 Shiping Liu1†
Shiping Liu1† Xiaoyu Fang1†Ruiyao Zhu2
Xiaoyu Fang1†Ruiyao Zhu2 Jing Zhang1
Jing Zhang1 Huijuan Wang1Jiaxi Lei1Chaoqun Wang3
Huijuan Wang1Jiaxi Lei1Chaoqun Wang3 Lu Wang1
Lu Wang1 Liying Zhan1*
Liying Zhan1*- 1Department of Critical Care Medicine, Renmin Hospital of Wuhan University, Wuhan, China
- 2Department of Infection Prevention and Control, Renmin Hospital of Wuhan University, Wuhan, China
- 3College of Life Sciences, Wuhan University, Wuhan, China
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS), the prime causes of morbidity and mortality in critically ill patients, are usually treated by general supportive treatments. Endoplasmic reticulum autophagy (ER-phagy) maintains cellular homeostasis by degrading damaged endoplasmic reticulum (ER) fragments and misfolded proteins. ER-phagy is crucial for maintaining ER homeostasis and improving the internal environment. ER-phagy has a particular role in some aspects, such as immunity, inflammation, cell death, pathogen infection, and collagen quality. In this review, we summarized the definition, epidemiology, and pathophysiology of ALI/ARDS and described the regulatory mechanisms and functions of ER-phagy as well as discussed the potential role of ER-phagy in ALI/ARDS from the perspectives of immunity, inflammation, apoptosis, pathogen infection, and fibrosis to provide a novel and effective target for improving the prognosis of ALI/ARDS.
1 Introduction
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) are associated with various lung injuries that often lead to life-threatening consequences. Developed from ALI, ARDS was first formally described in 1967 (1). Recently, ALI has been defined as mild or moderate ARDS (2). ARDS is an acute respiratory failure characterized by severe hypoxemia caused by noncardiogenic pulmonary edema. There are multiple etiologies of ARDS, classified as pulmonary endogenous and exogenous, and the most common causes are pneumonia and sepsis (3). The pathophysiology of ARDS is divided into exudative, proliferative, and fibrotic stages. During the exudative phase, reduced lung compliance leads to gas exchange impairment. The proliferation phase is the recovery period of the lung microvascular barrier, while the fibrosis phase leads to poor recovery of lung injury and increased ARDS mortality (4). Due to the poor effects of prevention and treatment, ALI/ARDS has exceptionally high morbidity and mortality. In 2016, a study on the incidence of ARDS in intensive care units (ICUs) in 50 countries showed that 10% of ICU patients and 23% of mechanically ventilated patients met the diagnostic criteria for ARDS (5). The mortality in hospitals for ARDS is 35%-45%, and a significant proportion of ARDS mortality is attributed to sepsis-associated ARDS (6). ALI progression is associated with various clinical disorders, leading to diverse outcomes. Sepsis is the main cause of ALI death, and the mortality rate is up to 43% (2). Moreover, the prognosis of ALI/ARDS is unsatisfactory, and most patients will face sequelae, such as muscle weakness, physical deterioration, and even cognitive impairment (7).
The ER, the largest organelle in the cell, is responsible for lipid and protein biosynthesis, and provides important sites for modifying and folding nascent integral membranes and secreted proteins. However, under some pathological stimuli, ER function is disrupted, leading to the accumulation of misfolded proteins, which triggers ER stress. During ER stress, cells activate a series of complementary mechanisms in response to changes in protein folding, a process known as the unfolded protein response (UPR) (8). UPR is a highly conserved mechanism mediated by three proteins sensors located in ER: protein kinase RNA (PKR)-like ER kinase (PERK), inositol-requiring enzyme-1 (IRE-1), and activating transcription factor-6 (ATF6). ER stress induces ER-phagy by activating the UPR pathway (9). This phenomenon was first discovered in a yeast experiment in 2007, in which researchers found that ER stress induces ER selective autophagy, degrading dysfunctional ER membranes and restoring ER homeostasis (10). ER-phagy contains two primary autophagy mechanisms, namely, macroautophagy and microautophagy. The mechanism of macroautophagy is that the autophagy bilayer membrane extends and wraps the endoplasmic reticulum fragments, finally binding to the lysosomes and being degraded. Microautophagy is the process by which the lysosomal membrane undergoes invaginations and extrudes part of the endoplasmic reticulum into the lysosome (11).
Restricted treatments lead to poor prognosis of ALI/ARDS, and clarification of the pathogenesis is critical for treatment. Therefore, an in-depth understanding of the pathogenesis of ALI/ARDS is essential. In this review, we discussed the possible role of ER-phagy in ALI/ARDS, which may provide new insights for treating ALI/ARDS, thus reducing ALI/ARDS incidence and improving ALI/ARDS prognosis.
2 Regulatory mechanism of ER-phagy
Several studies have suggested that ER-phagy receptors are the primary regulatory mechanism of ER-phagy. To induce ER-phagy, late-stage ER stress activates ER-phagy receptors, which interact with the light chain (LC3)/GABARAP (GABA type A receptor-associated protein)/autophagy protein (ATG)8 autopahgy protein complex. In addition, recent studies have also found that complexes of mitochondrial oxidative phosphorylation (OXPHOS) and DDRGK1-mediated ER surface UFMylation are closely associated with ER-phagy. Therefore, mitochondrial factors and DDRGK1 proteins are also involved in regulating ER-phagy.
2.1 ER-phagy receptors
ER-phagy receptors are an essential part of ER-phagy, including the family with sequence similarity 134 member B (FAM134B), reticulon-3L (RTN3L), atlastin GTPase 3 (ATL3), testis expressed 264 (TEX264), cell cycle progression 1 (CCPG1), and preprotein translocation factor SEC62, coiled-coil domain-containing protein 1 (CALCOCO1),which bind to autophagy-related proteins and mediate ER-phagy (Figure 1).
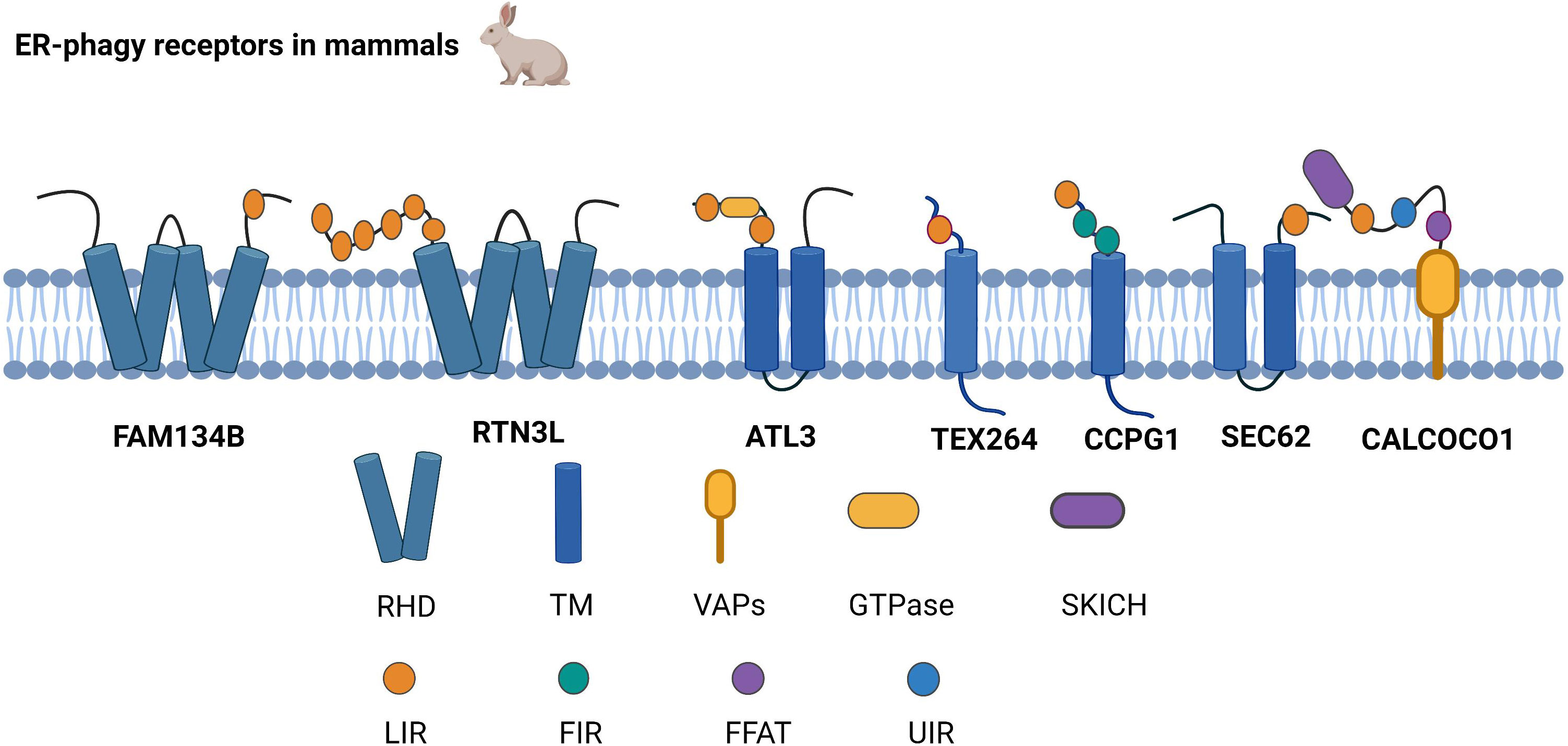
Figure 1 Structure of ER-phagy Receptors in Mammals. ER-phagy receptors, distributed in a specific ER domain, bind to autophagic proteins and then assemble into autophagosomes, thus recruiting the autophagy machinery. Mammalian ER-phagy receptors are mainly FAM134B, RTN3L, ATL3, TEX264, CCPG1, and SEC62, and the novel receptor CALCOCO1 has been found to mediate ER-phagy currently.
2.1.1 FAM134B
FAM134B is currently the most widely studied autophagy receptor, and it has two significant structures, LIR and RHD. LIR binds to the LC3/GABARAP family of autophagy proteins and is a typical structure of selective autophagy receptors (12, 13). RHD has two hairpin structures, namely, TM1-TM2 and TM3-TM4, which are responsible for anchoring the protein to the ER membrane, subsequently recognizing and inducing the bending of the ER membrane (11). Cinque et al. found that promoting TFEB/transcription factor 3 (TFE3) nuclear translocation by activating fibroblast growth factor (FGF) signaling facilitates the transcription of FAM134B (14). Because ATG5 and Beclin1 are valuable in FAM134B-mediated ER-phagy, it has been demonstrated that FAM134B depends on macro-ER-phagy (11).
