Portopulmonary Hypertension: From Bench to Bedside
 Christopher Thomas
Christopher Thomas Vladimir Glinskii
Vladimir Glinskii Vinicio de Jesus Perez
Vinicio de Jesus Perez Sandeep Sahay
Sandeep Sahay- 1Division of Pulmonary, Allergy & Critical Care Medicine, Stanford University School of Medicine, Stanford, CA, United States
- 2Houston Methodist Hospital Lung Center, Houston Methodist Hospital, Houston, TX, United States
Portopulmonary hypertension (PoPH) is defined as pulmonary arterial hypertension (PAH) associated with portal hypertension and is a subset of Group 1 pulmonary hypertension (PH). PoPH is a cause of significant morbidity and mortality in patients with portal hypertension with or without liver disease. Significant strides in elucidating the pathogenesis, effective screening algorithms, accurate diagnoses, and treatment options have been made in past 20 years. Survival of PoPH has remained poor compared to IPAH and other forms of PAH. Recently, the first randomized controlled trial was done in this patient population and showed promising results with PAH specific therapy. Despite positive effects on hemodynamics and functional outcomes, it is unclear whether PAH specific therapy has a beneficial effect on long term survival or transplant outcomes. In this review, we will discuss the epidemiology, pathophysiology, clinical and hemodynamic characteristics of PoPH. Additionally, this review will highlight the lacunae in our current management strategy, challenges faced and will provide direction to potentially useful futuristic management strategies.
Introduction and Background
Pulmonary arterial hypertension (PAH) is a chronic progressive disease characterized by elevated pulmonary artery pressure and pulmonary vascular resistance (PVR) eventually leading to right heart failure and premature death. PAH associated with portal hypertension is called portopulmonary hypertension (PoPH), and is a subset of Group 1 PH (1). Similar to other causes of PAH, PoPH is characterized by the presence of intimal proliferation, medial hypertrophy, adventitial fibrosis of the muscular arteries and plexiform arteriopathy, and is thought to be histopathologically indistinguishable from other PAH phenotypes (2–4). Importantly, there does not seem to be a link between the presence or severity of PoPH with the degree of underlying liver dysfunction or hepatic venous pressure gradient (5, 6).
Patients with PoPH may be treated with PAH specific therapies, which may decrease the severity of the disease, improve functional parameters and hemodynamics, and allow for liver transplant (LT) (7). Despite a rapidly evolving understanding of PoPH, challenges remain in the diagnosis, treatment and transplantation of patients with PoPH. In this review, we will discuss the latest expert opinion in diagnosis and management of PoPH, as well as identify key areas of future research focus.
Demographics and Clinical Characteristics of Patients With PoPH
The exact prevalence of PoPH is difficult to determine. The annual incidence of all types of PAH is <10 patients per million population, and PoPH patients are a subset of this group (8). PoPH was the third most common form of PAH in a population-based epidemiologic study in France (9). Historically, PoPH was thought to be 5–10% of all patients with PAH (10, 11). More recently, French authors have reported that the proportion of newly diagnosed (incident) patients with PoPH is as high as 15% of all patients with PAH, and is continuing to rise as wider screening practices are adopted (12). PoPH is thought to be present in anywhere between 2 and 10% of patients with portal hypertension (6, 13). Furthermore, while the vast majority of cases of PoPH are in patients with portal hypertension related to cirrhosis, non-cirrhotic causes of portal hypertension (including portal vein thrombosis, granulomatous disease, auto-immune disorders, drug reactions, infections, and congenital abnormalities) are also important contributors (14, 15). McDonnell et al. showed a prevalence of histopathologic changes of PAH of 0.61% in autopsies of patients with cirrhosis (16), and small cohort studies have shown prevalence of PoPH in patients with cirrhosis undergoing transplant evaluation to be between 5 and 6% (17–19).
Although the hemodynamic profile of patients with PoPH is better than in patients with idiopathic/familial PAH (IPAH/HPAH), their overall mortality is similar or worse (12, 20–22). The French Pulmonary Hypertension Registry (FHPR) shows a 1, 3, and 5-years survival of 84, 69, and 51%, respectively, for patients with PoPH (12), which is similar to the survival data for patients with IPAH, HPAH and anorexigen associated PAH (23). In contrast, analysis of the US based REVEAL registry showed that patients with PoPH had significantly worse survival when compared to patients with IPAH/FPAH: 67 vs. 85% at 2 years, and 40 vs. 64% at 5 years (Figure 1) (20). These findings were similar in the Spanish REHAP registry, where the 5 year mortality was 49 and 69%, for PoPH and IPAH/HPAH, respectively (Figure 2) (24, 25). Furthermore, severe PoPH is associated with significantly decreased survival in patients undergoing LT (7, 26), and severe PoPH precludes liver transplantation and significantly affects the course of hepatic failure in these patients (26, 27).
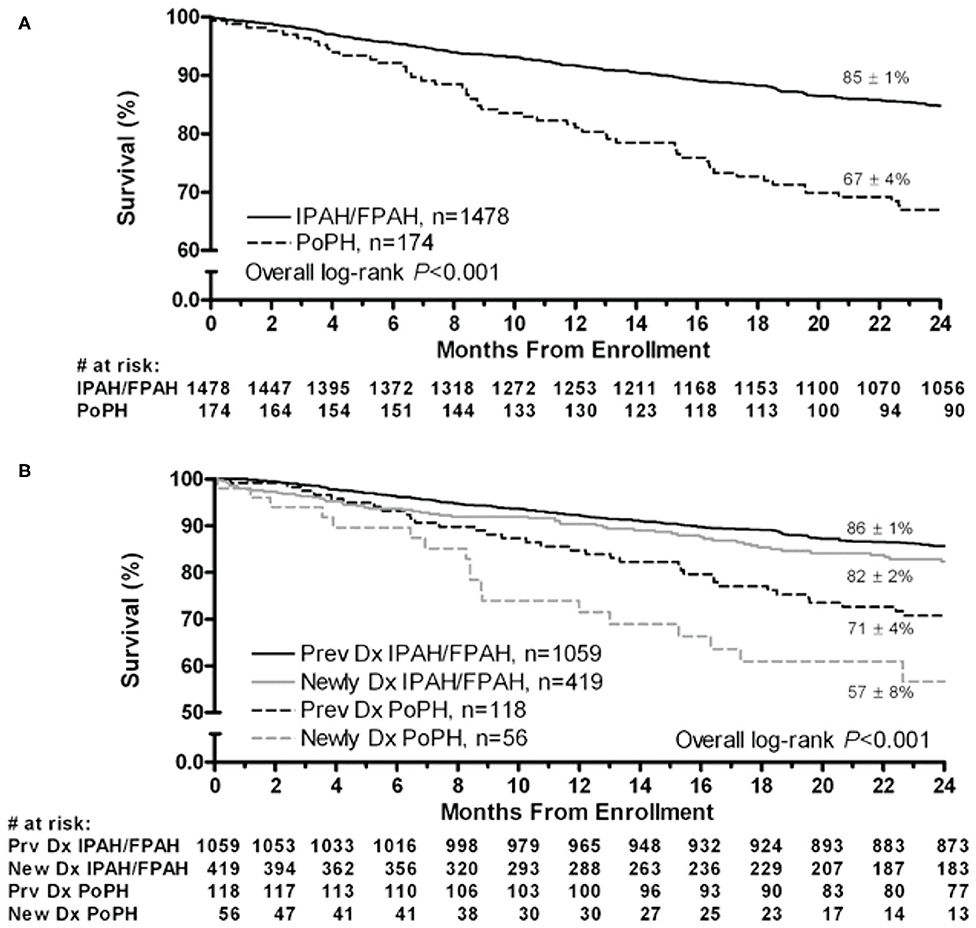
Figure 1. Two-year survival from enrollment. (A) Patients with PoPH and IPAH/FPAH. (B) Stratified by duration of disease from enrollment. Dx, diagnosis; FPAH, familial pulmonary arterial hypertension; IPAH, idiopathic pulmonary arterial hypertension; PoPH, portopulmonary hypertension. Reproduced with permission of Elsevier: Chest 141 (4) 906-915; doi: 10.1378/chest.11-0160 Epub 21 July 2011.
