Current Trends in Circulating Biomarkers for Melanoma Detection
 Nancy Huang
Nancy Huang  Katie J. Lee
Katie J. Lee Mitchell S. Stark
Mitchell S. Stark- The University of Queensland Diamantina Institute, The University of Queensland, Dermatology Research Centre, Brisbane, QLD, Australia
Melanomas have increased in global incidence and are the leading cause of skin cancer deaths. Whilst the majority of early-stage, non-metastatic melanomas can be cured with surgical excision alone, ~5% of patients with early melanomas will experience recurrence following a variable disease-free interval and progression to metastatic melanoma and ultimately death. This is likely because of primary tumor heterogeneity and progressive clonal divergency resulting in the growth of more aggressive tumor populations. Liquid biomarkers have the advantage of real-time, non-invasive longitudinal monitoring of tumor burden and heterogeneity over tissue markers. Currently, the only serological marker used in the staging and monitoring of melanoma is serum lactate dehydrogenase, which is not sufficiently specific or sensitive, and is not used routinely in all centers. An ideal melanoma biomarker would be used to identify patients who are at high-risk of primary melanoma, screen for relapse, detect early-stage melanoma, provide treatment outcomes to personalize systemic treatment, follow tumor heterogeneity, provide prognostic data before, during and after treatment, and monitor response to treatment. This review provides a summary of the current research in this field with a specific focus on circulating tumor cells, circulating tumor DNA, microRNA, and extracellular vesicles which may serve to suit these goals.
Introduction
Melanoma is an aggressive skin cancer with an increased global incidence over recent decades. Melanomas may be diagnosed following a clinical history and skin examination, sometimes with the assistance of total body photography or sequential digital dermatoscopic imaging in higher-risk patients (i.e., previous melanoma, family history, and phenotypic characteristics such as fair skin, high total body nevus count, etc.). The primary tumor is excised for dermatopathological examination, the gold standard for diagnosis and staging (1). If high-risk features are detected (e.g. >1 mm thickness), a sentinel lymph node (SLN) biopsy may be performed to assess for metastatic disease (2, 3).
Staging of melanoma is based on a combination of histological characteristics such as tumor thickness, ulceration, and mitotic rate, and metastasis to lymph nodes and distal sites (4). The American Joint Committee on Cancer TNM staging system is used to determine clinical management and as a guide for prognosis. Overall clinical stage is still a strong determinant of 5-year survival, with high mortality in advanced stages (5). Most melanomas are diagnosed in the early stage (stages I), which can often be completely cured by surgical excision, and thus have 97–99% 5-year survival (6, 7). Unfortunately, some early-stage melanomas recur following excision, and may develop metastases; thus, even melanomas within the same stage can vary in terms of progression and patient survival, likely due to tumor heterogeneity not detected on histopathology (8). SLN-positive individuals have increased treatment options including surgery, advanced therapeutics, and radiological imaging such as CT and PET scans to determine the extent of distal metastatic spread. Regular imaging is common to monitor tumor volume, location, and effectiveness of systemic therapies. This imaging is expensive and not available in all hospital centers.
In Australia, there is a disparity in melanoma incidence and survival between patients living in capital cities and those living in regional and rural areas, with higher incidence and mortality rates in the latter (6, 9). This may be due to a number of factors, such as lower socio-economic status and health literacy, lack of shade in public areas and increased occupational exposure (10). People living in remote and regional centers also have limited access to diagnostic and treatment services, resulting in reduced early detection of thin, easily treatable melanomas.
Systemic therapies for the management of metastatic melanoma (stages III and IV) include targeted therapies such as BRAF and MEK inhibitors, and immunotherapy using check-point inhibitors, including anti-CTLA-4 and anti-PD-1/PD-L1 (11, 12). Response rates to these treatments vary from 22 to 40%, with ~10–15% of patients exhibiting atypical response patterns such as pseudo-progression (12). Patients treated with targeted therapies must have melanomas with BRAF V600 mutations, currently determined using tumor biopsies (13).
Consequently, there is a need to identify biomarkers that can detect early-stage melanoma and stratify patients into melanoma recurrence risk groups. This will allow resource prioritization for screening high-risk groups which is particularly important in rural and regional areas, and allow intervention during early-stage disease, shown to be the biggest factor in improving overall survival (OS) (14).
Circulating Melanoma Biomarkers
A biomarker is a biological molecule that may be detected in bodily fluids such as blood, urine, saliva, or in tissues, and in comparison with a healthy individual or tissue, an increase/decrease of this biomarker is a sign of an abnormal process (15). Circulating biomarkers include tumor cells, nucleic acids (DNA, RNA, microRNA, etc.), extracellular vesicles (EVs), and metabolites, often called a liquid biopsy or a liquid biomarker. Liquid biopsies can have substantial benefits over tissue-based biomarkers as collection is minimally invasive, may allow for longitudinal tracking of a patient's tumor burden, detection of recurrence, and profiling tumor heterogeneity and clonal divergence (16). In addition, liquid biomarkers may be particularly useful when the primary melanoma lesion is limited (e.g. previously excised melanomas) or where tissue sampling may be difficult (13).