2.1.2 RTN3L
RTN3L is generally localized to the endoplasmic reticulum through the RHD structure. RTN3 interacts with LC3/GABARAP via the LIR. As an ER-phagy receptor, RTN3L in the oligomeric state promotes ER tubule fragment formation, and the functional LIR motif of RTN3L then promotes the transport of ER debris into lysosomes (15). Misfolded proteins escape the ER by damaging the ER membrane, while RTN3L protects ER membrane integrity based on ER-phagy (16). Furthermore, RTN3L maintains homeostasis in immunity and inflammation by disrupting polyubiquitination of K63 junctions and inhibiting the interferon regulatory factor 3 (IRF3) and nuclear factor kappa-B (NF-κB) pathways (17).
2.1.3 ATL3
ATL3 is a power-like GTPase that mainly regulates ER fusion; it is the primary tubular ER-phagy receptor in cells lacking RTN3L, and ATL3 deficiency suppresses tubular ER degradation. ATL3 interacts explicitly with the GABARAP of the ATG8 family through the interaction of two GABARAP motifs, resulting in the sequestration of tubular endoplasmic reticulum in autophagosomes, which are ultimately degraded by lysosomes (18). The process of the unc-51-like kinase 1 (ULK1) complex targeting the ER plays a key role during autophagosome formation, and ATL3 contributes to ULK1 fixation at the specific autophagosome formation site in the ER (19).
2.1.4 TEX264
The N and C termini of TEX264 are hydrophobic and relatively loose fragments, respectively, with a cytosolic gyrase inhibitor (Gyrl)-like structure between the N and C termini. TEX264 is mainly responsible for ER-phagy during starvation. Compared to other ER-phagy receptors, TEX264 is most strongly connected to the LC3, ATG8, and GABARAP autophagy proteins (20). The mechanism by which TEX264 initiates ER-phagy is by using its C-terminal LIR motif to interact with ATG8 in lipid form, and TEX264 then binds to LC3-positive phagosomes. Finally, the autophagosome fuses with lysosomes (21). Excessive ER ribosome volume causes some obstacles to ER-phagy. However, the unique structure of TEX264 solves this problem because its extended C-terminus assists the ER in binding to phagosomes by extending to the cytoplasm and binding to LC3 (22).
2.1.5 CCPG1
CCPG1 is an atypical endoplasmic autophagy receptor that interacts with ATG8 through the typical LIR motif in the cytoplasmic region and binds to FIP200 through the discrete motif (FIR) (23). Zhou et al. also found that the regulation of phosphorylation promotes the interaction of FIP200 with the FIR2 motif of CCPG1 (24). Therefore, it is likely that CCPG1 initiates ER-phagy upon interaction with both proteins. Researchers have found that in addition to the first two autophagy proteins, RB1-inducible coiled-coil (RB1CC1) is also localized to the FIR of CCPG1. Unlike other autophagy receptors, binding to RB1CC1 is required for CCPG1 to trigger ER-phagy (25).
2.1.6 SEC62
SEC62 is a component of the translocon complex and a critical ER-phagy receptor (26). Under ER stress induction, SEC62 binds to the ATG8 autophagy-associated protein to assist in the formation of autophagosomes containing misfolded or unfolded proteins and the ER. Autophagosomes are then transmitted to lysosomes (27). Recent studies have demonstrated that SEC62 not only participates in the UPR, but also improves stress tolerance during the ER stress recovery phase, indicating that SEC62 is a significant autophagy receptor during the ER stress recovery phase (26, 27). The bulk of secretory and membrane proteins is located in the ER and is translocated via the SEC61 protein transduction channel. Both SEC62 and SEC63 are associated with SEC61 channels, which form the SEC complex and regulate protein translocation (28, 29).
2.1.7 CALCOCO1
Recently, researchers have found that in addition to the six typical ER receptors in mammals, calcium-binding and CALCOCO1 also mediate ER-phagy as soluble receptors. CALCOCO1 consists of the N-terminal SKIP carboxyl homology (SKICH) domain, the intermediate helix-helix region (CC), and different carboxy-terminal (CT) domains. CALCOCO1 tends to interact with the GABARAP subfamily of ATG8 to mediate macroautophagy. During autophagy, the LIR docking site (LDS) and UIM‐docking site (UDS) are required to stabilize the GABARAP interaction with CALCOCO1, and the UDS sites do not interfere with the role of the LDS sites. CALCOCO1 acts with vesicle-associated membrane protein (vamp)-associated proteins to mediate ER-phagy via two phenylalanines (FFs) in an acidic tract (FFAT) motif (30).
2.2 Other novel regulatory mechanisms of ER-phagy
With a more comprehensive understanding of the regulatory mechanism of ER-phagy, researchers have found that OXPHOS and DDRGK1-mediated ER surface UFMylation also promote ER-phagy. OXPHOS is positively correlated with ER-phagy. Under starvation conditions, deficiency of OXPHOS severely impedes ER-phagy activation. UFM1-specific ligase 1 (UFL1) is recruited to the ER by DDRGK1, which subsequently impels the activation, conjugation, and ligation of UFM1 to the substrate ribophorin I (RPN1) and ribosomal protein L26 (RPL26) via ubiquitin-like modifier activating enzyme 5 (UBA5), ubiquitin-fold modifier conjugating enzyme 1 (UFC1), and the UFL1 cascade, thus triggering ER-phagy to ameliorate the intensity of ER stress (Figure 2) (31).
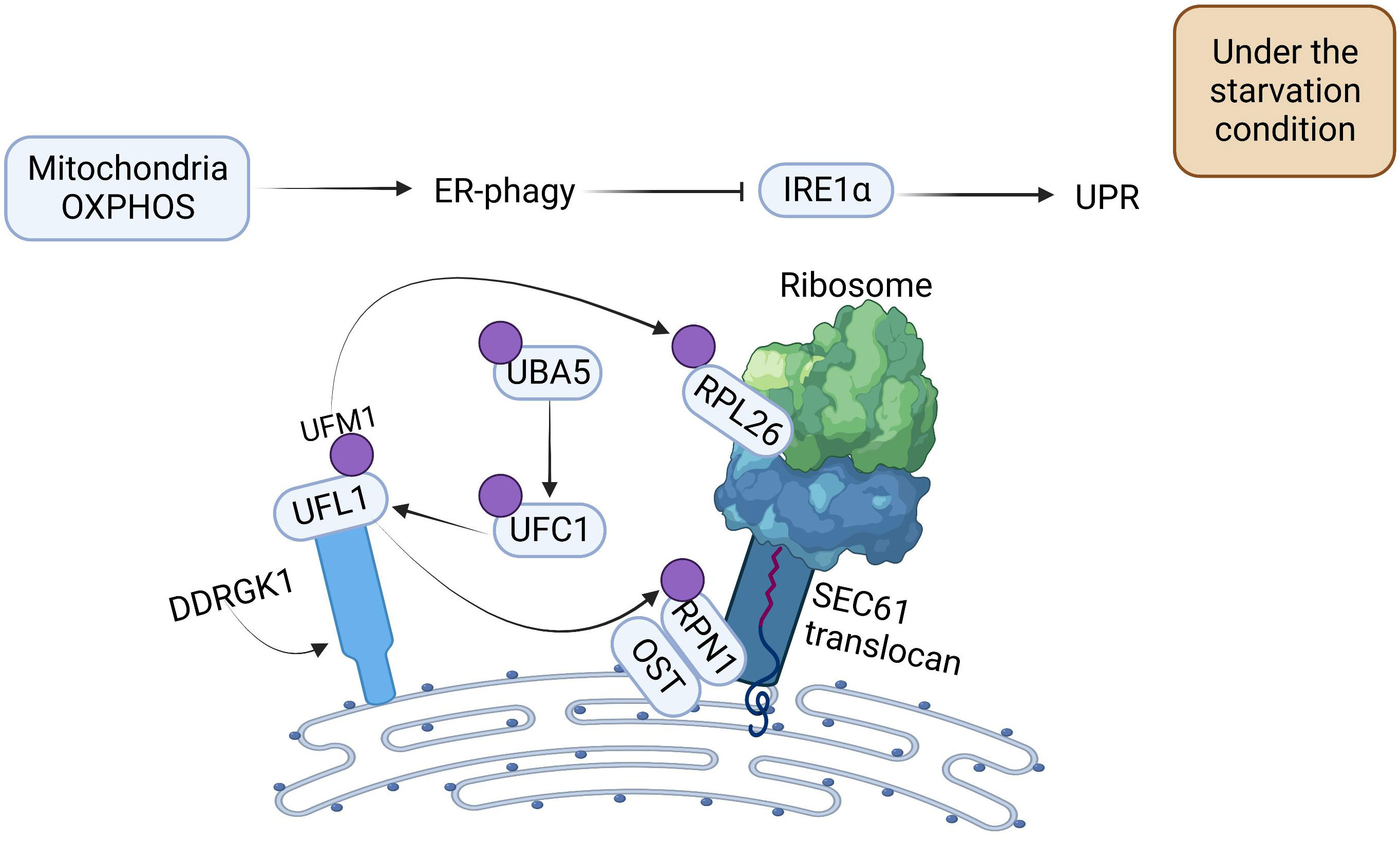
Figure 2 The Mechanism of Oxidative Phosphorylation and DDRGK1-mediated UFMylation Regulation for Propelling ER-phagy. Mitochondria OXPHOS can directly promote ER-phagy. DDRGK1-mediated UFMylation interacts together with E1, E2, and E3. UFL1 delivers UFM1 to the ER surface proteins RPL26 and RPN1, enabling the UFMylation of ER, which then initiates ER-phagy. ER-phagy inhibits IRE1 activity, which in turn blocks the UPR.
3 Potential roles and mechanisms of ER-phagy in ARDS
Autophagy is of great value in delaying ARDS exacerbation. ER-phagy, selective autophagy with high efficiency, plays a crucial role in autophagy. ER-phagy may prevent and ameliorate ARDS induced by different causes by regulating immunity, inflammation, apoptosis, pathogen invasion, and collagen synthesis (Figure 3).
