 Trestan Pillonel
Trestan Pillonel Claire Bertelli
Claire Bertelli Gilbert Greub
Gilbert Greub- Center for Research on Intracellular Bacteria, Institute of Microbiology, Centre Hospitalier Universitaire Vaudois, Lausanne, Switzerland
The Chlamydiae phylum exclusively encompasses bacteria sharing a similar obligate intracellular life cycle. Existing 16S rDNA data support a high diversity within the phylum, however genomic data remain scarce owing to the difficulty in isolating strains using culture systems with eukaryotic cells. Yet, Chlamydiae genome data extracted from large scale metagenomic studies might help fill this gap. This work compares 33 cultured and 27 environmental, uncultured chlamydial genomes, in order to clarify the phylogenetic relatedness of the new chlamydial clades and to investigate the genetic diversity of the Chlamydiae phylum. The analysis of published chlamydial genomes from metagenomics bins and single cell sequencing allowed the identification of seven new deeply branching chlamydial clades sharing genetic hallmarks of parasitic Chlamydiae. Comparative genomics suggests important biological differences between those clades, including loss of many proteins involved in cell division in the genus Similichlamydia, and loss of respiratory chain and tricarboxylic acid cycle in several species. Comparative analyses of chlamydial genomes with two proteobacterial orders, the Rhizobiales and the Rickettsiales showed that genomes of different Rhizobiales families are much more similar than genomes of different Rickettsiales families. On the other hand, the chlamydial 16S rRNAs exhibit a higher sequence conservation than their Rickettsiales counterparts, while chlamydial proteins exhibit increased sequence divergence. Studying the diversity and genome plasticity of the entire Chlamydiae phylum is of major interest to better understand the emergence and evolution of this ubiquitous and ancient clade of obligate intracellular bacteria.
Introduction
Bacteria of the phylum Chlamydiae are all obligate intracellular bacteria that multiply within eukaryotic host cells (Horn, 2015). Various chlamydial species were identified in mammals, birds, reptiles, fishes, arthropods, and unicellular eukaryotes (Taylor-Brown et al., 2015). Several Chlamydiae, such as Chlamydia trachomatis, Chlamydia abortus, and Chlamydia pneumoniae, are well-known important human and animal pathogens (Elwell et al., 2016). Other Chlamydia-related organisms such as Waddlia chondrophila, associated with abortion in cattle, are increasingly recognized as emerging pathogens posing a zoonotic or vector-borne risk (Taylor-Brown and Polkinghorne, 2017). Multiple species of the Simkaniaceae, “Candidatus Piscichlamydiaceae” and “Ca. Clavichlamydiaceae” are associated with epitheliocystis, a disease affecting the gill of fish. Rhabdochlamydiaceae spp. are also highly prevalent in ticks, the most common arthropod vector of human and animal disease (Pilloux et al., 2015). Analysis of environmental 16S rRNA sequences indicates that those organisms are highly diverse and are present in a wide variety of freshwater, marine and terrestrial environments (Lagkouvardos et al., 2014). However, there are currently only six validated families (Chlamydiaceae, Parachlamydiaceae, Simkaniaceae, Waddliaceae, Criblamydiaceae, and Rhabdochlamydiaceae) and three Candidatus families (Clavichlamydiaceae, Parilichlamydiaceae, and Piscichlamydiaceae) (Stride et al., 2013; Horn, 2015).
The last comparative analysis of the phylum Chlamydiae (Collingro et al., 2011) included genomic data from four families; a single species of Waddliaceae and Simkaniaceae, and multiple species of Chlamydiaceae and Parachlamydiaceae. This work reported a relatively large set of 560 genes conserved in all four chlamydial clades, and a large number of accessory genes restricted to families exhibiting much larger genomes than vertebrate parasites of the Chlamydia genus. Some virulence factors such as the Type III secretion system were shown to be conserved in all four families investigated. On the other hand, the F-like conjugative DNA transfer system identified by Greub et al. (2004) in Protochlamydia amobophila was restricted to Simkaniaceae and Parachlamydiaceae (Bertelli et al., 2016). Since then, multiple genomes belonging to the family Parachlamydiaceae were sequenced from strains in pure culture, including one representative of a new genus named Rubidus (Domman et al., 2014; Ishida et al., 2014; Yamaguchi et al., 2015; Yamane et al., 2015; Bertelli et al., 2016; Bou Khalil et al., 2016, 2017; Fukumoto et al., 2016). In addition, genome sequences representing two distinct genera of the family Criblamydiaceae were recently released (Bertelli et al., 2014, 2015).
Previously, technically challenging and time-consuming in vitro cultures were required to obtain sufficient sample material for genomic analysis of Chlamydiae (Jacquier et al., 2013). This difficulty was recently overcome by culture-independent genomic characterization of uncultivable Chlamydiae. In 2013, the genome of C. trachomatis could be recovered directly from clinical samples using a metagenomic approach (Andersson et al., 2013; Seth-Smith et al., 2013). More recently, the complete genome of “Candidatus Chlamydia sanzinia”—an uncultivated snake pathogen—was sequenced without culture (Taylor-Brown et al., 2016). The fish pathogen “Candidatus Similichlamydia epinepheli,” a representative of the Candidatus family Parilichlamydiaceae, was partially recovered using a similar approach (Taylor-Brown et al., 2017). Moreover, several “Candidatus Rhabdochlamydia spp.” genomes could also be directly sequenced from ticks samples (Pillonel et al., unpublished data). Single-cell genomics is another promising approach for the study of uncultivable organisms (Gawad et al., 2016). This technique allowed the recovery of partial genomes of three new chlamydial lineages from marine environments (Collingro et al., 2017).
Thus, from 2011 to 2017, a large number of additional chlamydial genomes have been made available but their diversity and their relative position in the phylogenetic tree of the phylum Chlamydiae remained so far unknown. Updated comparative genomic analyses will allow to clarify the phylogenetic relatedness of the new chlamydial clades and to improve our understanding of the evolution of shared and distinct genetic features in this diverse group of obligate intracellular bacteria.
Therefore, we performed here a comprehensive comparative analysis of the phylum Chlamydiae including all genomic data currently publicly available (as of June 2017). We have identified 7 putative novel family-level lineages retrieved from metagenomics bins. Extremely diverse genomic characteristics were observed, with genome size ranging from 1 to 3.4 Mbp and GC content ranging from 26.23 to 55.76%. Despite those variations, essential mechanisms involved in host-symbiont interactions such as the type III secretion apparatus and the ADP-ATP translocase (a transporter involved in energy parasitism) are conserved in all newly discovered lineages. On the other hand, fundamental biological differences such as distinct division mechanisms of novel and phylogenetically distantly-related Chlamydiae could be highlighted.
Methods
Data Retrieval
Genome assemblies were downloaded from the RefSeq database, and from Genbank for the assemblies absent from RefSeq (Table 1; O'Leary et al., 2016). All genome assemblies from the phylum Chlamydiae (NCBI taxonomy ID 204428) were included in this study with the exception of the Chlamydia genus. Since several Chlamydia species are well-studied pathogens with many sequenced genomes, a single representative genome assembly was considered for each of the 13 Chlamydia species out of a total of 326 genome assemblies (June 2017).
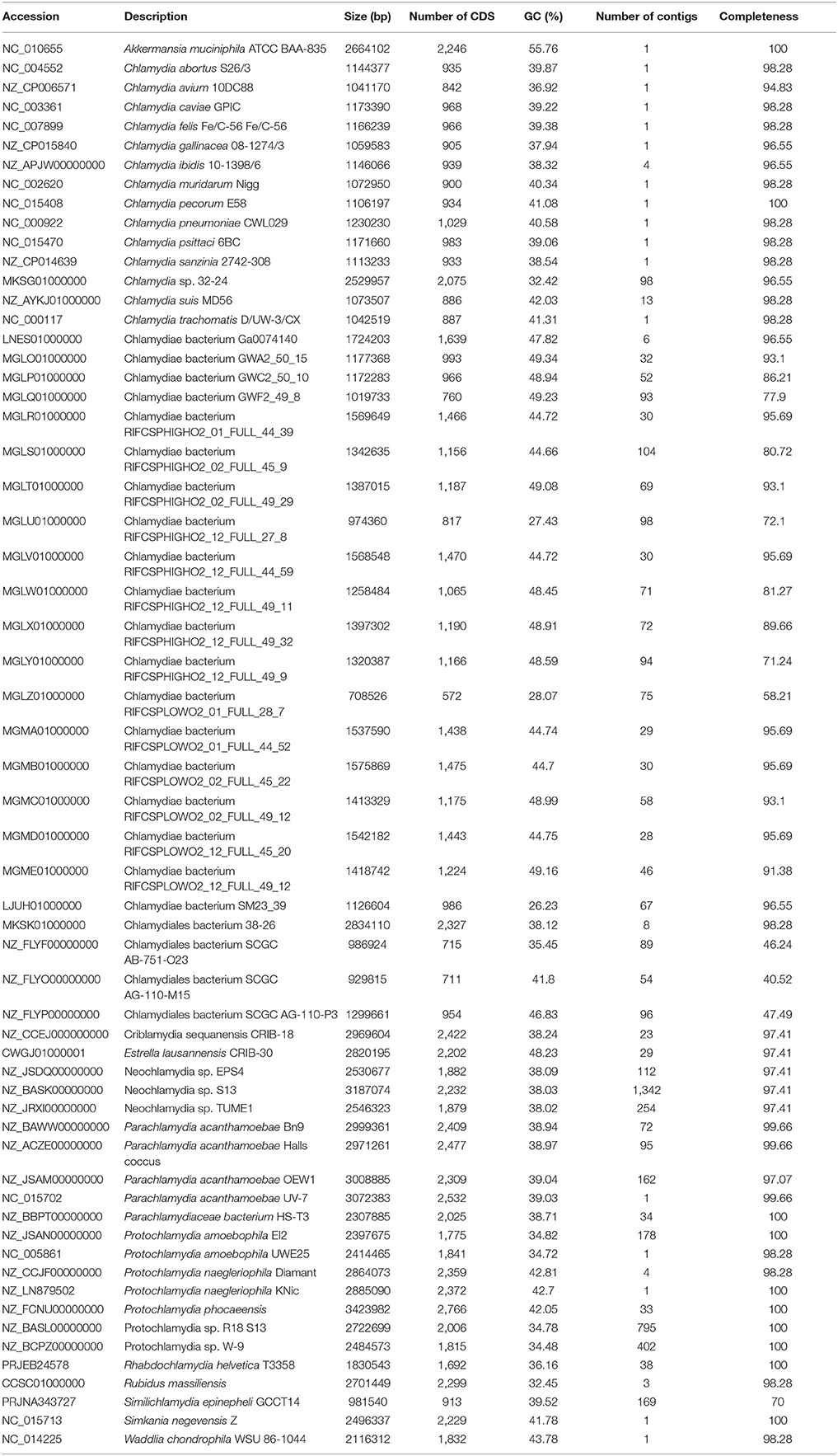
Table 1. List of genomes included in the analysis, with GC content (%), size (bp) and completeness as evaluated using checkM.
