 Jianan Zhao
Jianan Zhao Ping Jiang
Ping Jiang Shicheng Guo
Shicheng Guo Steven J. Schrodi
Steven J. Schrodi Dongyi He
Dongyi He- 1Guanghua Clinical Medical College, Shanghai University of Traditional Chinese Medicine, Shanghai, China
- 2Department of Rheumatology, Shanghai Guanghua Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai, China
- 3Department of Medical Genetics, School of Medicine and Public Health, University of Wisconsin-Madison, Madison, WI, United States
- 4Arthritis Institute of Integrated Traditional and Western Medicine, Shanghai Chinese Medicine Research Institute, Shanghai, China
Rheumatoid arthritis (RA) is a chronic inflammatory joint disease that can lead to clinical manifestations of systemic diseases. Its leading features include chronic synovial inflammation and degeneration of the bones and joints. In the past decades, multiple susceptibilities for rheumatoid arthritis have been identified along with the development of a remarkable variety of drugs for its treatment; which include analgesics, glucocorticoids, nonsteroidal anti-inflammatory medications (NSAIDs), disease-modifying anti-rheumatic drugs (DMARDs), and biologic response modifiers (bDMARDs). Despite the existence of many clinical treatment options, the prognosis of some patients remains poor due to complex mechanism of the disease. Programmed cell death (PCD) has been extensively studied and ascertained to be one of the essential pathological mechanisms of RA. Its dysregulation in various associated cell types contributes to the development of RA. In this review, we summarize the role of apoptosis, cell death-associated neutrophil extracellular trap formation, necroptosis, pyroptosis, and autophagy in the pathophysiology of RA to provide a theoretical reference and insightful direction to the discovery and development of novel therapeutic targets for RA.
Introduction
RA is an autoimmune disease that is usually accompanied by swelling, tenderness, and pain in the joints. It gradually leads to the degeneration of the synovium and joints, often causing disability and premature death (1). RA typically affects one percent of the population, with apparently higher incidences among women than in men (2). The contributing factors and pathogenesis of RA are multifaceted. Studies have shown that it is mainly the production of autoantigens, under the influence of genetic factors, smoking, dust (silica, textile dust), and microbial populations, that activates the adaptive and innate immune system responsible for the pathogenesis of RA. A variety of immune cells release several cytokines and mediators to instigate chronic inflammation of the synovial membrane and destruction of bones and joints during RA progression. These effects thus characterize the immune disorder of RA, which involves immune complex-mediated complement activation, immune response to the post-translationally modified protein autoantigen, dysregulation of cell factor network, and activation of bone destruction-related cells (3–5).
At present, targeted immunotherapy with specific mechanisms (biologically targeted agents and various kinase inhibitors) has made significant progress in the clinical treatment of RA, thereby significantly improving the clinical outcomes in patients. These therapies have been demonstrated to target key molecules and cell nodes that are enriched in the complex mechanism of the disease. However, the treatment failure experienced by some patients administered with immuno-targeted therapies suggests that renewed attention should be duly rewarded to other pathways, such as cell death, for their assessment as prospective targets of intervention (6). Cell death plays different roles under diverse physiological and pathological conditions. Stimulation of death of specific cell types plays specific role depending upon the environmental context. The consequences of cell death in diseases may serve destructive or protective functions, such as those accomplished by the release of inflammatory mediators or protection of host from “harmful or rogue cells”, respectively. For instance, induction of activation-induced cell death (AICD), by the CD44 mediated FasL expression on the surface of T cells through the activation of a tyrosine kinase, IP3 receptor-dependent Ca2+ mobilization, and actin cytoskeletal rearrangements; eliminates self-reactive T cells thereby, suppressing RA (7).
Multiple studies have demonstrated the connection between PCD and RA, especially in the context of various cell populations, including fibroblast-like synoviocytes (FLS), T cells, B cells, monocyte-macrophages, neutrophils, osteoblasts/chondrocytes, and osteoclasts. During the progression of RA, the antagonism of FLS cells to cell death results in excessive proliferation of synoviocytes triggering synovitis. Meanwhile, the imbalance in the cell death of autoimmune T and B cells increases autoimmune and inflammatory responses, whereas the imbalanced cell death of monocyte-macrophages and neutrophils contributes to a variety of pathological reactions associated with RA. On the other hand, enhanced cell death of osteoblasts/chondrocytes and osteoclasts is involved in the RA-associated bone destruction. Thus, the unbalanced cell death of multiple cell types, works in tandem to form a vicious circle that aggravates RA. Accordingly, this review aims to describe the various forms of PCD that may play critical role in the progression of RA, thereby providing insightful reference and direction for further analysis of disease mechanisms and targeted development of innovative therapies for clinical use.
