DOAB 1063T = CECT 30248T = LMG 32139T), isolated from necrotic wheat leaf tissues naturally infected by Xanthomonas translucens in Alberta, Canada. The DNA G + C content of the type strain 23L3CT is 65.7%.
Data Availability Statement
The seven whole-genome sequences generated in this study have been deposited at DDBJ/ENA/GenBank under the accession numbers: JACONR000000000 (strain 17L2C = DOAB1058), JACONS000000000 (15aL3B = DOAB1061), JACONT0000000 00 (23L3C = DOAB1063), JACONU000000000 (25L1A = DOAB1064), JACONV000000000 (32L3A = DOAB1067), JACONW000000000 (10L4B = DOAB1069), and JACONX00000 0000 (5L2A = DOAB1055). The version described here are the first versions: JACONR010000000, JACONS 010000000, JACONT010000000, JACONU010000000, JACONV 010000000, JACONW010000000, and JACONX010000000. Also, 175 DNA sequences of 16S rRNA and 245 housekeeping genes generated in this study are deposited in GenBank database under the accession numbers: MT878368-MT878416 (16S rRNA), MT941277-MT941292 (leuS), MT941293-MT941306, MT941327-MT941342 and MT941366-MT941372 (gyrB), MT941373-MT941388, MT941343-MT941358 and MT941389-MT941393(rpoB), MT941311-MT941326 (rpoD), MT941394-MT941402. (atpD), MT941359-MT941365 (ppkA), MT941308-MT941310 (recA), and MT941307 (fusA).
Author Contributions
JT received funding, contributed to the conception, design and data collection, and analysis and write-up. MH and GD collected the diseased leaf samples and did the isolation. RX contributed in MLSA gene sequencing. SG performed genome sequencing. DC and KH generated the electron microscopy data. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This study is funded by Agriculture and Agri-Food Canada through projects J-002272, J-001577 and J-000409.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to the staff of the Molecular Technologies Laboratory, Ottawa Research and Development Centre, Ottawa, Canada (MTL-ORDC) for the Illumina NextSeq sequencing; and to the Alberta Agriculture and Forestry for providing the resources to survey wheat fields. Finally, the authors are thankful to all the technicians and summer students who worked on these projects.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.666689/full#supplementary-material
Supplementary Figure 1 | Comparative analysis of matrix-assisted laser desorption/ionization-time-of-flight mass spectrometric profiles of Pseudomonas sp. nov. 32L3A (= DOAB 1067) and Sphingomonas sp. nov. 23L3C (= DOAB 1063) and their corresponding closest phylogenetic neighbors. Asterisks denote sets of seven (m/z) or eight peaks (m/z) that could be used to differentiate the novel strains from the closest known species of the respective genera. MALDI-TOF profiles were visualized using mMass 5.5.0 (Niedermeyer and Strohalm, 2012).
Supplementary Figure 2 | Phylogenetic trees inferred from nucleotide distances derived from genome sequences of representative strains using the Genome BLAST Distance Phylogeny (GBDP) approach: (A) Pseudomonas sp. 10L4B; (B) Pseudomonas sp. 32L3A; and (C) Sphingomonas sp. 23L3C The branch lengths are scaled in terms of GBDP distance formula d5. The numbers above branches are GBDP pseudo-bootstrap support values >50% from 100 replications, with an average branch support of 99.7%. Trees were inferred with FastME 2.1.6.1 (Farris, 1972) and rooted at the midpoint (Lefort et al., 2015).