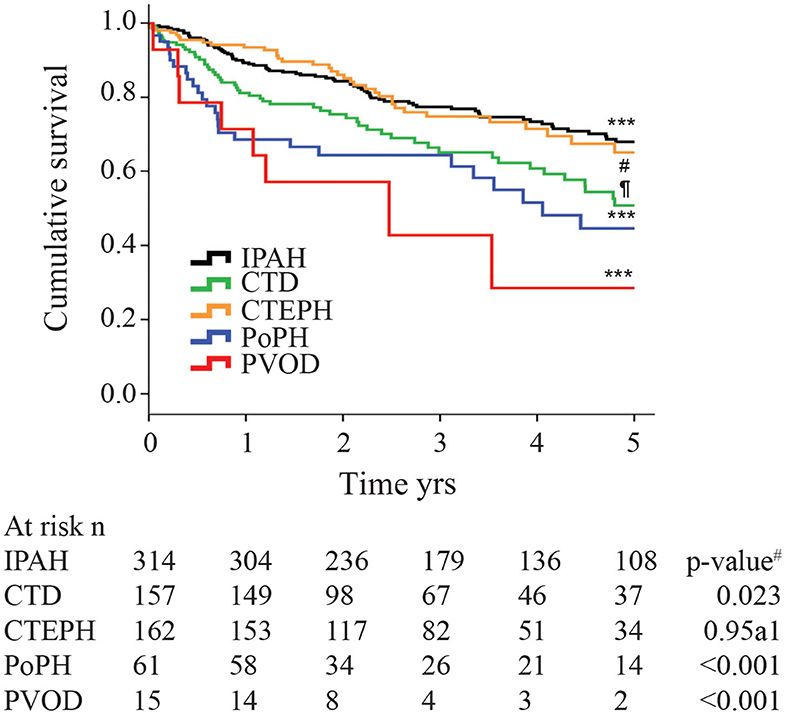
Figure 2. Kaplan-Meier estimates of 5-years survival from time of diagnosis in different pulmonary hypertension subtypes. IPAH, Idiopathic Pulmonary arterial hypertension; CTD, connective tissue disease; CTEPH, chronic thromboembolic pulmonary hypertension; PoPH, portopulmonary hypertension; PVOD, pulmonary veno-occlusive disease. The p-value for the overall comparison is <0.001. # compared with IPAH. Reproduced with permission of the © ERS 2020: European Respiratory Journal 40 (3) 596-603; doi: 10.1183/09031936.00101211 Published 31 August 2012.
The lack of better outcomes in patients with PoPH as compared to their IPAH/FPAH counterparts is not entirely clear, especially in light of their relatively better hemodynamic profile and functional status (20, 25). One possible explanation is increased deaths from liver related events, which account for 25–33% of deaths in patients with PoPH, as compared to only 5% in patients with IPAH (22, 25). In patients with liver disease, higher Child-Pugh Scores are associated with worse outcomes (14, 22), which makes it clear that the severity of liver disease (rather than PoPH) is the main factor in worse outcomes in this population. Another possibility is a relative delay in PAH-specific treatments within the PoPH cohort. PoPH patients started on PAH-specific therapy live longer, despite worse hemodynamics, when compared to PoPH patients not on therapy (25). However, the percentage of PoPH patients on pulmonary vasodilators at both time of enrollment and at 90-days post enrollment into the REVEAL registry was significantly lower than their IPAH/FPAH counterparts (20). The cause of this delay is likely multifactorial, and includes milder symptoms, better hemodynamics, and lack of definitive data to suggest the ideal agent in patients with PoPH.
Likewise, the difference in survival seen between different PoPH cohorts are also likely due to a variety of factors, which include differences in regional screening practices for the presence of PoPH. Patients in the US and Spain with cirrhosis are typically screened at time of liver transplant evaluation, which likely skews the diagnosis of PoPH to patients with more severe liver disease. In contrast, all patients with cirrhosis are screened for PoPH in France, which likely leads to increased rates of diagnosis of PoPH in patients with milder liver disease, which in turn increases the overall survival rates. Rates of referrals of patients to PH specific centers may differ by region (the REVEAL registry included PAH patients treated in the community in addition to academic PH-centers). There may also be differences in baseline characteristics of patients enrolled in each respective registry (including New York Heart Association Functional Class [NYHA FC] and severity of underlying liver disease at the time of enrollment). Additional prospective studies are needed as PoPH-specific management guidelines are established and standardized, to see if this gap can be improved.
Pathogenesis of PoPH
The underlying mechanisms for the development of PoPH are poorly understood and continue to be an area of active research. Liver cirrhosis and portal hypertension lead to splanchnic vasodilatation and formation of portosystemic shunts, which are thought to contribute to PoPH pathogenesis in multiple ways (Figure 3). Mechanically, increased intrahepatic resistance to flow from cirrhosis results in an increased portal pressure gradient and portosystemic collateralization through the reperfusion/dilation of preexisting vessels and through the generation of new vessels (28). On a molecular level, portosystemic shunting enables blood to bypass the liver, and thus evade hepatic metabolism of vasoactive substances. This direct reduction in the peripheral vascular resistance, combined with indirect vasodilation via intestinal vasoactive substances that are now able to bypass the liver and reach systemic circulation, ultimately result in a hyperdynamic state. This phenomenon has been seen in patients immediately after undergoing a transjugular intrahepatic portosystemic shunt (TIPS) placement, and persisted even a month out (29). Likewise, levels of vasoactive substances acting in the pulmonary circulation (including pulmonary vasodilators nitric oxide and prostacyclin, as well as pulmonary vasoconstrictors endothelin-1, thromboxane A2, and serotonin), are similarly dysregulated in cirrhosis (30–32). The imbalance of these vasoactive substances in the pulmonary vasculature results in net vasoconstriction and pulmonary vascular resistance elevation.
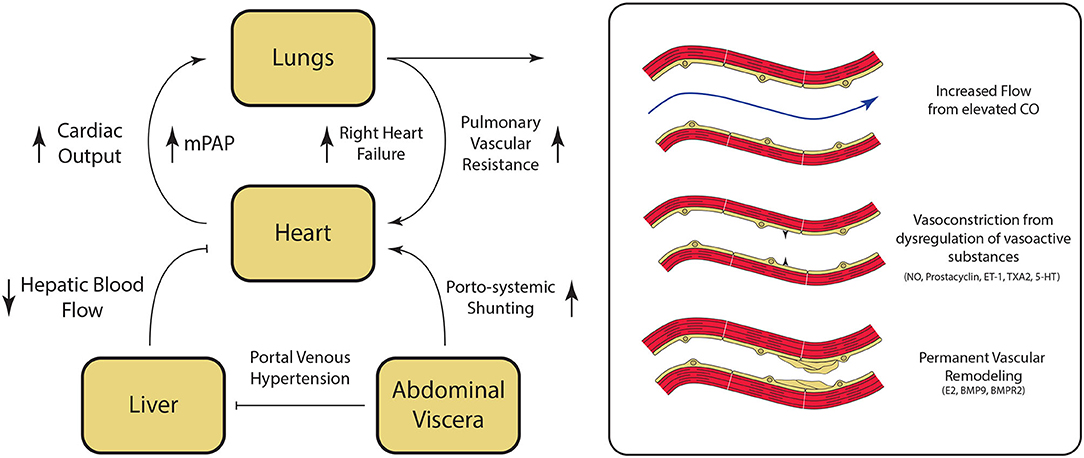
Figure 3. Pathogenesis of Portopulmonary Hypertension (PoPH). Hepatic fibrosis results in portal venous hypertension, increased resistance to hepatic blood flow, and splanchnic vasodilation. Splanchnic vasodilation results in increase in overal circulating volume. and the diversion of blood flow from the liver to the heart via porto-systemic shunting resulting in an overall hyperdynamic state. Dysregulation of key regulator of pulmonary vascular tone associated with cirrhosis results in pulmonary vasoconstriction. At the same time, damage to the pulmonary endothelium and the underlying smooth muscle results in permanent vascular remodeling, which ultimately leads to the development of pulmonary hypertension. CO, cardiac output; NO, nitric oxide; ET-1, endothelin-1; TXA2, thromboxane A2; 5-HT, serotonin; E2, estrogen; BMP9, Bone Morphogenic Protein 9; BMPR2, Bone Morphogenic Receptor 2. Please refer to the text for additional details.