Apoptosis in the Pathophysiology of Rheumatoid Arthritis
Apoptosis is a form of PCD that has been extensively studied and deliberated (8). Briefly, it essentially mediates cell death via two mechanisms involving the exogenous death receptor and the endogenous mitochondrial pathways. In the exogenous death receptor pathway, FasL binds to Fas, resulting in the recruitment of various proteins, and activation of downstream caspase-8, caspase-7, and caspase-3. Caspase-7 activates caspase-6 and the BH3 interacting-domain death agonist (BID) to crosstalk with the endogenous mitochondrial pathway. This results in the activation of a variety of mitochondrial pro-apoptotic proteins that causes accumulation of the B cell lymphoma/leukemia-2 (Bcl-2)-associated X protein (Bax), and Bcl-2 homologous antagonist/killer protein (Bak). Concomitantly, it also leads to the inhibition of anti-apoptotic proteins such as Bcl-2, which cause mitochondrial dysfunction and release of cytochrome c (Cyt c) to activate downstream caspase-9 and caspase-3 that mediate cell death. In addition, the endoplasmic reticulum and lysosomes mediate apoptosis through diverse mechanisms. There are many types of cells that are affected in the course of RA development. One of the important mechanisms contributing to the pathogenesis of RA involves the unbalanced regulation of apoptosis that leads to abnormal expansion or excessive apoptosis of specific cell types. Consequently, it is vital to clarify the relationship between apoptosis and RA especially in the context of various cell types involved.
Although synovial cells are capable to induce apoptosis and reduce cell proliferation through the Fas/FasL pathway, it is quite evident that this mechanism of action is defective during RA. Anti-Fas monoclonal antibody has been proved successful in inducing apoptosis of synovial cells in vitro. However, the pro-inflammatory factors such as tumor necrosis factor (TNF)-α and interleukin (IL)-1β have been shown to inhibit Anti-Fas monoclonal antibody-induced apoptosis, potentially by upregulating the Bcl-2-mediated survival, and downregulating the expression of CPP32 and ICH-1L (9). Studies have shown that activators of transcription 3 (STAT3) is an important transcription factor that plays critical role in the regulation of cell survival and apoptosis of various cell types involved in RA. For instance, overexpression of STAT3-YF, a dominant-negative mutant of STAT3, has been shown to inhibit the activation and target gene expression of endogenous STAT3, thereby promoting synovial cell apoptosis (10). Furthermore, heat-shock-protein-70 (HSP70) has been shown to inhibit the apoptotic signal of RA peripheral blood lymphocytes (PBL), resulting in incomplete activation of the CD95/Fas pathway. This phenomenon is characterized by caspase-3/7 activation but no DNA fragmentation, and may thus serve as an important mechanism in the progression of RA (11). Peptidylarginine deiminase IV (PADI4) is responsible for the conversion of peptidylarginine to citrulline. Interestingly, a specific PADI4 SNP haplotype has been reported to strongly induce apoptosis via multiple mitochondrial pathways through the downregulation of BCL-XL, upregulation of Bax, and the release of Cyt c into the cytoplasm; thereby providing a possible mechanism underlying the increase of SNP PADI4 activity and highlighting its critical role in the pathogenesis of RA (12). Inconsistently, studies have shown that although excessive TNF-α can attenuate Fas-mediated apoptosis of synovial cells, the effect may not be related to the expression of Fas and Bcl-2. Nevertheless, the benefits of anti-TNF-α therapy in the management of RA are undeniable (13).
At present, the commonly used drugs in clinical settings for the treatment of RA include methotrexate, nonsteroidal anti-inflammatory drugs (NSAIDs), and a variety of biological agents, all of which have been shown to be partially involved in the regulation of apoptosis. Methotrexate can increase the local concentration of extracellular adenosine, which can serve as a substrate for the elevated concentration of adenosine deaminase in the synovial fluid of RA patients. This elevated concentration of adenosine may therefore, serve to be beneficial for the treatment of RA by a mechanism that potentially involves the activation of caspase pathway to induce DNA fragmentation and promote FLS cell apoptosis (14). A variety of NSAIDs, such as indomethacin, diclofenac, oxaprozin, and zaltoprofen, can inhibit the proliferation of synovial cells and induce apoptosis. The mechanism underlying this effect may be related to the activation of peroxisome proliferator-activated receptor gamma (PPAR gamma) (15). As far as biological agents are concerned, infliximab is an anti-TNF-α monoclonal antibody that partially promotes caspase-dependent apoptosis and reduces the production of IL-10 and IL-12 (16). Additionally, rituximab reportedly binds to CD20 and induces B cell apoptosis through antibody- and complement- mediated cytotoxicity, thereby treating RA (17). Furthermore, peficitinib, a JAK inhibitor, has been documented to inhibit the autocrine phosphorylation of STAT3 and anti-apoptotic genes in a concentration-dependent manner to promote FLS cell apoptosis (18) (See Figure 1).
