 Woori Kwak1†
Woori Kwak1† Young-Hyun Han2†
Young-Hyun Han2† Donghyeok Seol3
Donghyeok Seol3 Hyaekang Kim3
Hyaekang Kim3 Hyeonju Ahn3
Hyeonju Ahn3 Misun Jeong1
Misun Jeong1 Jaeku Kang2,4Heebal Kim5
Jaeku Kang2,4Heebal Kim5 Tae Hyun Kim6,7*
Tae Hyun Kim6,7*- 1C&K Genomics, Songpa-gu, South Korea
- 2Priority Research Center, Myunggok Medical Research Institute, College of Medicine, Konyang University, Daejeon, South Korea
- 3Department of Agricultural Biotechnology and Research Institute of Agriculture and Life Sciences, Seoul National University, Seoul, South Korea
- 4Department of Pharmacology, College of Medicine, Konyang University, Daejeon, South Korea
- 5Research Institute of Agriculture and Life Sciences, Seoul National University, Seoul, South Korea
- 6Myunggok Medical Research Institute, College of Medicine, Konyang University, Daejeon, South Korea
- 7Department of Obstetrics and Gynecology, Konyang University Hospital, Daejeon, South Korea
Despite the importance of Lactobacillus iners and its unique characteristics for the study of vaginal adaption, its genome and genomic researches for identifying molecular backgrounds of these specific phenotypes are still limited. In this study, the first complete genome of L. iners was constructed using a cost-effective long-read sequencing platform, Flongle from Oxford Nanopore, and comparative genome analysis was conducted using a total of 1,046 strain genomes from 10 vaginal Lactobacillus species. Single-molecule sequencing using Flongle effectively resolved the limitation of the 2nd generation sequencing technologies in dealing with genomic regions of high GC contents, and comparative genome analysis identified three potential core genes (INY, ZnuA, and hsdR) of L. iners which was related to its specific adaption to the vaginal environment. In addition, we performed comparative prophage analysis for 1,046 strain genomes to further identify the species specificity. The number of prophages in L. iners genomes was significantly smaller than other vaginal Lactobacillus species, and one of the specific genes (hsdR) was suggested as the means for defense against bacteriophage. The first complete genome of L. iners and the three specific genes identified in this study will provide useful resources to further expand our knowledge of L. iners and its specific adaption to the vaginal econiche.
Introduction
It is well known that the commensal microbiome in the vaginal tract is closely related to vaginal health, and the healthy vaginal econiche is dominated by a limited number of Lactobacillus species in most women (Ravel et al., 2011; Spear et al., 2011). Previous studies identified that the vaginal microbial communities of healthy women were typically dominated by one or few species of lactobacilli (Lactobacillus crispatus, Lactobacillus iners, Lactobacillus gasseri, or Lactobacillus jensenii) (Hummelen et al., 2010; Srinivasan et al., 2010), and they could promote a healthy vaginal econiche by actively preventing growth and colonization of bacterial, fungal, and viral pathogens (Petrova et al., 2015). Among these Lactobacillus species, L. iners has been considered as the most prevalent Lactobacillus species (Hummelen et al., 2010; Ravel et al., 2011) and known to have very unique characteristics compared to other commensal Lactobacillus species in the vaginal econiche.
This organism is known to have rod-shaped cell morphology with Gram-positive characteristics, but unlike other Lactobacillus species, it is not always clearly stained as Gram-positive, and some isolates showed coccobacillary morphology rather than bacillary (De Backer et al., 2007; Lebeer et al., 2008). It is unable to grow on de Man-Rogosa-Sharpe agar, a selective culture medium for lactobacilli growth, and nutrient requirement of L. iners is known to be more complex than the other vaginal lactobacilli (Rampersaud et al., 2011). Along with its unique phenotypic traits, the genome of L. iners is also unusual compared to other Lactobacillus species. L. iners has the smallest single circular genome (1.3 Mb) among known lactobacilli determined so far, and its size is within the range of the genome sizes of human symbionts and parasites. This small genome size is considered as a result of large scale gene loss and genome reduction during the rapid evolution for specific adaption to the vaginal econiche (Macklaim et al., 2011). A previous genome study of L. iners identified that the genome reduction of L. iners included the loss of genes involved in the carbohydrate transport and energy metabolism which might be related to its complex nutrient requirements (Macklaim et al., 2011). As opposed to massive gene loss, unlike other lactobacilli, L. iners produce an unusual pore-forming cholesterol-dependent cytolysin (CDC) called Inerolysin (INY) which is typically found in Gram-positive pathogenic bacteria (Rampersaud et al., 2011). Unlike other human-specific CDCs such as vaginolysin (VLY) from Gardnerella vaginalis or intermedilysin (ILY), INY can be active in the acidic vaginal environment (PH range of 4.5–6.0), and it has a broad range of target species (Rampersaud et al., 2011). The adhesion ability to epithelial surfaces is also one of the important phenotypic traits of vaginal Lactobacillus species because it allows colonization and host interaction and excludes pathogens (Osset et al., 2001). Even though the genome of L. iners lacks most of the known adhesion factors of Lactobacillus species (Morris et al., 2012), it shows a strong adhesion ability to vaginal epithelial cells (McMillan et al., 2013). In addition, L. iners is known as the only vaginal Lactobacillus species that continued presence in the vagina with the normal or intermediate condition as well as with bacterial vaginosis (BV) as diagnosed by Nugent scores (Burton and Reid, 2002; Verhelst et al., 2005; Tamrakar et al., 2007), and it indicates that L. iners seems to be better adapted than other lactobacilli to dynamically changing vaginal environments.
However, in spite of these unique characteristics and small genome size of L. iners, high-quality genomes and genomic researches for identifying molecular backgrounds of these specific phenotypes are still limited. The reason why L. iners genome still has not been completely sequenced might stem from the fact that the GC contents of this species was significantly lower than the other lactobacilli, including other vaginal lactobacilli (Macklaim et al., 2011; Mendes-Soares et al., 2014). In this study, therefore, we conducted the first complete genome assembly of the L. iners species isolated from the healthy vagina of South Korean woman using a latest cost-effective long-read sequencing platform, Flongle from Oxford Nanopore technologies and performed large scale comparative genome analysis with 9 Lactobacillus species (L. crispatus, L. gasseri, L. helveticus, L. jensenii, L, johnsonii, L. plantarum, L, reuteri, L. salivarius, L. vaginalis) which are previously reported to inhibit the human vagina. We identified potential candidate genes that could be closely related to the unique phenotypes of L. iners for its specific adaption in the vaginal econiche, and the complete genome constructed in this study can provide useful resources for future studies.
Materials and Methods
Sample Preparation
To isolate L. iners, healthy vaginal flora was collected from visiting patients of the outpatient clinic of obstetrics and gynecology of Konyang University hospital. Diluted vaginal flora was cultured on Tryptic Soy Agar plates with 5% defibrinated sheep blood anaerobically using the BD BBL GasPack system (NY, United States) in 37°C during 48 h. Using Tryptic Soy Broth with 5% defibrinated sheep blood, single colonies were grown to get enough amount of DNA for sequencing. According to cell morphology and Gram-staining of isolates, candidate L. iners strain KY was selected, and it was confirmed using 16S rRNA sequencing using ABI 3730xl. 16S rRNA sequencing was conducted using 24F-AGAGTTTGATCMTGGCTCAG, 1492R-TACGGYTACCTTGTTACGACTT primers, and the generated forward and reverse reads were merged. Merged full-length 16S rRNA sequence was matched to reference RNA sequence database of NCBI (refseq_rna) (Yatsunyk et al., 2008) using BLASTn for species identification.