Evaluation of Genome Completeness and Quality
Many genomes included in this work are metagenomics bins that may be of unequal quality. Indeed, metagenomics bins can be an incomplete representation of the organism's genomes, can be a mixture of multiple genomes or include chimeric DNA sequences. To screen for contaminants, the completeness and redundancy of each genome was thus evaluated based on the identification of 104 nearly universal (and generally single copy) bacterial markers with checkM (Parks et al., 2015). In addition, translated predicted coding sequences (CDS) of each genome were compared to RefSeq database using PLAST, a fast sequence similarity search tool, with the following parameters: -M BLOSUM62 -s 45 -seeds-use-ratio 60 -G 11 -E 1 -F F -max-hit-per-query 100 -max-hsp-per-hit 1 (Van Nguyen and Lavenier, 2009). PLAST was favored over BLAST for its lower memory usage. The taxonomy of the first PLAST hit (excluding hits against RefSeq sequences classified as belonging to the same species) was investigated with help of the NCBI taxonomy database (Federhen, 2012).
Comparative Genome Analysis and Reconstruction of the Species Phylogeny
Orthologs were identified using OrthoFinder version 0.4 (Emms and Kelly, 2015). A reference phylogeny was built based on single copy orthologs conserved in at least 55 of the 60 studied genomes (Table 1). Core single copy orthologs were aligned using mafft version 7.058b (Katoh and Standley, 2013). The concatenated alignment was used for the reconstruction of the species tree using FastTree 2.1.9 with double precision (Price et al., 2010). Circular plots were drawn using Circos version 0.69 (Krzywinski et al., 2009), whereas other plots were made with R (R Core Team, 2016).
Genome Annotation and Identification of Secretion Systems, Flagellar Subunits, and Selected Metabolic Traits
All genomes were annotated using GhostKOALA (Kanehisa et al., 2016) and Interproscan version 5.23–62.0 (Mitchell et al., 2015). Orthologs of the type III and type IV secretion systems, known effectors, division proteins, membrane proteins, the Euo master regulator, respiratory chain complexes and enzymes involved in menaquinone biosynthesis and glycogen metabolism were identified based on OrthoFinder grouping into groups of orthologous proteins (see Table S1 for the detailed reference locus list). The ATP:ADP antiporters homologs were identified by InterProScan annotation (interpro accession: IPR004667). The flagellar apparatus subunits, proteins involved in peptidoglycan, purine, pyrimidine and ubiquinone biosynthesis as well as the glycolysis pathway, the citrate cycle (TCA), and the pentose phosphate pathway (PPP) were identified based on KEGG ortholog (KO) annotation. Reported counts for each pathway/module are the non-redundant number of identified KO. Predicted coding sequences were compared to COG database (version 2014) (Galperin et al., 2015) using BLASTP version 2.3.0+(Camacho et al., 2009) with an e-value cutoff of 1e−5, a minimal query coverage of 50% and a minimal identity of 20%.
Identification of Phylogenetic Markers and Evaluation of Genetic Relatedness
Proteins conserved between pairs of genomes were identified by pairwise protein sequences comparisons with BLASTP version 2.3.0+ (Camacho et al., 2009). The identity of all reciprocal best blast hit (RBBH) with a minimal e-value of 10−5 and 50% of both query and hit coverage were retained for the comparative analyses. 16S and 23S rRNA sequences were identified using barrnap (https://github.com/tseemann/barrnap). HMM profiles were built from 21 reference sequences for nine previously identified phylogenetically informative markers (Pillonel et al., 2015). These profiles were used to identify markers in all chlamydial genomes included in this study using hmmsearch v3.1 (Eddy, 2011). For each marker, a phylogenetic tree was reconstructed based on the best hmmsearch hit in each genome. Bitscore cut-offs were defined individually for each marker based on the density distribution of hmmsearch bit scores and gene tree topologies: DnaA (260), SucA (750), the hypothetical protein 325 (350), FabI (400), RpoN (300), FtsK (750), PepF (470), Adk (175), and HemL (360). Pairwise amino acid sequence identities were calculated based on pairwise Needleman-Wunsch global alignments made with Needle from the EMBOSS package version 6.6 (Rice et al., 2000). Gaps were not considered in identity calculations. The probability distributions of pairwise amino acid identity values were estimated using the statistical package R with kernel density estimations (R Core Team, 2016). Density plots were made using the ggplot2 package (Wickham, 2016).
Results
Twelve Nearly Complete Genome Assemblies from Multiple New Deeply Branching Chlamydiae Clades
In total, 60 genomes (including one outgroup) were compared in this study: 21 poorly characterized metagenomics bins classified as Chlamydiae were retrieved from Genbank. Most (n = 17) originate from a single study of groundwater microbial ecosystems (Anantharaman et al., 2016). Others were sequenced as part of the microbial community of a biologically active filter of a water treatment plant (n = 1) (Pinto et al., 2016), as part of an investigation of bacterial communities in estuary sediments (n = 1) (Baker et al., 2015), and from an experimental bioreactor used to treat contaminated goldmine water (n = 2) (Kantor et al., 2015). In addition, three single-cell amplified partial genomes from marine Chlamydiae (Collingro et al., 2017) and 25 previously published chlamydial genomes (Table 1) were studied. The latter contained a majority (n = 20) of draft assemblies. The verrucomicrobial genome of Akkermansia muciniphila was included as an outgroup.
While most Chlamydiae share a similar life cycle, their genetic diversity is considerable. Standard procedures for the classification of Chlamydiae rely on a small set of reference genes or protein sequences, including the 16S rRNA sequence (Pillonel et al., 2015; Greub, 2017). Given that metagenomic datasets frequently lack rRNA operons, pairwise comparisons of all protein sequences were undertaken to better characterize the genetic diversity of newly sequenced genomes. The protein sequences of the 59 chlamydial genomes included in this comparative analysis were clustered into 10,162 orthologous groups (Table S2, Figure S1A). Among those groups, 4,553 are restricted to a single genome (44.8%, Figure S1B). Only three single copy orthologs were conserved in all chlamydial genomes (the two ribosomal proteins S8 and L6, and the tRNA threonylcarbamoyladenosine biosynthesis protein TsaE), but 108 orthologous groups were conserved in more than 90% of the genomes (55 out of 60, including the outgroup genome.The list of 108 orthologous groups is reported in Table S3). The concatenated alignment of those 108 protein sequences was used to build a reference phylogeny of all representative genomes of the phylum Chlamydiae (Figure 1A).
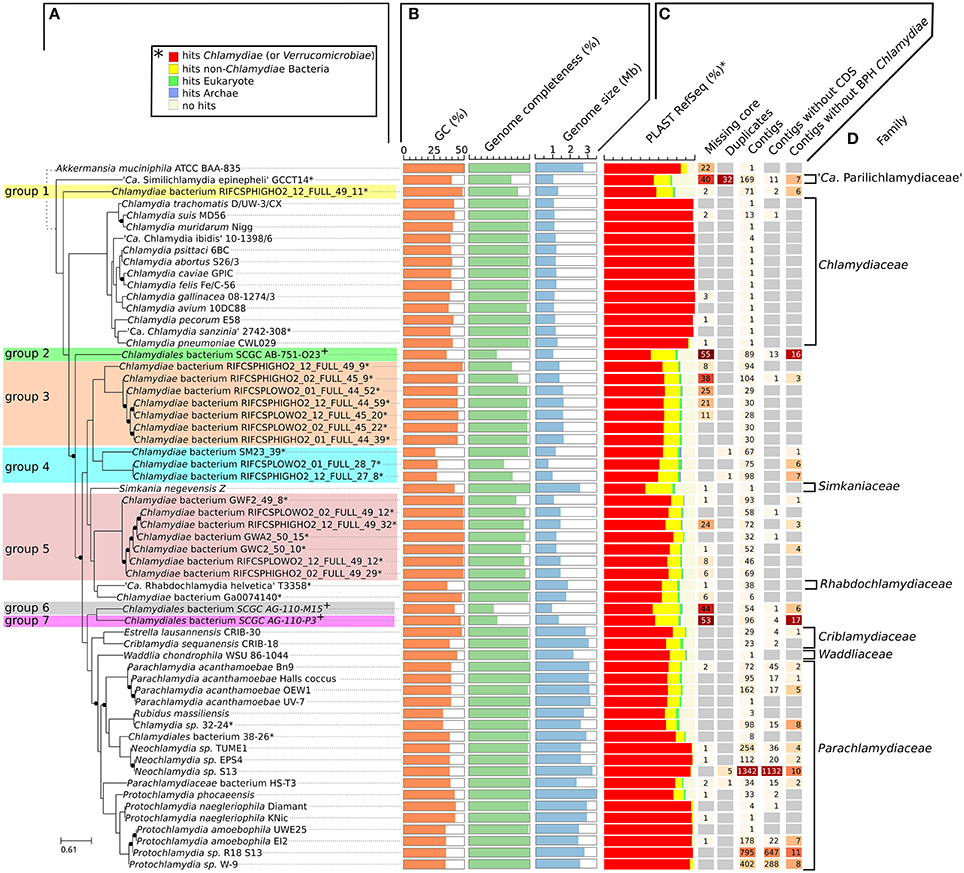
Figure 1. Diversity of the phylum Chlamydiae. (A) Phylogenetic tree of cultured and uncultured representative of the phylum Chlamydiae reconstructed based on the concatenated alignment of 108 single copy orthologs conserved in more than 90% of the genomes. Nodes with support values lower than 1 are indicated with black dots. The width of the black dots does not reflect a genetic distance (lines a shifted to accommodate the presence of the dots). *Genome sequenced using a shotgun metagenomics approach. +Single-cell amplified genomes (B) GC content, genome size and estimated genome completeness (C) quality measures: (i) number of missing core proteins (out of 108 proteins) (ii) number of duplicated core genes (iii) number of contigs in the assembly (iv) number of contigs without any CDS (v) number of contigs without any best PLAST hits against chlamydial sequences (RefSeq database version 81). (D) Described families of the Chlamydiae phylum.
The GC content of uncultured strains varies widely from 26.2 to 49.3% (Table 1, Figure 1B). Twelve of the metagenomics bins are predicted to be more than ninety percent complete, based on the identification of 104 nearly universal bacterial proteins (Table 1, Table S4, Figure 1B). Those nearly complete assemblies range from 1.42 to 2.83 Mb in size. The largest metagenomics bin (Chlamydia sp. 32–24), is phylogenetically related to Neochlamydia spp. The smallest one, Chlamydiae bacterium RIFCSPLOWO2_01_FULL_28_7, only comprises about 700 kb that were predicted to represent approximately 58% of the genome. The most incomplete genomes are the three genomes from marine Chlamydiae (Figure 1, groups 2, 6, and 7). Those genomes are amplified from a single cell. Such approach frequently yield incomplete genomes (Collingro et al., 2017). Chlamydiales bacterium SCGC AG-110-M15 missed 55 of the 108 core proteins used to build the species phylogeny (50.9% of the dataset, Figure 1C).
