 Mickael Lafond1†Alexandra S. Tauzin2†Laetitia Bruel1
Mickael Lafond1†Alexandra S. Tauzin2†Laetitia Bruel1 Elisabeth Laville2
Elisabeth Laville2 Vincent Lombard3Jérémy Esque2
Vincent Lombard3Jérémy Esque2 Isabelle André2Nicolas Vidal4
Isabelle André2Nicolas Vidal4 Frédérique Pompeo5Nathalie Quinson1
Frédérique Pompeo5Nathalie Quinson1 Josette Perrier1Michel Fons6
Josette Perrier1Michel Fons6 Gabrielle Potocki-Veronese2
Gabrielle Potocki-Veronese2 Thierry Giardina1*
Thierry Giardina1*- 1Aix-Marseille Université, CNRS, Centrale Marseille, iSm2, Marseille, France
- 2TBI, Université de Toulouse, CNRS, INRAE, INSA, Toulouse, France
- 3CNRS, Aix-Marseille Université, AFMB, Marseille, France
- 4Yelen Analytics, Aix-Marseille Université, ICR, Marseille, France
- 5Aix-Marseille Université, CNRS, IMM, LCB, Marseille, France
- 6Aix-Marseille Université, CNRS, IMM, BIP, Marseille, France
Plant α-galactosides belonging to the raffinose family oligosaccharides (RFOs) and considered as prebiotics, are commonly degraded by α-galactosidases produced by the human gut microbiome. In this environment, the Ruminococcus gnavus E1 symbiont–well-known for various benefit–is able to produce an original RgAgaSK bifunctional enzyme. This enzyme contains an hydrolytic α-galactosidase domain linked to an ATP dependent extra-domain, specifically involved in the α-galactoside hydrolysis and the phosphorylation of the glucose, respectively. However, the multi-modular relationships between both catalytic domains remained hitherto unexplored and has been, consequently, herein investigated. Biochemical characterization of heterologously expressed enzymes either in full-form or in separated domains revealed similar kinetic parameters. These results were supported by molecular modeling studies performed on the whole enzyme in complex with different RFOs. Further enzymatic analysis associated with kinetic degradation of various substrates followed by high pressure anionic exchange chromatography revealed that catalytic efficiency decreased as the number of D-galactosyl moieties branched onto the oligosaccharide increased, suggesting a preference of RgAgaSK for RFO’s short chains. A wide prevalence and abundance study on a human metagenomic library showed a high prevalence of the RgAgaSK encoding gene whatever the health status of the individuals. Finally, phylogeny and synteny studies suggested a limited spread by horizontal transfer of the clusters’ containing RgAgaSK to only few species of Firmicutes, highlighting the importance of these undispersed tandem activities in the human gut microbiome.
Introduction
Raffinose family oligosaccharides (RFOs) are mainly found in the seeds of vegetables. From a structural point of view, RFOs are alpha-galactosyl derivatives of sucrose, and the family mainly contains raffinose (i.e., one galactosyl unit attached to sucrose), stachyose (i.e., two D-galactosyl units attached to sucrose) and verbascose (i.e., three D-galactosyl units attached to sucrose) with a degree of polymerization (DP) from 3 to 5, respectively. In some plants, RFOs are formed by up to 15 galactosyl residues (Bachmann et al., 1994). No human enzyme is able to degrade these compounds. Dietary RFOs pass through the stomach and the small intestine without major changes to reach the colon almost unchanged before being fermented or degraded by anaerobic commensal bacteria (Martínez-Villaluenga et al., 2008). During decades, RFOs were considered to be antinutritional factors, as their consumption in large quantities leaded to negative effects such as abdominal pains, flatulence, bloating and diarrhea, mainly due to their fermentation in the colon (Martínez-Villaluenga et al., 2008). Nowadays, the vision of the RFOs is changing thanks to the in-depth studies carried out in the last decade and they are now considered as prebiotics (Collins and Reid, 2016) as their consumption at the right dose could promote the growth of beneficial bacteria including Bifidobacteria and Lactobacilli (Ejby et al., 2016; Collins et al., 2017; Zartl et al., 2018). In addition, in vitro studies have also shown that the presence of raffinose can reduce the growth of pathogens (Shoaf et al., 2006). Therefore, in the highly competitive ecosystem constituted by the gastrointestinal tract, RFOs represent a selective advantage for organisms capable of metabolizing them to the detriment of others, and for the host health.
In the CAZy database1, the bacterial enzymes responsible for the degradation of RFOs, α-galactosidases, are classified into the glycoside hydrolase (GH) families 4, 27, 36, 57, 97, and 110, but mainly gathered within the families GH27 and GH36 (Lombard et al., 2014). Over the last 15 years, the structures of several α-galactosidases have been determined within the GH27, GH36, and GH97 families (Fujimoto et al., 2003, 2009; Garman and Garboczi, 2004; Golubev et al., 2004; JCSG., 2005, 2013; Comfort et al., 2007; Lieberman, 2009; Okuyama et al., 2009; Fernández-Leiro et al., 2010; Guce et al., 2010; Vorobiev et al., 2010; Bruel et al., 2011; Fredslund et al., 2011; Merceron et al., 2012; Yu et al., 2014; Adamson et al., 2016; Kikuchi et al., 2017; Kytidou et al., 2018; Hobbs et al., 2019; Rowland et al., 2019). The α-galactosidases from GH27 and GH36 families share a common catalytic domain characterized by a (β/α)8-barrel folding type and a C-terminal domain composed of β-sheets. In contrast, α-galactosidases in the GH36 family have an N-terminal domain that is missing in GH27 family members (Comfort et al., 2007).
Remarkably, a bifunctional enzyme belonging to the GH36 family, the α-(1,6)-galactosidase/sucrose kinase (RgAgaSK) naturally produced by Ruminococcus gnavus E1, a human gut commensal isolated from healthy human feces (Ramare et al., 1993), was identified (Bruel et al., 2011). The RgAgaSK encoding gene, found highly transcribed in vivo, belongs to the Rgaga1 polysaccharide utilization locus (PUL)–suggested to be involved in RFOs metabolization–which includes one transcriptional regulator, three ABC transporter components and one sucrose 6Fructose-phosphate phosphorylase (RgSPP) encoding genes (Bruel et al., 2011; Tauzin et al., 2019). In addition, the study of the Rgaga1 PUL organization highlighted a high synteny with loci from Blautia hansenii DSM 20583, Eubacterium rectale ATCC 33656 and to a lesser extent Clostridium sp. L2-50 (Tauzin et al., 2019). Interestingly, RgAgaSK consists of two domains: one closely related to α-galactosidases GH36 family and the other one containing a nucleotide-binding motif (Walker A motif) (Bruel et al., 2011). The bi-functional RgAgaSK enzyme has shown a high sequence identity (i.e., more than 90%) with enzymes from B. hansenii DSM 20583 and E. rectale ATCC 33656, while the locus from Clostidium sp. L2-50 harbors separated α-galactosidase and kinase encoding genes, the latter being completely absent in other related loci from Lactobacillales (Tauzin et al., 2019). From a biochemical point of view, RgAgaSK showed its ability to hydrolyze soluble α-galactosides derived from sucrose (e.g., raffinose), and also to use ATP as co-substrate to phosphorylate specifically sucrose on the C6 position of glucose residue generating sucrose-6Glucose-phosphate (S6P). In addition, RgSPP, from the same PUL, showed a selective activity on sucrose 6Fructose-phosphate (S6FP) acting both in phosphorolysis releasing α-D-glucose-1-phosphate (G1P) and α-D-fructose-6-phosphate (F6P), and in reverse phosphorolysis from G1P and F6P to S6FP. These results bring out an original glycolytic pathway from a well-known gut bacterium due to a unique PUL involved in RFO metabolic pathways reinforcing the crucial role of the kinase domain (Bruel et al., 2011; Tauzin et al., 2019).
Here, we investigate the relationships between the kinase and the α-galactosidase GH36 domains using biochemical, enzymatic and molecular modeling approaches. A phylogenetic analysis of the whole GH36 family was constructed to gain insight into the α-galactosidase evolution–especially for the RgAga module–while a synteny study was carried out to examine the spread of the Rgaga1 PUL. Finally, the abundance and the prevalence of the RgAgaSK gene were quantified in the human gut metagenome to assess its importance in healthy or pathological contexts.
Results
Expression and Purification of the Recombinant RgAgaSK and Its Truncated Variants
To investigate the interactions between both domains of the RgAgaSK enzyme, we generated two variants consisting of each domain individually expressed in Escherichia coli BL21 cells, the N-terminal domain corresponding to the α-galactosidase domain (RgAga, from Ala-1 to Lys-720) and the C-terminal domain corresponding to the kinase domain (RgSK, from Arg-724 to Gln-935), respectively. The proteins were purified by Ni2+ affinity chromatography. The purified recombinant proteins showed major bands on SDS-PAGE at 80 and 25 kDa, respectively (Figures 1A,B, respectively). The molecular masses of these recombinant proteins were consistent with their theoretical molecular mass (81,245 and 23,898 Da, respectively). In each case, up to 10 mg.L–1 of pure recombinant proteins were obtained.
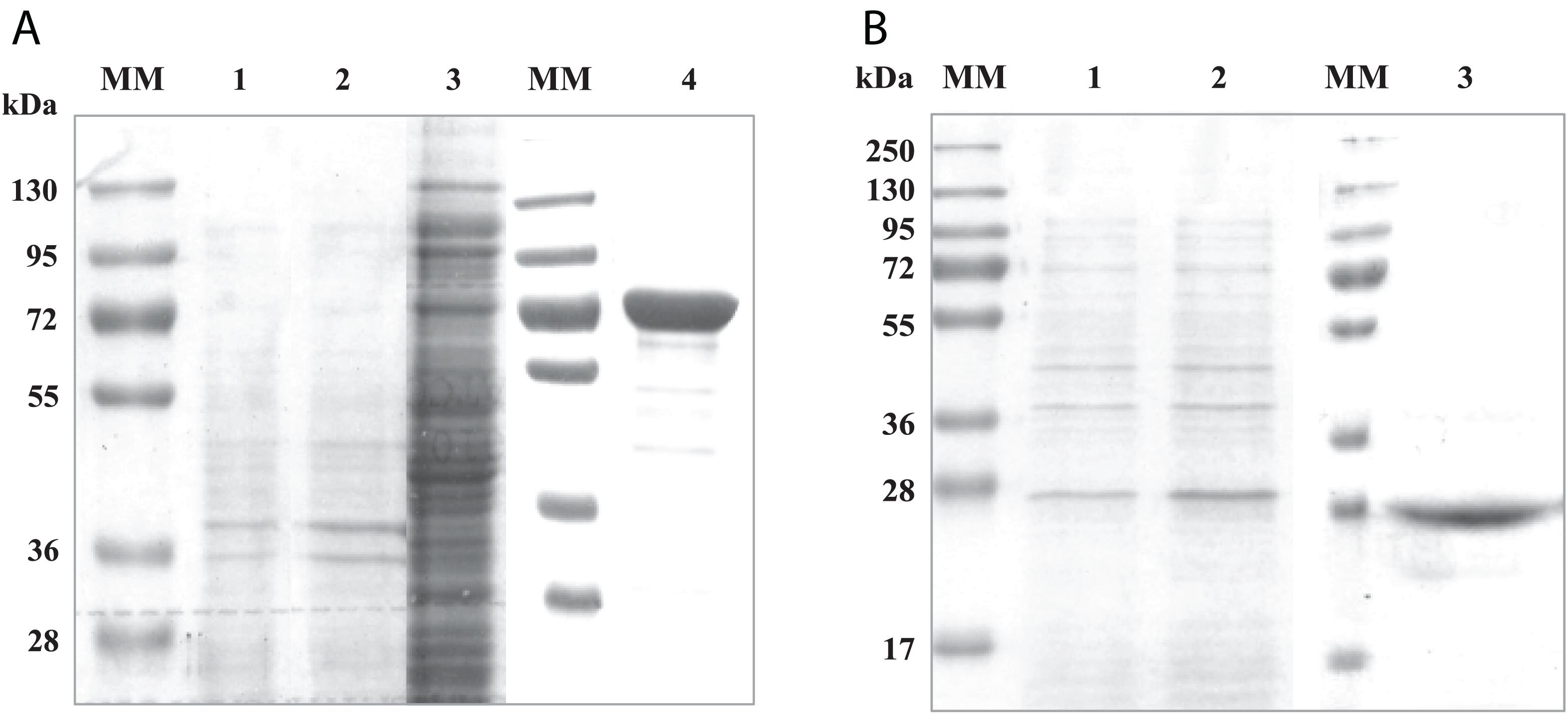
Figure 1. SDS-PAGE analysis of recombinant RgAga (A) and RgSK (B) expressed in Escherichia coli BL21 cells. (A) (1) Samples from cell extracts of E. coli BL21 pOPINE after induction, (2) cell extracts of E. coli BL21 pOPINE:rgaga before induction, (3) cell extracts of E. coli BL21 pOPINE:rgaga after induction (4) purified fraction of RgAga after elution from affinity chromatography and (MM) molecular weight markers. (B) (1) Samples from cell extracts of E. coli BL21 pOPINE:rgsk before induction, (2) cell extracts of E. coli BL21 pOPINE:rgsk after induction, (3) purified fraction of RgSK after elution from affinity chromatography and (MM) molecular weight markers.
To ensure the functionality of the isolated kinase domain, a kinase activity assay has been used in presence of sucrose and ATP, as previously described for RgAgaSK (Bruel et al., 2011; Figure 2). The migration profile of the phosphorylation reaction using RgSK showed the production of a phosphorylated-sucrose, product demonstrating the ability of RgSK to catalyze a phosphorylation reaction.