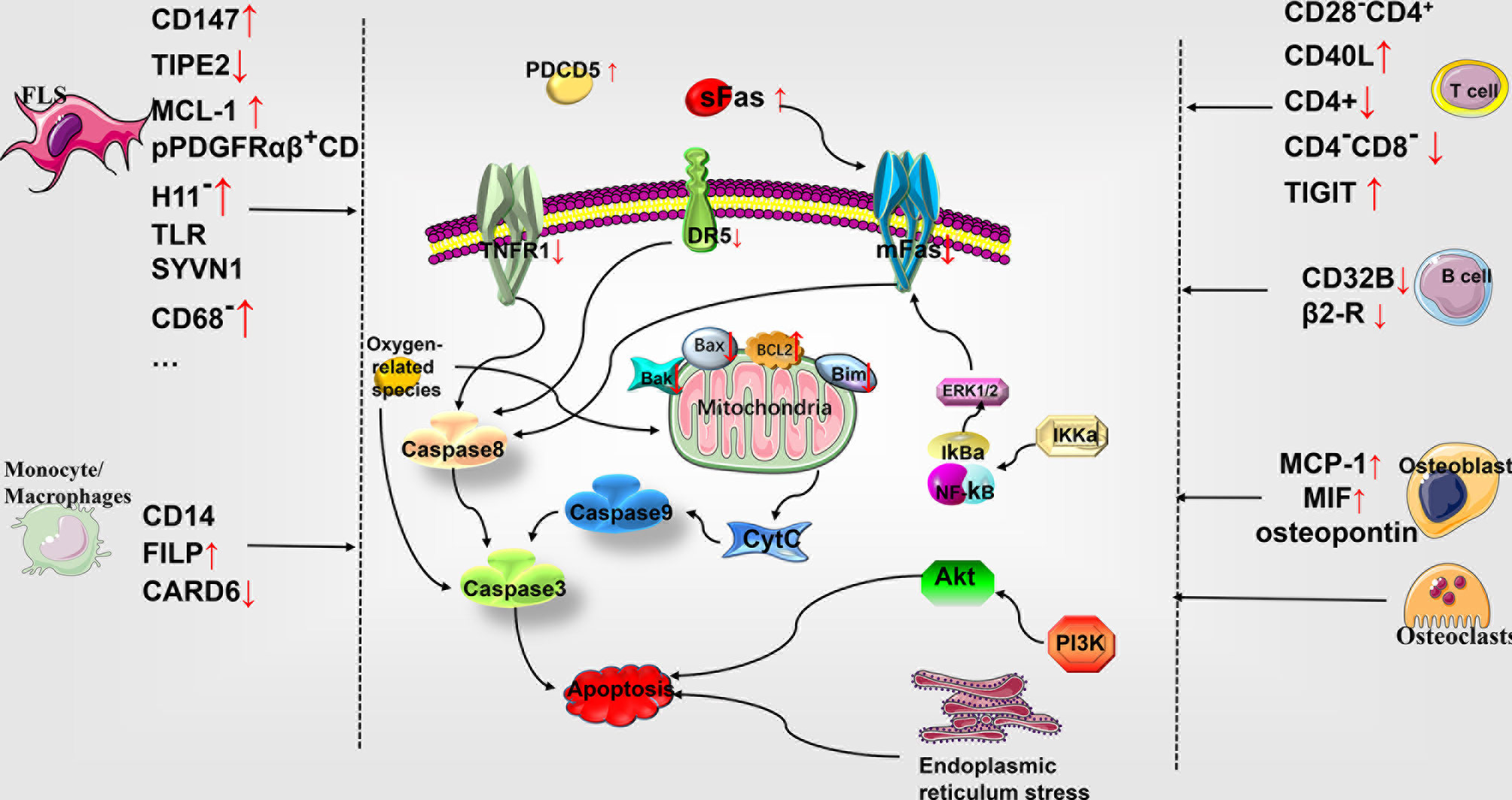
Figure 1 Role of apoptosis in the pathophysiology of rheumatoid arthritis. Multiple cellular subtypes express different molecules in RA, including FLS, monocyte-macrophages, T cells, B cells, osteoblasts, and chondrocytes. The differential gene expression influences the apoptosis of various cells through the molecules related to the exogenous death pathway and the endogenous mitochondrial pathway, thereby affecting the development of RA. MCL-1, myelogenous cell leukemia-1; SYVN1, the E3 ubiquitin ligase synoviolin; CARD6, caspase recruitment domain protein 6; PDCD5, programmed cell death 5; DR5, tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) receptor; mFas, membrane-bound Fas; Bcl-2, B cell lymphoma/leukemia -2; Bax, accumulation of Bcl-2–associated X protein; Bak, Bcl-2 homologous antagonist/killer; Bim, Bcl-2 interacting mediator of cell death; Cyt c, cytochrome c; ERK, extracellular signal-regulated kinase; IκBα, intracellular calcium, phosphorylated I-kappa-Bα; TNFR1, tumor necrosis factor receptor 1; PI3K, phosphatidylinositol 3-kinase; TLR, Toll-like receptor; FLS, fibroblast-like synoviocytes.