Shear stress from persistently high flows leads to endothelial cell injury and the activation and repression of genes that participate in the vascular remodeling process, and is believed to play a role in various forms of PAH (33–35). Exposure of and damage to the underlying arterial smooth muscle results in smooth muscle proliferation and thickening of the tunica intima, media, and adventitia within the pulmonary vasculature (4). The arterial wall thickening, in turn, can result in more sluggish pulmonary blood flow, platelet aggregation, and thrombus formation, which can then become recanalized over time (3). Autopsy data of patients with PoPH confirms that the end stage of this process is medial and intimal thickening, plexiform lesions and fibrotic venular obstruction (36). Together, these vascular lesions result in permanently elevated PVR that distinguishes PoPH from other causes of elevated mPAP in patients with portal hypertension (Figure 5).
Signaling mediated by bone morphogenic protein receptor 2 (BMPR2), a member of the TGF-β superfamily, has been shown to play a key role in familial pulmonary hypertension, and mutations in the gene encoding this receptor have also been found in 15–40% of idiopathic cases of PAH (37, 38). Bone morphogenic protein 9 (BMP9) is a circulating factor produced by hepatic stellate cells, which serves as a ligand for BMPR2 signaling, and has been shown to have a protecting effect against hepatic fibrosis (39). Recent studies have shown that patients with PoPH have significantly lower circulating levels of BMP9 as compared to control patients with advanced liver disease and no evidence of pulmonary hypertension (40). BMP9 levels were likewise significantly lower in patients with PoPH as compared to patients with other etiologies of group 1 PAH (41). Moreover, selective enhancement of endothelial BMPR2 with exogenous BMP9 has been shown to reverse the presence of PAH in multiple mouse models of PAH (42). Together, these findings suggest that BMP9 and BMPR2 associated downstream signaling pathways also likely play a role in the pathogenesis of PoPH.
More recently, activation of toll-like receptors (TLRs) by bacterial lipopolysaccharides (LPS) from bacterial translocation has been implicated in the pathogenesis of PAH. Mouse models deficient in BMPR2 exposed LPS were shown to develop evidence of pulmonary hypertension, which did not occur in their wild-type counterparts (43). The underlying mechanism for this is thought to be rooted in increased IL-6 production and activation of the STAT3 transcription factor, which is associated with increased expression of TLR4 (43). TLR4 is the main receptor for LPS and has been previously show to promote the development of PAH (44–46). Together, these data suggest that exposure to bacterial endotoxins, which occurs more frequently in cirrhotic patients due to recurrent gut bacterial translocations, may provide a “second hit” toward the development of PoPH in an otherwise predisposed host.
Lastly, there is growing evidence that altered sex hormone also contributes to the pathogenesis of PoPH. Estrogen binds to the promoter region of the BMPR2 gene and regulates its expression (47). Polymorphisms in CYP1B1 which metabolizes estrogen were associated with penetrance of PAH in women with BMPR2 mutations, but not men (48). Expression of CYP1B1 has also been shown to be increased in experimental models and human PAH (49). A candidate gene study to identify genetic risk factors in PoPH found single nucleotide polymorphisms (SNPs) in the genes coding for estrogen receptor 1 and aromatase, a critical enzyme in the estrogen metabolism (50). Recently, Al-Naamani et al. showed that in PoPH patients, the risk allele rs7175922 in CYP19A1 was associated with significantly higher levels of estradiol, urinary 2-hydroxyestrogen/16-α-hydroxyestrone (2-OHE/16α-OHE1), plasma levels of dehydroepiandrosterone-sulfate and plasma levels of 16-α-hydroxyestradiol (16α-OHE2) compared to the patients with liver disease but no PoPH (51). 16α-OHE1 leads to the development of PAH in animals via upregulation of miR-29 and has been associated with abnormal markers of insulin resistance (52). 16α-OHE1 has also been shown to increase oxidative-stress related proliferation in pulmonary arterial smooth muscle cells from PAH patients (53). Similarly, Sahay et al. performed qualitative immunohistochemical (IHC) staining of the explanted livers of PoPH patients and compared them with normal and cirrhotic livers. However, limited by the small sample size, they did not find any significant difference between the PoPH and cirrhotic livers, but aromatase expression was increased compared to the normal liver (54). More research is needed to explore the links between altered estrogen metabolism and the development of PoPH in patients with liver disease.
Screening and Diagnosis of Portopulmonary Hypertension
Screening for PoPH should begin with a detailed history and physical exam (Figure 4). Common symptoms of PoPH include unexplained dyspnea on exertion, fatigue, weakness, orthopnea, near-syncope, syncope, palpitations, and chest pain, with dyspnea being by far the most common. On physical exam, these patients often have an accentuated pulmonic component of the second heart sound (P2), a right sided S3, and a right sided S4. Additionally, the presence of a tricuspid regurgitant murmur or a pulmonic insufficiency murmur can be heard in 40% and 13% of patients, respectively (55). Other signs of right heart failure, including jugular venous distention, ascites, and bilateral lower extremity edema, may also be seen (56). Additional history looking for other known World Health Organization (WHO) Group 1 PAH risk factors should be taken. This includes use of drugs such as methamphetamines, use of dietary supplements, prior history of chemotherapy, and history of autoimmune diseases. Clinicians should also review the medical history for any other conditions known to be associated with PH, such as left heart disease, chronic lung disease, obstructive sleep apnea, venous thromboembolism, and hematologic disorders. If the history and physical arouses any suspicion for PoPH, the patient should undergo an echocardiogram.
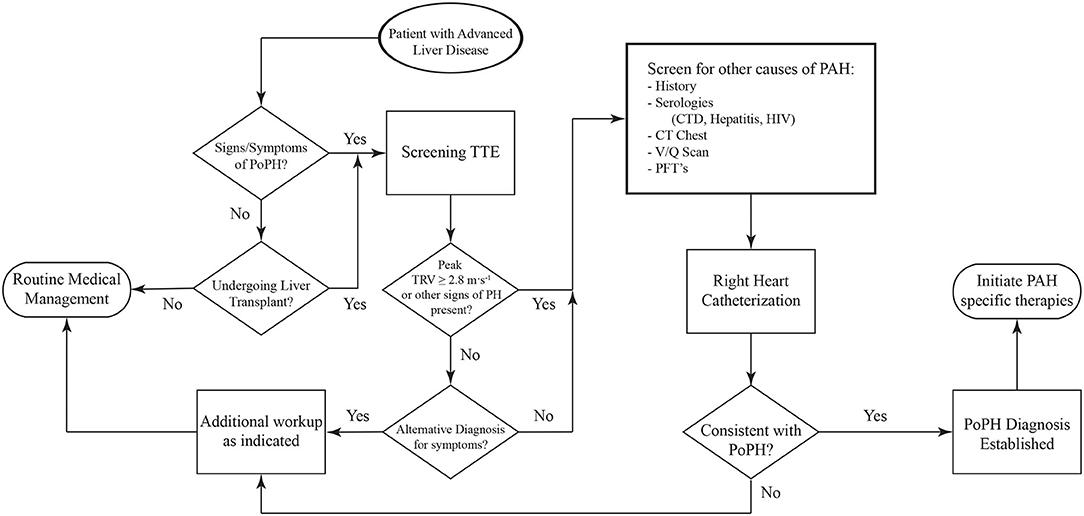
Figure 4. Algorithm for the diagnosis of PoPH. PoPH, Portopulmonary Hypertension; TTE, Transthoracic Echocardiogram; TRV, Tricuspid Regurgitant Velocity; CTD, Connective Tissue Diseases; PFT's, Pulmonary Function Tests.