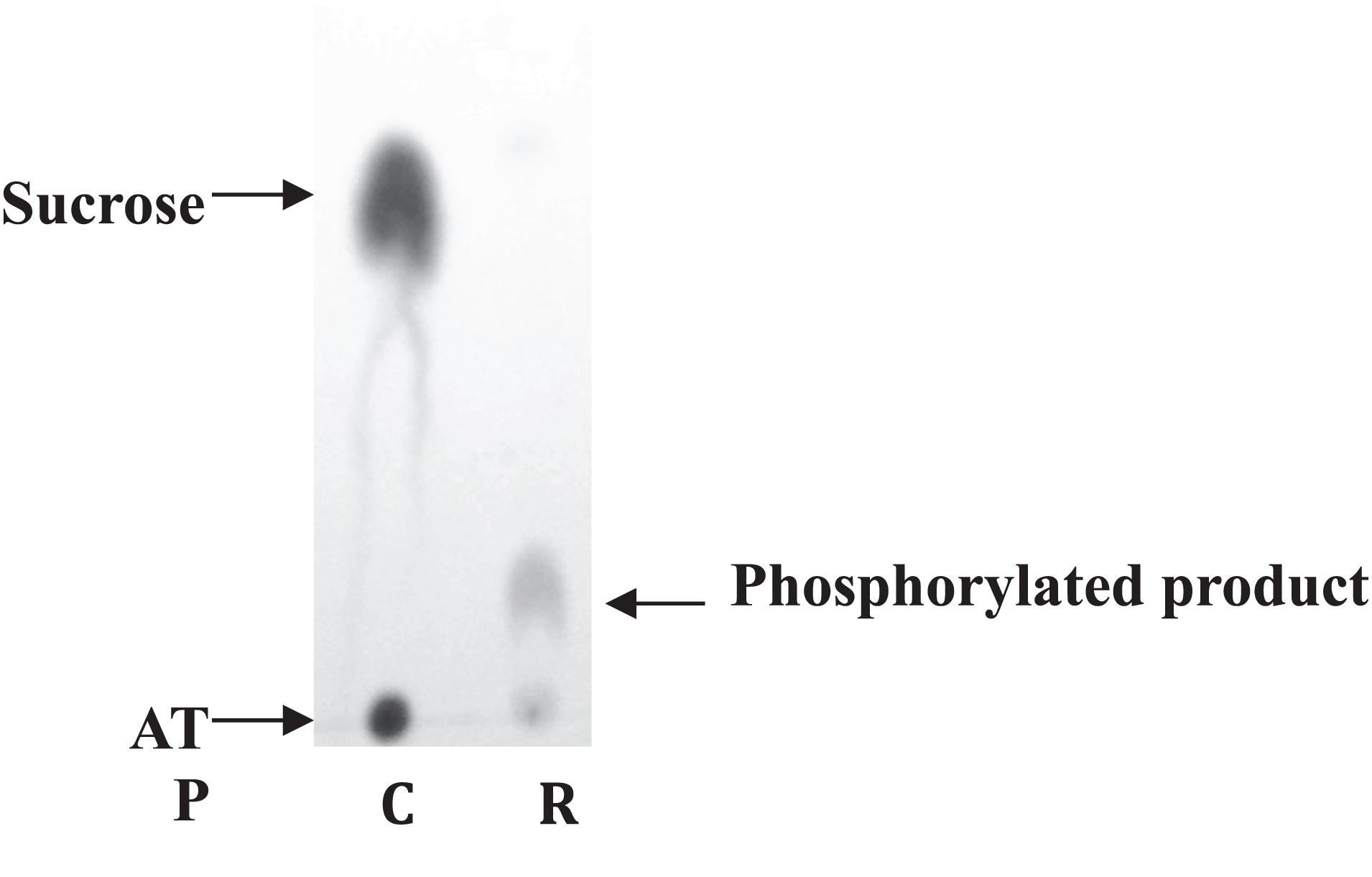
Figure 2. Sucrose kinase activity analysis of RgSK by TLC. Sucrose and ATP (R) were incubated together for 30 min at 30°C in presence of RgSK. The control (C) corresponded to the phosphorylation reaction in the presence of RgSK previously denatured.
Stability of RgAgaSK and RgSK
The stability of RgAgaSK and RgSK during storage at 4°C was tested, and protein degradation and sucrose kinase activity were monitored over time: 2 (D2), 8 (D8), and 15 (D15) days (Figure 3). The evaluation of the protein degradation on SDS-PAGE showed that RgSK was stable up to 8 days and completely degraded after 15 days, whereas RgAgaSK results in a partial degradation with the main band corresponding to the RgAga module alone (∼80 kDa).
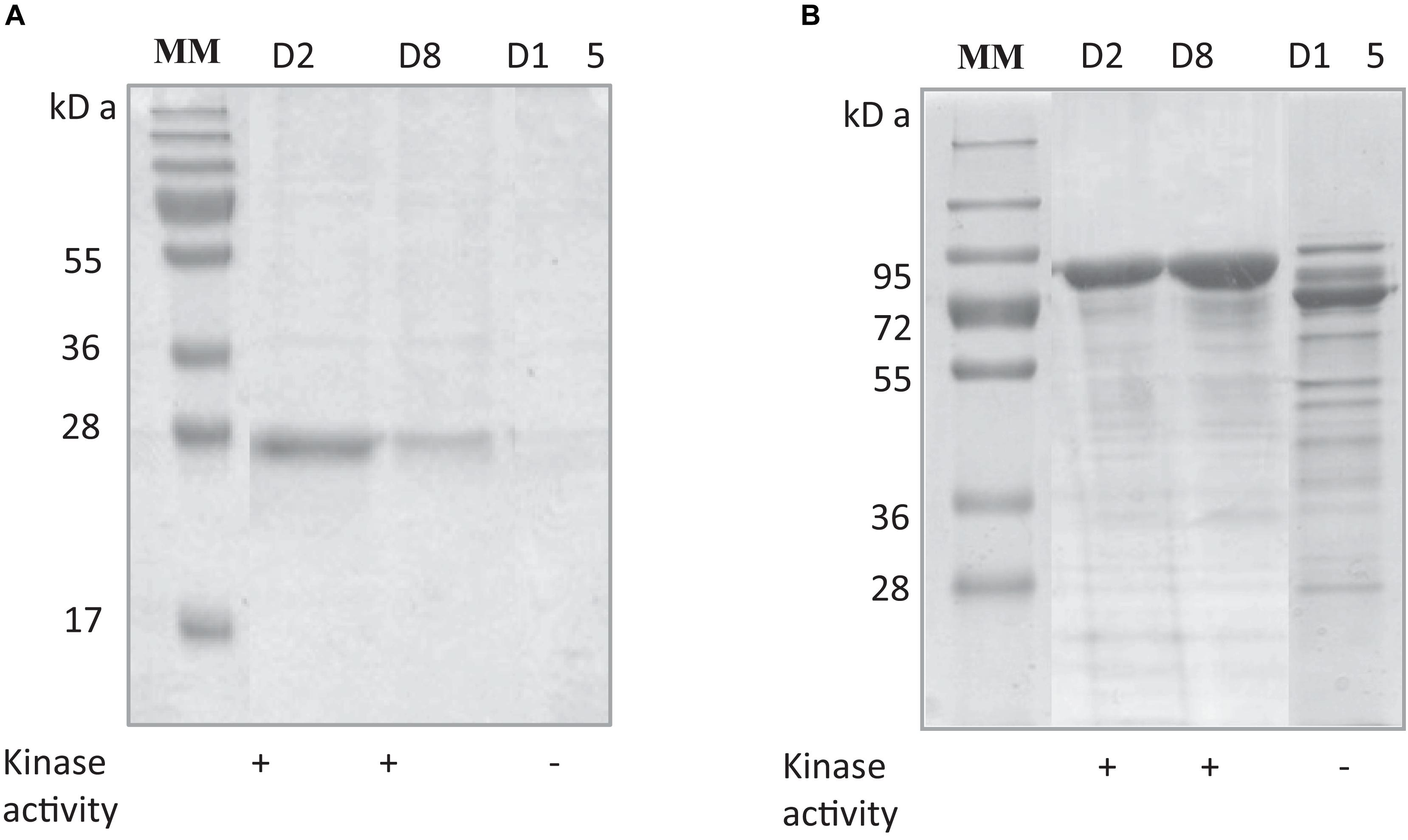
Figure 3. Comparison of protein degradation and sucrose kinase activity over time of (A) RgSK and (B) RgAgaSK. The proteins were stored at +4°C in HEPES buffer 50 mM at pH 7.0. The degradation of (A) RgSK and (B) RgAgaSK was evaluated by protein separation using SDS-PAGE and their respective kinase activity was evaluated on TLC, whose results are referenced in the tables under the gels. (+) means presence of sucrose kinase activity, whereas (–) correspond to absence of sucrose kinase activity.
RgAgaSK and RgAga: α-Galactosidase Activity Comparison on a Synthetic Substrate
The effects of pH and temperature on the enzyme stability were investigated for both recombinant enzymes, and no significant difference was found between RgAgaSK (whole enzyme) and RgAga (α-galactosidase domain). Indeed, enzymes displayed an optimum activity in the pH range from 5.0 to 7.0, whereas the activity was lost under pH 3.0 and over pH 8.0 (Figure S1). At pH 6.0, the optimum temperature was 42°C for both enzymes, and the activity drastically decreased above 55°C (Figure S1). The hydrolysis of pNPGal as substrate confirmed the α-exo-acting activity of the RgAga domain. Moreover, the kinetic parameters in optimum conditions showed that the absence of the kinase domain in RgAga did not alter the affinity (Km) with pNPGal substrate (1.2 ± 0.2 mM versus 1.3 ± 0,2 mM for RgAgaSK). The Vmax and the catalytic efficiency (kcat/Km) were the same order of magnitude (Table 1).
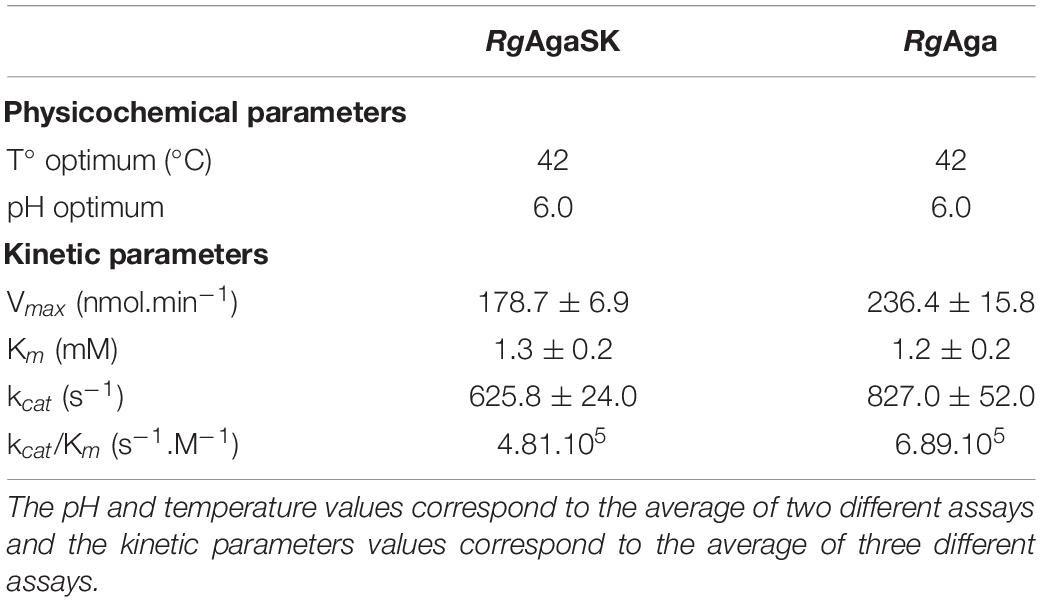
Table 1. Physicochemical and kinetic parameters for RgAgaSK and RgAga using pNPGal as substrate.
RgAgaSK and RgAga: α-Galactosidase Activity Comparison on Natural Substrates
Raffinose family oligosaccharides hydrolysis is usually measured using a global method (galactose oxidase or carbohydrate reducing end) which does not allow determining the hydrolysis kinetic when one of the products formed is itself a substrate. In order to analyze the α-galactosidase activity of RgAgaSK and RgAga toward natural substrates derived from plants [i.e., RFOs including raffinose (DP3), stachyose (DP4), and verbascose (DP5)], we followed RFOs hydrolysis using anionic exchange chromatography coupled with a pulsed amperometric detection (HPAEC-PAD) (Figure 4). Both RgAgaSK and RgAga showed similar hydrolysis profiles of raffinose (Figures 4A,B). In each case, 50% of the raffinose were consumed in 2 min reaching 80% after about 10 min. In parallel, the equimolar increase of galactose was a good indicator of the reliability of the method used (see blue and green line points in Figures 4A,B). Otherwise as observed in Figures 4C,D, stachyose was completely hydrolyzed after 30 min, and the profiles were similar whatever the protein modularity considered, i.e., with or without kinase domain. During the first 5 min, the hydrolysis of stachyose was concomitant with the production of raffinose, sucrose, and galactose. This result means that for two stachyose molecules in the medium, one was completely degraded (two galactoses and one sucrose) whereas the second was partially (one galactose and one raffinose), leading to a molar ratio of 3:1 for galactose and raffinose/sucrose, respectively. However, after 5 min, raffinose did not accumulate contrary to the sucrose, meaning that the majority of raffinose was completely degraded (two galactoses and one sucrose). Finally, for verbascose as for stachyose, no accumulation of intermediate products (i.e., stachyose and raffinose) was observed while the sucrose considered as a final product increased all over the hydrolysis reaction (Figures 4E,F).