Insensitivity to Apoptosis Leads to Abnormal Proliferation of FLS in RA
During the progression of RA, FLS exhibit resistance to apoptosis resulting in synovial hyperplasia that facilitates the release of related cytokines via multiple contributing mechanisms. CD147 is highly expressed in FLS during RA, and its extracellular site resists the TNF-α-induced apoptosis by promoting intracellular Ca2+ levels through the activation of nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB) signaling pathway (19). TNF-alpha-induced protein-8-like-2 (TIPE2) has been reported to be expressed in very low levels in adjuvant arthritis (AA) model. Moreover, it has been shown to increase the expression of TNF-related apoptosis-inducing ligand (TRAIL) receptor-2 (DR5), promote the activation of caspases, and inhibit the NF-κB signaling pathway to promote FLS cell apoptosis (20). Myelogenous cell leukemia-1 (MCL-1) is highly expressed in the synovial lining and sub-lining layers of FLS during RA. Its expression has been shown to increase in response to IL-1β stimulation and is related to the levels of TNF-α as well as the degree of inflammation. Furthermore, it reportedly inhibits FLS apoptosis by regulating the expression of mitochondrial proteins, such as Bax, Bak, and Bcl-2 interacting mediator of cell death (Bim) (21). Endoplasmic reticulum stress has been documented to exert a pro-inflammatory effect in RA. Studies have shown that FLS antagonizes endoplasmic reticulum stress-induced apoptosis during RA. Endoplasmic reticulum stress is accompanied by the activation of multiple TLR ligands and TLRs, which promotes the production of various cytokines and chemokines (22). Additionally, the E3 ubiquitin ligase synoviolin (SYVN1) has been reported to interact with inositol-requiring enzyme 1 (IRE1) to promote the ubiquitination and degradation of IRE1 and to inhibit FLS cell death in mice due to collagen-induced arthritis (CIA) triggered by endoplasmic reticulum stress. Remarkably, inhibition of SYVN1 reverses this effect (23).
Fas antigen expression is typically detectable in the initial stage of apoptosis, and the elevated expression of Bcl-2 is related to a decrease in apoptosis (24). In RA, the FLS subsets of pPDGFRαβ+CDH11- cells are present at a higher proportion in the sub-synovial layer and exhibit increased expression of Bcl-2, decreased expression of TNFR1, resistance to apoptosis, and abnormal proliferation (25). Studies have illuminated some of the ultrastructural characteristics of the apoptotic process in RA associated FLS and determined the modifications in related molecules. Evidently, the apoptotic FLS is present only in the deeper layer of the synovium, while FLS in the remaining layers neither undergo apoptosis nor express Fas antigen (24). Bcl-2 is highly expressed synovial tissues, especially in CD68-FLS cells during RA. Inhibition of Bcl-2 leads to changes in mitochondrial permeability, and Cyt c in addition to activation of caspase-9 and caspase-3, DNA fragmentation, and other apoptotic features (26). Soluble Fas (sFas) levels in RA joints have been reported to be elevated and inhibit membrane-bound Fas (mFas) by activating the extracellular signal-regulated kinase (ERK) 1/2, phosphatidylinositol 3-kinase (PI3K), caspase-8, and c-Jun N-terminal kinase (JNK) signaling pathways to enhance the anti-apoptotic effect of FLS. Moreover, it can also promote the cell migration of T cells via activation of PI3K. Remarkably, the injection of agonistic anti-Fas antibody has also been documented to evidently improve arthritis in mice with experimentally induced arthritis (27–29). RA patients exhibit higher levels of soluble FasL in the synovial fluid, which is negatively correlated with the levels of vascular endothelial growth factor 165 (VEGF165). This results in the induction of apoptosis in FLS while the reduction in the level of VEGF165 inhibits the migration and chemotaxis of endothelial cells (30). The activation of the ERK and AKT pathways apparently assist FLS in counteracting apoptosis during RA. Estrogen 17β-estradiol has been reported to activate the ERK1/2 signaling pathway to exacerbate RA, promote the TNF-α induced surge in matrix metalloproteinase (MMP)-3, and inhibit the TNF-α induced apoptosis of FLS (31). Imperatorin (IPT) has been demonstrated to reduce the survival of FLS influenced by IL-1β; promote the apoptosis of FLS by increasing the release of mitochondrial cytochrome c, the ratio of Bax/Bcl-2, and other mitochondrial pathways in RA; and alleviates