Laboratory workup should include serological screening tests for connective tissue diseases, hepatitis and HIV. Additional evaluation with pulmonary function tests (including spirometry, lung volumes, diffusion capacity, and a 6 min walk test) and a CT scan of the chest should be obtained to screen for the presence of significant lung disease, and to establish a baseline which can then be used to track disease progression (11, 57). A ventilation/perfusion (V/Q) scan should also be done to screen for the presence of chronic thromboembolic pulmonary hypertension (CTEPH) (especially in the setting of portal vein thrombosis). See Table 1 for more symptoms, physical exam and imaging findings.
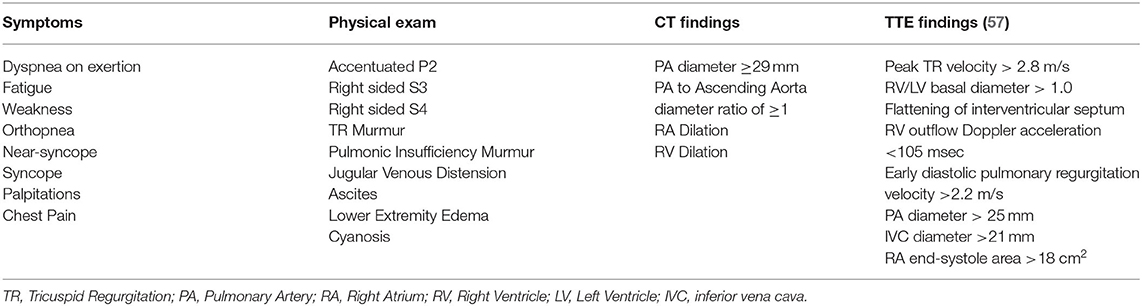
Table 1. Comon clinical and diagnostic findings in patients with Pulmonary Hypertension.
Transthoracic echocardiography (TTE) is the best screening test for PoPH, as it is for other forms of PAH. All patients with portal hypertension should be screened for PoPH with a TTE. A list of the most common echocardiographic findings that are seen in patients with pulmonary hypertension can be seen in Table 1. Although traditionally used throughout the literature to screen for the presence of pulmonary hypertension, right ventricular systolic pressure (RVSP) calculations using the simplified Bernoulli equation are prone to error, stemming from the difficulty in accurately assessing a patient's right atrial pressure. In their analysis of echocardiographic screen of patients undergoing liver transplant evaluation, Krowka et al. demonstrated only moderate correlation between the RVSP and the PA systolic pressures that were measured on right heart catheterization (RHC) (19). Moreover, this discrepancy increased with increasing values of RVSP. Likewise, cutoff values of RVSP used for ruling out the presence of PoPH have differed between studies, and the ideal threshold value for a positive screen has been a matter of debate. As such, it has been suggested that the use of RVSP should be replaced by continuous wave Doppler measurements of peak tricuspid regurgitant velocity, with cutoff values of 2.8 m·s−1 for intermediate and 3.4 m·s−1 for high probability for the presence of PH (58). Issues with calculating RVSP notwithstanding, current guidelines from the American Association for the Study of Liver Diseases (AASLD) recommend evaluation with right heart catheterization in all patients being considered for LT with an RVSP ≥45 mmHg on screening echocardiogram (59). The International Liver Transplant Society (ILTS) guidelines recommend a RHC in patients with RVSP > 50 mmHg and/or evidence of right ventricular (RV) hypertrophy or dysfunction on TTE (60). Clinicians should understand that when lower RVSP cutoffs are used, the sensitivity will approach 100%, but the specificity will decrease, resulting in a significant number of false positives (17). However, given the high mortality associated with severe PoPH and LT, performing an unnecessary RHC is preferable to “missing” PoPH. In practice, all patients in whom there are TTE findings suspicious for PoPH (elevated RVSP or signs of RV dysfunction) should undergo RHC.
Right Heart Catheterization Hemodynamics Interpretation in PoPH
The diagnosis of PoPH includes hemodynamic evaluation of both the pulmonary and hepatic circulations. Portal hypertension can be challenging to diagnose non-invasively. Esophageal varices and skin collaterals are suggestive of portal hypertension. On ultrasound, decreased portal vein flow velocity, and portal vein biphasic or flow reversal are suggestive of portal hypertension (61). A hepatic venous pressure gradient (HVPG) (wedge hepatic vein pressure [WHVP]—free hepatic vein pressure [FHVP]) of >5 mmHg is diagnostic of sinusoidal portal hypertension (62).
On RHC, the patient must have both an elevated mPAP and PVR and a normal pulmonary artery wedge pressure (PAWP) for a positive diagnosis of PoPH. During the most recent World Symposium on Pulmonary Hypertension, the hemodynamic criteria for the diagnosis of pre-capillary pulmonary hypertension (which includes WHO Group 1) were revised. With this change, patients with a mPAP > 20 mmHg (previously > 25 mmHg), a PVR of ≥ 3 Wood units (WU) (≥240 dynes), and a PAWP < 15 mmHg meet the diagnostic criteria for the presence of PAH (1).
Careful evaluation of a patient's hemodynamic profile on RHC is crucial in order to distinguish PoPH from other clinical conditions that are common in patients with liver disease that can also result in elevated mPAPs. These conditions can be classified into three broad categories: hyperdynamic state, volume overload and true portopulmonary hypertension (63). See Figure 5 for comparisons between these hemodynamic profiles.
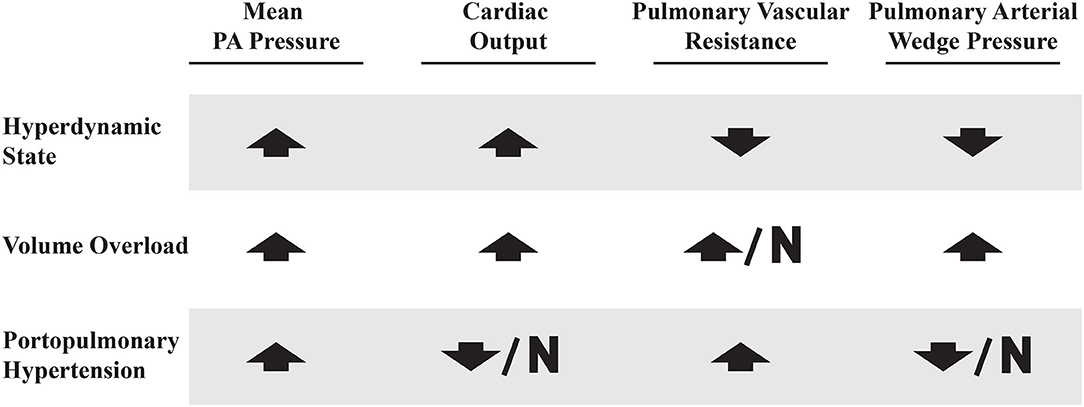
Figure 5. Right heart catheterization profiles in patients with elevated mean PA pressures. Up arrow, increased; Down arrow, decreased; N, normal.
Patients with decompensated liver disease may present with an elevated mPAP, PAWP, and PVR. This may be due to a combination of volume overload, hyperdynamic state, and PoPH (perhaps all three). Volume overload is managed with careful diuresis, as patients with liver disease are prone to kidney injury (64). A hyperdynamic state can be intrinsic to liver disease, but may also be due to other treatable medical comorbidities (such as anemia, arteriovenous shunts, and hyperthyroidism) (65–67). This common clinical scenario speaks to the challenge of diagnosing and treating PoPH, which is best done at expert centers which are most familiar with these complicated patients.