Footnotes
- ^ https://www.grainews.ca/features/detect-and-avoid-bacterial-leaf-streak/#post-50393
- ^ https://www.dsmz.de/services/online-tools/tygs
- ^ http://ggdc.dsmz.de
- ^ https://antismash.secondarymetabolites.org/#!/start
- ^ https://www.bruker.com
References
Abby, S. S., Neron, B., Menager, H., Touchon, M., and Rocha, E. P. (2014). MacSyFinder: a program to mine genomes for molecular systems with an application to CRISPR-Cas systems. PLoS One 9:e110726. doi: 10.1371/journal.pone.0110726
Abriouel, H., Casado Munoz, M. D. C., Lavilla Lerma, L., Perez Montoro, B., Bockelmann, W., Pichner, R., et al. (2015). New insights in antibiotic resistance of Lactobacillus species from fermented foods. Food Res. Int. 78, 465–481. doi: 10.1016/j.foodres.2015.09.016
Adhikari, T. B., Gurung, S., Hansen, J. M., and Bonman, J. M. (2012). Pathogenic and genetic diversity of Xanthomonas translucens pv. undulosa in North Dakota. Phytopathology 102, 390–402. doi: 10.1094/phyto-07-11-0201
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410.
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online at: http//www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed July 28, 2020).
Anguita-Maeso, M., Olivares-Garcia, C., Haro, C., Imperial, J., Navas-Cortes, J. A., and Landa, B. B. (2019). Culture-dependent and culture-independent characterization of the olive xylem microbiota: Effect of sap extraction methods. Front. Plant Sci. 10:1708.
Behrendt, U., Ulrich, A., and Schumann, P. (2003). Fluorescent pseudomonads associated with the phyllosphere of grasses; Pseudomonas trivialis sp. nov., Pseudomonas poae sp. nov. and Pseudomonas congelans sp. nov. Inter. J. Syst. Evol. Microbiol. 53, 1461–1469. doi: 10.1099/ijs.0.02567-0
Behrendt, U., Ulrich, A., Schumann, P., Meyer, J.-M., and Spröer, C. (2007). Pseudomonas lurida sp. nov., a fluorescent species associated with the phyllosphere of grasses. Inter. J. Syst. Evol. Microbiol. 57, 979–985. doi: 10.1099/ijs.0.64793-0
Bizzini, A., and Greub, G. (2010). Matrix-assisted laser desorption ionization time-of-flight mass spectrometry, a revolution in clinical microbial identification. Clin. Microbiol. Infect. 16, 1614–1619. doi: 10.1111/j.1469-0691.2010.03311.x
Blin, K., Shaw, S., Steinke, K., Villebro, R., Ziemert, N., Lee, S. Y., et al. (2019). antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 47, W81–W87.
Bodilis, J., Ghysels, B., Osayande, J., Matthijs, S., Pirnay, J. P., Denayer, S., et al. (2009). Distribution and evolution of ferripyoverdine receptors in Pseudomonas aeruginosa. Environ. Microbiol. 11, 2123–2135. doi: 10.1111/j.1462-2920.2009.01932.x
Borges-Walmsley, M. I., Beauchamp, J., Kelly, S. M., Jumel, K., Candlish, D., Harding, S. E., et al. (2003). Identification of oligomerization and drug-binding domains of the membrane fusion protein EmrA. J. Biol. Chem. 278, 12903–12912. doi: 10.1074/jbc.m209457200
Brady, C. L., Goszczynska, T., Venter, S. N., Cleenwerck, I., De Vos, P., Gitaitis, R. D., et al. (2011). Pantoea allii sp. nov., isolated from onion plants and seed. Int. J. Syst. Evol. Microbiol. 61, 932–937. doi: 10.1099/ijs.0.022921-0
Busse, H. J., Denner, E. B. M., Buczolits, S., Salkinoja-Salonen, M., Bennasar, A., and Kampfer, P. (2003). Sphingomonas aurantiaca sp. nov., Sphingomonas aerolata sp. nov. and Sphingomonas faeni sp. nov., air- and dustborne and Antarctic, orange-pigmented, psychrotolerant bacteria, and emended description of the genus Sphingomonas. Int. J. Syst. Evol. Microbiol. 53, 1253–1260. doi: 10.1099/ijs.0.02461-0
Camoez, M., Sierra, J. M., Dominguez, M. A., Ferrer-Navarro, M., Vila, J., and Roca, I. (2016). Automated categorization of methicillin-resistant Staphylococcus aureus clinical isolates into different clonal complexes by MALDI-TOF mass spectrometry. Clin. Microbiol. Infect. 22, e161–e161.