collagen-induced arthritis by reducing the expression of MMP1/3 (32). Similarly, tissue inhibitor of metalloproteinases-3 (TIMP-3) has been shown to inhibit matrix MMP and promote apoptosis. Furthermore, RA- associated FLS ectopically expressing TIMP3 gene exhibit a higher sensitivity to apoptosis by inhibiting the upregulation of Fas/CD95 and activation of NF-κB stimulated by TNF-α (33). The expression of cancerous inhibitor of protein phosphatase 2A (CIP2A) has been reported to activate Akt and inhibit caspase-3, caspase-9, and PARP for promoting the anti-apoptotic effect of RA associated FLS (34). Ceramide, a lipid medium, has been reported to inhibit the phosphorylation and activation of anti-apoptotic protein kinases Akt, MEK, and ERK1/2 in synovial cells stimulated by platelet-derived growth factor (PDGF) (35). TNF-α in RA patients has been documented to stimulate the hypoxia-inducible factor-1α (HIF-1α) activation in FLS through the ERK pathway in addition to transcriptional activation of B-cell activating factor (BAFF), which promotes the survival and inhibits the apoptosis of FLS (36). Lowered pH has been shown to activate the acid-sensing ion channel 3 (ASIC3) expressed in FLS, resulting in an increase in Ca2+ and a decrease in p-ERK levels, thereby inducing cell death through calcium-dependent pathways (37). Similarly, a pH 5.5 solution and capsaicin, a transient receptor potential vanilloid 1 (TRPV1) agonist, have been reported to increase the expression of TRPV1 mRNA, accompanied by increased Ca2+, reactive oxygen species (ROS), and depolarization of the mitochondrial membrane potential to promote synovial cell apoptosis (38), However, a pH 6.8 solution triggered activation of phospholipase C, intracellular release of Ca2+, nuclear translocation of NF-κB, and inhibition of ROS production, results in the mitigation of capsaicin-induced apoptosis of synovial cells (39). Thus, the decrease in the pH may prove to be beneficial in limiting the proliferation of the synovium and subsequent joint damage associated with RA.
There are many oxygen-related species in RA, including nitric oxide (NO), ROS, and their products, which affect the apoptosis of FLS through different mechanisms. NO is elevated in RA synovial fluid, and serum directly inhibits caspase-3 activation and defends the proliferation of synovial cells (40). In addition, studies have shown that 3-nitrotyrosine, a marker of reactive nitrogen species, induces chondrocyte mitochondrial dysfunction and caspase-independent apoptosis through a calcium-dependent mechanism (41). Menthol, a specific agonist of transient receptor potential melastatin subtype 8 (TRPM8), promotes Ca2+ influx and TRPM8 activation to generate ROS, which induces synovial cell apoptosis (42). In addition, a prolonged O2/Ca2+-supporting phototherapy hydrogel (POGel) system has been shown to enhance the direct killing effect of singlet oxygen on cells and promote articular cartilage regeneration. The resultant calcium peroxide (CaO2) induces mitochondrial Ca2+ overload and endoplasmic reticulum Ca2+ disorder, triggering Ca2+-related FLS apoptosis and immunogenic cell death to alleviate RA-related synovial proliferation and joint destruction (43). Mitomycin C has been demonstrated to inhibit FLS proliferation and promote FLS apoptosis by increasing ROS production, inducing mitochondrial dysfunction, and increasing the release of mitochondrial cytochrome c, the ratio of Bax/Bcl-2, caspase-9, caspase-3, and cleavage of poly(ADP-ribose) polymerase (44). Moreover, apigenin, a dietary plant-flavonoid, has been reported to induce FLS apoptosis and activate ERK1-2/caspase-3/caspase-7 signaling by generating excessive ROS (45). Similarly, hempseed oil has been shown to promote apoptosis of MH7A cells in a time- and dose- dependent manner, accompanied by lipid accumulation, increased ROS production, and elevated expression of an endoplasmic reticulum stress protein, the EBP-homologous protein (CHOP) (46). Furthermore, resveratrol, a phytoalexin, has been reported to induce MH7A cell apoptosis by stimulating the release of Cyt c, activating caspase-3 and caspase-9, up-regulating the transcription of the NAD-dependent deacetylase- sirtuin 1, and downregulating the transcription of survival factor- Bcl-xl (47). Interestingly, geldanamycin (GA) can promote ROS production in RA associated FLS and induce apoptosis in vitro (48). In addition, programmed cell death 5 (PDCD5) and P53 may also serve as critical apoptotic targets. In RA patients, PDCD5 levels