Despite the metagenomics approach to sequence Chlamydiae from more complex samples, only few genomes exhibited signs of contamination. The strongest evidence was present in the S. epinepheli genome where 32 of the 104 nearly universal proteins were present in more than one copy, indicating that it might be an admixture of two genomes (Figure 1C). Nevertheless, the 32 duplicated S. epinepheli markers were all monophyletic in phylogenetic reconstructions including all chlamydial homologs and their closest homologs in the RefSeq database, indicating that the assembly might be an admixture of two closely related strains. In order to further evaluate if the assemblies contained only fragments of Chlamydiae genomes, all CDS were compared to the RefSeq database. A majority of CDSs of each analyzed genome exhibited a best non-identity PLAST hit (excluding hits against RefSeq sequences classified as belonging to the same species) against chlamydial sequences present in the RefSeq database (Figure 1C). Assemblies reconstructed from metagenomics and single cell data exhibit between 55.77 and 75.93% of best PLAST hits against chlamydial sequences, which is higher than for S. negevensis (45.01%). S. negevensis is the only representative of the family Simkaniaceae currently available in RefSeq. The higher proportion of PLAST hits against non-chlamydial sequences (32.48%) and without any hits (22.51%) as compared to representative of other new families absent from RefSeq might be related to the much larger size of S. negevensis genome (Figure 1B). Several of the most fragmented assemblies harbored contigs without any best hits against chlamydial sequences (Figure 1C). Nevertheless, chlamydial hits were systematically distributed along the whole assembly, with only few small contigs exhibiting no best hits against chlamydial sequences, as can be seen on Figure 2 for the assembly RIFCSPHIGHO2_01_FULL_44_39. Overall, these results indicate that all assemblies contain mostly chlamydial DNA sequences, supporting the reliability of the analysis.
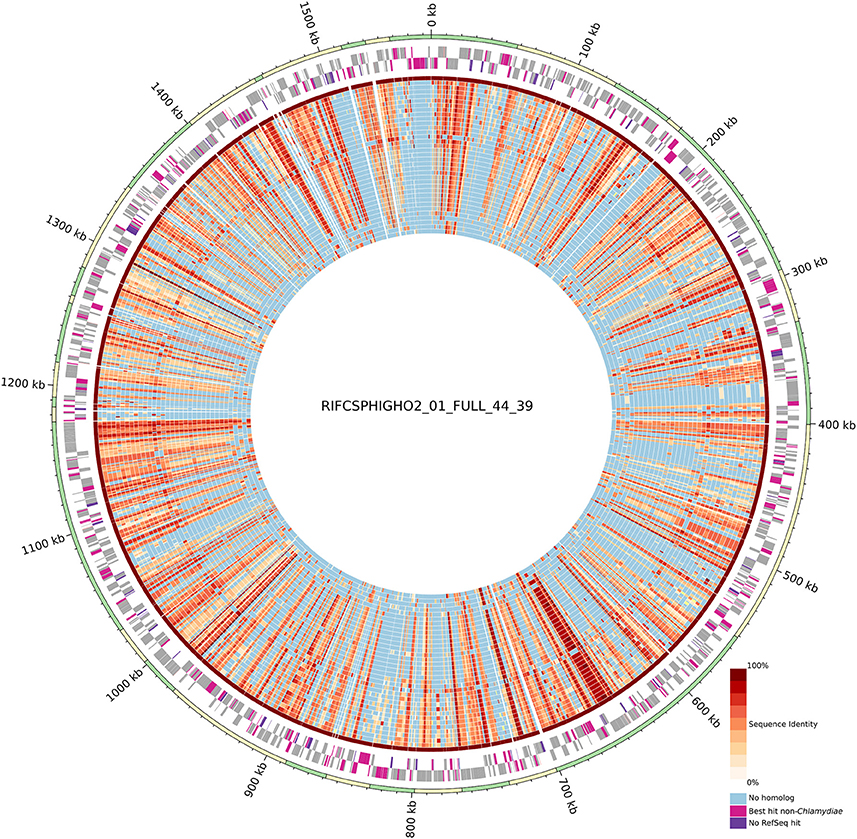
Figure 2. Circular representation of the uncultured genome RIFCSPHIGHO2_01_FULL_44_39 (accession: MGLR01000000). The outer circle indicates contig boundaries. The predicted open reading frames (ORFs) of the leading and lagging strands are reported in gray. Pink ORFs are proteins without a best hit against chlamydial sequences in the RefSeq database. Violet ORFs are proteins without any significant hit in the RefSeq database. The inner blue/red circles show the conservation of the closest identified orthologous protein (red scale) in the 37 other chlamydial species and one other genome of group 3 (Figure 1). Identity values were calculated based on the alignment of orthologous groups inferred using OrthoFinder. The absence of any ortholog is indicated in blue. Most contigs exhibit a majority of best PLAST hits against chlamydial sequences.
Classification and Diversity of New Chlamydial Genomes
We recently proposed a scheme for the classification of chlamydial genomes at various taxonomic levels (species, genus and family). This scheme was used here to update the classification of all unclassified chlamydial genomes (Figure 3). Briefly, the strain HS-T3 likely belongs to a new genus in the family Parachlamydiaceae (Figure S2). Chlamydia sp. 38–26 is a new Neochlamydia species (Figure S3) and Chlamydia sp. 32–24 is a Rubidus massiliensis strain (Figure S4). There are four Pr. amoebophila strains (Figure S5) and three strains of the same Neochlamydia species (S13, TUME1 and EPS4, Figure S6).
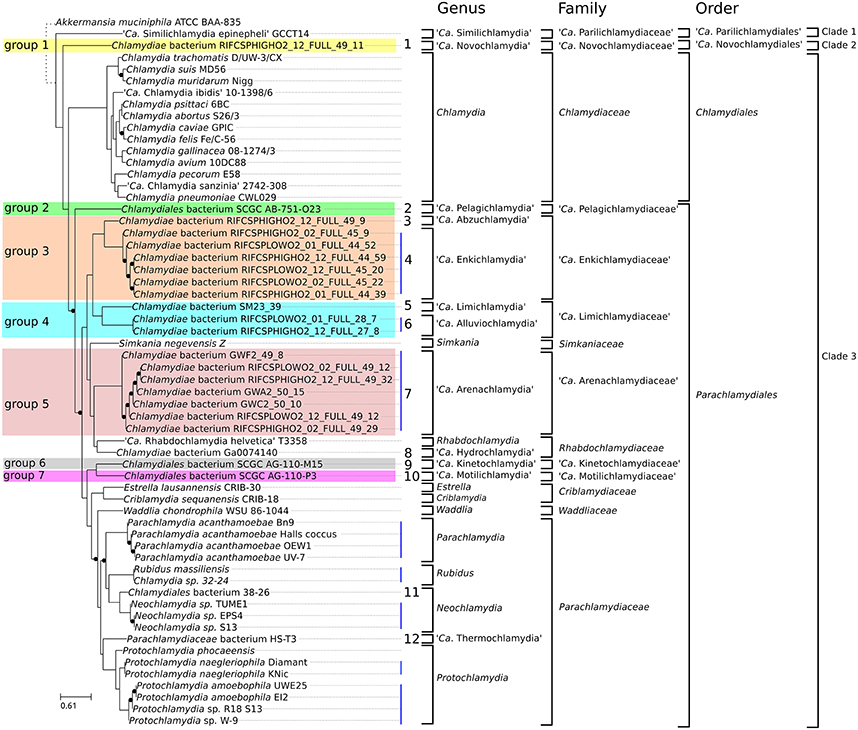
Figure 3. Updated classification of the order Chlamydiales based on genomic data. Proposed new Candidatus genera, families and orders are indicated in bold. Only three genera (Chlamydia, Neochlamydia and Protochlamydia) have more than one species. Vertical blue lines indicate genomes of the same species. Numbers from 1 to 12 indicate new candidate species. Candidate species were not named. Eleven new Candidatus genera are proposed. Novochlamydia (from Latin novus, new), Pelagichlamydia (from the Latin world pelagus, sea), Abzuchlamydia (from Abzu, the underground waters of the Aquifer in the Sumerian mythology), Enkichlamydia (from the Sumerian god Enki, lord of the Abzu), Limichlamydia (from the Latin world limus, silt, alluvium), Alluviochlamydia (from the Latin world alluvio, alluvium), Arenachlamydia (from the Latin world Arena, sand), Hydrochlamydia (from the Greek prefix hydro-, relating to water), Kinetochlamydia (from the Greek prefix kineto-, motion; movement), Motilichlamydia (from Latin world motus, past participle of movere, to move), Thermochlamydia (from the Greek world thermos, warm, hot).
Regarding the new deeply branching assemblies, Group 3 and Group 4 encompass each two candidate species from two different genera (Figures S7, S8). The seven assemblies of group 5 are part of the same candidate species (Figure S9). Altogether, the new genomes can be classified into 12 new candidate species (Figure 3). Half of them are only represented by <90% complete genomic data (Figure 1B, Table 1).
Most genome assemblies sequenced using a metagenomics approach lack rRNA operons: 15 lack 16S rRNA, whereas 17 lack 23S rRNA (Figure S2). This happens frequently as rRNA operons are too highly conserved to be assigned reliably to specific genome assemblies. In addition, most genomes lack one or several of the nine proposed taxonomic markers (Figures S2–S9; Pillonel et al., 2015). This is a major limitation for any scheme relying on a limited set of genes. Therefore, in order to evaluate the genus and family level diversity of the Chlamydiae phylum, the identity of reciprocal best blast hits (RBBH) was used to evaluate the divergence of chlamydial lineages at various taxonomic depths.
Figure 4 shows the distribution of RBBH identity values between pairs of genomes. As expected, the distribution of sequence identity values shifts continuously toward lower values with increased phylogenetic distance (Figure 4A). W. chondrophila and the different genera classified in the Parachlamydiaceae family show similar levels of divergence (Figure 4B). This is not the case with S. negevensis, exhibiting a median identity lower than 50% with P. amoebophila (Figure 4B). The median of the median pairwise identities among members of the Chlamydiales and Parachlamydiales (Figure 3) is of 44.44%, whereas Chlamydiaceae and the two deep branching clades 1 and 2 exhibit a median identity of respectively, 39.81 and 41.14% (using one representative genome per species, see detailed values in Figure S10). The high divergence of clade 1 and 2 is even more obvious on Figure 4A, with a clear shift of the distribution of the RBBH identities peaking at about 35% identity between C. trachomatis and clade 1 and 2. Altogether, the phylum exhibits at least three highly divergent clades, and may now include seven new candidate family-level lineages and 11 new genera (Figure 3, Figure S11).