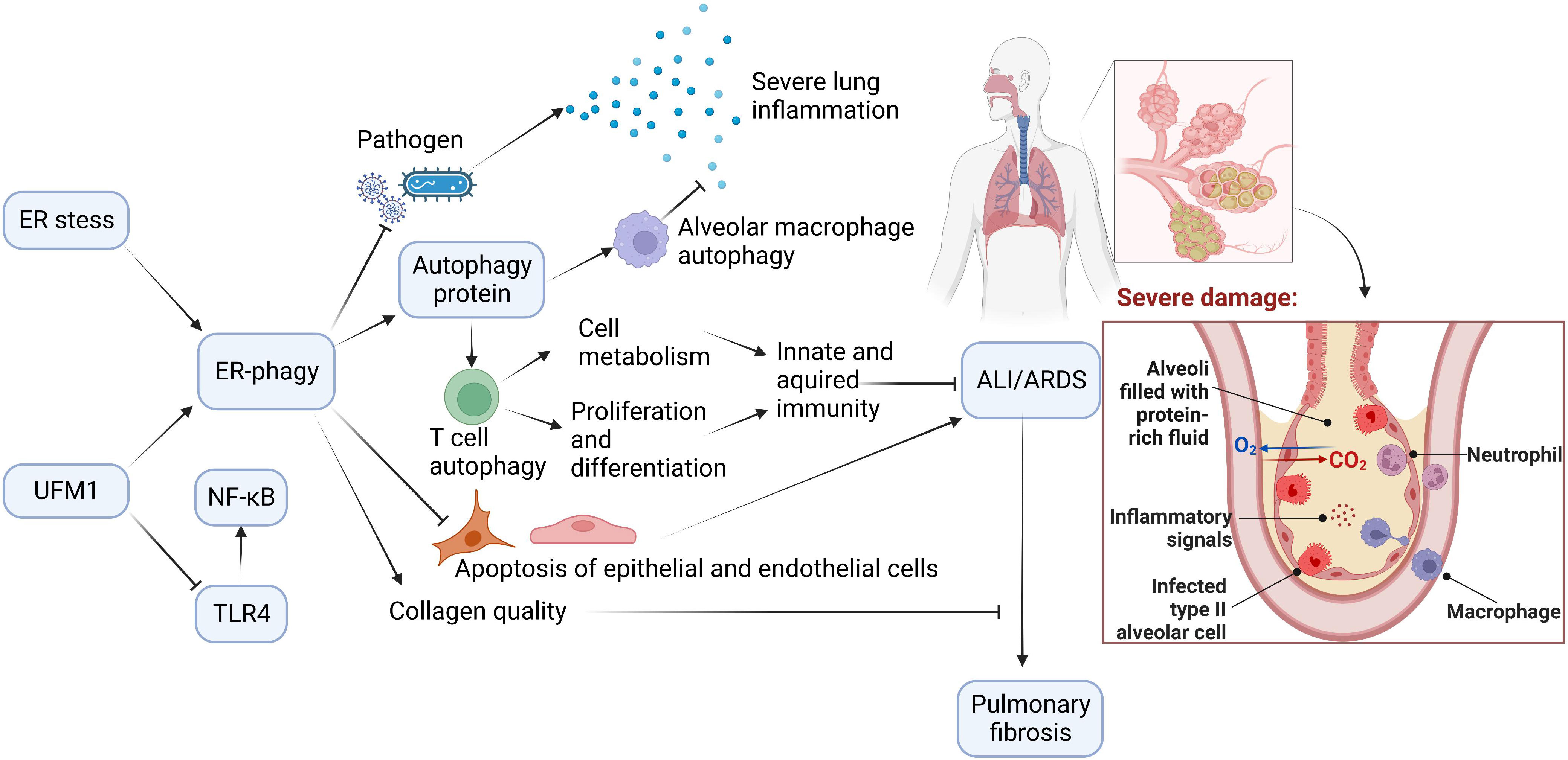
Figure 3 The Potential Role of ER-phagy in ALI/ARDS. Immunity and inflammation are closely associated with ALI/ARDS progression. During the ER-phagy, the autophagy proteins initiate autophagy of immune cells. Autophagy of macrophages reduces inflammation in the lung, and autophagy of T cells promotes cell metabolism, and enhances proliferation and differentiation of cells. ER-phagy can also reduce apoptosis and prevent pathogen infection, which prevent and improve ALI/ARDS. Furthermore, ER-phagy maintains collagen quality and avoids the progression of ALI/ARDS to pulmonary fibrosis.
3.1 ER-phagy regulates immune and inflammatory responses by regulating immune cells, controlling inflammatory material release, and inhibiting variant anti-trypsin and severe pancreatitis
The inflammatory response is the main pathophysiology of ALI/ARDS and the core of ALI/ARDS Pathogenesis (32). Dysregulation of inflammation caused by immune cells is associated with ARDS Pathogenesis (33), and macrophages have been demonstrated to regulate lung inflammation. Macrophages can be polarized into two subsets, namely, M1 and M2. M1 macrophages promote inflammatory responses, while M2 macrophages contribute to the repair of damaged tissue (34). Several studies have demonstrated that the polarization regulation of macrophages plays pivotal roles in various stages of ALI/ARDS (35, 36). Nevertheless, under different circumstances, the properties of diverse macrophage subsets are not rigorously inflexible in each phase of ALI/ARDS (34). Autophagy, responsible for the maintenance of homeostasis, is associated with the polarization of macrophages. Liu et al. found that autophagy promotes the downregulation of inflammation through modulating macrophage polarization in LPS-treated mice (37). Moreover, researchers found that certain drugs use the effect of autophagy on macrophage polarization, thus improving the prognosis of associated diseases (38, 39). For example, Araloside C ameliorates atherosclerosis by regulating macrophage polarization via autophagy (38). Whether ER-phagy is involved in the paradoxical regulation of macrophage polarization remains unresolved, whereas researchers suggested that the certain target at ER affects macrophage polarization by activating selective autophagy. For example, due to the enhancement of M2-like macrophage polarization by regulating autophagy via NOD-like receptor family, pyrin domain containing 3 (NLRP3) deubiquitination, ubiquitin-specific protease 19 (USP19), an ER-anchored deubiquitinating enzyme, may serve as an important target in treating inflammation-related diseases (40). Autophagy is also closely associated with ALI/ARDS. Lipopolysaccharide (LPS), a common factor inducing ALI/ARDS, may aggravate inflammatory responses via inhibition of autophagosome degradation (41). However, several traditional Chinese medicines suppress inflammatory pathway by promoting autophagosome degradation, such as Astragaloside IV, JFNE-A (42, 43). In addition, the certain drug may alleviate ALI/ARDS by modulating macrophage polarization via autophagy, such as Sirtuin 6 (SIRT6) (44). Be it autophagy itself or the regulation of macrophage polarization via autophagy, all of these mechanisms play critical roles in inflammation. Therefore, the essential contribution of autophagy to immunity is regulating inflammation, and modulating immune cells is involved in the whole process (45).
As a critical type of organelle autophagy, ER-phagy plays a crucial role in alleviating inflammation as well (46). ER-phagy may regulates immune and inflammatory responses by regulating immune cells. Unlike the traditional forms, Anti-PD-L1 (Programmed death ligand 1), a promising measure in treating ALI/ARDS, promotes autophagy by inhibiting the PI3K/Akt/mTOR pathway at the ER in neutrophils, thus attenuating inflammatory responses (47). USP19 anchored at ER alleviates inflammatory responses and damaged tissues by increasing autophagy flux in macrophages (40).Naive T cells differentiate into effector T cells and long-lived memory T cells, which play critical roles in immune responses. As a crucial regulator of T cell quiescence, fully understanding cellular metabolism may be required for developing novel approaches to modulate protective and pathological T cell responses in human diseases (48). T cells require functional autophagy to adapt to activated energy requirements. In T-cell activation and differentiation, autophagy promotes cellular metabolism by degrading cellular components, thus accommodating the energy requirements of activated T cells (49).A previous study has demonstrated that the initiation of ER-phagy by ER stress maintains homeostasis in mature T lymphocytes. Researchers have shown that ER-phagy regulates calcium influx by maintaining ER homeostasis, which subsequently improves T-cell function (50). Recently, the prevalent coronavirus disease 2019 (COVID-19) has been the critical cause of ARDS, and ICU patients with COVID-19 are associated with severe ARDS. Researchers have found that nonsurvivors have significantly increased neutrophils and significantly reduced T cells. Therefore, maintaining homeostasis of T cells is likely to be crucial for preventing and ameliorating ALI/ARDS (51). Although the researches of ER-phagy of immune cells in ALI/ARDS is not very clear, many researchers demonstrated that immune cells maintain homeostasis through autophagy, thus improving the prognosis of ALI/ARDS. Alveolar macrophages are the main immune cells in lung, and researchers have suggested that inducing autophagy in alveolar macrophages significantly alleviates lung inflammation (52). MicroRNAs are associated with the autophagy of alveolar macrophages in LPS-induced ALI/ARDS. Exosomal miR-384-5p target the Beclin-1 autophagy protein to maintain the stabilization of autophagy in alveolar macrophages, thus alleviating lung pathological changes and attenuating inflammatory responses (53). Due to the induction of autophagy of macrophages via inhibition of transforming growth factor-β-activated kinase-1-binding protein 2 (TAB2), miR155 becomes a potential candidate for anti-inflammatory therapy during septic lung Injury (54). Moreover, researches demonstrated that some drugs ameliorate ALI/ARDS by activated autophagy of macrophages to mitigate inflammation. SIRT6 induces autophagy of macrophages by activating the AMPK pathway, which leads to M2 macrophage polarization and attenuates inflammation in sepsis-induced ARDS (44). Lipoxins (LXs), synthesized by immune cells such as macrophages and neutrophils, promote anti-inflammatory activities. Researchers showed that LXA4 receptor agonist alleviates ALI-associated inflammation and lung injury by stimulating autophagy (55). Sophoridine may inhibit LPS-induced ALI by enhancing autophagy of macrophages and reducing inflammation (41). At present, the role of autophagy of immune cells in clinical practice is still under research, and these researches mainly focus on immune system diseases. A-synuclein, an autophagy-related marker of peripheral blood lymphocytes, is potentially applied to diagnosis of systemic lupus erythematosus in clinical practice (56). Due to inhibition of over-expansion of activated T lymphocytes via autophagy, Tripterygium glycoside fraction n2 (TGA2) provides an effective therapy for inflammatory bowel disease (IBD) (57). These findings also provide important values for exploring functions of ER-phagy of immune cells in clinical practice.
When defending against infection, some immune cells, including macrophages and neutrophils, often cause inflammatory responses in the lung. Pulmonary inflammation is mainly caused by inflammatory factors released from alveolar macrophages. However, the autophagy-related proteins, ATG and FIP200, have been found to maintain innate immune homeostasis, thus avoiding aggravating ALI/ARDS caused by dysregulated inflammation (58). Although ER-phagy effectively regulates lung inflammation, excessive ER-phagy produces side effects. Neutrophils are the first line of defense against pathogen invasion, and the neutrophil extracellular trap (NET) is released to control spread of the pathogen and then kill the pathogen (59). SARS-CoV-2 induces NET release from neutrophils, while excessive NET release causes apoptosis of lung epithelial cells and severe inflammation (60). However, more vigorous activity of autophagy proteins causes more NET release, suggesting that excessive ER-phagy may lead to COVID-19-induced ARDS (60, 61). Oxidative stress and the inflammatory response play vital roles in sepsis-induced ALI/ARDS. Bhattacharya et al. found that the lack of autophagy proteins alleviates oxidative stress and neutrophil-mediated inflammation (62). Therefore, regulation of ER-phagy is needed to alleviate sepsis-induced ALI/ARDS.
