
- 1Clinical Research Center, National Hospital Organization Nagoya Medical Center, Nagoya, Aichi, Japan
- 2Pathogen Genomics Center, National Institute of Infectious Diseases, Musashimurayama, Tokyo, Japan
- 3Department of Biotechnology, Graduate School of Engineering, Nagoya University, Nagoya, Aichi, Japan
- 4Department of AIDS Research, Graduate School of Medicine, Nagoya University, Nagoya, Aichi, Japan
Virus replication in the host proceeds by chains of interactions between viral and host proteins. The interactions are deeply influenced by host immune molecules and anti-viral compounds, as well as by mutations in viral proteins. To understand how these interactions proceed mechanically and how they are influenced by mutations, one needs to know the structures and dynamics of the proteins. Molecular dynamics (MD) simulation is a powerful computational method for delineating motions of proteins at an atomic-scale via theoretical and empirical principles in physical chemistry. Recent advances in the hardware and software for biomolecular simulation have rapidly improved the precision and performance of this technique. Consequently, MD simulation is quickly extending the range of applications in biology, helping to reveal unique features of protein structures that would be hard to obtain by experimental methods alone. In this review, we summarize the recent advances in MD simulations in the study of virus–host interactions and evolution, and present future perspectives on this technique.
Introduction
Proteins fluctuate spontaneously in solution (Ishima and Torchia, 2000). Accumulating evidence indicates that such fluctuations play key roles in the specific functions of proteins, such as catalytic reactions of enzymes (Nicholson et al., 1995; Lu et al., 1998; Eisenmesser et al., 2005; Henzler-Wildman et al., 2007; Abbondanzieri et al., 2008), interactions with other biomolecules (Thorpe and Brooks, 2007), and biomolecular motors and pumps (Astumian, 1997). Multiple experimental methods are available to characterize the protein dynamics (Figure 1). However, it is usually difficult to delineate motions of proteins at an atomic scale.
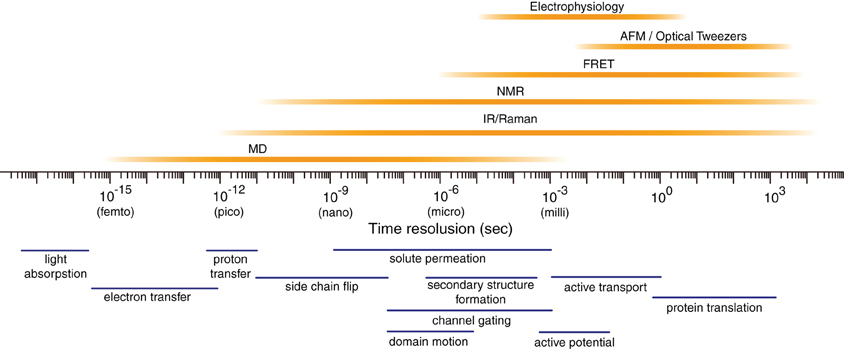
Figure 1. Temporal resolution of various biophysical techniques. The timescales of some fundamental atom- or molecule-scale motions are shown below. AFM, atomic force microscopy; FRET, fluorescence resonance energy transfer; IR, infrared radiation; NMR, nuclear magnetic resonance.
MD Simulation in Biology
Outline
Molecular dynamics (MD) simulation is a computational method to address the above issue (Figure 1) (Henzler-Wildman and Kern, 2007; Dror et al., 2010). This technique enables us to calculate movements of atoms in a molecular system, such as proteins in water, by numerically solving Newton's equations of motions (Karplus and Petsko, 1990; Adcock and McCammon, 2006). In a simple molecular system, all atoms and covalent bonds connecting the atoms are assumed to be the charged spheres and springs, respectively. Parameters of mathematical functions describing the potential energy of a system, termed the “force field,” are set to simulate the movements of atoms and molecules. Frequently used force fields for proteins, such as the “AMBER” (Pearlman et al., 1995; Case et al., 2005) and “CHARMM” (Brooks et al., 2009) force fields, have the formulae of covalent bonds, angles, dihedrals, van der Waals, and electrostatic potentials.
Performance and Consistency with Experimental Data
Application of MD simulation in the field of protein chemistry was first reported in 1977 (McCammon et al., 1977). Since then, the performance of this technique have been quickly improved quantitatively and qualitatively along with the rapid advances in hardware and software on biomolecular simulation (Lindorff-Larsen et al., 2012). The results of MD simulation are critically influenced by the force fields (Lindorff-Larsen et al., 2012). The qualities of parameters in the force fields, especially for dihedrals and electrostatic potentials, have been improved quantitatively and qualitatively over time by introducing improved approximation to the quantum ground-state potential energy surface. Recently, eight different protein force fields were evaluated on the basis of the consistency of simulations with the NMR data (Lindorff-Larsen et al., 2012). The study demonstrates that the most recent versions, while not perfect, provide results that are highly consistent with the experimental data (Lindorff-Larsen et al., 2012). In addition, explicit introduction of effects of the solvation has contributed to the qualitative improvement for the precision and performance of MD simulations (Adcock and McCammon, 2006).
MD in Structural Biology
MD simulation currently allows us to investigate the structural dynamics of proteins on timescales of nanoseconds to microseconds, and will probably allow investigation to milliseconds in the future (Figure 1) (Henzler-Wildman and Kern, 2007; Dror et al., 2010). This technique is widely used in the field of structural biology (Karplus and McCammon, 2002; Karplus and Kuriyan, 2005; Dodson et al., 2008). First, MD simulation is useful for refining the experimentally determined three-dimensional (3-D) structures of proteins (Autore et al., 2010; Ozen et al., 2011). Second, MD simulation is beneficial for constructing previously undescribed 3-D structures of proteins in combination with homology modeling techniques (Marti-Renom et al., 2000; Sanchez et al., 2000; Baker and Sali, 2001), when a reported structure of a homolog is available. Third and most importantly, MD simulation provides a unique tool to address the structural dynamics of proteins, i.e., the time evolution of conformations in solution, at timescales of nanoseconds to microseconds (Henzler-Wildman and Kern, 2007; Dror et al., 2010). The structural snapshots obtained during MD simulation are helpful for depicting the unique structural features of proteins (Karplus and McCammon, 2002; Karplus and Kuriyan, 2005; Dodson et al., 2008).
MD Simulation in Virology
To date, MD simulations have been applied in a range of virus researches, as shown in the following sections.