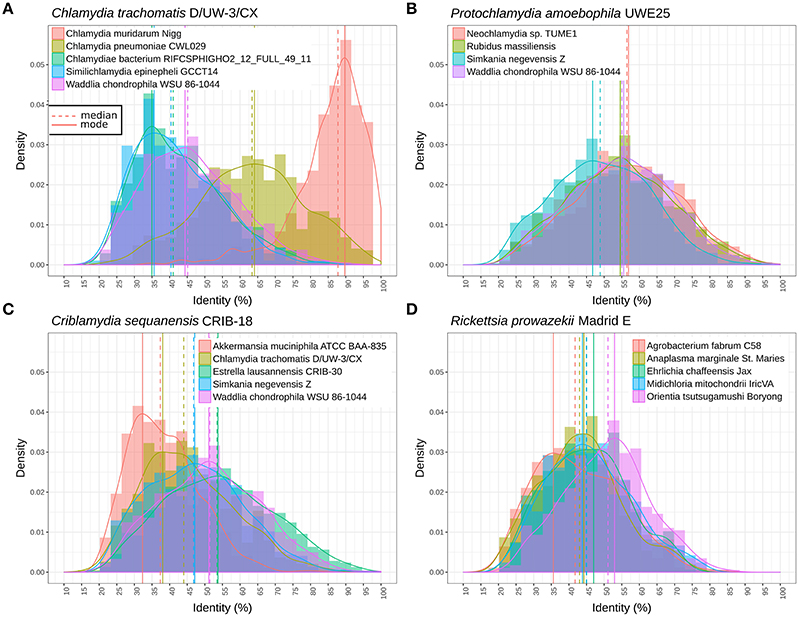
Figure 4. Distribution of the identity of reciprocal best blast hit between pairs of genomes. (A) Protein sequences exhibit similar levels of divergence between different Parachlamydiaceae genera and Waddlia chondrophila (different family), with a median amino acid identity around 55%. The distribution is clearly shifted for Simkania negevensis, with a median identity inferior to 50%. (B) Distribution of protein identities between C. trachomatis and the closely related C. muridarum genome, C. pneumoniae, W. chondrophila, and two deep branching taxa (S. epinepheli and one groundwater metagenomic bin). The continuous decline in sequence identity with divergence time is clearly visible here. The most distant chlamydial lineages are extremely divergent and skewed, with a lot of lowly conserved proteins and some highly conserved ones (mode of 35% sequence identity). (C) Distribution of sequence identities of C. sequenanesis and representatives of four recognized families and the outgroup, A. muciniphila. (D) Identical analyses comparing the obligate intracellular bacteria Rickettsia prowazekii (alpha-proteobacteria) with Orientia tsutsugamushi (Rickettsieae), Anaplasma marginale (Anaplasmataceae), Ehrlichia chafeensis (Anaplasmataceae), Midichloria mitochondrii (“Candidatus Midichloriaceae”), and Agrobacterium fabrum (free-living member of the Rhizobiales order).
Correlation between 16S rRNA Sequence Conservation and Whole Genome Relatedness
Gupta and colleagues recently disputed the 16S rRNA identity cutoffs proposed to delineate families of the phylum Chlamydiae, arguing that the number of chlamydial families was inflated by the high cutoff in use (Gupta et al., 2015). Nevertheless, different bacterial clades may exhibit variable rates of sequences evolution (Kuo and Ochman, 2009). In addition, the 16S rRNA gene may not evolve at a constant rate across the entire tree of life and may not necessarily be a reliable indicator of whole genome relatedness (Konstantinidis and Tiedje, 2005; Kuo and Ochman, 2009). In order to put the Chlamydiae phylum into a broader perspective, we compared the 16S rRNA pairwise sequence identity and the conservation of protein sequences with data from another diverse group of intracellular bacteria, the order Rickettsiales. Representatives of the Rhizobiales order were also included for comparison (Table S5).
Figure 5A shows the relationships of 16S rRNA and median RBBH identities for the three considered orders. For both measures, the Chlamydiales and Rickettsiales orders exhibit higher sequence divergence than the Rhizobiales order. The sequence divergence of different families belonging to the same order (see Tables S5, S6 for the detailed classification used here) reveals that the 16S rRNA gene of Rickettsiales families is more divergent than in the Chlamydiales families (Figure 5B). On the other hand, the median pairwise protein identity is also relatively low for most Chlamydiales (Figures S12A–D). Given the skewed shape of RBBH identity distributions (Figure 4), the median identity may not be the most appropriate summary metrics to estimate genome divergence. An alternative would be the maximum of the density distribution of RBBH identity rather than the median (Figures 5C,D). Using the maximum of the estimated distribution of amino acid identities, the Chlamydiae cloud shifted toward lower values, indicating that the distribution of RBBH identities in pairs of chlamydial genomes is more skewed than for Rickettsiales and Rhizobiales. This still holds true when comparing Parachlamydiales only (excluding Chlamydiaceae and Similichlamydiaceae).
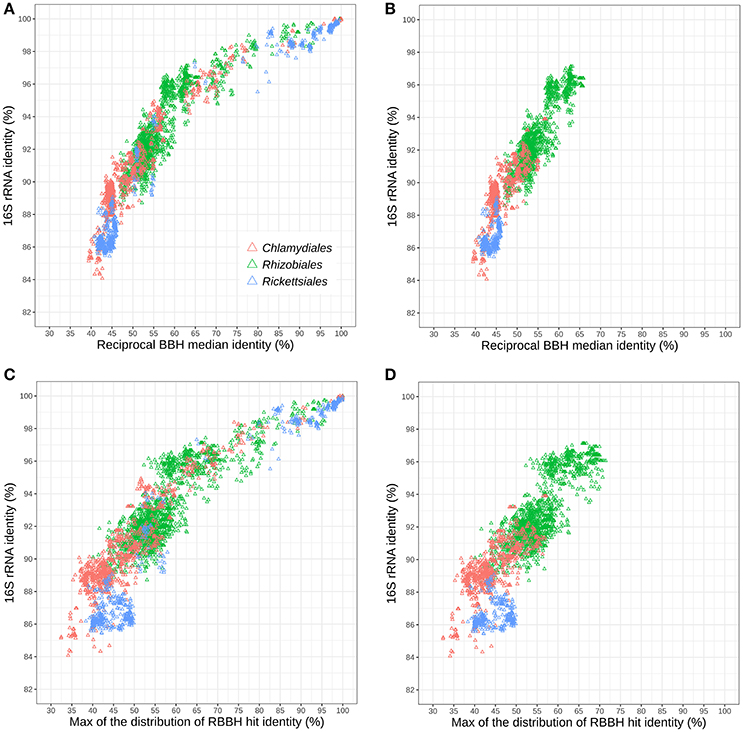
Figure 5. Relationships between 16S rRNA and protein sequences conservation within three distinct bacterial orders: Rhizobiales, Chlamydiales, and Rickettsiales (indicated with colors). (A) Relationships between 16S rRNA identity and the median protein identity within the three bacterial orders and (B) for different families of the same order. (C) Relationships between 16S rRNA and the modal identity within the three bacterial orders and (D) between different families of the same order.
The difference between Rickettsiales and Chlamydiales could be due to the fact that the Chlamydiales dataset includes incomplete genomes. Missing data might lead to spurious reciprocal best blast hits, leading to an undervaluation of whole genome relatedness. Nevertheless, the trend is still visible when only complete chlamydial genomes are considered (Figures S12E,F). In conclusion, chlamydial families exhibit higher conservation of 16S rRNA but a skewed distribution of whole genome RBBH identity toward values that are as low, or even lower than genomes exhibiting lower 16S rRNA identities.
Conservation of Mechanisms Involved in the Interaction with Eukaryotic Hosts and Other Microbial Cells
Several genomes included in this analysis are only a partial representation of the true chlamydial genome, which precludes any in-depth genome content comparisons. Nevertheless, the identification of even partial molecular machineries and metabolic pathways can provide first insights into the ancestrality and conservation of genomic features that may be essential to their unique lifestyle. The recent sequencing of S. epinepheli, the most diverging Chlamydiae described thus far, revealed the presence of a type III secretion system (T3SS) and the conservation of several key virulence factors (Taylor-Brown et al., 2017). T3SS components were identified in all new chlamydial clades (Figure 6, column T3SS), including the most incomplete ones. Several described T3SS effectors such as Mip and Nue are also widely distributed within the whole phylum (Figure S13). The entire clade that includes the Simkaniaceae, the Rhabdochlamydiaceae and several new lineages lack any homolog of Type II secretion system effector protease CPAF (Figure S13). In C. trachomatis, CPAF mutants display impaired generation of infectious elementary bodies (Snavely et al., 2014) and cpaf is therefore an essential gene. Our observations suggest that CPAF is dispensable in some chlamydial lineages, and its role in other Chlamydia-related organisms where it is conserved remains to be confirmed.

Figure 6. Comparative genomics of the phylum Chlamydiae. Identified homologs of proteins involved in nucleotide uptake (ntt), part of molecular machines (i.e., T3SS, Flagellum) and part of selected metabolic pathway. For KEGG pathways/modules, number indicates non redundant count of KEGG orthologs part of the corresponding pathway/module. Reference locus tag, KEGG and Pfam accessions are indicated in Table S1.
As obligate intracellular bacteria, Chlamydiae acquire essential nutrients from the cytosol of their host, including nucleotides and sugars such as ATP and glucose-6-phosphate. All but one partial genome encode at least two ADP/ATP antiporter homologs. These transporters are involved in nucleotide, ATP or nicotinamide adenine dinucleotide uptake from the host (Tjaden et al., 1999; Greub and Raoult, 2003; Haferkamp et al., 2004; Fisher et al., 2013). Furthermore, most genomes also encode a homolog of the glucose-6-phosphate transporter UhpC.
The genomes of Simkania negevensis and several Parachlamydiaceae strains encode an apparently complete type IV secretion system (T4SS) located either on the chromosome or on a plasmid (Bertelli et al., 2016) that may be involved in conjugative DNA transfer (Greub et al., 2004). Several strains of Parachlamydia acanthamoebae encode remnants of the T4SS in their genome (Greub et al., 2009; Bertelli et al., 2016). No traces of T4SS genes could be found in any other chlamydial lineages (Figure 6). The T4SS of S. negevensis, Protochlamydia naegleriophila KNic and R. massiliensis are located on a plasmid, which may have facilitated its loss or acquisition in the various chlamydial lineages. Assignment of plasmid sequences to specific organisms within the context of a metagenomic dataset is extremely difficult. Chlamydial plasmids may thus be missing in some assemblies, including for the genome of Chlamydia sp. 32–34.
It was recently suggested that some marine Chlamydiae may possess a flagellum regulated by a chemotaxis system (Collingro et al., 2017). The identification of several flagellar and chemotaxis genes in highly divergent chlamydial genomes retrieved by single cell-genomics was interpreted as an evidence that motility was an ancient feature of the Chlamydiae phylum (Collingro et al., 2017). Although most new uncultivated chlamydial clades were also sequenced from aquatic samples, none exhibited homologs of the flagellar genes identified in the genome assemblies SCGC AG-110-P3, AB-751-O23 and AG-110-M15 (Figure 6, group 2, 6, and 7), questioning whether this flagellar system is as old as Collingro et al. hypothesized. Two proteins of the type III secretion system that are homologous to flagellar proteins, fliQ/sctS (K02420) and fliN/sctQ (K02417), were sometimes annotated as part of the flagellum (Figure 6) but did not branch with SCGC Chlamydiae homologs in phylogenetic reconstructions (see for example Figure S14). A chemotaxis system is also encoded in several chlamydial genomes lacking flagellar genes including several Parachlamydiaceae (P. phocaeensis, P. naegleriophila, and P. acanthamoebae) and the two Criblamydiaceae genomes currently available. This system was thus proposed to be involved in signal transduction (Collingro et al., 2011). In the present analysis, chemotaxis genes were identified in the genome of Chlamydia sp. 38–26, which is closely related to Neochlamydia sp. strains that do not harbor a chemotaxis system. Overall, our data suggests that all Chlamydiae exhibit a similar obligate intracellular lifestyle, and that most are non-motile.
