 Kun Yang1,2*
Kun Yang1,2* Matthew Holt3Min Fan1,2
Matthew Holt3Min Fan1,2 Victor Lam4Yong Yang3Tuanzhu Ha1,2David L. Williams1,2
Victor Lam4Yong Yang3Tuanzhu Ha1,2David L. Williams1,2 Chuanfu Li1,2Xiaohui Wang1,2*
Chuanfu Li1,2Xiaohui Wang1,2*- 1Department of Surgery, James H. Quillen College of Medicine, East Tennessee State University, Johnson City, TN, United States
- 2Center of Excellence in Inflammation, Infectious Disease and Immunity, James H. Quillen College of Medicine, East Tennessee State University, Johnson City, TN, United States
- 3James H. Quillen College of Medicine, East Tennessee State University, Johnson City, TN, United States
- 4College of Arts and Science, New York University, New York City, NY, United States
Coronavirus disease 2019 (COVID-19), an infectious respiratory disease propagated by a new virus known as Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2), has resulted in global healthcare crises. Emerging evidence from patients with COVID-19 suggests that endothelial cell damage plays a central role in COVID-19 pathogenesis and could be a major contributor to the severity and mortality of COVID-19. Like other infectious diseases, the pathogenesis of COVID-19 is closely associated with metabolic processes. Lactate, a potential biomarker in COVID-19, has recently been shown to mediate endothelial barrier dysfunction. In this review, we provide an overview of cardiovascular injuries and metabolic alterations caused by SARS-CoV-2 infection. We also propose that lactate plays a potential role in COVID-19-driven endothelial cell injury.
Introduction
Coronavirus disease 2019 (COVID-19) is defined as an infectious respiratory disease propagated by a new virus labeled the Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2). COVID-19 was first identified in Wuhan, China in November of 2019 and has become a global health threat affecting more than 200 million people with a mortality rate of 2.3% due to its high contagiousness and lack of specific antiviral treatments (1). There are excellent reviews and articles on the clinical manifestation, hematology laboratory, and management of COVID-19 patients (1–3). By November 2021, over twenty COVID-19 vaccines have been approved in different parts of the world (4). Despite the COVID-19 vaccine, which has been quickly and successfully developed and employed to fight against the COVID-19 infection, the exact mechanisms by which the SARS-CoV-2 significantly causes dysfunction of several systems, including respiratory system, nerve system, and cardiovascular system have not been elucidated entirely.
Emerging clinical data has shown that the COVID-19 patients with cardiovascular diseases (CVDs) have a greater mortality (11%) than in total case mortality (2.3%) (5). On the other hand, COVID-19 infected patients exhibit cardiovascular disorders and heart attack symptoms (6–8). This evidence suggests that SARS-CoV-2 infection could cause cardiovascular dysfunction which is a major factor contributing to the mortality of a substantial proportion of the patients with severe COVID-19 infection. Importantly, the symptoms of severe COVID-19 infected patients resemble the clinical features of endothelial dysfunction (8, 9), indicating that SARS-CoV-2 could cause the endothelium damage. Indeed, electron microscopy analysis of post-mortem tissues showed that SARS-CoV-2 could infect pulmonary endothelial cells and induce endotheliitis in COVID-19 infected patients who are critically ill (9). In addition, SARS-CoV-2 could directly infect endothelial cells via angiotensin-converting enzyme 2 (ACE2), subsequently alter the vascular homeostasis, and induce clinical manifestations such as acute respiratory distress syndrome (ARDS) (9–11).
Recent studies highlight the role of metabolisms in the regulation of innate immune and inflammatory responses (12–15). Metabolic reprograming plays a critical role in innate immune and inflammatory responses (16). Severe COVID-19 infected patients usually exhibit the “cytokine storm”, indicating that the metabolisms in these patients have been altered. It is possible that aerobic glycolytic metabolism could be involved in the pathogenesis of the COVID-19 infection that induces severe conditions in those who are infected (17). Importantly, aerobic glycolytic metabolism not only regulates innate immune response (12), but also modulates endothelial cell function (14, 18). Generally, virus infections activate several immune cell types, such as dendritic cells, neutrophils and macrophages to produce pro-inflammatory cytokines that fight against virus invasions and maintain the tissues’ homeostasis (19). This process requires a rapid energy production to provide fuel for immune cell proliferation and inflammation (20). Recent evidence has shown that aerobic glycolytic metabolism and subsequent lactate production can be considered as an integral part of cellular signaling (12–14, 21). Furthermore, more recent findings in immunometabolism show that aerobic glycolysis can be a metabolic choice of immune cells and the function of aerobic glycolytic metabolism is not limited to supporting cell proliferation (22). Thus, it appears that cells can modulate their metabolism to adapt to different energy requirements and signaling events in pathophysiological situations.
Historically, lactate was the end product of aerobic glycolytic metabolism and was considered as a “waste” to be cleared from blood by the liver and kidney (23). Growing evidence suggests that lactate can be used as a sensitive and independent biomarker for critical illnesses, including sepsis (24), cardiovascular dysfunction (25, 26) and various types of cancer (27). Lactate could be a potent signaling molecule in vascular homeostasis, which is supported by a study showing that lactate disrupts vascular barrier function and increases vascular permeability of bone marrow during inflammation (14). Moreover, lactate dehydrogenase (LDH), a key enzyme in aerobic glycolysis, has been associated with worse outcomes in patients with viral infections, including COVID-19 (28–31). In addition, serum lactate levels in severe COVID-19 infected patients are significantly increased (32–35), suggesting increased aerobic glycolytic metabolism in COVID-19 infected patients (17). The important question is whether the COVID-19 infection alters cellular metabolisms that contribute to cardiovascular dysfunction. Based on current knowledge that aerobic glycolytic metabolism is involved in metabolic immune function (36) and cardiovascular dysfunction (37), understanding of the potential mechanisms by which SARS-CoV-2 causes endothelial cell barrier dysfunction could provide preventative and therapeutic solutions for severe COVID-19 patients. In this review, we summarize the association between the COVID-19 infection and cardiovascular dysfunction and discuss the potential role of aerobic glycolytic metabolism and SARS-CoV-2 induced endothelial cell barrier dysfunction, leading to multiple organ damage.
Cardiovascular Disorder in COVID-19 Patients
Cardiovascular Disease Is a High Risk for COVID-19 Infection
Cardiovascular disease (CVD) is a common comorbidity in the patients with Severe Acute Respiratory Syndrome (SARS) and was just as prevalent in patients who experienced Middle East Respiratory Syndrome (MERS) during the previous global pandemic (38). Numerous studies have shown that there are similar genetic identity (79.6%) and biological features shared between SARS-CoV-2 (for COVID-19) and SARS-CoV (for SARS) (39–41). One study that was conducted held a 12-year follow-up that consisted of 25 patients who had recovered from the SARS-CoV infection; among these patients, 44% of them exhibited cardiovascular system abnormalities (42). Once again, due to the similarity in structure between SARS-CoV and SARS-CoV-2, it is highly possible that Covid-19 may also cause similar future troubles for the myocardium. Therefore, it is no surprise that CVD is present in the patients with COVID-19 (with a prevalence of ~17%) (6, 43, 44). A study, including 416 hospitalized COVID-19 infected patients in Wuhan (China), by Shi et al. showed that patients with a history of CVDs had higher risk of in-hospital death (45). Similarly, reports involving 1,591 patients (with a mortality rate of 26%) with COVID-19 in the Lombardy (Italy) (46) and 393 patients with COVID-19 in New York City (USA) (47) showed pre-existing CVD rates of 21% and 14%, respectively. In addition, a meta-analysis of fifty-six studies including 159,698 COVID-19 patients revealed that 25% of ICU-admitted patients had CVD, and the pooled prevalence of acute cardiac injury by 33.6%, arrhythmia by 33.0%, heart failure by 20.4%, coronal artery disease 20.6% and hypertension by 43.6%, respectively (48). Another meta-analysis including both ICU and non-ICU COVID-19 infected patients in China (1,527 cases in total) showed that the proportions of hypotension, cardio-cerebrovascular disease and diabetes in COVID-19 patients were 17.1%, 16.4% and 9.7%, respectively (49). Among these patients, the incidences of hypertension, cardio-cerebrovascular disease and diabetes were at least twofold higher in ICU cases than in non-ICU counterparts (49), indicating that the patients with CVD are more susceptible to suffer severe condition and are at a higher risk of death. In a study from the National Health Commission of China (NHC), mortality data for Covid-19 was released, and it determined that 17% of the patients exhibited a history of coronary heart disease while 35% had a history of hypertension (50). These data led to the conclusion that who had any sort of underlying CVDs and were simultaneously infected with SARS-CoV-2 had a higher chance of experiencing more severe symptoms. Therefore, it can be inferred that CVD/CVD-related risk factors strongly affect the prognosis of the COVID-19 patients.
COVID-19 Infection Induces Cardiovascular Dysfunction
Importantly, COVID-19 infected patients who do not have CVD exhibit cardiovascular dysfunctions, including myocardial injury, cardiac arrhythmia, as well as thrombotic complications; this indicates that COVID-19 itself can induce cardiovascular disorders (6, 51). This mechanism of direct infection occurs when the virus immediately infects cardiomyocytes originating from induced pluripotent stem cells (iPSCs); as a result, SARS-CoV-2 infection in iPSCs induces morphological and cytotoxic effects characterized by detaching from neighboring cells and increased cell death, suggesting SARS-CoV-2 directly causes damages to cardiac tissue (52). This observation may explain myocardial complications in SARS-CoV-2 infection. Furthermore, in another study pertaining to iPSCs, data revealed that after 72 hours of exposure to the SARS-CoV-2 infection, apoptosis as well as cessation of beating will appear (53). To measure the severity of injury done to the myocardium, the use of serum troponin (troponin T or troponin I) level, which is a specific marker for cardiac injury, can be applied (54–56). A multicenter study showed that 278 (45.3%) of 614 COVID-19 infected patients had elevated serum levels of troponin (troponin T or troponin I) (56). Importantly, increased troponin levels, independent from concomitant cardiac disease, were associated with increased in-hospital mortality (56). A report from the National Health Commission of China showed that serum troponin I levels were increased and cardiac arrest occurred in 12% of COVID-19 infected patients who did not have CVDs previously during hospitalization (2, 44, 50). Data from autopsy analysis shows that SARS-CoV-2 virus was identified in 24 (61.5%) cardiac tissues of 39 patients with COVID-19 infection (57). A report from a single-center shows that cardiac arrhythmia was also a prevalent manifestation in 138 patients with COVID-19 infection (58). This study documented that arrhythmia occurred in 17% of hospitalized patients and 44% of ICU-admitted COVID-19 infected patients (58). With a broad range of laboratory coagulation parameter alterations including D-dimer, prothrombin time and fibrinogen in COVID-19 infected patients, coagulation dysfunction has been considered as a hallmark of SARS-CoV-2 infection. Tang et al. observed that, in 183 consecutive COVID-19 infected patients, non-survivors had higher D-dimer levels, fibrinogen degradation products and longer prothrombin time, when compared with survivors (59). A study by Klock et al. shows that the incidence of thrombotic complications was 31% in 184 ICU-admitted patients with COVID-19 infection in Dutch (60). This study also shows that venous thromboembolism, confirmed by CT pulmonary angiogram (CTPA) and ultrasonography, accounted for 87% of all thrombotic events (60). The thromboembolism in COVID-19 infected patients may result from excessive inflammation, hypoxia and diffuse intravascular coagulation (61, 62). However, anticoagulant therapy using prophylactic heparin in COVID-19 patients who developed sepsis-induced coagulopathy markedly reduced 28-day mortality from 64% to 40% (63). In addition, uncontrolled blood pressure is a risk factor for COVID-19 patients by causing acute kidney injury and chronic obstructive pulmonary disease (COPD) (64).
Endothelial Cell Dysfunction in COVID-19 Infected Patients
The endothelium is a layer of endothelial cells (ECs) that line the interior surface of blood vessels and plays a critical role in mediating vasomotor tone, maintaining blood fluidity, and balancing local inflammatory mediators (65, 66). The maladaptive response of ECs to acute inflammation contributes to the pathogenesis of various infectious diseases and multiple organ dysfunction syndrome (MODS) (67). Several recent studies investigated mechanisms by which SARS-Cov-2 induces EC dysfunctions, including inflammation, vasoconstriction, permeability, and coagulation (68–70). A study by Ackermann et al. shows that COVID-19 infection not only causes acute respiratory distress syndrome (ARDS), but also harms the vasculature (71). This pathologic study included seven lung tissues from COVID-19 infected patients, seven lungs from Influenza (H1N1) patients with ARDS, and 10 from age-matched uninfected controls. These lung tissues were examined with seven-color immunohistochemical analysis, micro-computed tomographic imaging, scanning electron microscopy, corrosion casting, and direct multiplexed measurement of gene expression. The authors compared the results between the groups focusing on the three distinct angiocentric features including: 1) severe endothelial injury associated with intracellular SARS-CoV-2 virus and disrupted endothelial cell membranes, 2) widespread vascular thrombosis with microangiopathy and occlusion of alveolar capillaries, and 3) significant new vessel growth through a mechanism of intussusceptive angiogenesis. The COVID-19 infected patients exhibited 9 times more alveolar capillary microthrombi (P<0.001) compared with H1N1 influenza patients. COVID-19 infected patients also presented with 2.7 greater of the amount of new vessel growth through intussusceptive angiogenesis (P<0.001) than in H1N1 influenza. In addition, endothelial cells from the COVID-19 patients exhibited cellular swelling, disrupted intracellular junctions, and a loss of contact with the basement membrane. An in vitro study by Robles and colleagues showed that the spike protein of SARS-CoV-2 promotes the expression of leukocyte adhesion molecules VCAM1 and ICAM1 upon binding to integrin α5β1 on ECs, resulting in increased leukocyte adhesion to ECs (72). A previous study shows that integrin α5β1 activates NF-κB in ECs to elicit inflammation (73). Consistently, SARS-CoV-2 protein treatment enhanced p65 nuclear accumulation and IL-6 expression in ECs (72, 74). To explore the mechanism of myocardial injury in COVID-19 infected patients, Feng et al. utilized a rhesus macaque model of SARS-CoV-2 respiration tract infection (75). They observed that increased infiltration of inflammatory cells in left ventricle tissues and elevated levels of inflammatory cytokines in infected macaques, suggesting the occurrence of viral myocarditis following SARS-CoV-2 infection (75). Notably, the expression of ICAM1 and VCAM1 in the left ventricle tissues was also upregulated in infected macaques as compared to healthy controls (75). These findings provided evidence showing that the endothelial cell damage may be attributed to direct SARS-CoV-2 infection and perivascular inflammation.