Some ER-phagy receptors also initiate ER-phagy to reduce lung inflammation, thus preventing and ameliorating ALI/ARDS. Variant antitryptases cause reduced lung tissue compliance and aggravate lung inflammation. Researchers have found that endoplasmic reticulum to lysosome-related degradation (ERLAD) removes these variant proteins, while the delivery of variant proteins to the lysosome requires LC3 lipidation. FAM134B-mediated ER-phagy degrades the variant of anti-trypsin by binding to lipidated LC3, thus relieving lung tissue inflammation and facilitating gas exchange (63). In the early stage of acute pancreatitis, the local pancreatic tissue releases large amounts of inflammatory cytokines into the bloodstream, which invade the intestine via blood flow and damage the intestinal barrier. Increased intestinal wall permeability allows bacteria and endotoxins to enter the bloodstream, causing severe inflammatory responses that disrupt the endothelial barrier (64). Nevertheless, Lahiri et al. found that the exocrine trypsin accumulated in the ER triggers ER stress, and CCPG1-mediated ER-phagy relieves pancreatic inflammation and ER stress, thus avoiding severe pancreatitis-induced ALI/ARDS (65).
In addition, NF-κB plays a fundamental role in inflammatory and immune responses, which is associated with pathogenesis of ARDS. Nimbolide protects against ARDS through suppressing NF-κB nuclear translocation (66). DDRGK1 and UFL1 mediate ER-phagy by interacting with UFM1. Some researches found that DDRGK1 and UFL1, UFM1 are involved in modulating NF-κB signaling. DDRGK1 degrades inhibitors of nuclear factor kappa-B (IκB), resulting in NF-κB nuclear translocation (67). In contrast, UFL1 and UFM1 abrogates NF-κB activation. For example, overexpression of UFM1 suppresses the LPS-induced toll-like receptor 4 (TLR4) pathway, preventing NF-κB nuclear translocation and alleviating the inflammatory response to avoid endothelial damage (68). However, the functional relevance of ER-phagy in NF-κB signaling requires further exploration, which may provide novel approaches to the treatment of ALI/ARDS.
3.2 ER-phagy inhibits the apoptosis of alveolar epithelial cells, lung capillary endothelial cells induced by ER stress/mitochondrial dysfunction
In the early stage, UPR maintains cellular homeostasis. However, if unstable ER is persistent, UPR will induce cell death through apoptotic mechanisms, such as PERK- C/EBP homologous protein (CHOP) (69), IRE1- apoptosis signal-regulating kinase (ASK)-Jun N-terminal kinases (JNK) (70). In addition, Calcium also plays a key role in apoptosis triggered by UPR (71). Similar to ER stress, autophagy is a cytoprotective mechanism under nonphysiological conditions. The relationship between autophagy and apoptosis is changeable. Under normal circumstances, autophagy can protect cells from apoptosis. However, prolonged autophagy induced by ER-stress may contribute to apoptosis. In some conditions, apoptosis induced by ER stress even turns to a protective autophagy in resistance to some drugs (72).
ER-phagy is closely linked to ER stress, and the UPR is involved in ER stress triggering ER-phagy (73). For example, Metformin promotes autophagosome formation by activating UPR (74). However, when ER stress loses its balance with ER-phagy, excessive ER stress and ER-phagy induce apoptosis, which exacerbates the progression of ALI/ARDS. Therefore, appropriate regulation of the interaction between ER-phagy and ER stress effectively reduces apoptosis, which may prevent and improve ALI/ARDS. It has been found that both the activation and inhibition of ER-phagy regulate the intensity of ER stress. Liu and colleagues found that Dexmedetomidine induces ER-phagy by activating FAM134B, which subsequently attenuated the ER stress (75). Globular adiponectin has been found to activate SEC62-mediated ER-phagy, thus mitigating ER stress-induced cardiomyocytes apoptosis (76).However, Lim et al. found that palmitate reduces ER stress by inhibiting ER-phagy (77). Persistent ER stress has been demonstrated to cause apoptosis of alveolar epithelial cells, whereas appropriate ER-phagy decreases apoptosis by alleviating ER stress. In acute lung injury models, 4-phenyl butyric acid (4-PBA) triggers ER-phagy to regulate excessive ER stress, attenuating lung inflammation and reducing apoptosis of alveolar epithelial cells (78). As a scavenger of reactive oxygen species (ROS) and inducer of antioxidant systems, Melatonin suppresses ER stress by restoring autophagic flux, thus attenuating apoptosis and lower infiltration of inflammatory cells in lung (79). Therefore, Melatonin may contribute to ameliorate ALI/ARDS by regulating interaction between ER stress and ER-phagy. In addition to persistent ER stress leading to apoptosis, inhibition of ER-phagy also activates the apoptosis pathway in certain conditions (80).
Mitochondrial dysfunction exacerbates hyperoxia-induced damage to alveolar epithelial cells (81). As the most important organelles in the cell, the ER and mitochondria have tight junction structures known as mitochondria-associated ER membranes (MAMs). Recently, the endoplasmic reticulum-mitochondria encounter structure (ERMES) has been demonstrated to connect the ER and the mitochondria, facilitating the exchange of material between the two organelles. However, misfolded proteins accumulated in the ERMES cause mitochondrial dysfunction (82). Kellner et al. found that heavily accumulated ROS induces oxidative stress, which later causes ALI/ARDS by increasing the apoptosis of lung capillary endothelial cells (83). Chen et al. found that impaired mitochondria produce excessive ROS. Nevertheless, ER-phagy has been demonstrated to inhibit apoptosis due to oxidative stress and ER stress in other tissues. In the model of mitochondrial injury, Panax notoginseng saponin (PNS) attenuates ROS-mediated ryanodine receptor 2 (RYR2) oxidation, subsequently inhibiting ER stress-induced apoptosis via ER-phagy (84). Luo et al. found that the accumulation of AGEs activates the ROS pathway, which initiates FAM134B-mediated ER-phagy and then impedes the apoptosis of nucleus pulposus cells (85).
Although the apoptotic effect of ER-phagy on ALI/ARDS has not yet been determined, researchers have demonstrated that ER-phagy is closely associated with lung diseases, which plays crucial roles in cellular survival. As inducers of ER-phagy, both FAM134B-1 and FAM134B-2 isoforms are positive in normal lung tissue, whereas a significant downregulation of FAM134B is observed in lung cancer (86). In addition, many researches proved that autophagy is involved in the progression of ALI/ARDS by regulating apoptosis. Epithelial cell dysfunction, such as Alveolar type 2 (AT2) cells, has emerged as a critical part of the pathophysiology of diffuse parenchymal diseases. Quality control systems, including UPR and autophagy, are essential to avoid the occurrence of ALI/ARDS. AT2 cells maintain homeostasis by selective removal of impaired cellular organelles via autophagy, such as ER or lysosome-related organelles (87). The oxidative stress is associated with ROS and autophagy, and autophagy is seen as a secondary defense of oxidative stress. Beyer et al. found that autophagy ameliorates dysfunctional mitochondria, thus restoring barrier integrity by regulating apoptosis of lung microvascular endothelial cells in ROS-induced lung injury (88). Certain drugs, such as Genipin, inhibit apoptosis by mitigating oxidative stress and mitochondrial damage via activation of autophagy in LPS-induced ALI (89).
Besides, apoptotic alveolar macrophages releases more ROS and exacerbates inflammatory cascading, resulting in apoptosis of alveolar epithelial cells and aggravated lung injury (52, 90). However, autophagy of alveolar macrophages contributes to maintain cellular homeostasis and ameliorate lung injury by regulating apoptosis. Fan et al. found that autophagy of alveolar macrophages reduces alveolar macrophage apoptosis by mitigating endoplasmic reticulum stress and oxidative stress (91). MiR−223−3p−loaded exosomes activates alveolar macrophage autophagy and restores anti-apoptotic effects by targeting serine/threonine kinase 39 (STK39) in the lung tissue of ALI mice (92). Meanwhile, the integrity of degradation is essential for the autophagy process. Under reduced lysosome conditions, LPS promotes the accumulation of autophagosomes in alveolar macrophages, leading to severe inflammation and apoptosis of alveolar macrophages (90). Certain drugs can alleviate ALI by suppressing apoptosis of alveolar macrophages via autophagy of alveolar macrophages, and some of them contributes to alleviate lung inflammation as well, including LXA4 receptor agonist (55). Nevertheless, Complement C5a exacerbates alveolar macrophage apoptosis via enhancement of autophagy in acute lung injury (93).
ER-phagy is triggered by mediating ER surface UFMylation via activated UFM1. IRE1α cleaves the X-box-binding protein 1 (XBP-1)-encoded mRNA via endonuclease to produce the active transcription factor XBP1, thus initiating the UPR. XBP-1 induces the upregulation of UFM1 expression by inhibiting vesicular trafficking (94). The translocation of UFM1 to the ER has been found to be dependent on UFM1-binding protein 1 containing a PCI domain (UFBP1). Under ER stress, UFM1 and UFBP1 are upregulated and reduce apoptosis, suggesting that UFM1-mediated ER-phagy may protect tissue injury by inhibition of apoptosis (95).
3.3 ER-phagy avoids the proliferation and transmission of pathogens
Pathogen infection is a common cause of ALI/ARDS, and ER-phagy is closely related to pathogenicmicrobial clearance. ER-phagy may prevent infection by activated ER-phagy receptors or regulating immune cells, thus preventing or ameliorating the development of ALI/ARDS.
ER-phagy receptors, including FAM134B, ATL3, SEC62, and others, control viral proliferation and transmission. Viral invasion of lung tissue leads to damaged vascular endothelium, the body then initiates FAM134B-mediated ER-phagy and reduces the supply of raw materials and vesicle membranes required for viral replication, thus inhibiting viral proliferation and transmission (13). ATL3 interacts with the viral capsid, preventing virus assembly and transport, and ATL3 cleaves the spike protein on the immature SARS-CoV-2 surface by activating the Flynn protease (96). Upon activation of SEC62 via viral infection, activated SEC62 drives IRE1α phosphorylation, which subsequently induces the IRE1-JNK pathway and delivers autophagosomes to lysosomes, thus mitigating virus-induced ER stress and impeding viral replication (97). However, RTN3 endures viral proliferation, facilitating the virus to escape from the ER to the cytoplasm by reshaping the ER membrane to make it more flexible (98). Usually, viruses are cleared by ER-phagy, but coronaviruses inhibit autophagosome formation through related proteases or their specialized structures, resulting in impaired ER-phagy (99). ER-phagy, mediated by ER-phagy receptors, is also associated with fungal infection. For example, in response to A.fumigatus stimulation, the levels of CCPG1 expression are increased to a large extent. CCPG1-mediated ER-phagy removes pathogens and suppresses excessive inflammatory responses, thus avoiding aggravating ALI/ARDS caused by pulmonary fungal infection in the immunocompromised setting (100).