Conservation of Division Mechanisms across the Phylum Chlamydiae
All described Chlamydiae exhibit a similar biphasic developmental cycle, including the most deeply branching clades (Abdelrahman and Belland, 2005; Steigen et al., 2013; Seth-Smith et al., 2017). The transcription factor Euo targets more than 100 promoters in W. chondrophila, and may be a key player in cell cycle regulation (Domman and Horn, 2015; de Barsy et al., 2016). It is conserved throughout the whole phylum, including S. epinepheli and “Ca. Novochlamydiaceae” (Figure 6), with exception of three of the most incomplete genomes probably due to the partial data available. Interestingly, the genome assembly of S. epinepheli lacks all proteins involved in peptidoglycan biosynthesis (Figure 6) and all but three proteins involved in chlamydial division (Figure S13). Remnants of those proteins could not be found in the raw assembly using tBLASTn. Although the genome of this strain is partially covered, peptidoglycan synthesis genes are located in various regions along the chromosome, which reinforces the likely absence of these genes from S. epinepheli. Indeed, this bacterium divides through a budding process (Seth-Smith et al., 2017), which suggests that the family Parilichlamydiaceae may present a significantly different division mechanism than other chlamydial lineages.
High Metabolic Diversity of the Chlamydiae Phylum
Menaquinones are electron shuttles that allow the transfer of electrons between membrane-bound protein complexes in the electron transport chain (Meganathan, 2001). The Chlamydiaceae synthesize menaquinone through the alternative futalosine pathway (Hiratsuka et al., 2008; Barta et al., 2014). Other clades, including the deep branching “Ca. Novochlamydiaceae” (Figure 6), encode the traditional pathway, supporting the hypothesis that the futalosine pathway was horizontally acquired by the Chlamydiaceae lineage (Barta et al., 2014; Subtil et al., 2014). No menaquinone biosynthesis enzymes could be identified in the S. epinepheli genome (Figure 6). A homolog of MenB was identified (Figure 6) but it exhibits more similarity with enoyl-CoA hydratase and did not cluster with other chlamydial sequences in phylogenetic reconstructions, thus likely having another enzymatic role than MenB (Figure S15). Most genomes encode the necessary components of a minimal respiratory chain except the seven genomes of “Ca. Arenachlamydiaceae” (Figure 6). Those 1.4 Mb genomes are predicted to be nearly complete but do not encode the menaquinone biosynthesis pathway, nor any component of the respiratory chain, suggesting that they use alternative respiration systems.
Overall, enzymes of the respiratory chain exhibit variable patterns of parallel losses across the entire phylum. The NADH-quinone oxidoreductase (complex I) is for instance restricted to the Parachlamydiaceae-Waddliaceae-Criblamydiaceae clade, and was lost at least three times independently in subclades (Figure 6). Few proteins homologous to complex I were identified in the low GC genomes recovered from estuary sediments and groundwater (“Ca. Limichlamydiaceae”). They encode a putative NADP-reducing hydrogenase exhibiting 44.5–56% amino acid identity to the hnd operon of Desulfovibrio fructosivorans (Malki et al., 1995; Figure S16). The F-type ATPase is also restricted to Parachlamydiaceae-Waddliaceae-Criblamydiaceae clade, as opposed to the V-type ATPase that is conserved in all sequenced chlamydial genomes.
The citrate cycle (TCA) is incomplete in all Chlamydiaceae due to the absence of three enzymes; citrate synthase (GltA), aconitase (Acn) and isocitrate dehydrogenase (Icd) (Omsland et al., 2014). Several Chlamydiae such as the three Neochlamydia sp. and the seven “Ca. Arenachlamydiaceae” genomes (repeatedly missing the same set of enzymes) exhibit an even less complete TCA (Figure 6). Indeed, no homologs of TCA enzymes could be identified in the S. epinepheli genome assembly. On the other hand, several homologs of the pentose phosphate pathway (PPP), glycolysis and glycogen metabolism were identified in all assemblies that are more than 90% complete (Figure 6).
The various Parachlamydiaceae species exhibit significant differences in their ability to synthetize amino acids, with reduced biosynthetic abilities in Protochlamydia amoebophila strains and Neochlamydia strains and extended metabolic abilities for Parachlamydia strains (Figure S13). The 3 closely related Neochlamydia genomes exhibit genomes larger than 2.5 Mbp, but overall highly reduced metabolic capacities (Figures S17, S18). On the other hand, they encode large numbers of mobile genetic elements and repeat proteins (Figure S17). S. epinepheli has the most reduced metabolic capacities, but its genome is only about 70% complete. While the metabolism of amino acids, nucleotides, cofactors and vitamins was not investigated in detail, we generally observed that all genomes had limited predicted biosynthetic capabilities for all those compounds and that marked differences exist within the phylum (Figure S13), as already observed in previous comparative analyses (Bertelli et al., 2010, 2015; Collingro et al., 2011; Omsland et al., 2014).
Discussion
Shotgun metagenomics allows the reconstruction of genomes from complex microbial communities. It can yield draft and nearly complete genomes without the need for cultivation (Alneberg et al., 2014), allowing to investigate the biology of uncultivable or difficult-to-culture organisms, including members of the phylum Chlamydiae. The comparative analysis of 21 metagenomic bins from public databases with previously described genomes allowed the identification of seven new candidate family level lineages, 11 new candidate genera and 12 new candidate species (Figure 3). Six of those new species exhibit nearly complete genomes (more than 90 % complete).
A reference phylogeny was reconstructed based on 108 single copy orthologs conserved in more than 90% of the genomes. Given the incomplete nature of the dataset and the high sequence composition bias of several genomes, the reconstructed phylogeny might not precisely reflect the true evolutionary relationships between the most deeply branching lineages. Nevertheless, this phylogeny is congruent with recently published phylogenies based on 16S rRNA and whole genome datasets (Figure 1; Gupta et al., 2015; Pillonel et al., 2015). While no information regarding host or lifestyle is available for the newly identified clades, genomic analyses support the hypothesis that they all share the same obligate intracellular lifestyle. They all encode a type III secretion system, homologs of known effector proteins and transporters involved in nutrient uptake. In addition, members of the Parilichlamydiaceae, the most deeply branching lineage in the current phylogeny, are fish pathogens that share the typical biphasic life cycle of other Chlamydiae (Steigen et al., 2013; Stride et al., 2013; Seth-Smith et al., 2017). Despite these similarities, some central mechanisms such as cell division might significantly differ in distantly-related clades such as S. epinepheli. No homologs of most proteins involved in peptidoglycan biosynthesis and cell division could be identified in the S. epinepheli assembly. This partial genome also shows particularly limited capacities for the synthesis of amino acids, effectors, vitamins and nucleotides. Pairwise protein sequences comparisons revealed that S. epinepheli is highly divergent as compared to previously sequenced members of the phylum. This high sequence divergence might impair the correct identification of homologs based on amino acid sequence comparisons. The discovery and sequencing of novel intermediate species should facilitate the identification of groups of orthologs among such distantly-related organisms. It should clarify the origin of these highly different biosynthetic abilities and further differences in core chlamydial mechanisms.
Several genomes predicted to be nearly complete showed little evidences of autonomous energy production capabilities. Those Chlamydiae might completely depend on their host for energy generation. Protein complexes of the respiratory chain are expressed at the end of the replication cycle (König et al., 2017). They might be essential at the extracellular stage. Chlamydial lineages which have lost the complete respiratory chain might not present the classical biphasic life-cycle of most Chlamydiae. Elementary bodies (EB) and cell lysis were never described for any of the three Acanthamoeba endosymbionts of the Neochlamydia genus (Ishida et al., 2014). The transition from a biphasic life-style to vertical transmission is expected to lead to major changes at the genomic level. Those three strains exhibit genomes more than twice larger than Chlamydiaceae spp., but highly reduced functional repertoires as compared to other Parachlamydiaceae (Figures S17, S18). They also harbor a high number of mobile genetic elements and repetitive elements (Figure S17; Domman et al., 2014). Similar genome features were observed in recently established insect endosymbionts such as Serratia symbiotica and Sodalis glossinidius (Toh et al., 2006; Lamelas et al., 2011). Gene losses strongly affect carbohydrate biosynthetic pathways and the production of energy, as illustrated by the loss of multiple components of the respiratory chain (Figure 6). The reduction of the electron transport chain is associated with the near complete loss of the tricarboxylic acid (TCA) cycle in at least two distinct lineages (“Ca. Arenachlamydiaceae” and the three Neochlamydia spp.).
The loss of glycogen metabolism is generally associated with parasitic bacteria (Henrissat et al., 2002). This is not the case for Chlamydiae (Omsland et al., 2014). Indeed, homologs of the enzymes involved in the biosynthesis of glycogen were identified in all new assemblies. One hypothesis to explain the conservation of glycogen biosynthesis in these obligate intracellular organisms is that these enzymes are effectors manipulating the host metabolism (Ball et al., 2013; Ball and Greub, 2015).
Two-fold variations in genome GC content can be observed across the phylum. The genomes of two related species of “Ca. Limichlamydiaceae” exhibit a GC content lower than 30%, similarly to some primary and secondary insects symbionts such as Buchnera and Spiroplasma (Moran et al., 2008; Lo et al., 2016). The increasing genomic coverage of the phylum, including the small low GC genome clades and five different genera of the Parachlamydiaceae, indicates that gene losses occur in parallel in different chlamydial lineages, and that highly reduced genomes are not restricted to the Chlamydiaceae family. Nevertheless, additional complete genome sequences will be necessary to gain further knowledge on the variable patterns of genome evolution across the phylum.
Family-level lineages of the phylum Chlamydiae exhibit a higher conservation of the 16S rRNA gene as compared to members of the Rickettsiales order. Yet, the comparison of complete and nearly complete genomes suggest that chlamydial protein sequences show similar level or increased level of divergence for the most distantly related families. The distribution of RBBH identities seems to be more skewed toward lower values in the phylum Chlamydiae. Endosymbiosis is characterized by an acceleration of the rate of evolution (Itoh et al., 2002; Woolfit and Bromham, 2003; Kuo and Ochman, 2009). The Buchnera synonymous rate of evolution was for instance evaluated to be about twice that of low-codon-bias genes of Escherichia coli and Salmonella typhimurium (Clark et al., 1999). This is particularly due to an accelerated accumulation of mutations by genetic drift due to the small effective population size of those bacteria (Moran, 1996), but also to the loss of DNA repair mechanisms (Moran and Bennett, 2014). Members of the Chlamydia genus lack homologs of several repair proteins such as MutT, MutM, and MutH (Stephens et al., 1998). Several attempts were made to evaluate the mutation rate and the impact of genetic drift on members of the Chlamydiaceae family. They report a rather high dN/dS ratio for C. trachomatis and C. pneumoniae, which is indicative of increased level of genetic drift (Rocha et al., 2006; Kuo et al., 2009; Joseph et al., 2012). Estimates of mutation rates differ by several folds between studies, and were recently estimated to be similar to free-living bacteria for C. trachomatis (Joseph et al., 2012; Hadfield et al., 2017). As several genomes are now available for several Chlamydia-related species, it would be interesting to estimate and compare the dN/dS ratio of other clades.