Genome Sequencing and Assembly
DNA was extracted from the cultured bacteria cells using Kit PureHelix Genomic DNA Prep Kit (Solution Type)-Bacteria with minor modification. Briefly, cell pellets were resuspended in 600 μl Cell Resuspension solution with 4 μl Lysozyme (100 mg/ml) and incubated at 37°C for 1 h for Gram-positive bacteria. The cell was re-collected by centrifuging at 12000 rpm for 2 min. The pellet was resuspended in 600 μl Cell Lysis solution containing 3 μl RNase A (4 mg/ml) by pipetting and then lysed at 37°C for 30 min. To remove protein, protein precipitation solution was added to lyse the sample. The clear supernatant after centrifuging was transferred into a new tube and DNA was precipitated by adding 0.8 Vol of Isopropanol. Isolated gDNA was quantified and qualified by gel electrophoresis, 260/280 nm absorbance ratio and Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen).
The library was prepared using the ONT 1D ligation Sequencing kit (SQK-LSK109) with the native barcoding expansion kit (EXP-NBD104) following the manufacturer’s protocol. In brief, genomic DNA was fragmented to target length using g-Tube (Covaris). Fragmented DNA was repaired using the NEBNext FFPE DNA Repair Mix and NEBNext Ultra II End Repair/dA-Tailing Module. The end-prepped DNA was individually barcoded with ONT native barcode by NEB Blunt/TA Ligase Master Mix. Barcoded DNA samples were pooled in equal molar amounts. It was ligated with the adapter using the NEBNext Quick Ligation Module. After every enzyme reaction, the DNA samples were purified using AMPure XP beads (Beckman Coulter). The final library was loaded onto Flongle flow cell (FLO-FLG001, R9.4.1), and sequencing was performed on a MinION MK1b and MinKNOW software (19.06.8).
Base-calling and de-multiplexing for generated fast5 from Flongle were conducted using Guppy (v3.4.3) from Oxford Nanopore technologies. To remove the sequencing artifact and chimeric read, Porechop (v0.2.4)1 was employed. Genome assembly was conducted using CANU (v1.8) (Koren et al., 2017) with genomeSize = 1.3 Mb parameter. To correct the errors in the assembled sequence, Nanopolish (v0.11.1) (Loman et al., 2015) was used repeatedly until no correction is available. In addition, Racon (Vaser et al., 2017), Rebaler2 and medaka3 were also employed to compare the polishing efficiency. Assembled CANU contig was corrected multiple times based on Racon using Rebaler, and two additional rounds of medaka polishing were conducted for reducing insertion and deletion error which can affect the gene prediction. To finish the circular genome of L. iners, Simple_circularise.py script4 and Circlator (v1.5.5) (Hunt et al., 2015) were used. After making assembled genome of L. iners KY to circular form using Simple_circularise.py script, Circlator was used to confirm the circularization, and the start position of the circularized genome was adjusted to dnaA gene manually. Assembled genome was submitted to NCBI database with accession GCA_010748955.1. To identify unassembled regions in the previous studies, 6 scaffold level genomes in Refseq database were mapped to the constructed genome using Minimap2 (Li, 2018).
Genome Annotation and Comparative Genome Analysis
Genome annotation was conducted using Prokka (v1.14.5) (Seemann, 2014) with –rfam option to enable search for ncRNA. Genomic map of constructed L. iners KY genome was constructed using CGVIEW (Grant and Stothard, 2008). For comparative genome analysis, a total of 1,045 available genomes for 10 Lactobacillus species in NCBI Refseq (Haft et al., 2017) data were used (L. crispatus: 111, L. gasseri: 43, L. helveticus: 57, L. iners: 23, L, jensenii: 37, L. johnsonii: 38, L. plantarum: 469, L. reuteri: 175, L. salivarius: 88, L. vaginalis: 4). Four genomes were randomly selected from each species to construct the gene cluster for 10 Lactobacillus species using OrthoMCL (Li et al., 2003), and then the identified gene clusters specifically existed in L. iners were confirmed in 1,045 genomes based on the Prokka annotation. Using all annotated proteins from 9 Lactobacillus species, uniqueness of L. iners specific genes was confirmed based on BLASTp (Subject Coverage > 0.5, Identity > 0.5, evalue < 1-e5).
Comparative Prophage Analysis
Existing prophages in 1,045 genomes of 10 Lactobacillus species and L. iners KY were identified using ProphET (Reis-Cunha et al., 2019), a standalone prophage detection program. To test the significance of the numbers of prophages in the genomes of the 10 Lactobacillus species, Kruskall–Wallis test was used, and the pairwise Wilcoxon Rank-Sum test was conducted to compare between each species with FDR correction. For identification of detected prophage from Lactobacillus genomes, 2,169 phage and prophage sequences were downloaded from NCBI refseq database, and detected prophage sequences were matched using BLASTn (Altschul et al., 1990).
Results and Discussion
Constructing the Complete Genome of L. iners KY
Table 1 shows the features of generated raw data from Flongle. About 89, 134, and 215 Mb were generated for 6, 10, and 20K insert size library, respectively. Based on the L. iners’s estimated genome size of 1.3 Mb, 68.48X, 103.72X, and 165.87X coverage data were retained from each library. We tried to generate the same amount of data for each library during data generation, but the amount of generated data was increased according to the increase of insert size. On the contrary, the median read length was decreased according to the increase of insert size. N50 length was 4,639 bp for 6K library, 6,727 bp for 10K library, and 7,778 bp for 20K library which was shorter than our expectation based on the insert size of each library. After removing the adapter and chimeric reads from raw data using Porechop, genome assembly using CANU was conducted. Table 2 shows the results of genome assemblies using CANU for each library. Among 3 libraries with different insert sizes, only 20K library which showed high coverage, and long N50 length was succeeded to construct the chromosome level assembly. Scaffold level assembly (8 scaffolds and 5 scaffolds, respectively) was resulted from CANU assemblies using 6K, 10K libraries, and the number of scaffolds was decreased in accordance with the coverage increase. After conducting Nanopolish for error correction in the assembled genome, total length was increased to 1,357,225 bp, and GC contents was decreased to 33.40%, and assembly statistics after circularization was 1,337,870 bp with 33.39% GC contents. Prokka annotation identified 1,684 CDSs in the polished genome. However, the number of predicted genes was much higher than previously reported genome assemblies of L. iners, and the polished assembly contained many pseudogenes originated from assembly errors. This indicated that assembly polishing using Nanopolish could not effectively resolve the remained errors in the assembly. Therefore, to achieve more improved L. iners genome assembly, we employed additional assembly polishing tools such as Racon, Rebaler, and Medaka which can be applicable only for Nanopore reads. After polishing using Racon (Reblaer) and two additional rounds with Medaka, the final assembly was 1,339,101 bp with 33.35% GC contents, and the number of predicted genes was decreased to 1,465 genes (CDS: 1,354, rRNA: 18, misc_RNA: 22, tRNA: 70, tmRNA: 1) with reduced pseudogenes. This result indicates that the quality of genome assembly using only Nanopore reads varies depending on the polishing tools, and Medaka, the polishing tool from its manufacturer showed better performance than any other tools applied in this study.
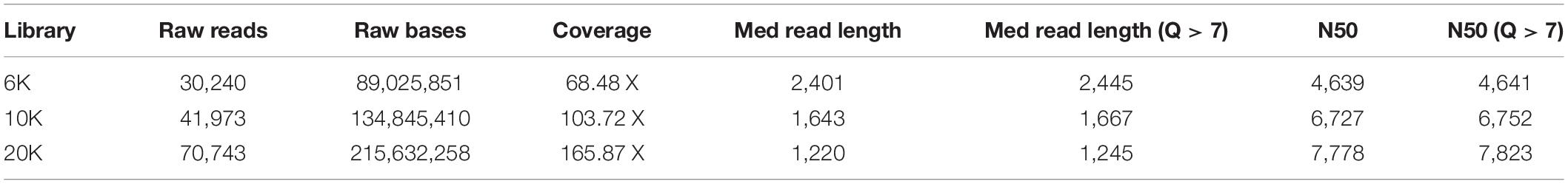
Table 1. Generated data information for three libraries constructed in this study.