Many researches have shown that autophagy of immune cells plays a vital role in improving the prognosis of ALI/ARDS, such as alveolar macrophages. Due to the effective removal of intracellular pathogens and subcellular organelles (including ER) through activation of autophagy, STING was initially viewed as a crucial molecule in immunity. However, a recent research has demonstrated that in alveolar macrophages, STING inhibits autophagic flux by perturbing lysosomal digestion, thus leading to sepsis-induce ALI (101). Autophagy dysfunction induced by prostaglandin E2 (PGE2) contributes to survival of P. aeruginosa in alveolar macrophages, thus increasing the occurrence of ALI in P. aeruginosa-infected bone marrow transplant (BMT) mice (102). Defective autophagy leads to depletion of ATP synthesis by severe mitochondrial dysfunction, then significantly decreases antimicrobial activity. However, researchers have found that AMPK- peroxisome proliferator-activated receptor-gamma, coactivator 1α (PPARGC1A) axis promotes antimicrobial host defense through activation of autophagy in alveolar macrophages (103). Although the direct effect of ER-phagy of immune cells on pathogen infection is not largely explored, ER-phagy may regulate the proliferation and differentiation of immune cells through autophagy proteins. Catechins activate CD8+ T cytotoxic lymphocytes by upregulating the protein levels of Beclin-1 and Atg5-Atg12, thus enhancing the adaptive immunity against viral infections (104). ATG proteins inhibit activated CD8+ T cells from converting into effector cells, whereas autophagy also promotes the transformation of effector cells into memory cells during this process, which improves the quality of memory cells and strengthens the host defense mechanisms (105). Pei and colleagues found that ATG proteins are associated with the proliferation and differentiation of NKT cells. The deletion of ATG proteins causes a corresponding decrease in NKT cells, which weakens both innate and acquired immune function. Due to the limitation of resisting pathogen infection effectively, infection-induced ALI/ARDS is more easily caused (106).
In addition to clearance of virus and fungus, ER-phagy is also responsible for eliminating bacteria. Bacteria survive and proliferate in host cells, which depend on a safe and nutritive environment. The ER has such conditions, which encourage bacteria to stabilize and multiply inside the cell (107). These bacteria are prone to trigger ER stress. As a result, ER-phagy is initiated, immediately eliminating bacteria and maintaining ER homeostasis (108). Researchers have demonstrated that when infected by gram-positive bacteria, the defensive machinery activates ER-phagy by inhibiting mTOR activity. ER-phagy reduces PERK and CHOP expression, and it improves the immune system to enhance the defensive function of the host (109).
3.4 ER-phagy prevents the progression of ALI/ARDS to pulmonary fibrosis by controlling collagen synthesis
If the pathological changes of ALI/ARDS are not corrected immediately, severe pulmonary fibrosis, the major cause of mortality in ARDS patients, will occur in the later stage to aggravate the development of the disease (110). Some researchers have attempted to restore the integrity of alveolar-capillary barrier by two molecules, platelet-endothelial cell adhesion molecule-1 (PECAM1) and wingless-related integration site (Wnt), thus attenuating diffuse alveolar damage and preventing fibroproliferative ARDS (111).
In addition to inducing apoptosis in lung epithelial cells, persistent ER stress also exacerbates lung fibrosis. ALI appears in bleomycin-treated mice, and palmitate increases apoptosis of lung epithelial cells, which exacerbates endoplasmic reticulum stress by upregulating the expression of UPR proapoptotic proteins, resulting in the progression of ALI/ARDS to pulmonary fibrosis (112). Moreover, researches have shown that ER stress promotes the development and progression of pulmonary fibrosis by the regulation of alveolar epithelial cell (AEC) apoptosis, epithelial–mesenchymal transition, and myofibroblast differentiation (113). Therefore, controlling the intensity of ER stress may be beneficial for the prognosis of ALI/ARDS through reduced pulmonary fibrosis.
ER stress is closely associated with autophagy, and some drugs-mediated protection against pulmonary fibrosis through regulation of ER stress and autophagy. For example, Spermidine has been shown to resolve fibrosis by inhibiting ER stress and enhancing autophagy. Thus, autophagy also plays a critical role in suppressing fibrotic lung. Recently, researches demonstrated that defective autophagy contributes to excess production of extracellular matrix and alteration of fibroblasts to myofibroblasts (114). Yang et al. found that Oridonin attenuates LPS-induced early pulmonary fibrosis by restoring impaired autophagy (115). Therefore, high-quality autophagy may prevent ALI/ARDS from progressing to pulmonary fibrosis. As a cellular organelle with efficient autophagy, ER has been demonstrated to be involved in a novel anti-fibrotic function (116). Excessive deposition of extracellular matrix, mainly collagen protein, is the main factor leading to pulmonary fibrosis. However, ER-phagy maintains procollagen quality to prevent fibrosis due to intense ER stress. Because collagen synthesis is complex and prone to errors, FAM134B-mediated ER-phagy degrades misfolded collagen, avoiding intense ER stress due to the accumulation of these proteins. Researchers found that the ER-resident lectin chaperone protein, CANX, and the ER phagocytic receptor, FAM134B, are pivotal in this process. CANX, a coreceptor for FAM134B-mediated ER-phagy, is responsible for binding and recognizing misfolded procollagen in the ER lumen. FAM134B then binds to the LC3 autophagic protein and transports the ER containing CANX and procollagen to lysosomes for degradation (117). FAM134A and FAM134C also function in maintaining collagen homeostasis. Therefore, deficiency of the FAM134 protein inhibits ER-phagy, thus causing a significant increase in collagen and leading to a dramatically increased incidence of pulmonary fibrosis (118).
Apart from the regulation of immunity, inflammation, apoptosis, and pathogen infection, autophagy of immune cells also plays an important role in pulmonary fibrosis. Aggregated macrophages in damaged lung areas may aggravate inflammation and accelerate fibrogenesis, whereas Dioscin decreases chemokines and proinflammatory cytokines secretion by inducing autophagy of alveolar macrophages, thus avoiding pulmonary fibrosis due to collagen deposition (52). Trehalose exerts antifibrotic effects by restoring lysosomal function, accelerating autophagic substrate degradation, and enhancing autophagic flux in Crystalline Silica (CS)-treated Alveolar Macrophages (119). Nevertheless, Liu et al. found that TP53-upregulated modulator of apoptosis (BBC3) triggers the release of pro-fibrotic cytokines by enhancing autophagy of alveolar macrophages (120).
4 Conclusions and perspectives
ALI/ARDS, an intractable inflammatory disease in ICU, has a high mortality rate. It is urgent to find the treatment for ALI/ARDS, and the key is to understand the pathogenesis. However, the pathogenesis of ARDS is not elucidated up to now.
ER-phagy was discovered in 2007, and researchers began intensively studying ER-phagy through ER-phagy receptors in 2015. Therefore, as an important organelle autophagy, ER-phagy is a relatively new field of research. Recently, researchers have conducted many studies on ER-phagy. When ER stress asts too long, and the proteasome degradation system is insufficient to compensate, ER-phagy is mediated by different ER-phagy receptors or other pathways, resulting in removal of damaged ER fragments, effectively improving the intracellular environment. In other damaged tissues, since ER-phagy is involved in the regulation of inflammation and apoptosis, it is regarded as an important therapeutic target of associated diseases.
ALI/ARDS is characterized by complex pathological changes, and inflammation and apoptosis play crucial roles in each pathological stage. Although the specific mechanisms of ER-phagy in ALI/ARDS have not yet be determined, ER stress and autophagy have been proved to be involved in the progression of ALI/ARDS. Researches demonstrated that ER stress is a vital pathogenesis leading to ALI/ARDS, and this damage can be reversed by autophagy via activation or inhibition of related pathways. ER-phagy may be involved in the different etiology-related ARDS by regulating immunity and inflammation, modulating apoptosis, preventing microbial infection, and preventing lung fibrosis. Besides, immunity and inflammation are the most important pathogenesis of ALI/ARDS, and immune cells are essential to the immune and inflammatory responses. Although ER-phagy of immune cells is not largely explored, the autophagy of immune cells does play critical roles in ALI/ARDS through affecting four aspects, including immunity and inflammation, apoptosis, microbial infection, and fibrosis. Many researches have shown that autophagy of immune cells can ameliorate ALI/ARDS. However, several studies suggested that under certain circumstances, autophagy has a detrimental role in ALI/ARDS pathogenesis, which is related to the primary etiology, the cell types, the stage of ALI/ARDS. Therefore, further study on ER-phagy of immune cells is a promising measure to resolve problems of therapies. As for the detail directions of researches, researchers can mainly focus on the relevance of ER-phagy in macrophage polarization and inflammatory signals in the future.
Existing studies have demonstrated that ER-phagy is pivotal for improving the prognosis of many diseases. Accordingly, conducting experiments on the effects of ER-phagy in ALI/ARDS may provide significant values in developing novel therapies for ALI/ARDS.
Author contributions
SL, XF, RZ, JZ, HW, JL, CW, LW, LZ wrote and reviewed the manuscript. SL prepared the figures. SL and XF discussed the ideas in the draft. LZ reviewed the manuscript and provided suggestions. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Key Research and Development Program of China (2021YFC2501800) and National Natural Science Foundation of China (82102245).
Acknowledgments
The authors would like to acknowledge the literatures that has contributed to this field but is not included here due to the length of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Butt Y, Kurdowska A, Allen TC. Acute lung injury: a clinical and molecular review. Arch Pathol Lab Med (2016) 140(4):345–50. doi: 10.5858/arpa.2015-0519-RA
2. Mowery NT, Terzian WTH, Nelson AC. Acute lung injury. Curr Probl Surg (2020) 57(5):100777. doi: 10.1016/j.cpsurg.2020.100777
3. Meyer NJ, Gattinoni L, Calfee CS. Acute respiratory distress syndrome. Lancet (2021) 398(10300):622–37. doi: 10.1016/s0140-6736(21)00439-6
4. Saguil A, Fargo MV. Acute respiratory distress syndrome: diagnosis and management. Am Fam Physician (2020) 101(12):730–8.