Neutralization Escape and Cell Tropism Switching of HIV-1 Mediated by an Electrostatic Mechanism
It is very important to clarify how viruses evade neutralization antibodies in order to understand the viral life cycle and evolution, and to develop vaccines. MD simulation is used to address this issue as it pertains to human immunodeficiency virus type 1 (HIV-1). The third variable (V3) loop of the HIV-1 envelope gp120 protein constitutes the major antibody epitopes of HIV-1 and the major determinants for the entry coreceptor use of HIV-1. By analyzing the 40,000 structural snapshots obtained from 10–30 ns of MD simulations of the identical gp120 outer domain carrying a distinct V3 loop with net charge of +3 or +7, Yokoyama and colleagues showed that the change in V3 net charge alone is sufficient to induce global changes in fluctuation and conformation of the loops involved in binding to CD4, coreceptor, and neutralizing antibodies (Naganawa et al., 2008; Yokoyama et al., 2012). Structural changes caused by a reduction in the V3 net charge via V3 mutations are tightly linked to viral CCR5 coreceptor tropism (Naganawa et al., 2008), as well as to a reduction in viral neutralization sensitivity to anti-V3 antibodies (Naganawa et al., 2008) and anti-CD4 binding site monoclonal antibodies (Yokoyama et al., 2012). These findings suggest a hitherto unrecognized mechanism, V3-mediated electrostatic modulation of the structure and dynamics of the gp120 interaction surface, for adjusting the relative replication fitness and evolution of HIV-1 (Yokoyama et al., 2012). In addition, they partly explain a virological mystery, i.e., why HIV-1 variants using CCR5, which carries a V3 loop with a lower level of positive net charge, predominantly persist before the onset of AIDS.
Mechanisms of Viral Escape from Host Defense Systems
Viruses also evade host defense systems other than neutralization antibodies (Figure 2). MD simulation is used to clarify the structural basis for viral escape from host defense systems by mutations. Mutations at the 120th amino acid in the HIV-2 capsid protein play a key role in evading tripartite motif-containing protein 5α (TRIM5α), an anti-retroviral cellular protein induced by interferon, both in vivo (Onyango et al., 2010) and in vitro (Song et al., 2007). An MD simulation study has revealed that the mutations could extensively influence the conformation and fluctuation of the interaction surface of capsid proteins by altering the probability of hydrogen bond formation between helices 4 and 5 (Miyamoto et al., 2011).
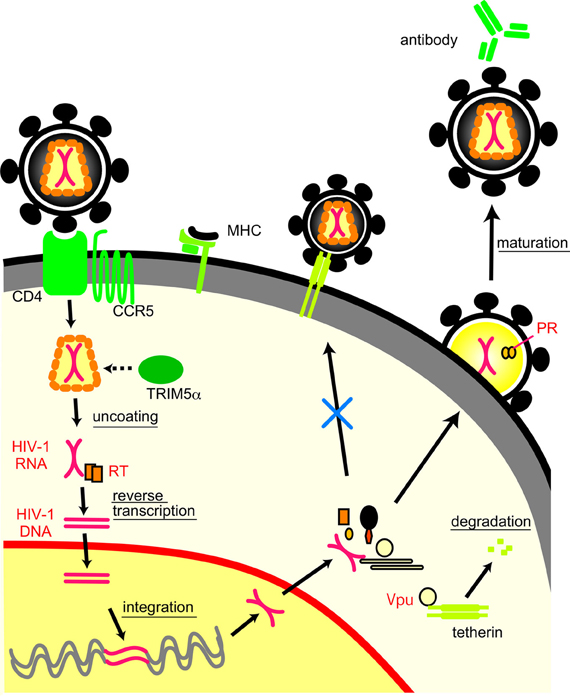
Figure 2. Life cycle of HIV-1 and interactions between viral proteins and host immune molecules.
HIV-1 Vpu antagonizes an antiviral cellular protein termed tetherin, also known as BST-2/CD317/HM1.24, by interaction with the transmembrane (TM) domain of tetherin and subsequent degradation (Douglas et al., 2010; Kobayashi et al., 2011). An MD simulation suggests that alignment of the four amino acid residues (I34, L37, L41, and T45) on the same helical face in the human tetherin TM domain is crucial for the Vpu-mediated antagonism against human tetherin (Kobayashi et al., 2011). The interface structure of the tetherin TM for the antagonism was also predicted by the MD simulation of another group (Zhou et al., 2012) and experimentally confirmed by an NMR study (Skasko et al., 2012).
MD simulation is also used to study the mechanisms of functional interactions between cytotoxic T lymphocyte (CTL) epitope and major histocompatibility complex (MHC) molecules (Reboul et al., 2012). An MD simulation study has revealed that a 13-mer epitope peptide from Epstein-Barr virus has the low structural flexibility in an MHC molecule that induces a CTL response but exhibits high flexibility in another MHC molecule that cannot induce a CTL response (Reboul et al., 2012). Thus, structural flexibility of CTL epitope region seems to be critical for the specific recognition by MHC molecules, and mutations that alter the flexibility may influence CTL response. There are other viral proteins and immune molecules involved in viral evasion from host defense systems (Neil and Bieniasz, 2009; Malim and Bieniasz, 2012). MD simulations should also be applicable for the studies of these molecules.
Structure and Function of Viral Enzymes
Viral enzymes are essential for viral replications and thus are important targets for anti-viral drug development. MD simulations are used to study the basis of the structural dynamics that allow the viral enzyme and its drug to function properly. Viral polymerase (Pol) is essential for viral genome replication in the viral life cycle. The Pol is composed of the fingers, palm, and thumb subdomains, which form a cavity for the substrate binding, as in eukaryotic Pol (Joyce and Steitz, 1994; Lamers et al., 2006; Cameron et al., 2009). MD simulations suggest that the finger and thumb domains of HIV-1 reverse transcriptase (RT) are especially mobile among the various regions of this enzyme (Zhou et al., 2005; Kirmizialtin et al., 2012). The mobility is severely attenuated by binding of allosteric non-nucleotide RT inhibitors (NNRTIs) (Zhou et al., 2005). Interestingly, a large conformational change of RT subdomains during millisecond timescale simulations can lock the correct nucleotide at the active site but promotes release of a mismatched nucleotide (Kirmizialtin et al., 2012). Furthermore, conformational dynamics leading to opening and closing motions of the substrate binding cleft are highly conserved among four RNA Pols in the picornavirus family, despite the amino acid identity being as low as 30–74% (Moustafa et al., 2011). These findings are consistent with each other and strongly suggest that the structural dynamics of viral Pol play a key role in the polymerization.
Viral protease (PR) plays a key role in viral propagation by catalyzing cleavages of viral precursor proteins (Pettit et al., 1994, 2002; Steven et al., 2005). HIV-1 PR and other retroviral PRs have unique regions termed the “flaps” outside the substrate binding clefts (Dunn et al., 2002). MD simulation studies suggest that the PR flaps in HIV-1 are intrinsically mobile, undergoing conversions between the “semiopen,” “open,” and “closed” conformations (Hornak et al., 2006; Deng et al., 2011). This movement is severely attenuated upon placement of the substrate or PR inhibitor in the binding cleft (Karthik and Senapati, 2011), suggesting that flap movement plays a critical role in PR function.