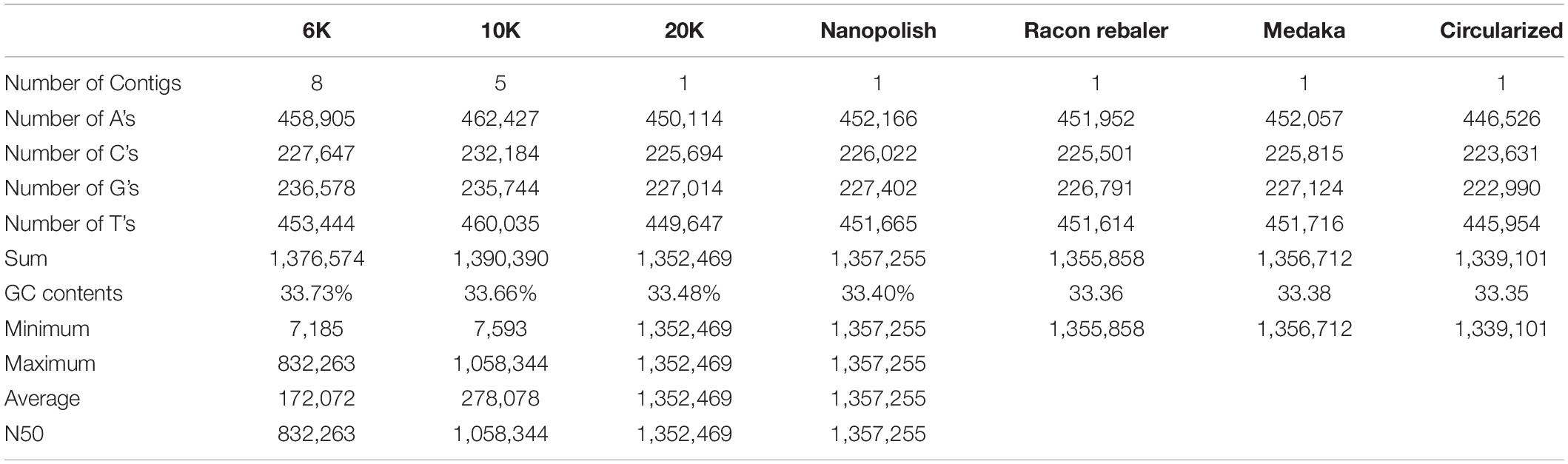
Table 2. Summary statistics for assemblies using three libraries with different insert size, polishing and circularization.
We compared the complete genome constructed in this study with the previously reported 6 scaffold level genomes to identify the unassembled region and its characteristics. Orange circles in Figure 1 indicate commonly unassembled regions in previous scaffolds level assemblies. GC contents of those regions were higher or lower than the average. L. iners has the smallest genome size among the known Lactobacillus species, but complete and chromosome level genomes are hardly available for this small genome. This indicates that GC contents can be one of the reasons for the failure of complete genome assembly in earlier studies. Previous genome assemblies for L. iners were conducted using 2nd generation sequencing platforms such as 454 from Roche and Hiseq from Illumina. Since these kinds of platforms use PCR (emulsion PCR for Roche and Bridge PCR for Illumina) for its data generation process, data generation for specific genomic regions with exceptionally high or low GC contents can be difficult. But single-molecule sequencing platforms such as Nanopore and Pacbio which do not employ PCR for its data generation can reduce this limitation, and another recent successful chromosome level assembly of L. iners LI335 using Pacbio platform showed the effectiveness of single-molecule sequencing. Especially, Flongle platform from Oxford Nanopore technologies is cheaper than any other sequencing platform currently available in the market, and it can generate about 1 Gb data of almost 200X coverage, based on 5 mb microbial genome. Even though a high error rate is still challenging, the future improvement in accuracy and data throughput of Flongle with proper error correction algorithms and tools can provide more cost-effective and easy ways for constructing complete genomes of various kinds of unknown microbes.
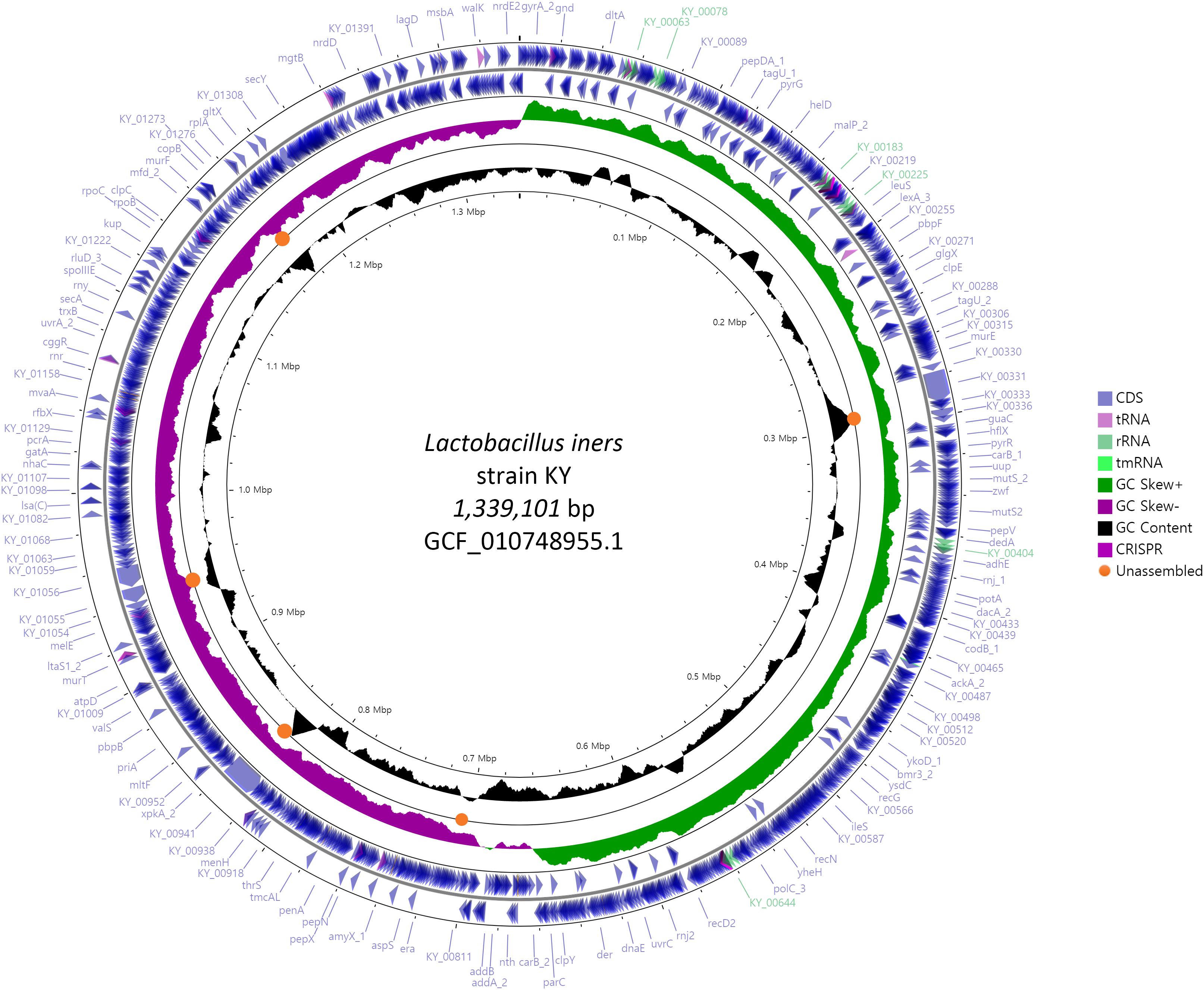
Figure 1. Circular genome map of L. iners KY using Prokka and CGVIEW. The arrow direction of CDS shows the location of the gene in the genome. Five colors including bright purple indicate annotated features from Prokka. Orange circles indicate the commonly unassembled region in six previous scaffold level assembly.