Treatment of PoPH
The importance of screening for PoPH was established by Krowka et al. in a retrospective review of case reports and case series, which looked at the relationship between the presence of untreated PoPH and cardiopulmonary related mortality in patients undergoing LT (7). In this seminal work, Krowka et al. examined 43 patients with PoPH (confirmed by RHC) who were not on medical treatment and underwent LT. The patients were grouped based on severity of pulmonary hypertension. Patients with a mPAP between 25 and 35 mmHg were considered to have mild disease, those with a mPAP between 35 and 50 mmHg were considered moderate, and those with a mPAP >50 mmHg were considered severe. In these groups, 100% of the patients with severe and 50% of the patients with moderate PoPH died due to cardiopulmonary related events (the majority died during the transplantation hospitalization). No mortality was reported among the patients with mild PoPH (mPAP < 35 mmHg). As a result of this study, and others, routine screening for the presence of pulmonary hypertension is now recommended in all patients undergoing evaluation for LT.
After this landmark study established that PoPH patients with higher mPAP are at high risk of mortality with LT, it prompted the question of whether medical therapy could mitigate some of those risks. Treatment of PoPH prior to or instead of liver transplantation remains challenging and is fraught with a paucity of data and large knowledge gaps. Randomized controlled trials of pulmonary hypertension specific medical therapies for patients with portopulmonary hypertension have been limited by concerns regarding adverse hepatic effects of PAH specific medications (68). We will review select clinical trials of PAH-specific therapy in PoPH, as well as the indications and contraindications for liver transplantation.
Retrospective data on treatment of PoPH is quite heterogenous, with some studies showing improvement in hemodynamics and outcomes, and others showing no treatment effect. There is higher quality data from prospective non-randomized studies, from which a few themes emerge. A recently published prospective cohort study from the French Pulmonary Hypertension Registry (FPHR) examined data on 637 patients with PoPH, 90% of whom were started on PAH-specific therapies (12). These patients experienced significant improved WHO functional class, 6-min walk distance (6MWD) and hemodynamic parameters. The ANGEL study is a recent single arm prospective trial of ambrisentan in 31 patients with PoPH (69). This trial showed improved hemodynamics (PVR, mPAP, cardiac index [CI]) and functional class, but no change in 6MWD. From these prospective studies, it is reasonable to conclude that PAH directed therapy has favorable effects on hemodynamics and functional outcomes, but the effects on mortality and candidacy for LT are unclear.
As previously stated, the vast majority of randomized controlled trials of PAH-specific therapies in PAH patients have excluded patients with PoPH. One notable exception is the PATENT-1 trial by Ghofrani et al. (phase 3, double-blind, randomized controlled trial), which randomized 443 patients with various etiologies of WHO group 1 PAH to receive either placebo or two different doses of riociguat (70). This trial is significant in that it included 13 patients with PoPH, 11 of whom received the highest dose of study medication (2.5 mg daily). The primary end point of the PATENT-1 trial was 6MWD, which was significantly improved in the riociguat 2.5 mg group. Secondary hemodynamic (PVR, mPAP, and CO) and functional (WHO functional class, Borg dyspnea scale and time to clinical worsening) endpoints were also significantly improved in the riociguat 2.5 mg daily group. The PATENT-2 trial was an extension of the PATENT-1 study, in which all eligible patients from the first study were given riociguat and followed for 2 years. PATENT-2 showed sustained improvements in functional class and 6MWD (71). A recently published subgroup analysis of the 13 patients with PoPH who were enrolled in PATENT-1 and 2 showed similar improvements in WHO functional class, 6MWD and PVR (72). Importantly, riociguat was well-tolerated by the PoPH patients.
The first ever randomized controlled trial of PAH specific therapy in patients with PoPH was published in 2019. The PORTICO study, which randomized 85 patients with PoPH to receive either macitentan or placebo for 12 weeks, found that the macitentan group had a 35% reduction in PVR, the primary endpoint (73). While there were significant improvements in some secondary endpoints (mPAP and CI), there were no significant improvements in WHO functional class and 6MWD. Importantly, macitentan was well-tolerated in this population, with no hepatic side effects.
A notable limitation of PORTICO is that it excluded patients with Child-Pugh class C liver disease and patients with a MELD score of >19, which are groups with known poor survival (12).
It is still unclear whether this significant reduction in PVR could translate into more meaningful outcomes for patients (improved functional capacity, decreased mortality) or alter candidacy for liver transplant. However, a recently published post-hoc analysis of the PORTICO study found that patients in the macitentan treatment group had significantly decreased waitlist and perioperative mortality risk categories as compared with the placebo arm (74).
Taken together, these studies show that PAH specific therapies can improve hemodynamics and functional outcomes in patients with PoPH. Importantly, these medications seem to be well-tolerated in patients with liver disease and PoPH. By extension, they can help patients become candidates for LT and potentially live longer. In the absence of PoPH specific treatment guidelines, clinicians should follow the general treatment principles of PAH, with special attention to the unique considerations for patients with PoPH. More research is needed to elucidate the nuances of PAH specific therapies in the PoPH population.
Liver Transplantation
As previously discussed, the severity of PoPH is an established risk factor for liver transplant waitlist mortality (75) and poor perioperative outcomes (7). As such, decisions about candidacy for liver transplantation in patients with PoPH are complex and best done by experienced centers.
The following general principles are recommended by the AASLD (59) for management of patients with liver disease who are being considered for LT. All patients who are undergoing evaluation for LT should be screened for PoPH with transthoracic echocardiography, and patients with RVSP > 45 mmHg should be further evaluated by RHC. Patients with mPAP < 35 mmHg do not require treatment with PAH specific therapies and can proceed with further evaluation for LT. Patients with mPAP > 35 mmHg should be evaluated by an experienced PH specialist and be considered for PAH specific therapy. If the mPAP can be lowered to <35 mmHg and the PVR can be lowered to <5 WU with medical therapy, LT can be pursued.
For some patients with mild liver disease and PoPH, their higher risk for waitlist mortality is not accurately captured by the MELD score (76). LT candidates with PoPH are eligible for MELD exception points if the following criteria are met: initial mPAP > 35 and PVR > 3 WU; patients are treated with PAH specific therapy; and post-treatment mPAP is lowered to <35 mmHg (76).
Giving MELD exception points to patients with PoPH is controversial for a number of reasons. The U.S. based Organ Procurement and Transplant Network (OPTN) requires a RHC and resubmission of full hemodynamic data every 3 months (76) to maintain the MELD exception points, which is burdensome and often impractical for both patients and institutions. Despite having to submit full hemodynamic data every 3 months, only mPAP < 35 mmHg is used to maintain eligibility MELD exception points. Some patients may achieve a lower PVR with PAH directed therapy, but continue to have an elevated mPAP from volume overload and/or a high flow state (60). In an analysis of patients with PoPH on the LT waitlist, initial PVR was an independent predictor of waitlist mortality, while pre- and post-treatment mPAP were not (75). This suggests that a broader hemodynamic picture should be used to maintain MELD exception points eligibility.
In one analysis of 155 patients on the LT waitlist with approved PoPH MELD exception points, less than half of the patients actually met the standardized OPTN MELD exception criteria. Furthermore, almost one third of patients in this study lacked sufficient hemodynamic data to diagnose PoPH or had hemodynamic data that was inconsistent with PoPH (77). These researchers found that overall mortality was higher for patients given PoPH MELD exception points (as compared to patients listed without exception points), which shows the need for more research into methods of risk stratification for patients with PoPH.
In the era of the AASLD/ILTS guidelines and MELD exception points, data on the natural course of PoPH after LT is limited. In one retrospective cohort study, hemodynamics and survival data for 35 patients with PoPH were examined (78). After LT, 6 month, 1 year, and 3 year survival rates were 80, 77, and 77%, respectively. Of the 27/35 patients who survived more than 6 months after LT, all were able to be weaned from intravenous epoprostenol. Furthermore, most of the patients had improvement in their hemodynamics after LT, with 30% having a mPAP < 25 mmHg at last follow up. This data suggests that carefully selected patients with PoPH who undergo LT will have significant improvement in their hemodynamics.
Challenges in the Diagnosis and Management of PoPH
There are a number of barriers to the effective diagnosis and treatment of patients with PoPH. As previously discussed, accurate diagnosis of PoPH is challenging, and the differential for an elevated mPAP in the setting of liver disease is broad. There is only one randomized controlled trial for PAH specific therapy in patients with PoPH. Advanced liver disease symptoms have significant overlap with the side effects of PAH specific therapies (e.g., nausea, fatigue, and fluid overload), which can make the initiation and maintenance of treatment challenging (67).