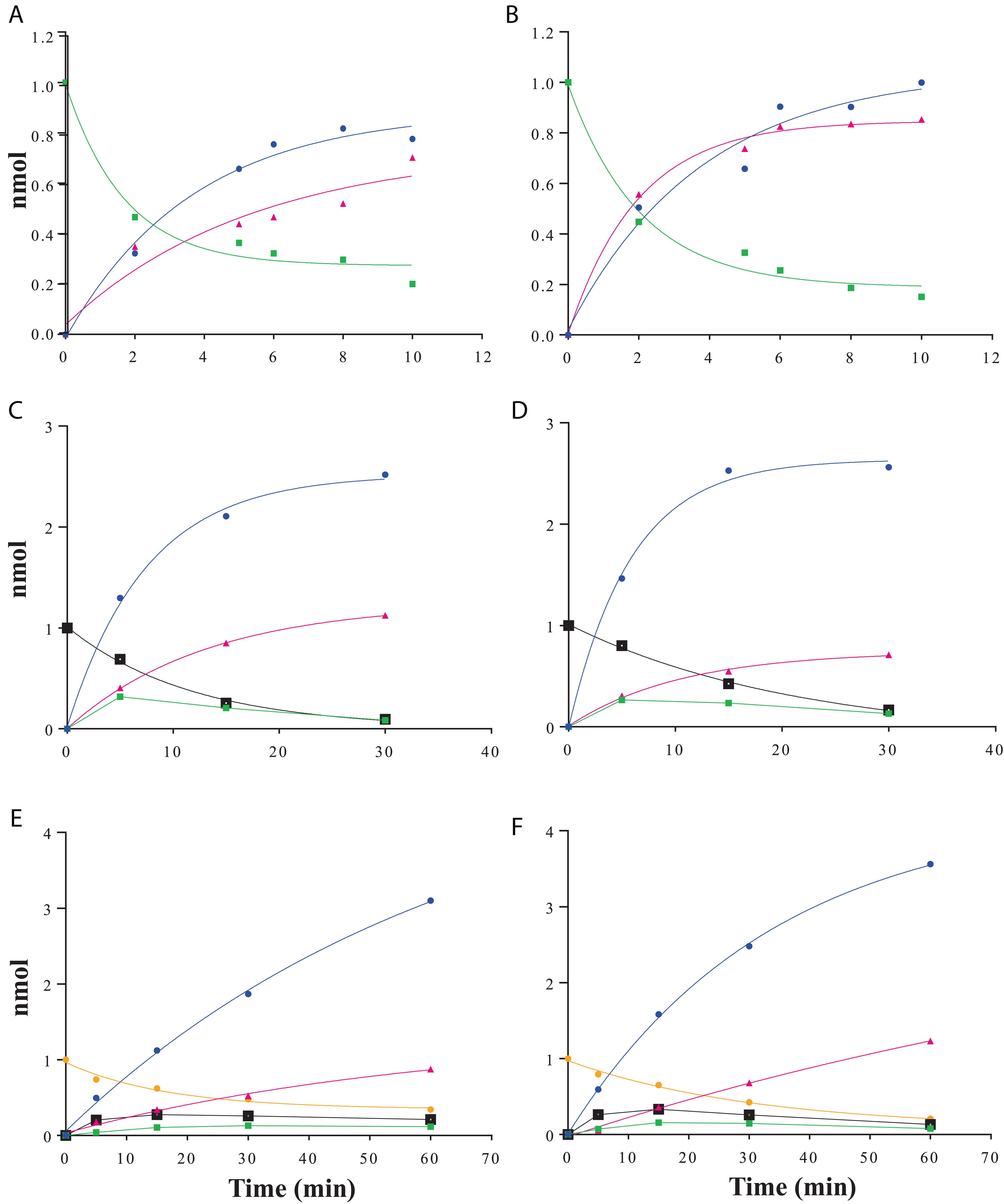
Figure 4. RFOs hydrolysis kinetics using HPAEC-PAD analysis. Raffinose hydrolysis by RgAgaSK (A) and RgAga (B); stachyose hydrolysis by RgAgaSK (C) and RgAga (D) and verbascose hydrolysis by RgAgaSK (E) and RgAga (F). Galactose appears in blue, sucrose in pink, raffinose in green, stachyose in black, and verbascose in yellow. Recombinant proteins (25 nM) were incubated at 42°C with 100 μM of stachyose. The values correspond to the averages of two isolated tests.
Subsequently, the catalytic efficiencies (kcat/Km) of RgAgaSK and RgAga toward the different RFOs were determined using the Matsui equation (Matsui et al., 1991; Figure 5). No substantial difference was observed between the two enzymes whatever the substrate used, i.e., raffinose, stachyose, and verbascose. Interestingly, for both enzymes, the catalytic efficiency is dependent on the substrate length. Indeed, this value decreases when the substrate length – the number of galactosyl residues – increases (i.e., DP3 to DP5), as shown in Figure 5.
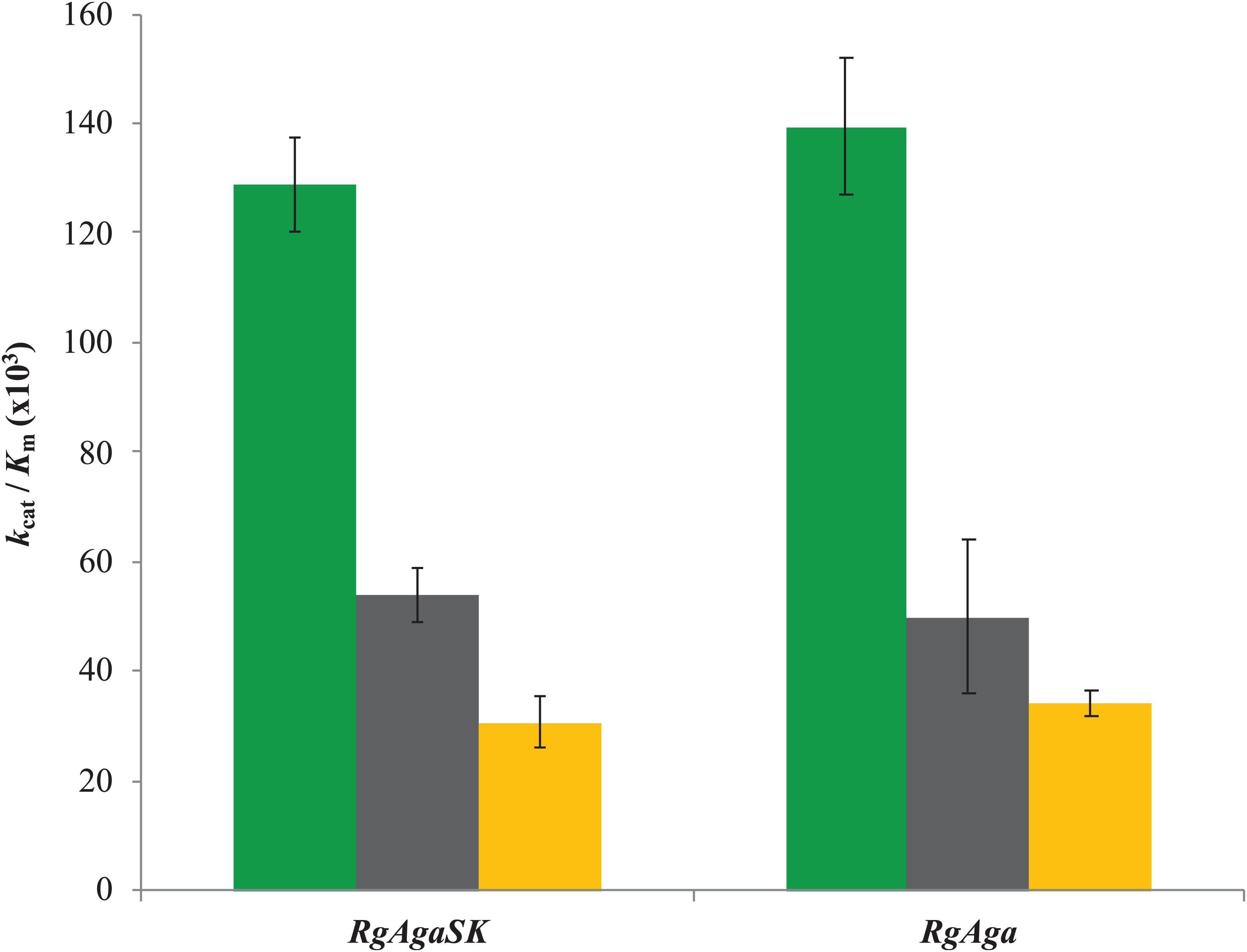
Figure 5. Catalytic efficiencies of RgAgaSK and RgAga toward different RFOs. Raffinose appears in green, stachyose in black, and verbascose in yellow. Catalytic efficiencies were calculated from results obtained between 0 and 10 min for raffinose hydrolysis and between 0 and 5 min for stachyose and verbascose hydrolysis. The values corresponded to the average of two different assays.
RgAgaSK and RgSK: Hydrolytic and Kinase Activities Comparison on Natural Substrates
In order to investigate the influence of the α-galactosidase domain on kinase activity, the kinetic parameters of RgAgaSK and RgSK using sucrose as substrate were analyzed and compared.
Kinetic Parameters of Kinase Activity
The kinase activity of RgAgaSK and RgSK was evaluated following ATP consumption using bioluminescence assay. Under these basal conditions, different enzymatic activity analyses were performed for both RgAgaSK and RgSK using different concentrations of ATP from 0 to 10 mM (Figure 6). Regarding ATP hydrolysis, the results showed that in both cases the enzymes presented Michaelian kinetics (Figure 6). The Michaelis constant (Km) and the catalytic constant (kcat) were similar as those of RgAgaSK (8.4 ± 1.9 mM and 38.1 ± 4.8 s–1, respectively), and for RgSK (6.3 ± 1.8 mM and 27.9 ± 4.3 s–1, respectively). In addition, the catalytic efficiencies (kcat/Km) calculated for RgAgaSK and RgSK were 4.53.103 and 4.42.103 s–1.M–1, respectively (Table 2). To conclude, as observed above, the absence of the α-galactosidase domain does not seem to affect the kinase activity.
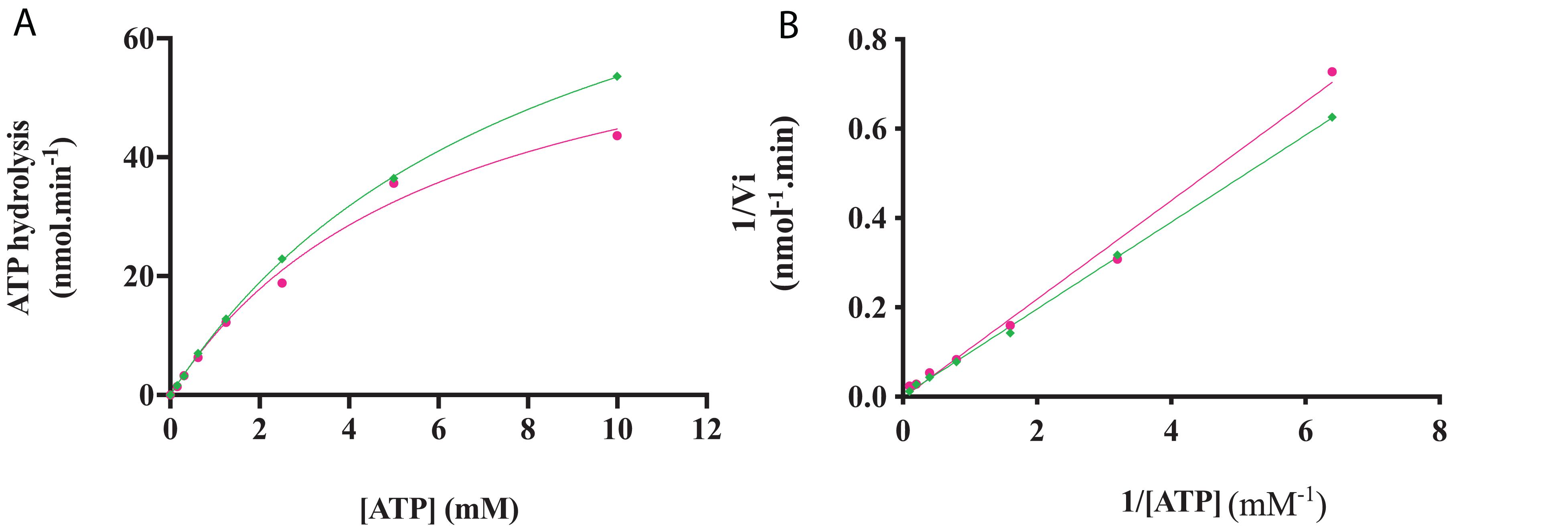
Figure 6. RgAgaSK and RgSK kinetics toward ATP. The phosphorylation rate of ATP by RgAgaSK (in green) and RgSK (in pink) was evaluated by using the representation of Michaelis-Menten (A) and Lineweaver Burk (B) plot. The proteins (1.3 μM) were incubated with varying concentrations of ATP ranging from 0 to 10 mM for 3 min at 30°C and pH 8.0. The values correspond to the average of three different assays.