Comparative Genome Analysis and Specific Genes in L. iners
Using OrthoMCL with 40 Lactobacillus genomes (4 randomly selected from each 10 vaginal Lactobacillus species), a total of 7,453 gene clusters were constructed. We started with 40 genomes because OrthoMCL could not handle whole 1,046 Lactobacillus genome used in this study. Among 7,453 gene clusters, 174 clusters were specifically identified in L. iners genomes, and most of them encoded hypothetical proteins. And then, further filtering was conducted based on the Prokka annotation results of 1,046 strain genomes to identify unique core genes of all L. iners genomes among vaginal Lactobacillus species, and three specific genes uniquely existed in all L. iners genomes remained. Table 3 shows three specific genes (INY, ZnuA, and hsdR) which commonly and uniquely existed in the L. iners genomes, and the detailed sequence information for each gene is summarized in Supplementary Table 1.

Table 3. Lactobacillus iners specific genes cluster identified in comparative genome analysis.
Inerolysin (INY) is a well known pore-forming toxin that is specifically produced by L. iners, and it is included in the cholesterol-dependent cytolysins (CDCs) (Rampersaud et al., 2011; Christie et al., 2018). It binds to the cell membrane and forms oligomeric complexes inserted into the lipid bilayer to make aqueous pores (Flanagan et al., 2009). According to the Black Queen Hypothesis, organisms tend to lose the capacity to synthesize metabolites if they are provided by their hosts or community members, and it can account for gene loss and genome reduction (Morris et al., 2012). During the adaption process to the vaginal econiche, large scale gene loss with horizontal gene acquisition occurred in L. iners (Macklaim et al., 2011), and it is highly possible that remaining unique core genes are closely related to the specific adaption of L. iners to the vaginal econiche. The vaginal environment is not simple because the fluctuation of hormone can affect mucus and glycogen production, PH, and microbial species which might provide essential nutrients for L. iners. The previous study showed that genomes of 11 L. iners strains commonly contain this pore-forming toxin gene (Rampersaud et al., 2011), and our study using all available L. iners genomes also showed that 24 L. iners genomes also commonly had this gene, and this specific type of CDC only existed in L. iners among 10 Lactobacillus species. This indicates that inerolysin may be one of the essential genes for L. iners to stably obtain essential nutrients from host and live in dynamically changing vaginal echoniche. Therefore, it can be used as a potential target gene for specific modulation of L. iners and related microbe species in the vaginal flora.
High-affinity zinc uptake system binding-protein ZnuA Type I (ZnuA) is one of the components of ZnuABC, the high-affinity transporter specialized for transporting zinc ions (Patzer and Hantke, 1998; Yatsunyk et al., 2008). ZnuA gene, one of the core genes of L. iners identified in this study, is highly conserved between strains of L. iners. In the moderate conditions when zinc is abundant, zinc uptake is mediated by the low-affinity permease ZupT, a member of ZIP family transporters (Hantke, 2005). However, in environments with very low zinc availability, zinc import is ensured by ZnuABC, and it is one of the important parts of the systems for metal ion homeostasis in bacteria (Patzer and Hantke, 1998; Yatsunyk et al., 2008). In addition, ZnuA is also known to be closely related to the adhesion ability to the epithelial cells in the host, and it is considered as a virulence factor (Gabbianelli et al., 2011; Li et al., 2015). Li et al. (2015) showed that ZnuA was also significantly important for Pseudomonas aeruginosa to adhere to polystyrene plates and HeLa cells, and Gabbianelli et al. (2011) showed that inactivation of ZnuA dramatically decreased the adhering ability of E. coli O157:H7 to Caco-2 cells. Therefore, given that L. iners the only vaginal Lactobacillus species that possesses ZnuA gene, this gene may be one of the essential genes for their adaption to the vaginal econiche and the potential key mediator for strong adhesion to the vaginal epithelial cells, such as previously reported fibronectin (Fn)-binding protein (McMillan et al., 2013).
Type I restriction enzyme R protein (hsdR) is one of three components in the type I R/M (restriction and modification) system (Loenen et al., 2013), and this system combines the functions of site-specific methylation and restriction activity in one large multimeric protein. Genes of this system typically form operon, but each component from different operons or single genes can be intermixed in combination. This system can provide protection against invading DNA such as foreign plasmids or the DNA of bacteriophage, and it is known to be one of the phage resistance mechanisms for some specific lactic acid bacteria (Allison and Klaenhammer, 1998). For Lactobacillus species, L. helveticus was reported to have a plasmid-linked R/M system (Clara et al., 1990), and a recent study showed that the phage resistance strain of L. helveticus used Type I R/M system as a defense mechanism for bacteriophage invasion (Zago et al., 2017). Based on the Prokka annotation of 1,046 vaginal Lactobacillus, we identified some strains of each species also had hsdR gene (L. crispatus: 16.2%, L. gasseri: 9.3%. L. helveticus: 28.1%, L. jensenii: 94.6%, L. johnsonii: 26.3%, L. plantarum: 61.0%, L. reuteri: 39.8%, L. salivarius: 70.5%, L. vaginalis: 75.0%). However, the hsdR gene sequence of L. iners was very unique compared to that of other vaginal Lactobacillus species, which had high sequence similarity with Staphylococcus genus. Meanwhile, hsdR genes from all L. iners genomes had high sequence similarity with E. coli. Also, most of the L. iners hsdR protein sequences were much shorter (300–600 a.a) than the previously known hsdR genes (about 1080 a.a), and they contained c-terminal domain of Type I restriction R subunit. It is well known that the number of vaginal Lactobacillus species decreases during the progress of bacterial vaginosis (BV), and it is replaced with anaerobic bacteria such as Gardnerella species and genital mycoplasmas (Sobel, 2000). Previous studies strongly suggested that bacteriophage was one of the reasons for sudden Lactobacillus decrease during BV (Pavlova et al., 1997; Kiliç et al., 2001), and meta-transcriptome study of L. iners showed that CRISPR anti-bacteriophage defense system and restriction-modification system were highly upregulated during BV (Macklaim et al., 2013). Only a small number of L. iners genomes are known to have cas proteins (Macklaim et al., 2013), and this indicates that the L. iners specific hsdR gene is one of the potential core genes providing L. iners resistance and viability against bacteriophage infection during BV. Further studies on these genes will provide more understanding about the specific adaption of L. iners to the vaginal econiche.