Many of the drugs used to treat PoPH are metabolized in the liver, and drug metabolism is altered by liver disease and other medications that patients may be taking (67). Additionally, patients may have hepatic encephalopathy or lack the social support needed to safely administer parenteral therapy or take medications multiple times per day (67). More generally, many of the treatment barriers that apply to all patients with PAH apply to patients with PoPH: late referrals, lack of an expert center in close proximity to where the patient lives, and insurance issues. There is significant variability in the screening and management practices of PoPH, discordance between published guidelines and actual practice patterns, and disagreements about the role of LT in PoPH (79).
Conclusions and Areas of Future Focus
PoPH remains a challenging disease entity with many facets in need of further research. More research is needed into which types of PAH specific therapies will benefit patients with varying severities of PoPH. The first randomized control trial in PoPH was recently published, which showed that macitentan significantly lowered PVR in patients with PoPH. It unfortunately remains unclear if hemodynamics improvements translate into successful LT or substantial survival benefit. While it is clear that patients with severe disease have high mortality with or without LT, their long-term response to treatment is unknown. It is unknown if mild PoPH needs to be treated or given MELD exception points, and the risk for progression to more severe disease is unknown.
Pre-operative risk stratification of patients prior to LT remains a challenge, as the traditional cut off of a mPAP of 35 mmHg is somewhat arbitrary and is based on a small retrospective review of case reports and series. There are no published guidelines on initiation of PAH specific therapies in PoPH, or whether to continue PoPH treatment post-LT. Finally, there is no centralized national registry of PoPH patients who undergo LT; this would serve as a foundation for focusing on the aforementioned areas of need.
While there are indeed many barriers to and challenges in caring for patients with PoPH, it remains a field with many areas of active investigation. As the mechanisms and treatment continue to be explored, we are confident that care for these complex patients will continue to improve.
Author Contributions
CT and VG: primarily responsible for writing the manuscript. VJ and SS: editing and revision and mentoring. All authors contributed to the article and approved the submitted version.
Funding
SS has grant funding from ACCP CHEST unrelated to the current work.
Conflict of Interest
SS is a speaker and consultant for J&J, Bayer, United Therapeutics, Advisor for Liquidia, and Gossamer biotech. SS has received prior and current research grants from ACCP CHEST, and is a clinical trial end point adjudication committee member in a trial sponsored by GSK.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. (2019) 53:1801913. doi: 10.1183/13993003.01913-2018
2. Jonigk D, Golpon H, Bockmeyer CL, Maegel L, Hoeper MM, Gottlieb J, et al. Plexiform lesions in pulmonary arterial hypertension composition, architecture, and microenvironment. Am J Pathol. (2011) 179:167–79. doi: 10.1016/j.ajpath.2011.03.040
3. Edwards BS, Weir EK, Edwards WD, Ludwig J, Dykoski RK, Edwards JE. Coexistent pulmonary and portal hypertension: morphologic and clinical features. J Am Coll Cardiol. (1987) 10:1233–8. doi: 10.1016/S0735-1097(87)80123-7
4. Humbert M, Guignabert C, Bonnet S, Dorfmuller P, Klinger JR, Nicolls MR, et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J. (2019) 53:1801887. doi: 10.1183/13993003.01887-2018
5. Krowka MJ. Portopulmonary hypertension. Semin Respir Crit Care Med. (2012) 33:17–25. doi: 10.1055/s-0032-1301731
6. Hadengue A, Benhayoun MK, Lebrec D, Benhamou JP. Pulmonary hypertension complicating portal hypertension: prevalence and relation to splanchnic hemodynamics. Gastroenterology. (1991) 100:520–8. doi: 10.1016/0016-5085(91)90225-A
7. Krowka MJ, Plevak DJ, Findlay JY, Rosen CB, Wiesner RH, Krom RA. Pulmonary hemodynamics and perioperative cardiopulmonary-related mortality in patients with portopulmonary hypertension undergoing liver transplantation. Liver Transpl. (2000) 6:443–50. doi: 10.1053/jlts.2000.6356
8. Peacock AJ, Murphy NF, McMurray JJ, Caballero L, Stewart S. An epidemiological study of pulmonary arterial hypertension. Eur Respir J. (2007) 30:104–9. doi: 10.1183/09031936.00092306
9. Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med. (2006) 173:1023–30. doi: 10.1164/rccm.200510-1668OC
10. Badesch DB, Raskob GE, Elliott CG, Krichman AM, Farber HW, Frost AE, et al. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest. (2010) 137:376–87. doi: 10.1378/chest.09-1140
11. Zelniker TA, Huscher D, Vonk-Noordegraaf A, Ewert R, Lange TJ, Klose H, et al. The 6MWT as a prognostic tool in pulmonary arterial hypertension: results from the COMPERA registry. Clin Res Cardiol. (2018) 107:460–70. doi: 10.1007/s00392-018-1207-5
12. Savale L, Guimas M, Ebstein N, Fertin M, Jevnikar M, Renard S, et al. Portopulmonary hypertension in the current era of pulmonary hypertension management. J Hepatol. (2020) 73:130–9. doi: 10.1016/j.jhep.2020.02.021
13. Chen HS, Xing SR, Xu WG, Yang F, Qi XL, Wang LM, et al. Portopulmonary hypertension in cirrhotic patients: Prevalence, clinical features and risk factors. Exp Ther Med. (2013) 5:819–24. doi: 10.3892/etm.2013.918
14. Sithamparanathan S, Nair A, Thirugnanasothy L, Coghlan JG, Condliffe R, Dimopoulos K, et al. National Pulmonary Hypertension Service Research Collaboration of the United, and Ireland, Survival in portopulmonary hypertension: Outcomes of the United Kingdom National Pulmonary Arterial Hypertension Registry. J Heart Lung Transplant. (2017) 36:770–9. doi: 10.1016/j.healun.2016.12.014
15. Schouten JN, Garcia-Pagan JC, Valla DC, Janssen HL. Idiopathic noncirrhotic portal hypertension. Hepatology. (2011) 54:1071–81. doi: 10.1002/hep.24422
16. McDonnell PJ, Toye PA, Hutchins GM. Primary pulmonary hypertension and cirrhosis: are they related? Am Rev Respir Dis. (1983) 127:437–41. doi: 10.1164/arrd.1983.127.4.437
17. Colle IO, Moreau R, Godinho E, Belghiti J, Ettori F, Cohen-Solal A, et al. Diagnosis of portopulmonary hypertension in candidates for liver transplantation: a prospective study. Hepatology. (2003) 37:401–9. doi: 10.1053/jhep.2003.50060
18. Kawut SM, Taichman DB, Ahya VN, Kaplan S, Archer-Chicko CL, Kimmel SE, et al. Hemodynamics and survival of patients with portopulmonary hypertension. Liver Transpl. (2005) 11:1107–11. doi: 10.1002/lt.20459
19. Krowka MJ, Swanson KL, Frantz RP, McGoon MD, Wiesner RH. Portopulmonary hypertension: Results from a 10-year screening algorithm. Hepatology. (2006) 44:1502–10. doi: 10.1002/hep.21431
20. Krowka MJ, Miller DP, Barst RJ, Taichman D, Dweik RA, Badesch DB, et al. Portopulmonary hypertension: a report from the US-based REVEAL Registry. Chest. (2012) 141:906–15. doi: 10.1378/chest.11-0160
21. Swanson KL, Wiesner RH, Nyberg SL, Rosen CB, Krowka MJ. Survival in portopulmonary hypertension: Mayo Clinic experience categorized by treatment subgroups. Am J Transplant. (2008) 8:2445–53. doi: 10.1111/j.1600-6143.2008.02384.x
22. Le Pavec J, Souza R, Herve P, Lebrec D, Savale L, Tcherakian C, et al. Portopulmonary hypertension: survival and prognostic factors. Am J Respir Crit Care Med. (2008) 178:637–43. doi: 10.1164/rccm.200804-613OC
23. Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. (2010) 122:156–63. doi: 10.1161/CIRCULATIONAHA.109.911818