in the serum and synovial fluid are elevated, which may be attributed to the negative association of reduced sensitivity of FLS to apoptosis and its interaction with CRP, ESR, IL-17, and TNF-α (49, 50). The expression and nuclear translocation of PDCD5 have been reported elevated in the synovial tissue and FLS in RA. Overexpression of PDCD5 causes triptolide to promote caspase-3 expression and apoptosis in FLS (51). In addition, daphnetin has been shown to reduce the expression of DNMT1, DNMT3a, and DNMT3b and mediate the demethylation of pro-apoptotic genes- DR3, PDCD5, FasL, and P53, resulting in augmented apoptosis of synovial cells in CIA (52). The p53 upregulated modulator of apoptosis (PUMA) has been documented to promote apoptosis of FLS cells in a potentially P53 independent manner. Thus, gene therapy targeting PUMA can also serve as a promising treatment strategy for RA (53). However, studies have shown that the abnormal function of P53 also contributes to the abnormal proliferation of FLS (54), and calpain inhibitor 1 (ALLN) can induce P53 protein expression in synovial cells and cooperate with hydrogen peroxide to promote the apoptosis of synoviocytes (55). Furthermore, TNF-α depletes CREB binding protein (CBP)-induced P53 acetylation state, thereby attenuating its transcriptional activity. Consequently, the overexpression of CBP assists in activating and restoring the transcriptional activity of P53, which ultimately increases the TNF-α-induced apoptosis of synoviocytes in RA (56).
A synthetic second mitochondria-derived activator of caspase (Smac 066) has been reported to inhibit the cell survival promoting inhibitors of apoptosis proteins (IAPs), thereby demonstrating a robust pro-apoptotic effect on FLS in RA. Its mechanism may involve the activation of caspase-3 and caspase-8, the downregulation of IAPs, and the upregulation of IGFBP-5 (57). Jüngel et al. have shown that trichostatin increases the in vitro sensitivity of FLS to TRAIL-induced apoptosis by upregulating p21Waf1/Cip1 (58). Thus, targeting the differentially expressed apoptosis promoting molecules and promoting the generation of oxygen-related species may serve as a potential measure to reduce the proliferation of RA synovium and increase the sensitivity of FLS to apoptosis. However, it is worth noting that FLS with a high proliferation state is significantly less sensitive to Fas-induced apoptosis than FLS with a low proliferation rate. Additionally, the sensitivity of FLS in the S or G2/M phase to TRAIL-induced apoptosis has been reported to be substantially lower than that in the G0/G1 phase (59).
Abnormal Expansion of Autoimmune T Cells Mediates Inflammation of the Synovium
In RA, elaborate studies have shown that a large number of T cells infiltrate the synovium; interact with other cells, including dendritic cells (DC), monocytes, macrophages, and FLS; release a large number of inflammatory and a small number of anti-inflammatory mediators; and jointly mediate the chronic inflammation of the synovium. Evidently, this RA associated abnormal expansion of autoimmune T cells may be caused by increased homing as well as retention, and/or an imbalanced regulation of apoptotic pathway in these cells (60). Among the CD4+ T lymphocytes in RA patients, CD28-CD4+ T cells highly express Bcl-2, which leads to atypical clonal expansion of autoimmune T cells [60]. In addition, the circulating T cell populations of RA patients highly express CD40L and interact with CD40 to activate B cells and decrease CD32B expression, which inhibits the apoptosis of autoimmune memory B cells. Furthermore, this effect can be enhanced by IL-4, IL-10, and IL-21 (61). The RA auto-antibody B7-H1 exerts a dual regulatory effect on CD4+ T cells in promoting proliferation as well as apoptosis. It has been reported to stimulate the proliferation of T cells and the release of IL-10, accompanied by the upregulation of TNF apoptosis-inducing ligand and caspase-3, resulting in abnormal responses of T cells associated with aggravated RA (62). Treg cells can regulate a variety of immune cells to prevent autoimmune destruction by various mechanisms. Rapetti et al. have shown that failure to inhibit CD4+ T cell activation is concomitant with the failure of Treg cells in inhibiting the B cell activation and production of pro-inflammatory factors and autoantibodies in RA. Thus, the reduced ability of Treg cells in regulating the apoptosis of B cells decreases the expression of Fas in B cells, rendering them resistant to apoptosis (63).