ACE-2 and Endothelial Cell Dysfunction in COVID-19 Infection
Angiotensin-Converting Enzyme 2 (ACE2) is a type-I transmembrane glycoprotein that negatively regulates the renin-angiotensin system (RAS) by degrading Ang II to the heptapeptide Ang 1-7 (76, 77). The protein was initially identified as a homolog to ACE in 2000 by Tipnis et al. (78). Besides its peptidase-dependent actions in regulating RAS, ACE2 was then identified as an essential receptor for SARS coronavirus in 2003 (79). Although ACE2 is shown as a protective molecule against lethal lung injury in SARS, the expression of ACE2 is not limited to respiratory system (80). Instead, a recent immunohistochemical analysis showed that ACE2 has limited expression in respiratory tracts compared to other tissues/cells, including enterocytes, renal tubules, gallbladder, cardiomyocytes, male reproductive cells, placental trophoblasts, ductal cells, eye, and vasculature (76). Intriguingly, ACE2 is expressed in arterial and venous endothelial cells and arterial smooth muscle cells in various human organs (81). It is suggested that ACE2 is required to maintain the endothelial integrity inside the vessels (11). Indeed, existing data for SARS-CoV-1 in 2002 SARS pandemic indicate that virus binding can reduce ACE2 levels, which may lead to endothelial dysfunction (80).
Recent evidence suggests that ACE2 is a functional receptor for SARS-CoV-2 to enter host target cells (82, 83). The infection of SARS-CoV-2 begins with SARS-CoV-2 cleaving its S protein through transmembrane protease serine 2 (TMPRSS-2) and attaching to the ACE-2 receptor (84–86). This ongoing infection produces significant endotheliitis, a robust immune response and a subsequent increase in pro-inflammatory cytokines, vasoactive molecules, and immune cells like neutrophils, macrophages, monocytes, and lymphocytes which all play a role in propagating a response known as the cytokine storm (71, 84, 87, 88). In vitro study using engineered human blood vessel organoids showed that SARS-CoV-2 can directly infect endothelial cells via ACE2 (89). Varga et al. reported that the presence of viral inclusion structures was detected in endothelial cells in COVID-19 patients (9). Intriguingly, neutralization of ACE2 using soluble human ACE2 decreased virus-infected endothelial cells in vivo (89). These pieces of evidence highlight the role of ACE2 in mediating of SARS-CoV-2 induced endothelial cell injury. Indeed, the endothelium is a vulnerable target by SARS-CoV-2 infection, and these infected endothelial cells exhibit dramatic changes in morphology and function (71). Therefore, endothelial cell dysfunction could be an important and potential pathogenesis of COVID-19 infection induced multiple organ dysfunction. In the following sections, we step beyond our focus on the virus and discuss the role of aerobic glycolytic metabolism in COVID-19-driven endothelial cell injury in order to better understand the potential mechanisms that cause the endothelial cell damage in COVID-19 infected patients.
Switching Metabolism in COVID-19 Infection
Previous studies have shown that virus infection dramatically modified the cellular metabolism of host cells (90). It is hypothesized that the virus-driven metabolic process in a host cell is to provide macromolecules needed for virion replication and assembly (20, 90). Thomas et al. observed that the levels of glucose and free fatty acid in the serum of COVID-19 infected patients were significantly increased providing fuels for viral proliferation (91). Similarly, Shen et al. reported that the serum glucose levels were elevated in the severe patients infected with COVID-19, when compared with control groups (92). In addition, patients with pre-existing metabolic diseases, including diabetes, have greater risk of developing severe conditions (93). A study including 174 COVID-19 infected patients implies that diabetic patients without other comorbidities were at high risk of severe pneumonia, excessive inflammation responses and hypercoagulable state (94). To understand the potential mechanism by which uncontrolled diabetes is a risk factor for severe COVID-19, Codo et al. investigated the correlation between glycolysis and SARS-CoV-2 replication and found that glucose enhanced SARS-CoV-2 load in monocytes in a dose dependent manner (95). In agreement, a retrospective observational study, including 2433 COVID-19 patients admitted to the Houshen Shan hospital in Wuhan between February and April in 2020, indicates that elevated glucose level could be a predictive maker for the disease progression and the fatality of COVID-19 patients (96). Moreover, He et al. reported that COVID-19 infected patients without pre-existing diabetes also presented high blood glucose levels (97), indicating that SARS-CoV-2 infection may change metabolic profiles in these patients. Indeed, most viruses tested to date can induce aerobic glycolytic metabolism to favor their replication (20), which seems to be the same case for SARS-CoV-2 infection. Thus, when the virus enters a diabetic patient, especially a Type II diabetic patient, the high glucose levels within the host results in a disrupted glucose metabolism. This disruption favors SARS-CoV-2 replication and cytokine production while simultaneously dampening the proper effects of the immune system (T-cell response/function is worsened), prompting a more severe inflammatory response (cytokine storm) within this demographic (95).
Mitochondria are essential cellular organelles in regulating cellular energy, metabolism, and host innate immunity (98–100). Transcriptomic study by Mooamalla et al. shows that SARS-CoV-2 infection downregulated tricarboxylic acid cycle (TCA) and oxidative phosphorylation in several human respiratory cell lines, indicating mitochondrial dysfunction (101). Indeed, emerging evidence shows that SARS-CoV-2 highjacks mitochondria and replicates in mitochondria, leading to impaired mitochondrial dynamics and cell death (102). It is proposed that aerobic glycolytic metabolism is enhanced when mitochondrial defect occurs (103, 104). Mooamalla and colleagues also found that the expression of lactate dehydrogenase (LDHA), which is a dispensable enzyme for aerobic glycolysis, was increased and lactate production was elevated in SARS-CoV-2-infected human respiratory cell lines (101). Notably, similar observation is made in peripheral blood mononuclear cells (PBMCs) isolated from COVID-19 patients, in which the rate of glycolysis was increased, and the mitochondrial respiration was impaired (105).
It has been reported that SARS-CoV-2 affects both the upper and lower respiratory tract, which, in many cases, results in hypoxemia (106, 107). In addition to virus-driven metabolic changes, lack of oxygen may also be a determinant in regulating metabolism in patients with COVID-19 infection. Inadequate oxygen supply shifts oxidative phosphorylation to aerobic glycolysis, leading to increased production of lactate and extracellular acidification. It is demonstrated that lactate is a natural suppressor for antiviral signaling though inhibiting retinoic acid-inducible gene (RIG) (21). Collectively, COVID-19 infection could induce a metabolic switch from oxidative phosphorylation to aerobic glycolysis which does not only benefit to virus replication, but also priming innate immunity mediated pro-inflammatory cytokine production (95, 108). In addition, the intermediates of aerobic glycolytic metabolism could play an important role in the regulation of pro-inflammatory response and endothelial cell dysfunction (12, 14).
Aerobic Glycolytic Metabolism and Endothelial Cell Injury in COVID-19 Infection
As mentioned above, growing evidence shows that COVID-19 infection switches metabolisms from oxidative phosphorylation to aerobic glycolytic metabolism (17, 105), which allows the rapid production of energy and other substrates for viral replication (20). Lactate is the end product of aerobic glycolysis and serves as an important diagnostic biomarker for critical illnesses, such as sepsis/septic shock (24). It has been shown that severe COVID-19 patients developed typical symptoms that are similar to septic shock, such as vascular microthrombosis, multi-organ dysfunction syndrome (MODS), coagulopathy, high cytokine production (109). Considering the parallels in the pathophysiology of sepsis and COVID-19, it is proposed that viral sepsis is crucial to the pathogenetic mechanisms of COVID-19 (110). In this case, lactate generated from aerobic glycolytic metabolism may be also applied as a biomarker for diagnosis and prognosis of COVID-19 infected patients. Velavan et al. showed that hospitalized patients with moderate to severe COVID-19 (N = 18) had significantly higher blood lactate levels than mild ambulatory COVID-19 patients (N = 16) (33). In addition, a retrospective study including 45 ICU-admitted patients with COVID-19 showed that sequential organ failure assessment (SOFA) score and initial blood lactate levels were significantly higher in non-survivors (N = 11) compared to survivors (N = 34), indicating that blood lactate level mirrors organ dysfunction and is associated with poor clinical outcomes of COVID-19 ICU patients (32). In consistent with this observation, Metkus et al. reported that non-survivors (N = 88) had significantly elevated blood lactate levels than survivors (N = 155) of COVID-19 patients (3.6 mmol/L vs. 2.0 mmol/L, P = 0.005) (111). Moreover, blood lactate levels positively and independently correlate with troponin (troponin I or troponin T) levels in COVID-19 patients (N = 243, P = 0.007), suggesting that lactate may serve as predictor for myocardia injury in COVID-19 patients (111). These pieces of evidence suggest that elevated lactate levels could correlate with both severity and mortality of COVID-19. Of note, a pooled analysis, including 1,532 COVID-19 patients, reported that increased lactate dehydrogenase (LDH) levels were associated with a 6-fold increase in odds of severe COVID-19 and 16-fold increase in odds of COVID-19 mortality (112). Given that LDH is involved in lactate production, it is advisable that lactate consumption might be also increased.
Although lactate was considered as a waste in the past decades (23), growing evidence has shown that lactate may exert important regulatory roles in various pathophysiological processes, including immunosuppression (12, 113–115), cell signaling (13, 116) and gene transcription (117–119). Recent studies further reveal that lactate can directly induce permeability in inflammatory bone marrow endothelium by downregulation of VE-cadherin expression (14), indicating that lactate could contribute to the pathophysiologic mechanisms of cardiovascular injury in COVID-19 infection. As a result, the implication of serum lactate may be able to present us with an improved method of measuring clinical severity and observe clinical treatment response in the context of COVID-19. In the sections below, we discuss the possible mechanisms of lactate-mediated endothelial injuries in the pathogenesis of COVID-19 infection.
Lactate and SARS-CoV-2 Infection-Induced Endothelial Cell Injury
The integrity of endothelium is required for maintaining the vascular homeostasis (120). SARS-CoV-2 infects the host cells using the ACEs receptor (85), which is expressed by endothelial cells. Established evidence suggests that SARS-CoV-2 hijacks the endothelial cells and causes significant changes in endothelial cell morphology, i.e. disruption of intercellular junctions and cell swelling in COVID-19 infected patients (9, 71). Several lines of in vitro and in vivo evidence also demonstrate that endothelial cells are highly susceptible to SARS-CoV-2 infection (89, 121–123). It has been reported that SARS-CoV-2 proliferation in endothelial cells directly induces apoptosis in COVID-19 patients (9). In addition, circulating endothelial cells (CECs) have been considered as a marker for damaged endothelium in various vascular diseases (124–126). Importantly, COVID-19 infected patients have higher numbers of CECs than healthy controls, indicating the occurrence of endothelium damages in COVID-19 patients due to direct virus infection (127).
Infection of SARS-CoV-2 in the pulmonary tissues impairs gas exchange leading systemic hypoxia and enhanced glycolysis metabolism in endothelial cells and immune cells by stabilizing hypoxia-inducible factor-1 (HIF-1) (128). HIF-1 is a powerful inducer of glycolysis via upregulation of enzymes involved in glycolysis, including hexokinase (HK), pyruvate kinase 2 (PKM2), LDHA/LDHB and pyruvate dehydrogenase kinase (PDK) in COVID-19 (129–131) (Figure 1). In addition, SARS-CoV-2 infection triggers mitochondrial ROS production, leading HIF1 stabilization and consequently promotes glycolysis (95) (Figure 1). Notably, it is proposed that lactate can induce the activation of hypoxia-inducible factor-1 (HIF-1), which further enhances aerobic glycolysis and promotes SARS-CoV-2 infection and replication (95, 132, 133). Indeed, inhibition of lactate production by 2-DG or oxamate suppressed aerobic glycolysis efficiently and reduced viral load in human monocytes (95). Previous studies demonstrate that endothelial cells rely heavily on aerobic glycolysis for ATP production while having little glucose oxidation (134, 135). This may make endothelial cells more susceptible to SARS-CoV-2 infection. Therefore, it is possible that lactate generated from aerobic glycolysis could be beneficial to SARS-CoV-2 proliferation and mediation of endothelial cell injury.