Cassago, A., Ferreira, A. P., Ferreira, I. M., Fornezari, C., Gomes, E. R., Greene, K. S., et al. (2012). Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proc. Natl. Acad. Sci. U S A 109, 1092–1097. doi: 10.1073/pnas.1112495109
Cezard, C., Farvacques, N., and Sonnet, P. (2015). Chemistry and biology of pyoverdines, Pseudomonas primary siderophores. Curr. Med. Chem. 22, 165–186. doi: 10.2174/0929867321666141011194624
Chen, H. (2012). Population structure and species description of aquatic Sphingomonadaceae. Ludwig-Maximilians-University Munich: Faculty of Biology, 122. Ph.D. dissertation.
Cherif-Silini, H., Thissera, B., Bouket, A. C., Saadaoui, N., Silini, A., Eshelli, M., et al. (2019). Durum Wheat stress tolerance induced by endophyte Pantoea agglomerans with genes contributing to plant functions and secondary metabolite arsenal. Int. J. Mol. Sci. 2019:20.
Curland, R. D., Gao, L., Bull, C. T., Vinatzer, B. A., Dill-Macky, R., Van Eck, L., et al. (2018). Genetic diversity and virulence of wheat and barley strains of Xanthomonas translucens from the upper midwestern United States. Phytopathology 108, 443–453. doi: 10.1094/phyto-08-17-0271-r
Darriba, D., Posada, D., Kozlov, A. M., Stamatakis, A., Morel, B., and Flouri, T. (2020). ModelTest-NG: A new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 37, 291–294. doi: 10.1093/molbev/msz189
Deletoile, A., Decre, D., Courant, S., Passet, V., Audo, J., Grimont, P., et al. (2009). Phylogeny and identification of Pantoea species and typing of Pantoea agglomerans strains by multilocus gene sequencing. J. Clin. Microbiol. 47, 300–310. doi: 10.1128/jcm.01916-08
Denner, E. B., Paukner, S., Kampfer, P., Moore, E. R., Abraham, W. R., Busse, H. J., et al. (2001). Sphingomonas pituitosa sp. nov., an exopolysaccharide-producing bacterium that secretes an unusual type of sphingan. Int. J. Syst. Evol. Microbiol. 51, 827–841. doi: 10.1099/00207713-51-3-827
Duveiller, E. (1990). Seed detection of Xanthomonas campestris pv. undulosa using a modification of Wilbrink’s agar medium. Parasitica 1990:3–17.
Duveiller, E., Bragard, C., Rudolph, K., and Fucikovsky, L. (1997). “General concepts and methods for the identification of pathogenic bacteria of wheat,” in The bacterial diseases of wheat: Concepts and methods of disease management, eds E. Duveiller, L. Fucikovsky, and K. Rudolph (Mexico, D.F: CIMMYT).
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Farris, J. S. (1972). Estimating phylogenetic trees from distance matrices. Am. Natural. 1972, 645–667. doi: 10.1086/282802
Felsenstein, J. (1985). Confidence limits on phylogenies: An approach using the bootstrap. Evolution 39, 783–791. doi: 10.2307/2408678
Ferreira, M., Bonneau, S., Briand, M., Cesbron, S., Portier, P., Darrasse, A., et al. (2019). Xanthomonas citri pv. viticola affecting grapevine in Brazil: Emergence of a successful monomorphic pathogen. Front. Plant Sci. 10:489.
Fischer-Le Saux, M., Bonneau, S., Essakhi, S., Manceau, C., and Jacques, M. A. (2015). Aggressive Emerging pathovars of Xanthomonas arboricola represent widespread epidemic clones distinct from poorly pathogenic strains, as revealed by Multilocus Sequence Typing. Appl. Environ. Microbiol. 81, 4651–4668. doi: 10.1128/aem.00050-15
Ftayeh, R. M., von Tiedemann, A., and Rudolph, K. W. (2011). A new selective medium for isolation of Clavibacter michiganensis subsp. michiganensis from tomato plants and seed. Phytopathology 101, 1355–1364. doi: 10.1094/phyto-02-11-0045
Gause, G. F., and Brazhnikova, M. G. (1944). Gramicidin S and its use in the treatment of infected wounds. Nature 1944:154.
