 Nadja Schubert1
Nadja Schubert1 Tina Schumann1Elena Daum1
Tina Schumann1Elena Daum1 Karolin Flade1Yan Ge1Lara Hagedorn1Winfried Edelmann2
Karolin Flade1Yan Ge1Lara Hagedorn1Winfried Edelmann2 Luise Müller1
Luise Müller1 Marc Schmitz1,3,4Gunnar Kuut5Veit Hornung5
Marc Schmitz1,3,4Gunnar Kuut5Veit Hornung5 Rayk Behrendt1,6
Rayk Behrendt1,6 Axel Roers1,7*
Axel Roers1,7*- 1Institute for Immunology, Medical Faculty Carl Gustav Carus, University of Technology (TU) Dresden, Dresden, Germany
- 2Department of Cell Biology, Albert Einstein College of Medicine, Bronx, NY, United States
- 3National Center for Tumor Diseases (NCT), University Hospital Carl Gustav Carus, University of Technology (TU) Dresden, Dresden, Germany
- 4German Cancer Consortium (DKTK), Partner Site Dresden, and German Cancer Research Center (DKFZ), Heidelberg, Germany
- 5Gene Center and Department of Biochemistry, Ludwig-Maximilians-Universität München, Munich, Germany
- 6Institute for Clinical Chemistry and Clinical Pharmacology, University Hospital Bonn, Bonn, Germany
- 7Institute for Immunology, University Hospital Heidelberg, Heidelberg, Germany
Innate DNA sensors detect foreign and endogenous DNA to induce responses to infection and cellular stress or damage. Inappropriate activation by self-DNA triggers severe autoinflammatory conditions, including Aicardi-Goutières syndrome (AGS) that can be caused by defects of the cytosolic DNase 3’repair exonuclease 1 (TREX1). TREX1 loss-of-function alleles are also associated with systemic lupus erythematosus (SLE). Chronic activation of innate antiviral immunity in TREX1-deficient cells depends on the DNA sensor cGAS, implying that accumulating TREX1 DNA substrates cause the inflammatory pathology. Retrotransposon-derived cDNAs were shown to activate cGAS in TREX1-deficient neuronal cells. We addressed other endogenous sources of cGAS ligands in cells lacking TREX1. We find that induced loss of TREX1 in primary cells induces a rapid IFN response that requires ongoing proliferation. The inflammatory phenotype of Trex1-/- mice was partially rescued by additional knock out of exonuclease 1, a multifunctional enzyme providing 5’ flap endonuclease activity for Okazaki fragment processing and postreplicative ribonucleotide excision repair. Our data imply genome replication as a source of DNA waste with pathogenic potential that is efficiently degraded by TREX1.
Introduction
Aberrant activation of innate antiviral immunity is a key pathogenetic principle in a spectrum of autoinflammatory and autoimmune diseases (1–3). Innate sensing of virus infection largely relies on detection of viral nucleic acids. This bears the problem of avoiding sensor activation by endogenous DNA and RNA, a difficult task that the immune system can fail to meet for many different reasons, resulting in chronic expression of type I interferon (IFN) as well as proinflammatory cytokines, and severe, often fatal, inflammatory diseases (1–4). A growing number of distinct human conditions have been reported, currently about 40, that are caused by immune responses to endogenous nucleic acids or other means of inappropriate nucleic acid sensor activation, and are collectively addressed as type I interferonopathies (2, 5).
Double-stranded DNA activates the sensor cGAS in a sequence-independent manner (6). Upon ligand binding, cGAS catalyzes the formation of the second messenger 2’3’-cGAMP which activates STING, resulting in IRF3 and NF-κB activation (7, 8). The cGAS/STING axis is essential for defense against numerous viral and non-viral pathogens (9). However, cGAS does not discriminate between microbial and endogenous DNA and is readily activated by self-DNA (6). Thus, effective safeguard mechanisms are key to avoid uncontrolled activation. cGAS is expressed in the cytosol and in the nucleus (10, 11). Sensor functions of cGAS in the nucleus are incompletely understood. Most nuclear cGAS is tightly tethered to nucleosomes, by binding to the acidic patch formed by nucleosomal core histones and is thereby kept inactive (reviewed in (10, 11). Chromatin structure, nuclear proteins competing with cGAS for the histone acidic patch or DNA, cofactors required for DNA binding, and posttranslational modifications of histones or cGAS, all contribute to regulation of nuclear cGAS DNA sensing (11). Disturbance of histone stoichiometry can result in pathogenic activation of nuclear cGAS by chromosomal DNA (12). Cytosolic cGAS is not exposed to relevant amounts of DNA in healthy, uninfected cells due to efficient cytosolic DNA waste disposal ensured by 3’ repair exonuclease 1 (TREX1, DNase III). This ER-anchored non-processive exonuclease degrades ssDNA, but also dsDNA with overhangs, which it can unwind (13, 14).
Trex1 is one of several genes, defects of which can each cause the mendelian autoimmune disease Aicardi-Goutières syndrome (AGS) characterized by debilitating encephalitis and inflammation of other organs (15, 16). While homozygous loss of Trex1 results in AGS, heterozygous Trex1 loss-of-function alleles are associated with systemic lupus erythematosus (SLE), contributing to the polygenic predisposition for this severe autoimmune disease (17). AGS and SLE overlap clinically and also share key biochemical features, including chronic activation of type I IFN signaling and antinuclear auto-antibodies (18–21).
TREX1-deficient mice reproduce chronic type I IFN response, auto-antibody formation and multi-organ inflammation (22–24), most prominently inflammation of the myocardium, that is fatal by the age of few months, whereas the CNS is only discretely affected (25). Trex1-/- pathology is largely rescued by additional inactivation of cGAS, STING, IRF3 or IFNAR expression (22, 24, 26). Likewise, IFN expression of human TREX1-deficient cells is abrogated by additional inactivation of the cGAS/STING axis (27). These findings indicate inappropriate cGAS activation by some endogenous TREX1 DNA substrate as the key pathogenetic event.
TREX1 degrades HIV cDNA and limits innate responses to the virus (28). Endogenous retrovirus cDNA can serve as activating ligands for cGAS (29), and de-repression of endogenous retroelements in cells with defective histone methylation is associated with induction of type I IFN signaling (30). TREX1 may serve to continuously degrade replication intermediates of endogenous retroelements (24, 31). While we observed no amelioration of the Trex1-/- phenotype by treatment with reverse transcriptase inhibitors (32), human TREX1-deficient neurons and astrocytes, however, were shown to accumulate DNA originating from endogenous retroelements in the cytosol and their spontaneous IFN response was inhibited by antiretroviral drugs. Moreover, chronic IFN signaling of AGS patients was ameliorated by treatment with reverse transcriptase inhibitors (27, 33).
Herein, we address other sources of immunostimulatory DNA in cells lacking TREX1. DNA damage can lead to chromosomal aberrations resulting in problems of mitotic chromosome segregation and formation of micronuclei. Collapse of the unstable micronuclear envelope allows cGAS to access micronuclear chromatin. This chain of events triggers the chronic interferon response in AGS caused by defects of RNase H2 (34). Besides retroelement replication intermediates or mislocalized chromatin, the complex metabolism required to ensure genome maintenance, in particular a multitude of repair pathways, but also DNA replication, might contribute activating cGAS ligands. Oligonucleotide DNA waste released during repair and/or replication could stimulate cGAS in TREX1-deficient cells. As oligonucleotides equilibrate between nucleus and cytosol, TREX1-mediated degradation might also reduce nuclear DNA waste and prevent activation of nuclear cGAS (35). We demonstrate that DNA replication is essential for the cGAS/STING response of TREX1-deficient cells and identify exonuclease (Exo)1, an enzyme providing 5’flap-endonuclease activity during Okazaki fragment maturation and postreplicative ribonucleotide excision repair, as an important contributor to the cell-intrinsic innate immune response upon loss of TREX1.
Results
Spontaneous DNA Damage, but No Effect of p53-Mediated DNA Damage Responses on Chronic cGAS/STING Activation in Trex1-/- Mice
Trex1-/- cells feature tonic DNA damage checkpoint signaling (36), increased numbers of strand breaks and spontaneous phosphorylation of p53 (35). In AGS caused by defects of RNase H2 (37), extensive genome damage results in micronucleus formation which drives the chronic IFN response of these patients (34). This response is counter-regulated by p53-dependent DNA damage responses which eliminate cells with high damage load and prevent them from prolonged IFN production (38). Micronuclear DNA or chromatin bridges were shown to trigger an IFN response in cancer cells treated with genotoxic drugs (39, 40), and TREX1 was reported to limit cGAS activation in genetically unstable cells by degradation of micronuclear DNA (41). Micronucleated cells can be quantified by flow cytometric analysis of erythrocytes, as erythrocyte precursors expel the main nucleus but frequently retain micronuclei (42). We found increased numbers of micronucleated erythrocytes in Trex1-/- mice (Figure 1A and Figure S1), reflecting spontaneous DNA damage and chromosomal aberrations. However, compared to a 30-fold increase in mice lacking RNase H2 in the hematopoietic system (manuscript in preparation), micronucleated cells were only mildly (2-fold) increased in Trex1-/- mice. To address whether cells carrying genome damage are an important source of type I IFN in Trex1-/- mice, we crossed Trex1-/- to Trp53-/- animals (43) and compared the IFN response of double knock out mice to that of Trex1-/- littermates. Additional loss of p53 further increased fractions of micronucleated erythrocytes (Figure 1A), but did not increase transcript levels of type I IFN-stimulated genes (ISGs) in total blood cells (Figure 1B) nor enhance inflammation of the myocardium in TREX1-deficient mice (Figure 1C). These data strongly argue against genome damage and cytosolic chromatin as the trigger of cGAS activation in the absence of TREX1 and lead us to investigate alternative sources of TREX1-sensitive cGAS activating DNA ligands.
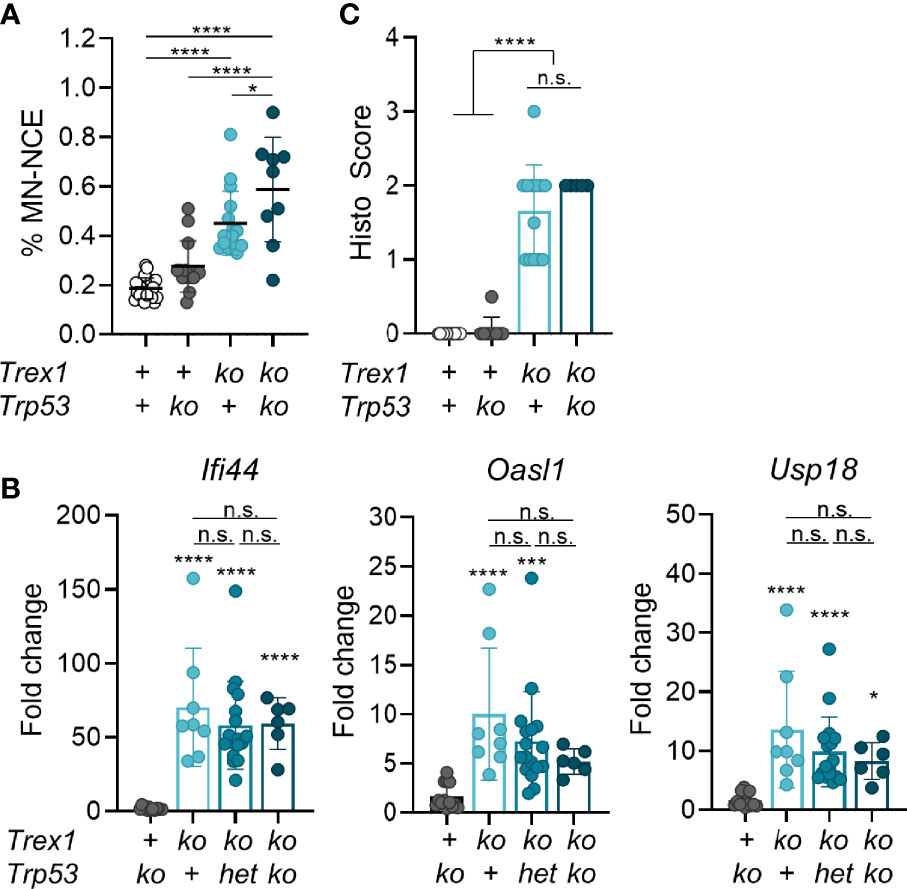
Figure 1 Mild genome damage, but no amelioration of the inflammatory phenotype of Trex1-/- mice lacking also p53. (A) Flow cytometric quantification of micronucleated erythrocytes from 6 week-old mice of the indicated genotypes (WT n = 22, Trp53-/- n = 14, Trex1-/- n = 16, Trp53-/-Trex1-/- n = 9). See Figure S1 for gating strategy. The Trex1 null allele published by Morita et al. was used (23). (B) ISG transcript levels in total blood cells quantified by qRT-PCR. Fold change compared with mean of WT controls (n = 16) is shown. Trp53-/- n = 13, Trex1-/- n = 8, Trex1-/- Trp53WT/- n = 17, Trex1-/-Trp53-/- n = 6. (C) Quantification of myocarditis intensity in H&E-stained sections of heart tissue from 6-10 week-old mice of the indicated genotypes (WT n = 11, Trp53-/- n = 9, Trex1-/- n = 12, Trex1-/-Trp53-/- n = 5; see M&M for heart inflammation score). ****p < 0.0001, ***p < 0.001, *p < 0.05, n.s., not significant.