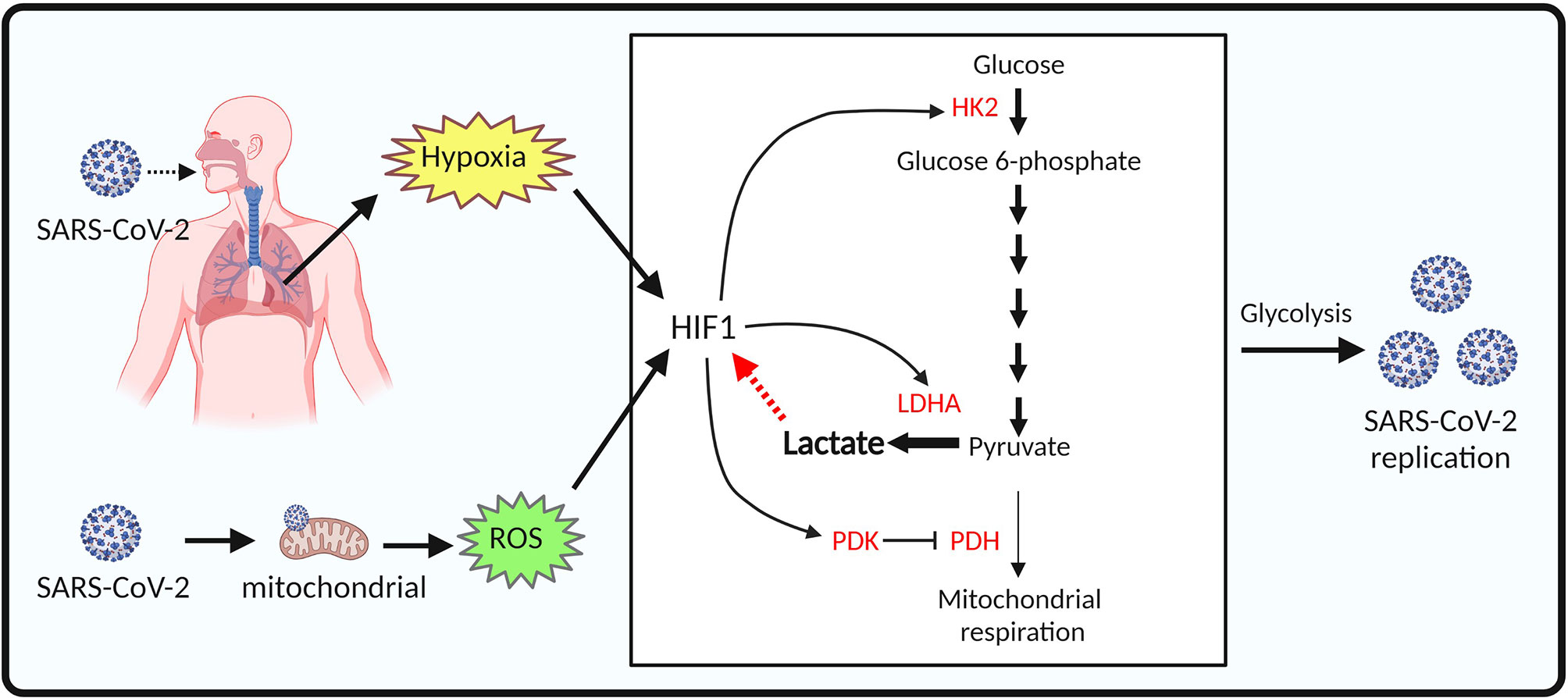
Figure 1 Proposed model of aerobic glycolysis activation in SARS-CoV-2 infected endothelial cells. SARS-CoV-2 infection of pulmonary tissues leads to hypoxia. SARS-CoV-2 infection also causes mitochondrial dysfunction and reactive oxygen species (ROS) production in endothelial cells. Both hypoxia and ROS mediate HIF-1 stabilization. Enzymes involved in glycolysis, including hexokinase (HK), pyruvate kinase 2 (PKM2), lactate dehydrogenase (LDH) are upregulated by HIF-1 signaling, resulting in increased lactate production and SARS-CoV-2 replication in endothelial cells.
Lactate and Endothelium Permeability in COVID-19 Infection
Endothelium hyperpermeability contributes to tissue fluid overload (edema) and the persistent hypotension in critically ill patients. Prolonged edema may lead to multiple organ failure and ultimately death (136). Clinical data suggests that COVID-19 infected patients with severe conditions exhibit lower values of serum albumin, indicating the presence of vascular permeability (137). Wu and colleagues provided histological evidence showing that ICU-admitted COVID-19 infected patients who were characterized with hypoalbuminemia had disrupted inter-endothelial junctional complex in the lung tissues (138). It is well known that disarrangement of junctional proteins in the plasma membrane of adjacent endothelial cells increases vascular permeability (139, 140). Vascular endothelial cadherin (VE-cadherin) is one of the determinants of endothelial cell contact integrity (141). A recent study by Flores-Pliego et al. showed that the expression of VE-cadherin, as well as Claudin 5, decreased in the endothelium of decidua and chorionic villi of placentas derived from women with severe COVID-19, when compared to healthy controls (142). Similarly, Feng and colleagues utilized a rhesus macaque model of SARS-CoV-2 respiratory tract infection and observed that SARS-CoV-2 infection significantly reduced VE-cadherin levels in the heart of rhesus macaques when compared to uninfected controls (75). In agreement with these in vivo observations, several in vitro studies demonstrated that SARS-CoV-2 spike proteins can also disorganize the VE-cadherin complex and decrease VE-cadherin levels in cultured endothelial cells (69, 72, 74). Therefore, SARS-CoV-2 infection can disrupt VE-cadherin largely responsible for the vascular permeability in COVID-19 patients.
Notably, a recent study by Khatib-Massalha et al. showed that lactate directly decreases VE-cadherin expression in endothelial cells, which contributes to the hyperpermeability of bone marrow (BM) endothelium (14). G protein-couple receptor 81 (GPR81) is a lactate receptor (143). Activation of GPR81 by its agonist (3,5-DHBA) has similar effects as lactate on reducing the expression of VE-cadherin in endothelial cells (14). In contrast, knockout of GPR81 attenuated lactate-induced BM vascular permeability, demonstrating that GPR81 is essential for lactate-induced vascular permeability (14). In addition, it is reported that SARS-CoV-2 infection activates pyroptotic signaling in lungs and promotes interleukin-1β (IL-1β) release, which results in downregulation of VE-cadherin on lung endothelial cells (144). IL-1β-induced downregulation of VE-cadherin contributes to lung vascular injury following SARS-CoV-2 infection (144). The underlying mechanism for IL-1β-induced downregulation of VE-cadherin in SARS-CoV-2-infected endothelial cells could be mediated by cAMP response element binding protein (CREB)-mediated suppression of VE-cadherin transcription (145). Therefore, it is conceivable that the action of lactate in promoting vascular permeability is mediated not only by favoring SARS-CoV-2 replication cells, but also by directly disrupting VE-cadherin and suppressing VE-cadherin transcription in endothelial cells upon GPR81 activation (Figure 2).
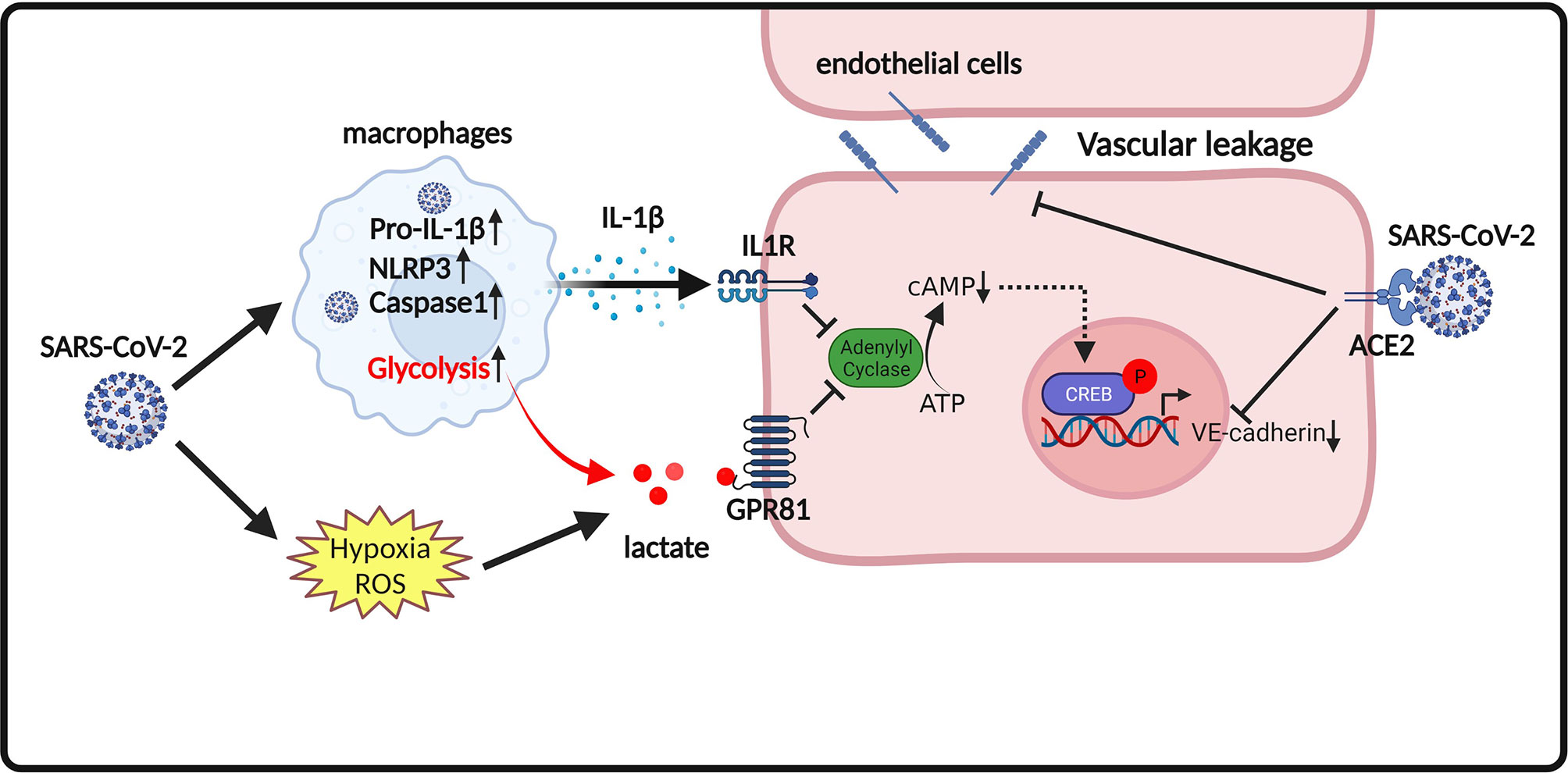
Figure 2 Proposed model of endothelium permeability induced by lactate/GPR81 signaling and SARS-CoV-2 infection. SARS-CoV-2 infection promotes the release of the pro-inflammatory cytokine IL-1β. IL-1β suppresses cAMP formation and CREB-mediated transcription of VE-cadherin in endothelial cells. SARS-CoV-2 infection also increases lactate production. Lactate activates GPR81 and reduces cAMP generation and CREB-mediated transcription of VE-cadherin in endothelial cells. In addition, SARS-CoV-2 spike proteins directly disorganize VE-cadherin complex and suppress VE-cadherin transcription in endothelial cells. Disruption of VE-cadherin complex is responsible for vascular permeability in COVID-19.
Lactate and Coagulation in COVID-19 Infection
COVID-19-induced multiple organ damage is associated with an abnormal coagulation (146). COVID-19 patient autopsies have revealed thrombi in the microvasculature (147). All-cause mortality in COVID-19 patients with thrombotic events is significantly higher than those without thrombotic events (148). Several hospital-based studies in Wuhan (China) reveal that some of the COVID-19 patients had elevated serum levels of pro-coagulation factors, including prothrombin (PT) and D-dimer, while the levels of fibrinogen and platelet are normal, representing the risk of thrombosis (59, 149, 150). Mechanistic studies reveal that SARS-Cov-2 spike protein directly binds platelet ACE2 and induces phosphorylation of ERK, p38 and JUK to activate platelets, which promotes thrombosis in COVID-19 (151). In addition, a recent study shows that SARS-Cov-2 virions can be internalized by platelets causing programmed cell death of platelets and extracellular vesicle release from platelets (152) (Figure 3). Moreover, inflammation and metabolism changes caused by SARS-Cov-2 infection are also considered as major contributors to coagulopathy in infected patients (153). However, with currently unknown mechanisms, clinical management of thrombosis with standard anti-coagulation dose of heparin failed to show satisfying outcomes (154–156). Two other plausible methods of managing coagulation include RAS inhibitors and statins. It is reported that the implementation of either substance has beneficial effects on COVID-19 clinical symptoms (157–159).
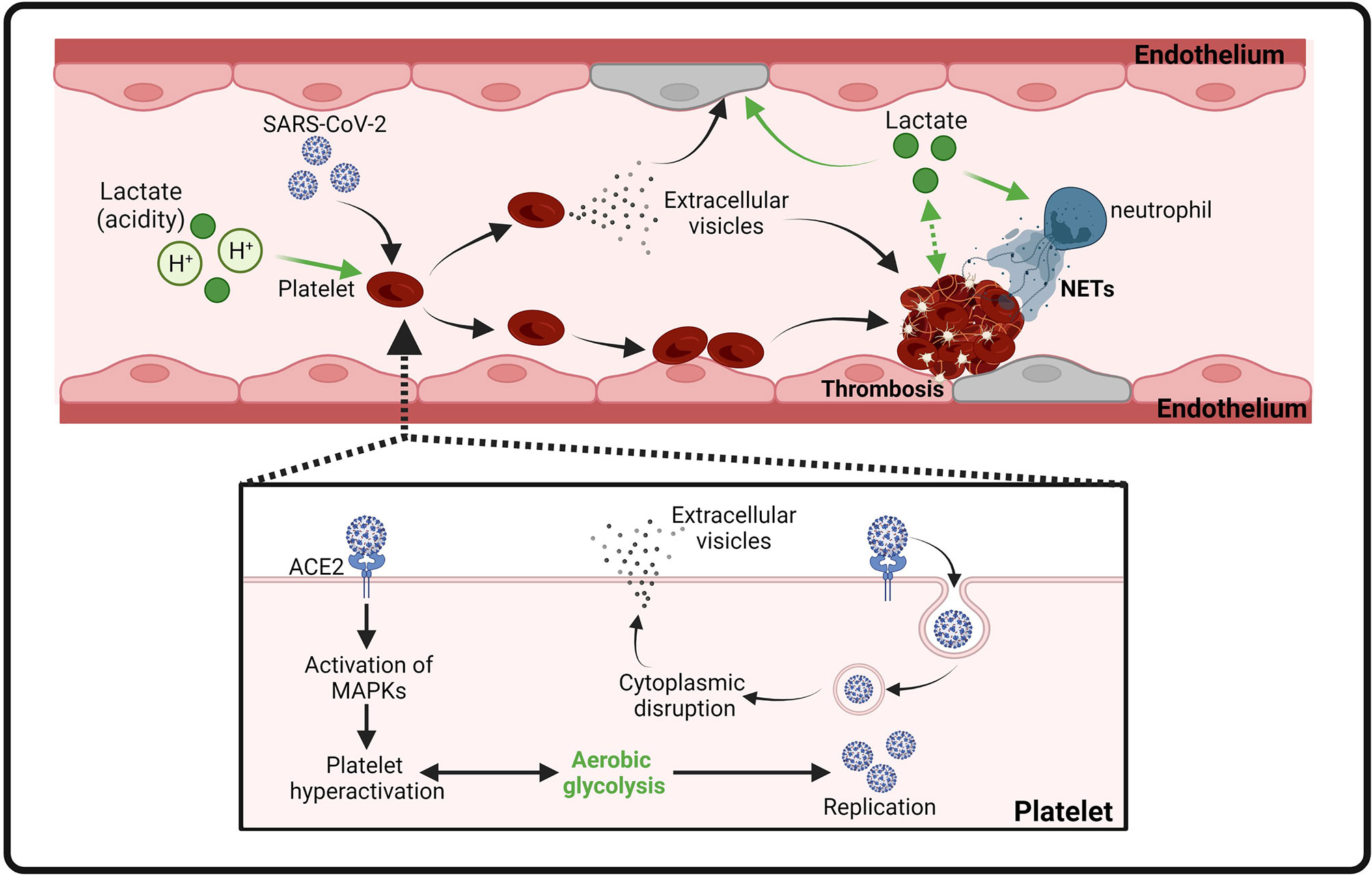
Figure 3 Proposed model of platelet activation, thrombosis and endothelial cell injury induced by SARS-CoV-2 infection and lactate. Binding of SARS-CoV-2 spike protein to ACE2 leads to MAPK signaling activation and subsequent platelet activation. Activated platelets release coagulation factors and cytokines to promote thrombosis. Internalization of SARS-CoV-2 virions induces the release of extracellular vesicles from platelets to facilitate thrombosis. In addition, lactate (acidity) also contributes to thrombosis by promoting activation of platelets, endothelial cells, and NETs.