Multiple serological protein markers have been explored, with lactate dehydrogenase (LDH) currently the only marker with some clinical value for monitoring treatment response and prognosis; however, its sensitivity and specificity are lacking (3, 4, 17–19). Other markers such as S100B, MIA protein, CRO, PD-L1, IL-8, TIL, osteopontin, and YKL-40 have been explored; however, their clinical utility is limited as many of these markers are associated with other biological processes such as inflammation, infection, autoimmune conditions or other cancers (17, 19). Aside from LDH and S100B, there are no additional clinically validated liquid biomarkers that can be used to detect early-stage melanoma, pre-treatment prognosis, expected treatment outcomes, or monitor treatment outcomes in metastatic melanoma (12, 18).
An ideal biomarker would be sensitive and specific for melanoma to screen high-risk populations and monitor for relapse in early-stage, asymptomatic melanoma patients, and be used to personalize treatments in advanced-stage melanoma (e.g. selecting optimal treatment, identifying when to add adjuvant therapies) to reduce healthcare costs and adverse effects such as toxicity (12, 16). Other favorable qualities would be a test that is low cost, has high throughput capacity, and is readily accessible in rural and regional settings.
Cellular Profiling
Whole blood analysis is standard practice for melanoma patients to measure blood cell counts. This review will not discuss these analyses, and instead focus on circulating tumor cells.
Circulating Tumor Cells (CTC)
CTCs are shed from solid tumors into the circulation, and are considered a vital step in the establishment of distant metastases (16, 20). They have a short half-life due to physical and oxidative stress, anoikis, absence of growth factors and cytokines, and cell loss from implantation into capillary beds (16, 21). Detection of CTCs remains challenging despite advances in cell isolation, enrichment, and analysis methods, with detection rates in advanced melanoma patients ranging from 28 to 87% (16, 22). This is due to several factors, including low concentrations in circulation, marker heterogeneity, and lack of standardized method for isolation (20, 22–26).
Patients with early-stage melanoma commonly have only single positive melanoma markers on CTCs, whilst patients with advanced stages not only had higher CTC counts, but also a greater variety of markers (25). Reverse transcription polymerase chain reaction (RT-PCR) is the most commonly used method for detecting CTC markers (sensitivity 1–10 CTCs/mL whole blood, specificity 99.9%); most detection techniques rely on one to two known markers (25, 27–29). Aya-Bonilla et al. compared isolation and detection methods, finding individual detection rates of 28% for multi-marker immunostaining, 42% for a 5-gene panel via RT-PCR, and 53% for a 19-gene droplet digital PCR (ddPCR) assay; however, by combining all three methods, the detection rate was bolstered to 72% (22). Commonly-used CTC markers include tyrosinase, Melan-A/MART-1, MAGE3, MCAM, gp-100, MITF, and GalNac-T, which have specificities ranging from 85 to 100% but sensitivities from 6 to 95% (Table 1) (24, 27). Other isolation methods include microchips, antibody-coated immunomagnetic beads, and combining RT-PCR with selective fluorescent imaging (26). Therefore, it may be worthwhile adopting a multi-marker approach to improve clinical utility of any one biomarker.

Table 1. CTC markers of melanoma.
CTCs have also been detected in early-stage melanoma including melanoma in-situ (MIS) (29), with Voit et al. reporting a positive association between detectable CTC counts and progression-free survival (PFS) in stage I-III patients (33). Lucci et al. noted that the detection of one or more melanoma CTCs (per 7.5 mL peripheral blood) in stage III patients was significantly associated with disease relapse within 6 months (24). Fluctuations in CTC counts have been associated with treatment response, disease progression, and OS (16, 29).
New technological advances to enrich and isolate CTCs, and standardization in isolation techniques will likely improve the clinical utility of CTCs to define heterogeneity and occult invasiveness, assess tumor burden, direct treatment decisions, and monitor response to treatment (21, 29).
Molecular Profiling
Molecular profiling aims to detect changes in signaling and presence of abnormal genetic material that contribute to the development and progression of melanoma. There are numerous other circulation molecules under investigation (including long non-coding RNA), however, this review will focus on circulating tumor DNA, microRNA, and EVs.
Circulating Tumor DNA
Circulating tumor DNA (ctDNA) are fragments of single or double-stranded DNA released at low concentrations by tumor cells into the circulation, likely by cell apoptosis and necrosis, although the precise mechanism is uncertain (3, 16–18, 34). ctDNA can be measured in plasma or serum, but it has a short half-life as a result of rapid clearance though the kidneys, liver and spleen (16, 34). This timeframe can be extended if with specific collection tubes which preserve the ctDNA at room temperature up to 7 days (35). ctDNA levels are associated with tumor burden, location, and vascularity, and the rate of cellular turnover (16–18). As patients with early-stage or MIS have low tumor burden and vascularity, ctDNA levels are often not detectable in these groups using conventional technology (3, 16, 17). There are also technical issues in detecting and analyzing ctDNA. Firstly, ctDNA only constitutes a small fraction of the total background cell-free DNA (cfDNA), which itself is only present in small quantities in the circulation (16, 34). Ongoing advances in technology, such as the use of ddPCR, are sensitive at detecting low-level ctDNA, and are readily reproducible (36). The proportion of cfDNA also increases with leukocyte lysis, which can occur during blood collection and clotting, reducing the ability to detect ctDNA (16, 37). Consequently, plasma is the preferred source of ctDNA (16). There is however a lack of consistency in blood collection processes, including types of tubes used, time to plasma separation and processing, and storage and transport conditions (16, 37). These differences make analysis and comparison across different study groups troublesome; thus, cut-off values have not been established, reducing the clinical utility of ctDNA (3, 17). Finally, to distinguish ctDNA from cfDNA, specific somatic mutations in the melanoma tumor must first be identified (3). Tumor heterogeneity thereby continues to hinder detection rates, as ~25% of melanoma patients do not have identifiable tumor-derived ctDNA mutations present in the circulation (3, 36).