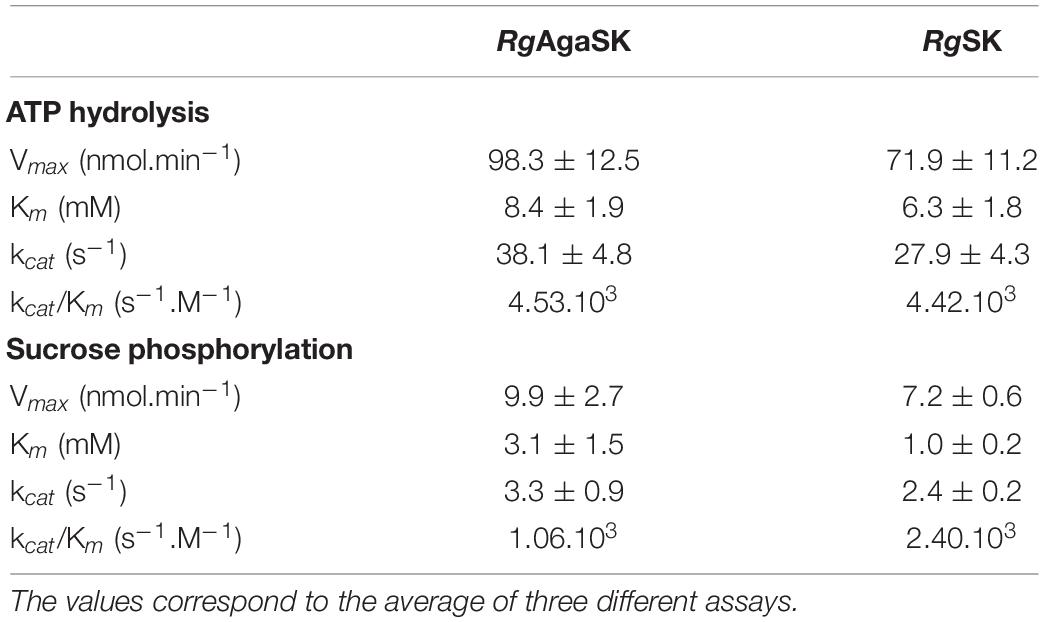
Table 2. Comparison of RgAgaSK and RgSK ATP hydrolysis kinetic parameters and phosphorylation kinetic parameters using sucrose as substrate.
Kinetic Parameters of Phosphorylation Activity
The characterization of the phosphorylation activity on sucrose by RgAgaSK and RgSK was assessed by HPAEC-PAD. Analysis of the phosphorylation reaction profiles for each form showed the formation of a single product eluted at 22 min (Figure 7) and identified as S6P by mass spectrometric analysis (Figure S2). Then, in order to determine and compare the kinetic parameters of RgAgaSK and RgSK for sucrose phosphorylation reaction, enzymatic activity analyses were performed in initial rate conditions with different sucrose concentrations (Figure 8). The results obtained showed that both RgAgaSK and RgSK had a Michaelian sucrose phosphorylation kinetics with a Michaelis constant (Km) and a catalytic constant (kcat) for RgAgaSK of 3.1 ± 1.5 mM and 3.3 ± 0.9 s–1 and for RgSK of 1.0 ± 0.2 mM and 2.4 ± 0.2 s–1, respectively (Figure 8). The analysis of specificity constants allowed to calculate a similar affinity for sucrose between the two proteins but a two times faster phosphorylation rate for RgSK. The calculation of the catalytic efficiency (kcat/Km), which reflects the specificity of the enzyme with respect to its substrate, makes it possible to compare the activities of the two proteins between them with 1.06.103 and 2.40. 103 s–1.M–1 for RgAgaSK and RgSK, respectively (Table 2). Thus, conversely to the results observed above between RgAgaSK and RgSK where the SK domain has no impact to the α-galactosidase activity, here the absence of the α-galactosidase domain seems slightly affecting the kinase activity leading to an increase of the phosphorylation activity. This observation could be explained by the topology of the domain interactions where the RgSK domain could not interact with the RgAga active site due to the length of the linker, rather than RgSK seems more dependent of the linker flexibility and of the RgAga domain movement on the bottom (see Figure 9 below).
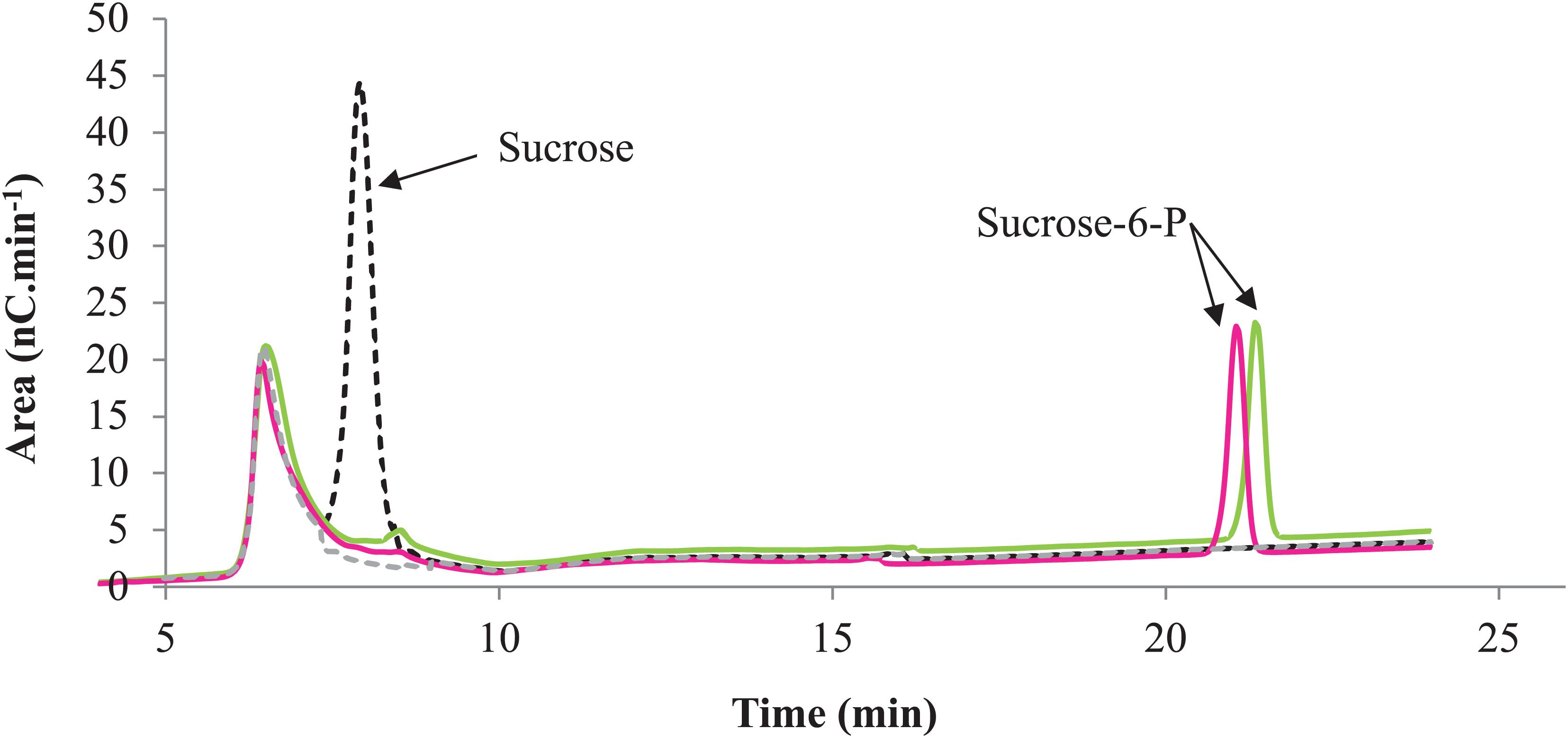
Figure 7. Phosphorylation profile of sucrose by RgAgaSK and RgSK. HPAEC-PAD chromatograms superimposition with RgAgaSK reaction appearing in green, with RgSK appearing in pink. The reaction control (black dash line) was the reaction with denatured enzyme, and the buffer control (gray dash line) was the buffer only without substrates and enzymes.
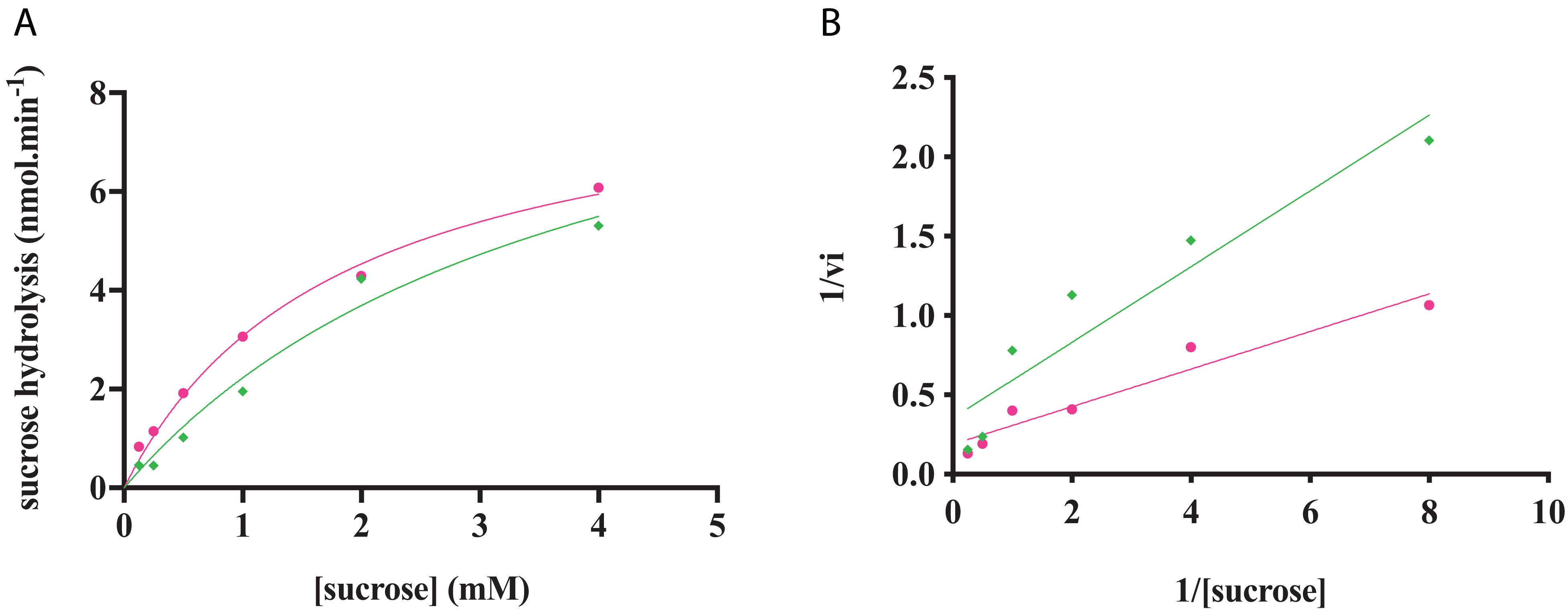
Figure 8. RgAgaSK and RgSK kinetics toward sucrose. The phosphorylation rate of sucrose by RgAgaSK (in green) and RgSK (in pink) was evaluated by using the representation of Michaelis-Menten (A) and Lineweaver Burk (B) plot. The proteins (50 nM) were incubated with varying concentrations of sucrose ranging from 0 to 4 mM for 5 min at 30°C. The values are the average of three different assays.
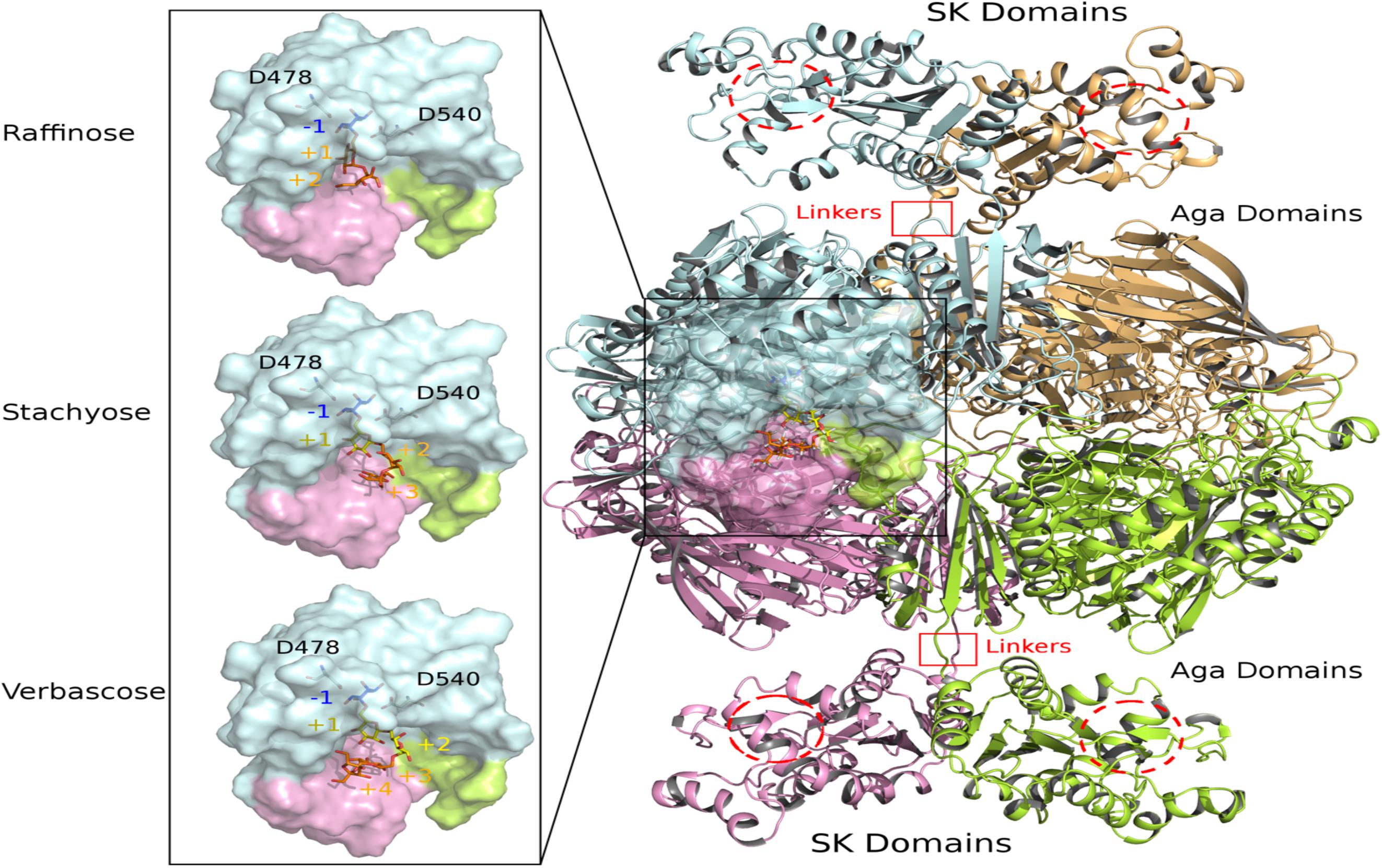
Figure 9. View of modeled quaternary assembly of RgAgaSK-RFO complexes. The tetrameric RgAgaSK structure is shown as carton on the right. Each subunit is highlighted according to a specific color (pale cyan, bright orange, lemon, and pink). SK Domains are located on the top and the bottom of the complex with putative active sites indicated with red dashed circles. Aga domains are viewed as an assembly in the middle, connected to the SK domains by linkers highlighted by red squares. The active site of one Aga subunit is shown as transparent surface. A zoom on this region is displayed on the left with three complexed ligands (raffinose, stachyose, and verbascose) docked independently in Aga active site. For reference, the catalytic residues are shown in ball & sticks at subsite –1. The galactosyl bound in the subsite –1 is colored in blue whereas the sucrose moiety at the end chain is colored in orange. Additional galactosyl units within stachyose and verbascose are colored in yellow.