5. Bellani G, Laffey JG, Pham T, Fan E, Brochard L, Esteban A, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. Jama (2016) 315(8):788–800. doi: 10.1001/jama.2016.0291
6. Auriemma CL, Zhuo H, Delucchi K, Deiss T, Liu T, Jauregui A, et al. Acute respiratory distress syndrome-attributable mortality in critically ill patients with sepsis. Intensive Care Med (2020) 46(6):1222–31. doi: 10.1007/s00134-020-06010-9
7. Herridge MS, Tansey CM, Matté A, Tomlinson G, Diaz-Granados N, Cooper A, et al. Functional disability 5 years after acute respiratory distress syndrome. N Engl J Med (2011) 364(14):1293–304. doi: 10.1056/NEJMoa1011802
8. Yao RQ, Ren C, Xia ZF, Yao YM. Organelle-specific autophagy in inflammatory diseases: a potential therapeutic target underlying the quality control of multiple organelles. Autophagy (2021) 17(2):385–401. doi: 10.1080/15548627.2020.1725377
9. Song S, Tan J, Miao Y, Zhang Q. Crosstalk of er stress-mediated autophagy and er-phagy: involvement of upr and the core autophagy machinery. J Cell Physiol (2018) 233(5):3867–74. doi: 10.1002/jcp.26137
10. Bernales S, Schuck S, Walter P. Er-phagy: selective autophagy of the endoplasmic reticulum. Autophagy (2007) 3(3):285–7. doi: 10.4161/auto.3930
11. Mo J, Chen J, Zhang B. Critical roles of Fam134b in er-phagy and diseases. Cell Death Dis (2020) 11(11):983. doi: 10.1038/s41419-020-03195-1
12. Chiramel AI, Dougherty JD, Nair V, Robertson SJ, Best SM. Fam134b, the selective autophagy receptor for endoplasmic reticulum turnover, inhibits replication of Ebola virus strains makona and mayinga. J Infect Dis (2016) 214(suppl 3):S319–s25. doi: 10.1093/infdis/jiw270
13. Lennemann NJ, Coyne CB. Dengue and zika viruses subvert reticulophagy by Ns2b3-mediated cleavage of Fam134b. Autophagy (2017) 13(2):322–32. doi: 10.1080/15548627.2016.1265192
14. Cinque L, De Leonibus C, Iavazzo M, Krahmer N, Intartaglia D, Salierno FG, et al. Mit/Tfe factors control er-phagy Via transcriptional regulation of Fam134b. EMBO J (2020) 39(17):e105696. doi: 10.15252/embj.2020105696
15. Grumati P, Morozzi G, Hölper S, Mari M, Harwardt MI, Yan R, et al. Full length Rtn3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. Elife (2017) 6. doi: 10.7554/eLife.25555
16. Chen YJ, Williams JM, Arvan P, Tsai B. Reticulon protects the integrity of the er membrane during er escape of Large macromolecular protein complexes. J Cell Biol (2020) 219(2). doi: 10.1083/jcb.201908182
17. Yang Z, Wang J, He B, Zhang X, Li X, Kuang E. Rtn3 inhibits rig-I-Mediated antiviral responses by impairing Trim25-mediated K63-linked polyubiquitination. Elife (2021) 10. doi: 10.7554/eLife.68958
18. Chen Q, Teng J, Chen J. Atl3, a cargo receptor for reticulophagy. Autophagy (2019) 15(8):1465–6. doi: 10.1080/15548627.2019.1609862
19. Liu N, Zhao H, Zhao YG, Hu J, Zhang H. Atlastin 2/3 regulate er targeting of the Ulk1 complex to initiate autophagy. J Cell Biol (2021) 220(7). doi: 10.1083/jcb.202012091
20. Delorme-Axford E, Popelka H, Klionsky DJ. Tex264 is a major receptor for mammalian reticulophagy. Autophagy (2019) 15(10):1677–81. doi: 10.1080/15548627.2019.1646540
21. An H, Ordureau A, Paulo JA, Shoemaker CJ, Denic V, Harper JW. Tex264 is an endoplasmic reticulum-resident Atg8-interacting protein critical for er remodeling during nutrient stress. Mol Cell (2019) 74(5):891–908.e10. doi: 10.1016/j.molcel.2019.03.034
22. Fielden J, Popović M, Ramadan K. Tex264 at the intersection of autophagy and DNA repair. Autophagy (2022) 18(1):40–9. doi: 10.1080/15548627.2021.1894059
23. Smith MD, Harley ME, Kemp AJ, Wills J, Lee M, Arends M, et al. Ccpg1 is a non-canonical autophagy cargo receptor essential for er-phagy and pancreatic er proteostasis. Dev Cell (2018) 44(2):217–32.e11. doi: 10.1016/j.devcel.2017.11.024
24. Zhou Z, Liu J, Fu T, Wu P, Peng C, Gong X, et al. Phosphorylation regulates the binding of autophagy receptors to Fip200 claw domain for selective autophagy initiation. Nat Commun (2021) 12(1):1570. doi: 10.1038/s41467-021-21874-1
25. Smith MD, Wilkinson S. Ccpg1, a cargo receptor required for reticulophagy and endoplasmic reticulum proteostasis. Autophagy (2018) 14(6):1090–1. doi: 10.1080/15548627.2018.1441473
26. Hu S, Ye H, Cui Y, Jiang L. Atsec62 is critical for plant development and is involved in er-phagy in arabidopsis thaliana. J Integr Plant Biol (2020) 62(2):181–200. doi: 10.1111/jipb.12872
27. Fumagalli F, Noack J, Bergmann TJ, Cebollero E, Pisoni GB, Fasana E, et al. Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat Cell Biol (2016) 18(11):1173–84. doi: 10.1038/ncb3423
28. Jung SJ, Kim H. Emerging view on the molecular functions of Sec62 and Sec63 in protein translocation. Int J Mol Sci (2021) 22(23). doi: 10.3390/ijms222312757
29. Mandon EC, Trueman SF, Gilmore R. Protein translocation across the rough endoplasmic reticulum. Cold Spring Harb Perspect Biol (2013) 5(2). doi: 10.1101/cshperspect.a013342
30. Nthiga TM, Kumar Shrestha B, Sjøttem E, Bruun JA, Bowitz Larsen K, Bhujabal Z, et al. Calcoco1 acts with vamp-associated proteins to mediate er-phagy. EMBO J (2020) 39(15):e103649. doi: 10.15252/embj.2019103649
31. Liang JR, Lingeman E, Luong T, Ahmed S, Muhar M, Nguyen T, et al. A genome-wide er-phagy screen highlights key roles of mitochondrial metabolism and er-resident ufmylation. Cell (2020) 180(6):1160–77.e20. doi: 10.1016/j.cell.2020.02.017
32. Huppert LA, Matthay MA, Ware LB. Pathogenesis of acute respiratory distress syndrome. Semin Respir Crit Care Med (2019) 40(1):31–9. doi: 10.1055/s-0039-1683996
33. Yang CY, Chen CS, Yiang GT, Cheng YL, Yong SB, Wu MY, et al. New insights into the immune molecular regulation of the pathogenesis of acute respiratory distress syndrome. Int J Mol Sci (2018) 19(2):588. doi: 10.3390/ijms19020588
34. Chen X, Tang J, Shuai W, Meng J, Feng J, Han Z. Macrophage polarization and its role in the pathogenesis of acute lung Injury/Acute respiratory distress syndrome. Inflammation Res (2020) 69(9):883–95. doi: 10.1007/s00011-020-01378-2
35. Wang J, Li R, Peng Z, Zhou W, Hu B, Rao X, et al. Gts-21 reduces inflammation in acute lung injury by regulating M1 polarization and function of alveolar macrophages. Shock (2019) 51(3):389–400. doi: 10.1097/shk.0000000000001144
36. Pinheiro NM, Santana FP, Almeida RR, Guerreiro M, Martins MA, Caperuto LC, et al. Acute lung injury is reduced by the A7nachr agonist pnu-282987 through changes in the macrophage profile. FASEB J (2017) 31(1):320–32. doi: 10.1096/fj.201600431R
37. Liu K, Zhao E, Ilyas G, Lalazar G, Lin Y, Haseeb M, et al. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy (2015) 11(2):271–84. doi: 10.1080/15548627.2015.1009787
38. Luo Y, Lu S, Gao Y, Yang K, Wu D, Xu X, et al. Araloside c attenuates atherosclerosis by modulating macrophage polarization Via Sirt1-mediated autophagy. Aging (Albany NY) (2020) 12(2):1704–24. doi: 10.18632/aging.102708
39. Kawano A, Ariyoshi W, Yoshioka Y, Hikiji H, Nishihara T, Okinaga T. Docosahexaenoic acid enhances M2 macrophage polarization Via the P38 signaling pathway and autophagy. J Cell Biochem (2019) 120(8):12604–17. doi: 10.1002/jcb.28527
40. Liu T, Wang L, Liang P, Wang X, Liu Y, Cai J, et al. Usp19 suppresses inflammation and promotes M2-like macrophage polarization by manipulating Nlrp3 function Via autophagy. Cell Mol Immunol (2021) 18(10):2431–42. doi: 10.1038/s41423-020-00567-7
41. Liang J, Liu J, Tang Y, Peng Q, Zhang L, Ma X, et al. Sophoridine inhibits endotoxin-induced acute lung injury by enhancing autophagy of macrophage and reducing inflammation. J Leukoc Biol (2022) 112(1):115–25. doi: 10.1002/jlb.3ma0322-428r
42. Rao Z, Zeng J, Li X, Peng L, Wang B, Luan F, et al. Jfne-a isolated from jing-fang n-butanol extract attenuates lipopolysaccharide-induced acute lung injury by inhibiting oxidative stress and the nf-Kb signaling pathway Via promotion of autophagy. Phytomedicine (2022) 96:153891. doi: 10.1016/j.phymed.2021.153891
43. Pei C, Wang F, Huang D, Shi S, Wang X, Wang Y, et al. Astragaloside iv protects from Pm2.5-induced lung injury by regulating autophagy Via inhibition of Pi3k/Akt/Mtor signaling in vivo and in vitro. J Inflammation Res (2021) 14:4707–21. doi: 10.2147/jir.S312167
44. Wang QL, Yang L, Liu ZL, Peng Y, Gao M, Deng LT, et al. Sirtuin 6 regulates macrophage polarization to alleviate sepsis-induced acute respiratory distress syndrome Via dual mechanisms dependent on and independent of autophagy. Cytotherapy (2022) 24(2):149–60. doi: 10.1016/j.jcyt.2021.09.001
45. Deretic V, Levine B. Autophagy balances inflammation in innate immunity. Autophagy (2018) 14(2):243–51. doi: 10.1080/15548627.2017.1402992
46. Li T, Chen Y, Li Y, Yao Z, Liu W. Fam134b-mediated endoplasmic reticulum autophagy protects against sepsis myocardial injury in mice. Aging (Albany NY) (2021) 13(10):13535–47. doi: 10.18632/aging.202786