Despite the challenges, monitoring ctDNA levels in metastatic melanoma patients is non-invasive, with potential clinical utility demonstrated in determining pre-treatment prognosis, monitoring of treatment response, assessment of clonal evolution and minimal residual disease (MRD), and tracking disease progression (16–18, 34). Monitoring MRD is particularly important to confirm complete responders to systemic therapies due to detection limits of radiological imaging (38).
Low or undetectable pre-treatment levels of ctDNA in patients with metastatic melanoma was associated with lower tumor burden and increased PFS and OS, independent of LDH and tumor stage (16–18, 36). However, if detectable levels of ctDNA following surgical excision was detected, this was associated with a poor PFS and OS (34). In patients who had detectable baseline ctDNA, there was a correlation with tumor burden and LDH, suggesting that ctDNA may reveal tumor burden in the absence of imaging (13, 18, 36). In addition, elevated baseline ctDNA levels correlate with reduced overall response rate, and shortened PFS and OS in melanoma patients treated with targeted or immune therapies (16, 17, 34, 39).
There are however limitations to using ctDNA as a predictive biomarker for patients undertaking second-line treatment immunotherapy after failed initial targeted treatment, as there was no association between low ctDNA levels and PFS (39). Additionally, in patients with brain metastases, the levels of ctDNA may be artificially low as there is less ctDNA released into the circulation (40); accurate detection relies on more invasive collection of cerebral spinal fluid (41).
MicroRNA
MicroRNA (miRNA) are short, non-coding RNA fragments which regulate gene transcription and expression, thereby affecting cell differentiation, proliferation, migration, and apoptosis (3, 16, 42, 43). They are secreted into the circulation, usually in lipid-bound vesicles or in a complex bound by protective proteins and are highly stable even under harsh conditions such as extremes of pH and temperature, multiple freeze-thaw cycles, and long-term storage (3, 14, 16).
Detection of miRNA relies on sequence-matching miRNA molecules (probes, oligos, etc.), and selective and sensitive amplification, but standard techniques such as real-time quantitative RT-PCR can be used. Often low concentrations of miRNA in blood and different expression profiles between serum and plasma samples, and across individuals, can make detection challenging (3, 16), and differences in sample collection and preparation, RNA extraction, normalization methods and storage, limit comparisons between studies (3). Many miRNAs have however been linked to melanoma and metastasis (3).
For example, miR-221 has a role in maintaining cell cycle regulation and proliferation, and so its dysregulation promotes melanoma progression (3, 44, 45). Serum levels of miR-221 were generally undetectable in controls and patients with MIS; however, they were significantly elevated in most patients with stage I melanoma, and associated with increasing clinical stage and tumor thickness (46). Other miRNA regulating cell cycle with altered expression in melanoma include miR-9, miR-150, miR-155, miR-205, and miR-222 (3, 45).
miR-199a-5p promotes melanoma metastasis and angiogenesis, and has been shown to be up-regulated 10-fold in patients with stage III melanoma compared to stage Ia disease (47). Even patients with stage Ib to IIb disease had a 6-fold increase in expression compared to healthy and stage Ia groups (47, 48). Up-regulation of miRNA-199a-5p in conjunction with up-regulated miR-877-3p, 1228-3p, 3613-5p and down-regulated miR-182-5p was associated with higher melanoma stage (stage III or higher) at the time of primary melanoma excision (47). Patients who did not have down-regulated miRNA-182-5p were usually within stages Ib-IIb (47). Additionally, dysregulation of miR-199a-5p, miR-150, miR-15b, miR-33a, and miR-424 increased the risk of melanoma recurrence, with increasing levels associated with tumor burden (16).
These studies are a few examples of dysregulated circulating miRNAs associated with differing melanoma stages relative to healthy controls. When used in isolation, individual miRNAs may not yet be able to be used clinically due to inconsistent expression across patients. The use of a collection or panel of miRNA may result in a more sensitive and specific biomarker (3). Margue et al. (49) showed that miR-301-3p, miR-200c-3p, miR-126-5p, miR-374a-5p, and miR-211-5p, when used in a combination, could differentiate patients with stage I and II melanoma from healthy controls. van Laar et al. also used a combination of 38 miRNAs (MEL38), to differentiate healthy controls from patients who had stage I-IV melanoma (50). We too have identified a panel of seven “melanoma-related” miRNA biomarkers that have prognostic value as a liquid biopsy measured in serum (51). In treatment naive stage IV patients, members of this MELmiR-7 panel were able to detect an increase in tumor burden in 100% of cases and the ability of the panel to define OS proved to be superior to LDH and S100B levels (delta log-likelihood = 11, p < 0.001) (51). Levels of circulating miRNA may fluctuate with systemic treatments used to treat late-stage melanoma patients so the use of miRNAs in this context requires further validation.