Giblot-Ducray, D., Marefat, A., Gillings, M. R., Parkinson, N. M., Bowman, J. P., Ophel-Keller, K., et al. (2009). Proposal of Xanthomonas translucens pv. pistaciae pv. nov., pathogenic to pistachio (Pistacia vera). Syst. Appl. Microbiol. 32, 549–557. doi: 10.1016/j.syapm.2009.08.001
Gomila, M., Pena, A., Mulet, M., Lalucat, J., and Garcia-Valdes, E. (2015). Phylogenomics and systematics in Pseudomonas. Front. Microbiol. 6:214.
Gonçalves, R. M., Balbi-Peña, M. I., Soman, J. M., Maringoni, A. C., Taghouti, G., Fischer-Le Saux, M., et al. (2019). Genetic diversity of Curtobacterium flaccumfaciens revealed by multilocus sequence analysis. Eur. J. Plant Pathol. 2019, 189–202. doi: 10.1007/s10658-018-01648-0
Grady, K. L., Sorensen, J. W., Stopnisek, N., Guittar, J., and Shade, A. (2019). Assembly and seasonality of core phyllosphere microbiota on perennial biofuel crops. Nat. Commun. 10:4135.
Hersemann, L., Wibberg, D., Blom, J., Goesmann, A., Widmer, F., Vorholter, F. J., et al. (2017). Comparative genomics of host adaptive traits in Xanthomonas translucens pv. graminis. BMC Genomics 18:35.
Hobbs, E. C., Yin, X., Paul, B. J., Astarita, J. L., and Storz, G. (2012). Conserved small protein associates with the multidrug efflux pump AcrB and differentially affects antibiotic resistance. Proc. Natl. Acad. Sci. U S A 109, 16696–16701. doi: 10.1073/pnas.1210093109
Jacques, M. A., Durand, K., Orgeur, G., Balidas, S., Fricot, C., Bonneau, S., et al. (2012). Phylogenetic analysis and polyphasic characterization of Clavibacter michiganensis strains isolated from tomato seeds reveal that nonpathogenic strains are distinct from C. michiganensis subsp. michiganensis. Appl. Environ. Microbiol. 78, 8388–8402. doi: 10.1128/aem.02158-12
Jain, C., Rodriguez, R. L., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114.
Kampfer, P., Denner, E. B., Meyer, S., Moore, E. R., and Busse, H. J. (1997). Classification of “Pseudomonas azotocolligans” Anderson 1955, 132, in the genus Sphingomonas as Sphingomonas trueperi sp. nov. Int. J. Syst. Bacteriol. 47, 577–583. doi: 10.1099/00207713-47-2-577
Kobayashi, Y., Inagawa, H., Kohchi, C., Kazumura, K., Tsuchiya, H., Miwa, T., et al. (2018). Oral administration of Pantoea agglomerans-derived lipopolysaccharide prevents development of atherosclerosis in high-fat diet-fed apoE-deficient mice via ameliorating hyperlipidemia, pro-inflammatory mediators and oxidative responses. PLoS One 13:e0195008. doi: 10.1371/journal.pone.0195008
Kondejewski, L. H., Farmer, S. W., Wishart, D. S., Kay, C. M., Hancock, R. E., and Hodges, R. S. (1996). Modulation of structure and antibacterial and hemolytic activity by ring size in cyclic gramicidin S analogs. J. Biol. Chem. 271, 25261–25268. doi: 10.1074/jbc.271.41.25261
Konstantinidis, K. T., and Tiedje, J. M. (2007). Prokaryotic taxonomy and phylogeny in the genomic era: advancements and challenges ahead. Curr. Opin. Microbiol. 10, 504–509. doi: 10.1016/j.mib.2007.08.006
Koster, M., van de Vossenberg, J., Leong, J., and Weisbeek, P. J. (1993). Identification and characterization of the pupB gene encoding an inducible ferric-pseudobactin receptor of Pseudomonas putida WCS358. Mol. Microbiol. 8, 591–601. doi: 10.1111/j.1365-2958.1993.tb01603.x
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B., and Stamatakis, A. (2019). RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453–4455. doi: 10.1093/bioinformatics/btz305
Lane, D. J. (1991). 16S/23S rRNA Sequencing. Pages 115-175, in: Nucleic acid techniques in bacterial systematic. E. Stackebrandt and M. Goodfellow, eds. New York, NY: John Wiley and Sons.