Prophages in Lactobacillus Genomes and Adaption to Vaginal Econiche
Among various possible reasons contribute to the decrease of vaginal Lactobacillus species during BV, bacteriophage was suggested as one, and Pavlova and Tao (2000) showed that it could be induced from prophage in the Lactobacillus genomes. In the comparative gene family analysis using 10 vaginal Lactobacillus species, we suggested the L. iners specific hsdR gene and related Type I R/M system which was upregulated in BV might be one of the key elements for further defense against altered environmental phage load. Therefore, we conducted a prophage analysis for 1,046 genomes of vaginal Lactobacillus species to identify the evidence of this hypothesis. Figure 2 shows a scatter plot of the number of identified prophages and the genome size of each species used in this study. There was no significant correlation between the genome size and the number of identified prophages and this indicates that specific defense mechanisms are involved for defense against phage infection. Figure 3A shows a boxplot of the number of prophages that exists in 10 vaginal Lactobacillus species. The average numbers of prophage of each 10 vaginal Lactobacillus species were different (Kruskall–Wallis test, p < 0.05), and genomes of two species (L. iners and L. helveticus) had significantly smaller numbers of prophage compared to other 8 Lactobacillus species (Figure 3B, all pairwise Wilcoxon Rank-Sum test, FDR < 0.05). Two Lactobacillus species, L. iners and L. helveticus, have a significantly smaller number of detected prophages, and they are known to have a type I RM system which can be useful for the defense against bacteriophage invasion. But comparative gene analysis showed that not only these two Lactobacillus species but also some strains of other vaginal Lactobacillus species used in this study had hsdR gene. This result indicates that induced bacteriophage may not be the main reason for the sudden decrease of Lactobacillus species during BV. Because even though L. helveticus also seemed to have an effective defense system for bacteriophage but it is not the predominant Lactobacillus in vaginal econiche and it cannot retain its abundance during BV. Therefore, we can expect more complex reasons for the sudden reduction of vaginal Lactobacillus species during the progression of BV and more specific adaption mechanisms in L. iners are involved in retaining its abundance during the dynamic environmental change during BV. However, even though bacteriophage may not be the main reason for the sudden reduction of Lactobacillus species during BV, it can accelerate the reduction of Lactobacillus species and defense systems such as Type I RM system and CRISPR can be useful for viability in changing vaginal environment. Previous transcriptome study observed upregulation of the hsdR gene expression in L. iners during BV (Miller-Ensminger et al., 2018), and suggested the important role of the hsdR gene in some of L. iners strains without CRISPR system. Further study will be necessary to identify the role and relatedness of its unique hsdR gene for the defense against bacteriophage infection. Figure 4 shows the number of detected and identified prophage in 1,046 genomes. Same as previous study (Miller-Ensminger et al., 2018), most predicted prophage sequences could not be identified its origin even though blast filtering criteria was set to very low (22%: sum of S.Cov > 10%, 5.9%: sum of S.Cov > 50%). Two candidate reasons were expected for this result. Integrated prophage sequences can be weathered during bacterial genome evolution and the current public sequence database does not contain enough sequence information for bacteriophages and prophages. Because bacteriophage is known to have an extraordinary diversity, low similarity (Grose and Casjens, 2014; Pope et al., 2015), 2,169 genomes of bacteriophages and prophages are not enough to fully identify its origin. All identified phages and prophages were matched to Lactobacillus phage and prophage, such as Lactobacillus phage AQ113, KC5a, Lv-1, phi jlb1, phig1e, Sha1, and Lactobacillus prophage Lj928, Lj965, phiadh, etc., and Lactobacillus phages are known to have a wide host range for various Lactobacillus species (Kiliç et al., 2001). Detected prophages from L. iners, L. salivarius and L. vaginalis were not matched to previously known phage sequence. All identified prophages in L. crispatus (0.88%) and L. helveticus (27.45%) were Lactobacillus phage AQ113. Lactobacillus phage AQ113 was isolated from L. helveticus in dairy product and it is included in Myoviridae (Zago et al., 2013). In case of L. jensenii, all identified prophages were Lactobacillus phage Lv-1 (38.71%), and it is only found in the L. jensenii genomes. Lactobacillus phage Lv-1 was isolated from vaginal L. jensenii, and it is included in Siphoviridae (Martín Rosique et al., 2010). Identified prophages of L. plantarum (33.74%) were Lactobacillus phage SHA1 (Yoon et al., 2011) and phig1e (Kakikawa et al., 1996), and they are all isolated from L. plantarum in previous study. 6 and 4 types of prophages were identified in L. gasseri (48.15%) and L. johnsonii (38.71%) genomes, and prophages of two Lactobacillus species shared its source of origin (Sechaud et al., 1988). Two Lactobacillus phage, Lactobacillus phage SHA1 and Lj965 from L. plantarum and L. johnsonii (Ventura et al., 2003), were identified in L. reuteri genomes (4.94%). This result suggests that the host range of Lactobacillus phage varies according to its type and lineage, and it might be more narrow than previously reported. However, this result has limitations because it was based on the small number of previously reported phage genome sequences, and most of the detected prophage sequences are still unknown. Lactobacillus phage is known to be one of the important driving forces of genome evolution (Baugher et al., 2014), and previous study about phageome related to the schizophrenia showed Lactobacillus phage is closely related to human health and disease (Yolken et al., 2015). Therefore, accumulation of phage researches and its sequences is necessary for future research and it will help to further expand our knowledge of L. iners specific adaption to vaginal econiche and therapeutic modulation using Lactobacillus phage.
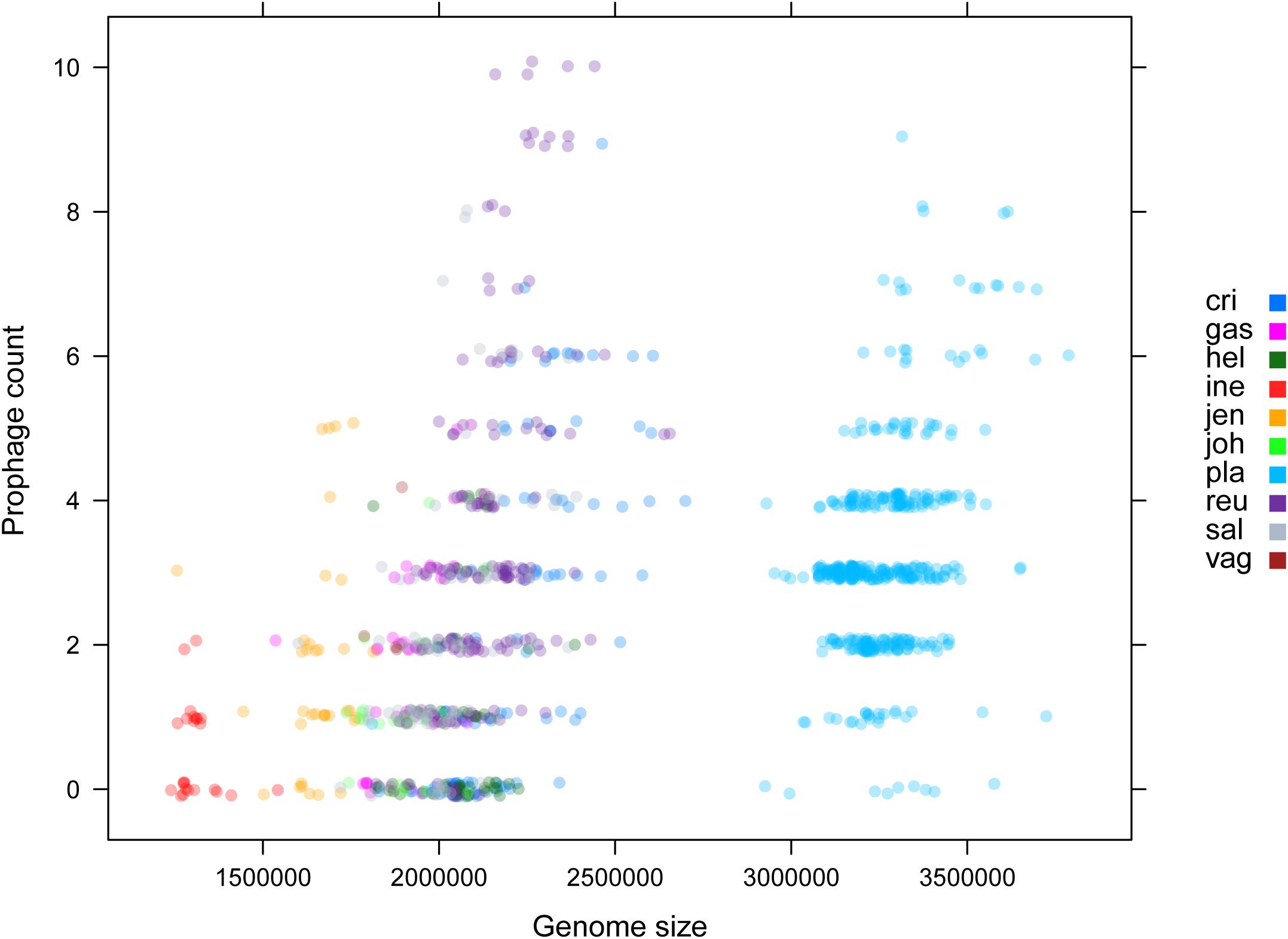
Figure 2. Scatter plot for genome size and identified prophage count using ProphET. Color indicates each vaginal Lactobacillus species used in this study.