Several studies have used experimentally induced arthritis animal models to study the relationship between T cells and apoptosis and delineate the possible underlying mechanisms. Studies have shown that activation of DCs by the receptor activator of nuclear factor (NF)-κB ligand (RANKL) is accompanied by a decrease in CD4+ T, B, and CD4-CD8- double negative T cells in MRL/LPR experimental autoimmune mice. Further, the TNF-related apoptosis-inducing ligand-receptor 2 (TRAIL-R2) of T cells has been shown to directly interact with TRAIL on DC cells to induce Fas-independent apoptosis as a new mechanism for maintaining peripheral immune tolerance (64). Overexpression of the Galectin-1 and downregulation of Galectin-3 has been reported to inhibit arthritis in the CIA mice and significantly reduce multiple disease activity scores, T cell infiltration, and microvessel density by inducing lymphatic T cell apoptosis (65). The P2RX7 in the K/BxN autoimmune arthritis model has been reported to antagonize the production of autoimmune B cells and autoantibodies by promoting cell death of T follicular helper cells. In contrast, the activation of TIGIT, a T cell exhaustion marker, has been demonstrated to upregulate the resistance factor of apoptosis and promote T cell survival, thereby counteracting the effect of P2RX7 (66). The arthritis resistance of HLA-DRB1*0402 expressing mice has been documented to be possibly related to the loss of autoimmune T cells, a higher number of regulatory T cells, and increased activation-induced apoptosis in these mice (67). In addition, the CD4-/- transgenic mice with HLA-DQ8 are resistant to CIA, whereas the CD8-/- HLA-DQ8 harboring mice exhibit a higher disease incidence and severity than the control group. These results indicate that CD4 is a necessary factor for initiating CIA in DQ8 transgenic mice. Although CD8+ T cells fail to induce CIA in DQ8 transgenic mice, they exert a protective effect against apoptosis (68). Thus, inducing autoimmune T cell apoptosis may serve as a potent strategy to treat RA. Galactoxylomannan (GalXM) acts on CD45RO T cells in RA to inhibit the interaction of CD45 phosphatase with T cells, the production of IL-17A, and the activation of STAT3 and caspase-3 to promote apoptosis of memory T cells (60). Furthermore, lentiviral particles facilitated expression of mFasL has been shown to activate caspase-3 and caspase-9 and release Cyt c by binding to Fas on the surface of T cells to induce apoptosis (69). Moreover, a new type of recombinant fusion protein scFvCD7, developed on the pretext that CD7 targets sFasL that specifically binds to T cells, can selectively activate the Fas/FasL signaling pathways in Th1 and Treg cells to induce autoimmune T cell apoptosis and thus, possesses a promising therapeutic value for RA (70).
Imbalanced Regulation of Apoptosis in Osteoblasts/Chondrocytes and Osteoclasts Mediates Bone Destruction in RA
Many RA associated mechanisms lead to excessive apoptosis of osteoblasts or chondrocytes, and the antagonism of apoptosis demonstrated by osteoclasts contributes significantly to bone destruction in RA. Therefore, protecting osteoblasts or chondrocytes and promoting osteoclast apoptosis may serve as promising treatment strategies for RA. Incubation of human chondrocytes with the synovial fluid of RA leads to increased secretion of MCP-1 and MIF, which promotes chondrocyte apoptosis (71). The expression of FasL in activated peripheral blood mononuclear cells combined with Fas expressed on the human osteoblast cell line MG63 and primary osteoblasts has been shown to result in osteoblast apoptosis and induction of bone loss in RA (72). Furthermore, the chondrocyte apoptosis in mouse arthritis induced by anti-type II collagen antibodies and lipopolysaccharide (mAbs/LPS) has been shown to be potentially mediated by osteopontin. Additionally, the inhibition of osteopontin results in the downregulation of TNF-α induced up-regulation of caspase-3, while the overexpression of osteopontin reverses this effect (73). Excessive ROS generation in the synovial fluid or plasma of RA patient leads to protein oxidation resulting in the generation of advanced oxidation protein products, which function as pro-inflammatory mediators. These products have been reported to exert pro-apoptotic effects on chondrocytes in vitro via the RAGE-nicotinamide adenine dinucleotide phosphate (NADPH) oxidase/poly (ADP-ribose) polymerase-1 (PARP-1)-mediated redox-dependent pathway (74). TNF-α can inhibit the apoptosis-promoting effect of NO on chondrocytes through the NF-κB/COX2 pathway (75). Evidently, YS-51S, a synthetic isoquinoline alkaloid, inhibits the oxidation of peroxynitrite on Cyt c, the expression of inducible nitric oxide synthase (iNOS) and NO-mediated apoptosis of osteoblasts (76). Furthermore, the extracts of Emblica officinalis have been shown to induce osteoclast apoptosis and compete with NF-κB binding, resulting in decreased secretion of IL-6 (77). Additionally, fraxetin has been reported to inhibit the TNF-α and IL-1β mediated expression of Fas, upregulate the expression of FLIP, and inhibit caspase-3 and caspase-8 by inhibiting osteoblast apoptosis (78).