The 16S rRNA, a gene under high purifying selection, is more conserved than protein sequences in the phylum Chlamydiae. This difference is larger in the phylum Chlamydiae than in other intracellular bacteria such as the Rickettsiales and might indicate that the accelerated rate of evolution of Chlamydiae is at least partly due to an increase in mutation rate rather than genetic drift alone. Drift might also be counter balanced by the strong selection pressure on rRNA (Woolfit and Bromham, 2003). Those parameters might nevertheless vary within the phylum itself. Members of the Parachlamydiales, which infect free-living amoebae, exhibit larger genomes and are expected to be less sensitive to genetic drift owing to their larger effective population size and reduced transmission bottlenecks as compared to vertebrate parasites of the Chlamydia and Similichlamydia genera. The skewed identity distribution is indeed more accentuated when members of the Chlamydiaceae and S. epinepheli are included in the comparison, two clades exhibiting genomes of highly reduced size. Considering the relative high divergence of Chlamydiae genomes as compared to the 16S rRNA sequence, dedicated cutoffs are needed to properly interpret 16S rRNA based surveys of chlamydial diversity. Such cutoffs specific for members of the Chlamydiae phylum have been previously proposed (Pillonel et al., 2015).
Gupta and colleagues recently proposed to split the phylum Chlamydiae into the order Chlamydiales and Parachlamydiales (Figure 3; Gupta et al., 2015). They also recognized the existence of the deep branching “Ca. Parilichlamydiaceae” clade, but included it in the Parachlamydiales order. The current work confirms the high level of divergence of the “Ca. Parilichlamydiaceae,” in addition to a potential new deeply branching lineage (“Ca. Novochlamydiaceae”). In addition, they appear to exhibit fundamental differences in division mechanisms that underline the need to create additional orders in the taxonomical classification for those deep branching lineages. The data presented is this study support the division of the Chlamydiae phylum into four orders: the Parachlamydiales, Chlamydiales, “Candidatus Parilichlamydiales” and “Candidatus Novochlamydiales.” While much work is still needed to fully comprehend the biology and diversity of members of the phylum Chlamydiae, this work provides a first insight into the metabolic and genetic diversity of what could be the most ancient and diverse clade of intracellular parasites of eukaryotes.
Author Contributions
TP designed the study, performed the analyses and wrote the manuscript. TP and CB contributed to the interpretation of the results and the redaction of the manuscript. GG contributed to the design of the study and the redaction of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank David Pasche for the helpful discussions regarding chlamydial taxonomy. The computations were performed at the Vital-IT Center for high-performance computing of the Swiss Institute of Bioinformatics (SIB, Lausanne, http://www.vital-it.ch).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00079/full#supplementary-material
Figure S1. Evolution of the pan and core-genome size of the phylum Chlamydiae. (A) Evolution of the size of the pan and core-genome with the addition of new genomes (based on 10 random sampling of genomes up to the total of genomes available). (B) Barplot showing the number of orthogroups identified in one to the 59 genomes included in the analysis. A large majority of orthologous groups are restricted to one or only few genomes.
Figure S2. Percentage of nucleotide and amino acid identitiy of the nine taxonomic markers as compared to Parachlamydiaceae sp. HS-T3. Sequence conservation suggest that strains with green rows are part of the same family (Parachlamydiaceae). The low conservation of DnaA, SucA, protein 325 and FabI supports classification of Parachlamydiaceae sp. HS T3 as the unique representative of a new Parachlamydiaceae genus.
Figure S3. Percentage of nucleotide and amino acid identitiy of the nine taxonomic markers as compared to Chlamydiales sp. 38 26. The 16S and 23S rRNA sequences were missing from the metagenomic bin, but the relatively high conservation of DnaA, SucA, protein 325 and FabI and low conservation of species-level markers (RpoN, FtsK, PepF, Adk, and HemL) suggest that the Chlamydiales sp. 38 26 is a new species of the genus Neochlamydia.
Figure S4. Percentage of nucleotide and amino acid identity of the nine taxonomic markers as compared to Rubidus massiliensis. Sequence conservation of both genus and species-level taxonomic markers indicate that Chlamydia sp. 32 24 is a new R. massiliensis strain.
Figure S5. Percentage of nucleotide and amino acid identity of the nine taxonomic markers as compared to Protochlamydia amoebophila. Sequence conservation of DnaA, SucA, protein 325 and FabI indicates that Protochlamydia sp. R18 S13 and Protochlamydia sp. W 9 are P. amoebophila strains.
Figure S6. Percentage of nucleotide and amino acid identity of the nine taxonomic markers as compared to Neochlamydia sp. TUME1. Sequence conservation of both genus and species-level taxonomic markers indicate that the three Neochlamydia assemblies are representatives of the same species.
Figure S7. Percentage of nucleotide and amino acid identity of the nine taxonomic markers as compared to Group 3 (“Ca. Enkichlamydiaceae”). The conservation of RpoN, PepF, Adk, and HemL indicates that all six assemblies of group 3 (“Ca. Enkichlamydiaceae”) are part of the same species. The conservation of protein 325 and FabI provide contradictory results regarding the classification of Chlamydiae sp. RIFCSPHIGHO2 12 FULL 49 9 as part of the same genus or not.
Figure S8. Percentage of nucleotide and amino acid identity of the nine taxonomic markers as compared to Chlamydiae bacterium SM23 39. The conservation of DnaA, protein 325 and FabI indicate that group 4 (“Ca. Limichlamydiaceae”) can be subdivided in two genera.
Figure S9. Percentage of nucleotide and amino acid identity of the nine taxonomic markers as compared to Group 5. The high level of conservation of RpoN, FtsK, and PepF indicates that all 7 group 5 (“Ca. Arenachlamydiaceae”) assemblies are part of the same species. The absence of 16S and 23S rRNA sequences precludes family-level classification based on the published scheme.
Figure S10. RBBH identity distribution of newly identified clades. (A) The two clades of marine Chlamydiae exhibit a median pairwise RBBH identity lower than 50%. Of note, those genomes are <50% complete. S. negevensis and W. chondrohila were added for comparison. (B) Comparison of representatives of the two genera of group 3. S. negevensis and Chlamydia sp. SM 23–39 were added for comparison. (C) The divergence of the two putative genus of group 4 is nearly as important as with other families (D) Chlamydia sp. 28–36 most closely related to Neochlamydia sp. (E) Comparison of the density distribution for different species of the Protochlamydia genus and comparison with representatives if two other genus. (F) The Parachlamydia sp. HS-T3 shows high sequence divergence (median RBBH identity of about 55%) with representatives of other Parachlamydiaceae genus.
Figure S11. Median RBBH identity heat map. Median identity of reciprocal best blast hits calculated between one representative of each species and Candidatus species of the Phylum Chlamydiae.
Figure S12. Density median RBBH and modal identity between families and orders, and complete chlamydia genomes comparisons. For this comparison, only a single order for the phylum Chlamydiae was considered. (A) Distribution of the median identity between families within three different orders. (B) Distribution of the median identity between families within three different orders. (C) Distribution of the median identity between the Rhizobiales and Rickettsiales orders (alpha-proteobacteria). (D) Distribution of the modal identity between the Rhizobiales and Rickettsiales orders (alpha-proteobacteria). (E) The relationship between 16S rRNA sequence identity and the median RBBH identity between families within three distinct bacterial orders. Only complete genomes were included. (F) The relationship between 16S rRNA sequence identity and the modal RBBH identity between families within three distinct bacterial orders. Only complete genomes were included.
Figure S13. Identified homologs of effectors/inclusion membrane proteins, Membrane, and division proteins, as well as number of homologs identified in selected biosynthesis pathways. Data based on GhostKOALA annotation and clustering into orthologous groups with Orthofinder. Reference locus tags and KEGG accessions are indicated in Table S1.
Figure S14. Phylogeny of the EscS/YscS/HrcS family type III secretion system export apparatus protein. The phylogeny include the 2 closest RefSeq and SwissProt hits of each chlamydial sequence. Amino acid sequences were aligned with mafft version 7.058b and the phylogeny was reconstructed using Fasttree 2.1.9 with default parameters.
Figure S15. Phylogeny of the orthologous group including menB. The phylogeny includes the 2 best UniProt and 4 best RefSeq hits of each sequence of each protein of the orthologous groups. The menB homolog of Chlamydiae and their homologs in the genomes of S. epinepheli are not monophyletic. Amino acid sequences were aligned with mafft version 7.058b and the phylogeny was reconstructed using Fasttree 2.1.9 with default parameters.
Figure S16. Phylogeny of the orthologous group including NADH-quinone oxidoreductase subunit E. The phylogeny includes the 2 best UniProt and 4 best RefSeq hits of each sequence of the orthologous groups. The NADH-quinone oxidoreductase subunit E orthologs from Chlamydiae and their homologs in low GC chlamydial genomes are not monophyletic. Amino acid sequences were aligned with mafft version 7.058b and the phylogeny was reconstructed using Fasttree 2.1.9 with default parameters.
Figure S17. Classification of protein into COG categories. The red gradient reflect the number of annotated proteins with the corresponding COG category and was normalized for each column (each COG category). The three Neochlamydia spp. encode a relatively large number of CDS (1,879–2,232), but a reduction in most COG categories except for mobile genetic elements (X) and transcription (K) as compared to closely related Parachlamydiaceae. The large increase in the transcription category in the 3 Neochlamydia genomes is due to the presence of a very large number of Leucine-rich repeat (LRR) proteins.
Figure S18. Comparison of KEGG functional categories for the Neochlamydia strains and their closest phylogenetic neigbour. Counts indicates the non-redundant number of Kegg Orthologs (KO) identified for each functional category. Gene losses are particularly important in categories such as ATP synthesis, amino acid metabolism and carbohydrate metabolism.
Table S1. Reference locus, KEGG pathways and modules found in Figure 1, and Figure S13.
Table S2. Complete list of OrthoFinder orthologous groups (one group per row). CDS are identified with Genbank or RefSeq locus tags.
Table S3. List of core orthologous groups conserved in 90% of the genomes (55 out of 60 genomes). The RefSeq/Genbank locus tags of the CDS of each orthologous groups can be found in Table S2.
Table S4. Detailed checkM results.
Table S5. List and detailed taxonomy of Rickettsiales and Rizhobiales genomes used in this study.
Table S6. Detailed Chlamydiales classification used in this study.