MD simulations are also used to study the structural dynamics of the substrates of viral PR. Peptides corresponding to cleavage junctions of viral precursor proteins of HIV-1 are intrinsically unstructured in aqueous solution (Datta et al., 2011; Ode et al., 2011). However, the folding preference of the junction peptides may be different among the junctions and related to the efficiency of substrate binding and cleavage reaction by PR (Ode et al., 2011). Furthermore, peptides at the capsid-p2 junction can adopt a helical conformation when the polarity of the environment is reduced (Datta et al., 2011). The MD simulation of PR and its substrates will help to clarify how the viral precursor is processed orderly during viral maturation.
Drug-Resistance Mechanisms
Antiviral drug resistance is a major clinical problem for the treatment of virus-infected individuals (Cortez and Maldarelli, 2011; van der Vries et al., 2011). Viral resistance to antiviral drugs is primarily caused by genetic mutations that eventually lead to a reduction in the drug affinity of drug target viral proteins. MD simulations are used to examine how viral mutations cause the drug resistance at the atomic level.
A reduction in the binding affinity of the PR inhibitors to HIV-1 PR can be caused by a reduction in hydrophobic interactions (Kagan et al., 2005; Wittayanarakul et al., 2005; Sadiq et al., 2007; Chen et al., 2010; Dirauf et al., 2010), reduction in electrostatic interactions (Ode et al., 2005, 2006, 2007a; Chen et al., 2010), changes in flexibility at the flap of the PR (Piana et al., 2002; Perryman et al., 2004; Chang et al., 2006; Foulkes-Murzycki et al., 2007), and changes in the shape of the inhibitor-binding pocket (Ode et al., 2005, 2006, 2007b). Reduction in binding affinity of the nucleotide/nucleoside RT inhibitors (NRTIs) to HIV-1 RT can be caused by a distinct conformational preference of NRTIs in the substrate/NRTI-binding site compared to normal substrates (Carvalho et al., 2006) or enhancement of ATP-mediated excision of misincorporated nucleotide analogs via increased accessibility of ATP to the terminus of extending DNA (White et al., 2004; Carvalho et al., 2007). Reduction in the binding affinity of the NNRTIs to HIV-1 RT can be attained by occlusion of the NNRTI-entry pathway (Rodriguez-Barrios and Gago, 2004; Rodriguez-Barrios et al., 2005) or restoration of the proper flexibility of the RT even with NNRTIs (Zhou et al., 2005).
A change in volume of the binding site of influenza virus (IFV) M2 channel blockers has been shown to reduce the blockers' binding affinity (Gu et al., 2011; Leonov et al., 2011; Wang et al., 2011). Disruption of the proper guidance of IFV neuraminidase (NA) inhibitors into their binding pocket is proposed as a possible mechanism for the reduction in the binding affinity of the inhibitors (Le et al., 2010; Kasson, 2012). MD simulations are also used to study how the genetic differences of HIV variants around the world can influence the efficacy of antiviral inhibitors (Batista et al., 2006; Ode et al., 2007a; Matsuyama et al., 2010; Soares et al., 2010; Kar and Knecht, 2012). Thus, MD simulation will be valuable to assist in the study of drug efficacy when genetic information on the drug target proteins is available (Shenderovich et al., 2003; Stoica et al., 2008; Sadiq et al., 2010; Wright and Coveney, 2011).
Antiviral Drug Discovery and Development
MD simulations are used to assist in the discovery and development of antiviral drugs (Durrant and McCammon, 2011; Borhani and Shaw, 2012). MD simulations allow sampling snapshots of fluctuated protein structures, which include their short-lived conformations as well as stable conformations. This is beneficial for searching conformations of a protein on ligand-binding, since ligand-binding can stabilize conformation of a protein that is not the most stable at ligand-free state (Tobi and Bahar, 2005; Xu et al., 2008). Thus, the MD simulations are used to improve the enrichment performance of molecular docking during in silico drug screening by taking accounts of multiple docking poses (Okimoto et al., 2009). The method is also applied for identifying concealed drug-binding sites, which are apparently masked and not evident from the X-ray crystal structures, by considering the structural flexibility of proteins. For example, MD simulations have been used to find a trench adjacent to the active site of HIV-1 integrase (Schames et al., 2004). A site-directed mutagenesis study provided evidence that the trench indeed plays key roles in ligand-binding (Lee and Robinson, 2006). These findings have been used to design HIV-1 integrase inhibitors with potent antiviral effects (Durrant and McCammon, 2011).
Likewise, MD simulations are used to assist in the development of antiviral drugs against IFV. Using this method, a universal cavity adjacent to the binding site of natural substrate has been reported with NA proteins of human 2009 pandemic H1N1, avian H5N1, and human H2N2 strains (Amaro et al., 2011). MD simulations were also used to construct a 3-D structure model of CCR5, a major coreceptor of HIV-1 (Maeda et al., 2008; Da and Wu, 2011).
Virion Structure
It is essential to clarify the structure of virions in order to understand the mechanisms of viral infection and assembly. MD simulation is used to address this issue. Using a super computer, Freddolino et al. performed 50-nanosecond-timescale MD simulations of the symmetric structure of a complete satellite tobacco mosaic virus (STMV) particle containing about 1 million atoms (Arkhipov et al., 2006) (Figure 3). Thus, far, this is one of the largest systems among the MD simulations reported in all biological fields. Notably, the virion with viral RNA was stable during the simulations, whereas the one without the RNA was unstable, suggesting that viral RNA plays a key role in stabilizing the STMV virion (Arkhipov et al., 2006). The study is consistent with the experimental data (Day et al., 2001) and therefore provides a set of rationale conditions for performing the MD simulation of virion. Likewise, Larsson et al. reported about 1-microsecond-timescale MD simulations of the satellite tobacco necrosis virus (STNV) (Larsson et al., 2012). Their study reproduced the biochemical phenomenon of the STNV virion in solution (Unge et al., 1986), i.e., the swelling of capsid upon Ca2+ removal by EDTA treatment. These findings will provide a structural basis for identifying the key regulators of assembly and infections and for illustrating how they function mechanically. Although MD simulation of virions composed of very large numbers of atoms is still difficult in most cases, progress in the hardware and software for the simulation, together with the accumulation of biological and physicochemical information on virions, will help us to overcome these limitations in the MD simulation of virions.
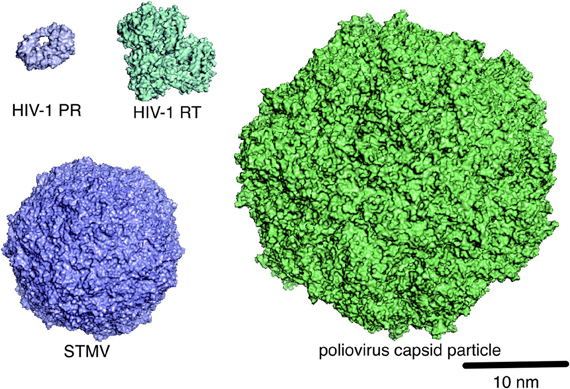
Figure 3. Molecular scales of viral proteins and capsid particles. The structures of HIV-1 PR (PDB code: 1HHP), HIV-1 RT (PDB code: 1RTD), STMV (PDB code: 1A34), and poliovirus capsid particle (PDB code: 1HXS), which are deposited in the Protein Data Bank (PDB) (http://www.rcsb.org/pdb/home/home.do) or the VIPERdb (http://viperdb.scripps.edu/), are shown in surface representation.