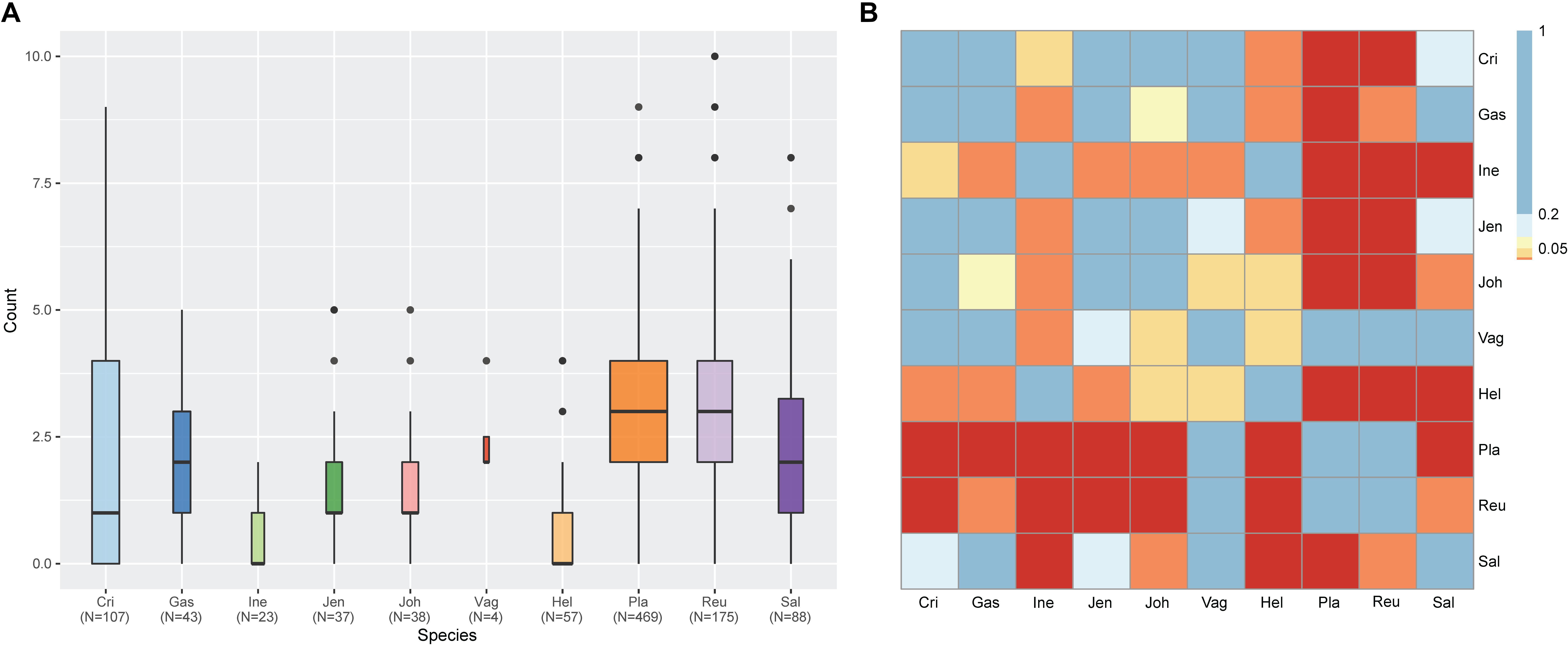
Figure 3. Result of prophage count analysis for 1,046 vaginal Lactobacillus species using ProphET. (A) Boxplot for the number of prophage in each strain genome. Band width indicate the number of genomes used for plot. (B) Heatmap for the result of the pairwise Wilcoxon Rank-Sum test. Colors indicate the significance level with FDR correction. (Blue > 0.2 < Sky blue > 0.1 < Yellow > 0.05 < Orange > 0.01 > Red).
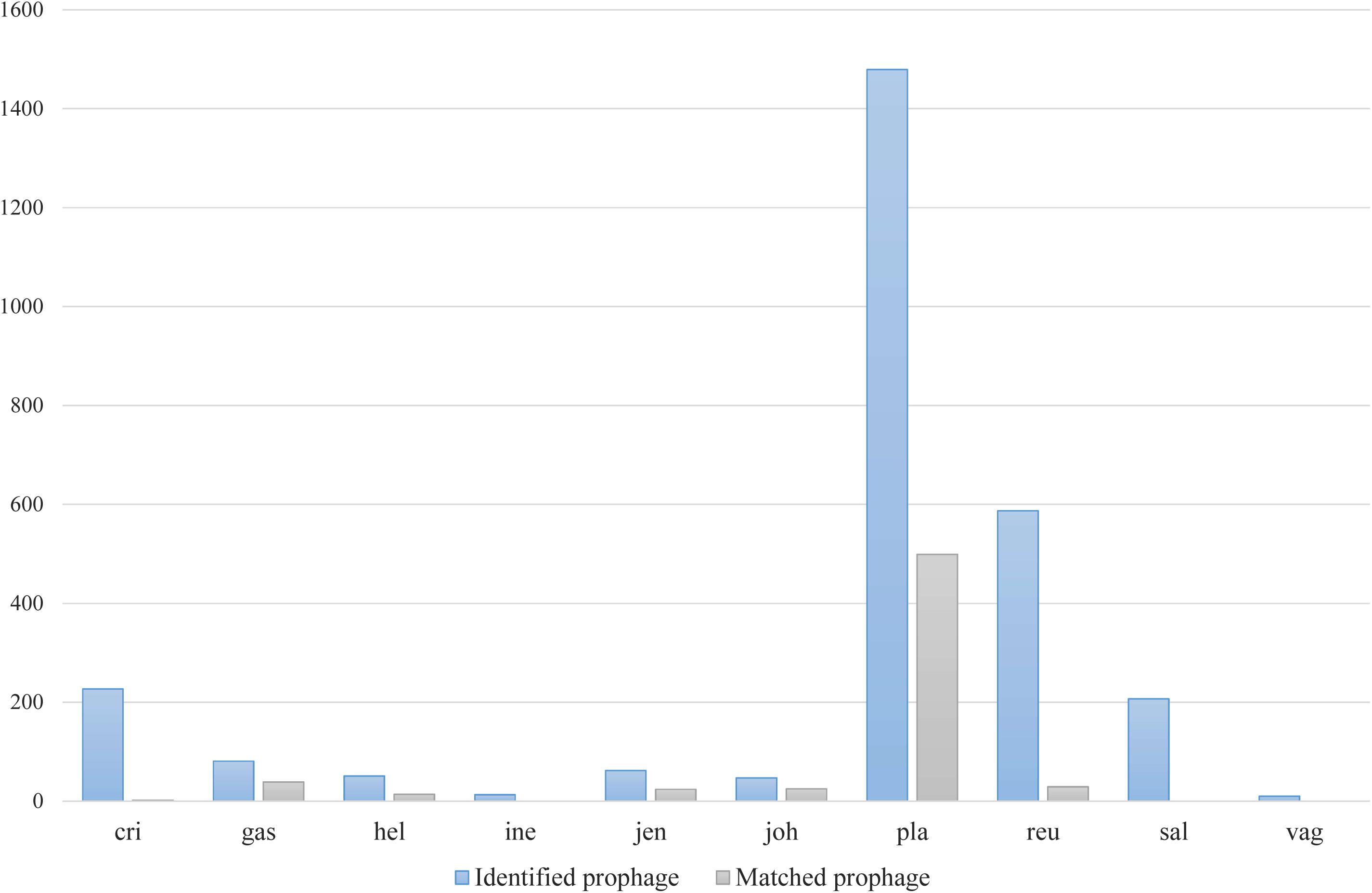
Figure 4. Number of detected and identified prophages using previously known phage and prophage genome sequences.