Neither Nucleotide Excision Repair Nor the Structure-Specific Endonuclease MUS81 Are a Relevant Source of Immunostimulatory Oligonucleotide Waste Accumulating in Trex1-/- Cells
Primary TREX1-deficient fibroblasts from AGS patients not exposed to experimental genotoxic hazards (35, 36) showed DNA staining in the cytosol, suggesting leakage of DNA from the nucleus or mitochondria in healthy cells. Higher amounts of DNA seem to leak into the cytosol of genetically unstable cancer cells, in particular after exposure to genotoxic hazards (44–48). This DNA, likely a waste from DNA repair (44), seems to overwhelm TREX1 exonuclease activity (44, 48). Whether the same mechanisms generating large amounts of TREX1 substrates in genome-damaged cells also contribute to release of DNA from the nucleus in healthy cells is unknown. Several DNA repair pathways involve release of oligonucleotides that could, upon hybridization, form partially double-stranded structures, e.g. Y-DNA (29), capable of ligating cGAS. Nucleotide excision repair (NER) removes bulky DNA lesions by excision of short single-stranded patches containing the lesions (49). High amounts of these, 22-30 nt-long flaps may be released also in the absence of exogenous genotoxic stress, since NER is triggered already by minimal distortions of undamaged DNA resulting in low-level DNA turnover in healthy cells termed ‘gratuitous repair’ (50, 51). Moreover, we had earlier found that in contrast to cell-intrinsic activation of transcriptional responses to loss of TREX1 in several different cell types (25), this study and unpublished), inactivation of TREX1 in B cells had no detectable effect on the transcriptome of murine B cells (25). Since B cells were reported to be NER-deficient (52), our observation would be plausibly explained by NER being a source of the cGAS ligand accumulating in Trex1-/- mice. We therefore generated mice double-deficient for TREX1 and XPA, an essential NER protein (49, 53), using CRISPR/Cas9-mediated mutagenesis in embryos (Figure S2A). As expected, Xpa-/- animals (Figure 2A) showed a loss of repair synthesis in UV-irradiated cells (Figure S2B), indicating absence of NER function. We compared ISG expression in blood cells of mice lacking TREX1 or XPA and of Trex1-/-Xpa-/- animals (Figure 2B). Xpa-/- mice proficient for TREX1 did not show enhanced ISG expression compared to control mice. IFN signaling was not reduced in the double mutants compared to Trex1-/- mice. Moreover, the myocarditis typical of TREX1-deficient mice was not ameliorated by additional inactivation of NER (Figure 2C).
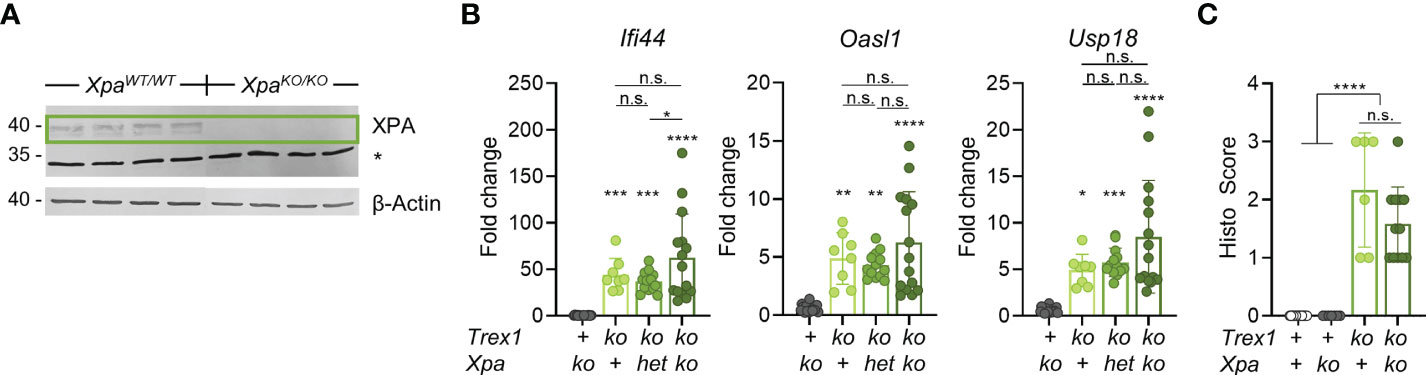
Figure 2 Additional inactivation of NER has no impact on the spontaneous IFN response of Trex1-/- mice. (A) Western blot analysis of XPA expression in protein extracts from ear tissue from control and XPA-/- mice. * non-specific band at 34 kDa. (B) ISG transcript levels in total blood cells from 6 week-old mice of the indicated genotypes quantified by qRT-PCR. The Trex1Δ478 null allele generated in this study was used. Fold change compared with mean of WT controls (n = 16) is shown (Trex1-/- n = 8, Xpa-/- n = 18, Trex1-/-XpaWT/- n = 13, Trex1-/-Xpa-/- n = 15. (C) Quantification of myocarditis intensity in H&E-stained sections of heart tissue from 9-11 week-old mice of the indicated genotypes (WT n = 15, Xpa-/- n = 7, Trex1-/- n = 6, Trex1-/-Xpa-/- n = 12). See M&M for heart inflammation score. ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05, n.s., not significant.
MUS81 is a DNA repair factor reported to be a source of cytoplasmic DNA in cancer cells (54). This structure-specific endonuclease is essential for genome integrity as it is required for resolution of Holiday junctions (55) and prevention of chromosome bridges in anaphase (56), as well as repair of stalled and collapsed replication forks (57). MUS81 function has important impact on the activity of the type I IFN system [reviewed in (58)]. Loss of the enzyme results in pronounced spontaneous IFN production (59). As MUS81 deficiency leads to chromosomal instability, a likely scenario is that micronuclear DNA is the key stimulator of this IFN response. Due to this spontaneous IFN response of MUS81-deficient cells, the question whether MUS81 contributes to production of the immunostimulatory cGAS ligand accumulating in otherwise undamaged cells in the absence of TREX1, cannot be directly addressed by inactivation of MUS81 in Trex1-/- cells or mice. However, in case the IFN response of cells lacking TREX1 depended on DNA waste produced by MUS81, no synergistic IFN induction by TREX1 deficiency and MUS81 deficiency would be expected. We inactivated TREX1 and/or MUS81 in THP1 cells (Figure 3A). The robust spontaneous ISG expression of TREX1-/- cells was rescued by reconstitution with functional TREX1 (Fig S3A&B). As expected, also MUS81-/- cells featured prominent type I IFN signaling (Figure 3B). The double deficient cells, however, showed synergistically enhanced ISG expression (Figure 3B). This result argues that MUS81 is not required for generation of the cGAS ligand accumulating as a result of TREX1 deficiency. Collectively, our data rule out NER as a dominant source of the endogenous immunostimulatory DNA accumulating in mouse cells lacking TREX1 and argue against a significant contribution of the structure-specific endonuclease MUS81 to generation of this pathogenic cGAS ligand.
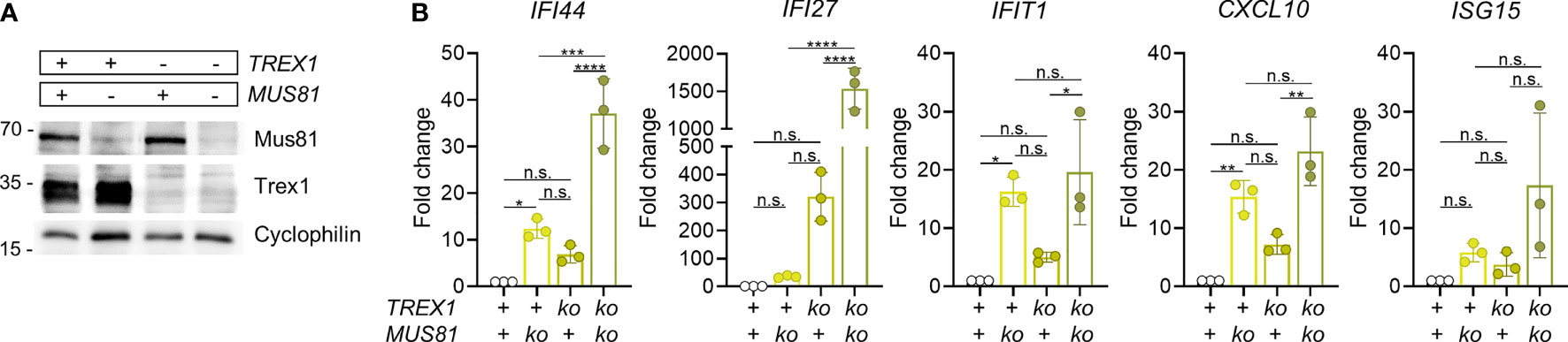
Figure 3 Simultaneous inactivation of Trex1 and of Mus81 result in synergistically enhanced IFN response. (A) Western blot analysis of MUS81 expression by polyclonal cultures resulting from lentiviral transduction of control or Trex1-/- THP1 cells (see Figure S3) with lentiCRISPR v2 plasmid encoding Mus81-specific guide RNA (TCTGAAATACGAAGCGCGTG) and selection for the presence of the plasmid. (B) ISG transcript levels in THP1 cells of the indicated genotypes quantified by qRT-PCR. Fold change compared with mean of WT controls is shown. Graph summarizes 3 independent experiments. ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05, n.s., not significant.