Langlois, P. A., Snelling, J., Hamilton, J. P., Bragard, C., Koebnik, R., Verdier, V., et al. (2017). Characterization of the Xanthomonas translucens complex using draft genomes, comparative genomics, phylogenetic analysis and diagnostic LAMP assays. Phytopathology 2017, 519–527. doi: 10.1094/phyto-08-16-0286-r
Lassalle, F., Dastgheib, S. M. M., Zhao, F. J., Zhang, J., Verbarg, S., Fruhling, A., et al. (2020). Phylogenomics reveals the basis of adaptation of Pseudorhizobium species to extreme environments and supports a taxonomic revision of the genus. Syst. Appl. Microbiol. 44:126165. doi: 10.1016/j.syapm.2020.126165
Latasa, C., Roux, A., Toledo-Arana, A., Ghigo, J. M., Gamazo, C., Penades, J. R., et al. (2005). BapA, a large secreted protein required for biofilm formation and host colonization of Salmonella enterica serovar Enteritidis. Mol. Microbiol. 58, 1322–1339. doi: 10.1111/j.1365-2958.2005.04907.x
Lefort, V., Desper, R., and Gascuel, O. (2015). FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 32, 2798–2800. doi: 10.1093/molbev/msv150
Li, Y., Erickson, J. W., Stalnecker, C. A., Katt, W. P., Huang, Q., Cerione, R. A., et al. (2016). Mechanistic basis of glutaminase activation: A key enzyme that promotes glutamine metabolism in cancer cells. J. Biol. Chem. 291, 20900–20910.
Lindh, E., Kjaeldgaard, P., Frederiksen, W., and Ursing, J. (1991). Phenotypical properties of Enterobacter agglomerans (Pantoea agglomerans) from human, animal and plant sources. APMIS 99, 347–352. doi: 10.1111/j.1699-0463.1991.tb05160.x
Liu, W., Li, L., Khan, M. A., and Zhu, F. (2012). Popular molecular markers in bacteria. Mol. Gen. Mikrobiol. Virusol. 2012, 14–17.
Lomovskaya, O., and Lewis, K. (1992). Emr, an Escherichia coli locus for multidrug resistance. Proc. Natl. Acad. Sci. U S A 89, 8938–8942. doi: 10.1073/pnas.89.19.8938
Maier, T., Klepel, S., Renner, U., and Kostrzewa, M. (2006). Fast and reliable MALDI-TOF MS–based microorganism identification. Nat. Methods 3:3.
Malette, A., Xu, R., Gerdis, S., Chi, S. I., Daniels, G. C., Harding, M. W., et al. (2020). Analysis of draft genome sequences of two new Pantoea strains associated with wheat leaf necrotic tissues caused by Xanthomonas translucens reveals distinct species. Microbiol. Resour. Announc. 2020:9.
McMullen, M., and Adhikari, T. (2011). Bacterial leaf streak and black chaff of wheat. Plant disease management. Fargo, ND: North Dakota State University Extension Publication, 1566.