Modeling of Enzyme Assemblies and Complexes With RFOs
The 3D model of RgAgaSK was built using the 3D X-ray structure structure of α-galactosidase domain in its tetrameric assembly [PDB: 2YFN; (Bruel et al., 2011)], which was then fused to the RgSK domain modeled by threading techniques due to the lack of structural templates covering the full sequence. The spatial constraints imposed by the quaternary organization of RgAga and the C-terminal orientation of the four chains led to the introduction of the RgSK domains oriented toward the exterior of the tetrameric assembly (Figure 9). The proposed 3D model of RgAgaSK suggests that interface between RgSK domains involves a network of stabilizing interactions. It also reveals that such RgSK domains are not located in the proximity of RgAga active sites, although conformational rearrangements of the linkers could bring closer RgAga and RgSK domains without creating interferences in cross-talking of active sites as suggested previously.
Detailed inspection of the 3D organization of RgAga domains reveals that substrate binding pocket is formed by loop regions of the catalytic (β/α)8-barrel and also from N- and C-terminal regions of the two adjacent subunits (Figure 9), as earlier reported (Bruel et al., 2011). Catalytic dyad, formed by the nucleophile Asp478 and the acid/base Asp540, is buried at the bottom of RgAga active site pocket (subsite −1), properly oriented with respect to the anomeric center of the galactosyl residue at the non-reducing end of the RFOs (see blue sticks in Figure 9). Molecular surface representation of RgAga domains also reveals an active site of about 12Å depth with subsites +2, +3 rather exposed to the solvent whereas subsite −1 is buried within the protein (Figure 9). α-galactosidases from Geobacillus stearothermophilus (GsAgaA and GsAgaB), which both present 46% of sequence identity with the R. gnavus E1 α-galactosidase domain, also share a highly similar quaternary organization (Merceron et al., 2012).
The crystallographic structure of the inactivated GsAgaA was earlier determined in complex with raffinose and stachyose bound in subsites −1, +1, +2, and +3, enabling a detailed mapping of the network of stabilizing interactions (Merceron et al., 2012). This network of interactions reported to stabilize sugars bound at subsites −1, +1, and +2 is found to be maintained in complexes of RgAga with docked RFOs. While binding interactions are definitely more extensive at subsite −1 involving amino acid residues Trp336, Asp366, Trp411, Arg443, Lys476, Trp537, positive subsites progressively involve fewer interactions with bound sugars mostly involving amino acid residues from adjacent subunit such as Asp52, Phe55, Arg66 (Figure 10). All amino acid residues interacting with RFOs are highly conserved with the exception of His199 from subsite +1.
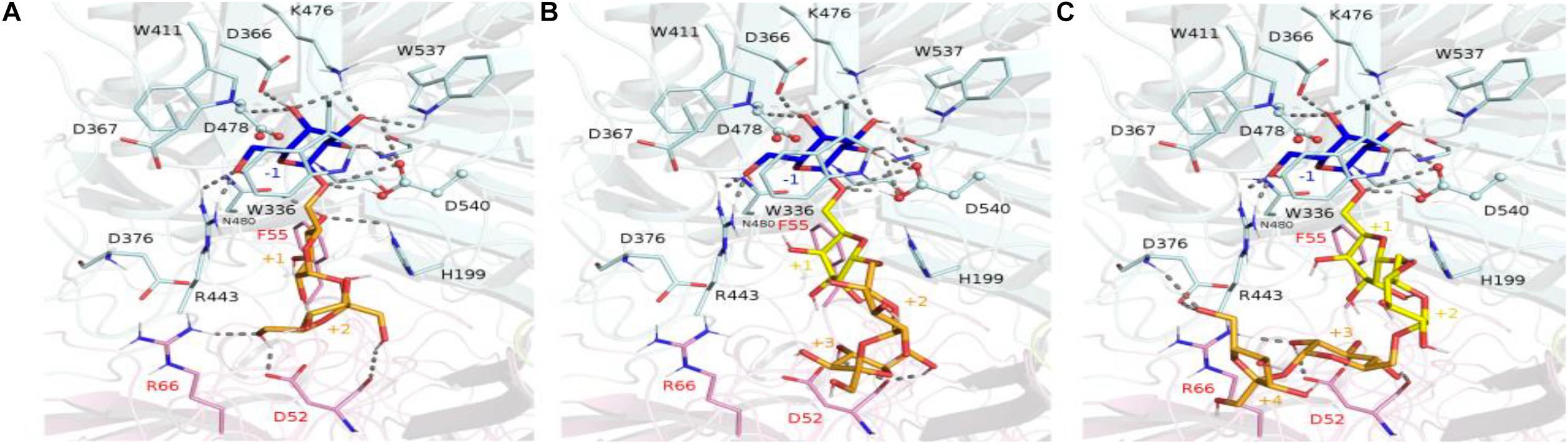
Figure 10. Molecular view of RFOs docked in RgAga active site. The interacting amino acid residues and the ligands, i.e., raffinose (A), stachyose (B), and verbascose (C), are shown in sticks and colored with the same color code as in Figure 9. Dashed lines represent the potential hydrogen bonds between any ligand with their surrounding residues. The binding subsites are labeled as –1, +1, +2, +3, and +4.
Phylogenetic Analysis of the GH36 Family
In order to gain more insight into the phylogenetic diversity of the GH36 family, a phylogenetic tree has been constructed, using the 843 sequences extracted from the CAZy database (Figure 11A; Lombard et al., 2014). GH36 enzymes are distributed in diverse phylogenetic groups from different ecosystems like soil, marine or human gut microbiome and covering the three super-kingdoms. The present phylogenetic and taxonomic analysis reveals five main clades containing characterized α-galactosidase enzymes (i.e., Firmicutes, Actinobacteria, Proteobacteria, Bacteroidetes, and Eukaryota). The distribution of the 75 characterized GH36 enzymes is globally well spread over the phylogenetic tree, with an α-galactosidase activity found for most of them. Nevertheless, specificities are not restricted to galactosidases. Indeed, α-N-acetylgalactosaminidase, β-L-arabinopyranosidase, raffinose synthase or even stachyose synthase activities are also identified mainly in Eukaryota clade. Therefore, we observed that RgAgaSK belongs to a Firmicutes clade constituted by almost only bacterium from the intestinal microbiome (e.g., Blautia, Clostridium, Eubacterium, Roseburia, Coprococcus) (Figure 11B). Interestingly, the SK domain appeared linked with α-galactosidase domain only for some of species found in this Firmicutes clade including RgAgaSK (Figure 11C).
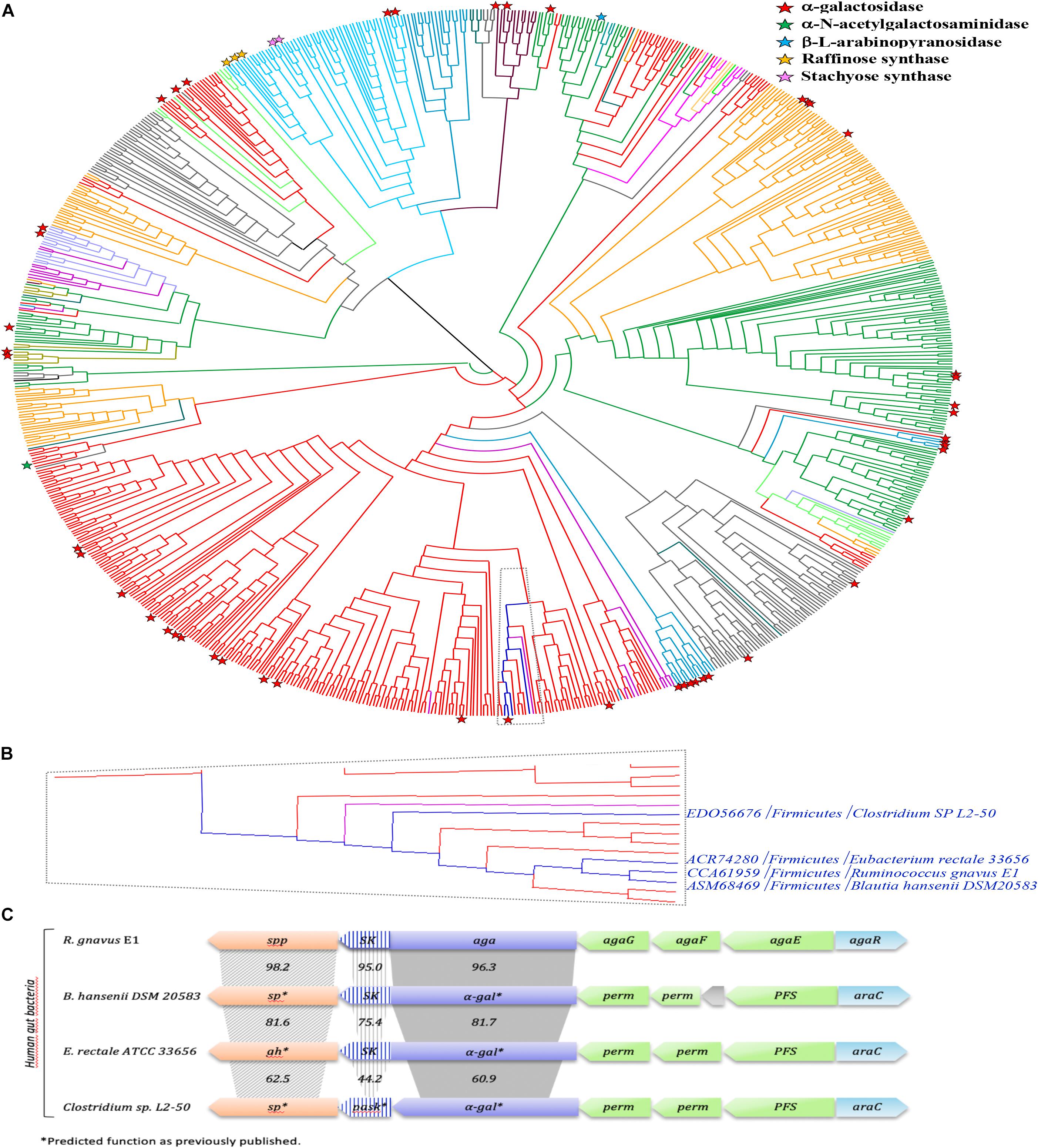
Figure 11. Phylogenetic study of the GH36 family and RgAgaSK genomic environment. (A) A Phylogenic tree of the GH36 family was constructed including all the GH36 referenced in the CAZy database at 11/06/2019. Members sharing full synteny with RgAgaSK are indicated in deep blue. The tree has been colored according to the phylogenetic classification of the organisms. Indeed, Firmicutes appeared in red, Bacteroidetes in gray, Proteobacteria in orange, Actinobacteria in deep green, Acidobacteria in light green, Eukaryota in light blue, Fungi in medium blue, Archaebacteria in deep violet, Spirochaetes in light violet, Thermotogae in light purple, Verrucomicrobia in water-green, Deinococcus in kaki, and Chloroflexi in pink. Characterized enzymes are labeled with a red star for their α-galactosidase activity, a green star for their α-N-acetylgalactosaminidase activity, a blue star for their β-L-arabinopyranosidase activity, an orange star for their raffinose synthase activity and a pink star for their stachyose synthase activity. (B) Zoom of the clade which includes RgAgaSK and members sharing full synteny with RgAgaSK (in deep blue) and other sequences from human gut bacteria. (C) Genomic environment of the RgAgaSK encoding gene. The values indicate the percentage of identities between the encoding genes according to their modularity i.e., kinase, α-gal and spp or sp.