Partial Rescue of the Trex1-/- Phenotype by Additional Inactivation of Exonuclease 1
We next turned our attention to another repair factor that had been shown to contribute to release of TREX1-sensitive DNA into the cytosol of genome-damaged cells (44), exonuclease 1 (EXO1). Homology-directed repair of double-strand breaks requires long-range resection of the 5’-end in order to produce the long single-stranded 3’-overhangs that are the substrates for strand invasion (60). EXO1 is a multifunctional enzyme with 5’-3’ exonuclease activity, but also 5’-flap structure-specific endonuclease activity, involved in end processing in preparation for homology-directed repair as well as in telomere homeostasis, mismatch repair, and Okazaki fragment maturation during lagging strand synthesis (61). Upon exposure of cells to genotoxic irradiation or drugs inducing double-strand breaks, 5’-end resection, performed by EXO1 and other endonucleases, was shown to be a source of DNA waste triggering a type I IFN response (44). TREX1-mediated degradation limited the innate immune response (44). We therefore asked, whether end resection by the EXO1 endonuclease activity might yield DNA waste also during steady-state genome repair. In addition, the EXO1 5’-flap cleavage occurring during genome replication might also release immunostimulatory DNA waste. We crossed Exo1-/- mice (62) to TREX1-deficient mice and compared littermates of different genotypes for weight gain and spontaneous type I IFN response. The conspicuous failure to thrive typical of Trex1-/- mice was largely, albeit not completely, rescued by the additional inactivation of EXO1 (Figures 4A, B). Even heterozygosity for the Exo1 knock out allele resulted in a significant rescue of Trex1-/- weight gain. Survival of Trex1-/- mice was significantly improved by additional loss of EXO1 (Figure 4C). Correspondingly, type I IFN signaling was clearly reduced in Trex1-/- mice lacking functional Exo1, as determined by quantification of ISG transcript levels in blood cells (Figure 4D and Figure S4A). For most ISGs, the increase in transcript levels seen in Trex1-/- mice was blunted to about half in Trex1-/-Exo1-/- animals (Figure 4D and Figure S4A). Type I IFN bioactivity was not elevated above the background of control animals in serum of Trex1-/- or Trex1-/-Exo1-/- mice (Figure S4B). A control experiment demonstrated that STING responses of EXO1-deficient cells are of normal intensity (Figure S4C). The reduced innate response was not reflected in amelioration of myocardial inflammation in the double vs. the Trex1 single mutants as assessed by histological analysis of inflammatory infiltration on H&E- (Figure 4E), anti-CD3- (Figure 4F) and van Gieson-stained sections (not shown). Collectively, we show that additional loss of EXO1 significantly ameliorates the spontaneous innate antiviral response of Trex1-/- animals, suggesting a role for EXO1 endonuclease activity in generation of cGAS-activating ligands in TREX1-deficient cells.
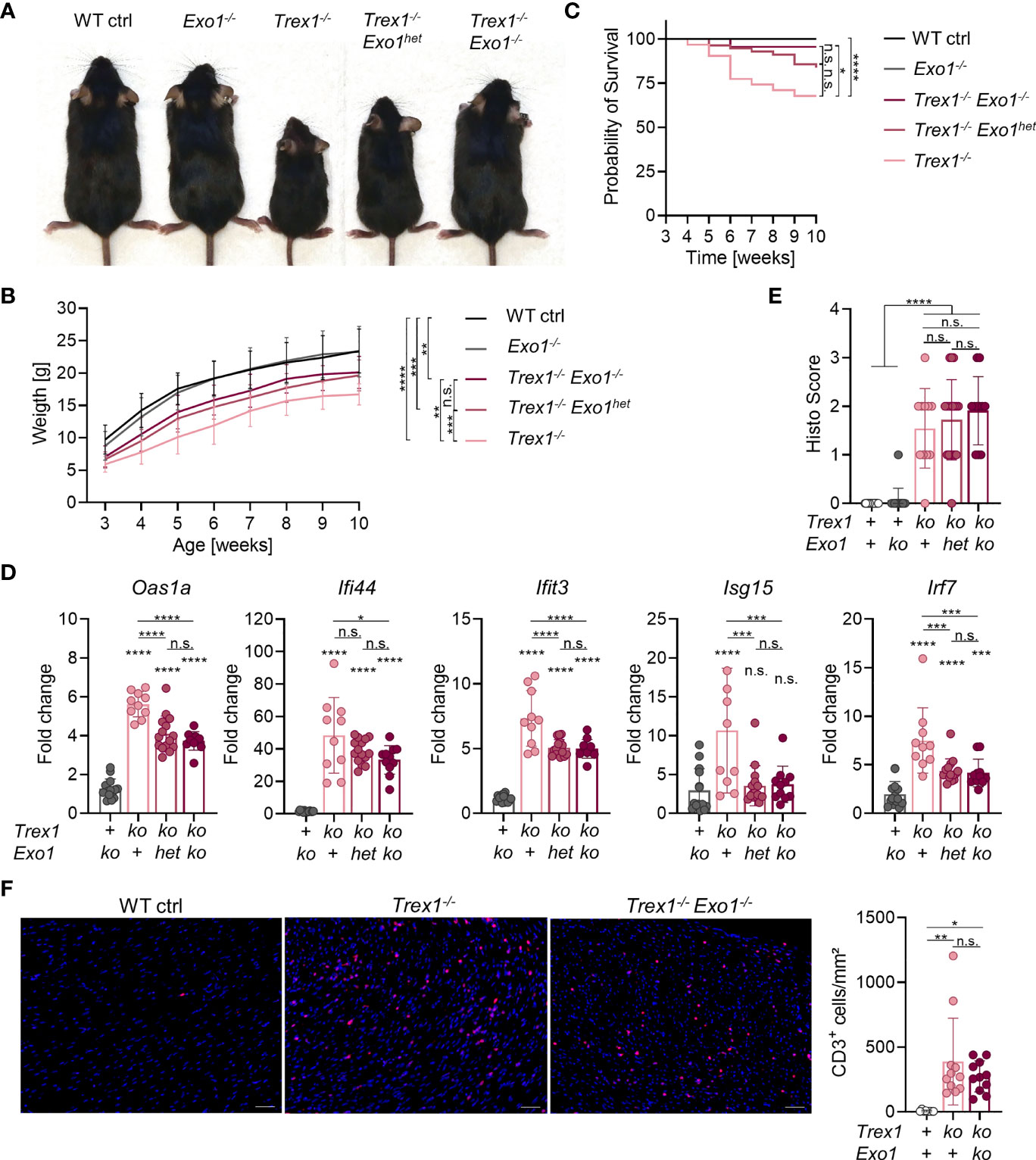
Figure 4 Partial rescue of the Trex1 inflammatory phenotype by additional inactivation of Exo1. (A) Macroscopic phenotype of 7-8 week-old mice of the indicated genotype. (B) Body weight of animals of the indicated genotype (WT n = 32, Exo1-/- n = 20, Trex1-/- n = 26, Trex1-/-Exo1+/- n = 46, Trex1-/-Exo1-/- n = 15). Statistical analysis was performed for the comparison of body weight at the age of 10 weeks. (C) Survival of mice of the indicated genotype until the age of 10 weeks (WT n = 46, Exo1-/- n = 31, Trex1-/- n = 31, Trex1-/-Exo1+/- n = 56, Trex1-/-Exo1-/- n = 22). (D) ISG transcript levels in total blood cells from 6 week-old mice of the indicated genotypes quantified by qRT-PCR. Fold change compared with mean of WT controls (n = 15) is shown (Trex1-/- n = 10, Exo1-/- n = 14, Trex1-/-Exo1WT/- n = 15, Trex1-/-Exo1-/- n = 11). See Figure S4 for expression of additional ISGs. (E) Quantification of inflammatory infiltration on H&E-stained sections of heart tissue from 10-12 week-old mice of the indicated genotypes. (F) Immunohistochemical staining of CD3 on sections of heart tissue from 10-12 week-old mice of the indicated genotypes (left). Scale bar 50 µm. Quantification of CD3+ cells per area of heart muscle by investigator-blinded automated image analysis (right). ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05, n.s., not significant.
Rapid Onset of Type I IFN and NF-κB Responses Upon Induced Inactivation of TREX1 in Primary Cells
To elucidate the contribution of EXO1 to generation of TREX1 substrates, we first established an in vitro model of induced loss of TREX1 expression. We differentiated macrophages from bone marrow (BMDMs, Figure S5A) of Trex1FL/FL R26CreERT2 mice (25) and induced Cre activity in Trex1FL/FL R26CreERT2 BMDMs by 4-hydroxytamoxifen (4-OHT, Figure 5A). This resulted in efficient deletion of the loxP-flanked Trex1 allele one day after the end of 4-OHT exposure, as determined by PCR (Figure S5B). Western blot analysis of cells harvested at daily intervals after 4-OHT induction (Figure 5A) revealed rapid decline of TREX1 protein which was almost completely lost 3 days after the end of induction and was undetectable on day 4 (Figure 5B and Figure S5C). Increased type I IFN bioactivity was detectable (not significant) in the culture supernatant already on day 2 after end of induction, but was very prominently increased on days 3 and 4 (Figure 5C). Transcript levels of the ISG Ifi44 were significantly increased already on day 2 after 4-OHT and were 10-fold up-regulated on day 3 (Figure 5D). RNA sequencing of 4-OHT-induced BMDMs harvested on days 1 to 4 revealed robust up-regulation of ISGs (Figure 5E and Figures S5D, E). Enrichment of the gene set ‘IFNα response’ was not significant on day 1 after end of 4-OHT, but very much so on days 2 to 4 (Figure 5E and Figures S5D, E). In addition to ISG induction, loss of TREX1 was reflected in up-regulation of NF-κB and TNF-induced genes on day 2, as expected for STING responses (Figure 5E). Gene signatures associated with cell cycle progression, including e.g. expression of proliferation marker MKI67 were significantly downregulated, most prominently on day 4, while expression of CDKN1A encoding cell cycle inhibitor p21 was upregulated (not shown), most likely reflecting IFN-induced cell cycle arrest (Figure 5E). Similar results were obtained in Trex1FL/FL R26CreERT2 embryonic fibroblasts (MEFs) (Figure S5F).