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H. P., and Goker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60. doi: 10.1186/1471-2105-14-60
Meyer, J. M. (2000). Pyoverdines: pigments, siderophores and potential taxonomic markers of fluorescent Pseudomonas species. Arch. Microbiol. 174, 135–142. doi: 10.1007/s002030000188
Miyoshi, T., and Tachibana, Y. (1994). A selective medium for isolation of Pseudomonas syringae, the pathogen of bacterial blossom blight of kiwifruit. J. Phytopathol. 60, 729–734. doi: 10.3186/jjphytopath.60.729
Moore, E. R., and Rossello-Mora, R. (2011). MALDI-TOF MS: a return to phenotyping in microbial identification? Syst. Appl. Microbiol. 34:1. doi: 10.1016/j.syapm.2011.01.001
Mulet, M., Lalucat, J., and Garcia-Valdes, E. (2010). DNA sequence-based analysis of the Pseudomonas species. Environ. Microbiol. 12, 1513–1530.
Niedermeyer, T. H., and Strohalm, M. (2012). mMass as a software tool for the annotation of cyclic peptide tandem mass spectra. PLoS One 7:e44913. doi: 10.1371/journal.pone.0044913
Ogier, J. C., Pages, S., Galan, M., Barret, M., and Gaudriault, S. (2019). rpoB, a promising marker for analyzing the diversity of bacterial communities by amplicon sequencing. BMC Microbiol. 19:171.
Ondov, B. D., Treangen, T. J., Melsted, P., Mallonee, A. B., Bergman, N. H., Koren, S., et al. (2016). Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 17:132.
Osdaghi, E., Young, A. J., and Harveson, R. M. (2020). Bacterial wilt of dry beans caused by Curtobacterium flaccumfaciens pv. flaccumfaciens: A new threat from an old enemy. Mol. Plant Pathol. 21, 605–621. doi: 10.1111/mpp.12926
Oyaizu, H., and Komagata, K. (1983). Grouping of Pseudomonas species on the basis of cellular fatty acid composition and the quinone system with special reference to the existence of 3-hydroxy fatty acids. J. Gen. Appl. Microbiol. 1983, 17–40. doi: 10.2323/jgam.29.17
Parkinson, N., Cowie, C., Heeney, J., and Stead, D. (2009). Phylogenetic structure of Xanthomonas determined by comparison of gyrB sequences. Int. J. Syst. Evol. Microbiol. 59, 264–274.
Parks, D. H., Rinke, C., Chuvochina, M., Chaumeil, P. A., Woodcroft, B. J., Evans, P. N., et al. (2018). Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 3:253. doi: 10.1038/s41564-017-0083-5
Paul, B., Dixit, G., Murali, T. S., and Satyamoorthy, K. (2019). Genome-based taxonomic classification. Genome 62, 45–52. doi: 10.1139/gen-2018-0072
Peng, Z., Hu, Y., Zhang, J., Huguet-Tapia, J. C., Block, A. K., Park, S., et al. (2019). Xanthomonas translucens commandeers the host rate-limiting step in ABA biosynthesis for disease susceptibility. Proc. Natl. Acad. Sci. U S A 116, 20938–20946. doi: 10.1073/pnas.1911660116
Rahimi-Khameneh, S., Hsieh, S., Xu, R., Avis, T. J., Li, S., Smith, D., et al. (2019). Pathogenicity and a TaqMan Real-Time PCR for Specific Detection of Pantoea allii, a Bacterial Pathogen of Onions. Plant Dis. 103, 3031–3040. doi: 10.1094/pdis-03-19-0563-re
Regalado, J., Lundberg, D. S., Deusch, O., Kersten, S., Karasov, T., Poersch, K., et al. (2020). Combining whole-genome shotgun sequencing and rRNA gene amplicon analyses to improve detection of microbe-microbe interaction networks in plant leaves. ISME J. 14, 2116–2130. doi: 10.1038/s41396-020-0665-8
Ryu, E. (1940). A simple method of differentiation between gram-positive and gram-negative organisms without staining. Kitasato Archives Exp. Med. J. 1940, 58–63.