47. Zhu CL, Xie J, Zhao ZZ, Li P, Liu Q, Guo Y, et al. Pd-L1 maintains neutrophil extracellular traps release by inhibiting neutrophil autophagy in endotoxin-induced lung injury. Front Immunol (2022) 13:949217. doi: 10.3389/fimmu.2022.949217
48. Chapman NM, Boothby MR, Chi H. Metabolic coordination of T cell quiescence and activation. Nat Rev Immunol (2020) 20(1):55–70. doi: 10.1038/s41577-019-0203-y
49. Yang G, Song W, Postoak JL, Chen J, Martinez J, Zhang J, et al. Autophagy-related protein Pik3c3/Vps34 controls T cell metabolism and function. Autophagy (2021) 17(5):1193–204. doi: 10.1080/15548627.2020.1752979
50. Jia W, Pua HH, Li QJ, He YW. Autophagy regulates endoplasmic reticulum homeostasis and calcium mobilization in T lymphocytes. J Immunol (2011) 186(3):1564–74. doi: 10.4049/jimmunol.1001822
51. Luo XH, Zhu Y, Mao J, Du RC. T Cell immunobiology and cytokine storm of covid-19. Scand J Immunol (2021) 93(3):e12989. doi: 10.1111/sji.12989
52. Du S, Li C, Lu Y, Lei X, Zhang Y, Li S, et al. Dioscin alleviates crystalline silica-induced pulmonary inflammation and fibrosis through promoting alveolar macrophage autophagy. Theranostics (2019) 9(7):1878–92. doi: 10.7150/thno.29682
53. Liu X, Gao C, Wang Y, Niu L, Jiang S, Pan S. Bmsc-derived exosomes ameliorate lps-induced acute lung injury by mir-384-5p-Controlled alveolar macrophage autophagy. Oxid Med Cell Longev (2021) 2021:9973457. doi: 10.1155/2021/9973457
54. Liu F, Nie C, Zhao N, Wang Y, Liu Y, Li Y, et al. Mir-155 alleviates septic lung injury by inducing autophagy Via inhibition of transforming growth factor-B-Activated binding protein 2. Shock (2017) 48(1):61–8. doi: 10.1097/shk.0000000000000839
55. Liu H, Zhou K, Liao L, Zhang T, Yang M, Sun C. Lipoxin A4 receptor agonist bml-111 induces autophagy in alveolar macrophages and protects from acute lung injury by activating mapk signaling. Respir Res (2018) 19(1):243. doi: 10.1186/s12931-018-0937-2
56. Colasanti T, Vomero M, Alessandri C, Barbati C, Maselli A, Camperio C, et al. Role of alpha-synuclein in autophagy modulation of primary human T lymphocytes. Cell Death Dis (2014) 5(5):e1265. doi: 10.1038/cddis.2014.211
57. Yang YQ, Wu YF, Xu FF, Deng JB, Wu LL, Han XD, et al. Tripterygium glycoside fraction N2: alleviation of dss-induced colitis by modulating immune homeostasis in mice. Phytomedicine (2019) 58:152855. doi: 10.1016/j.phymed.2019.152855
58. Lu Q, Yokoyama CC, Williams JW, Baldridge MT, Jin X, DesRochers B, et al. Homeostatic control of innate lung inflammation by vici syndrome gene Epg5 and additional autophagy genes promotes influenza pathogenesis. Cell Host Microbe (2016) 19(1):102–13. doi: 10.1016/j.chom.2015.12.011
59. Hidalgo A, Libby P, Soehnlein O, Aramburu IV, Papayannopoulos V, Silvestre-Roig C. Neutrophil extracellular traps: from physiology to pathology. Cardiovasc Res (2021) 118(13):2737–53. doi: 10.1093/cvr/cvab329
60. Veras FP, Pontelli MC, Silva CM, Toller-Kawahisa JE, de Lima M, Nascimento DC, et al. Sars-Cov-2-Triggered neutrophil extracellular traps mediate covid-19 pathology. J Exp Med (2020) 217(12). doi: 10.1084/jem.20201129
61. Park SY, Shrestha S, Youn YJ, Kim JK, Kim SY, Kim HJ, et al. Autophagy primes neutrophils for neutrophil extracellular trap formation during sepsis. Am J Respir Crit Care Med (2017) 196(5):577–89. doi: 10.1164/rccm.201603-0596OC
62. Bhattacharya A, Wei Q, Shin JN, Abdel Fattah E, Bonilla DL, Xiang Q, et al. Autophagy is required for neutrophil-mediated inflammation. Cell Rep (2015) 12(11):1731–9. doi: 10.1016/j.celrep.2015.08.019
63. Fregno I, Fasana E, Bergmann TJ, Raimondi A, Loi M, Soldà T, et al. Er-to-Lysosome-Associated degradation of proteasome-resistant atz polymers occurs Via receptor-mediated vesicular transport. EMBO J (2018) 37(17). doi: 10.15252/embj.201899259
64. Liang XY, Jia TX, Zhang M. Intestinal bacterial overgrowth in the early stage of severe acute pancreatitis is associated with acute respiratory distress syndrome. World J Gastroenterol (2021) 27(15):1643–54. doi: 10.3748/wjg.v27.i15.1643
65. Lahiri V, Klionsky DJ. Ccpg1 is a noncanonical autophagy cargo receptor essential for reticulophagy and pancreatic er proteostasis. Autophagy (2018) 14(7):1107–9. doi: 10.1080/15548627.2018.1467199
66. Pooladanda V, Thatikonda S, Bale S, Pattnaik B, Sigalapalli DK, Bathini NB, et al. Nimbolide protects against endotoxin-induced acute respiratory distress syndrome by inhibiting tnf-A mediated nf-Kb and hdac-3 nuclear translocation. Cell Death Dis (2019) 10(2):81. doi: 10.1038/s41419-018-1247-9
67. Xi P, Ding D, Zhou J, Wang M, Cong YS. Ddrgk1 regulates nf-Kb activity by modulating iκbα stability. PloS One (2013) 8(5):e64231. doi: 10.1371/journal.pone.0064231
68. Li YY, Zhang GY, He JP, Zhang DD, Kong XX, Yuan HM, et al. Ufm1 inhibits lps-induced endothelial cell inflammatory responses through the nf-Kb signaling pathway. Int J Mol Med (2017) 39(5):1119–26. doi: 10.3892/ijmm.2017.2947
69. Rozpedek W, Pytel D, Mucha B, Leszczynska H, Diehl JA, Majsterek I. The role of the Perk/Eif2α/Atf4/Chop signaling pathway in tumor progression during endoplasmic reticulum stress. Curr Mol Med (2016) 16(6):533–44. doi: 10.2174/1566524016666160523143937
70. Nishida T, Hattori K, Watanabe K. The regulatory and signaling mechanisms of the ask family. Adv Biol Regul (2017) 66:2–22. doi: 10.1016/j.jbior.2017.05.004
71. Groenendyk J, Agellon LB, Michalak M. Calcium signaling and endoplasmic reticulum stress. Int Rev Cell Mol Biol (2021) 363:1–20. doi: 10.1016/bs.ircmb.2021.03.003
72. Shen Y, Yang J, Zhao J, Xiao C, Xu C, Xiang Y. The switch from er stress-induced apoptosis to autophagy Via ros-mediated Jnk/P62 signals: a survival mechanism in methotrexate-resistant choriocarcinoma cells. Exp Cell Res (2015) 334(2):207–18. doi: 10.1016/j.yexcr.2015.04.010
73. Liu Y, Burgos JS, Deng Y, Srivastava R, Howell SH, Bassham DC. Degradation of the endoplasmic reticulum by autophagy during endoplasmic reticulum stress in arabidopsis. Plant Cell (2012) 24(11):4635–51. doi: 10.1105/tpc.112.101535
74. Kokott-Vuong A, Jung J, Fehr AT, Kirschfink N, Noristani R, Voigt A, et al. Increased post-hypoxic oxidative stress and activation of the perk branch of the upr in Trap1-deficient drosophila melanogaster is abrogated by metformin. Int J Mol Sci (2021) 22(21):11586. doi: 10.3390/ijms222111586
75. Liu Y, Wang S, Wang Z, Ding M, Li X, Guo J, et al. Dexmedetomidine alleviated endoplasmic reticulum stress Via inducing er-phagy in the spinal cord of neuropathic pain model. Front Neurosci (2020) 14:90. doi: 10.3389/fnins.2020.00090
76. Zhang Q, Zhang X, Ding N, Ge L, Dong Y, He C, et al. Globular adiponectin alleviates chronic intermittent hypoxia-induced H9c2 cardiomyocytes apoptosis Via er-phagy induction. Cell Cycle (2020) 19(22):3140–53. doi: 10.1080/15384101.2020.1836438
77. Lim Y, Kim S, Kim EK. Palmitate reduces starvation-induced er stress by inhibiting er-phagy in hypothalamic cells. Mol Brain (2021) 14(1):65. doi: 10.1186/s13041-021-00777-8
78. Zeng M, Sang W, Chen S, Chen R, Zhang H, Xue F, et al. 4-pba inhibits lps-induced inflammation through regulating er stress and autophagy in acute lung injury models. Toxicol Lett (2017) 271:26–37. doi: 10.1016/j.toxlet.2017.02.023
79. Fernández A, Ordóñez R, Reiter RJ, González-Gallego J, Mauriz JL. Melatonin and endoplasmic reticulum stress: relation to autophagy and apoptosis. J Pineal Res (2015) 59(3):292–307. doi: 10.1111/jpi.12264
80. Viswanath P, Radoul M, Izquierdo-Garcia JL, Ong WQ, Luchman HA, Cairncross JG, et al. 2-Hydroxyglutarate-Mediated autophagy of the endoplasmic reticulum leads to an unusual downregulation of phospholipid biosynthesis in mutant Idh1 gliomas. Cancer Res (2018) 78(9):2290–304. doi: 10.1158/0008-5472.Can-17-2926
81. Hernández-Cuervo H, Soundararajan R, Sidramagowda Patil S, Breitzig M, Alleyn M, Galam L, et al. Bmi1 silencing induces mitochondrial dysfunction in lung epithelial cells exposed to hyperoxia. Front Physiol (2022) 13:814510. doi: 10.3389/fphys.2022.814510
82. Cortés Sanchón A, Santhosh Kumar H, Mantovani M, Osinnii I, Mateos JM, Kaech A, et al. Er-misfolded proteins become sequestered with mitochondria and impair mitochondrial function. Commun Biol (2021) 4(1):1350. doi: 10.1038/s42003-021-02873-w
83. Kellner M, Noonepalle S, Lu Q, Srivastava A, Zemskov E, Black SM. Ros signaling in the pathogenesis of acute lung injury (Ali) and acute respiratory distress syndrome (Ards). Adv Exp Med Biol (2017) 967:105–37. doi: 10.1007/978-3-319-63245-2_8
84. Chen J, Li L, Bai X, Xiao L, Shangguan J, Zhang W, et al. Inhibition of autophagy prevents panax notoginseng saponins (Pns) protection on cardiac myocytes against endoplasmic reticulum (Er) stress-induced mitochondrial injury, Ca(2+) homeostasis and associated apoptosis. Front Pharmacol (2021) 12:620812. doi: 10.3389/fphar.2021.620812