Thachil et al. has recently discussed that hypoxia could be a mechanism of heparin resistance in COVID-19 patients (61). Indeed, oxygen deprivation has long been associated with thrombosis by triggering the pro-coagulation pathway (160). Both hypoxia and infection can result in enhanced aerobic glycolysis and consequent accumulation of lactate. It is noteworthy that metabolome analysis of venous thrombus from rabbits revealed that lactate is one of the most abundant metabolites in the thrombus (161, 162). Activated platelets, together with endothelial cells, are critical mediators of arterial thrombosis (163). Regardless of the nature of their stimulus, activated platelets switch their metabolism to aerobic glycolysis and produces a significant amount of lactic acid (164, 165). Increased extracellular lactate levels and acidity may further induce the continuous activation of Na+/H+ exchanger (NHE) in platelets and vascular endothelium, leading to the development of thrombosis (166, 167). In addition, elevated lactate levels in pulmonary embolism (PE) patients have been shown to correlate with impaired plasma fibrinolytic capacity and increase thrombin generation and neutrophil extracellular trap (NET) formation (168) (Figure 3). Importantly, pharmacological inhibition of aerobic glycolysis, which suppresses lactate production, efficiently reduced thrombosis in mice (165).
Lactate and HMGB1 in COVID-19 Infected Patients
High mobility group box 1 (HMGB1) is a chromatin-linked small protein that has nuclear, cytosolic and extracellular functions in various pathophysiological processes (169–174). Accumulating evidence shows that serum HMGB1 level is a potential biomarker for COVID-19 infected patients (175). A retrospective study, including 121 COVID-19 patients, shows that circulating HMGB1 and S100A8/A9 levels were significantly elevated in ICU-admitted COVID-19 patients (N = 40) compared to non-ICU COVID-19 patients (N = 81) (176). A similar observation was made by Chen et al. showing that severe COVID-19 patients (N = 11) had significantly higher levels of HMGB1 than non-severe COVID-19 patients (N = 29) (175). Gowda et al. reported that overexpression of SARS-Cov-2 spike protein in respiratory epithelial cells increased HMGB1 levels (52). In addition, SARS-Cov-2 spike protein caused cell death of epithelial cells, which may be responsible for subsequent release of HMGB1 (52) (Figure 4).
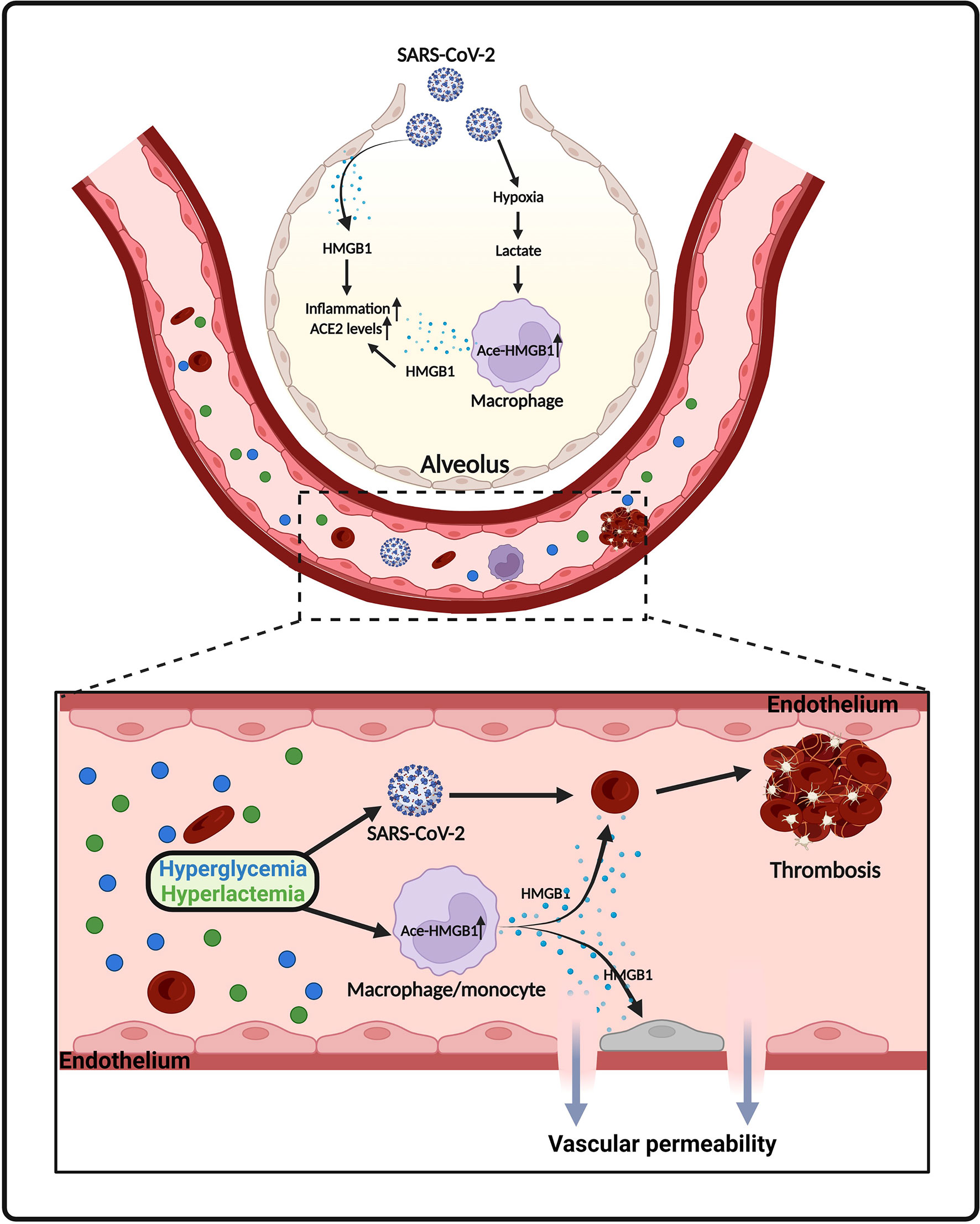
Figure 4 Proposed model of HMGB1 release in SARS-CoV-2 infection. SARS-CoV-2 infection causes death of epithelial cells and release of HMGB1. Lactate, derived from aerobic glycolysis, also promotes HMGB1 acetylation and release from macrophages/monocytes. Elevated levels of HMGB1 further promotes inflammatory responses, ACE2 expression, endothelium permeability and thrombosis in COVID-19.
We and others have shown that adaption to aerobic glycolysis in immune cells promotes the acetylation of HMGB1, leading to its extracellular release during infection (13, 177). HMGB1 acetylation is a concisely regulated process that involves various signaling pathways. Lu et al. shows that activation of JAK/STAT1 signaling is sufficient for LPS-induced HMGB1 hyperacetylation and cytoplasmic accumulation in macrophages (178). In addition, HMGB1 acetylation and release can be regulated by poly(ADP-ribose) polymerase-1 (PARP-1) in activated immune cells (179, 180). Moreover, previous studies indicate that HMGB1 acetylation is part of a general acetylation wave controlled by histone lysine acetylases and deacetylases (181–183). Interestingly, lactate is a potential inhibitor of histone lysine deacetylases (117). Indeed, our recent study demonstrated that lactate significantly increased nuclear translocation of histone lysine acetylases CBP and p300, while suppressed the expression of histone lysine deacetylase SIRT1 in macrophages (13). This regulatory role of lactate tilts the balance of acetylation/deacetylation of HMGB1 towards acetylation (13). Acetylated HMGB1 mainly localized in cytoplasm and subsequently released into the extracellular environment. In an in vitro endothelium barrier injury model, Zhou et al. observed that HMGB1 disrupted endothelium integrity and increased endothelium permeability (184). Consistently, we observed that lactate promoted HMGB1 secretion via exosome release and induced endothelium barrier dysfunction (13). In addition, it has been stated that hyperglycemia is common in hospitalized COVID-19 patients and is strongly associated with worse outcomes (185–188). COVID-19 patients with early-onset hyperglycemia, defined as blood glucose > 180 mg/dl during the first 2 days after ICU admission, had higher levels of lactate than patients without hyperglycemia (186). In diabetes hyperglycemia promotes the release of HMGB1 and upregulates receptor for advanced glycation end products (RAGE) (189) (Figure 4). Notably, numerous studies show that HMGB1 facilitates thrombosis via promoting platelet activation and NET formation (190–192) (Figure 4). HMGB1 can also induce the expression and activation of tissue factor (TF), which is involved in inflammation-related thrombosis, in endothelial cells in a concentration dependent manner (193). Moreover, in vitro treatment of alveolar epithelial cells with exogenous HMGB1 increased the expression of SARS-CoV-2 entry receptor ACE2 (175) (Figure 4).
Conclusions
SARS-CoV-2 infection causes metabolic reprogramming, such as increased glucose consumption and lactate production, which plays a role in the severity and mortality of COVID-19. Lactate is not only a valuable biomarker but also a critical signaling molecule in critical illness, including COVID-19. Thus, it is proposed that both reduced lactate production and inhibition of lactate-mediated signaling could improve COVID-19 (194). In this context, application of glycolysis inhibitors, such as 2-deoxy-D-glucose (2-DG), may have beneficial effects on COVID-19-infected patients. 2-DG is a glucose analogue which competitively inhibits the production of glucose-6-phosphate and consequently suppresses the glycolytic pathway (195). Our previous studies have demonstrated that at non-toxic dosages 2-DG markedly decreased lactate production and improved cardiac function in polymicrobial sepsis mice (196). Notably, the emergent use of 2-DG as an adjunct therapy in COVID-19 patients has been granted in India (197). In addition, animal studies show that lactate activates GPR81, a lactate specific receptor, to promote endothelial injury and immune cell dysfunction, which can be reversed by GPR81 inhibitors (12–14). Lactate can also be taken up by various cells through monocarboxylate transporter 1 (MCT1) (13, 198). We recently reported that blocking lactate influx by MCT1 inhibitor, as well as suppression of GPR81 signaling, decreased HMGB1 release from macrophages (13). Therefore, similar therapeutic strategies, either inhibition of lactate/GPR81 signaling or block lactate influx by MCT inhibitors, could also be used to abolish the detrimental effects of lactate in SARS-CoV-2 infection (194). On the other hand, priming the immune system with immunomodulatory components such as glucans may protect cardiovascular dysfunction in COVID-19. HMGB1 is a potential biomarker and may serve as a therapeutic target in severe COVID-19 (175). Our group previously reported that glucan phosphate improved cardiac function and suppressed HMGB1 translocation to the cytoplasm during sepsis (199, 200). This mode of action of glucan may counteract the effect of lactate in promoting HMGB1 release during SARS-CoV-2 infection (13). Importantly, a recent study has shown that glucans and mannans can be used as adjuvants to enhance the magnitude and durability of COVID-19 vaccines (201–203).
Since the outbreak of COVID-19, significant effort has been made to understand the pathogenesis of this new disease. With evidence collected from histological studies and biomedical tests, there has been increasing recognition that endothelial cell injury is one of the major contributors to the severity and mortality of COVID-19 infected patients. Recent evidence highlights the role of metabolism switching in the regulation of innate immune and inflammatory responses, which is observed in COVID-19 infected patients (204, 205). This review summarizes the potential role of aerobic glycolysis-derived lactate in the COVID-19 infection. In this mechanism of infection, lactate serves as an mediator that facilitates SARS-CoV-2 infection of endothelial cells, which leads to endothelial cell injury and multiple organ dysfunction. It is clear that growing evidence shows that lactate is involved in SARS-CoV-2-mediated endothelial cell death, vascular permeability, and coagulopathy. On the other hand, elevation of lactate levels, due to enhanced glycolysis, could also contribute to endothelial injury by altering immune cell function. Further basic science research is needed to validate whether targeting aerobic glycolytic metabolism could be beneficial for patients with COVID-19 infection.