Sapkota, S., Mergoum, M., and Liu, Z. (2020). The translucens group of Xanthomonas translucens: Complicated and important pathogens causing bacterial leaf streak on cereals. Mol. Plant Pathol. 21, 291–302. doi: 10.1111/mpp.12909
Sasser, M. (1990). Identification of bacteria by gas chromatography of cellular fatty acids. Newark, DE: MIDI Technical Note 101 MIDI Inc.
Sauget, M., Valot, B., Bertrand, X., and Hocquet, D. (2017). Can MALDI-TOF Mass Spectrometry Reasonably Type Bacteria? Trends Microbiol. 25, 447–455. doi: 10.1016/j.tim.2016.12.006
Sennhauser, G., Amstutz, P., Briand, C., Storchenegger, O., and Grutter, M. G. (2007). Drug export pathway of multidrug exporter AcrB revealed by DARPin inhibitors. PLoS Biol. 5:e7. doi: 10.1371/journal.pbio.0050007
Shore, A. C., Brennan, O. M., Ehricht, R., Monecke, S., Schwarz, S., Slickers, P., et al. (2010). Identification and characterization of the multidrug resistance gene cfr in a Panton-Valentine leukocidin-positive sequence type 8 methicillin-resistant Staphylococcus aureus IVa (USA300) isolate. Antimicrob Agents Chemother. 54, 4978–4984. doi: 10.1128/aac.01113-10
Simpson, J. T., Wong, K., Jackman, S. D., Schein, J. E., Jones, S. J., and Birol, I. (2009). ABySS: a parallel assembler for short read sequence data. Genome Res. 19, 1117–1123. doi: 10.1101/gr.089532.108
Singh, P., Santoni, S., Weber, A., This, P., and Peros, J. P. (2019). Understanding the phyllosphere microbiome assemblage in grape species (Vitaceae) with amplicon sequence data structures. Sci. Rep. 9:14294.
Smith, E. F., Jones, L. R., and Reddy, C. S. (1919). The black chaff of wheat. Science 50:48. doi: 10.1126/science.50.1280.48
Sneath, P. H. A. (1989). Analysis and interpretation of sequence data for bacterial systematics: the view of a numerical taxonomist. Syst. Appl. Microbiol. 1989, 15–23. doi: 10.1016/s0723-2020(89)80036-0
Spitaels, F., Wieme, A., Balzarini, T., Cleenwerck, I., Van Landschoot, A., De Vuyst, L., et al. (2014). Gluconobacter cerevisiae sp. nov., isolated from the brewery environment. Int. J. Syst. Evol. Microbiol. 64, 1134–1141. doi: 10.1099/ijs.0.059311-0
Stackebrandt, E. (2003). The richness of prokaryotic diversity: there must be a species somewhere. Food Technol. Biotechnol. 2003, 17–22.
Takeuchi, M., Hamana, K., and Hiraishi, A. (2001). Proposal of the genus Sphingomonas sensu stricto and three new genera, Sphingobium, Novosphingobium and Sphingopyxis, on the basis of phylogenetic and chemotaxonomic analyses. Int. J. Syst. Evol. Microbiol. 51, 1405–1417. doi: 10.1099/00207713-51-4-1405
Tambong, J. T. (2019). Taxogenomics and systematics of the genus Pantoea. Front. Microbiol. 10:2463.
Tambong, J. T., Xu, R., and Bromfield, E. S. P. (2017). Pseudomonas canadensis sp. nov., a biological control agent isolated from a field plot under long-term mineral fertilization. Int. J. Syst. Evol. Microbiol. 67, 889–895. doi: 10.1099/ijsem.0.001698
Tambong, J. T., de Cock, A. W., Tinker, N. A., and Levesque, C. A. (2006). Oligonucleotide array for identification and detection of Pythium species. Appl. Environ. Microbiol. 72, 2691–2706. doi: 10.1128/aem.72.4.2691-2706.2006
Tambong, J. T., Xu, R., Kaneza, C. A., and Nshogozabahizi, J. C. (2014). An in-depth analysis of a multilocus phylogeny identifies leuS as a reliable phylogenetic marker for the genus Pantoea. Evol Bioinform. 10, 115–125.