The use of miRNA as a melanoma biomarker is promising, and whilst several studies focused on an individual miRNA, further research into groups of miRNAs may provide a more sensitive and specific tool to differentiate early-stage melanoma from healthy controls and provide better pre-treatment prognosis.
Extracellular Vesicles
Extracellular vesicles are membrane-bound particles secreted by all cells, present in all body fluids, and transport cargo (DNA, RNA, miRNA, proteins, lipids, and metabolites) that are representative of the original cell (7, 16, 52, 53). The EVs secreted by tumor cells can deliver tumor-specific molecules to other body sites that may assist tumor survival and proliferation (16). Exosomes, which are nano-sized subset of EVs, released by melanoma cells may also stimulate the migration of endothelial cells, induce angiogenesis to promote distal metastasis, contribute to drug resistance, and may have immunosuppressive actions (54, 55).
Exosomes can be isolated based on physical characteristics such as size, morphology, concentration, cell surface markers or cargo (16, 52, 56, 57). Detection of tumor-derived exosomes with current technologies is difficult, due to their size, low quantities of cargo within each exosome, heterogeneity, and abundance of non-target EVs (7).
In melanoma patients relative to healthy controls, 4–20-fold higher levels of plasma tumor-derived exosomes were detected which persisted despite surgical resection of the primary lesion with no clinical evidence of residual disease (58–60). Exosomes are known to be involved in establishing pre-metastatic niches in cancers including melanoma (61). This persistence of the exosomes, post excision of primary lesions, may be a reason why in some patients, recurrence of metastatic disease occurs many years after initial primary diagnosis. Recently, Wang et al. compared the expression of EV surface markers MCSP, MCAM, CD61, and CD63 in healthy controls and patients who had a history of early-stage melanoma (including MIS) (7). Surface enhanced raman spectroscopy (SERS; an ultra-sensitive detection method) signatures and mapping images showed a distinct separation between the two groups, with higher levels of SERS signals observed in the melanoma patients (7).
Discussion
The identification and validation of liquid biomarkers has seen significant advancement over the past two decades. The biomarkers discussed in this review all hold great promise to improve early melanoma detection and disease relapse, as well as a prognostic marker at various stages of disease and treatment, and predictive markers to identify patients who would benefit most from systemic therapies, to personalize treatment options (Table 2) (12, 16, 34).
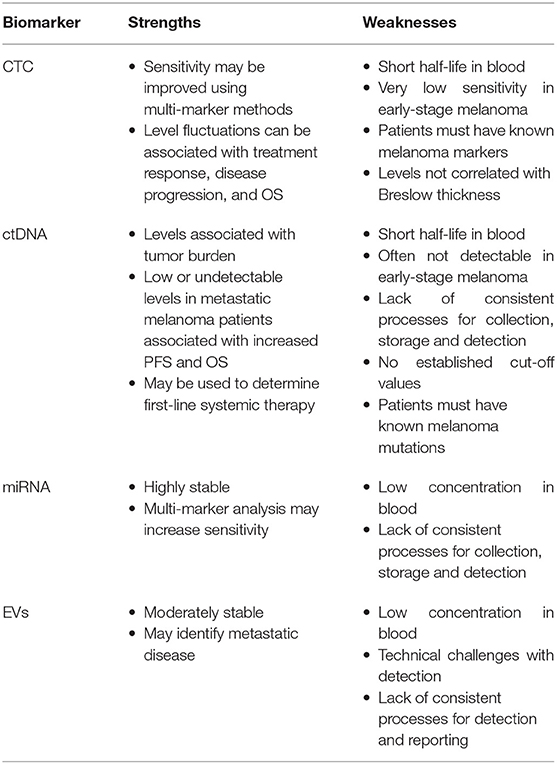
Table 2. Strengths and weaknesses of reviewed melanoma biomarkers.
CTCs are highly specific melanoma biomarkers as they rely on the detection of known melanoma markers, however this can come as a disadvantage for patients with tumor populations that do not display these markers (25). Whilst some detection methods have reported positive results in patients with early-stage melanoma, overall, the sensitivity remains quite low, likely as a result of more homogenous tumor populations and low circulating concentrations of CTCs in these patients (16, 29, 33). CTCs fluctuate with tumor burden in metastatic melanoma and can thus be used to monitor treatment outcomes and disease progression (16, 29). In metastatic melanoma, CTC counts are also associated with risk of disease recurrence and PFS/OS, and thus may assist in identifying high-risk patients who require more intensive screening and provide prognostic information (16, 24, 29).
ctDNA may have less clinical utility as an early-stage melanoma biomarker, especially in rural and regional settings. This is due to their low sensitivities in early-stage disease, their instability, need for specialized sample tubes, and highly specific processing requirements and storage conditions to prevent sample degradation (3, 16, 17, 37). On the other hand, ctDNA levels can be used to independently predict pre-treatment PFS and OS, personalize treatment options based on expected outcomes, and monitor response to treatment and disease progression (11, 13, 16–18, 34, 36, 39). Detection methods for ctDNA, such as ddPCR, are cost-effective, and next-generation sequencing can allow for high throughput testing (16).
miRNA may be a promising future melanoma biomarker due to its stability, monitoring of clonal divergence, and ability to differentiate healthy controls from patients with early-stage melanoma (3, 14, 16, 46, 51). Certain groups of miRNA markers were able to provide information on risk of recurrence, detect early metastasis, and prognosis (3, 16, 43, 51). RT-PCR is the predominant method used for miRNA detection, which is cost-effective and widely-available, however discrepancies in sequence matching may reduce the sensitivity (3, 16).