As previously mentioned, the RgAgaSK gene is located into the Rgaga1 PUL described by Bruel et al. (2011), and detailed by Tauzin et al. (2019) (Figure 11C). To figure out the accurate role of such Gram positive PUL in the human gut microbiome, we performed a synteny study based on all the prokaryotic strains containing GH13_18 and GH36 neighborhood enzymes coupled with a deep mining of R. gnavus E1 genome, that led us to propose a new model of sucrose, S6FP and RFOs metabolic pathways (Figure 11C; Tauzin et al., 2019). As represented in this last figure, the loci alignment shows (i) only four syntenies with high conservation of this usual gram positive PUL organization model (i.e., with B. hansenii, E. rectale and with a lesser extent C. spL2-50), (ii) high identities between α-gal or sk genes (i.e., more than 60%), and (iii) high identities between spp genes (i.e., more than 62%) which are always in downstream position of those encoding the bi-functional kinase and α-galactosidase enzymes. Nevertheless, the GH36 sequences comparative analysis showed a synteny limited to these four loci, and that not all sequences found in the same clade as RgAgaSK, or even in the whole phylogenetic tree, are included in similar PULs (Figure 11). This observation actually suggests that there is no common ancestor, but that dissemination has occurred by horizontal evolution to only a few species within the human digestive tract.
Prevalence and Abundance in the Human Gut Microbiome
Then, we searched the microbial gene richness of the four known GH36 associated to a kinase whether in the form of a bifunctional protein or in the form of two distinct proteins in the microbiome of 760 European subjects, by recovering prevalence and abundance of the homologous sequences in the frequency matrix of genes (MetaHIT Consortium, Li et al., 2014; Figure 12A). The four GH36s found in syntenic PULs belong to gram-positive Firmicutes listed in the most frequent components of the human indigenous intestinal microbiome (MetaHIT Consortium, Qin et al., 2010). The bimodular GH36 of R. gnavus E1, B. hansenii DSM 20583 and E. rectale ATCC 33656 (accession numbers CCA61959, ASM68469, and ACR74280, respectively), are highly prevalent whatever the medical status of the subjects. The gene of E. rectale was the most abundant. Besides, the separated GH36 and SK genes of Clostridium sp. L2-50 (accession numbers EDO56676 and WP_008399817.1, respectively) present the same richness profiles. They are less prevalent in IBD subjects than in healthy ones.
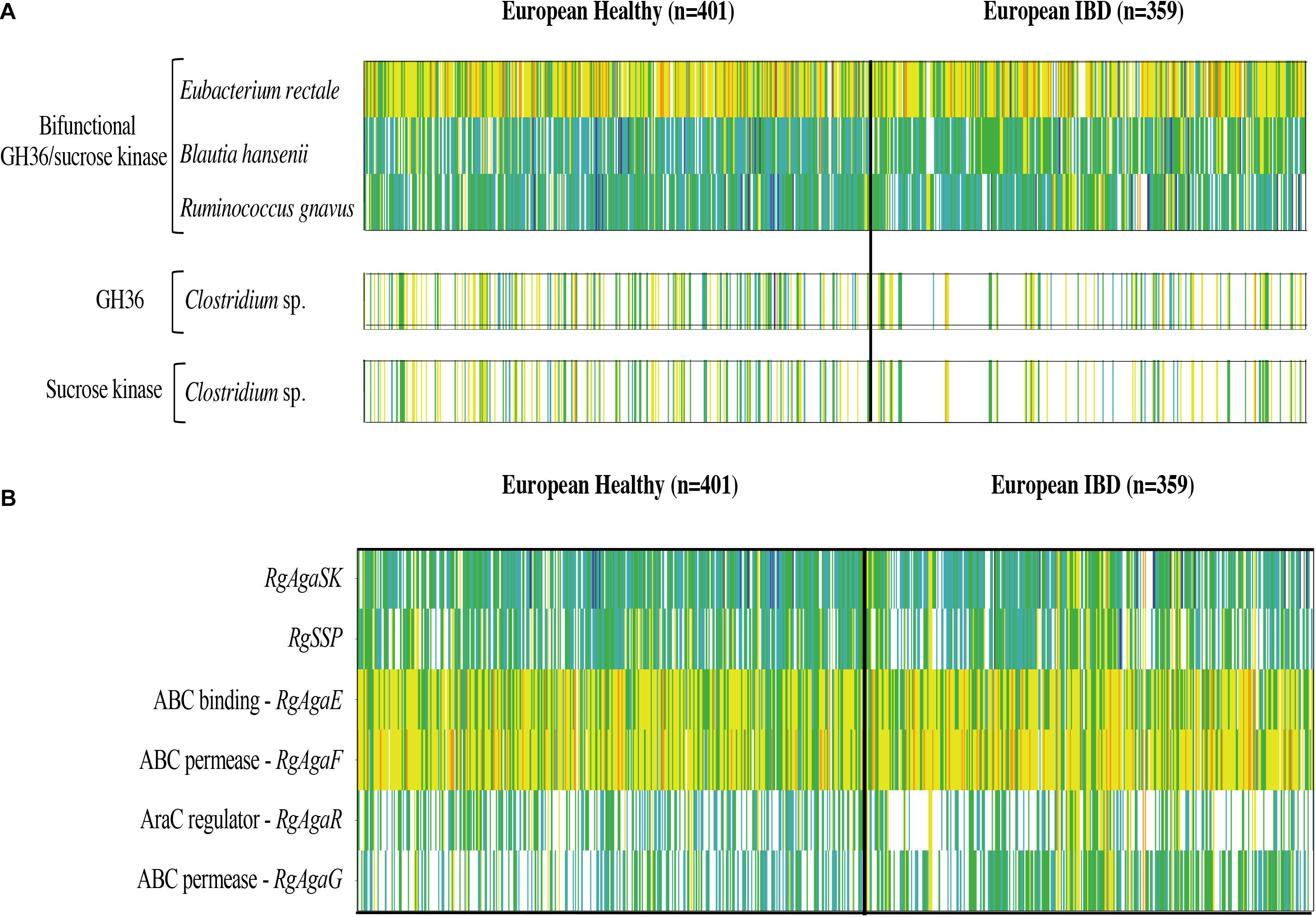
Figure 12. Abundance and prevalence of the GH36 and sucrose kinase encoding genes targeted in this study (A), and of the genes of the R. gnavus Rgaga1 locus (B) in the human gut microbiome of European healthy and IBD affected individuals. Gene abundance in the gut metagenome of 760 European subjects is represented by a color scale: white, not detected; blue, turquoise, green, yellow, and orange, increasing abundance with a 10-fold change between colors.
In addition, we examined the prevalence and abundance of the genes of R. gnavus Rgaga1 locus previously described in Bruel et al. (2011) and in Tauzin et al. (2019). We found six homologous genes in the human gut catalog. The prevalence and abundance profiles of the six genes of the cluster are different, with a higher abundance of AgaE and AgaF genes involved in substrate binding and transport (Figure 12B).
Discussion
Here, the function of each domains of the RgAgaSK, RgAga, and RgSK, have been investigated. The two domains RgAga and RgSK have been cloned and expressed separately in E. coli cells. Analysis of the physico-chemical properties of the α-galactosidase domain revealed no difference with the whole protein, whereas according to the kinetic parameters, RgAga appears slightly more efficient for pNPGal hydrolysis compared to RgAgaSK. However, these differences were not observed with the tested RFOs: raffinose, stachyose and verbascose. Thus, the RgAga activity on natural substrates was not significantly disturbed by the presence nor absence of the kinase domain, leading us to suggest that pNPGal had probably a non-specific interaction with the kinase domain.
The hydrolysis of RFOs (raffinose, stachyose, and verbascose) by RgAgaSK and RgAga was analyzed by following substrate consumption and products release using HPAEC-PAD analysis. This method was essential, as the substrates of interest are α-galactosylated derivatives of sucrose which means that some of the products, resulting from the hydrolysis of RFOs by α-galactosidase, are themselves substrates. Indeed, in most of the studies found in the literature, hydrolysis is followed by a global method that doses all of the released galactose residues–using galactose oxidase (Tauzin et al., 2019) or carbohydrate reducing ends using 3,5-dinitrosalicylic acid (McLauchlan et al., 1999)–without knowing exactly from which molecule present in the medium the galactose was derived. For stachyose and verbascose, chromatogram analysis showed no product corresponding to galactobio-oligosaccharides [(galactose-α(1,6)-galactose)n], confirming the α-exo-acting specificity of the RgAgaSK. All kinetics obtained show that during RFOs hydrolysis, there was no accumulation of intermediate molecules, i.e., raffinose or stachyose, suggesting that the hydrolysis of the different RFOs was done randomly and the degradation was complete until obtention of sucrose as end product. Analysis of the hydrolysis profiles for each RFO showed no significant difference between the kinetics observed with RgAgaSK or RgAga, suggesting that the presence of the kinase domain did not interfere the hydrolysis process for natural substrates. The analysis of the literature data reinforces this conclusion with similar kcat/Km values for raffinose with RgAgaSK (12.9.104 s–1.M–1) as for Thermotoga maritima (18.7.104 s–1.M–1) and Thermotoga neapolitana (10.6.104 s–1.M–1) in which none kinase domain is associated to the α-galactosidase catalytic module (King et al., 1998; Miller et al., 2001).
Overall, our proposed 3D modeling supports experimental results that indicate that the presence of RgSK domains does not affect the activity of the RgAga enzyme on RFOs. Nevertheless, for both enzyme modularities–with or without the RgSK domain–and depending on the RFOs assayed, catalytic efficiencies decreased as the number of galactosyl residues attached to the oligosaccharide increased. This observation could be explained by the 3D model which shows a high exposition to the surface of the longer RFO leading to a flexibility, and probably less affinity so either Km increases due to this flexibility, either the kcat decreases because the flexibility affects the good orientation for the catalysis.
Considering now the RgAgaSK and RgSK modularities, their characterization on ATP hydrolysis ability was evaluated by bioluminescence. In both cases, the analysis of affinity (Km) toward ATP and hydrolysis catalytic efficiency (kcat/Km) did not show significant differences, suggesting that the presence of the RgAga domain on the one hand does not interfere with ATP binding as affinities were nearly similar and, on the other hand, that its position did not affect accessibility to the ATP binding domain resulting in a comparable hydrolysis rate.
The phosphorylation activity of sucrose by RgAgaSK and RgSK was then assessed by HPAEC-PAD analysis, which allowed separating the different carbohydrates according to their degree of ionization. Phosphate groups being highly ionizable, the interaction of phosphorylated sucrose with the resin is therefore be stronger and the retention time longer than that of sucrose. Analysis of the affinity of both recombinant proteins toward sucrose showed no significant differences, suggesting that the presence of the RgAga domain does not affect sucrose binding in the active site of the kinase domain. However, the phosphorylation of sucrose in terms catalytic efficiency (kcat/Km) has almost doubled between RgAgaSK (1.06.103 s–1.M–1) and RgSK (2.40.103 s–1.M–1), which suggests that the presence of the RgAga domain slightly affects the phosphorylation activity of the kinase module. This hypothesis can be supported by the 3D model. Indeed, according to the topology of the assembly and the orientation of each domain and active site, the probability to affect the SK active site is higher.
Independently, the TLC analysis of sucrose phosphorylation in the presence of ATP revealed that protein degradation is associated with a loss of kinase activity after 15 days of storage for both RgSK and RgAgaSK. These observations showed (i) functionality of the kinase domain alone, which suggested that the functional folding of the ATP binding site is done independently of the presence of the α-galactosidase domain, and (ii) proved that RgAgaSK instability is related to its kinase domain.
Our phylogenic study reveals that the 75 characterized GH36s so far are spread all over the tree covering relatively well the family. Some of the members were found in the Firmicutes groups, known as the second main phyla in the human gut microbiome (Gentile and Weir, 2018). It is noteworthy that RgAgaSK is the only characterized member of a clade comprising solely bacteria from the intestinal microbiome (e.g., Blautia, Clostridium, Eubacterium, Roseburia, Coprococcus).
Nevertheless, in a previous study focusing on the characterization of the GH13 (RgSPP) from the Rgaga1 locus, we observed that the whole Rgaga1 locus (see Figure 11C) showed a high synteny with loci from B. hansenii, E. rectale and C. sp. L2-50 (Tauzin et al., 2019). This gene organization was not present for other GH36s clustered in the same clade or even in the whole family. This observation leads to the conclusion that the Rgaga1 PUL dissemination has occurred by horizontal evolution and was restricted to only few species within the human digestive tract.
We searched for homolog to the genes of the Rgaga1 locus in the human gut metagenomic gene catalog and retrieved their occurrence frequency values from the gene frequency table in 760 European subjects.
The genes encoding for the bimodular kinase/GH36 from R. gnavus E1, B. hansenii DSM 20583, and E. rectale ATCC 33656 are highly prevalent (Figure 12A). When both modules are encoded by separated genes, as found in Clostridium sp. L2-50, both genes display similar prevalence/abundance profiles suggesting a close association of these genes in microorganisms and probably synergistic activity in the metabolic pathway as a bimodular enzyme. In contrast, the six genes of the Rgaga1 locus have very different abundance profiles, especially a higher abundance of AgaE and AgaF genes involved in substrate binding and transport (Figure 12B). This suggests that the locus is not conserved in many gut bacterial genomes, and that each of its components could operate independently following different metabolic paths from different substrates (raffinose, sucrose, and S6FP) as presented in Tauzin et al. (2019).