Author Contributions
KY, MH, MF, VL, YY were involved in the literature search, and drafting and preparation of the manuscript. KY, CL and XW were involved in the idea generation. KY, TH and DW were involved in checking the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Institutes of Health grants HL071837 (CL), HL153270 (CL), GM083016 (CL, DW), GM119197 (DW), American Heart Association Postdoctoral Fellowship 916710 (MF), and C06RR0306551 (ETSU).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Gavriatopoulou M, Ntanasis-Stathopoulos I, Korompoki E, Fotiou D, Migkou M, Tzanninis IG, et al. Emerging Treatment Strategies for COVID-19 Infection. Clin Exp Med (2021) 21(2):167–79. doi: 10.1007/s10238-020-00671-y
2. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical Features of Patients Infected With 2019 Novel Coronavirus in Wuhan, China. Lancet (2020) 395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5
3. Terpos E, Ntanasis-Stathopoulos I, Elalamy I, Kastritis E, Sergentanis TN, Politou M, et al. Hematological Findings and Complications of COVID-19. Am J Hematol (2020) 95(7):834–47. doi: 10.1002/ajh.25829
4. Mohamed K, Rzymski P, Islam MS, Makuku R, Mushtaq A, Khan A, et al. COVID-19 Vaccinations: The Unknowns, Challenges, and Hopes. J Med Virol (2022) 94(4):1336–49. doi: 10.1002/jmv.27487
5. Dan S, Pant M, Upadhyay SK. The Case Fatality Rate in COVID-19 Patients With Cardiovascular Disease: Global Health Challenge and Paradigm in the Current Pandemic. Curr Pharmacol Rep (2020) p:1–10. doi: 10.1007/s40495-020-00239-0
6. Nishiga M, Wang DW, Han Y, Lewis DB, Wu JC. COVID-19 and Cardiovascular Disease: From Basic Mechanisms to Clinical Perspectives. Nat Rev Cardiol (2020). doi: 10.1038/s41569-020-0413-9
7. Bansal M. Cardiovascular Disease and COVID-19. Diabetes Metab Syndr (2020) 14(3):247–50. doi: 10.1016/j.dsx.2020.03.013
8. Mitrani RD, Dabas N, Goldberger JJ. COVID-19 Cardiac Injury: Implications for Long-Term Surveillance and Outcomes in Survivors. Heart Rhythm (2020) 17(11):1984–90. doi: 10.1016/j.hrthm.2020.06.026
9. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial Cell Infection and Endotheliitis in COVID-19. Lancet (2020) 395(10234):1417–8. doi: 10.1016/S0140-6736(20)30937-5
10. Gheblawi M, Wang K, Viveiros A, Nguyen Q, Zhong JC, Turner AJ, et al. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System: Celebrating the 20th Anniversary of the Discovery of ACE2. Circ Res (2020) 126(10):1456–74. doi: 10.1161/CIRCRESAHA.120.317015
11. Kumar A, Narayan RK, Kumari C, Faiq MA, Kulandhasamy M, Kant K, et al. SARS-CoV-2 Cell Entry Receptor ACE2 Mediated Endothelial Dysfunction Leads to Vascular Thrombosis in COVID-19 Patients. Med Hypotheses (2020) 145:110320. doi: 10.1016/j.mehy.2020.110320
12. Yang K, Xu J, Fan M, Tu F, Wang X, Ha T, et al. Lactate Suppresses Macrophage Pro-Inflammatory Response to LPS Stimulation by Inhibition of YAP and NF-kappaB Activation via GPR81-Mediated Signaling. Front Immunol (2020) 11:587913. doi: 10.3389/fimmu.2020.587913
13. Yang K, Fan M, Wang X, Xu J, Wang Y, Tu F, et al. Lactate Promotes Macrophage HMGB1 Lactylation, Acetylation, and Exosomal Release in Polymicrobial Sepsis. Cell Death Differ (2021) 29(1):133–46. doi: 10.1038/s41418-021-00841-9
14. Khatib-Massalha E, Bhattacharya S, Massalha H, Biram A, Golan K, Kollet O, et al. Lactate Released by Inflammatory Bone Marrow Neutrophils Induces Their Mobilization via Endothelial GPR81 Signaling. Nat Commun (2020) 11(1):3547. doi: 10.1038/s41467-020-17402-2
15. Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, et al. Itaconate Links Inhibition of Succinate Dehydrogenase With Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab (2016) 24(1):158–66. doi: 10.1016/j.cmet.2016.06.004
16. Sun L, Yang X, Yuan Z, Wang H. Metabolic Reprogramming in Immune Response and Tissue Inflammation. Arterioscler Thromb Vasc Biol (2020) 40(9):1990–2001. doi: 10.1161/ATVBAHA.120.314037
17. Icard P, Lincet H, Wu Z, Coquerel A, Forgez P, Alifano M, et al. The Key Role of Warburg Effect in SARS-CoV-2 Replication and Associated Inflammatory Response. Biochimie (2021) 180:169–77. doi: 10.1016/j.biochi.2020.11.010
18. Eelen G, de Zeeuw P, Simons M, Carmeliet P. Endothelial Cell Metabolism in Normal and Diseased Vasculature. Circ Res (2015) 116(7):1231–44. doi: 10.1161/CIRCRESAHA.116.302855
19. Rouse BT, Sehrawat S. Immunity and Immunopathology to Viruses: What Decides the Outcome? Nat Rev Immunol (2010) 10(7):514–26. doi: 10.1038/nri2802
20. Sanchez EL, Lagunoff M. Viral Activation of Cellular Metabolism. Virology (2015) 479-480:609–18. doi: 10.1016/j.virol.2015.02.038
21. Zhang W, Wang G, Xu ZG, Tu H, Hu F, Dai J, et al. Lactate Is a Natural Suppressor of RLR Signaling by Targeting MAVS. Cell (2019) 178:176–189.e15. doi: 10.1016/j.cell.2019.05.003
22. Jones W, Bianchi K. Aerobic Glycolysis: Beyond Proliferation. Front Immunol (2015) 6:227. doi: 10.3389/fimmu.2015.00227
23. Rabinowitz JD, Enerback S. Lactate: The Ugly Duckling of Energy Metabolism. Nat Metab (2020) 2(7):566–71. doi: 10.1038/s42255-020-0243-4
24. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). Jama-Journal Am Med Assoc (2016) 315(8):801–10. doi: 10.1001/jama.2016.0287
25. Lazzeri C, Valente S, Chiostri M, Gensini GF. Clinical Significance of Lactate in Acute Cardiac Patients. World J Cardiol (2015) 7(8):483–9. doi: 10.4330/wjc.v7.i8.483
26. Zymlinski R, Biegus J, Sokolski M, Siwolowski P, Nawrocka-Millward S, Todd J, et al. Increased Blood Lactate is Prevalent and Identifies Poor Prognosis in Patients With Acute Heart Failure Without Overt Peripheral Hypoperfusion. Eur J Heart Fail (2018) 20(6):1011–8. doi: 10.1002/ejhf.1156
27. Baltazar F, Afonso J, Costa M, Granja S. Lactate Beyond a Waste Metabolite: Metabolic Affairs and Signaling in Malignancy. Front Oncol (2020) 10:231. doi: 10.3389/fonc.2020.00231
28. Chen XY, Huang MY, Xiao ZW, Yang S, Chen XQ. Lactate Dehydrogenase Elevations is Associated With Severity of COVID-19: A Meta-Analysis. Crit Care (2020) 24(1):459. doi: 10.1186/s13054-020-03161-5
29. Li X, Xu S, Yu M, Wang K, Tao Y, Zhou Y, et al. Risk Factors for Severity and Mortality in Adult COVID-19 Inpatients in Wuhan. J Allergy Clin Immunol (2020) 146(1):110–8. doi: 10.1016/j.jaci.2020.04.006
30. Wu M-Y, Yao L, Wang Y, Zhu X-Y, Wang X-F, Tang P-J, et al. Clinical Evaluation of Potential Usefulness of Serum Lactate Dehydrogenase (LDH) in 2019 Novel Coronavirus (COVID-19) Pneumonia. Respir Res (2020) 21:171. doi: 10.1186/s12931-020-01427-8
31. Han Y, Zhang H, Mu S, Wei W, Jin C, Tong C, et al. Lactate Dehydrogenase, an Independent Risk Factor of Severe COVID-19 Patients: A Retrospective and Observational Study. Aging (Albany NY) (2020) 12(12):11245–58. doi: 10.18632/aging.103372
32. Vassiliou AG, Jahaj E, Ilias I, Markaki V, Malachias S, Vrettou C, et al. Lactate Kinetics Reflect Organ Dysfunction and Are Associated With Adverse Outcomes in Intensive Care Unit Patients With COVID-19 Pneumonia: Preliminary Results From a GREEK Single-Centre Study. Metabolites (2020) 10(10):386. doi: 10.3390/metabo10100386
33. Velavan TP, Kieu Linh LT, Kreidenweiss A, Gabor J, Krishna S, Kremsner PG. Longitudinal Monitoring of Lactate in Hospitalized and Ambulatory COVID-19 Patients. Am J Trop Med Hyg (2021) 104(3):1041–4. doi: 10.4269/ajtmh.20-1282
34. Nardi G, Sanson G, Tassinari L, Guiotto G, Potalivo A, Montomoli J, et al. Lactate Arterial-Central Venous Gradient Among COVID-19 Patients in ICU: A Potential Tool in the Clinical Practice. Crit Care Res Pract (2020) 2020:4743904. doi: 10.1101/2020.05.08.20095042
35. Iepsen UW, Plovsing RR, Tjelle K, Foss NB, Meyhoff CS, Ryrso CK, et al. The Role of Lactate in Sepsis and COVID-19: Perspective From Contracting Skeletal Muscle Metabolism. Exp Physiol (2021). doi: 10.1113/EP089474
36. Ganeshan K, Chawla A. Metabolic Regulation of Immune Responses. Annu Rev Immunol (2014) 32:609–34. doi: 10.1146/annurev-immunol-032713-120236
37. Tran DH, Wang ZV. Glucose Metabolism in Cardiac Hypertrophy and Heart Failure. J Am Heart Assoc (2019) 8(12):e012673. doi: 10.1161/JAHA.119.012673
38. Madjid M, Safavi-Naeini P, Solomon SD, Vardeny O. Potential Effects of Coronaviruses on the Cardiovascular System: A Review. JAMA Cardiol (2020) 5(7):831–40. doi: 10.1001/jamacardio.2020.1286
39. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A Pneumonia Outbreak Associated With a New Coronavirus of Probable Bat Origin. Nature (2020) 579(7798):270–3. doi: 10.1038/s41586-020-2012-7
40. Xu J, Zhao S, Teng T, Abdalla AE, Zhu W, Xie L, et al. Systematic Comparison of Two Animal-To-Human Transmitted Human Coronaviruses: SARS-CoV-2 and SARS-CoV. Viruses (2020) 12(2):244. doi: 10.3390/v12020244
41. Maldonado LL, Bertelli AM, Kamenetzky L. Molecular Features Similarities Between SARS-CoV-2, SARS, MERS and Key Human Genes Could Favour the Viral Infections and Trigger Collateral Effects. Sci Rep (2021) 11(1):4108. doi: 10.1038/s41598-021-83595-1
42. Wu Q, Zhou L, Sun X, Yan Z, Hu C, Wu J, et al. Altered Lipid Metabolism in Recovered SARS Patients Twelve Years After Infection. Sci Rep (2017) 7(1):9110. doi: 10.1038/s41598-017-09536-z
43. Dorjee K, Kim H, Bonomo E, Dolma R. Prevalence and Predictors of Death and Severe Disease in Patients Hospitalized Due to COVID-19: A Comprehensive Systematic Review and Meta-Analysis of 77 Studies and 38,000 Patients. PloS One (2020) 15(12):e0243191. doi: 10.1371/journal.pone.0243191
44. Clerkin KJ, Fried JA, Raikhelkar J, Sayer G, Griffin JM, Masoumi A, et al. COVID-19 and Cardiovascular Disease. Circulation (2020) 141(20):1648–55. doi: 10.1161/CIRCULATIONAHA.120.046941
45. Shi S, Qin M, Shen B, Cai Y, Liu T, Yang F, et al. Association of Cardiac Injury With Mortality in Hospitalized Patients With COVID-19 in Wuhan, China. JAMA Cardiol (2020) 5(7):802–10. doi: 10.1001/jamacardio.2020.0950
46. Grasselli G, Zangrillo A, Zanella A, Antonelli M, Cabrini L, Castelli A, et al. Baseline Characteristics and Outcomes of 1591 Patients Infected With SARS-CoV-2 Admitted to ICUs of the Lombardy Region, Italy. JAMA (2020) 323(16):1574–81. doi: 10.1001/jama.2020.4031
47. Goyal P, Choi JJ, Pinheiro LC, Schenck EJ, Chen R, Jabri A, et al. Clinical Characteristics of Covid-19 in New York City. N Engl J Med (2020) 382(24):2372–4. doi: 10.1056/NEJMc2010419
48. Hessami A, Shamshirian A, Heydari K, Pourali F, Alizadeh-Navaei R, Moosazadeh M, et al. Cardiovascular Diseases Burden in COVID-19: Systematic Review and Meta-Analysis. Am J Emerg Med (2021) 46:382–91. doi: 10.1016/j.ajem.2020.10.022
49. Li B, Yang J, Zhao F, Zhi L, Wang X, Liu L, et al. Prevalence and Impact of Cardiovascular Metabolic Diseases on COVID-19 in China. Clin Res Cardiol (2020) 109(5):531–8. doi: 10.1007/s00392-020-01626-9
50. Zheng YY, Ma YT, Zhang JY, Xie X. COVID-19 and the Cardiovascular System. Nat Rev Cardiol (2020) 17(5):259–60. doi: 10.1038/s41569-020-0360-5
51. Saed Aldien A, Ganesan GS, Wahbeh F, Al-Nassr N, Altarawneh H, Al Theyab L, et al. Systemic Inflammation may Induce Cardiac Injury in COVID-19 Patients Including Children and Adolescents Without Underlying Cardiovascular Diseases: A Systematic Review. Cardiovasc Revasc Med (2021) 35:169–78. doi: 10.1016/j.carrev.2021.04.007