Whilst the technical challenges associated with detection of EVs and poorly standardized reporting may limit its use as a melanoma biomarker, steps are being taken to address these issues, and some technologies for detection may allow for high volume testing (7, 16). Multiple EVs are able to differentiate healthy controls from patients with advanced-stage melanoma, and may provide prognostic information (16, 55, 57). The findings from current studies suggest that EVs will be a promising melanoma marker.
Conclusion
Non-invasive methods such as total body photography for high-risk patients and CT and PET scans for late-stage patients and those suspected of lymph metatasis with enlarged nodes, are available to screen patients, however there are usually cost and significant access barriers present, especially in rural and regional areas (43). On the other hand, obtaining a blood sample to assess for liquid biomarkers is easier, more accessible, and may be more cost-effective and allow for high throughput testing (16, 43). At present, there are currently no clinically-validated liquid biomarkers to distinguish these patients, and thus a melanoma biomarker that is AJCC stage-specific is urgently required.
Author Contributions
NH and MS conceived the paper and wrote the first draft of the manuscript. NH, KL, and MS contributed to manuscript revision, read, and approved the final submission.
Funding
MS is supported by the Merchant Charitable Foundation. KL is supported by an NHMRC Dora Lush Basic Research Scholarship. This research was conducted with the support of the NHMRC Centre of Research Excellence in Skin Imaging and Precision Diagnosis (APP2006551) and the ACRF Australian Centre of Excellence in Melanoma Imaging & Diagnosis.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
This work was undertaken at the Translational Research Institute.
Abbreviations
AJCC, American Joint Committee on Cancer; cfDNA, cell-free DNA; CT, computerized tomography; CTC, circulating tumor cells; ctDNA, circulating tumor DNA; ddPCR, digital droplet polymerase chain reaction; EV, extracellular vesicle; LDH, lactate dehydrogenase; miRNA, microRNA; MIS, melanoma in-situ; MRD, minimal residual disease; OS, overall survival; PFS, progression-free survival; RT-PCR, reverse transcriptase polymerase chain reaction; PET, positron emission tomography; SERS, surface enhanced raman spectroscopy; SLN, sentinel lymph node; TNM, tumor (T), node (N), and metastasis (M).
References
1. Shoo BA, Sagebiel RW, Kashani-Sabet M. Discordance in the histopathologic diagnosis of melanoma at a melanoma referral center. J Am Acad Dermatol. (2009) 62:751–6. doi: 10.1016/j.jaad.2009.09.043
2. Gonzalez A. Sentinel lymph node biopsy: past and present implications for the management of cutaneous melanoma with nodal metastasis. Am J Clin Dermatol. (2018) 19:24–30. doi: 10.1007/s40257-018-0379-0
3. Mumford SL, Towler BP, Pashler AL, Gilleard O, Martin Y, Newbury SF. Circulating microRNA biomarkers in melanoma: Tools and challenges in personalised medicine. Biomolecules. (2018) 8:1–25. doi: 10.3390/biom8020021
4. Gershenwald JE, Scolyer RA, Hess KR, Sondak VK, et al. Melanoma staging: evidence-based changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J Clin. (2017) 67:472–92. doi: 10.3322/caac.21409
5. Balch CM, Buzaid AC, Soong S, Atkins MB, Cascinelli N, Coit DG, et al. Final version of the American Joint Committee on cancer staging system for cutaneous melanoma. J Clin Oncol. (2001) 27:6199–206. doi: 10.1200/JCO.2001.19.16.3635
6. Australian Institute of Health Welfare. Cancer Data in Australia. Canberra, ACT: Australian Government(2021). Available online at: https://www.aihw.gov.au/reports/cancer/cancer-data-in-australia/contents/cancer-summary-data-visualisation (accessed June 8, 2021).
7. Wang J, Kao Y, Zhou Q, Wuethrich A, Stark MS, Schaider H, et al. An integrated microfluidic-SERS platform enables sensitive phenotyping of serum extracellular vesicles in early stage melanomas. Adv Funct Mater. (2021) 32:2010296. doi: 10.1002/adfm.202010296
8. Cheng Y, Lu J, Chen G, Ardekani GS, Rotte A, Martinka M, et al. Stage-specific prognostic biomarkers in melanoma. Oncotarget. (2015) 6:4180–9. doi: 10.18632/oncotarget.2907
9. Australian Institute of Health Welfare. Cancer Rates Higher in Rural and Remote Areas. Canberra, ACT: Australian Government (2007). Available online at: https://www.aihw.gov.au/news-media/media-releases/2007/jun/cancer-rates-higher-in-rural-and-remote-areas (accessed June 28, 2007).