85. Luo R, Li S, Li G, Lu S, Zhang W, Liu H, et al. Fam134b-mediated er-phagy upregulation attenuates ages-induced apoptosis and senescence in human nucleus pulposus cells. Oxid Med Cell Longev (2021) 2021:3843145. doi: 10.1155/2021/3843145
86. Keles U, Iscan E, Yilmaz HE, Karakülah G, Suner A, Bal E, et al. Differential expression of full-length and Nh(2) terminally truncated Fam134b isoforms in normal physiology and cancer. Am J Physiol Gastrointest Liver Physiol (2020) 319(6):G733–g47. doi: 10.1152/ajpgi.00094.2020
87. Katzen J, Beers MF. Contributions of alveolar epithelial cell quality control to pulmonary fibrosis. J Clin Invest (2020) 130(10):5088–99. doi: 10.1172/jci139519
88. Beyer AM, Norwood Toro LE, Hughes WE, Young M, Clough AV, Gao F, et al. Autophagy, tert, and mitochondrial dysfunction in hyperoxia. Am J Physiol Heart Circ Physiol (2021) 321(5):H985–h1003. doi: 10.1152/ajpheart.00166.2021
89. Zhang Z, Wang X, Ma C, Li Z, Chen H, Zhang Z, et al. Genipin protects rats against lipopolysaccharide-induced acute lung injury by reinforcing autophagy. Int Immunopharmacol (2019) 72:21–30. doi: 10.1016/j.intimp.2019.03.052
90. Chen S, Yuan J, Yao S, Jin Y, Chen G, Tian W, et al. Lipopolysaccharides may aggravate apoptosis through accumulation of autophagosomes in alveolar macrophages of human silicosis. Autophagy (2015) 11(12):2346–57. doi: 10.1080/15548627.2015.1109765
91. Fan T, Chen L, Huang Z, Mao Z, Wang W, Zhang B, et al. Autophagy decreases alveolar macrophage apoptosis by attenuating endoplasmic reticulum stress and oxidative stress. Oncotarget (2016) 7(52):87206–18. doi: 10.18632/oncotarget.13560
92. He N, Tan H, Deng X, Shu L, Qing B, Liang H. Mir-223-3p-Loaded exosomes from bronchoalveolar lavage fluid promote alveolar macrophage autophagy and reduce acute lung injury by inhibiting the expression of Stk39. Hum Cell (2022) 35(6):1736–51. doi: 10.1007/s13577-022-00762-w
93. Hu R, Chen ZF, Yan J, Li QF, Huang Y, Xu H, et al. Complement C5a exacerbates acute lung injury induced through autophagy-mediated alveolar macrophage apoptosis. Cell Death Dis (2014) 5(7):e1330. doi: 10.1038/cddis.2014.274
94. Zhang Y, Zhang M, Wu J, Lei G, Li H. Transcriptional regulation of the Ufm1 conjugation system in response to disturbance of the endoplasmic reticulum homeostasis and inhibition of vesicle trafficking. PloS One (2012) 7(11):e48587. doi: 10.1371/journal.pone.0048587
95. Lemaire K, Moura RF, Granvik M, Igoillo-Esteve M, Hohmeier HE, Hendrickx N, et al. Ubiquitin fold modifier 1 (Ufm1) and its target Ufbp1 protect pancreatic beta cells from er stress-induced apoptosis. PloS One (2011) 6(4):e18517. doi: 10.1371/journal.pone.0018517
96. Neufeldt CJ, Cortese M, Scaturro P, Cerikan B, Wideman JG, Tabata K, et al. Er-shaping atlastin proteins act as central hubs to promote flavivirus replication and virion assembly. Nat Microbiol (2019) 4(12):2416–29. doi: 10.1038/s41564-019-0586-3
97. Wu J, Zhang Z, Teng Z, Abdullah SW, Sun S, Guo H. Sec62 regulates endoplasmic reticulum stress and autophagy balance to affect foot-and-Mouth disease virus replication. Front Cell Infect Microbiol (2021) 11:707107. doi: 10.3389/fcimb.2021.707107
98. Aktepe TE, Liebscher S, Prier JE, Simmons CP, Mackenzie JM. The host protein reticulon 3.1a is utilized by flaviviruses to facilitate membrane remodelling. Cell Rep (2017) 21(6):1639–54. doi: 10.1016/j.celrep.2017.10.055
99. Chen X, Wang K, Xing Y, Tu J, Yang X, Zhao Q, et al. Coronavirus membrane-associated papain-like proteases induce autophagy through interacting with Beclin1 to negatively regulate antiviral innate immunity. Protein Cell (2014) 5(12):912–27. doi: 10.1007/s13238-014-0104-6
100. Wang LM, Chen XM, Yan HJ, Yan S, Sun XY, Zhang DW, et al. Ccpg1 involved in corneal aspergillus fumigatus infection. Int J Ophthalmol (2022) 15(4):541–6. doi: 10.18240/ijo.2022.04.03
101. Liu Q, Wu J, Zhang X, Li X, Wu X, Zhao Y, et al. Circulating mitochondrial DNA-triggered autophagy dysfunction Via sting underlies sepsis-related acute lung injury. Cell Death Dis (2021) 12(7):673. doi: 10.1038/s41419-021-03961-9
102. Martínez-Colón GJ, Taylor QM, Wilke CA, Podsiad AB, Moore BB. Elevated prostaglandin E(2) post-bone marrow transplant mediates interleukin-1β-Related lung injury. Mucosal Immunol (2018) 11(2):319–32. doi: 10.1038/mi.2017.51
103. Yang CS, Kim JJ, Lee HM, Jin HS, Lee SH, Park JH, et al. The ampk-Ppargc1a pathway is required for antimicrobial host defense through activation of autophagy. Autophagy (2014) 10(5):785–802. doi: 10.4161/auto.28072
104. Yang CC, Wu CJ, Chien CY, Chien CT. Green tea polyphenol catechins inhibit coronavirus replication and potentiate the adaptive immunity and autophagy-dependent protective mechanism to improve acute lung injury in mice. Antioxid (Basel) (2021) 10(6):928. doi: 10.3390/antiox10060928
105. Xu X, Araki K, Li S, Han JH, Ye L, Tan WG, et al. Autophagy is essential for effector Cd8(+) T cell survival and memory formation. Nat Immunol (2014) 15(12):1152–61. doi: 10.1038/ni.3025
106. Pei B, Zhao M, Miller BC, Véla JL, Bruinsma MW, Virgin HW, et al. Invariant nkt cells require autophagy to coordinate proliferation and survival signals during differentiation. J Immunol (2015) 194(12):5872–84. doi: 10.4049/jimmunol.1402154
107. Celli J, Tsolis RM. Bacteria, the endoplasmic reticulum and the unfolded protein response: friends or foes? Nat Rev Microbiol (2015) 13(2):71–82. doi: 10.1038/nrmicro3393
108. Dikic I. Open questions: why should we care about er-phagy and er remodelling? BMC Biol (2018) 16(1):131. doi: 10.1186/s12915-018-0603-7
109. Moretti J, Roy S, Bozec D, Martinez J, Chapman JR, Ueberheide B, et al. Sting senses microbial viability to orchestrate stress-mediated autophagy of the endoplasmic reticulum. Cell (2017) 171(4):809–23.e13. doi: 10.1016/j.cell.2017.09.034
110. Bao X, Liu X, Liu N, Zhuang S, Yang Q, Ren H, et al. Inhibition of Ezh2 prevents acute respiratory distress syndrome (Ards)-associated pulmonary fibrosis by regulating the macrophage polarization phenotype. Respir Res (2021) 22(1):194. doi: 10.1186/s12931-021-01785-x
111. Villar J, Zhang H, Slutsky AS. Lung repair and regeneration in Ards: role of Pecam1 and wnt signaling. Chest (2019) 155(3):587–94. doi: 10.1016/j.chest.2018.10.022
112. Chu SG, Villalba JA, Liang X, Xiong K, Tsoyi K, Ith B, et al. Palmitic acid-rich high-fat diet exacerbates experimental pulmonary fibrosis by modulating endoplasmic reticulum stress. Am J Respir Cell Mol Biol (2019) 61(6):737–46. doi: 10.1165/rcmb.2018-0324OC
113. Baek AR, Hong J, Song KS, Jang AS, Kim DJ, Chin SS, et al. Spermidine attenuates bleomycin-induced lung fibrosis by inducing autophagy and inhibiting endoplasmic reticulum stress (Ers)-induced cell death in mice. Exp Mol Med (2020) 52(12):2034–45. doi: 10.1038/s12276-020-00545-z
114. Araya J, Kojima J, Takasaka N, Ito S, Fujii S, Hara H, et al. Insufficient autophagy in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol (2013) 304(1):L56–69. doi: 10.1152/ajplung.00213.2012
115. Yang H, Wang L, Yang M, Hu J, Zhang E, Peng L. Oridonin attenuates lps-induced early pulmonary fibrosis by regulating impaired autophagy, oxidative stress, inflammation and emt. Eur J Pharmacol (2022) 923:174931. doi: 10.1016/j.ejphar.2022.174931
116. Zuo S, Wang B, Liu J, Kong D, Cui H, Jia Y, et al. Er-anchored Crth2 antagonizes collagen biosynthesis and organ fibrosis Via binding Larp6. EMBO J (2021) 40(16):e107403. doi: 10.15252/embj.2020107403
117. Forrester A, De Leonibus C, Grumati P, Fasana E, Piemontese M, Staiano L, et al. A selective er-phagy exerts procollagen quality control via a calnexin-Fam134b complex. EMBO J (2019) 38(2):e99847. doi: 10.15252/embj.201899847
118. Reggio A, Buonomo V, Berkane R, Bhaskara RM, Tellechea M, Peluso I, et al. Role of Fam134 paralogues in endoplasmic reticulum remodeling, er-phagy, and collagen quality control. EMBO Rep (2021) 22(9):e52289. doi: 10.15252/embr.202052289
119. He X, Chen S, Li C, Ban J, Wei Y, He Y, et al. Trehalose alleviates crystalline silica-induced pulmonary fibrosis via activation of the tfeb-mediated autophagy-lysosomal system in alveolar macrophages. Cells (2020) 9(1):122. doi: 10.3390/cells9010122
Keywords: ER-phagy, ER-phagy receptor, ALI, ARDS, immunity, inflammation, ER stress
Citation: Liu S, Fang X, Zhu R, Zhang J, Wang H, Lei J, Wang C, Wang L and Zhan L (2023) Role of endoplasmic reticulum autophagy in acute lung injury. Front. Immunol. 14:1152336. doi: 10.3389/fimmu.2023.1152336
Received: 30 January 2023; Accepted: 03 May 2023;
Published: 17 May 2023.
Edited by:
Rizgar A. Mageed, Queen Mary University of London, United KingdomReviewed by:
Xiaoping Zhang, Tongji University, ChinaVenkata Ramireddy Narala, Yogi Vemana University, India
Copyright © 2023 Liu, Fang, Zhu, Zhang, Wang, Lei, Wang, Wang and Zhan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liying Zhan, zhanliying@whu.edu.cn
†These authors have contributed equally to this work and share first authorship