Tchagang, C. F., Xu, R., Doumbou, C. L., and Tambong, J. T. (2018). Genome analysis of two novel Pseudomonas strains exhibiting differential hypersensitivity reactions on tobacco seedlings reveals differences in nonflagellar T3SS organization and predicted effector proteins. MicrobiologyOpen 7:e00553. doi: 10.1002/mbo3.553
Thompson, C. C., Chimetto, L., Edwards, R. A., Swings, J., Stackebrandt, E., and Thompson, F. L. (2013). Microbial genomic taxonomy. BMC Genomics 14:913. doi: 10.1186/1471-2164-14-913
van den Mooter, Steenackers, M., Maertens, M., Gossele, C., De Vos, F., and Swings, P. (1987). Differentiation between Xanthomonas campestris pv. graminis ISPP L 1980, pv. phleipratensis ISPP List 1980 emend., pv. poae Egli an Schmidt 1982, and pv. arrhenatheris Egli and Schmidt 1982, by numerical analysis of phenotypic features and protein gel electrophoresis. J. Phytopathol. 1987, 135–156. doi: 10.1111/j.1439-0434.1987.tb00442.x
Vauterin, L., Hoste, B., Kersters, K., and Swings, J. (1995). Reclassification of Xanthomonas Inter. J. Syst.Bacteriol. 1995, 472–489.
Vauterin, L., Yang, P., Hoste, B., Pot, B., Swings, J., and Kersters, K. (1992). Taxonomy of xanthomonads from cereals and grasses based on SDS-PAGE of proteins, fatty acid analysis and DNA hybridization. J. Gen. Microbiol. 1992, 1467–1477. doi: 10.1099/00221287-138-7-1467
Waleron, M., Misztak, A., Waleron, M., Franczuk, M., Jonca, J., Wielgomas, B., et al. (2019). Pectobacterium zantedeschiae sp. nov. a new species of a soft rot pathogen isolated from Calla lily (Zantedeschia spp.). Syst. Appl. Microbiol 42, 275–283. doi: 10.1016/j.syapm.2018.08.004
Wattam, A. R., Abraham, D., Dalay, O., Disz, T. L., Driscoll, T., Gabbard, J. L., et al. (2014). PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 42, D581–D591.
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Xu, L., Dong, Z., Fang, L., Luo, Y., Wei, Z., Guo, H., et al. (2019). OrthoVenn2: a web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 47, W52–W58.
Keywords: wheat bacterial leaf streak disease, Xanthomonas translucens, cultivable bacteria, Genome-based DNA-DNA hybridization (gDDH), Average Nucleotide Identity (ANI), Multilocus sequence analysis (MLSA), MALDI-TOF
Citation: Tambong JT, Xu R, Gerdis S, Daniels GC, Chabot D, Hubbard K and Harding MW (2021) Molecular Analysis of Bacterial Isolates From Necrotic Wheat Leaf Lesions Caused by Xanthomonas translucens, and Description of Three Putative Novel Species, Sphingomonas albertensis sp. nov., Pseudomonas triticumensis sp. nov. and Pseudomonas foliumensis sp. nov. Front. Microbiol. 12:666689. doi: 10.3389/fmicb.2021.666689
Received: 10 February 2021; Accepted: 22 March 2021;
Published: 19 May 2021.
Edited by:
Svetlana N. Dedysh, Winogradsky Institute of Microbiology, Russian Academy of Sciences (RAS), RussiaReviewed by:
Jillian Lang, Colorado State University, United StatesPortier Perrine, INRA Centre Angers-Nantes Pays de la Loire, France
Copyright © 2021 M.W. Harding, G.C. Daniels, and Her Majesty the Queen in Right of Canada, as represented by the Minister of Agriculture and Agri-Food Canada for the contribution of James T. Tambong, Renlin Xu, Suzanne Gerdis, Denise Chabot and Keith Hubbard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James T. Tambong, james.tambong@canada.ca