10. National Rural Health Alliance Inc. Submission to House of Representatives Standing Committee on Health. Skin Cancer in Australia: Awareness, Early Diagnosis and Management. Deakin West, ACT: National Rural Health Alliance Inc. (2014).
11. Gandini S, Zanna I, Pietro De Angelis S, Cocorocchio E, Queirolo P, Lee JH, et al. Circulating tumour DNA and melanoma survival: A systematic literature review and meta-analysis. Critic Rev Oncol Haematol. (2020) 157:103187. doi: 10.1016/j.critrevonc.2020.103187
12. Rozeman EA, Dekker TJA, Haanen JBAG, Blank CU. Advanced melanoma: current treatment options, biomarkers, and future perspectives. Am J Clin Dermatol. (2018) 19:303–17. doi: 10.1007/s40257-017-0325-6
13. Santiago-Walker A, Gagnon R, Mazumdar J, Casey M, Long GV, Schadendorf D, et al. Correlation of BRAF mutation status in circulating-free DNA and tumor and association with clinical outcomes across four BRAFi and MEKi clinical trials. Clin Cancer Res. (2015) 22:567–74. doi: 10.1158/1078-0432.CCR-15-0321
14. Carpi S, Polini B, Fogli S, Podestà A, Ylösmäki E, Cerullo V, et al. Circulating microRNAs as biomarkers for early diagnosis of cutaneous melanoma. Expert Rev Mol Diagn. (2020) 20:19–30. doi: 10.1080/14737159.2020.1696194
15. National Cancer Institute. Dictionary of Cancer Terms. Bethesda, MD: National Institutes of Health. Available online at: https://www.cancer.gov/publications/dictionaries/cancer-terms/def/biomarker (accessed February 14, 2022).
16. Lim SY, Lee JH, Diefenback RJ, Kefford RF, Rizos H. Liquid biomarkers in melanoma: detection and discovery. Mol Cancer. (2018) 17:1–14. doi: 10.1186/s12943-018-0757-5
17. Feng SN, Cen XT, Tan R, Wei SS, Sun LD. The prognostic value of circulating tumor DNA in patients with melanoma: a systematic review and meta-analysis. Transl Oncol. (2021) 14:101072. doi: 10.1016/j.tranon.2021.101072
18. Syeda MM, WIggins JMC CB, Long GV, Flaherty KT, Schadendorf D, et al. Circulating tumour DNA in patients with advanced melanoma treated with dabrafenib or dabrafenib plus trametinib: a clinical validation study. Lancet Oncol. (2021) 22:370–80. doi: 10.1016/S1470-2045(20)30726-9
19. Garbe C, Leiter U, Ellwanger U, Blaheta H, Meier F, Rassner G, et al. Diagnostic value and prognostic significance of protein S-100β, melanoma-inhibitory activity, and tyrosinase/MART-1 reverse transcription-polymerase chain reaction in the follow-up of high-risk melanoma patients. Cancer. (2003) 97:1737–45. doi: 10.1002/cncr.11250
20. Khoja L, Lorigan P, Dive C, Keiholz U, Fusi A. Circulating tumour cells as tumour biomarkers in melanoma: Detection methods and clinical relevance. Annals of Oncology. (2015) 26:33–9. doi: 10.1093/annonc/mdu207
21. Micalizzi DS, Maheswaran S, Haber DA. A conduit to metastasis: circulating tumor cell biology. Genes Dev. (2017) 31:1827–40. doi: 10.1101/gad.305805.117
22. Aya-Bonilla CA, Morici M, Hong X, McEvoy AC, Sullivan RJ, Freeman J, et al. Detection and prognostic role of heterogeneous populations of melanoma circulating tumour cells. Br J Cancer. (2020) 122:1059–67. doi: 10.1038/s41416-020-0750-9
23. Lin SY, Chang S, Lam S, Ramos RI, Tran K, Ohe S, et al. Prospective molecular profiling of circulating tumor cells from patients with melanoma receiving combinatorial immunotherapy. Clin Chem. (2020) 66:169–77. doi: 10.1373/clinchem.2019.307140
24. Lucci A, Hall CS, Patel SP, Narendran B, Bauldry JB, Royal RE, et al. Circulating tumor cells and early relapse in node-positive melanoma. Clin Cancer Res. (2020) 26:1886–95. doi: 10.1158/1078-0432.CCR-19-2670
25. Marsavela G, Aya-Bonilla CA, Warkiani ME, Gray ES, Ziman M. Melanoma circulating tumor cells: benefits and challenges required for clinical application. Cancer Lett. (2018) 424:1–8. doi: 10.1016/j.canlet.2018.03.013
26. De Souza LM, Robertson BM, Robertson GP. Future of circulating tumor cells in the melanoma clinical and research laboratory settings. Cancer Lett. (2017) 392:60–70. doi: 10.1016/j.canlet.2017.01.023
27. Rodic S, Mihalcioiu C, Saleh RR. Detection methods of circulating tumor cells in cutaneous melanoma: A systematic review. Critic Rev Oncol Haematol. (2014) 91:74–92. doi: 10.1016/j.critrevonc.2014.01.007