Perspective
Since the processing speed of computers is still doubling approximately every two years according to Moore's law, MD studies will be extended to simulations of larger and more complex system at longer timescales. This will then lead to a better understanding of the structures and dynamics of macromolecules involved in virus–host interactions.
Coarse-Grained (CG) MD Simulations
MD simulations of macromolecules consisting of large molecular systems, such as oligomeric proteins, macromolecular complexes, and membrane proteins in a lipid bilayer, and virions are desired to better understand viruses. However, such simulations require unrealistically long analytical times and high-performance computers at present, and thereby are still limited mostly to the small molecules (Henzler-Wildman and Kern, 2007; Dror et al., 2010). To cope with this issue and to improve the practicability of long timescale MD simulation, a “coarse-grained (CG) MD” simulation has been developed (Merchant and Madura, 2011; Takada, 2012). The CG-MD simulation employs “pseudo-atoms” that consist of several atoms in a group and calculates the movement of these “pseudo-atoms” rather than the movement of “individual atoms,” thereby greatly reducing the calculation time (Merchant and Madura, 2011; Takada, 2012). CG-MD simulations have been used to study helicases of hepatitis C virus (HCV) and simian virus 40 and have successfully reproduced enzyme motions, such as “ratcheting inchworm translocation” and “spring-loaded DNA unwinding” (Flechsig and Mikhailov, 2010; Yoshimoto et al., 2010). Briefly, the ratcheting inchworm translocation is the unidirectional motion of the HCV NS3 helicase during translocation that occurs by the step size of one base per ATP hydrolysis cycle (Gu and Rice, 2010). Meanwhile, the spring-loaded DNA unwinding is the discrete steps of unwinding of DNA by the HCV NS3 helicase that occurs periodically via a burst of 3-bp unwinding during NS3 translocation consuming ATPs (Myong et al., 2007).
CG-MD has also been applied to the study of the structural characteristics and stabilities of the capsid particle and virion (Figure 3). Such studies have been used to investigate small plant viruses (~28 nanometer in diameter), such as the three satellite plant viruses STMV, STNV, and the satellite panicum mosaic virus (SPMV), as well as the brome mosaic virus (BMV) (Arkhipov et al., 2006, 2009), and more complex capsids such as poliovirus (Arkhipov et al., 2006, 2009), asymmetric, conical-shaped HIV-1 capsid particles (Krishna et al., 2010), and the immature HIV-1 virion (Ayton and Voth, 2010). These studies have predicted various molecular interactions that can be tested experimentally. Thus, CG-MD may play a pivotal role in the MD study of micrometer-sized systems at millisecond timescale (Merchant and Madura, 2011; Takada, 2012) and therefore may uncover novel characteristics of the interactions in virus–host relationships.
Intrinsically Disordered Proteins
Some eukaryotic proteins have no stable 3-D structure under physiological conditions (Dunker et al., 2002, 2008; Dyson and Wright, 2005). These proteins are referred to as intrinsically disordered, natively unfolded, or intrinsically unstructured proteins. They undergo structural transition from a disordered to an ordered state upon binding to target molecules such as proteins, DNA, and small molecules (Dunker et al., 2005; Sandhu and Dash, 2007). They are often related to the “hub proteins” that have many binding partners and control important biological processes (Iakoucheva et al., 2002; Haynes et al., 2006; Sandhu, 2009). Interestingly, viral proteins or portions of viral proteins are often intrinsically disordered. These include genome-linked protein VPg protein of plant viruses (Grzela et al., 2008; Rantalainen et al., 2008; Jiang and Laliberte, 2011; Rantalainen et al., 2011), HIV-1 Tat (Shojania and O'Neil, 2010), and Vif proteins (Reingewertz et al., 2010), and paramyxovirus nucleoproteins and phosphoproteins (Habchi and Longhi, 2012). It has been proposed that the disordered structure is beneficial for viruses to gain multiple functions in the viral life cycle with limited genome size (Rantalainen et al., 2011; Habchi and Longhi, 2012; Xue et al., 2012). Clarifying the folding landscape of viral proteins by standard MD and CG-MD simulations may help in understanding the structural principles by which viral proteins execute multiple functions in the viral life cycle.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The MD studies of our laboratory described in this article are supported by grants from the Ministry of Health, Labor and Welfare for HIV/AIDS research and by a Grant-in-Aid for JSPS Fellows.
References
Abbondanzieri, E. A., Bokinsky, G., Rausch, J. W., Zhang, J. X., Le Grice, S. F., and Zhuang, X. (2008). Dynamic binding orientations direct activity of HIV reverse transcriptase. Nature 453, 184–189.
Adcock, S. A., and McCammon, J. A. (2006). Molecular dynamics: survey of methods for simulating the activity of proteins. Chem. Rev. 106, 1589–1615.
Amaro, R. E., Swift, R. V., Votapka, L., Li, W. W., Walker, R. C., and Bush, R. M. (2011). Mechanism of 150-cavity formation in influenza neuraminidase. Nat. Commun. 2, 388.
Arkhipov, A., Freddolino, P. L., and Schulten, K. (2006). Stability and dynamics of virus capsids described by coarse-grained modeling. Structure 14, 1767–1777.
Arkhipov, A., Roos, W. H., Wuite, G. J., and Schulten, K. (2009). Elucidating the mechanism behind irreversible deformation of viral capsids. Biophys. J. 97, 2061–2069.
Autore, F., Bergeron, J. R., Malim, M. H., Fraternali, F., and Huthoff, H. (2010). Rationalisation of the differences between APOBEC3G structures from crystallography and NMR studies by molecular dynamics simulations. PLoS ONE 5:e11515. doi: 10.1371/journal.pone.0011515
Ayton, G. S., and Voth, G. A. (2010). Multiscale computer simulation of the immature HIV-1 virion. Biophys. J. 99, 2757–2765.
Baker, D., and Sali, A. (2001). Protein structure prediction and structural genomics. Science 294, 93–96.
Batista, P. R., Wilter, A., Durham, E. H., and Pascutti, P. G. (2006). Molecular dynamics simulations applied to the study of subtypes of HIV-1 protease common to Brazil, Africa, and Asia. Cell Biochem. Biophys. 44, 395–404.
Borhani, D. W., and Shaw, D. E. (2012). The future of molecular dynamics simulations in drug discovery. J. Comput. Aided. Mol. Des. 26, 15–26.