References
Abdelrahman, Y. M., and Belland, R. J. (2005). The chlamydial developmental cycle. FEMS Microbiol. Rev. 29, 949–959. doi: 10.1016/j.femsre.2005.03.002
Alneberg, J., Bjarnason, B. S., de Bruijn, I., Schirmer, M., Quick, J., Ijaz, U. Z., et al. (2014). Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146. doi: 10.1038/nmeth.3103
Anantharaman, K., Brown, C. T., Hug, L. A., Sharon, I., Castelle, C. J., Probst, A. J., et al. (2016). Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat. Commun. 7:13219. doi: 10.1038/ncomms13219
Andersson, P., Klein, M., Lilliebridge, R. A., and Giffard, P. M. (2013). Sequences of multiple bacterial genomes and a Chlamydia trachomatis genotype from direct sequencing of DNA derived from a vaginal swab diagnostic specimen. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 19, E405–E408. doi: 10.1111/1469-0691.12237
Baker, B. J., Lazar, C. S., Teske, A. P., and Dick, G. J. (2015). Genomic resolution of linkages in carbon, nitrogen, and sulfur cycling among widespread estuary sediment bacteria. Microbiome 3:1. doi: 10.1186/s40168-015-0077-6
Ball, S. G., and Greub, G. (2015). Blurred pictures from the crime scene: the growing case for a function of Chlamydiales in plastid endosymbiosis. Microbes Infect. 17, 723–726. doi: 10.1016/j.micinf.2015.09.007
Ball, S. G., Subtil, A., Bhattacharya, D., Moustafa, A., Weber, A. P. M., Gehre, L., et al. (2013). Metabolic effectors secreted by bacterial pathogens: essential facilitators of plastid endosymbiosis? Plant Cell 25, 7–21. doi: 10.1105/tpc.112.101329
Barta, M. L., Thomas, K., Yuan, H., Lovell, S., Battaile, K. P., Schramm, V. L., et al. (2014). Structural and biochemical characterization of Chlamydia trachomatis hypothetical protein CT263 supports that menaquinone synthesis occurs through the futalosine pathway. J. Biol. Chem. 289, 32214–32229. doi: 10.1074/jbc.M114.594325
Bertelli, C., Aeby, S., Chassot, B., Clulow, J., Hilfiker, O., Rappo, S., et al. (2015). Sequencing and characterizing the genome of Estrella lausannensis as an undergraduate project: training students and biological insights. Front. Microbiol. 6:101. doi: 10.3389/fmicb.2015.00101
Bertelli, C., Cissé, O. H., Rusconi, B., Kebbi-Beghdadi, C., Croxatto, A., Goesmann, A., et al. (2016). CRISPR system acquisition and evolution of an obligate intracellular chlamydia-related bacterium. Genome Biol. Evol. 8, 2376–2386. doi: 10.1093/gbe/evw138
Bertelli, C., Collyn, F., Croxatto, A., Rückert, C., Polkinghorne, A., Kebbi-Beghdadi, C., et al. (2010). The waddlia genome: a window into chlamydial biology. PLoS ONE 5:e10890. doi: 10.1371/journal.pone.0010890
Bertelli, C., Goesmann, A., and Greub, G. (2014). Criblamydia sequanensis harbors a megaplasmid encoding arsenite resistance. Genome Announc. 2:e00949-14. doi: 10.1128/genomeA.00949-14
Bou Khalil, J. Y., Benamar, S., Baudoin, J.-P., Croce, O., Blanc-Tailleur, C., Pagnier, I., et al. (2016). Developmental cycle and genome analysis of “Rubidus massiliensis,” a new vermamoeba vermiformis pathogen. Front. Cell. Infect. Microbiol. 6:31. doi: 10.3389/fcimb.2016.00031
Bou Khalil, J. Y., Benamar, S., Di Pinto, F., Blanc-Tailleur, C., Raoult, D., and La Scola, B. (2017). Protochlamydia phocaeensis sp. nov., a new Chlamydiales species with host dependent replication cycle. Microbes Infect. 19, 343–350. doi: 10.1016/j.micinf.2017.02.003
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Clark, M. A., Moran, N. A., and Baumann, P. (1999). Sequence evolution in bacterial endosymbionts having extreme base compositions. Mol. Biol. Evol. 16, 1586–1598. doi: 10.1093/oxfordjournals.molbev.a026071
Collingro, A., Köstlbacher, S., Mussmann, M., Stepanauskas, R., Hallam, S. J., and Horn, M. (2017). Unexpected genomic features in widespread intracellular bacteria: evidence for motility of marine chlamydiae. ISME J. 11, 2334-2344. doi: 10.1038/ismej.2017.95
Collingro, A., Tischler, P., Weinmaier, T., Penz, T., Heinz, E., Brunham, R. C., et al. (2011). Unity in variety—the pan-genome of the chlamydiae. Mol. Biol. Evol. 28, 3253–3270. doi: 10.1093/molbev/msr161
de Barsy, M., Frandi, A., Panis, G., Théraulaz, L., Pillonel, T., Greub, G., et al. (2016). Regulatory (pan-)genome of an obligate intracellular pathogen in the PVC superphylum. ISME J. 10, 2129–2144. doi: 10.1038/ismej.2016.23
Domman, D., Collingro, A., Lagkouvardos, I., Gehre, L., Weinmaier, T., Rattei, T., et al. (2014). Massive expansion of ubiquitination-related gene families within the Chlamydiae. Mol. Biol. Evol. 31, 2890–2904. doi: 10.1093/molbev/msu227
Domman, D., and Horn, M. (2015). Following the footsteps of chlamydial gene regulation. Mol. Biol. Evol. 32, 3035–3046. doi: 10.1093/molbev/msv193
Eddy, S. R. (2011). Accelerated Profile HMM Searches. PLoS Comput. Biol. 7:e1002195. doi: 10.1371/journal.pcbi.1002195
Elwell, C., Mirrashidi, K., and Engel, J. (2016). Chlamydia cell biology and pathogenesis. Nat. Rev. Microbiol. 14, 385–400. doi: 10.1038/nrmicro.2016.30
Emms, D. M., and Kelly, S. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16:157. doi: 10.1186/s13059-015-0721-2
Federhen, S. (2012). The NCBI Taxonomy database. Nucleic Acids Res. 40, D136–D143. doi: 10.1093/nar/gkr1178
Fisher, D. J., Fernández, R. E., and Maurelli, A. T. (2013). Chlamydia trachomatis transports NAD via the Npt1 ATP/ADP translocase. J. Bacteriol. 195, 3381–3386. doi: 10.1128/JB.00433-13
Fukumoto, T., Matsuo, J., Okubo, T., Nakamura, S., Miyamoto, K., Oka, K., et al. (2016). Acanthamoeba containing endosymbiotic chlamydia isolated from hospital environments and its potential role in inflammatory exacerbation. BMC Microbiol. 16:292. doi: 10.1186/s12866-016-0906-1
Galperin, M. Y., Makarova, K. S., Wolf, Y. I., and Koonin, E. V. (2015). Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 43, D261–D269. doi: 10.1093/nar/gku1223
Gawad, C., Koh, W., and Quake, S. R. (2016). Single-cell genome sequencing: current state of the science. Nat. Rev. Genet. 17, 175–188. doi: 10.1038/nrg.2015.16
Greub, G. (2017). International Committee on Systematics of Prokaryotes Subcommittee on the taxonomy of Chlamydiae. Minutes of the closed meeting, 31 March 2015, New Orleans, U. S. A. Int. J. Syst. Evol. Microbiol. 67, 512–513. doi: 10.1099/ijsem.0.001528
Greub, G., Collyn, F., Guy, L., and Roten, C.-A. (2004). A genomic island present along the bacterial chromosome of the Parachlamydiaceae UWE25, an obligate amoebal endosymbiont, encodes a potentially functional F-like conjugative DNA transfer system. BMC Microbiol. 4:48. doi: 10.1186/1471-2180-4-48
Greub, G., Kebbi-Beghdadi, C., Bertelli, C., Collyn, F., Riederer, B. M., Yersin, C., et al. (2009). High throughput sequencing and proteomics to identify immunogenic proteins of a new pathogen: the dirty genome approach. PLoS ONE 4:e8423. doi: 10.1371/journal.pone.0008423
Greub, G., and Raoult, D. (2003). History of the ADP/ATP-translocase-encoding gene, a parasitism gene transferred from a Chlamydiales ancestor to plants 1 billion years ago. Appl. Environ. Microbiol. 69, 5530–5535. doi: 10.1128/AEM.69.9.5530-5535.2003
Gupta, R. S., Naushad, S., Chokshi, C., Griffiths, E., and Adeolu, M. (2015). A phylogenomic and molecular markers based analysis of the phylum Chlamydiae: proposal to divide the class Chlamydiia into two orders, Chlamydiales and Parachlamydiales ord. nov., and emended description of the class Chlamydiia. Antonie Van Leeuwenhoek 108, 765–781. doi: 10.1007/s10482-015-0532-1
Hadfield, J., Harris, S. R., Seth-Smith, H. M. B., Parmar, S., Andersson, P., Giffard, P. M., et al. (2017). Comprehensive global genome dynamics of Chlamydia trachomatis show ancient diversification followed by contemporary mixing and recent lineage expansion. Genome Res. 27, 1220-1229. doi: 10.1101/gr.212647.116
Haferkamp, I., Schmitz-Esser, S., Linka, N., Urbany, C., Collingro, A., Wagner, M., et al. (2004). A candidate NAD+ transporter in an intracellular bacterial symbiont related to Chlamydiae. Nature 432, 622–625. doi: 10.1038/nature03131
Henrissat, B., Deleury, E., and Coutinho, P. M. (2002). Glycogen metabolism loss: a common marker of parasitic behaviour in bacteria? Trends Genet. TIG 18, 437–440. doi: 10.1016/S0168-9525(02)02734-8
Hiratsuka, T., Furihata, K., Ishikawa, J., Yamashita, H., Itoh, N., Seto, H., et al. (2008). An alternative menaquinone biosynthetic pathway operating in microorganisms. Science 321, 1670–1673. doi: 10.1126/science.1160446
Horn, M. (2015). “Chlamydiae,” in Bergey's Manual of Systematics of Archaea and Bacteria (John Wiley & Sons, Inc.). doi: 10.1002/9781118960608.pbm00005
Ishida, K., Sekizuka, T., Hayashida, K., Matsuo, J., Takeuchi, F., Kuroda, M., et al. (2014). Amoebal endosymbiont neochlamydia genome sequence illuminates the bacterial role in the defense of the host amoebae against Legionella pneumophila. PLoS ONE 9:e95166. doi: 10.1371/journal.pone.0095166
Itoh, T., Martin, W., and Nei, M. (2002). Acceleration of genomic evolution caused by enhanced mutation rate in endocellular symbionts. Proc. Natl. Acad. Sci. U.S.A. 99, 12944–12948. doi: 10.1073/pnas.192449699
Jacquier, N., Aeby, S., Lienard, J., and Greub, G. (2013). Discovery of new intracellular pathogens by amoebal coculture and amoebal enrichment approaches. J. Vis. Exp. e51055 doi: 10.3791/51055
Joseph, S. J., Didelot, X., Rothschild, J., de Vries, H. J. C., Morré, S. A., Read, T. D., et al. (2012). Population genomics of Chlamydia trachomatis: insights on drift, selection, recombination, and population structure. Mol. Biol. Evol. 29, 3933–3946. doi: 10.1093/molbev/mss198
Kanehisa, M., Sato, Y., and Morishima, K. (2016). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Kantor, R. S., van Zyl, A. W., van Hille, R. P., Thomas, B. C., Harrison, S. T. L., and Banfield, J. F. (2015). Bioreactor microbial ecosystems for thiocyanate and cyanide degradation unravelled with genome-resolved metagenomics. Environ. Microbiol. 17, 4929–4941. doi: 10.1111/1462-2920.12936
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment Software Version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
König, L., Siegl, A., Penz, T., Haider, S., Wentrup, C., Polzin, J., et al. (2017). Biphasic metabolism and host interaction of a chlamydial symbiont. MSystems 2:e00202-16. doi: 10.1128/mSystems.00202-16
Konstantinidis, K. T., and Tiedje, J. M. (2005). Towards a genome-based taxonomy for prokaryotes. J. Bacteriol. 187, 6258–6264. doi: 10.1128/JB.187.18.6258-6264.2005
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kuo, C.-H., Moran, N. A., and Ochman, H. (2009). The consequences of genetic drift for bacterial genome complexity. Genome Res. 19, 1450–1454. doi: 10.1101/gr.091785.109
Kuo, C.-H., and Ochman, H. (2009). Inferring clocks when lacking rocks: the variable rates of molecular evolution in bacteria. Biol. Direct 4:35. doi: 10.1186/1745-6150-4-35
Lagkouvardos, I., Weinmaier, T., Lauro, F. M., Cavicchioli, R., Rattei, T., and Horn, M. (2014). Integrating metagenomic and amplicon databases to resolve the phylogenetic and ecological diversity of the Chlamydiae. ISME J. 8, 115–125. doi: 10.1038/ismej.2013.142
Lamelas, A., Gosalbes, M. J., Manzano-Marín, A., Peretó, J., Moya, A., and Latorre, A. (2011). Serratia symbiotica from the aphid cinara cedri: a missing link from facultative to obligate insect endosymbiont. PLoS Genet. 7:e1002357. doi: 10.1371/journal.pgen.1002357
Lo, W.-S., Huang, Y.-Y., and Kuo, C.-H. (2016). Winding paths to simplicity: genome evolution in facultative insect symbionts. FEMS Microbiol. Rev. 40, 855–874. doi: 10.1093/femsre/fuw028
Malki, S., Saimmaime, I., De Luca, G., Rousset, M., Dermoun, Z., and Belaich, J. P. (1995). Characterization of an operon encoding an NADP-reducing hydrogenase in Desulfovibrio fructosovorans. J. Bacteriol. 177, 2628–2636. doi: 10.1128/jb.177.10.2628-2636.1995
Meganathan, R. (2001). Biosynthesis of menaquinone (vitamin K2) and ubiquinone (coenzyme Q): a perspective on enzymatic mechanisms. Vitam. Horm. 61, 173–218. doi: 10.1016/S0083-6729(01)61006-9
Mitchell, A., Chang, H.-Y., Daugherty, L., Fraser, M., Hunter, S., Lopez, R., et al. (2015). The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 43, D213–D221. doi: 10.1093/nar/gku1243
Moran, N. A. (1996). Accelerated evolution and Muller's rachet in endosymbiotic bacteria. Proc. Natl. Acad. Sci. U.S.A. 93, 2873–2878.