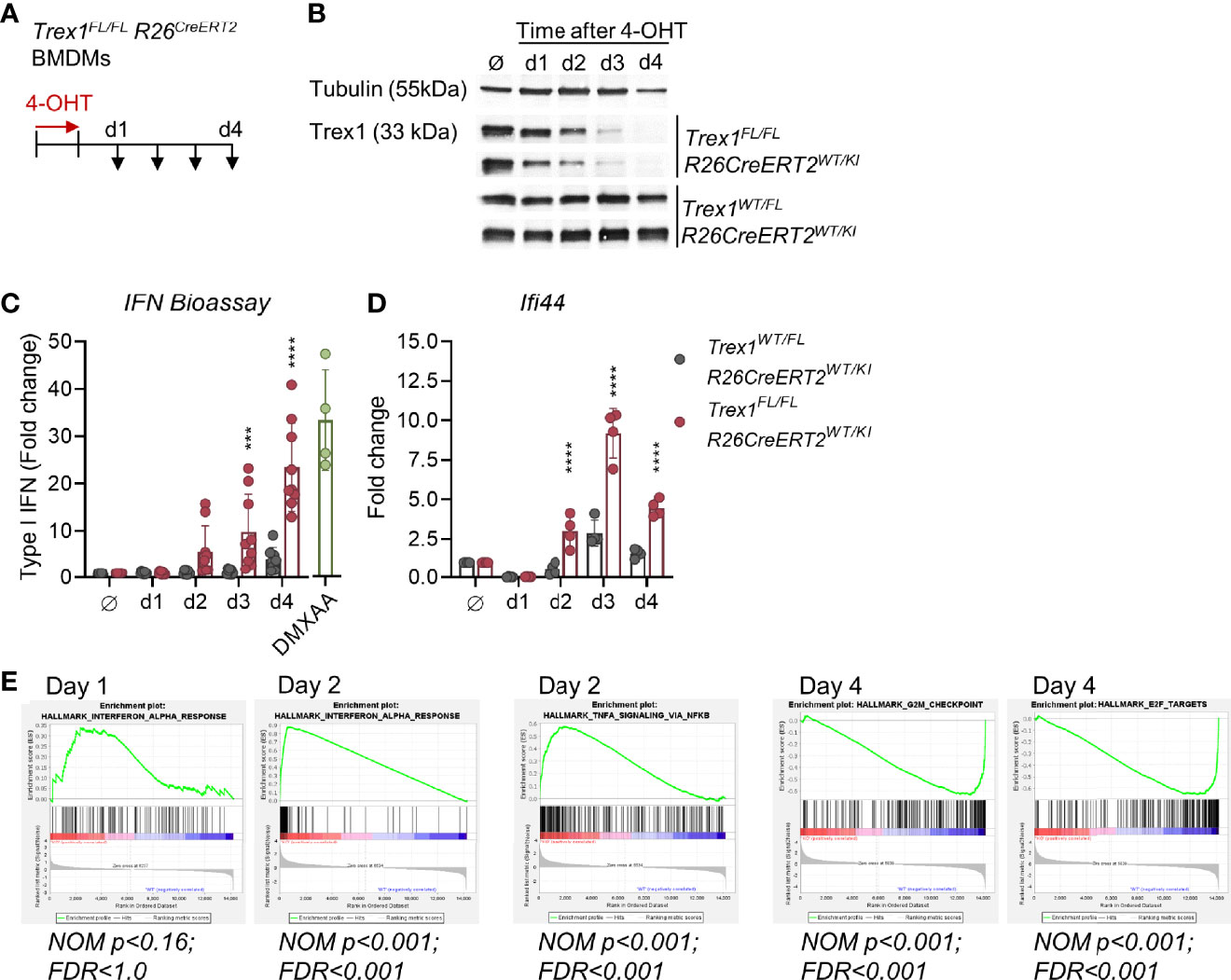
Figure 5 Rapid loss of TREX1 and onset of type I IFN responses upon induced inactivation of Trex1 in vitro. (A) Inactivation of the Trex1 gene was induced in macrophages differentiated from Trex1FL/FLR26CreERT2 bone marrow (BMDMs) by culture in the presence of 4-OHT for 1 day. 4-OHT treated BMDMs from Trex1WT/FLR26CreERT2 mice as well as non-4-OHT-treated cells from both genotypes were used as controls. Samples were obtained on days 1-4 after end of 4-OHT induction. (B) Western blot analysis of TREX1 expression in BMDMs at the indicated time points after 4-OHT induction, representative of 6 animals per genotype. (C) Assay for type I IFN bioactivity in supernatant of BMDMs sampled at the indicated time points after 4-OHT induction. Supernatant was added to 1x104 LL171 luciferase IFN-reporter cells followed by quantification of luciferase activity (Trex1FL/FLR26CreERT2 n = 9, control n = 8). Supernatant of WT BMDMs treated with DMXAA served as high control. (D) Transcript levels of the ISG Ifi44 determined by qRT-PCR in BMDMs sampled at the indicated time points after 4-OHT induction. Fold change compared to untreated control cells (n = 4) is shown. (E) Gene set enrichment analysis of RNA sequencing data from Trex1FL/FLR26CreERT2 vs. control (Trex1WT/FL R26CreERT2) BMDMs sampled at the indicated time points after 4-OHT induction. ****p < 0.0001, ***p < 0.001.
Collectively, our finding that the IFN response is detectable already at day 2 after 4-OHT induction, a time point at which large amounts of TREX1 were still present, indicates that even slight reductions of TREX1 expression levels result in accumulation of the cGAS ligand and activation of cGAS/STING responses. Thus, steady-state TREX1 expression levels of healthy cells are just sufficient to degrade its DNA substrate. This finding is compatible with ongoing large-scale production of this substrate by at least a fraction of the cells in the culture at any time point.
The Type I IFN Response of TREX1-Deficient Cells Requires Ongoing Proliferation
Large amounts of oligonucleotide waste might originate from genome replication. Millions of Okazaki fragments are precisely ligated during lagging strand synthesis. Okazaki fragment synthesis by Polδ continues beyond the 5’-end of the previous Okazaki fragment, displacing a 5’-flap that requires endonucleolytic trimming (63). Nick translation by iterative rounds of strand displacement synthesis and flap cleavage not only remove the RNA primer but also the 5’-portion of primer initiator DNA of the previous Okazaki fragment synthesized by POLα. In addition to FEN1, the major flap endonuclease involved in Okazaki fragment maturation, also DNA2 and EXO1 contribute. A recent study suggested that primarily FEN1 and EXO1 compete for Okazaki fragment flap cleavage, with DNA2 playing only a minor role (64). EXO1 overexpression in yeast can partially compensate loss of FEN1 (65).
Whether DNA waste originating from Okazaki fragment maturation can trigger cGAS/STING responses is unknown. To approach this question, we asked whether the IFN response of cells lacking TREX1 requires DNA replication. This is not easily addressed by experiments aiming to arrest the cell cycle of constitutively TREX1-deficient cells, because immunostimulatory cGAS ligands, formed before halting proliferation, may be stable in the absence of TREX1 and maintain the IFN response. We therefore induced inactivation of Trex1 in cells in vitro and simultaneously arrested the cell cycle. Treatment of BMDM cultures with the CDK4/6 blocker Palbociclib, arresting cells in G1 (66), for 3 days, reduced the fraction of cells in S-phase 10-fold (Figures 6A, B). Palbociclib administration in parallel to 4-OHT-induced knock out of Trex1 (Figure S6A) abrogated the IFN response triggered by loss of TREX1, as determined by quantification of type I IFN bioactivity in culture supernatant (Figure 6C) and ISG transcript levels (Figure 6D). A control experiment demonstrated that BMDMs arrested by Palbociclib are capable of mounting cGAS/STING responses (Figure S6B). To independently confirm that the IFN response to induced loss of TREX1 depends on proliferation, we repeated the experiment in a different cell type, MEFs derived from Trex1FL/FL R26CreERT2 mice, using a different cell cycle inhibitor, Mimosine, that is chemically unrelated to Palbociclib and arrests cells in late G1 by activating checkpoint signaling independent of DNA damage (67). 4-OHT-induced inactivation of Trex1 resulted in an IFN response that was blunted by Mimosine-mediated G1 arrest (Figure 6E and Figure S6C). A control experiment demonstrated unimpaired IFN responses of Mimosine-exposed cells to STING activation by DMXAA (Figure S6C). Collectively, the IFN response of TREX1-deficient cells requires ongoing proliferation and functional EXO1, compatible with oligonucleotides released by flap endonucleases during lagging strand synthesis as the pathogenic ligand for cGAS.
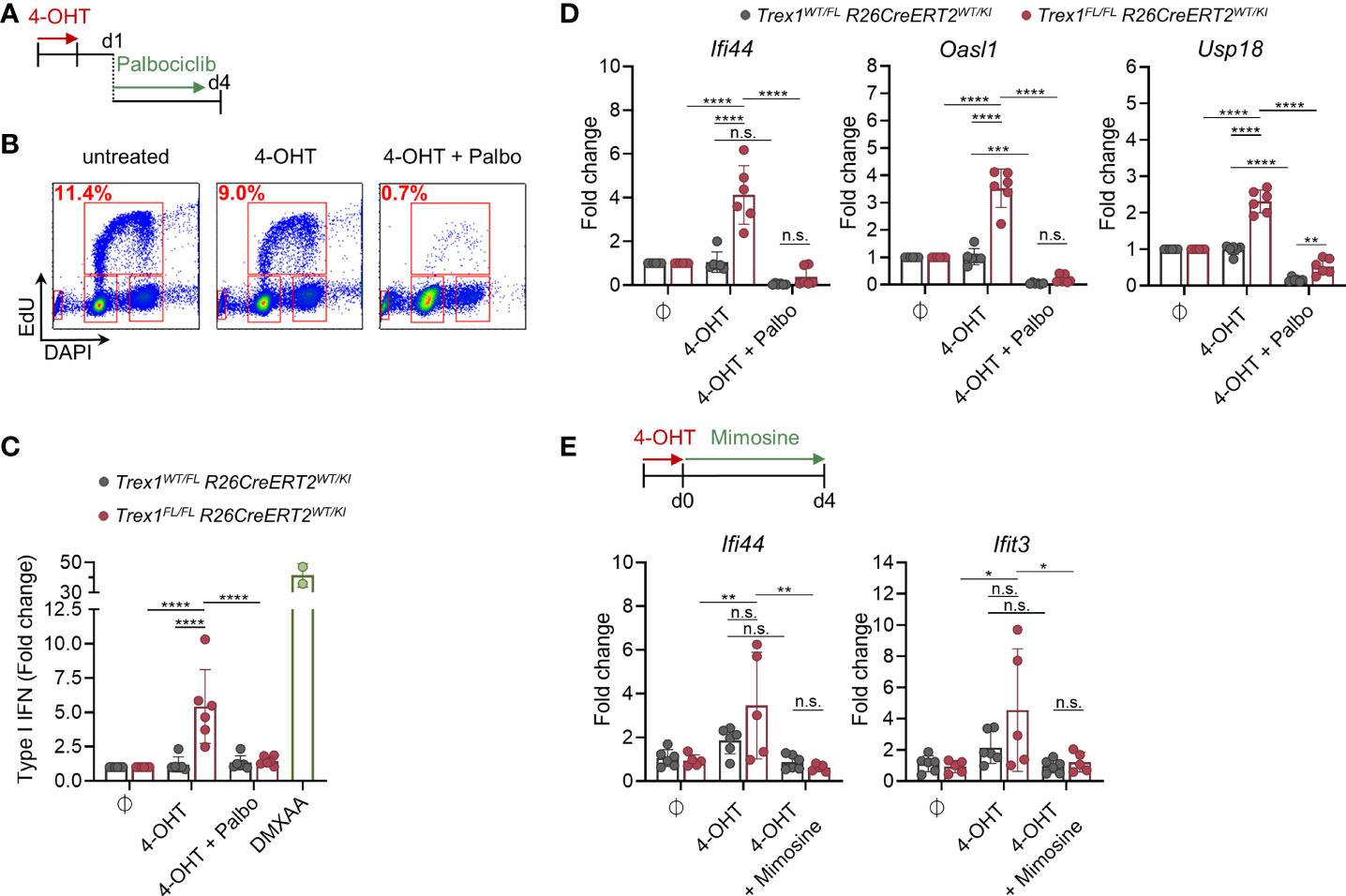
Figure 6 IFN response upon induced loss of TREX1 in macrophage cultures requires ongoing proliferation. (A) Trex1FL/FLR26CreERT2 and control BMDMs were treated with 4-OHT in vitro for 22h. 1 day after end of 4-OHT induction, CDK4/6 inhibitor Palbociclib was added to arrest the cells in G1. (B) Verification of proliferative arrest after 72h of Palbociclib treatment (d4 after end of 4-OHT) as determined by flow cytometric quantification of EdU incorporation and DNA content (DAPI). Representative of 3 independent experiments. (C) Quantification of type I IFN bioactivity in supernatant from BMDMs treated with 4-OHT and Palbociclib (n = 6 each genotype, pooled from 3 independent experiments). Supernatant was added to 1x104 LL171 luciferase IFN-reporter cells followed by quantification of luciferase activity. Supernatant of WT BMDMs treated with DMXAA served as high control. (D) ISG transcript levels in BMDMs after 72h of Palbociclib treatment (d4 after end of 4-OHT) as determined by qRT-PCR (n = 6 for each genotype, pooled from 3 independent experiments). Fold change compared to mean of untreated control cells (n = 6) is shown. (E) Trex1FL/FLR26CreERT2 and control BMDMs were treated with 4-OHT in vitro for 22h. At the end of 4-OHT induction, Mimosine was added to arrest the cells in G1. ISG transcript levels in MEFs after 96h of Mimosine treatment (d4 after end of 4-OHT) as determined by qRT-PCR (Trex1WT/FLR26CreERT2 n = 6, Trex1FL/FLR26CreERT2 n = 5, pooled from 2 independent experiments). ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05, n.s., not significant.