Brooks, B. R., Brooks, C. L. 3rd., Mackerell, A. D. Jr., Nilsson, L., Petrella, R. J., Roux, B., Won, Y., Archontis, G., Bartels, C., Boresch, S., Caflisch, A., Caves, L., Cui, Q., Dinner, A. R., Feig, M., Fischer, S., Gao, J., Hodoscek, M., Im, W., Kuczera, K., Lazaridis, T., Ma, J., Ovchinnikov, V., Paci, E., Pastor, R. W., Post, C. B., Pu, J. Z., Schaefer, M., Tidor, B., Venable, R. M., Woodcock, H. L., Wu, X., Yang, W., York, D. M., and Karplus, M. (2009). CHARMM: the biomolecular simulation program. J. Comput. Chem. 30, 1545–1614.
Cameron, C. E., Moustafa, I. M., and Arnold, J. J. (2009). Dynamics: the missing link between structure and function of the viral RNA-dependent RNA polymerase? Curr. Opin. Struct. Biol. 19, 768–774.
Carvalho, A. T., Fernandes, P. A., and Ramos, M. J. (2006). Insights on resistance to reverse transcriptase: the different patterns of interaction of the nucleoside reverse transcriptase inhibitors in the deoxyribonucleotide triphosphate binding site relative to the normal substrate. J. Med. Chem. 49, 7675–7682.
Carvalho, A. T., Fernandes, P. A., and Ramos, M. J. (2007). The excision mechanism in reverse transcriptase: pyrophosphate leaving and fingers opening are uncoupled events with the analogues AZT and d4T. J. Phys. Chem. B 111, 12032–12039.
Case, D. A., Cheatham, T. E. 3rd, Darden, T., Gohlke, H., Luo, R., Merz, K. M. Jr., Onufriev, A., Simmerling, C., Wang, B., and Woods, R. J. (2005). The amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688.
Chang, C. E., Shen, T., Trylska, J., Tozzini, V., and McCammon, J. A. (2006). Gated binding of ligands to HIV-1 protease: brownian dynamics simulations in a coarse-grained model. Biophys. J. 90, 3880–3885.
Chen, J., Zhang, S., Liu, X., and Zhang, Q. (2010). Insights into drug resistance of mutations D30N and I50V to HIV-1 protease inhibitor TMC-114, free energy calculation and molecular dynamic simulation. J. Mol. Model. 16, 459–468.
Cortez, K. J., and Maldarelli, F. (2011). Clinical management of HIV drug resistance. Viruses 3, 347–378.
Da, L. T., and Wu, Y. D. (2011). Theoretical studies on the interactions and interferences of HIV-1 glycoprotein gp120 and its coreceptor CCR5. J. Chem. Inf. Model. 51, 359–369.
Datta, S. A., Temeselew, L. G., Crist, R. M., Soheilian, F., Kamata, A., Mirro, J., Harvin, D., Nagashima, K., Cachau, R. E., and Rein, A. (2011). On the role of the SP1 domain in HIV-1 particle assembly: a molecular switch? J. Virol. 85, 4111–4121.
Day, J., Kuznetsov, Y. G., Larson, S. B., Greenwood, A., and McPherson, A. (2001). Biophysical studies on the RNA cores of satellite tobacco mosaic virus. Biophys. J. 80, 2364–2371.
Deng, N. J., Zheng, W., Gallicchio, E., and Levy, R. M. (2011). Insights into the dynamics of HIV-1 protease: a kinetic network model constructed from atomistic simulations. J. Am. Chem. Soc. 133, 9387–9394.
Dirauf, P., Meiselbach, H., and Sticht, H. (2010). Effects of the V82A and I54V mutations on the dynamics and ligand binding properties of HIV-1 protease. J. Mol. Model. 16, 1577–1583.
Dodson, G. G., Lane, D. P., and Verma, C. S. (2008). Molecular simulations of protein dynamics: new windows on mechanisms in biology. EMBO Rep. 9, 144–150.
Douglas, J. L., Gustin, J. K., Viswanathan, K., Mansouri, M., Moses, A. V., and Fruh, K. (2010). The great escape: viral strategies to counter BST-2/tetherin. PLoS Pathog. 6:e1000913. doi: 10.1371/journal.ppat.1000913
Dror, R. O., Jensen, M. O., Borhani, D. W., and Shaw, D. E. (2010). Exploring atomic resolution physiology on a femtosecond to millisecond timescale using molecular dynamics simulations. J. Gen. Physiol. 135, 555–562.
Dunker, A. K., Brown, C. J., Lawson, J. D., Iakoucheva, L. M., and Obradovic, Z. (2002). Intrinsic disorder and protein function. Biochemistry 41, 6573–6582.
Dunker, A. K., Cortese, M. S., Romero, P., Iakoucheva, L. M., and Uversky, V. N. (2005). Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 272, 5129–5148.
Dunker, A. K., Silman, I., Uversky, V. N., and Sussman, J. L. (2008). Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 18, 756–764.
Dunn, B. M., Goodenow, M. M., Gustchina, A., and Wlodawer, A. (2002). Retroviral proteases. Genome Biol. 3, Reviews3006.
Durrant, J. D., and McCammon, J. A. (2011). Molecular dynamics simulations and drug discovery. BMC Biol. 9, 71.
Dyson, H. J., and Wright, P. E. (2005). Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell. Biol. 6, 197–208.
Eisenmesser, E. Z., Millet, O., Labeikovsky, W., Korzhnev, D. M., Wolf-Watz, M., Bosco, D. A., Skalicky, J. J., Kay, L. E., and Kern, D. (2005). Intrinsic dynamics of an enzyme underlies catalysis. Nature 438, 117–121.
Flechsig, H., and Mikhailov, A. S. (2010). Tracing entire operation cycles of molecular motor hepatitis C virus helicase in structurally resolved dynamical simulations. Proc. Natl. Acad. Sci. U.S.A. 107, 20875–20880.
Foulkes-Murzycki, J. E., Scott, W. R., and Schiffer, C. A. (2007). Hydrophobic sliding: a possible mechanism for drug resistance in human immunodeficiency virus type 1 protease. Structure 15, 225–233.
Grzela, R., Szolajska, E., Ebel, C., Madern, D., Favier, A., Wojtal, I., Zagorski, W., and Chroboczek, J. (2008). Virulence factor of potato virus Y, genome-attached terminal protein VPg, is a highly disordered protein. J. Biol. Chem. 283, 213–221.
Gu, M., and Rice, C. M. (2010). Three conformational snapshots of the hepatitis C virus NS3 helicase reveal a ratchet translocation mechanism. Proc. Natl. Acad. Sci. U.S.A. 107, 521–528.
Gu, R. X., Liu, L. A., Wei, D. Q., Du, J. G., Liu, L., and Liu, H. (2011). Free energy calculations on the two drug binding sites in the M2 proton channel. J. Am. Chem. Soc. 133, 10817–10825.
Habchi, J., and Longhi, S. (2012). Structural disorder within paramyxovirus nucleoproteins and phosphoproteins. Mol. Biosyst. 8, 69–81.
Haynes, C., Oldfield, C. J., Ji, F., Klitgord, N., Cusick, M. E., Radivojac, P., Uversky, V. N., Vidal, M., and Iakoucheva, L. M. (2006). Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput. Biol. 2:e100. doi: 10.1371/journal.pcbi.0020100
Henzler-Wildman, K. A., Lei, M., Thai, V., Kerns, S. J., Karplus, M., and Kern, D. (2007). A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature 450, 913–916.