Moran, N. A., and Bennett, G. M. (2014). The tiniest tiny genomes. Annu. Rev. Microbiol. 68, 195–215. doi: 10.1146/annurev-micro-091213-112901
Moran, N. A., McCutcheon, J. P., and Nakabachi, A. (2008). Genomics and evolution of heritable bacterial symbionts. Annu. Rev. Genet. 42, 165–190. doi: 10.1146/annurev.genet.41.110306.130119
O'Leary, N. A., Wright, M. W., Brister, J. R., Ciufo, S., Haddad, D., McVeigh, R., et al. (2016). Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745. doi: 10.1093/nar/gkv1189
Omsland, A., Sixt, B. S., Horn, M., and Hackstadt, T. (2014). Chlamydial metabolism revisited: interspecies metabolic variability and developmental stage-specific physiologic activities. FEMS Microbiol. Rev. 38, 779–801. doi: 10.1111/1574-6976.12059
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Pillonel, T., Bertelli, C., Salamin, N., and Greub, G. (2015). Taxogenomics of the order Chlamydiales. Int. J. Syst. Evol. Microbiol. 65, 1381–1393. doi: 10.1099/ijs.0.000090
Pilloux, L., Aeby, S., Gaümann, R., Burri, C., Beuret, C., and Greub, G. (2015). The high prevalence and diversity of chlamydiales DNA within Ixodes ricinus ticks suggest a role for ticks as reservoirs and vectors of chlamydia-related bacteria. Appl. Environ. Microbiol. 81, 8177–8182. doi: 10.1128/AEM.02183-15
Pinto, A. J., Marcus, D. N., Ijaz, U. Z., Bautista-de lose Santos, Q. M., Dick, G. J., and Raskin, L. (2016). Metagenomic evidence for the presence of comammox nitrospira-like bacteria in a drinking water system. MSphere 1, e00054–e00015. doi: 10.1128/mSphere.00054-15
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS ONE 5:e9490. doi: 10.1371/journal.pone.0009490
R Core Team (2016). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rice, P., Longden, I., and Bleasby, A. (2000). EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. TIG 16, 276–277. doi: 10.1016/S0168-9525(00)02024-2
Rocha, E. P. C., Smith, J. M., Hurst, L. D., Holden, M. T. G., Cooper, J. E., Smith, N. H., et al. (2006). Comparisons of dN/dS are time dependent for closely related bacterial genomes. J. Theor. Biol. 239, 226–235. doi: 10.1016/j.jtbi.2005.08.037
Seth-Smith, H. M. B., Harris, S. R., Skilton, R. J., Radebe, F. M., Golparian, D., Shipitsyna, E., et al. (2013). Whole-genome sequences of Chlamydia trachomatis directly from clinical samples without culture. Genome Res. 23, 855–866. doi: 10.1101/gr.150037
Seth-Smith, H. M. B., Katharios, P., Dourala, N., Mateos, J. M., Fehr, A. G. J., Nufer, L., et al. (2017). Ca. Similichlamydia in epitheliocystis co-infection of gilthead seabream gills: unique morphological features of a deep branching chlamydial family. Front. Microbiol. 8:508. doi: 10.3389/fmicb.2017.00508
Snavely, E. A., Kokes, M., Dunn, J. D., Saka, H. A., Nguyen, B. D., Bastidas, R. J., et al. (2014). Reassessing the role of the secreted protease CPAF in Chlamydia trachomatis infection through genetic approaches. Pathog. Dis. 71, 336–351. doi: 10.1111/2049-632X.12179
Steigen, A., Nylund, A., Karlsbakk, E., Akoll, P., Fiksdal, I. U., Nylund, S., et al. (2013). ‘Cand. Actinochlamydia clariae’ gen. nov., sp. nov., a unique intracellular bacterium causing epitheliocystis in catfish (Clarias gariepinus) in Uganda. PLoS ONE 8:e66840. doi: 10.1371/journal.pone.0066840
Stephens, R. S., Kalman, S., Lammel, C., Fan, J., Marathe, R., Aravind, L., et al. (1998). Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 282, 754–759. doi: 10.1126/science.282.5389.754
Stride, M. C., Polkinghorne, A., Miller, T. L., Groff, J. M., LaPatra, S. E., and Nowak, B. F. (2013). Molecular characterization of “Candidatus Parilichlamydia carangidicola,” a novel chlamydia-like epitheliocystis agent in yellowtail kingfish, seriola lalandi (Valenciennes), and the proposal of a new family, “Candidatus Parilichlamydiaceae” fam. nov. (Order Chlamydiales). Appl. Environ. Microbiol. 79, 1590–1597. doi: 10.1128/AEM.02899-12
Subtil, A., Collingro, A., and Horn, M. (2014). Tracing the primordial Chlamydiae: extinct parasites of plants? Trends Plant Sci. 19, 36–43. doi: 10.1016/j.tplants.2013.10.005
Taylor-Brown, A., Bachmann, N. L., Borel, N., and Polkinghorne, A. (2016). Culture-independent genomic characterisation of Candidatus Chlamydia sanzinia, a novel uncultivated bacterium infecting snakes. BMC Genomics 17:710. doi: 10.1186/s12864-016-3055-x
Taylor-Brown, A., Pillonel, T., Bridle, A., Qi, W., Bachmann, N. L., Miller, T. L., et al. (2017). Culture-independent genomics of a novel chlamydial pathogen of fish provides new insight into host-specific adaptations utilized by these intracellular bacteria. Environ. Microbiol. 19, 1899–1913. doi: 10.1111/1462-2920.13694
Taylor-Brown, A., and Polkinghorne, A. (2017). New and emerging chlamydial infections of creatures great and small. New Microbes New Infect. 18, 28–33. doi: 10.1016/j.nmni.2017.04.004
Taylor-Brown, A., Vaughan, L., Greub, G., Timms, P., and Polkinghorne, A. (2015). Twenty years of research into Chlamydia-like organisms: a revolution in our understanding of the biology and pathogenicity of members of the phylum Chlamydiae. Pathog. Dis. 73, 1–15. doi: 10.1093/femspd/ftu009
Tjaden, J., Winkler, H. H., Schwöppe, C., Van Der Laan, M., Möhlmann, T., and Neuhaus, H. E. (1999). Two nucleotide transport proteins in Chlamydia trachomatis, one for net nucleoside triphosphate uptake and the other for transport of energy. J. Bacteriol. 181, 1196–1202.
Toh, H., Weiss, B. L., Perkin, S. A. H., Yamashita, A., Oshima, K., Hattori, M., et al. (2006). Massive genome erosion and functional adaptations provide insights into the symbiotic lifestyle of Sodalis glossinidius in the tsetse host. Genome Res. 16, 149–156. doi: 10.1101/gr.4106106
Van Nguyen, H., and Lavenier, D. (2009). PLAST: parallel local alignment search tool for database comparison. BMC Bioinformatics 10:329. doi: 10.1186/1471-2105-10-329
Wickham, H. (2016). ggplot2: Elegant Graphics for Data Analysis. New York, NY: Springer-Verlag. doi: 10.1007/978-0-387-98141-3
Woolfit, M., and Bromham, L. (2003). Increased rates of sequence evolution in endosymbiotic bacteria and fungi with small effective population sizes. Mol. Biol. Evol. 20, 1545–1555. doi: 10.1093/molbev/msg167
Yamaguchi, H., Matsuo, J., Yamazaki, T., Ishida, K., and Yagita, K. (2015). Draft genome sequence of high-temperature-adapted Protochlamydia sp. HS-T3, an amoebal endosymbiotic bacterium found in acanthamoeba isolated from a hot spring in Japan. Genome Announc. 3:e01507-14. doi: 10.1128/genomeA.01507-14
Yamane, C., Yamazaki, T., Nakamura, S., Matsuo, J., Ishida, K., Yamazaki, S., et al. (2015). Amoebal endosymbiont Parachlamydia acanthamoebae Bn9 can grow in immortal human epithelial HEp-2 cells at low temperature; an in vitro model system to study chlamydial evolution. PLoS ONE 10:e0116486. doi: 10.1371/journal.pone.0116486
Keywords: Chlamydiae, comparative genomics, bacterial taxonomy, chlamydial metabolism, endosymbiont evolution, obligate intracellular
Citation: Pillonel T, Bertelli C and Greub G (2018) Environmental Metagenomic Assemblies Reveal Seven New Highly Divergent Chlamydial Lineages and Hallmarks of a Conserved Intracellular Lifestyle. Front. Microbiol. 9:79. doi: 10.3389/fmicb.2018.00079
Received: 26 September 2017; Accepted: 12 January 2018;
Published: 20 February 2018.
Edited by:
Michael Thomas-Poulsen, University of Copenhagen, DenmarkReviewed by:
William C. Nelson, Pacific Northwest National Laboratory, United StatesKelly Brayton, Washington State University, United States
Copyright © 2018 Pillonel, Bertelli and Greub. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gilbert Greub, gilbert.greub@chuv.ch