52. Gowda P, Patrick S, Joshi SD, Kumawat RK, Sen E. Glycyrrhizin Prevents SARS-CoV-2 S1 and Orf3a Induced High Mobility Group Box 1 (HMGB1) Release and Inhibits Viral Replication. Cytokine (2021) 142:155496. doi: 10.1016/j.cyto.2021.155496
53. Sharma A, Garcia G Jr, Wang Y, Plummer JT, Morizono K, Arumugaswami V, et al. Human iPSC-Derived Cardiomyocytes Are Susceptible to SARS-CoV-2 Infection. Cell Rep Med (2020) 1(4):100052. doi: 10.1016/j.xcrm.2020.100052
54. Majure DT, Gruberg L, Saba SG, Kvasnovsky C, Hirsch JS, Jauhar R, et al. Usefulness of Elevated Troponin to Predict Death in Patients With COVID-19 and Myocardial Injury. Am J Cardiol (2021) 138:100–6. doi: 10.1016/j.amjcard.2020.09.060
55. Manocha KK, Kirzner J, Ying X, Yeo I, Peltzer B, Ang B, et al. Troponin and Other Biomarker Levels and Outcomes Among Patients Hospitalized With COVID-19: Derivation and Validation of the HA2T2 COVID-19 Mortality Risk Score. J Am Heart Assoc (2021) 10(6):e018477. doi: 10.1161/JAHA.120.018477
56. Lombardi CM, Carubelli V, Iorio A, Inciardi RM, Bellasi A, Canale C, et al. Association of Troponin Levels With Mortality in Italian Patients Hospitalized With Coronavirus Disease 2019: Results of a Multicenter Study. JAMA Cardiol (2020) 5(11):1274–80. doi: 10.1001/jamacardio.2020.3538
57. Lindner D, Fitzek A, Brauninger H, Aleshcheva G, Edler C, Meissner K, et al. Association of Cardiac Infection With SARS-CoV-2 in Confirmed COVID-19 Autopsy Cases. JAMA Cardiol (2020) 5(11):1281–5. doi: 10.1001/jamacardio.2020.3551
58. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA (2020) 323(11):1061–9. doi: 10.1001/jama.2020.1585
59. Tang N, Li D, Wang X, Sun Z. Abnormal Coagulation Parameters are Associated With Poor Prognosis in Patients With Novel Coronavirus Pneumonia. J Thromb Haemost (2020) 18(4):844–7. doi: 10.1111/jth.14768
60. Klok FA, Kruip M, van der Meer NJM, Arbous MS, Gommers D, Kant KM, et al. Incidence of Thrombotic Complications in Critically Ill ICU Patients With COVID-19. Thromb Res (2020) 191:145–7. doi: 10.1016/j.thromres.2020.04.013
61. Thachil J. Hypoxia - an Overlooked Trigger for Thrombosis in COVID-19 and Other Critically Ill Patients. J Thromb Haemost (2020) 18(11):3109–10. doi: 10.1111/jth.15029
62. Sriram K, Insel PA. Inflammation and Thrombosis in COVID-19 Pathophysiology: Proteinase-Activated and Purinergic Receptors as Drivers and Candidate Therapeutic Targets. Physiol Rev (2021) 101(2):545–67. doi: 10.1152/physrev.00035.2020
63. Tang N, Bai H, Chen X, Gong J, Li D, Sun Z, et al. Anticoagulant Treatment is Associated With Decreased Mortality in Severe Coronavirus Disease 2019 Patients With Coagulopathy. J Thromb Haemost (2020) 18(5):1094–9. doi: 10.1111/jth.14817
64. Ran J, Song Y, Zhuang Z, Han L, Zhao S, Cao P, et al. Blood Pressure Control and Adverse Outcomes of COVID-19 Infection in Patients With Concomitant Hypertension in Wuhan, China. Hypertens Res (2020) 43(11):1267–76. doi: 10.1038/s41440-020-00541-w
65. Pober JS, Sessa WC. Evolving Functions of Endothelial Cells in Inflammation. Nat Rev Immunol (2007) 7(10):803–15. doi: 10.1038/nri2171
66. Sun HJ, Wu ZY, Nie XW, Bian JS. Role of Endothelial Dysfunction in Cardiovascular Diseases: The Link Between Inflammation and Hydrogen Sulfide. Front Pharmacol (2019) 10:1568. doi: 10.3389/fphar.2019.01568
67. Vallet B. Bench-To-Bedside Review: Endothelial Cell Dysfunction in Severe Sepsis: A Role in Organ Dysfunction? Crit Care (2003) 7(2):130–8. doi: 10.1186/cc1864
68. Raghavan S, Kenchappa DB, Leo MD. SARS-CoV-2 Spike Protein Induces Degradation of Junctional Proteins That Maintain Endothelial Barrier Integrity. Front Cardiovasc Med (2021) 8:687783. doi: 10.3389/fcvm.2021.687783
69. Nader D, Fletcher N, Curley GF, Kerrigan SW. SARS-CoV-2 Uses Major Endothelial Integrin Alphavbeta3 to Cause Vascular Dysregulation in-Vitro During COVID-19. PloS One (2021) 16(6):e0253347. doi: 10.1371/journal.pone.0253347
70. Huertas A, Montani D, Savale L, Pichon J, Tu L, Parent F, et al. Endothelial Cell Dysfunction: A Major Player in SARS-CoV-2 Infection (COVID-19)? Eur Respir J (2020) 56(1):2001634. doi: 10.1183/13993003.01634-2020
71. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N Engl J Med (2020) 383(2):120–8. doi: 10.1056/NEJMoa2015432
72. Robles JP, Zamora M, de la Escalera GM, Clapp C. The Spike Protein of SARS-CoV-2 Induces Endothelial Inflammation Through Integrin α5β1 and NF-κB. bioRxiv (2021) 298(3):101695. doi: 10.1101/2021.08.01.454605
73. Klein S, de Fougerolles AR, Blaikie P, Khan L, Pepe A, Green CD, et al. Alpha 5 Beta 1 Integrin Activates an NF-Kappa B-Dependent Program of Gene Expression Important for Angiogenesis and Inflammation. Mol Cell Biol (2002) 22(16):5912–22. doi: 10.1128/MCB.22.16.5912-5922.2002
74. Rauti R, Shahoha M, Leichtmann-Bardoogo Y, Nasser R, Tamir R, Miller V, et al. Effect of SARS-CoV-2 Proteins on Vascular Permeability. bioRxiv (2021) 10:e69314. doi: 10.7554/eLife.69314
75. Feng Y, Song X, Huang Y, Deng W, Li M, Guo X, et al. SARS-CoV-2 Leads to Myocardial Injury in Rhesus Macaque. Signal Transduct Target Ther (2021) 6(1):338. doi: 10.1038/s41392-021-00747-5
76. Hikmet F, Mear L, Edvinsson A, Micke P, Uhlen M, Lindskog C. The Protein Expression Profile of ACE2 in Human Tissues. Mol Syst Biol (2020) 16(7):e9610. doi: 10.15252/msb.20209610
77. Kuba K, Imai Y, Ohto-Nakanishi T, Penninger JM. Trilogy of ACE2: A Peptidase in the Renin-Angiotensin System, a SARS Receptor, and a Partner for Amino Acid Transporters. Pharmacol Ther (2010) 128(1):119–28. doi: 10.1016/j.pharmthera.2010.06.003
78. Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A Human Homolog of Angiotensin-Converting Enzyme. Cloning and Functional Expression as a Captopril-Insensitive Carboxypeptidase. J Biol Chem (2000) 275(43):33238–43. doi: 10.1074/jbc.M002615200
79. Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, et al. Angiotensin-Converting Enzyme 2 is a Functional Receptor for the SARS Coronavirus. Nature (2003) 426(6965):450–4. doi: 10.1038/nature02145
80. Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A Crucial Role of Angiotensin Converting Enzyme 2 (ACE2) in SARS Coronavirus-Induced Lung Injury. Nat Med (2005) 11(8):875–9. doi: 10.1038/nm1267
81. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue Distribution of ACE2 Protein, the Functional Receptor for SARS Coronavirus. A First Step in Understanding SARS Pathogenesis. J Pathol (2004) 203(2):631–7. doi: 10.1002/path.1570
82. Zamorano Cuervo N, Grandvaux N. ACE2: Evidence of Role as Entry Receptor for SARS-CoV-2 and Implications in Comorbidities. Elife (2020) 9:e61390. doi: 10.7554/eLife.61390
83. Shang J, Wan Y, Luo C, Ye G, Geng Q, Auerbach A, et al. Cell Entry Mechanisms of SARS-CoV-2. Proc Natl Acad Sci U S A (2020) 117(21):11727–34. doi: 10.1073/pnas.2003138117
84. Pons S, et al. The Vascular Endothelium: The Cornerstone of Organ Dysfunction in Severe SARS-CoV-2 Infection. Critical Care (2020) 24(1):353. doi: 10.1186/s13054-020-03062-7
85. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell (2020) 181:271–280.e8. doi: 10.1016/j.cell.2020.02.052
86. Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, et al. A Novel Angiotensin-Converting Enzyme-Related Carboxypeptidase (ACE2) Converts Angiotensin I to Angiotensin 1-9. Circ Res (2000) 87(5):E1–9. doi: 10.1161/01.RES.87.5.e1
87. Teuwen LA, Geldhof V, Pasut A, Carmeliet P. Covid-19: The Vasculature Unleashed. Nat Rev Immunol (2020) 20(7):389–91. doi: 10.1038/s41577-020-0343-0
88. Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, et al. Clinical and Immunological Features of Severe and Moderate Coronavirus Disease 2019. J Clin Invest (2020) 130:2620–9. doi: 10.1172/JCI137244
89. Monteil V, Kwon H, Prado P, Hagelkruys A, Wimmer RA, Stahl M, et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human Ace2. Cell (2020) 181(4):905–13.e7. doi: 10.1016/j.cell.2020.04.004
90. Thaker SK, Ch'ng J, Christofk HR. Viral Hijacking of Cellular Metabolism. BMC Biol (2019) 17(1):59. doi: 10.1186/s12915-019-0678-9
91. Thomas T, Stefanoni D, Reisz JA, Nemkov T, Bertolone L, Francis RO, et al. COVID-19 Infection Alters Kynurenine and Fatty Acid Metabolism, Correlating With IL-6 Levels and Renal Status. JCI Insight (2020) 5(14):e140327. doi: 10.1172/jci.insight.140327
92. Shen B, Yi X, Sun Y, Bi X, Du J, Zhang C, et al. Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. Cell (2020) 182(1):59–72.e15. doi: 10.1016/j.cell.2020.05.032
93. Costa FF, Rosario WR, Ribeiro Farias AC, de Souza RG, Duarte Gondim RS, Barroso WA. Metabolic Syndrome and COVID-19: An Update on the Associated Comorbidities and Proposed Therapies. Diabetes Metab Syndr (2020) 14(5):809–14. doi: 10.1016/j.dsx.2020.06.016
94. Guo W, Li M, Dong Y, Zhou H, Zhang Z, Tian C, et al. Diabetes is a Risk Factor for the Progression and Prognosis of COVID-19. Diabetes Metab Res Rev (2020) e3319. doi: 10.1002/dmrr.3319
95. Codo AC, Davanzo GG, Monteiro LB, de Souza GF, Muraro SP, Virgilio-da-Silva JV, et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and Monocyte Response Through a HIF-1alpha/Glycolysis-Dependent Axis. Cell Metab (2020) 32(3):498–9. doi: 10.2139/ssrn.3606770
96. Wang W, Shen M, Tao Y, Fairley CK, Zhong Q, Li Z, et al. Elevated Glucose Level Leads to Rapid COVID-19 Progression and High Fatality. BMC Pulm Med (2021) 21(1):64. doi: 10.1186/s12890-021-01413-w
97. He B, Wang J, Wang Y, Zhao J, Huang J, Tian Y, et al. The Metabolic Changes and Immune Profiles in Patients With COVID-19. Front Immunol (2020) 11:2075. doi: 10.3389/fimmu.2020.02075
98. Yu SB, Pekkurnaz G. Mechanisms Orchestrating Mitochondrial Dynamics for Energy Homeostasis. J Mol Biol (2018) 430(21):3922–41. doi: 10.1016/j.jmb.2018.07.027
99. Tzameli I. The Evolving Role of Mitochondria in Metabolism. Trends Endocrinol Metab (2012) 23(9):417–9. doi: 10.1016/j.tem.2012.07.008
100. Burtscher J, Cappellano G, Omori A, Koshiba T, Millet GP. Mitochondria: In the Cross Fire of SARS-CoV-2 and Immunity. iScience (2020) 23(10):101631. doi: 10.1016/j.isci.2020.101631
101. Moolamalla STR, Balasubramanian R, Chauhan R, Priyakumar UD, Vinod PK. Host Metabolic Reprogramming in Response to SARS-CoV-2 Infection: A Systems Biology Approach. Microb Pathog (2021) 158:105114. doi: 10.1016/j.micpath.2021.105114
102. Ganji R, Reddy PH. Impact of COVID-19 on Mitochondrial-Based Immunity in Aging and Age-Related Diseases. Front Aging Neurosci (2020) 12:614650. doi: 10.3389/fnagi.2020.614650
103. Senyilmaz D, Teleman AA. Chicken or the Egg: Warburg Effect and Mitochondrial Dysfunction. F1000Prime Rep (2015) 7:41. doi: 10.12703/P7-41
104. Vaupel P, Multhoff G. Revisiting the Warburg Effect: Historical Dogma Versus Current Understanding. J Physiol (2021) 599(6):1745–57. doi: 10.1113/JP278810
105. Ajaz S, McPhail MJ, Singh KK, Mujib S, Trovato FM, Napoli S, et al. Mitochondrial Metabolic Manipulation by SARS-CoV-2 in Peripheral Blood Mononuclear Cells of Patients With COVID-19. Am J Physiol Cell Physiol (2021) 320(1):C57–65. doi: 10.1152/ajpcell.00426.2020
106. Jahani M, Dokaneheifard S, Mansouri K. Hypoxia: A Key Feature of COVID-19 Launching Activation of HIF-1 and Cytokine Storm. J Inflamm (Lond) (2020) 17:33. doi: 10.1186/s12950-020-00263-3
107. Brosnahan SB, Jonkman AH, Kugler MC, Munger JS, Kaufman DA. COVID-19 and Respiratory System Disorders: Current Knowledge, Future Clinical and Translational Research Questions. Arterioscler Thromb Vasc Biol (2020) 40(11):2586–97. doi: 10.1161/ATVBAHA.120.314515