28. Brownbridge GG, Gold J, Edward M, Mackie RM. Evaluation of the use of tyrosinase- specific and MelanA/MART-1-specific reverse transcriptase coupled-polymerase chain reaction to detect melanoma cells in peripheral blood samples from 299 patients with malignant melanoma. Br J Dermatol. (2000) 144:279–87. doi: 10.1046/j.1365-2133.2001.04015.x
29. Kiniwa Y, Nakamura K, Mikoshiba A, Ashida A, Akiyama Y, Moriomoto A, et al. Usefulness of monitoring circulating tumor cells as a therapeutic biomarker in melanoma with BRAF mutation. BMC Cancer. (2021) 21:1–8. doi: 10.1186/s12885-021-08016-y
30. Proebstle TM, Jiang W, Högel J, Keilholz U, Weber L, Voit C. Correlation of positive RT-PCR for tyrosinase in peripheral blood of malignant melanoma patients with clinical stage, survival and other risk factors. Br J Cancer. (2000) 82:118–23. doi: 10.1054/bjoc.1998.0887
31. Schmidt H, Sorensen BS, Sjogren P, Christensen IJ, Fode K, Larsen J, et al. Circulating tyrosinase and MART-1 mRNA does not independently predict relapse or survival in patients with AJCC stage I–II melanoma. J Investig Dermatol. (2006) 126:849–54. doi: 10.1038/sj.jid.5700139
32. Reid A, Millward M, Pearce R, Lee M, Frank MH, Ireland A, et al. Markers of circulating tumour cells in the peripheral blood of patients with melanoma correlate with disease recurrence and progression. Br J Dermatol. (2013) 168:85–92. doi: 10.1111/bjd.12057
33. Voit C, Kron M, Rademaker J, Schwürzer-Voit M, Sterry W, Weber L. Molecular staging in stage II and III melanoma patients and its effect on long-term survival. J Clin Oncol. (2005) 23:1218–27. doi: 10.1200/JCO.2005.04.098
34. Weinstein D, Leininger J, Hamby C, Safai B. Diagnostic and prognostic biomarkers in melanoma. J Clin Aesthet Dermatol. (2014) 7:13–24. Available online at: https://pubmed.ncbi.nlm.nih.gov/25013535/
35. Parackal S, Zou D, Day R, Black M, Guilford P. Comparison of Roche Cell-Free DNA collection Tubes ((R)) to Streck Cell-Free DNA BCT ((R)) s for sample stability using healthy volunteers. Pract Lab Med. (2019) 16:e00125. doi: 10.1016/j.plabm.2019.e00125
36. Lee JH, Long GV, Boyd S, Lo S, Menzies AM, Tembe V, et al. Circulating tumour DNA predicts response to anti-PD1 antibodis in metastatic melanoma. Ann Oncol. (2017) 28:1130–6. doi: 10.1093/annonc/mdx026
37. Risberg B, Tsui DWY, Biggs H, Ruiz-Valdepenas Martin de. Almagro A, Dawson S, Hodgkin C, et al. Effects of collection and processing procedures on plasma circulating cell-free DNA from cancer patients. J Mol Diagn. (2018) 20:883–92. doi: 10.1016/j.jmoldx.2018.07.005
38. Stark MS. Melanoma treatment guided by a panel of microRNA biomarkers. Melanoma Manag. (2017) 4:75–7. doi: 10.2217/mmt-2017-0006
39. Marsavela G, Lee J, Calapre L, Wong SQ, Pereira MR, McEvoy AC, et al. Circulating tumor DNA predicts outcome from first-, but not second-line treatment and indentifies melanoma patients who may benefit from combination immunotherapy. Clin Cancer Res. (2020) 26:5926–33. doi: 10.1158/1078-0432.CCR-20-2251
40. McEvoy AC, Warburton L, Al-Ogaili Z, Celliers L, Calapre L, Pereira MR, et al. Correlation between circulating tumour DNA and metabolic tumour burden in metastatic melanoma patients. BMC Cancer. (2018) 18:726. doi: 10.1186/s12885-018-4637-6
41. De Mattos-Arruda L, Mayor R, Ng CKY, Weigelt B, Martinez-Ricarte F, Torrejon D, et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat Commun. (2015) 6:8839. doi: 10.1038/ncomms9839
42. Hulstaert E, Bronchez L, Volders P, Vandesompele J, Mestdagh P. Long non-coding RNAs in cutaneous melanoma: Clinical perspectives. Oncotarget. (2017) 8:43470–80. doi: 10.18632/oncotarget.16478
43. Shiiyama R, Fukushima S, Jinnin M, Yamashita J, Miyashita A, Nakahara S, et al. Sensitive detection of melanoma metastasis using circulating microRNA expression profiles. Melanoma Res. (2013) 23:366–72. doi: 10.1097/CMR.0b013e328363e485
44. Thyagarajan A, Tsai KY, Sahu RP. MicroRNA heterogeneity in melanoma progression. Semin Cancer Biol. (2019) 59:208–20. doi: 10.1016/j.semcancer.2019.05.021
45. Nakahara S, Fukushima S, Okada E, Morinaga J, Kubo Y, Tokuzumi A, et al. MicroRNAs that predict the effectiveness of anti-PD-1 therapies in patients with advanced melanoma. J Dermatol Sci. (2020) 97:77–9. doi: 10.1016/j.jdermsci.2019.11.010