Discussion
The concept that endogenous DNA waste poses a major threat to immune homoeostasis, can potentially trigger lethal disease, and is therefore continuously eliminated by efficient DNA waste disposal in healthy mammals, was fueled by the discovery that defects of DNases in different compartments cause autoinflammatory nucleic acid sensor activation associated with chronic type I IFN responses and consecutive autoimmunity. Defects of the endonuclease DNase I that degrades chromatin debris in the circulation and other extracellular compartments result in Lupus-like disease in mice (68, 69) and low serum DNase I activity is associated with SLE (70, 71). Lysosomal DNase II is key to degrade chromatin in macrophages that phagocytose nuclei expelled by erythrocyte precursors and to clearance of damaged nuclear DNA by autophagy (72). Loss of this enzyme triggers IFN-driven disease in mouse and man (73, 74) Likewise, mutations compromising removal of dead cells and efficient disposal of their chromatin, including e.g. defects of the complement system, are linked to IFN-driven pathology (75). AGS caused by TREX1 mutations was paradigmatic for monogenic autoinflammatory and autoimmune conditions resulting from uncontrolled nucleic acid sensor activation and inspired coining of the term ‘interferonopathies’ (2). The observation that defects of the cytosolic exonuclease TREX1 cause monogenic cGAS/STING-driven AGS and are associated with SLE extended the concept of DNA waste disposal to the cytosolic and/or nuclear compartments and raised questions about nature and origin of the immunogenic TREX1 DNA substrates that trigger disease in the absence of the nuclease.
We show that the spontaneous type I IFN response of Trex1 knock out mice is reduced by additional absence of functional EXO1 and that IFN production of TREX1-deficient cells in vitro required ongoing proliferation. These findings are compatible with oligonucleotide waste generated by the flap endonucleases during lagging strand Okazaki fragment maturation as an important source of the pathogenic cGAS ligand accumulating in TREX1-deficient cells. In contrast to long Okazaki fragments in prokaryotes, mammalian Okazaki fragments are only 150-200 nt in length (76), and about 107 fragments are processed per S phase in mammalian cells. Maturation of each fragment involves displacement of its 5’-RNA end by polymerase δ elongating the next fragment. Strand displacement synthesis usually halts after displacement of only one nucleotide followed by cleavage of this one-nucleotide 5’-flap by flap endonuclease FEN1. Iterative cycles of strand displacement and flap cleavage result in removal of the Okazaki fragment RNA primer and the subsequent DNA sequence that was synthesized by error-prone polymerase α. This ‘nick translation’ maintains a ligatable nick that is eventually sealed by DNA ligase I. In A/T-rich sequences, much longer 5’-flaps can be displaced which are rapidly covered by RPA and are no longer substrates for FEN1 (63). These long flaps are cleaved by the flap endonuclease DNA2 in yeast (63). This ‘two nuclease model’ of Okazaki fragment processing was recently extended to include a contribution by EXO1 flap endonuclease activity. Overexpression of EXO1 can compensate for a loss of FEN1 (RAD27) or DNA2 in yeast (65). RAD27 and EXO1 were shown to redundantly process lagging strand flaps and the inactivation of the Rad27 or the Exo1 gene reduced strand displacement synthesis by Polδ (64).
Upon accumulation in large amounts in the absence of TREX1, long flaps originating from lagging strand synthesis are likely to form cGAS ligands by random hybridization, considering that 2/3 of mammalian genomes are comprised of repetitive sequence, including tandem repeats and transposable elements (77, 78). Moreover, the capacity of single strand-binding proteins that might prevent hybridization was shown to be exhausted in TREX1-deficient cells (35). Hybridization of oligonucleotides will not efficiently form higher order structures promoting cGAS activation by phase separation (79). However, random hybridization will frequently yield Y-DNA structures, i.e. double-stranded DNA with unpaired ends, which were shown to activate cGAS, provided unpaired guanosines are contained in the single-stranded overhangs (29). Flaps originating from Okazaki fragment maturation can include the Okazaki fragment RNA primer and form RNA:DNA hybrids upon hybridization, which were shown to be suboptimal cGAS ligands, but do activate the sensor (80). The role for EXO1 in generation of an immunogenic waste of flaps during lagging strand replication could be either in actual cleavage of long flaps, but also in ensuring strand displacement synthesis to proceed, thereby allowing generation of long flap structures. In yeast, loss of EXO1 or FEN1 resulted in reduced strand displacement synthesis (64). In line with a concept of DNA replication as a source of immunogenic DNA waste, Yang et al. observed cytosolic accumulations of BrdU-labeled single-stranded oligonucleotides in BrdU-pulsed TREX1-deficient fibroblasts (36).
Of note, replication-dependent and EXO1-dependent generation of immunogenic oligonucleotide waste on a large scale might occur not only during lagging strand synthesis but also during postreplicative ribonucleotide excision repair (81). Millions of single ribonucleotides are incorporated into the genomic DNA during each S phase and their rapid removal involves incision by RNase H2 followed by strand displacement synthesis and flap cleavage by FEN1 and EXO1 (81). EXO1 also contributes to other repair pathways, including repair of double-strand breaks by homologous recombination repair (HRR). In this pathway EXO1 is involved in end resection in preparation for strand invasion. While end resection was shown to be a source of DNA accumulating in the cytosol of genetically unstable tumor cells and even more so after additional experimental genome damage (44), we posit that steady state repair of double-strand breaks in genetically stable cells not exposed to genotoxic hazards does not occur on a large scale and therefore does not yield oligonucleotide waste in amounts sufficient to form cGAS ligands by random hybridization. In contrast, flap cleavage occurs on a massive scale during genome replication.
While our findings strongly suggest Okazaki fragment processing and/or postreplicative repair as an important source of immunogenic DNA waste in mice, Thomas et al. demonstrated that LINE1 retrotransposons are a major source of cytosolic DNA in human TREX1-deficient neuronal cells (27). Neurons and astrocytes differentiated from pluripotent stem cell lines rendered TREX1-deficient by CRISPR-mediated targeted mutagenesis, accumulated LINE1-derived, reverse transcribed DNA in their cytosol and mounted IFN responses that were inhibited by reverse transcriptase inhibitors (27). Type I IFN produced by astrocytes exerted toxic effects on neurons (27). In line with this observation, Rice et al. reduced the IFN and ISG expression of AGS patients by treatment with antiretroviral drugs (33). Both, replication-derived DNA waste and retroelement-derived cDNA may contribute to cGAS activation in TREX1-deficient mammals in vivo, with differential relevance in different species and cell types. Replication-derived waste plausibly plays no role in postmitotic neurons and astrocytes. Transposable elements evolved to be expressed and mobile in embryonic tissues, particularly in the germ-track, but to be largely inactive in somatic cells (82). However, as exceptions to this principle, some somatic tissues, including the CNS, show prominent transposon activity, likely reflecting an advantage to organismal function by somatic genetic diversification (83, 84). Transposable element activity also differs significantly between mouse and man (85). In addition to efficient reduction of IFN responses in the CNS as determined by IFNα levels in cerebrospinal fluid, antiretroviral treatment also reduced serum IFN and IFN signaling in blood cells (33). Whether this amelioration of IFN responses outside the CNS might partially be due to blockade of P2X7-mediated caspase activation and reduction of inflammatory cell death by the nucleoside RTIs (86) is currently unclear.
Genome damage and chromosomal translocations trigger cGAS/STING responses by micronuclear chromatin. TREX1-deficient cells exhibit mild genome damage and TREX1 was shown to contribute to degradation of micronuclear DNA (41), in line with the slight increase in numbers of micronucleated cells we observed in Trex1-/- mice. If micronuclear DNA was an important cGAS ligand in TREX1-deficient cells, however, IFN signaling would be expected to increase upon additional loss of p53, whereby cells carrying severe genome damage would be allowed to survive longer and continue to produce IFN. As IFN signaling was not enhanced in Trex1-/-Trp53-/- mice compared to p53-proficient Trex1-/- animals, we conclude that TREX1-mediated elimination of micronuclear chromatin and control of STING activation by micronuclei or chromatin bridges is relevant in genetically unstable, genome-damaged cells, rather than in TREX1-deficient, but otherwise healthy, primary cells.
Collectively, we demonstrate that activation of innate antiviral immunity of TREX1-deficient cells depends on their active proliferation and that additional loss of EXO1 significantly reduces the type I IFN response. While hyperactivity of EXO1 exonuclease activity in DNA mismatch-repair-deficient cancer cells was shown to activate the cGAS/STING pathway by induction of genome instability and consecutive mislocalization of chromatin to the cytosol (87), our findings strongly suggest 5’-flap cleavage during Okazaki fragment processing and/or during postreplicative ribonucleotide excision repair as the source of the pathogenic cGAS ligand accumulating in the absence of TREX1. We propose that genome replication of mammalian cells is invariably associated with production of immunogenic DNA waste triggering severe autoinflammation unless efficiently degraded.
Material and Methods
Mice
For reasons of clarity, we collectively refer to both, the Trex1 knock out line published by Morita et al. (23) and the Trex1Δ478/Δ478 line generated in this study, as ‘Trex1-/-’. Trex1Δ478/Δ478 mice, which phenocopy the published line, were used in experiments of Figure 2, and Figure S2. Trex1Δ478/Δ478XpaΔ11/Δ11 mice were generated by microinjection of a mixture of four individual Cas9 RNPs into zygotes of C57BL/6 mice allowing simultaneous targeting of Trex1 (GTGACTTCGGGCCGAGACGA and CGGTGCTTGCCAGTACAGGC) and Xpa (GTGTTTATCATCAGCATCTC and GAACCCACGCCATTCACAGTG). The injection mix contained 1.73 µM of each crRNA (IDT), 6.9 µM tracrRNA (IDT) and 6.9 µM Cas9 protein (ToolGen). Trex1FL/FL Rosa26-CreERT2WT/KI mice were described earlier (25). Mice were housed under specific pathogen-free conditions at the Experimental Center, Medical Faculty Carl Gustav Carus, TU Dresden. All procedures were in accordance with institutional guidelines on animal welfare and were approved by the Landesdirektion Dresden (permit number TVV-88/2017).
Induced Inactivation of Trex1 in Primary Cells In Vitro
Bone marrow cells from Trex1FL/FLR26CreERT2 or Trex1WT/FL R26CreERT2 control mice were cultured overnight in RPMI 1640 medium supplemented with 10% heat-inactivated FCS, 1% Pen/Strep, 2 mM L-Alanyl-L-glutamine and 1 mM sodium pyruvate (all Biochrom). Non-adherent cells were transferred to 10 cm petri dishes and differentiated for six days in RPMI medium (supplemented as described) containing 30 % L929 supernatant. Fresh differentiation medium was added after three days. BMDMs were harvested by trypsinization and scraping. For experiments addressing the kinetics of loss of TREX1, 2.5x105 cells were seeded onto 6-well plates and treated with 2 µg/ml (Z)-4-Hydroxytamoxifen (Sigma, Cat#H7904) for 22 h. To address effects of cell cycle arrest on the IFN response after induced knock out of Trex1, 2x106 cells were seeded onto 10 cm petri dishes and induced with 4-OHT. One day after washing away 4-OHT, cells from 10 cm petri dishes were split to 6 well plates at a density of 2.5x105 and maintained in the presence of 5 µM Palbociclib (Selleckchem, Cat#S1116) for 72 h. Efficiency of Palbociclib-induced cell cycle arrest was verified by pulsing with 10 µM EdU for 4h at 37°C. Upon harvest, incorporated EdU was quantified flow cytometrically using the Click-iT™ EdU Alexa Fluor™ 647 Flow Cytometry Assay Kit (Invitrogen, Cat#C10419) following the manufacturer’s instructions.