108. Soto-Heredero G, Gomez de Las Heras MM, Gabande-Rodriguez E, Oller J, Mittelbrunn M. Glycolysis - a Key Player in the Inflammatory Response. FEBS J (2020) 287(16):3350–69. doi: 10.1111/febs.15327
109. Olwal CO, Nganyewo NN, Tapela K, Djomkam Zune AL, Owoicho O, Bediako Y, et al. Parallels in Sepsis and COVID-19 Conditions: Implications for Managing Severe COVID-19. Front Immunol (2021) 12:602848. doi: 10.3389/fimmu.2021.602848
110. Li H, Liu L, Zhang D, Xu J, Dai H, Tang N. Sars-Cov-2 and Viral Sepsis: Observations and Hypotheses. Lancet (2020) 395(10235):1517–20. doi: 10.1016/S0140-6736(20)30920-X
111. Metkus TS, Sokoll LJ, Barth AS, Czarny MJ, Hays AG, Lowenstein CJ, et al. Myocardial Injury in Severe COVID-19 Compared With Non-COVID-19 Acute Respiratory Distress Syndrome. Circulation (2021) 143(6):553–65. doi: 10.1161/CIRCULATIONAHA.120.050543
112. Henry BM, Aggarwal G, Wong J, Benoit S, Vikse J, Plebani M, et al. Lactate Dehydrogenase Levels Predict Coronavirus Disease 2019 (COVID-19) Severity and Mortality: A Pooled Analysis. Am J Emerg Med (2020) 38(9):1722–6. doi: 10.1016/j.ajem.2020.05.073
113. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional Polarization of Tumour-Associated Macrophages by Tumour-Derived Lactic Acid. Nature (2014) 513(7519):559–63. doi: 10.1038/nature13490
114. Nolt B, Tu F, Wang X, Ha T, Winter R, Williams DL, et al. Lactate and Immunosuppression in Sepsis. Shock (2018) 49(2):120–5. doi: 10.1097/SHK.0000000000000958
115. Comito G, Iscaro A, Bacci M, Morandi A, Ippolito L, Parri M, et al. Lactate Modulates CD4(+) T-Cell Polarization and Induces an Immunosuppressive Environment, Which Sustains Prostate Carcinoma Progression via TLR8/miR21 Axis. Oncogene (2019) 38(19):3681–95. doi: 10.1038/s41388-019-0688-7
116. Cerda-Kohler H, Henriquez-Olguin C, Casas M, Jensen TE, Llanos P, Jaimovich E, et al. Lactate Administration Activates the ERK1/2, Mtorc1, and AMPK Pathways Differentially According to Skeletal Muscle Type in Mouse. Physiol Rep (2018) 6(18):e13800. doi: 10.14814/phy2.13800
117. Latham T, Mackay L, Sproul D, Karim M, Culley J, Harrison DJ, et al. Lactate, a Product of Glycolytic Metabolism, Inhibits Histone Deacetylase Activity and Promotes Changes in Gene Expression. Nucleic Acids Res (2012) 40(11):4794–803. doi: 10.1093/nar/gks066
118. Zhao Y, Zhao B, Wang X, Guan G, Xin Y, Sun YD, et al. Macrophage Transcriptome Modification Induced by Hypoxia and Lactate. Exp Ther Med (2019) 18(6):4811–9. doi: 10.3892/etm.2019.8164
119. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic Regulation of Gene Expression by Histone Lactylation. Nature (2019) 574(7779):575–80. doi: 10.1038/s41586-019-1678-1
120. Sandoo A, van Zanten JJ, Metsios GS, Carroll D, Kitas GD, et al. The Endothelium and its Role in Regulating Vascular Tone. Open Cardiovasc Med J (2010) 4:302–12. doi: 10.2174/1874192401004010302
121. Liu F, Han K, Blair R, Kenst K, Qin Z, Upcin B, et al. SARS-CoV-2 Infects Endothelial Cells In Vivo and In Vitro. Front Cell Infect Microbiol (2021) 11:701278. doi: 10.3389/fcimb.2021.701278
122. Bernard I, Limonta D, Mahal LK, Hobman TC, et al. Endothelium Infection and Dysregulation by SARS-CoV-2: Evidence and Caveats in COVID-19. Viruses (2020) 13(1):29. doi: 10.3390/v13010029
123. Fox SE, Li G, Akmatbekov A, Harbert JL, Lameira FS, Brown JQ, et al. Unexpected Features of Cardiac Pathology in COVID-19 Infection. Circulation (2020) 142(11):1123–5. doi: 10.1161/CIRCULATIONAHA.120.049465
124. Bertolini F, Shaked Y, Mancuso P, Kerbel RS. The Multifaceted Circulating Endothelial Cell in Cancer: Towards Marker and Target Identification. Nat Rev Cancer (2006) 6(11):835–45. doi: 10.1038/nrc1971
125. Farinacci M, Krahn T, Dinh W, Volk HD, Dungen HD, Wagner J, et al. Circulating Endothelial Cells as Biomarker for Cardiovascular Diseases. Res Pract Thromb Haemost (2019) 3(1):49–58. doi: 10.1002/rth2.12158
126. Yoo JW, Moon JY, Hong SB, Lim CM, Koh Y, Huh JW. Clinical Significance of Circulating Endothelial Cells in Patients With Severe Sepsis or Septic Shock. Infect Dis (Lond) (2015) 47(6):393–8. doi: 10.3109/00365548.2014.1001999
127. Chioh FW, Fong SW, Young BE, Wu KX, Siau A, Krishnan S, et al. Convalescent COVID-19 Patients are Susceptible to Endothelial Dysfunction Due to Persistent Immune Activation. Elife (2021) 10:e64909. doi: 10.7554/eLife.64909
128. Kumar A, Vaish M, Karuppagounder SS, Gazaryan I, Cave JW, Starkov AA, et al. HIF1alpha Stabilization in Hypoxia is Not Oxidant-Initiated. Elife (2021) 10:e72873. doi: 10.7554/eLife.72873
129. Qi F, Zhang W, Huang J, Fu L, Zhao J. Single-Cell RNA Sequencing Analysis of the Immunometabolic Rewiring and Immunopathogenesis of Coronavirus Disease 2019. Front Immunol (2021) 12:651656. doi: 10.3389/fimmu.2021.651656
130. Kim JW, Gao P, Liu YC, Semenza GL, Dang CV. Hypoxia-Inducible Factor 1 and Dysregulated C-Myc Cooperatively Induce Vascular Endothelial Growth Factor and Metabolic Switches Hexokinase 2 and Pyruvate Dehydrogenase Kinase 1. Mol Cell Biol (2007) 27(21):7381–93. doi: 10.1128/MCB.00440-07
131. Lum JJ, Bui T, Gruber M, Gordan JD, DeBerardinis RJ, Covello KL, et al. The Transcription Factor HIF-1alpha Plays a Critical Role in the Growth Factor-Dependent Regulation of Both Aerobic and Anaerobic Glycolysis. Genes Dev (2007) 21(9):1037–49. doi: 10.1101/gad.1529107
132. De Saedeleer CJ, Copetti T, Porporato PE, Verrax J, Feron O, Sonveaux P. Lactate Activates HIF-1 in Oxidative But Not in Warburg-Phenotype Human Tumor Cells. PloS One (2012) 7(10):e46571. doi: 10.1371/journal.pone.0046571
133. Tian M, Liu W, Li X, Zhao P, Shereen MA, Zhu C, et al. HIF-1alpha Promotes SARS-CoV-2 Infection and Aggravates Inflammatory Responses to COVID-19. Signal Transduct Target Ther (2021) 6(1):308. doi: 10.1038/s41392-021-00726-w
134. Parra-Bonilla G, Alvarez DF, Al-Mehdi AB, Alexeyev M, Stevens T. Critical Role for Lactate Dehydrogenase A in Aerobic Glycolysis That Sustains Pulmonary Microvascular Endothelial Cell Proliferation. Am J Physiol Lung Cell Mol Physiol (2010) 299(4):L513–22. doi: 10.1152/ajplung.00274.2009
135. Parra-Bonilla G, Alvarez DF, Alexeyev M, Vasauskas A, Stevens T. Lactate Dehydrogenase a Expression is Necessary to Sustain Rapid Angiogenesis of Pulmonary Microvascular Endothelium. PloS One (2013) 8(9):e75984. doi: 10.1371/journal.pone.0075984
136. Gustot T. Multiple Organ Failure in Sepsis: Prognosis and Role of Systemic Inflammatory Response. Curr Opin Crit Care (2011) 17(2):153–9. doi: 10.1097/MCC.0b013e328344b446
137. Aziz M, Fatima R, Lee-Smith W, Assaly R. The Association of Low Serum Albumin Level With Severe COVID-19: A Systematic Review and Meta-Analysis. Crit Care (2020) 24(1):255. doi: 10.1186/s13054-020-02995-3
138. Wu MA, Fossali T, Pandolfi L, Carsana L, Ottolina D, Frangipane V, et al. COVID-19: The Key Role of Pulmonary Capillary Leakage. An Observational Cohort Study. medRxiv (2020), 2020.05.17.20104877.
139. Duong CN, Vestweber D. Mechanisms Ensuring Endothelial Junction Integrity Beyond VE-Cadherin. Front Physiol (2020) 11:519. doi: 10.3389/fphys.2020.00519
140. Wallez Y, Huber P. Endothelial Adherens and Tight Junctions in Vascular Homeostasis, Inflammation and Angiogenesis. Biochim Biophys Acta (2008) 1778(3):794–809. doi: 10.1016/j.bbamem.2007.09.003
141. Gavard J. Endothelial Permeability and VE-Cadherin: A Wacky Comradeship. Cell Adh Migr (2014) 8(2):158–64. doi: 10.4161/cam.29026
142. Flores-Pliego A, Miranda J, Vega-Torreblanca S, Valdespino-Vazquez Y, Helguera-Repetto C, Espejel-Nunez A, et al. Molecular Insights Into the Thrombotic and Microvascular Injury in Placental Endothelium of Women With Mild or Severe COVID-19. Cells (2021) 10(2):364. doi: 10.3390/cells10020364
143. Kuei C, Yu J, Zhu J, Wu J, Zhang L, Shih A, et al. Study of GPR81, the Lactate Receptor, From Distant Species Identifies Residues and Motifs Critical for GPR81 Functions. Mol Pharmacol (2011) 80(5):848–58. doi: 10.1124/mol.111.074500
144. Xiong S, Zhang L, Richner JM, Class J, Rehman J, Malik AB. Interleukin-1ra Mitigates SARS-CoV-2-Induced Inflammatory Lung Vascular Leakage and Mortality in Humanized K18-hACE-2 Mice. Arterioscler Thromb Vasc Biol (2021) 41(11):2773–85. doi: 10.1161/ATVBAHA.121.316925
145. Xiong S, Hong Z, Huang LS, Tsukasaki Y, Nepal S, Di A, et al. IL-1beta Suppression of VE-Cadherin Transcription Underlies Sepsis-Induced Inflammatory Lung Injury. J Clin Invest (2020) 130(7):3684–98. doi: 10.1172/JCI136908
146. Ortega-Paz L, Capodanno D, Montalescot G, Angiolillo DJ. Coronavirus Disease 2019-Associated Thrombosis and Coagulopathy: Review of the Pathophysiological Characteristics and Implications for Antithrombotic Management. J Am Heart Assoc (2021) 10(3):e019650. doi: 10.1161/JAHA.120.019650
147. Wichmann D, Sperhake JP, Lutgehetmann M, Steurer S, Edler C, Heinemann A, et al. Autopsy Findings and Venous Thromboembolism in Patients With COVID-19: A Prospective Cohort Study. Ann Intern Med (2020) 173(4):268–77. doi: 10.7326/L20-1206
148. Bilaloglu S, Aphinyanaphongs Y, Jones S, Iturrate E, Hochman J, Berger JS. Thrombosis in Hospitalized Patients With COVID-19 in a New York City Health System. JAMA (2020) 324(8):799–801. doi: 10.1001/jama.2020.13372
149. Wang L, He WB, Yu XM, Hu DL, Jiang H. Prolonged Prothrombin Time at Admission Predicts Poor Clinical Outcome in COVID-19 Patients. World J Clin Cases (2020) 8(19):4370–9. doi: 10.12998/wjcc.v8.i19.4370
150. Long H, Nie L, Xiang X, Li H, Zhang X, Fu X, et al. D-Dimer and Prothrombin Time Are the Significant Indicators of Severe COVID-19 and Poor Prognosis. BioMed Res Int (2020) 2020:6159720. doi: 10.1155/2020/6159720
151. Zhang S, Liu Y, Wang X, Yang L, Li H, Wang Y, et al. SARS-CoV-2 Binds Platelet ACE2 to Enhance Thrombosis in COVID-19. J Hematol Oncol (2020) 13(1):120. doi: 10.1186/s13045-020-00954-7
152. Koupenova M, Corkrey HA, Vitseva O, Tanriverdi K, Somasundaran M, Liu P, et al. SARS-CoV-2 Initiates Programmed Cell Death in Platelets. Circ Res (2021) 129(6):631–46. doi: 10.1161/CIRCRESAHA.121.319117
153. Connors JM, Levy JH. COVID-19 and its Implications for Thrombosis and Anticoagulation. Blood (2020) 135(23):2033–40. doi: 10.1182/blood.2020006000
154. REMAP-CAP Investigators, ACTIV-4a Investigators, ATTACC Investigators, Goligher EC, Bradbury CA, McVerry BJ, et al. Therapeutic Anticoagulation With Heparin in Critically Ill Patients With Covid-19. N Engl J Med (2021) 385(9):777–89. doi: 10.1056/NEJMoa2103417
155. Thomas W, White D, Cox-Morton S, MacDonald S, Besser M. Heparin Failure and COVID-19: Should We Explore Other Anticoagulants? An Observational Report Regarding in-Vitro Recovery of Anticoagulant Action in COVID-19 Patients in Intensive Care. Thromb Res (2020) 195:226–7. doi: 10.1016/j.thromres.2020.08.010
156. White D, MacDonald S, Bull T, Hayman M, de Monteverde-Robb D, Sapsford D, et al. Correction to: Heparin Resistance in COVID19 Patients in the Intensive Care Unit. J Thromb Thrombolysis (2020) 50(2):478. doi: 10.1007/s11239-020-02196-3
157. Daniels LB, Sitapati AM, Zhang J, Zou J, Bui QM, Ren J, et al. Relation of Statin Use Prior to Admission to Severity and Recovery Among COVID-19 Inpatients. Am J Cardiol (2020) 136:149–55. doi: 10.1016/j.amjcard.2020.09.012