Hornak, V., Okur, A., Rizzo, R. C., and Simmerling, C. (2006). HIV-1 protease flaps spontaneously open and reclose in molecular dynamics simulations. Proc. Natl. Acad. Sci. U.S.A. 103, 915–920.
Iakoucheva, L. M., Brown, C. J., Lawson, J. D., Obradovic, Z., and Dunker, A. K. (2002). Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 323, 573–584.
Jiang, J., and Laliberte, J. F. (2011). The genome-linked protein VPg of plant viruses-a protein with many partners. Curr. Opin. Virol. 1, 347–354.
Joyce, C. M., and Steitz, T. A. (1994). Function and structure relationships in DNA polymerases. Annu. Rev. Biochem. 63, 777–822.
Kagan, R. M., Shenderovich, M. D., Heseltine, P. N., and Ramnarayan, K. (2005). Structural analysis of an HIV-1 protease I47A mutant resistant to the protease inhibitor lopinavir. Protein Sci. 14, 1870–1878.
Kar, P., and Knecht, V. (2012). Origin of decrease in potency of darunavir and two related antiviral inhibitors against HIV-2 compared to HIV-1 protease. J. Phys. Chem. B 116, 2605–2614.
Karplus, M., and Kuriyan, J. (2005). Molecular dynamics and protein function. Proc. Natl. Acad. Sci. U.S.A. 102, 6679–6685.
Karplus, M., and McCammon, J. A. (2002). Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 9, 646–652.
Karplus, M., and Petsko, G. A. (1990). Molecular dynamics simulations in biology. Nature 347, 631–639.
Karthik, S., and Senapati, S. (2011). Dynamic flaps in HIV-1 protease adopt unique ordering at different stages in the catalytic cycle. Proteins 79, 1830–1840.
Kasson, P. M. (2012). Receptor binding by influenza virus: using computational techniques to extend structural data. Biochemistry 51, 2359–2365.
Kirmizialtin, S., Nguyen, V., Johnson, K. A., and Elber, R. (2012). How conformational dynamics of DNA polymerase select correct substrates: experiments and simulations. Structure 20, 618–627.
Kobayashi, T., Ode, H., Yoshida, T., Sato, K., Gee, P., Yamamoto, S. P., Ebina, H., Strebel, K., Sato, H., and Koyanagi, Y. (2011). Identification of amino acids in the human tetherin transmembrane domain responsible for HIV-1 Vpu interaction and susceptibility. J. Virol. 85, 932–945.
Krishna, V., Ayton, G. S., and Voth, G. A. (2010). Role of protein interactions in defining HIV-1 viral capsid shape and stability: a coarse-grained analysis. Biophys. J. 98, 18–26.
Lamers, M. H., Georgescu, R. E., Lee, S. G., O'Donnell, M., and Kuriyan, J. (2006). Crystal structure of the catalytic alpha subunit of E. coli replicative DNA polymerase III. Cell 126, 881–892.
Larsson, D. S., Liljas, L., and van der Spoel, D. (2012). Virus capsid dissolution studied by microsecond molecular dynamics simulations. PLoS Comput. Biol. 8:e1002502. doi: 10.1371/journal.pcbi.1002502
Le, L., Lee, E. H., Hardy, D. J., Truong, T. N., and Schulten, K. (2010). Molecular dynamics simulations suggest that electrostatic funnel directs binding of Tamiflu to influenza N1 neuraminidases. PLoS Comput. Biol. 6:e1000939. doi: 10.1371/journal.pcbi.1000939
Lee, D. J., and Robinson, W. E. Jr. (2006). Preliminary mapping of a putative inhibitor-binding pocket for human immunodeficiency virus type 1 integrase inhibitors. Antimicrob. Agents Chemother. 50, 134–142.
Leonov, H., Astrahan, P., Krugliak, M., and Arkin, I. T. (2011). How do aminoadamantanes block the influenza M2 channel, and how does resistance develop? J. Am. Chem. Soc. 133, 9903–9911.
Lindorff-Larsen, K., Maragakis, P., Piana, S., Eastwood, M. P., Dror, R. O., and Shaw, D. E. (2012). Systematic validation of protein force fields against experimental data. PLoS ONE 7:e32131. doi: 10.1371/journal.pone.0032131
Lu, H. P., Xun, L., and Xie, X. S. (1998). Single-molecule enzymatic dynamics. Science 282, 1877–1882.
Maeda, K., Das, D., Yin, P. D., Tsuchiya, K., Ogata-Aoki, H., Nakata, H., Norman, R. B., Hackney, L. A., Takaoka, Y., and Mitsuya, H. (2008). Involvement of the second extracellular loop and transmembrane residues of CCR5 in inhibitor binding and HIV-1 fusion: insights into the mechanism of allosteric inhibition. J. Mol. Biol. 381, 956–974.
Malim, M. H., and Bieniasz, P. D. (2012). HIV restriction factors and mechanisms of evasion. Cold Spring Harb. Perspect. Med. 2, a006940.
Marti-Renom, M. A., Stuart, A. C., Fiser, A., Sanchez, R., Melo, F., and Sali, A. (2000). Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 29, 291–325.
Matsuyama, S., Aydan, A., Ode, H., Hata, M., Sugiura, W., and Hoshino, T. (2010). Structural and energetic analysis on the complexes of clinically isolated subtype C HIV-1 proteases and approved inhibitors by molecular dynamics simulation. J. Phys. Chem. B 114, 521–530.
McCammon, J. A., Gelin, B. R., and Karplus, M. (1977). Dynamics of folded proteins. Nature 267, 585–590.
Merchant, B. A., and Madura, J. D. (2011). A review of coarse-grained molecular dynamics techniques to access extended spatial and temporal scales in biomolecular simulations. Annu. Rep. Comput. Chem. 7, 67–87.
Miyamoto, T., Yokoyama, M., Kono, K., Shioda, T., Sato, H., and Nakayama, E. E. (2011). A single amino acid of human immunodeficiency virus type 2 capsid protein affects conformation of two external loops and viral sensitivity to TRIM5alpha. PLoS ONE 6:e22779. doi: 10.1371/journal.pone.0022779
Moustafa, I. M., Shen, H., Morton, B., Colina, C. M., and Cameron, C. E. (2011). Molecular dynamics simulations of viral RNA polymerases link conserved and correlated motions of functional elements to fidelity. J. Mol. Biol. 410, 159–181.
Myong, S., Bruno, M. M., Pyle, A. M., and Ha, T. (2007). Spring-loaded mechanism of DNA unwinding by hepatitis C virus NS3 helicase. Science 317, 513–516.