MEFs were generated by standard procedures. Briefly, after caesarean section, E14.5 mouse embryos were decapitated and internal organs were removed. Tissue was cut into small pieces, digested with 1x trypsin (0.25%, Invitrogen) for 30 min at 37°C and disaggregated by pipetting. The cell suspension was seeded into 10 cm petri dishes with DMEM (Gibco) supplemented with 10% heat inactivated FCS, 1% Pen/Strep, 1x nonessential amino acids and 100 µM β-mercaptoethanol. Induction of Cre activity was done as described above. To address effects of cell cycle inhibition on the IFN response after induced knock out of Trex1, 1.1x105 cells were seeded onto 6 cm petri dishes and induced with 4-OHT. On the next day, 4-OHT-containing medium was removed and fresh supplemented DMEM containing 200µM L-Mimosine (Sigma-Aldrich, Cat# M0253) was added to the cells for four days.
Quantitative RT-PCR
Total RNA was isolated using NucleoSpin RNA Kit (Macherey-Nagel) and reverse transcribed into cDNA using PrimeScript RT Reagent Kit (Takara). qRT-PCR was performed using Luna® Universal qPCR Master Mix (New England BioLabs) on a CFX384 Touch Real-Time PCR Detection System (Bio-Rad). Primer Sequences are listed in Supplementary Table S1. Target gene expression was normalized to the housekeeping gene Tbp1.
Quantification of Micronucleated Erythrocytes
Blood was sampled using retrobulbar puncture and mixed with heparin/PBS (250 units/ml, Biochrom). 20 µl of the blood/heparin/PBS mixture was added to 2 ml ice-cold Methanol, inverted and stored at -80°C until further use. For quantification of micronucleated erythrocytes, the protocol published by Balmus et al. was followed (42). Briefly, fixed blood cells were washed in bicarbonate buffer and stained against CD71 and Ter119 in the presence of RNase A. After washing and addition of PI, cells were analysed by flow cytometry and gated for single Ter119+ CD71- PI+ micronucleated normochromatic erythrocytes (MN-NCE). 5x105 cells in P1 SSC/FSC gate were acquired.
Assay for Nucleotide Excision Repair Activity
To detect ‘unscheduled’ DNA synthesis occurring in non-S phase cells repairing UV-induced genome damage (‘repair synthesis’), 5x106 splenocytes from XpaWT/WT and XpaKO/KO mice were seeded onto 10 cm petri dishes in 3 ml PBS and irradiated with 20 J/m² UVC (xy min) using an OSRAM HNS 15W OFR 3224 lamp. 6 ml of serum-free RPMI containing 1% Pen/Strep, 1x NEA and 10 µM EdU was added for 3h. Cells were harvested and incorporated EdU was quantified flow cytometrically using the Click-iT™ EdU Alexa Fluor™ 647 Flow Cytometry Assay Kit (Invitrogen, Cat#C10419) following the manufacturer’s instructions.
Western Blot Analysis
Ears of XpaWT/WT and XpaKO/KO animals were homogenized and lysed in RIPA buffer [50 mM Tris HCl, 150 mM NaCl, 1% NP-40, 0,1% SDS, 0,5% sodium deoxycholate, 1x Complete protease inhibitor (Roche)] for 30 min on ice. Lysates were cleared by centrifugation (16.000 xg, 10 min, 4°C) and supernatant was incubated with 6x Laemmli sample buffer at 95°C for 5 min. Cell pellets were directly lysed in 2x Laemmli sample buffer and incubated at 95°C for 5 min. Proteins were separated on 12% SDS polyacrylamide gels and transferred to a nitrocellulose membrane (Amersham Hybond-ECL, GE Healthcare). Membranes were blocked in 5% low fat dried milk/TBS-T or 1x Roti®Block and incubated overnight with the respective primary antibody. After washing in TBS-T, membranes were incubated with secondary antibodies for 1 h at room temperature and washed again in TBS-T. For signal detection, membranes were incubated in ECL (Amersham) or AP buffer following incubation in NBT/BCIP substrate (Roche).
The following primary antibodies were used: β-Tubulin (9F3, Cell Signaling Technology, Cat#2128), β-actin (13E5, Cell Signaling Technology, Cat#4970), Xpa (abcam, Cat#ab58464, 1:500), mTrex1 (C-11, Santa Cruz, Cat#sc-133112, 1:250), hTrex1 (E-6, Santa Cruz, Cat#sc-271870, 1:250), Mus81 (MTA30 2G10/3, Santa Cruz, Cat#58382, 1:250). Secondary antibodies used were AP-coupled anti-rabbit (Dako) and anti-mouse (Cell Signaling Technology).
Histology
Hearts were fixed in 4 % formalin and embedded in paraffin. 3 µm paraffin sections were stained with hematoxylin and eosin. Inflammation was quantified by an experienced histopathologist in a blinded fashion. A histological score for H&E staining was given depending on the grade of inflammation (0 – no sign of inflammation, 1 - slight diffuse increase in inflammatory cell infiltration, 2 - few small dense focal inflammatory infiltrates, 3 - large inflammatory infiltrates extending through more than half of the thickness of the ventricular muscle plus focal destruction of myocardial structure). To judge the effect of additional loss of EXO1 on the intensity of myocarditis of Trex1-/- mice, density of T cell infiltration on sections stained for CD3 and tissue fibrosis on Masson’s trichrome-stained sections were analyzed. CD3 immunohistochemical staining was performed on a Ventana Discovery Ultra Instrument. In brief, antigen retrieval using cell conditioning 1 solution (Ventana Medical Systmes) was performed at 95°C for 32 min followed by incubation with anti-CD3 antibody (SP7, abcam, Cat#ab16669, 1:200) at 36°C for 32 min, anti-rabbit OmniMap HRP-coupled secondary antibody (Ventana Medical Systems) for 12 min and finally Opal 650 TSA fluorophore (Akoya Biosciences, 1:100) at RT for 8 min. Primary and secondary antibodies were removed by denaturation at 100°C for 24 min in cell conditioning 2 buffer (Ventana Mediacl Systems) before counterstaining with DAPI. Sections were scanned (100x magnification) and images aquired (x200 magnification) by Vectra 3.0 Automated Imaging System (Akoya Biosciences). CD3+ cells were quantified using inForm (Akoya Biosciences) and R software. Masson’s trichrome staining was done according to standard protocols (Sigma-Aldrich, Procedure No.HT15).
Transcriptome Analysis
5 ng/µl RNA were sequenced on Illumina NovaSeq 6000 and data were demultiplexed using JE demultiplexer tool. Read quality was checked by FastQC. Transcript abundance was quantified using Salmon tool (88) and tximport (89) was used to generate gene level count matrices. The murine genome GRCm39 (90) was used as the reference. All post mapping analysis and visualization was conducted in R unless specifically stated otherwise. Normalization, exploratory analysis and differential expression analysis was performed using DESeq2 (91). Genes were considered significantly differentially expressed at FDR <0.01 and absolute log fold change >1.5. GGplot2 (92) was used to visualize results. List of significant DE genes was used to conduct a further gene set enrichment analysis (GSEA) (93, 94).
Statistical Analysis
Unless stated otherwise, significance was calculated by One-way ANOVA with Tukey’s post-hoc test (Figures 1–4) or Two-way ANOVA with Sidak’s post-hoc test (Figures 5, 6).
Author’s Note
While our manuscript was under review, Kim et al. published that TREX1 indeed degrades oligonucleotide waste released upon UV irradiation by the nucleotide excision repair (NER) machinery (95), supporting our initial hypothesis that the IFN response of TREX1-deficient cells might be driven by NER-derived DNA waste. Such DNA might contribute to UV-induced inflammation, but not to STING activation in unirradiated TREX1-deficient cells, as we found no amelioration of the IFN response of Trex1-/-Xpa-/- mice compared to Trex1-/- mice.
Data Availability Statement
The original contributions presented in the study are publicly available. This data can be found here: https://www.ncbi.nlm.nih.gov/search/all/?term=GSE197291.