Data Availability Statement
The datasets generated for this study can be found in the NCBI, under accession number PRJNA603871.
Ethics Statement
The studies involving human participants were reviewed and approved by Konyang University Hospital (IRB FILE No: 2017-12-021-010). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
WK and TK designed the experiments, interpreted the data, drafted the manuscript, and supervised the study. DS, HA, and JK performed the bioinformatic analyses and interpreted the data. Y-HH, HKK, and MJ performed the experiments. HBK contributed to the revision. All authors reviewed the manuscript.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2017R1C1B5076693) and Konyang University Myunggok Research Fund of 2017.
Conflict of Interest
WK and MJ were employed by C&K Genomics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all the volunteers involved in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01048/full#supplementary-material
Footnotes
- ^ https://github.com/w1bw/Porechop
- ^ https://github.com/rrwick/Rebaler
- ^ https://nanoporetech.github.io/medaka/
- ^ https://github.com/Kzra/Simple-Circularise
References
Allison, G. E., and Klaenhammer, T. R. (1998). Phage resistance mechanisms in lactic acid bacteria. Int. Dairy J. 8, 207–226.
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410.
Baugher, J., Durmaz, E., and Klaenhammer, T. (2014). Spontaneously induced prophages in Lactobacillus gasseri contribute to horizontal gene transfer. Appl. Environ. Microbiol. 80, 3508–3517. doi: 10.1128/AEM.04092-13
Burton, J. P., and Reid, G. (2002). Evaluation of the bacterial vaginal flora of 20 postmenopausal women by direct (Nugent score) and molecular (polymerase chain reaction and denaturing gradient gel electrophoresis) techniques. J. Infect. Dis. 186, 1770–1780.
Christie, M. P., Johnstone, B. A., Tweten, R. K., Parker, M. W., and Morton, C. J. (2018). Cholesterol-dependent cytolysins: from water-soluble state to membrane pore. Biophys. Rev. 10, 1337–1348. doi: 10.1007/s12551-018-0448-x
Clara, G., Limsowtin, G. K., Séchaud, L., Veaux, M., and Accolas, J.-P. (1990). Evidence for a plasmid-linked restriction-modification system in Lactobacillus helveticus. Appl. Environ. Microbiol. 56, 3412–3419.
De Backer, E., Verhelst, R., Verstraelen, H., Alqumber, M. A., Burton, J. P., Tagg, J. R., et al. (2007). Quantitative determination by real-time PCR of four vaginal Lactobacillus species, Gardnerella vaginalis and Atopobium vaginae indicates an inverse relationship between L. gasseri and L. iners. BMC Microbiol. 7:115. doi: 10.1186/1471-2180-7-115
Flanagan, J. J., Tweten, R. K., Johnson, A. E., and Heuck, A. P. (2009). Cholesterol exposure at the membrane surface is necessary and sufficient to trigger perfringolysin O binding. Biochemistry 48, 3977–3987. doi: 10.1021/bi9002309
Gabbianelli, R., Scotti, R., Ammendola, S., Petrarca, P., Nicolini, L., and Battistoni, A. (2011). Role of ZnuABC and ZinT in Escherichia coli O157: H7 zinc acquisition and interaction with epithelial cells. BMC Microbiol. 11:36. doi: 10.1186/1471-2180-11-36
Grant, J. R., and Stothard, P. (2008). The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 36(Suppl._2), W181–W184. doi: 10.1093/nar/gkn179
Grose, J. H., and Casjens, S. R. (2014). Understanding the enormous diversity of bacteriophages: the tailed phages that infect the bacterial family Enterobacteriaceae. Virology 468, 421–443. doi: 10.1016/j.virol.2014.08.024
Haft, D. H., DiCuccio, M., Badretdin, A., Brover, V., Chetvernin, V., O’Neill, K., et al. (2017). RefSeq: an update on prokaryotic genome annotation and curation. Nucleic Acids Res. 46, D851–D860. doi: 10.1093/nar/gkx1068
Hummelen, R., Fernandes, A. D., Macklaim, J. M., Dickson, R. J., Changalucha, J., Gloor, G. B., et al. (2010). Deep sequencing of the vaginal microbiota of women with HIV. PLoS One 5:e12078. doi: 10.1371/journal.pone.0012078
Hunt, M., De Silva, N., Otto, T. D., Parkhill, J., Keane, J. A., and Harris, S. R. (2015). Circlator: automated circularization of genome assemblies using long sequencing reads. Genome Biol. 16:294. doi: 10.1186/s13059-015-0849-0
Kakikawa, M., Oki, M., Tadokoro, H., Nakamura, S., Taketo, A., and Kodaira, K.-I. (1996). Cloning and nucleotide sequence of the major capsid proteins of Lactobacillus bacteriophage Φgle. Gene 175, 157–165.
Kiliç, A. O., Pavlova, S. I., Alpay, S., Kiliç, S. S., and Tao, L. (2001). Comparative study of vaginalLactobacillus phages isolated from women in the United States and Turkey: prevalence, morphology, host range, and DNA homology. Clin. Diagn. Lab. Immunol. 8, 31–39.
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Lebeer, S., Vanderleyden, J., and De Keersmaecker, S. C. (2008). Genes and molecules of lactobacilli supporting probiotic action. Microbiol. Mol. Biol. Rev. 72, 728–764. doi: 10.1128/MMBR.00017-08
Li, B., Wang, B., Gao, X., Gao, L., Liang, Y., Duan, K., et al. (2015). “The zinc uptake system (znuA Locus) is important for bacterial adhesion and virulence in Pseudomonas aeruginosa,” in Biomedical Engineering and Environmental Engineering: Proceedings of the 2014 2nd International Conference on Biomedical Engineering and Environmental Engineering (ICBEEE 2014), December 24–25, 2014, (Wuhan: CRC Press), 135.
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. doi: 10.1093/bioinformatics/bty191
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189.
Loenen, W. A., Dryden, D. T., Raleigh, E. A., and Wilson, G. G. (2013). Type I restriction enzymes and their relatives. Nucleic Acids Res. 42, 20–44. doi: 10.1093/nar/gkt847
Loman, N. J., Quick, J., and Simpson, J. T. (2015). A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods 12:733. doi: 10.1038/nmeth.3444
Macklaim, J. M., Fernandes, A. D., Di Bella, J. M., Hammond, J.-A., Reid, G., and Gloor, G. B. (2013). Comparative meta-RNA-seq of the vaginal microbiota and differential expression by Lactobacillus iners in health and dysbiosis. Microbiome 1:12. doi: 10.1186/2049-2618-1-12
Macklaim, J. M., Gloor, G. B., Anukam, K. C., Cribby, S., and Reid, G. (2011). At the crossroads of vaginal health and disease, the genome sequence of Lactobacillus iners AB-1. Proc. Natl. Acad. Sci. 108(Suppl. 1), 4688–4695. doi: 10.1073/pnas.1000086107
Martín Rosique, R., Escobedo Martín, S., and Suárez Fernández, J. E. (2010). Induction, structural characterization, and genome sequence of Lv1, a prophage from a human vaginal Lactobacillus jensenii strain. Int. Microbiol. 13, 113–121.