Finally, we noticed that the kinase and GH36 genes of Clostridium sp. L2-50 are less prevalent in the IBD subjects which is in accordance with the finding of Manichanh (2006) showing that the fecal microbiota of patients with Crohn’s disease contains a markedly reduced diversity of Firmicutes of which some Clostridium species are less prevalent and abundant. Although some Clostridia groups possess pathogenic species, most of them have commensal relationships with the host, playing an important role in the metabolic welfare of epithelial cells of the colon by releasing butyrate as an end-product of fermentation of monosaccharides (Lopetuso et al., 2013).
To conclude, we confirmed that the RgAgaSK is perfectly tuned to face both α-galactosidase and kinase activities. Indeed, the Rgaga1-like PULs code for two proteins (i.e., RgSPP and RgAgaSK), yielding three complementary enzyme activities involved in the metabolism of sucrose and its derivatives (RFOs) and providing an evolutionary advantage to certain human gut bacteria to colonize their competitive environment.
Materials and Methods
Cloning, Expression and Purification of RgAgaSK, RgAga, and RgSK
RgAgaSK was produced as previously described by Bruel et al. (2011). The rgaga and rgsk genes were amplified from plasmid containing full RgAgaSK gene with PCR reaction primers (Table S1). Recombinant proteins, with C-terminal His6-tag, were synthesized from E. coli BL21 cells (DE3) grown in LB broth, containing 50 mg.L–1 ampicillin and 1 mM IPTG, during an overnight culture at 23°C. Bacterial lysis were carried out with “Cell Disruptor” (Constant System LTD) in the binding buffer for affinity chromatography (50 mM HEPES at pH 7.7, 150 mM NaCl and 10 mM imidazole) at 1.37 kbar. After centrifugation at 10,000 × g during 20 min at 4°C, the soluble fractions were loaded onto a Ni-NTA column and the recombinant proteins were eluted with 125 mM imidazole in 50 mM HEPES at pH 7.7 and 150 mM NaCl. Fractions containing proteins were pooled, dialyzed against 50 mM HEPES buffer at pH 7.0 and concentrated with 10,000 molecular weight cutoff (MWCO) filter using Amicon Ultra centrifugal filter devices. The concentrated proteins were injected in HiPrep 16/60 SephacrylTM S-300, using 50 mM HEPES at pH 7.0 and 200 mM NaCl with a flow rate of 1 mL.min–1 and purified to near homogeneity (>90%). The active fractions were pooled and dialyzed against HEPES buffer 50 mM at pH 7.0. The purity of the protein was checked by SDS-PAGE (12%, Laemmli, 1970). The protein concentration was determined using the Bio-Rad protein assay kit with bovine serum albumin as standard or measuring the absorbance at 280 nm. The molar extinction coefficient used were: 142,740, 119,695, and 23,045 M–1.cm–1 for RgAgaSK, RgAga, and RgSK, respectively.
Galactosidase Assay
Galactosidase activity was measured using the synthetic substrate pNPGal. The enzyme (48 nM) was incubated with 2 mM pNPGal in McIlvaine’s buffer (100 mM citric acid, 200 mM Na2HPO4, pH 6.0) in 96 microtiter wells (200 μl), and the increase in absorbance at 405 nm was monitored for 10 min at 42°C in a microplate reader [KRL test (Spiral patent); Kirial International]. One unit of enzyme activity was defined as the amount of protein that released 1 μmol of pNP/min at 42°C and pH 6.0. The extinction coefficient for the pNP under this condition was 1,582.72 M–1.cm–1.
Temperature, pH, and Kinetic Parameters
The optimal pH was determined on pNPGal (20 mM) in McIlvaine’s buffer in a pH range of 3.0 to 8.0. The optimal temperature was determined at temperatures ranging from 10 to 70°C. For determination of the apparent Michaelis-Menten constants, the initial velocities of the enzymes were measured at 42°C in McIlvaine’s buffer at pH 6.0, with pNPGal concentrations ranging from 0.078 to 10 mM. The kinetic parameters were estimated using non-linear regression with GraphPad Prism software (GraphPad Prism version 3.00 for Windows 95; GraphPad Software, San Diego, CA, United States).
Bioluminescence Kinase Activity Assay
The characterization of ATP hydrolysis activity by RgAgaSK and RgSK was evaluated by bioluminescence, which allowed quantity determination of ATP present in the reaction assay using luciferin/luciferase reagent according to manufacturer’s instructions (YELEN Analytics, France) and luminometer reader (Tecan, France) described in Razafimanjato et al. (2011). Thus, for RgAgaSK and RgSK the initial rate conditions were determined at pH 8.0 and 30°C, in saturated sucrose condition (5 mM), with 1.3 μM of enzyme for three minutes. Under these conditions, different enzymatic activity analyses were performed for both RgAgaSK and RgSK using different concentrations of ATP (0 to 10 mM) and maintaining sucrose concentration to 5 mM.
RFO Product Hydrolysis Assay
The hydrolysis of RFOs was performed under optimal temperature and pH conditions with 25 nM of enzyme and 100 μM of substrate. The reaction was stopped by addition of 130 mM NaOH. The samples were centrifuged for 3 min at 13,000 × g and injected (20 μL) on the HPAEC system (ICS-5000®, Thermo Fisher Scientific, Courtaboeuf, France). Elution was achieved by a linear gradient of sodium acetate (0 to 130 mM) in 130 mM NaOH for 19 min at a flow rate of 1 mL.min–1. The elution of mono-, di- and oligosaccharides was monitored by an electrochemical detector (Pulse Amperometric Detection, PAD). The calibration curves ranging from 0 to 100 μM were designed with the different substrates used and products formed (galactose, sucrose, raffinose, stachyose, and verbascose), which allowed their release or consumption to be quantified using the Chromeleon® software (Dionex). All analyses were duplicated.
To evaluate α-galactosidase activity on RFOs, the initial slopes of the regression lines are used. The catalytic efficiency (kcat/Km) of the reactions is calculated using the Matsui equation: [E]∗(kcat/Km)∗t = ln([S0]/[St]), with[E] the enzyme concentration used in nM, (kcat/Km) the catalytic efficiency in min–1.M–1, t time in minutes, [S0] and [St] substrate concentration at time 0 min and time t in nM, respectively (Matsui et al., 1991).
3D Modeling of RgAgaSK
The full sequence of RgAgaSK from R. gnavus E1 was extracted from UniProt (G4T4R7). The α-galactosidase domain comprising the first 719 amino acids at the N-terminal of RgAgaSK was determined by X-ray crystallography (PDB-ID: 2YFN, Bruel et al., 2011) with a resolution of 1.45Å. The kinase domain, corresponding to the last 215 amino acid residues of the C-terminal domain of RgAgaSK, was modeled by threading techniques using the I-TASSER webserver (Zhang, 2008; Roy et al., 2010; Yang et al., 2015) with a C-score of −0.05 and using mainly as template the crystallographic structure of Thermus thermophilus HB8 uridine kinase (PDB code: 3ASY, Tomoike et al., 2011) Modeler 9.19 (Webb and Sali, 2016) was subsequently used to merge the RgAga and RgSK domains into RgAgaSK and build the tetrameric assembly. For each step, ten models were generated using automodel class and fixing RgAga domain coordinates. Finally, the models with lowest DOPE score (Shen and Sali, 2006) were chosen for modeling RgAgaSK-RFOs complexes.
3D Modeling of RgAgaSK-RFO Complexes
The binding mode of three distinct RFOs in RgAgaSK active site was then investigated. Three complexes were built, RgAgaSK-RAF (complex with raffinose), RgAgaSK-STACH (complex with stachyose) and RgAgaSK-VERB (complex with verbascose). RgAgaSK-RAF and RgAgaSK-STACH complexes were built after superimposition onto the crystal structure of G. stearothermophilus α-galactosidase co-crystallized in complex with raffinose (PDB-ID: 4FNT) and with stachyose (PDB-ID: 4FNU). The complex with verbascose (RgAgaSK-VERB) was built using GLYCAM_06j-1 Force Field (Kirschner et al., 2008) and the program tleap of AMBERTOOLS17 (Case et al., 2017). Then, geometry of verbascose ligand was fitted onto stachyose binding mode. Finally, all complexes were minimized in vacuum with a cut-off of 99Å and 500 steps of steepest descent then 500 steps of conjugated gradient. All graphics were prepared using PyMOL 1.7 (Schrödinger).
Phylogenetic Analysis of the GH36 Family
A phylogenetic analysis of the GH36 family was performed on CAZy release (November 06th, 2019) considering the bounded modules of 6,125 sequences in the public database. Signal sequences and additional modules were removed to isolate the catalytic modules for bioinformatics analysis. The amino acid sequences were aligned with MAFFT v7.271 (Katoh, 2005) and clustered above 95% identities using CD-HIT version 4.6 reducing the number of sequences considered to 843. We used the option–maxiterate 1000 and–localpair to obtain high accuracy where–maxiterate # indicates the maximum number of iterative refinement and localpair forces local pairwise alignment. To generate the tree, we built a matrix of maximum likelihood distances based on model of substitution LG (Le and Gascuel, 2008). Finally, the phylogeny reconstruction tree was performed using BIONJ (Gascuel, 1997). Evolutionary tree construction was conducted using Dendroscope software (Huson et al., 2007).
Cluster analysis was based on the neighbor-joining method with the closely related bacterium R. gnavus E1 as the out-group root. Synteny blocks were analyzed by the MaGE platform that allows the comparison of CDS predicted from genomic DNA of R. gnavus E1 to those predicted from genomic DNA present in the bases PkGDB (Prokaryotic Genome DataBase) and NCBI RefSeq (collection of Raw sequences from whole genome sequencing). Beyond a simple sequence comparison, this interface allows to analyze the synteny between two chromosomes. Only, strictly conserved synteny has been considered here and a multiple sequence alignment was produced using Muscle program (Edgar, 2004) to evaluate the sequence identities percentage between closely modules i.e., between α-galactosidases, SKs, or SPPs.
The prevalence and abundance of the GH36/sucrose kinase genes in the human fecal microbiome were determined by searching for the homolog sequences in the translated catalog of 9.9 million reference genes using BLASTP, E-value = 0, identity ≥ 90% (MetaHIT Consortium, Li et al., 2014). The gene richness in the human gut was assessed by recovering the occurrence frequency data of the homolog sequences identified in the catalog from the gene frequency table in 760 European subjects consisting of two cohorts, one of healthy individuals and the other of individuals in remission from inflammatory bowel disease (Li et al., 2014).
Data Availability Statement
The ORFs and protein sequences of RgAgaSK protein were previously deposited in GenBank under the accession number FQ790379.1 and CCA61959.1, respectively (Bruel et al., 2011).
Author Contributions
ML and AT were involved in data acquisition, data interpretation, and manuscript writing. LB was involved in biochemical and enzymatic analysis and data interpretation. EL and GP-V was involved the prevalence analysis. VL was involved in the sequences extraction from CAZy database and phylogenic analysis. NV was involved in the bioluminescence assay. NQ, MF, and JP were involved in the data interpretation. JE and IA planned and performed molecular modeling studies and contributed to manuscript writing. FP was involved in data acquisition. TG was the coordinator of the study and was involved in the manuscript writing and editing. All authors read and approved the final manuscript.
Funding
This work was co-funded, in part, by the “Aix-Marseille Université,” “CNRS,” and the “Ministère Français de l’Enseignement Supérieur et de la Recherche,” and by the European Union’s framework program Horizon 2020 (MSCA-IF-2015 707457, CaTSYS).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to the “Analyse et Valorisation de la Biodiversité” platform for the access to the Cell Disruptor and the IBiSA “Biopolymères, Biologie Structurale” (BiBS) platform (INRAE,Nantes) for the mass spectrometry analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.579521/full#supplementary-material
Footnotes
References
Adamson, C., Pengelly, R. J., Shamsi Kazem, Abadi, S., Chakladar, S., Draper, J., et al. (2016). Structural Snapshots for Mechanism-Based Inactivation of a Glycoside Hydrolase by Cyclopropyl Carbasugars. Angew. Chem. Int. Ed. 55, 14978–14982. doi: 10.1002/anie.201607431
Bachmann, M., Matile, P., and Keller, F. (1994). Metabolism of the Raffinose Family Oligosaccharides in Leaves of Ajuga reptans L. (Cold Acclimation. Translocation, and Sink to Source Transition: Discovery of Chain Elongation Enzyme). Plant Physiol. 105, 1335–1345. doi: 10.1104/pp.105.4.1335
Bruel, L., Sulzenbacher, G., Cervera Tison, M., Pujol, A., Nicoletti, C., Perrier, J., et al. (2011). α-Galactosidase/Sucrose Kinase (AgaSK), a Novel Bifunctional Enzyme from the Human Microbiome Coupling Galactosidase and Kinase Activities. J. Biol. Chem. 286, 40814–40823. doi: 10.1074/jbc.M111.286039
Case, D. A., Cerutti, D. S., Cheatham, T. E., Darden, T. A., Duke, R. E., Giese, T. J., et al. (2017). Amber 2017, University of California, San Francisco. San Francisco: University of California.