24. Escribano-Subias P, Blanco I, Lopez-Meseguer M, Lopez-Guarch CJ, Roman A, Morales P, et al. Survival in pulmonary hypertension in Spain: insights from the Spanish registry. Eur Respir J. (2012) 40:596–603. doi: 10.1183/09031936.00101211
25. Lázaro Salvador M, Quezada Loaiza C, Rodríguez Padial L, Barberá J, López-Meseguer M, López-Reyes R, et al. Portopulmonary hypertension: prognosis and management in the current treatment era. Results from the REHAP Registry. Intern Med J. (2020). doi: 10.1111/imj.14751
26. Krowka MJ, Mandell MS, Ramsay MA, Kawut SM, Fallon MB, Manzarbeitia C, et al. Hepatopulmonary syndrome and portopulmonary hypertension: a report of the multicenter liver transplant database. Liver Transpl. (2004) 10:174–82. doi: 10.1002/lt.20016
27. Sussman N, Kaza V, Barshes N, Stribling R, Goss J, O'Mahony C, et al. Successful liver transplantation following medical management of portopulmonary hypertension: a single-center series. Am J Transplant. (2006) 6:2177–82. doi: 10.1111/j.1600-6143.2006.01432.x
28. Bolognesi M, Di Pascoli M, Verardo A, Gatta A. Splanchnic vasodilation and hyperdynamic circulatory syndrome in cirrhosis. World J Gastroenterol. (2014) 20:2555–63. doi: 10.3748/wjg.v20.i10.2555
29. Van der Linden P, Le Moine O, Ghysels M, Ortinez M, Deviere J. Pulmonary hypertension after transjugular intrahepatic portosystemic shunt: effects on right ventricular function. Hepatology. (1996) 23:982–7. doi: 10.1002/hep.510230507
30. Huertas A, Guignabert C, Barbera JA, Bartsch P, Bhattacharya J, Bhattacharya S, et al. Pulmonary vascular endothelium: the orchestra conductor in respiratory diseases: highlights from basic research to therapy. Eur Respir J. (2018) 51:1700745. doi: 10.1183/13993003.00745-2017
31. Shenoda B, Boselli J. Vascular syndromes in liver cirrhosis. Clin J Gastroenterol. (2019) 12:387–97. doi: 10.1007/s12328-019-00956-0
32. Porres-Aguilar M, Altamirano JT, Torre-Delgadillo A, Charlton MR, Duarte-Rojo A. Portopulmonary hypertension and hepatopulmonary syndrome: a clinician-oriented overview. Eur Respir Rev. (2012) 21:223–33. doi: 10.1183/09059180.00007211
33. Rodriguez-Roisin R, Krowka MJ, Herve P, Fallon MB, E.R.S.T.F.P.-H.Committee VDS. Pulmonary-Hepatic vascular Disorders (PHD). Eur Respir J. (2004) 24:861–80. doi: 10.1183/09031936.04.00010904
34. Baeyens N, Bandyopadhyay C, Coon BG, Yun S, Schwartz MA. Endothelial fluid shear stress sensing in vascular health and disease. J Clin Invest. (2016) 126:821–8. doi: 10.1172/JCI83083
35. Pascall E, Tulloh RM. Pulmonary hypertension in congenital heart disease. Future Cardiol. (2018) 14:343–53. doi: 10.2217/fca-2017-0065
36. Furuta M, Sato T, Tsujino I, Tanino M, Watanabe T, Nishimura M. An autopsy case of portopulmonary hypertension associated with idiopathic portal hypertension. Eur Resp J. (2013) 42:P2649. Available online at: https://erj.ersjournals.com/content/42/Suppl_57/P2649
37. P.International PHC, Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, et al. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet. (2000) 26:81–4. doi: 10.1038/79226
38. Thomson JR, Machado RD, Pauciulo MW, Morgan NV, Humbert M, Elliott GC, et al. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-beta family. J Med Genet. (2000) 37:741–5. doi: 10.1136/jmg.37.10.741
39. Desroches-Castan A, Tillet E, Ricard N, Ouarne M, Mallet C, Belmudes L, et al. Bone morphogenetic protein 9 is a paracrine factor controlling liver sinusoidal endothelial cell fenestration and protecting against hepatic fibrosis. Hepatology. (2019) 70:1392–408. doi: 10.1002/hep.30655
40. Rochon ER, Krowka MJ, Bartolome S, Heresi GA, Bull T, Roberts K, et al. Pulmonary vascular complications of liver disease 2 study, bmp 9/10 in pulmonary vascular complications of liver disease. Am J Respir Crit Care Med. (2020) 201:1575–8. doi: 10.1164/rccm.201912-2514LE
41. Nikolic I, Yung LM, Yang P, Malhotra R, Paskin-Flerlage SD, Dinter T, et al. Bone morphogenetic protein 9 is a mechanistic biomarker of portopulmonary hypertension. Am J Respir Crit Care Med. (2019) 199:891–902. doi: 10.1164/rccm.201807-1236OC
42. Long L, Ormiston ML, Yang X, Southwood M, Graf S, Machado RD, et al. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat Med. (2015) 21:777–85. doi: 10.1038/nm.3877
43. Soon E, Crosby A, Southwood M, Yang P, Tajsic T, Toshner M, et al. Bone morphogenetic protein receptor type II deficiency and increased inflammatory cytokine production. A gateway to pulmonary arterial hypertension. Am J Respir Crit Care Med. (2015) 192:859–72. doi: 10.1164/rccm.201408-1509OC
44. Bauer EM, Chanthaphavong RS, Sodhi CP, Hackam DJ, Billiar TR, Bauer PM. Genetic deletion of toll-like receptor 4 on platelets attenuates experimental pulmonary hypertension. Circ Res. (2014) 114:1596–600. doi: 10.1161/CIRCRESAHA.114.303662
45. Bauer EM, Shapiro R, Zheng H, Ahmad F, Ishizawar D, Comhair SA, et al. High mobility group box 1 contributes to the pathogenesis of experimental pulmonary hypertension via activation of Toll-like receptor 4. Mol Med. (2013) 18:1509–18. doi: 10.2119/molmed.2012.00283
46. Young KC, Hussein SM, Dadiz R, deMello D, Devia C, Hehre D, et al. Toll-like receptor 4-deficient mice are resistant to chronic hypoxia-induced pulmonary hypertension. Exp Lung Res. (2010) 36:111–9. doi: 10.3109/01902140903171610
47. Austin ED, Hamid R, Hemnes AR, Loyd JE, Blackwell T, Yu C, et al. BMPR2 expression is suppressed by signaling through the estrogen receptor. Biol Sex Differ. (2012) 3:6. doi: 10.1186/2042-6410-3-6
48. Austin ED, Cogan JD, West JD, Hedges LK, Hamid R, Dawson EP, Wheeler LA, Parl FF, Loyd JE Phillips J3rd A Alterations in oestrogen metabolism: implications for higher penetrance of familial pulmonary arterial hypertension in females. Eur Respir J (2009) 34:1093-9. doi: 10.1183/09031936.00010409
49. White K, Johansen AK, Nilsen M, Ciuclan L, Wallace E, Paton L, et al. Activity of the estrogen-metabolizing enzyme cytochrome P450 1B1 influences the development of pulmonary arterial hypertension. Circulation. (2012) 126:1087–98. doi: 10.1161/CIRCULATIONAHA.111.062927
50. Roberts KE, Fallon MB, Krowka MJ, Brown RS, Trotter JF, Peter I, et al. Pulmonary Vascular Complications of Liver Disease Study, Genetic risk factors for portopulmonary hypertension in patients with advanced liver disease. Am J Respir Crit Care Med. (2009) 179:835–42. doi: 10.1164/rccm.200809-1472OC
51. Al-Naamani N, Krowka MJ, Forde KA, Krok KL, Feng R, Heresi GA, et al. Estrogen signaling and portopulmonary hypertension: the pulmonary vascular complications of liver disease study (PVCLD2). Hepatology. (2020). doi: 10.1002/hep.31314
52. Chen X, Talati M, Fessel JP, Hemnes AR, Gladson S, French J, et al. Estrogen metabolite 16alpha-hydroxyestrone exacerbates bone morphogenetic protein receptor type II-associated pulmonary arterial hypertension through microrna-29-mediated modulation of cellular metabolism. Circulation. (2016) 133:82–97. doi: 10.1161/CIRCULATIONAHA.115.016133
53. Hood KY, Montezano AC, Harvey AP, Nilsen M, MacLean MR, Touyz RM. Nicotinamide adenine dinucleotide phosphate oxidase-mediated redox signaling and vascular remodeling by 16alpha-hydroxyestrone in human pulmonary artery cells: implications in pulmonary arterial hypertension. Hypertension. (2016) 68:796–808. doi: 10.1161/HYPERTENSIONAHA.116.07668
54. Sahay S, Barrios R, Deavers M, Ren Y, Frost AE. aromatase expression of liver in portopulmonary hypertension, A63. In: Ride the Lightning: Basic and Translational Studies in Pulmonary Hypertension. San Diego, CA (2018). p. A2102–A2102.
55. Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med. (1987) 107:216–23. doi: 10.7326/0003-4819-107-2-216
56. Robalino BD, Moodie DS. Association between primary pulmonary hypertension and portal hypertension: Analysis of its pathophysiology and clinical, laboratory and hemodynamic manifestations. J Am Coll Cardiol. (1991) 17:492–8. doi: 10.1016/S0735-1097(10)80121-4
57. Frost A, Badesch D, Gibbs JSR, Gopalan D, Khanna D, Manes A, et al. Diagnosis of pulmonary hypertension. Eur Respir J. (2019) 53. doi: 10.1183/13993003.01904-2018
58. Lau EM, Tamura Y, McGoon MD, Sitbon O. The 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: a practical chronicle of progress. Eur Respir J. (2015) 46:879–82. doi: 10.1183/13993003.01177-2015
59. Martin P, DiMartini A, Feng S, Brown R Jr, Fallon M. Evaluation for liver transplantation in adults: 2013 practice guideline by the American Association for the Study of Liver Diseases and the American Society of Transplantation. Hepatology. (2014) 59:1144–65. doi: 10.1002/hep.26972
60. Krowka MJ, Fallon MB, Kawut SM, Fuhrmann V, Heimbach JK, Ramsay MA, et al. International liver transplant society practice guidelines: diagnosis and management of hepatopulmonary syndrome and portopulmonary hypertension. Transplantation. (2016) 100:1440–52. doi: 10.1097/TP.0000000000001229
61. Hernández DB, Serrano MA, Gabutti JA, Casanova IE, Guerrero M. Portal hypertension. What, when and how of portal gradients, TIPS and Neo-porta. Europ Congress Radiol. (2018). doi: 10.1594/ecr2018/C-3153
62. de Franchis R, Baveno VIF. Expanding consensus in portal hypertension: report of the Baveno VI Consensus Workshop: stratifying risk and individualizing care for portal hypertension. J Hepatol. (2015) 63:743–52. doi: 10.1016/j.jhep.2015.05.022
63. Cartin-Ceba R, Krowka MJ. Portopulmonary hypertension. Clin Liver Dis. (2014) 18:421–38. doi: 10.1016/j.cld.2014.01.004
64. Garcia-Tsao G, Parikh CR, Viola A. Acute kidney injury in cirrhosis. Hepatology. (2008) 48:2064–77. doi: 10.1002/hep.22605
65. Park SC, Beerman LB, Gartner JC, Zitelli BJ, Malatack JJ, Fricker FJ, et al. Echocardiographic findings before and after liver transplantation. Am J Cardiol. (1985) 55:1373–8. doi: 10.1016/0002-9149(85)90507-7
66. Reddy YNV, Melenovsky V, Redfield MM, Nishimura RA, Borlaug BA. High-output heart failure: a 15-year experience. J Am Coll Cardiol. (2016) 68:473–82. doi: 10.1016/j.jacc.2016.05.043
67. AbuHalimeh B, Krowka MJ, Tonelli AR. Treatment barriers in portopulmonary hypertension. Hepatology. (2019) 69:431–43. doi: 10.1002/hep.30197
68. Sitbon O, O'Callaghan DS, Savale L. Portopulmonary hypertension: light at the end of the tunnel? Chest. (2012) 141:840–2. doi: 10.1378/chest.11-2378
69. Preston IR, Burger CD, Bartolome S, Safdar Z, Krowka M, Sood N, et al. Ambrisentan in portopulmonary hypertension: A multicenter, open-label trial. J Heart Lung Transplant. (2020) 39:464–72. doi: 10.1016/j.healun.2019.12.008
70. Ghofrani HA, Galie N, Grimminger F, Grunig E, Humbert M, Jing ZC, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. (2013) 369:330–40. doi: 10.1056/NEJMoa1209655
71. Rubin LJ, Galie N, Grimminger F, Grunig E, Humbert M, Jing ZC, et al. Riociguat for the treatment of pulmonary arterial hypertension: a long-term extension study (PATENT-2). Eur Respir J. (2015) 45:1303–13. doi: 10.1183/09031936.00090614
72. Cartin-Ceba R, Halank M, Ghofrani HA, Humbert M, Mattson J, Fritsch A, et al. Riociguat treatment for portopulmonary hypertension: a subgroup analysis from the PATENT-1/-2 studies. Pulm Circ. (2018) 8:2045894018769305. doi: 10.1177/2045894018769305
73. Sitbon O, Bosch J, Cottreel E, Csonka D, de Groote P, Hoeper MM, et al. Macitentan for the treatment of portopulmonary hypertension (PORTICO): a multicentre, randomised, double-blind, placebo-controlled, phase 4 trial. Lancet Respir Med. (2019) 7:594–604. doi: 10.1016/S2213-2600(19)30091-8
74. Krowka M, Cottreel E, Hoeper MM, Kim NH, Martin N, Sitbon O, et al. Macitentan improves risk categorization for liver transplant mortality in patients with portopulmonary hypertension: a portico study post hoc analysis. Liver Transpl. (2020) 26:935–40. doi: 10.1002/lt.25747
75. DuBrock HM, Goldberg DS, Sussman NL, Bartolome SD, Kadry Z, Salgia RJ, et al. Predictors of Waitlist Mortality in Portopulmonary Hypertension. Transplantation. (2017) 101:1609–15. doi: 10.1097/TP.0000000000001666
76. Guidance to Liver Transplant Programs and the National Liver Review Board for: Adult MELD Exception Review.
77. Goldberg DS, Batra S, Sahay S, Kawut SM, Fallon MB. MELD exceptions for portopulmonary hypertension: current policy and future implementation. Am J Transplant. (2014) 14:2081–7. doi: 10.1111/ajt.12783
78. Savale L, Sattler C, Coilly A, Conti F, Renard S, Francoz C, et al. Long-term outcome in liver transplantation candidates with portopulmonary hypertension. Hepatology. (2017) 65:1683–92. doi: 10.1002/hep.28990
Keywords: portopulmonary hypertension, pulmonary arterial hypertension, portal hypertension, liver transplant, MELD exception
Citation: Thomas C, Glinskii V, de Jesus Perez V and Sahay S (2020) Portopulmonary Hypertension: From Bench to Bedside. Front. Med. 7:569413. doi: 10.3389/fmed.2020.569413
Received: 04 June 2020; Accepted: 23 September 2020;
Published: 03 November 2020.
Edited by:
Laurent Pierre Nicod, University of Lausanne, SwitzerlandReviewed by:
Olivier Sitbon, Université Paris-Saclay, FranceJohn-David Aubert, Centre Hospitalier Universitaire Vaudois (CHUV), Switzerland
Copyright © 2020 Thomas, Glinskii, de Jesus Perez and Sahay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sandeep Sahay, ssahay@houstonmethodist.org
†These authors have contributed equally to this work