46. Kanemaru H, Fukushima S, Yamashita J, Honda N, Oyama R, Kakimoto A, et al. The circulating microRNA-221 level in patients with malignant melanoma as a new tumor marker. J Dermatol Sci. (2011) 61:187–93. doi: 10.1016/j.jdermsci.2010.12.010
47. Sánchez-Sendra B, García-Giménez JL, González-Muñoz JF, Navarro L, Murgui A, Terrádez L, et al. Circulating miRNA expression analysis reveals new potential biomarkers for human cutanous melanoma staging. Eur Acad Dermatol Venerol. (2020) 34:e126–9. doi: 10.1111/jdv.16060
48. Leidinger P, Keller A, Borries A, Reichrath J, Rass K, Jager SU, et al. High-throughput miRNA profiling of human melanoma blood samples. BioMed Central Cancer. (2010) 10:262. doi: 10.1186/1471-2407-10-262
49. Margue C, Reinsbach S, Philippidou D, Beaume N, Walters C, Schneider JG, et al. Comparison of a healthy miRNome with melanoma patient miRNomes: Are microRNAs suitable serum biomarkers for cancer. Oncotarget. (2015) 6:12110–27. doi: 10.18632/oncotarget.3661
50. van Laar R, Lincoln M, van Laar B. Development and validation of a plasma-based melanoma biomarker suitable for clinical use. Br J Cancer. (2018) 118:857–66. doi: 10.1038/bjc.2017.477
51. Stark MS, Klein K, Weide B, Haydu LE, Pflugfelder A, Tang YH, et al. The Prognostic and Predictive Value of Melanoma-related MicroRNAs Using Tissue and Serum: A MicroRNA Expression Analysis. EBioMedicine. (2015) 2:671–80. doi: 10.1016/j.ebiom.2015.05.011
52. Bollard SM, Casalou C, Goh CY, Tobin DJ, Kelly P, McCann A, et al. Circulating melanoma-derived extracellular vesicles: Impact on melanoma diagnosis, progression monitoring, and treatment response. Pharmaceuticals (Basel, Switzerland). (2020) 13:1–18. doi: 10.3390/ph13120475
53. Mondal SK, Whiteside TL. Proteomic profiles of melanoma cell-derived exosomes in plasma: discovery of potential biomarkers of melanoma progression. Melanoma Res. (2021) 31:472–5. doi: 10.1097/CMR.0000000000000762
54. Mirzaei H, Gholamin S, Shahidsales S, Sahebkar A, Jaafari MR, Mirzaei HR, et al. MicroRNAs as potential diagnostic and prognostic biomarkers in melanoma. Eur J Cancer. (2016) 53:25–32. doi: 10.1016/j.ejca.2015.10.009
55. Alegre E, Zubiri L, L P-GJ, González-Cao M, Soria L, Martín-Algarra S, et al. Circulating melanoma exosomes as diagnostic and prognosis biomarkers. Clin Chimira Acta. (2015) 454:28–32. doi: 10.1016/j.cca.2015.12.031
56. Mears R, Craven RA, Hanrahan S, Totty N, Upton C, Young SL, et al. Proteomic analysis of melanoma-derived exosomes by two-dimensional polyacrylamide gel electrophoresis and mass spectrometry. Proteomics (Weinheim). (2004) 4:4019–31. doi: 10.1002/pmic.200400876
57. Mannavola F, D'Oronzo S, Cives M, Stucci LS, Ranieri G, Silvestris F, et al. Extracellular vesicles and epigenetic modifications are hallmarks of melanoma progression. Int J Mol Sci. (2019) 21:1–16. doi: 10.3390/ijms21010052
58. Sharma P, Diergaarde B. S, Kirkwood JM, Whiteside TL. Melanoma cell-derived exosomes in plasma of melanoma patients suppress functions of immune effector cells. Sci Rep. (2020) 10:92. doi: 10.1038/s41598-019-56542-4
59. Lee J, Eberhardt M, Blume K, Vera J, Baur AS. Evidence for liver and peripheral immune cells secreting tumor- suppressive extracellular vesicles in melanoma patients. EBioMedicine. (2020) 62:103119. doi: 10.1016/j.ebiom.2020.103119
60. Logozzi M, De Milito A, Lugini L, Borghi M, Calabrò L, Spada M, et al. High levels of exosomes expressing CD63 and caveolin-1 in plasma of melanoma patients. PLoS ONE. (2009) 4:e5219. doi: 10.1371/journal.pone.0005219
Keywords: melanoma, biomarker, CTC (circulation tumor cells), ctDNA (circulating tumor DNA), miRNA—microRNA, extracellular vesicles (EVs)
Citation: Huang N, Lee KJ and Stark MS (2022) Current Trends in Circulating Biomarkers for Melanoma Detection. Front. Med. 9:873728. doi: 10.3389/fmed.2022.873728
Received: 11 February 2022; Accepted: 07 March 2022;
Published: 05 April 2022.
Edited by:
Andreas Recke, University of Lübeck, GermanyReviewed by:
Nicola Pimpinelli, University of Florence, ItalyCopyright © 2022 Huang, Lee and Stark. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mitchell S. Stark, m.stark@uq.edu.au