McMillan, A., Macklaim, J. M., Burton, J. P., and Reid, G. (2013). Adhesion of Lactobacillus iners AB-1 to human fibronectin: a key mediator for persistence in the vagina? Reproduct. Sci. 20, 791–796. doi: 10.1177/1933719112466306
Mendes-Soares, H., Suzuki, H., Hickey, R. J., and Forney, L. J. (2014). Comparative functional genomics of Lactobacillus spp. reveals possible mechanisms for specialization of vaginal lactobacilli to their environment. J. Bacteriol. 196, 1458–1470. doi: 10.1128/JB.01439-13
Miller-Ensminger, T., Garretto, A., Brenner, J., Thomas-White, K., Zambom, A., Wolfe, A. J., et al. (2018). Bacteriophages of the urinary microbiome. J. Bacteriol. 200, e738–e717. doi: 10.1128/JB.00738-17
Morris, J. J., Lenski, R. E., and Zinser, E. R. (2012). The Black Queen Hypothesis: evolution of dependencies through adaptive gene loss. mBio 3:e00036-12. doi: 10.1128/mBio.00036-12
Osset, J., Bartolomé, R. M., García, E., and Andreu, A. (2001). Assessment of the capacity of Lactobacillus to inhibit the growth of uropathogens and block their adhesion to vaginal epithelial cells. J. Infect. Dis. 183, 485–491.
Patzer, S. I., and Hantke, K. (1998). The ZnuABC high-affinity zinc uptake system and its regulator zur in Escherichia coli. Mol. Microbiol. 28, 1199–1210.
Pavlova, S. I., Kiliç, A. O., Mou, S. M., and Tao, L. (1997). Phage infection in vaginal lactobacilli: an in vitro study. Infect. Dis. Obst. Gynecol. 5, 36–44.
Pavlova, S. I., and Tao, L. (2000). Induction of vaginal Lactobacillus phages by the cigarette smoke chemical benzo [a] pyrene diol epoxide. Mutat. Res. 466, 57–62.
Petrova, M. I., Lievens, E., Malik, S., Imholz, N., and Lebeer, S. (2015). Lactobacillus species as biomarkers and agents that can promote various aspects of vaginal health. Front. Physiol. 6:81. doi: 10.3389/fphys.2015.00081
Pope, W. H., Bowman, C. A., Russell, D. A., Jacobs-Sera, D., Asai, D. J., Cresawn, S. G., et al. (2015). Whole genome comparison of a large collection of mycobacteriophages reveals a continuum of phage genetic diversity. eLife 4:e06416. doi: 10.7554/eLife.06416
Rampersaud, R., Planet, P. J., Randis, T. M., Kulkarni, R., Aguilar, J. L., Lehrer, R. I., et al. (2011). Inerolysin, a cholesterol-dependent cytolysin produced by Lactobacillus iners. J. Bacteriol. 193, 1034–1041. doi: 10.1128/JB.00694-10
Ravel, J., Gajer, P., Abdo, Z., Schneider, G. M., Koenig, S. S., McCulle, S. L., et al. (2011). Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. U.S.A. 108(Suppl. 1), 4680–4687. doi: 10.1073/pnas.1002611107
Reis-Cunha, J. L., Bartholomeu, D. C., Manson, A. L., Earl, A. M., and Cerqueira, G. C. (2019). ProphET, prophage estimation tool: a stand-alone prophage sequence prediction tool with self-updating reference database. PLoS One 14:e223364. doi: 10.1371/journal.pone.0223364
Sechaud, L., Cluzel, P.-J., Rousseau, M., Baumgartner, A., and Accolas, J.-P. (1988). Bacteriophages of lactobacilli. Biochimie 70, 401–410.
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Spear, G. T., Gilbert, D., Landay, A. L., Zariffard, R., French, A. L., Patel, P., et al. (2011). Pyrosequencing of the genital microbiotas of HIV-seropositive and-seronegative women reveals Lactobacillus iners as the predominant Lactobacillus species. Appl. Environ. Microbiol. 77, 378–381. doi: 10.1128/AEM.00973-10
Srinivasan, S., Liu, C., Mitchell, C. M., Fiedler, T. L., Thomas, K. K., Agnew, K. J., et al. (2010). Temporal variability of human vaginal bacteria and relationship with bacterial vaginosis. PLoS One 5:e10197. doi: 10.1371/journal.pone.0010197
Tamrakar, R., Yamada, T., Furuta, I., Cho, K., Morikawa, M., Yamada, H., et al. (2007). Association between Lactobacillus species and bacterial vaginosis-related bacteria, and bacterial vaginosis scores in pregnant Japanese women. BMC Infect. Dis. 7:128. doi: 10.1186/1471-2334-7-128
Vaser, R., Sović, I., Nagarajan, N., and Šikić, M. (2017). Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746. doi: 10.1101/gr.214270.116
Ventura, M., Canchaya, C., Pridmore, D., Berger, B., and Brüssow, H. (2003). Integration and distribution of Lactobacillus johnsonii prophages. J. Bacteriol. 185, 4603–4608.
Verhelst, R., Verstraelen, H., Claeys, G., Verschraegen, G., Van Simaey, L., De Ganck, C., et al. (2005). Comparison between Gram stain and culture for the characterization of vaginal microflora: definition of a distinct grade that resembles grade I microflora and revised categorization of grade I microflora. BMC Microbiol. 5:61. doi: 10.1186/1471-2180-5-61
Yatsunyk, L. A., Easton, J. A., Kim, L. R., Sugarbaker, S. A., Bennett, B., Breece, R. M., et al. (2008). Structure and metal binding properties of ZnuA, a periplasmic zinc transporter from Escherichia coli. JBIC 13, 271–288.
Yolken, R. H., Severance, E. G., Sabunciyan, S., Gressitt, K. L., Chen, O., Stallings, C., et al. (2015). Metagenomic sequencing indicates that the oropharyngeal phageome of individuals with schizophrenia differs from that of controls. Schizophrenia Bull. 41, 1153–1161. doi: 10.1093/schbul/sbu197
Yoon, B. H., Jang, S. H., and Chang, H.-I. (2011). Sequence analysis of the Lactobacillus temperate phage Sha1. Arch. Virol. 156, 1681–1684. doi: 10.1007/s00705-011-1048-2
Zago, M., Orrù, L., Rossetti, L., Lamontanara, A., Fornasari, M. E., Bonvini, B., et al. (2017). Survey on the phage resistance mechanisms displayed by a dairy Lactobacillus helveticus strain. Food Microbiol. 66, 110–116. doi: 10.1016/j.fm.2017.04.014
Keywords: vaginal microbe, Lactobacillus iners, long-read assembly, Oxford nanopore, genomic adaptation
Citation: Kwak W, Han Y-H, Seol D, Kim H, Ahn H, Jeong M, Kang J, Kim H and Kim TH (2020) Complete Genome of Lactobacillus iners KY Using Flongle Provides Insight Into the Genetic Background of Optimal Adaption to Vaginal Econiche. Front. Microbiol. 11:1048. doi: 10.3389/fmicb.2020.01048
Received: 30 January 2020; Accepted: 28 April 2020;
Published: 26 May 2020.
Edited by:
Vasco Ariston De Carvalho Azevedo, Federal University of Minas Gerais, BrazilReviewed by:
Sylwia Bloch, University of Gdańsk, PolandAlicja Węgrzyn, Polish Academy of Sciences, Poland
Patrice François, Geneva University Hospitals (HUG), Switzerland
Marcus Vinicius Canário Viana, Federal University of Pará, Brazil
Copyright © 2020 Kwak, Han, Seol, Kim, Ahn, Jeong, Kang, Kim and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tae Hyun Kim, th2580@kyuh.ac.kr; th2580.kim@gmail.com
†These authors share first authorship