158. De Spiegeleer A, Bronselaer A, Teo JT, Byttebier G, De Tre G, Belmans L, et al. The Effects of ARBs, ACEis, and Statins on Clinical Outcomes of COVID-19 Infection Among Nursing Home Residents. J Am Med Dir Assoc (2020) 21(7):909–914 e2. doi: 10.1016/j.jamda.2020.06.018
159. Baral R, Tsampasian V, Debski M, Moran B, Garg P, Clark A, et al. Association Between Renin-Angiotensin-Aldosterone System Inhibitors and Clinical Outcomes in Patients With COVID-19: A Systematic Review and Meta-Analysis. JAMA Netw Open (2021) 4(3):e213594. doi: 10.1001/jamanetworkopen.2021.3594
160. Yan SF, Mackman N, Kisiel W, Stern DM, Pinsky DJ. Hypoxia/Hypoxemia-Induced Activation of the Procoagulant Pathways and the Pathogenesis of Ischemia-Associated Thrombosis. Arterioscler Thromb Vasc Biol (1999) 19(9):2029–35. doi: 10.1161/01.ATV.19.9.2029
161. Franczyk B, Gluba-Brzozka A, Lawinski J, Rysz-Gorzynska M, Rysz J. Metabolomic Profile in Venous Thromboembolism (VTE). Metabolites (2021) 11(8):495. doi: 10.3390/metabo11080495
162. Maekawa K, Sugita C, Yamashita A, Moriguchi-Goto S, Furukoji E, Sakae T, et al. Higher Lactate and Purine Metabolite Levels in Erythrocyte-Rich Fresh Venous Thrombus: Potential Markers for Early Deep Vein Thrombosis. Thromb Res (2019) 177:136–44. doi: 10.1016/j.thromres.2019.03.011
163. Koupenova M, Kehrel BE, Corkrey HA, Freedman JE. Thrombosis and Platelets: An Update. Eur Heart J (2017) 38(11):785–91. doi: 10.1093/eurheartj/ehw550
164. Cesar JM, Pallares E, Rubi J, Navarro JL. Lactate Production by Thrombin-Activated Platelets of Patients With Primary Thrombocythemia. Thromb Res (2006) 118(3):335–9. doi: 10.1016/j.thromres.2005.09.008
165. Kulkarni PP, Tiwari A, Singh N, Gautam D, Sonkar VK, Agarwal V, et al. Aerobic Glycolysis Fuels Platelet Activation: Small-Molecule Modulators of Platelet Metabolism as Anti-Thrombotic Agents. Haematologica (2019) 104(4):806–18. doi: 10.3324/haematol.2018.205724
166. Cure E, Cure MC. COVID-19 May Predispose to Thrombosis by Affecting Both Vascular Endothelium and Platelets. Clin Appl Thromb Hemost (2020) 26:1076029620933945. doi: 10.1177/1076029620933945
167. Chang HB, Gao X, Nepomuceno R, Hu S, Sun D. Na(+)/H(+) Exchanger in the Regulation of Platelet Activation and Paradoxical Effects of Cariporide. Exp Neurol (2015) 272:11–6. doi: 10.1016/j.expneurol.2014.12.023
168. Zabczyk M, Natorska J, Janion-Sadowska A, Malinowski KP, Janion M, Undas A. Elevated Lactate Levels in Acute Pulmonary Embolism Are Associated With Prothrombotic Fibrin Clot Properties: Contribution of NETs Formation. J Clin Med (2020) 9(4):953. doi: 10.3390/jcm9040953
169. Zhu X, Messer JS, Wang Y, Lin F, Cham CM, Chang J, et al. Cytosolic HMGB1 Controls the Cellular Autophagy/Apoptosis Checkpoint During Inflammation. J Clin Invest (2015) 125(3):1098–110. doi: 10.1172/JCI76344
170. Kozlova AL, Valieva ME, Maluchenko NV, Studitsky VM. [HMGB Proteins as DNA Chaperones That Modulate Chromatin Activity]. Mol Biol (Mosk) (2018) 52(5):737–49. doi: 10.1134/S0026893318050096
171. Reeves R. Nuclear Functions of the HMG Proteins. Biochim Biophys Acta (2010) 1799(1-2):3–14. doi: 10.1016/j.bbagrm.2009.09.001
172. Andersson U, Tracey KJ. HMGB1 in Sepsis. Scand J Infect Dis (2003) 35(9):577–84. doi: 10.1080/00365540310016286
173. Kang R, Zhang Q, Zeh HJ 3rd, Lotze MT, Tang D. HMGB1 in Cancer: Good, Bad, or Both? Clin Cancer Res (2013) 19(15):4046–57. doi: 10.1158/1078-0432.CCR-13-0495
174. Li W, Sama AE, Wang H. Role of HMGB1 in Cardiovascular Diseases. Curr Opin Pharmacol (2006) 6(2):130–5. doi: 10.1016/j.coph.2005.10.010
175. Chen R, Huang Y, Quan J, Liu J, Wang H, Billiar TR, et al. HMGB1 as a Potential Biomarker and Therapeutic Target for Severe COVID-19. Heliyon (2020) 6(12):e05672. doi: 10.1016/j.heliyon.2020.e05672
176. Chen L, Long X, Xu Q, Tan J, Wang G, Cao Y, et al. Elevated Serum Levels of S100A8/A9 and HMGB1 at Hospital Admission are Correlated With Inferior Clinical Outcomes in COVID-19 Patients. Cell Mol Immunol (2020) 17(9):992–4. doi: 10.1038/s41423-020-0492-x
177. Yang L, Xie M, Yang M, Yu Y, Zhu S, Hou W, et al. PKM2 Regulates the Warburg Effect and Promotes HMGB1 Release in Sepsis. Nat Commun (2014) 5:4436. doi: 10.1038/ncomms5436
178. Lu B, Antoine DJ, Kwan K, Lundback P, Wahamaa H, Schierbeck H, et al. JAK/STAT1 Signaling Promotes HMGB1 Hyperacetylation and Nuclear Translocation. Proc Natl Acad Sci U S A (2014) 111(8):3068–73. doi: 10.1073/pnas.1316925111
179. Yang Z, Li L, Chen L, Yuan W, Dong L, Zhang Y, et al. PARP-1 Mediates LPS-Induced HMGB1 Release by Macrophages Through Regulation of HMGB1 Acetylation. J Immunol (2014) 193(12):6114–23. doi: 10.4049/jimmunol.1400359
180. Ditsworth D, Zong WX, Thompson CB. Activation of Poly(ADP)-Ribose Polymerase (PARP-1) Induces Release of the Pro-Inflammatory Mediator HMGB1 From the Nucleus. J Biol Chem (2007) 282(24):17845–54. doi: 10.1074/jbc.M701465200
181. Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, et al. Monocytic Cells Hyperacetylate Chromatin Protein HMGB1 to Redirect it Towards Secretion. EMBO J (2003) 22(20):5551–60. doi: 10.1093/emboj/cdg516
182. Xu S, Zeng Z, Zhao M, Huang Q, Gao Y, Dai X, et al. Evidence for Sirt1 Mediated Hmgb1 Release from Kidney Cells in the Early Stages of Hemorrhagic Shock. Front Physiol (2019) 10:854. doi: 10.3389/fphys.2019.00854
183. Bi X, Xu M, Li J, Huang T, Jiang B, Shen L, et al. Heat Shock Protein 27 Inhibits HMGB1 Translocation by Regulating CBP Acetyltransferase Activity and Ubiquitination. Mol Immunol (2019) 108:45–55. doi: 10.1016/j.molimm.2019.02.008
184. Zhou H, Jin C, Cui L, Xing H, Liu J, Liao W, et al. HMGB1 Contributes to the Irradiation-Induced Endothelial Barrier Injury Through Receptor for Advanced Glycation Endproducts (RAGE). J Cell Physiol (2018) 233(9):6714–21. doi: 10.1002/jcp.26341
185. Reiterer M, Rajan M, Gomez-Banoy N, Lau JD, Gomez-Escobar LG, Ma L, et al. Hyperglycemia in Acute COVID-19 is Characterized by Insulin Resistance and Adipose Tissue Infectivity by SARS-CoV-2. Cell Metab (2021) 33(11):2174–2188 e5. doi: 10.1016/j.cmet.2021.09.009
186. Mazori AY, Bass IR, Chan L, Mathews KS, Altman DR, Saha A, et al. Hyperglycemia is Associated With Increased Mortality in Critically Ill Patients With COVID-19. Endocr Pract (2021) 27(2):95–100. doi: 10.1016/j.eprac.2020.12.015
187. Charoenngam N, Alexanian SM, Apovian CM, Holick MF. Association Between Hyperglycemia at Hospital Presentation and Hospital Outcomes in COVID-19 Patients With and Without Type 2 Diabetes: A Retrospective Cohort Study of Hospitalized Inner-City COVID-19 Patients. Nutrients (2021) 13(7):2199. doi: 10.3390/nu13072199
188. Xiao F, Zhou YC, Zhang MB, Chen D, Peng SL, Tang HN, et al. Hyperglycemia and Blood Glucose Deterioration are Risk Factors for Severe COVID-19 With Diabetes: A Two-Center Cohort Study. J Med Virol (2021). doi: 10.1002/jmv.27556
189. Wu H, Chen Z, Xie J, Kang LN, Wang L, Xu B. High Mobility Group Box-1: A Missing Link Between Diabetes and Its Complications. Mediators Inflamm (2016) 2016:3896147. doi: 10.1155/2016/3896147
190. Dyer MR, Chen Q, Haldeman S, Yazdani H, Hoffman R, Loughran P, et al. Deep Vein Thrombosis in Mice is Regulated by Platelet HMGB1 Through Release of Neutrophil-Extracellular Traps and DNA. Sci Rep (2018) 8(1):2068. doi: 10.1038/s41598-018-20479-x
191. Stark K, Philippi V, Stockhausen S, Busse J, Antonelli A, Miller M, et al. Disulfide HMGB1 Derived From Platelets Coordinates Venous Thrombosis in Mice. Blood (2016) 128(20):2435–49. doi: 10.1182/blood-2016-04-710632
192. Vogel S, Bodenstein R, Chen Q, Feil S, Feil R, Rheinlaender J, et al. Platelet-Derived HMGB1 is a Critical Mediator of Thrombosis. J Clin Invest (2015) 125(12):4638–54. doi: 10.1172/JCI81660
193. Lv B, Wang H, Tang Y, Fan Z, Xiao X, Chen F. High-Mobility Group Box 1 Protein Induces Tissue Factor Expression in Vascular Endothelial Cells via Activation of NF-kappaB and Egr-1. Thromb Haemost (2009) 102(2):352–9. doi: 10.1160/TH08-11-0759
194. AbdelMassih AF, Menshawey R, Hozaien R, Kamel A, Mishriky F, Husseiny RJ, et al. The Potential Use of Lactate Blockers for the Prevention of COVID-19 Worst Outcome, Insights From Exercise Immunology. Med Hypotheses (2021) 148:110520. doi: 10.1016/j.mehy.2021.110520
195. Pajak B, Siwiak E, Soltyka M, Priebe A, Zielinski R, Fokt I, et al. 2-Deoxy-D-Glucose and Its Analogs: From Diagnostic to Therapeutic Agents. Int J Mol Sci (2019) 21(1):234. doi: 10.3390/ijms21010234
196. Zheng Z, Ma H, Zhang X, Tu F, Wang X, Ha T, et al. Enhanced Glycolytic Metabolism Contributes to Cardiac Dysfunction in Polymicrobial Sepsis. J Infect Dis (2017) 215(9):1396–406. doi: 10.1093/infdis/jix138
197. Sahu KK, Kumar R. Role of 2-Deoxy-D-Glucose (2-DG) in COVID-19 Disease: A Potential Game-Changer. J Family Med Prim Care (2021) 10(10):3548–52. doi: 10.4103/jfmpc.jfmpc_1338_21
198. McCullagh KJ, Poole RC, Halestrap AP, O'Brien M, Bonen A. Role of the Lactate Transporter (MCT1) in Skeletal Muscles. Am J Physiol (1996) 271(1 Pt 1):E143–50. doi: 10.1152/ajpendo.1996.271.1.E143
199. Li C, Ha T, Kelley J, Gao X, Qiu Y, Kao RL, et al. Modulating Toll-Like Receptor Mediated Signaling by (1–>3)-Beta-D-Glucan Rapidly Induces Cardioprotection. Cardiovasc Res (2004) 61(3):538–47. doi: 10.1016/j.cardiores.2003.09.007
200. Ha T, Ha T, Kelley J, Gao X, Qiu Y, Kao RL, et al. Glucan Phosphate Attenuates Myocardial HMGB1 Translocation in Severe Sepsis Through Inhibiting NF-kappaB Activation. Am J Physiol Heart Circ Physiol (2011) 301(3):H848–55. doi: 10.1152/ajpheart.01007.2010
201. Borriello F, Poli V, Shrock E, Spreafico R, Liu X, Pishesha N, et al. An Adjuvant Strategy Enabled by Modulation of the Physical Properties of Microbial Ligands Expands Antigen Immunogenicity. Cell (2022) 185(4):614–629 e21. doi: 10.1016/j.cell.2022.01.009
202. Pulendran B, P SA, O'Hagan DT. Emerging Concepts in the Science of Vaccine Adjuvants. Nat Rev Drug Discov (2021) 20(6):454–75. doi: 10.1038/s41573-021-00163-y
203. Cordova-Martinez A, Caballero-Garcia A, Roche E, Noriega DC. Beta-Glucans Could Be Adjuvants for SARS-CoV-2 Virus Vaccines (COVID-19). Int J Environ Res Public Health (2021) 18(23):12636. doi: 10.3390/ijerph182312636
204. Andrade Silva M, da Silva A, do Amaral MA, Fragas MG, Camara NOS. Metabolic Alterations in SARS-CoV-2 Infection and Its Implication in Kidney Dysfunction. Front Physiol (2021) 12:624698. doi: 10.3389/fphys.2021.624698
Keywords: COVID-19, aerobic glycolytic metabolism, lactate, endothelial cell, cardiovascular dysfunction, HMGB1 (High mobility group box 1), thrombosis, vascular permeability
Citation: Yang K, Holt M, Fan M, Lam V, Yang Y, Ha T, Williams DL, Li C and Wang X (2022) Cardiovascular Dysfunction in COVID-19: Association Between Endothelial Cell Injury and Lactate. Front. Immunol. 13:868679. doi: 10.3389/fimmu.2022.868679
Received: 03 February 2022; Accepted: 01 March 2022;
Published: 23 March 2022.
Edited by:
Guo-Chang Fan, University of Cincinnati, United StatesReviewed by:
Tianqing Peng, Western University, CanadaXianzhong Meng, University of Colorado Denver, United States
Qun Sophia Zang, Loyola University Chicago, United States
Copyright © 2022 Yang, Holt, Fan, Lam, Yang, Ha, Williams, Li and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kun Yang, Yangk1@etsu.edu; Xiaohui Wang, Wangx3@etsu.edu