Collins, S. L., McMillan, A., Seney, S., van der Veer, C., Kort, R., Sumarah, M. W., et al. (2017). Promising Prebiotic Candidate Established by Evaluation of Lactitol, Lactulose, Raffinose, and Oligofructose for Maintenance of a Lactobacillus -Dominated Vaginal Microbiota. Appl. Environ. Microbiol. 84, e2200–e2217. doi: 10.1128/AEM.02200-17
Collins, S., and Reid, G. (2016). Distant Site Effects of Ingested Prebiotics. Nutrients 8:523. doi: 10.3390/nu8090523
Comfort, D. A., Bobrov, K. S., Ivanen, D. R., Shabalin, K. A., Harris, J. M., Kulminskaya, A. A., et al. (2007). Biochemical Analysis of Thermotoga maritima GH36 α-Galactosidase (Tm GalA) Confirms the Mechanistic Commonality of Clan GH-D Glycoside Hydrolases †. Biochemistry 46, 3319–3330. doi: 10.1021/bi061521n
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Ejby, M., Fredslund, F., Andersen, J. M., Vujičić Žagar, A., Henriksen, J. R., Andersen, T. L., et al. (2016). An ATP Binding Cassette Transporter Mediates the Uptake of α-(1,6)-Linked Dietary Oligosaccharides in Bifidobacterium and Correlates with Competitive Growth on These Substrates. J. Biol. Chem. 291, 20220–20231. doi: 10.1074/jbc.M116.746529
Fernández-Leiro, R., Pereira-Rodríguez, Á, Cerdán, M. E., Becerra, M., and Sanz-Aparicio, J. (2010). Structural Analysis of Saccharomyces cerevisiae α-Galactosidase and Its Complexes with Natural Substrates Reveals New Insights into Substrate Specificity of GH27 Glycosidases. J. Biol. Chem. 285, 28020–28033. doi: 10.1074/jbc.M110.144584
Fredslund, F., Abou Hachem, M., Jonsgaard Larsen, R., Gerd Sørensen, P., Coutinho, P. M., Lo Leggio, L., et al. (2011). Crystal Structure of α-Galactosidase from Lactobacillus acidophilus NCFM: Insight into Tetramer Formation and Substrate Binding. J. Mol. Biol. 412, 466–480. doi: 10.1016/j.jmb.2011.07.057
Fujimoto, Z., Kaneko, S., Kim, W.-D., Park, G.-G., Momma, M., and Kobayashi, H. (2009). The tetramer structure of the glycoside hydrolase family 27 alpha-galactosidase I from Umbelopsis vinacea. Biosci. Biotechnol. Biochem. 73, 2360–2364. doi: 10.1271/bbb.90604
Fujimoto, Z., Kaneko, S., Momma, M., Kobayashi, H., and Mizuno, H. (2003). Crystal Structure of Rice α-Galactosidase Complexed with D-Galactose. J. Biol. Chem. 278, 20313–20318. doi: 10.1074/jbc.M302292200
Garman, S. C., and Garboczi, D. N. (2004). The Molecular Defect Leading to Fabry Disease: Structure of Human α-Galactosidase. J. Mol. Biol. 337, 319–335. doi: 10.1016/j.jmb.2004.01.035
Gascuel, O. (1997). BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 14, 685–695. doi: 10.1093/oxfordjournals.molbev.a025808
Gentile, C. L., and Weir, T. L. (2018). The gut microbiota at the intersection of diet and human health. Science 362, 776–780. doi: 10.1126/science.aau5812
Golubev, A. M., Nagem, R. A. P., Brandão Neto, J. R., Neustroev, K. N., Eneyskaya, E. V., Kulminskaya, A. A., et al. (2004). Crystal Structure of α-Galactosidase from Trichoderma reesei and Its Complex with Galactose: Implications for Catalytic Mechanism. J. Mol. Biol. 339, 413–422. doi: 10.1016/j.jmb.2004.03.062
Guce, A. I., Clark, N. E., Salgado, E. N., Ivanen, D. R., Kulminskaya, A. A., Brumer, H., et al. (2010). Catalytic Mechanism of Human α-Galactosidase. J. Biol. Chem. 285, 3625–3632. doi: 10.1074/jbc.M109.060145
Hobbs, J. K., Meier, E. P. W., Pluvinage, B., Mey, M. A., and Boraston, A. B. (2019). Molecular analysis of an enigmatic Streptococcus pneumoniae virulence factor: The raffinose-family oligosaccharide utilization system. J. Biol. Chem. 294, 17197–17208. doi: 10.1074/jbc.RA119.010280
Huson, D. H., Richter, D. C., Rausch, C., Dezulian, T., Franz, M., and Rupp, R. (2007). Dendroscope: An interactive viewer for large phylogenetic trees. BMC Bioinformatics 8:460. doi: 10.1186/1471-2105-8-460
JCSG. (2005). Crystal structure of Alpha-galactosidase (EC 3.2.1.22) (Melibiase) (tm1192) from Thermotoga maritima at 2.34 A resolution. Europe: PDB.
JCSG. (2013). Crystal structure of a putative alpha-galactosidase (BF1418) from Bacteroides fragilis NCTC 9343 at 1.57 A resolution. Europe: PDB.
Katoh, K. (2005). MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33, 511–518. doi: 10.1093/nar/gki198
Kikuchi, A., Okuyama, M., Kato, K., Osaki, S., Ma, M., Kumagai, Y., et al. (2017). A novel glycoside hydrolase family 97 enzyme: Bifunctional β- l -arabinopyranosidase/α-galactosidase from Bacteroides thetaiotaomicron. Biochimie 142, 41–50. doi: 10.1016/j.biochi.2017.08.003
King, M. R., Yernool, D. A., Eveleigh, D. E., and Chassy, B. M. (1998). Thermostable α-galactosidase from Thermotoga neapolitana : cloning, sequencing and expression. FEMS Microbiol. Lett. 163, 37–42. doi: 10.1111/j.1574-6968.1998.tb13023.x
Kirschner, K. N., Yongye, A. B., Tschampel, S. M., González-Outeiriño, J., Daniels, C. R., Foley, B. L., et al. (2008). GLYCAM06: A generalizable biomolecular force field. Carbohydrates: GLYCAM06. J. Comput. Chem. 29, 622–655. doi: 10.1002/jcc.20820
Kytidou, K., Beekwilder, J., Artola, M., van Meel, E., Wilbers, R. H. P., Moolenaar, G. F., et al. (2018). Nicotiana benthamiana α-galactosidase A1.1 can functionally complement human α-galactosidase A deficiency associated with Fabry disease. J. Biol. Chem. 293, 10042–10058. doi: 10.1074/jbc.RA118.001774
Laemmli, U. K. (1970). Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 227, 680–685. doi: 10.1038/227680a0
Le, S. Q., and Gascuel, O. (2008). An improved general amino acid replacement matrix. Mol. Biol. Evol. 25, 1307–1320. doi: 10.1093/molbev/msn067
Li, J., Jia, H., Cai, X., Zhong, H., Feng, Q., Sunagawa, S., et al. (2014). An integrated catalog of reference genes in the human gut microbiome. Nat. Biotech 32, 834–841.
Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M., and Henrissat, B. (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495. doi: 10.1093/nar/gkt1178
Lopetuso, L. R., Scaldaferri, F., Petito, V., and Gasbarrini, A. (2013). Commensal Clostridia: leading players in the maintenance of gut homeostasis. Gut Pathog. 5:23. doi: 10.1186/1757-4749-5-23
Manichanh, C. (2006). Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 55, 205–211. doi: 10.1136/gut.2005.073817
Martínez-Villaluenga, C., Frias, J., and Vidal-Valverde, C. (2008). Alpha-Galactosides: Antinutritional Factors or Functional Ingredients? Crit. Rev. Food Sci. Nutr. 48, 301–316. doi: 10.1080/10408390701326243
Matsui, I., Ishikawa, K., Matsui, E., Miyairi, S., Fukui, S., and Honda, K. (1991). Subsite Structure of Saccharomycopsis α-Amylase Secreted from Saccharomyces cerevisiae. J. Biochem. 109, 566–569. doi: 10.1093/oxfordjournals.jbchem.a123420
McLauchlan, W. R., Garcia-Conesa, M. T., Williamson, G., Roza, M., Ravestein, P., and Maat, J. (1999). A novel class of protein from wheat which inhibits xylanases. Biochem. J. 338(Pt 2), 441–446. doi: 10.1042/0264-6021:3380441
Merceron, R., Foucault, M., Haser, R., Mattes, R., Watzlawick, H., and Gouet, P. (2012). The Molecular Mechanism of Thermostable α-Galactosidases AgaA and AgaB Explained by X-ray Crystallography and Mutational Studies. J. Biol. Chem. 287, 39642–39652. doi: 10.1074/jbc.M112.394114
MetaHIT Consortium, Li, J., Jia, H., Cai, X., Zhong, H., Feng, Q., et al. (2014). An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 32, 834–841. doi: 10.1038/nbt.2942
MetaHIT Consortium, Qin, J., Li, R., Raes, J., Arumugam, M., Burgdorf, K. S., et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65. doi: 10.1038/nature08821
Miller, E. S., Parker, K. N., Liebl, W., Lam, D., Callen, W., Snead, M. A., et al. (2001). αa-D-Galactosidases from Thermotoga species. Methods Enzymol. 330, 246–260. doi: 10.1016/S0076-6879(01)30380-4
Okuyama, M., Kitamura, M., Hondoh, H., Kang, M.-S., Mori, H., Kimura, A., et al. (2009). Catalytic Mechanism of Retaining α-Galactosidase Belonging to Glycoside Hydrolase Family 97. J. Mol. Biol. 392, 1232–1241. doi: 10.1016/j.jmb.2009.07.068
Ramare, F., Nicoli, J., Dabard, J., Corring, T., Ladire, M., Gueugneau, A. M., et al. (1993). Trypsin-dependent production of an antibacterial substance by a human Peptostreptococcus strain in gnotobiotic rats and in vitro. Appl. Environ. Microbiol. 59, 2876–2883. doi: 10.1128/aem.59.9.2876-2883.1993
Razafimanjato, H., Benzaria, A., Taïeb, N., Guo, X.-J., Vidal, N., Di Scala, C., et al. (2011). The ribotoxin deoxynivalenol affects the viability and functions of glial cells. Glia 59, 1672–1683. doi: 10.1002/glia.21214
Rowland, R. J., Wu, L., and Davies, G. J. (2019). Crystal structure of human alpha-galactosidase A in complex with alpha-galactose configured cyclosulfamidate ME763. Europe: PDB.
Roy, A., Kucukural, A., and Zhang, Y. (2010). I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738. doi: 10.1038/nprot.2010.5
Shen, M., and Sali, A. (2006). Statistical potential for assessment and prediction of protein structures. Protein Sci. 15, 2507–2524. doi: 10.1110/ps.062416606
Shoaf, K., Mulvey, G. L., Armstrong, G. D., and Hutkins, R. W. (2006). Prebiotic Galactooligosaccharides Reduce Adherence of Enteropathogenic Escherichia coli to Tissue Culture Cells. Infect. Immun. 74, 6920–6928. doi: 10.1128/IAI.01030-06
Tauzin, A. S., Bruel, L., Laville, E., Nicoletti, C., Navarro, D., Henrissat, B., et al. (2019). Sucrose 6F-phosphate phosphorylase: a novel insight in the human gut microbiome. Microb. Genomics 5:e000253. doi: 10.1099/mgen.0.000253
Tomoike, F., Nakagawa, N., Kuramitsu, S., and Masui, R. (2011). A Single Amino Acid Limits the Substrate Specificity of Thermus thermophilus Uridine-Cytidine Kinase to Cytidine. Biochemistry 50, 4597–4607. doi: 10.1021/bi102054n
Vorobiev, S., Chen, Y., Seetharaman, J., Belote, R., Sahdev, S., Xiao, R., et al. (2010). Crystal structure of the alpha-galactosidase from Lactobacillus brevis, Northeast Structural Genomics Consortium Target LbR11. Europe: PDB.
Webb, B., and Sali, A. (2016). Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinformatic 54, 5.6.1–5.6.37. doi: 10.1002/cpbi.3
Yang, J., Yan, R., Roy, A., Xu, D., Poisson, J., and Zhang, Y. (2015). The I-TASSER Suite: protein structure and function prediction. Nat. Methods 12, 7–8. doi: 10.1038/nmeth.3213
Yu, Y., Mena-Barragán, T., Higaki, K., Johnson, J. L., Drury, J. E., Lieberman, R. L., et al. (2014). Molecular Basis of 1-Deoxygalactonojirimycin Arylthiourea Binding to Human α-Galactosidase A: Pharmacological Chaperoning Efficacy on Fabry Disease Mutants. ACS Chem. Biol. 9, 1460–1469. doi: 10.1021/cb500143h
Zartl, B., Silberbauer, K., Loeppert, R., Viernstein, H., Praznik, W., and Mueller, M. (2018). Fermentation of non-digestible raffinose family oligosaccharides and galactomannans by probiotics. Food Funct. 9, 1638–1646. doi: 10.1039/C7FO01887H
Keywords: raffinose oligosaccharide family, sucrose, α-galactosidase, sucrose-kinase, human gut microbiome, GH36 family
Citation: Lafond M, Tauzin AS, Bruel L, Laville E, Lombard V, Esque J, André I, Vidal N, Pompeo F, Quinson N, Perrier J, Fons M, Potocki-Veronese G and Giardina T (2020) α-Galactosidase and Sucrose-Kinase Relationships in a Bi-functional AgaSK Enzyme Produced by the Human Gut Symbiont Ruminococcus gnavus E1. Front. Microbiol. 11:579521. doi: 10.3389/fmicb.2020.579521
Received: 02 July 2020; Accepted: 19 October 2020;
Published: 12 November 2020.
Edited by:
Hauke Smidt, Wageningen University & Research, NetherlandsReviewed by:
Diego Fabian Gomez-Casati, National University of Rosario, ArgentinaEvelyne Forano, INRAE Clermont-Auvergne-Rhône-Alpes, France
Copyright © 2020 Lafond, Tauzin, Bruel, Laville, Lombard, Esque, André, Vidal, Pompeo, Quinson, Perrier, Fons, Potocki-Veronese and Giardina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thierry Giardina, thierry.giardina@univ-amu.fr
†These authors have contributed equally to this work