Naganawa, S., Yokoyama, M., Shiino, T., Suzuki, T., Ishigatsubo, Y., Ueda, A., Shirai, A., Takeno, M., Hayakawa, S., Sato, S., Tochikubo, O., Kiyoura, S., Sawada, K., Ikegami, T., Kanda, T., Kitamura, K., and Sato, H. (2008). Net positive charge of HIV-1 CRF01_AE V3 sequence regulates viral sensitivity to humoral immunity. PLoS ONE 3:e3206. doi: 10.1371/journal.pone.0003206
Neil, S., and Bieniasz, P. (2009). Human immunodeficiency virus, restriction factors, and interferon. J. Interferon Cytokine Res. 29, 569–580.
Nicholson, L. K., Yamazaki, T., Torchia, D. A., Grzesiek, S., Bax, A., Stahl, S. J., Kaufman, J. D., Wingfield, P. T., Lam, P. Y. S., Jadhav, P. K., Hodge, C. N., Domaille, P. J., and Chang, C.-H. (1995). Flexibility and function in HIV-1 protease. Nat. Struct. Biol. 2, 274–280.
Ode, H., Matsuyama, S., Hata, M., Hoshino, T., Kakizawa, J., and Sugiura, W. (2007a). Mechanism of drug resistance due to N88S in CRF01_AE HIV-1 protease, analyzed by molecular dynamics simulations. J. Med. Chem. 50, 1768–1777.
Ode, H., Matsuyama, S., Hata, M., Neya, S., Kakizawa, J., Sugiura, W., and Hoshino, T. (2007b). Computational characterization of structural role of the non-active site mutation M36I of human immunodeficiency virus type 1 protease. J. Mol. Biol. 370, 598–607.
Ode, H., Neya, S., Hata, M., Sugiura, W., and Hoshino, T. (2006). Computational simulations of HIV-1 proteases - multi-drug resistance due to nonactive site mutation L90M. J. Am. Chem. Soc. 128, 7887–7895.
Ode, H., Ota, M., Neya, S., Hata, M., Sugiura, W., and Hoshino, T. (2005). Resistant mechanism against nelfinavir of human immunodeficiency virus type 1 proteases. J. Phys. Chem. B 109, 565–574.
Ode, H., Yokoyama, M., Kanda, T., and Sato, H. (2011). Identification of folding preferences of cleavage junctions of HIV-1 precursor proteins for regulation of cleavability. J. Mol. Model. 17, 391–399.
Okimoto, N., Futatsugi, N., Fuji, H., Suenaga, A., Morimoto, G., Yanai, R., Ohno, Y., Narumi, T., and Taiji, M. (2009). High-performance drug discovery: computational screening by combining docking and molecular dynamics simulations. PLoS Comput. Biol. 5:e1000528. doi: 10.1371/journal.pcbi.1000528
Onyango, C. O., Leligdowicz, A., Yokoyama, M., Sato, H., Song, H., Nakayama, E. E., Shioda, T., de Silva, T., Townend, J., Jaye, A., Whittle, H., Rowland-Jones, S., and Cotten, M. (2010). HIV-2 capsids distinguish high and low virus load patients in a West African community cohort. Vaccine 28(Suppl. 2), B60–B67.
Ozen, A., Haliloglu, T., and Schiffer, C. A. (2011). Dynamics of preferential substrate recognition in HIV-1 protease: redefining the substrate envelope. J. Mol. Biol. 410, 726–744.
Pearlman, D. A., Case, D. A., Caldwell, J. W., Ross, W. S., Cheatham, T. E. I., DeBolt, S., Ferguson, D., Seibel, G., and Kollman, P. (1995). AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comp. Phys. Commun. 91, 1–41.
Perryman, A. L., Lin, J. H., and McCammon, J. A. (2004). HIV-1 protease molecular dynamics of a wild-type and of the V82F/I84V mutant: possible contributions to drug resistance and a potential new target site for drugs. Protein Sci. 13, 1108–1123.
Pettit, S. C., Henderson, G. J., Schiffer, C. A., and Swanstrom, R. (2002). Replacement of the P1 amino acid of human immunodeficiency virus type 1 Gag processing sites can inhibit or enhance the rate of cleavage by the viral protease. J. Virol. 76, 10226–10233.
Pettit, S. C., Moody, M. D., Wehbie, R. S., Kaplan, A. H., Nantermet, P. V., Klein, C. A., and Swanstrom, R. (1994). The p2 domain of human immunodeficiency virus type 1 Gag regulates sequential proteolytic processing and is required to produce fully infectious virions. J. Virol. 68, 8017–8027.
Piana, S., Carloni, P., and Rothlisberger, U. (2002). Drug resistance in HIV-1 protease: flexibility-assisted mechanism of compensatory mutations. Protein Sci. 11, 2393–2402.
Rantalainen, K. I., Eskelin, K., Tompa, P., and Makinen, K. (2011). Structural flexibility allows the functional diversity of potyvirus genome-linked protein VPg. J. Virol. 85, 2449–2457.
Rantalainen, K. I., Uversky, V. N., Permi, P., Kalkkinen, N., Dunker, A. K., and Makinen, K. (2008). Potato virus A genome-linked protein VPg is an intrinsically disordered molten globule-like protein with a hydrophobic core. Virology 377, 280–288.
Reboul, C. F., Meyer, G. R., Porebski, B. T., Borg, N. A., and Buckle, A. M. (2012). Epitope flexibility and dynamic footprint revealed by molecular dynamics of a pMHC-TCR complex. PLoS Comput. Biol. 8:e1002404. doi: 10.1371/journal.pcbi.1002404
Reingewertz, T. H., Shalev, D. E., and Friedler, A. (2010). Structural disorder in the HIV-1 Vif protein and interaction-dependent gain of structure. Protein Pept. Lett. 17, 988–998.
Rodriguez-Barrios, F., Balzarini, J., and Gago, F. (2005). The molecular basis of resilience to the effect of the Lys103Asn mutation in non-nucleoside HIV-1 reverse transcriptase inhibitors studied by targeted molecular dynamics simulations. J. Am. Chem. Soc. 127, 7570–7578.
Rodriguez-Barrios, F., and Gago, F. (2004). Understanding the basis of resistance in the irksome Lys103Asn HIV-1 reverse transcriptase mutant through targeted molecular dynamics simulations. J. Am. Chem. Soc. 126, 15386–15387.
Sadiq, S. K., Wan, S., and Coveney, P. V. (2007). Insights into a mutation-assisted lateral drug escape mechanism from the HIV-1 protease active site. Biochemistry 46, 14865–14877.
Sadiq, S. K., Wright, D. W., Kenway, O. A., and Coveney, P. V. (2010). Accurate ensemble molecular dynamics binding free energy ranking of multidrug-resistant HIV-1 proteases. J. Chem. Inf. Model. 50, 890–905.
Sanchez, R., Pieper, U., Melo, F., Eswar, N., Marti-Renom, M. A., Madhusudhan, M. S., Mirkovic, N., and Sali, A. (2000). Protein structure modeling for structural genomics. Nat. Struct. Biol. 7(Suppl.), 986–990.
Sandhu, K. S. (2009). Intrinsic disorder explains diverse nuclear roles of chromatin remodeling proteins. J. Mol. Recognit. 22, 1–8.