Ethics Statement
The animal study was reviewed and approved by Landesdirektion Dresden. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author Contributions
NS designed and performed experiments; TS, ED, and KF performed experiments; GK, LH, and YG analyzed transcriptome data; WE generated Exo1-/- mice; LM and MS performed and analyzed immunohistochemistry; VH and RB contributed to the design of the study and discussed data; AR conceived and supervised the study; NS and AR wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by DFG grants Collaborative Research Center 237 Nucleic Acid Immunity, project B17 to AR, project B19 to RB and project A09 to VH, MedDrive Grant 60.496 of the Faculty of Medicine Carl Gustav Carus, TU Dresden to NS.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Excellent technical assistance by Madeleine Rickauer and Christa Haase is gratefully acknowledged.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.880413/full#supplementary-material
References
1. Roers A, Hiller B, Hornung V. Recognition of Endogenous Nucleic Acids by the Innate Immune System. Immunity (2016) 44(4):739–54. doi: 10.1016/j.immuni.2016.04.002
2. Crow YJ, Stetson DB. The Type I Interferonopathies: 10 Years on. Nat Rev Immunol (2021). doi: 10.1038/s41577-021-00633-9
3. Schlee M, Hartmann G. Discriminating Self From Non-Self in Nucleic Acid Sensing. Nat Rev Immunol (2016) 16(9):566–80. doi: 10.1038/nri.2016.78
4. Uggenti C, Lepelley A, Crow YJ. Self-Awareness: Nucleic Acid-Driven Inflammation and the Type I Interferonopathies. Annu Rev Immunol (2019) 37:247–67. doi: 10.1146/annurev-immunol-042718-041257
5. Rodero MP, Crow YJ. Type I Interferon-Mediated Monogenic Autoinflammation: The Type I Interferonopathies, a Conceptual Overview. J Exp Med (2016) 213(12):2527–38. doi: 10.1084/jem.20161596
6. Hopfner KP, Hornung V. Molecular Mechanisms and Cellular Functions of Cgas-Sting Signalling. Nat Rev Mol Cell Biol (2020) 21(9):501–21. doi: 10.1038/s41580-020-0244-x
7. Ablasser A, Chen ZJ. Cgas in Action: Expanding Roles in Immunity and Inflammation. Science (2019) 363(6431). doi: 10.1126/science.aat8657
8. Zhang X, Bai XC, Chen ZJ. Structures and Mechanisms in the Cgas-Sting Innate Immunity Pathway. Immunity (2020) 53(1):43–53. doi: 10.1016/j.immuni.2020.05.013
9. Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, et al. Pan-Viral Specificity of Ifn-Induced Genes Reveals New Roles for Cgas in Innate Immunity. Nature (2014) 505(7485):691–5. doi: 10.1038/nature12862
10. Volkman HE, Cambier S, Gray EE, Stetson DB. Tight Nuclear Tethering of Cgas Is Essential for Preventing Autoreactivity. Elife (2019) 8. doi: 10.7554/eLife.47491
11. de Oliveira Mann CC, Hopfner KP. Nuclear Cgas: Guard or Prisoner? EMBO J (2021) 40(16):e108293. doi: 10.15252/embj.2021108293
12. Uggenti C, Lepelley A, Depp M, Badrock AP, Rodero MP, El-Daher MT, et al. Cgas-Mediated Induction of Type I Interferon Due to Inborn Errors of Histone Pre-Mrna Processing. Nat Genet (2020) 52(12):1364–72. doi: 10.1038/s41588-020-00737-3
13. Mazur DJ, Perrino FW. Identification and Expression of the Trex1 and Trex2 Cdna Sequences Encoding Mammalian 3'–>5' Exonucleases. J Biol Chem (1999) 274(28):19655–60. doi: 10.1074/jbc.274.28.19655
14. Grieves JL, Fye JM, Harvey S, Grayson JM, Hollis T, Perrino FW. Exonuclease Trex1 Degrades Double-Stranded DNA to Prevent Spontaneous Lupus-Like Inflammatory Disease. Proc Natl Acad Sci USA (2015) 112(16):5117–22. doi: 10.1073/pnas.1423804112
15. Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, et al. Mutations in the Gene Encoding the 3'-5' DNA Exonuclease Trex1 Cause Aicardi-Goutieres Syndrome at the Ags1 Locus. Nat Genet (2006) 38(8):917–20. doi: 10.1038/ng1845
16. Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, et al. Characterization of Human Disease Phenotypes Associated With Mutations in Trex1, Rnaseh2a, Rnaseh2b, Rnaseh2c, Samhd1, Adar, and Ifih1. Am J Med Genet A (2015) 167A(2):296–312. doi: 10.1002/ajmg.a.36887
17. Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, et al. Mutations in the Gene Encoding the 3'-5' DNA Exonuclease Trex1 Are Associated With Systemic Lupus Erythematosus. Nat Genet (2007) 39(9):1065–7. doi: 10.1038/ng2091
18. Nehar-Belaid D, Hong S, Marches R, Chen G, Bolisetty M, Baisch J, et al. Mapping Systemic Lupus Erythematosus Heterogeneity at the Single-Cell Level. Nat Immunol (2020) 21(9):1094–106. doi: 10.1038/s41590-020-0743-0
19. Ramantani G, Kohlhase J, Hertzberg C, Innes AM, Engel K, Hunger S, et al. Expanding the Phenotypic Spectrum of Lupus Erythematosus in Aicardi-Goutieres Syndrome. Arthritis Rheum (2010) 62(5):1469–77. doi: 10.1002/art.27367
20. Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, et al. Interferon and Granulopoiesis Signatures in Systemic Lupus Erythematosus Blood. J Exp Med (2003) 197(6):711–23. doi: 10.1084/jem.20021553
21. Rice GI, Forte GM, Szynkiewicz M, Chase DS, Aeby A, Abdel-Hamid MS, et al. Assessment of Interferon-Related Biomarkers in Aicardi-Goutieres Syndrome Associated With Mutations in Trex1, Rnaseh2a, Rnaseh2b, Rnaseh2c, Samhd1, and Adar: A Case-Control Study. Lancet Neurol (2013) 12(12):1159–69. doi: 10.1016/S1474-4422(13)70258-8
22. Gall A, Treuting P, Elkon KB, Loo YM, Gale M Jr, Barber GN, et al. Autoimmunity Initiates in Nonhematopoietic Cells and Progresses Via Lymphocytes in an Interferon-Dependent Autoimmune Disease. Immunity (2012) 36(1):120–31. doi: 10.1016/j.immuni.2011.11.018
23. Morita M, Stamp G, Robins P, Dulic A, Rosewell I, Hrivnak G, et al. Gene-Targeted Mice Lacking the Trex1 (Dnase Iii) 3'–>5' DNA Exonuclease Develop Inflammatory Myocarditis. Mol Cell Biol (2004) 24(15):6719–27. doi: 10.1128/MCB.24.15.6719-6727.2004
24. Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 Prevents Cell-Intrinsic Initiation of Autoimmunity. Cell (2008) 134(4):587–98. doi: 10.1016/j.cell.2008.06.032
25. Peschke K, Achleitner M, Frenzel K, Gerbaulet A, Ada SR, Zeller N, et al. Loss of Trex1 in Dendritic Cells Is Sufficient to Trigger Systemic Autoimmunity. J Immunol (2016) 197(6):2157–66. doi: 10.4049/jimmunol.1600722
26. Ablasser A, Hemmerling I, Schmid-Burgk JL, Behrendt R, Roers A, Hornung V. Trex1 Deficiency Triggers Cell-Autonomous Immunity in a Cgas-Dependent Manner. J Immunol (2014) 192(12):5993–7. doi: 10.4049/jimmunol.1400737
27. Thomas CA, Tejwani L, Trujillo CA, Negraes PD, Herai RH, Mesci P, et al. Modeling of Trex1-Dependent Autoimmune Disease Using Human Stem Cells Highlights L1 Accumulation as a Source of Neuroinflammation. Cell Stem Cell (2017) 21(3):319–31.e8. doi: 10.1016/j.stem.2017.07.009
28. Yan N, Regalado-Magdos AD, Stiggelbout B, Lee-Kirsch MA, Lieberman J. The Cytosolic Exonuclease Trex1 Inhibits the Innate Immune Response to Human Immunodeficiency Virus Type 1. Nat Immunol (2010) 11(11):1005–13. doi: 10.1038/ni.1941
29. Herzner AM, Hagmann CA, Goldeck M, Wolter S, Kubler K, Wittmann S, et al. Sequence-Specific Activation of the DNA Sensor Cgas by Y-Form DNA Structures as Found in Primary Hiv-1 Cdna. Nat Immunol (2015) 16(10):1025–33. doi: 10.1038/ni.3267
30. Kato M, Takemoto K. Shinkai YA Somatic Role for the Histone Methyltransferase Setdb1 in Endogenous Retrovirus Silencing. Nat Commun (2018) 9(1):1683. doi: 10.1038/s41467-018-04132-9
31. Beck-Engeser GB, Eilat D, Wabl M. An Autoimmune Disease Prevented by Anti-Retroviral Drugs. Retrovirology (2011) 8:91. doi: 10.1186/1742-4690-8-91
32. Achleitner M, Kleefisch M, Hennig A, Peschke K, Polikarpova A, Oertel R, et al. Lack of Trex1 Causes Systemic Autoimmunity Despite the Presence of Antiretroviral Drugs. J Immunol (2017) 199(7):2261–9. doi: 10.4049/jimmunol.1700714
33. Rice GI, Meyzer C, Bouazza N, Hully M, Boddaert N, Semeraro M, et al. Reverse-Transcriptase Inhibitors in the Aicardi-Goutieres Syndrome. N Engl J Med (2018) 379(23):2275–7. doi: 10.1056/NEJMc1810983
34. Reijns MA, Rabe B, Rigby RE, Mill P, Astell KR, Lettice LA, et al. Enzymatic Removal of Ribonucleotides From DNA Is Essential for Mammalian Genome Integrity and Development. Cell (2012) 149(5):1008–22. doi: 10.1016/j.cell.2012.04.011
35. Wolf C, Rapp A, Berndt N, Staroske W, Schuster M, Dobrick-Mattheuer M, et al. Rpa and Rad51 Constitute a Cell Intrinsic Mechanism to Protect the Cytosol From Self DNA. Nat Commun (2016) 7:11752. doi: 10.1038/ncomms11752
36. Yang YG, Lindahl T, Barnes DE. Trex1 Exonuclease Degrades Ssdna to Prevent Chronic Checkpoint Activation and Autoimmune Disease. Cell (2007) 131(5):873–86. doi: 10.1016/j.cell.2007.10.017
37. Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, et al. Mutations in Genes Encoding Ribonuclease H2 Subunits Cause Aicardi-Goutieres Syndrome and Mimic Congenital Viral Brain Infection. Nat Genet (2006) 38(8):910–6. doi: 10.1038/ng1842
38. Hiller B, Hoppe A, Haase C, Hiller C, Schubert N, Muller W, et al. Ribonucleotide Excision Repair Is Essential to Prevent Squamous Cell Carcinoma of the Skin. Cancer Res (2018) 78(20):5917–26. doi: 10.1158/0008-5472.CAN-18-1099
39. Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic Progression Following DNA Damage Enables Pattern Recognition Within Micronuclei. Nature (2017) 548(7668):466–70. doi: 10.1038/nature23470
40. Flynn PJ, Koch PD, Mitchison TJ. Chromatin Bridges, Not Micronuclei, Activate Cgas After Drug-Induced Mitotic Errors in Human Cells. Proc Natl Acad Sci USA (2021) 118(48). doi: 10.1073/pnas.2103585118
41. Mohr L, Toufektchan E, von Morgen P, Chu K, Kapoor A, Maciejowski J. Er-Directed Trex1 Limits Cgas Activation at Micronuclei. Mol Cell (2021) 81(4):724–38.e9. doi: 10.1016/j.molcel.2020.12.037
42. Balmus G, Karp NA, Ng BL, Jackson SP, Adams DJ. McIntyre RE. A High-Throughput in Vivo Micronucleus Assay for Genome Instability Screening in Mice. Nat Protoc (2015) 10(1):205–15. doi: 10.1038/nprot.2015.010
43. Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, et al. Tumor Spectrum Analysis in P53-Mutant Mice. Curr Biol (1994) 4(1):1–7. doi: 10.1016/s0960-9822(00)00002-6
44. Erdal E, Haider S, Rehwinkel J, Harris AL. McHugh PJ. A Prosurvival DNA Damage-Induced Cytoplasmic Interferon Response Is Mediated by End Resection Factors and Is Limited by Trex1. Genes Dev (2017) 31(4):353–69. doi: 10.1101/gad.289769.116
45. Ahn J, Xia T, Konno H, Konno K, Ruiz P, Barber GN. Inflammation-Driven Carcinogenesis Is Mediated Through Sting. Nat Commun (2014) 5:5166. doi: 10.1038/ncomms6166
46. Shen YJ, Le Bert N, Chitre AA, Koo CX, Nga XH, Ho SS, et al. Genome-Derived Cytosolic DNA Mediates Type I Interferon-Dependent Rejection of B Cell Lymphoma Cells. Cell Rep (2015) 11(3):460–73. doi: 10.1016/j.celrep.2015.03.041