Sandhu, K. S., and Dash, D. (2007). Dynamic alpha-helices: conformations that do not conform. Proteins 68, 109–122.
Schames, J. R., Henchman, R. H., Siegel, J. S., Sotriffer, C. A., Ni, H., and McCammon, J. A. (2004). Discovery of a novel binding trench in HIV integrase. J. Med. Chem. 47, 1879–1881.
Shenderovich, M. D., Kagan, R. M., Heseltine, P. N., and Ramnarayan, K. (2003). Structure-based phenotyping predicts HIV-1 protease inhibitor resistance. Protein Sci. 12, 1706–1718.
Shojania, S., and O'Neil, J. D. (2010). Intrinsic disorder and function of the HIV-1 Tat protein. Protein Pept. Lett. 17, 999–1011.
Skasko, M., Wang, Y., Tian, Y., Tokarev, A., Munguia, J., Ruiz, A., Stephens, E. B., Opella, S. J., and Guatelli, J. (2012). HIV-1 Vpu antagonizes the innate restriction factor BST-2 via lipid-embedded helix-helix interactions. J. Biol. Chem. 287, 58–67.
Soares, R. O., Batista, P. R., Costa, M. G., Dardenne, L. E., Pascutti, P. G., and Soares, M. A. (2010). Understanding the HIV-1 protease nelfinavir resistance mutation D30N in subtypes B and C through molecular dynamics simulations. J. Mol. Graph. Model. 29, 137–147.
Song, H., Nakayama, E. E., Yokoyama, M., Sato, H., Levy, J. A., and Shioda, T. (2007). A single amino acid of the human immunodeficiency virus type 2 capsid affects its replication in the presence of cynomolgus monkey and human TRIM5alphas. J. Virol. 81, 7280–7285.
Steven, A. C., Heymann, J. B., Cheng, N., Trus, B. L., and Conway, J. F. (2005). Virus maturation: dynamics and mechanism of a stabilizing structural transition that leads to infectivity. Curr. Opin. Struct. Biol. 15, 227–236.
Stoica, I., Sadiq, S. K., and Coveney, P. V. (2008). Rapid and accurate prediction of binding free energies for saquinavir-bound HIV-1 proteases. J. Am. Chem. Soc. 130, 2639–2648.
Takada, S. (2012). Coarse-grained molecular simulations of large biomolecules. Curr. Opin. Struct. Biol. 22, 130–137.
Thorpe, I. F., and Brooks, C. L. 3rd. (2007). Molecular evolution of affinity and flexibility in the immune system. Proc. Natl. Acad. Sci. U.S.A. 104, 8821–8826.
Tobi, D., and Bahar, I. (2005). Structural changes involved in protein binding correlate with intrinsic motions of proteins in the unbound state. Proc. Natl. Acad. Sci. U.S.A. 102, 18908–18913.
Unge, T., Montelius, I., Liljas, L., and Ofverstedt, L. G. (1986). The EDTA-treated expanded satellite tobacco necrosis virus: biochemical properties and crystallization. Virology 152, 207–218.
van der Vries, E., Schutten, M., and Boucher, C. A. (2011). The potential for multidrug-resistant influenza. Curr. Opin. Infect. Dis. 24, 599–604.
Wang, J., Ma, C., Fiorin, G., Carnevale, V., Wang, T., Hu, F., Lamb, R. A., Pinto, L. H., Hong, M., Klein, M. L., and DeGrado, W. F. (2011). Molecular dynamics simulation directed rational design of inhibitors targeting drug-resistant mutants of influenza A virus M2. J. Am. Chem. Soc. 133, 12834–12841.
White, K. L., Chen, J. M., Margot, N. A., Wrin, T., Petropoulos, C. J., Naeger, L. K., Swaminathan, S., and Miller, M. D. (2004). Molecular mechanisms of tenofovir resistance conferred by human immunodeficiency virus type 1 reverse transcriptase containing a diserine insertion after residue 69 and multiple thymidine analog-associated mutations. Antimicrob. Agents Chemother. 48, 992–1003.
Wittayanarakul, K., Aruksakunwong, O., Saen-oon, S., Chantratita, W., Parasuk, V., Sompornpisut, P., and Hannongbua, S. (2005). Insights into saquinavir resistance in the G48V HIV-1 protease: quantum calculations and molecular dynamic simulations. Biophys. J. 88, 867–879.
Wright, D. W., and Coveney, P. V. (2011). Resolution of discordant HIV-1 protease resistance rankings using molecular dynamics simulations. J. Chem. Inf. Model. 51, 2636–2649.
Xu, Y., Colletier, J. P., Jiang, H., Silman, I., Sussman, J. L., and Weik, M. (2008). Induced-fit or preexisting equilibrium dynamics? lessons from protein crystallography and MD simulations on acetylcholinesterase and implications for structure-based drug design. Protein Sci. 17, 601–605.
Xue, B., Mizianty, M. J., Kurgan, L., and Uversky, V. N. (2012). Protein intrinsic disorder as a flexible armor and a weapon of HIV-1. Cell. Mol. Life Sci. 69, 1211–1259.
Yokoyama, M., Naganawa, S., Yoshimura, K., Matsushita, S., and Sato, H. (2012). Structural dynamics of HIV-1 envelope gp120 outer domain with V3 loop. PLoS ONE 7:e37530. doi: 10.1371/journal.pone.0037530
Yoshimoto, K., Arora, K., and Brooks, C. L. 3rd. (2010). Hexameric helicase deconstructed: interplay of conformational changes and substrate coupling. Biophys. J. 98, 1449–1457.
Zhou, J., Zhang, Z., Mi, Z., Wang, X., Zhang, Q., Li, X., Liang, C., and Cen, S. (2012). Characterization of the interface of the bone marrow stromal cell antigen 2-Vpu protein complex via computational chemistry. Biochemistry 51, 1288–1296.
Keywords: MD simulation, viral protein, three-dimensional structure, protein dynamics, coarse-grained MD
Citation: Ode H, Nakashima M, Kitamura S, Sugiura W and Sato H (2012) Molecular dynamics simulation in virus research. Front. Microbio. 3:258. doi: 10.3389/fmicb.2012.00258
Received: 08 June 2012; Paper pending published: 20 June 2012;
Accepted: 02 July 2012; Published online: 19 July 2012.
Edited by:
Masaru Yokoyama, National Institute of Infectious Diseases, JapanReviewed by:
Hiroyuki Toh, National Institute of Advanced Industrial Science and Technology, JapanYasuyuki Miyazaki, The University of Tokushima Graduate School, Japan
Copyright © 2012 Ode, Nakashima, Kitamura, Sugiura and Sato. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Hirotaka Ode, Department of Infectious Diseases and Immunology, Clinical Research Center, National Hospital Organization Nagoya Medical Center, 4-1-1 Sannomaru, Naka-ku, Nagoya, Aichi, 460-0001, Japan. e-mail: odehir@nih.go.jp