47. Takahashi A, Loo TM, Okada R, Kamachi F, Watanabe Y, Wakita M, et al. Downregulation of Cytoplasmic Dnases Is Implicated in Cytoplasmic DNA Accumulation and Sasp in Senescent Cells. Nat Commun (2018) 9(1):1249. doi: 10.1038/s41467-018-03555-8
48. Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, et al. DNA Exonuclease Trex1 Regulates Radiotherapy-Induced Tumour Immunogenicity. Nat Commun (2017) 8:15618. doi: 10.1038/ncomms15618
49. Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JH. Understanding Nucleotide Excision Repair and Its Roles in Cancer and Ageing. Nat Rev Mol Cell Biol (2014) 15(7):465–81. doi: 10.1038/nrm3822
50. Branum ME, Reardon JT, Sancar A. DNA Repair Excision Nuclease Attacks Undamaged DNA. A Potential Source of Spontaneous Mutations. J Biol Chem (2001) 276(27):25421–6. doi: 10.1074/jbc.M101032200
51. Hanawalt PC. Subpathways of Nucleotide Excision Repair and Their Regulation. Oncogene (2002) 21(58):8949–56. doi: 10.1038/sj.onc.1206096
52. Hyka-Nouspikel N, Lemonidis K, Lu WT, Nouspikel T. Circulating Human B Lymphocytes Are Deficient in Nucleotide Excision Repair and Accumulate Mutations Upon Proliferation. Blood (2011) 117(23):6277–86. doi: 10.1182/blood-2010-12-326637
53. de Vries A, van Oostrom CT, Hofhuis FM, Dortant PM, Berg RJ, de Gruijl FR, et al. Increased Susceptibility to Ultraviolet-B and Carcinogens of Mice Lacking the DNA Excision Repair Gene Xpa. Nature (1995) 377(6545):169–73. doi: 10.1038/377169a0
54. Ho SS, Zhang WY, Tan NY, Khatoo M, Suter MA, Tripathi S, et al. The DNA Structure-Specific Endonuclease Mus81 Mediates DNA Sensor Sting-Dependent Host Rejection of Prostate Cancer Cells. Immunity (2016) 44(5):1177–89. doi: 10.1016/j.immuni.2016.04.010
55. Schwartz EK, Heyer WD. Processing of Joint Molecule Intermediates by Structure-Selective Endonucleases During Homologous Recombination in Eukaryotes. Chromosoma (2011) 120(2):109–27. doi: 10.1007/s00412-010-0304-7
56. Naim V, Wilhelm T, Debatisse M, Rosselli F. Ercc1 and Mus81-Eme1 Promote Sister Chromatid Separation by Processing Late Replication Intermediates at Common Fragile Sites During Mitosis. Nat Cell Biol (2013) 15(8):1008–15. doi: 10.1038/ncb2793
57. Hanada K, Budzowska M, Davies SL, van Drunen E, Onizawa H, Beverloo HB, et al. The Structure-Specific Endonuclease Mus81 Contributes to Replication Restart by Generating Double-Strand DNA Breaks. Nat Struct Mol Biol (2007) 14(11):1096–104. doi: 10.1038/nsmb1313
58. Li T, Chen ZJ. The Cgas-Cgamp-Sting Pathway Connects DNA Damage to Inflammation, Senescence, and Cancer. J Exp Med (2018) 215(5):1287–99. doi: 10.1084/jem.20180139
59. Laguette N, Bregnard C, Hue P, Basbous J, Yatim A, Larroque M, et al. Premature Activation of the Slx4 Complex by Vpr Promotes G2/M Arrest and Escape From Innate Immune Sensing. Cell (2014) 156(1-2):134–45. doi: 10.1016/j.cell.2013.12.011
60. Symington LS, Gautier J. Double-Strand Break End Resection and Repair Pathway Choice. Annu Rev Genet (2011) 45:247–71. doi: 10.1146/annurev-genet-110410-132435
61. Keijzers G, Liu D, Rasmussen LJ. Exonuclease 1 and Its Versatile Roles in DNA Repair. Crit Rev Biochem Mol Biol (2016) 51(6):440–51. doi: 10.1080/10409238.2016.1215407
62. Schaetzlein S, Chahwan R, Avdievich E, Roa S, Wei K, Eoff RL, et al. Mammalian Exo1 Encodes Both Structural and Catalytic Functions That Play Distinct Roles in Essential Biological Processes. Proc Natl Acad Sci USA (2013) 110(27):E2470–9. doi: 10.1073/pnas.1308512110
63. Burgers PMJ, Kunkel TA. Eukaryotic DNA Replication Fork. Annu Rev Biochem (2017) 86:417–38. doi: 10.1146/annurev-biochem-061516-044709
64. Kahli M, Osmundson JS, Yeung R, Smith DJ. Processing of Eukaryotic Okazaki Fragments by Redundant Nucleases Can Be Uncoupled From Ongoing DNA Replication In Vivo. Nucleic Acids Res (2019) 47(4):1814–22. doi: 10.1093/nar/gky1242
65. Tishkoff DX, Boerger AL, Bertrand P, Filosi N, Gaida GM, Kane MF, et al. Identification and Characterization of Saccharomyces Cerevisiae Exo1, a Gene Encoding an Exonuclease That Interacts With Msh2. Proc Natl Acad Sci USA (1997) 94(14):7487–92. doi: 10.1073/pnas.94.14.7487
66. Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, et al. Specific Inhibition of Cyclin-Dependent Kinase 4/6 by Pd 0332991 and Associated Antitumor Activity in Human Tumor Xenografts. Mol Cancer Ther (2004) 3(11):1427–38. doi: 10.1158/1535-7163.1427.3.11
67. Kubota S, Fukumoto Y, Ishibashi K, Soeda S, Kubota S, Yuki R, et al. Activation of the Prereplication Complex Is Blocked by Mimosine Through Reactive Oxygen Species-Activated Ataxia Telangiectasia Mutated (Atm) Protein Without DNA Damage. J Biol Chem (2014) 289(9):5730–46. doi: 10.1074/jbc.M113.546655
68. Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Moroy T. Features of Systemic Lupus Erythematosus in Dnase1-Deficient Mice. Nat Genet (2000) 25(2):177–81. doi: 10.1038/76032
69. Kenny EF, Raupach B, Abu Abed U, Brinkmann V, Zychlinsky A. Dnase1-Deficient Mice Spontaneously Develop a Systemic Lupus Erythematosus-Like Disease. Eur J Immunol (2019) 49(4):590–9. doi: 10.1002/eji.201847875
70. Chitrabamrung S, Rubin RL, Tan EM. Serum Deoxyribonuclease I and Clinical Activity in Systemic Lupus Erythematosus. Rheumatol Int (1981) 1(2):55–60. doi: 10.1007/BF00541153
71. Yasutomo K, Horiuchi T, Kagami S, Tsukamoto H, Hashimura C, Urushihara M, et al. Mutation of Dnase1 in People With Systemic Lupus Erythematosus. Nat Genet (2001) 28(4):313–4. doi: 10.1038/91070
72. Lan YY, Londono D, Bouley R, Rooney MS, Hacohen N. Dnase2a Deficiency Uncovers Lysosomal Clearance ofVia Autophagy. Cell Rep (2014) 9(1):180–92. doi: 10.1016/j.celrep.2014.08.074
73. Rodero MP, Tesser A, Bartok E, Rice GI, Della Mina E, Depp M, et al. Type I Interferon-Mediated Autoinflammation Due to Dnase Ii Deficiency. Nat Commun (2017) 8(1):2176. doi: 10.1038/s41467-017-01932-3
74. Okabe Y, Kawane K, Akira S, Taniguchi T, Nagata S. Toll-Like Receptor-Independent Gene Induction Program Activated by Mammalian DNA Escaped From Apoptotic DNA Degradation. J Exp Med (2005) 202(10):1333–9. doi: 10.1084/jem.20051654
75. Manderson AP, Botto M, Walport MJ. The Role of Complement in the Development of Systemic Lupus Erythematosus. Annu Rev Immunol (2004) 22:431–56. doi: 10.1146/annurev.immunol.22.012703.104549
76. Smith DJ, Whitehouse I. Intrinsic Coupling of Lagging-Strand Synthesis to Chromatin Assembly. Nature (2012) 483(7390):434–8. doi: 10.1038/nature10895
77. de Koning AP, Gu W, Castoe TA, Batzer MA, Pollock DD. Repetitive Elements May Comprise Over Two-Thirds of the Human Genome. PloS Genet (2011) 7(12):e1002384. doi: 10.1371/journal.pgen.1002384
78. Padeken J, Zeller P, Gasser SM. Repeat DNA in Genome Organization and Stability. Curr Opin Genet Dev (2015) 31:12–9. doi: 10.1016/j.gde.2015.03.009
79. Du M, Chen ZJ. DNA-Induced Liquid Phase Condensation of Cgas Activates Innate Immune Signaling. Science (2018) 361(6403):704–9. doi: 10.1126/science.aat1022
80. Mankan AK, Schmidt T, Chauhan D, Goldeck M, Honing K, Gaidt M, et al. Cytosolic RNA:DNA Hybrids Activate the Cgas-Sting Axis. EMBO J (2014) 33(24):2937–46. doi: 10.15252/embj.201488726
81. Sparks JL, Chon H, Cerritelli SM, Kunkel TA, Johansson E, Crouch RJ, et al. Rnase H2-Initiated Ribonucleotide Excision Repair. Mol Cell (2012) 47(6):980–6. doi: 10.1016/j.molcel.2012.06.035
82. Haig D. Transposable Elements: Self-Seekers of the Germline, Team-Players of the Soma. Bioessays (2016) 38(11):1158–66. doi: 10.1002/bies.201600125
83. Baillie JK, Barnett MW, Upton KR, Gerhardt DJ, Richmond TA, De Sapio F, et al. Somatic Retrotransposition Alters the Genetic Landscape of the Human Brain. Nature (2011) 479(7374):534–7. doi: 10.1038/nature10531
84. Upton KR, Gerhardt DJ, Jesuadian JS, Richardson SR, Sanchez-Luque FJ, Bodea GO, et al. Ubiquitous L1 Mosaicism in Hippocampal Neurons. Cell (2015) 161(2):228–39. doi: 10.1016/j.cell.2015.03.026
85. Huang CR, Burns KH, Boeke JD. Active Transposition in Genomes. Annu Rev Genet (2012) 46:651–75. doi: 10.1146/annurev-genet-110711-155616
86. Fowler BJ, Gelfand BD, Kim Y, Kerur N, Tarallo V, Hirano Y, et al. Nucleoside Reverse Transcriptase Inhibitors Possess Intrinsic Anti-Inflammatory Activity. Science (2014) 346(6212):1000–3. doi: 10.1126/science.1261754
87. Guan J, Lu C, Jin Q, Lu H, Chen X, Tian L, et al. Mlh1 Deficiency-Triggered DNA Hyperexcision by Exonuclease 1 Activates the Cgas-Sting Pathway. Cancer Cell (2021) 39(1):109–21.e5. doi: 10.1016/j.ccell.2020.11.004
88. Roberts A, Pachter L. Streaming Fragment Assignment for Real-Time Analysis of Sequencing Experiments. Nat Methods (2013) 10(1):71–3. doi: 10.1038/nmeth.2251
89. Soneson C, Love MI, Robinson MD. Differential Analyses for Rna-Seq: Transcript-Level Estimates Improve Gene-Level Inferences. F1000Res (2015) 4:1521. doi: 10.12688/f1000research.7563.2
90. Howe KL, Contreras-Moreira B, De Silva N, Maslen G, Akanni W, Allen J, et al. Ensembl Genomes 2020-Enabling Non-Vertebrate Genomic Research. Nucleic Acids Res (2020) 48(D1):D689–D95. doi: 10.1093/nar/gkz890
91. Love MI, Huber W, Anders S. Moderated Estimation of Fold Change and Dispersion for Rna-Seq Data With Deseq2. Genome Biol (2014) 15(12):550. doi: 10.1186/s13059-014-0550-8
92. Wickham H. Ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag New York (2016). doi: 10.1007/978-3-319-24277-4
93. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc Natl Acad Sci USA (2005) 102(43):15545–50. doi: 10.1073/pnas.0506580102
94. Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. Pgc-1alpha-Responsive Genes Involved in Oxidative Phosphorylation Are Coordinately Downregulated in Human Diabetes. Nat Genet (2003) 34(3):267–73. doi: 10.1038/ng1180
Keywords: Trex1, type I interferon, Exo1, replication, cytosolic DNA, interferonopathy
Citation: Schubert N, Schumann T, Daum E, Flade K, Ge Y, Hagedorn L, Edelmann W, Müller L, Schmitz M, Kuut G, Hornung V, Behrendt R and Roers A (2022) Genome Replication Is Associated With Release of Immunogenic DNA Waste. Front. Immunol. 13:880413. doi: 10.3389/fimmu.2022.880413
Received: 21 February 2022; Accepted: 05 April 2022;
Published: 11 May 2022.
Edited by:
Frédéric Rieux-Laucat, U1163 Institut Imagine (INSERM), FranceReviewed by:
Nadia Jeremiah, Institut Curie, FranceGeneviève Pépin, Université du Québec à Trois-Rivières, Canada
Copyright © 2022 Schubert, Schumann, Daum, Flade, Ge, Hagedorn, Edelmann, Müller, Schmitz, Kuut, Hornung, Behrendt and Roers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Axel Roers, axel.roers@uni-heidelberg.de