Decreased Apoptosis of Pro-Inflammatory Cell Subtypes Exacerbates the RA Associated Inflammation and Bone Destruction
It is well-established that there is an increase in the pro-inflammatory cell populations during RA. As has been discussed so far, increasing the sensitivity of these “harmful” cell populations to apoptosis can be helpful in alleviating RA. CD14 monocytes in the peripheral blood and synovial fluid of RA patients have been reported to be resistant to spontaneous apoptosis, which may be due to the increased expression of miR-155 resulting in the inhibition of apoptotic factors- caspase-10 and APAF1 (79). Moreover, the upregulation of the survival protein FLIP in macrophages has been shown to protect macrophages by inhibiting Fas-mediated apoptosis, thereby contributing to inflammatory diseases (80). Encouragingly, thalidomide has been shown to induce monocyte apoptosis through endogenous mitochondrial pathways by promoting the release of Cyt c, activating caspase-3, caspase-9, and NF-κB; and inhibiting Akt-1 activity thereby facilitating the treatment of RA (81). In addition to the effects of sex hormones on FLS, androgens have been documented to increase Bax, Fas, and PARP expression in the THP-1 mononuclear macrophage cells. Multiple studies have suggested that sex hormones can regulate apoptosis of macrophage-like cells implicating their potential therapeutic effect on RA (82, 83). Short-term treatment with 100-300 ng/ml Cyclosporin A, an immunosuppressant of RA, has been shown to induce apoptosis of THP-1 monocyte cells (84). Moreover, the LPS-induced expression of caspase recruitment domain protein 6 (CARD6) is found to be suppressed in macrophages which results in the augmented release of pro-inflammatory factors. In contrast, overexpression of CARD6 results in decreased expression of pro-inflammatory factors and chemokines in addition to inhibition of TNFR1/tumor necrosis factor receptor-associated factor-2 (TRAF2)/NF-κB signaling pathway, which alleviates not only the TNF-α induced inflammation and apoptosis of macrophages but also the level of bone destruction in CIA mice. Furthermore, it inhibits the activation of cleaved caspase-3, indicating that CARD6 plays an anti-inflammatory and anti-apoptotic role in RA (85). The high expression of SH3s may be involved in the protection and survival of synovial cells as well as stimulation of cell proliferation (86). Chronic inflammation in RA causes neutrophils to activate and release hydrogen peroxide along with the enzyme myeloperoxidase. These phenomena ultimately results in the formation of hypochlorous acid (HOCl), which is found to be elevated in RA synovial fluid. HOCl has been reported to induce apoptosis of human mesenchymal progenitor cells (MPCs) by increasing Bax-dependent mitochondrial permeability and the AIF-/EndoG- dependent pathway (87). Cell death and cartilage destruction in RA synovium are potential sources of mitochondrial antigen release that induce local antigen-driven IgG2/lambda B cell and inflammatory responses (88). The expression of β2-R in the lymphatic B cells of patients with RA has been documented to be significantly reduced as compared to that in healthy controls. The expression of β2-R has been reported to be negatively correlated with disease progression, which may be related to the lowered induction of cAMP and cell death (89). In addition, studies have shown that the expression of sphingosine 1-phosphate in the lymphoblastic B cell lines of RA patients increases through the G protein-coupled receptor-mediated activation of PI3K signaling, leading to the antagonism of Fas-mediated apoptosis (90).
Autophagy in the Pathophysiology of Rheumatoid Arthritis
Autophagy is a self-eating phenomenon responsible for the elimination of impaired organelles, abnormal and old nonfunctional proteins, and other intracellular substances via lysosomal degradation; for their recycle and efficient use in important cellular functions, especially in response to stressful physiological conditions (91). The detailed molecular mechanisms and characteristics of autophagy have been widely reviewed, and mainly involve the mTOR- mediated formation of autophagosomes and autophagolysosomes and the reutilization of resultant products (92, 93). The expression of autophagy associated proteins in the synovial tissues of RA patients has been reported to be remarkably increased (Beclin1, ATG5, LC3) and significantly correlated with the serum levels of inflammatory markers (CRP, ESR) and autoantibodies (cyclic citrullinated peptide, CCP; and rheumatoid factor, RF). Moreover, the anti-TNF-α therapy and IL-6R inhibitor treatment have been documented to result in decreased levels of autophagy (94, 95). Hydroxychloroquine and chloroquine, the commonly used therapeutic drugs for RA, have also been demonstrated to inhibit autophagy by preventing immune activation and cytokine production of various cells and regulating the expression of CD154 on T cells (96). Thus, the phenomenon of autophagy is closely related to the progression of RA. Accordingly we have primarily discussed the relationship between autophagy and various cell subtypes, including FLS, T cells, antigen-presenting cells, osteoblasts, osteoclasts, macrophages, and neutrophils (See Figure 2).
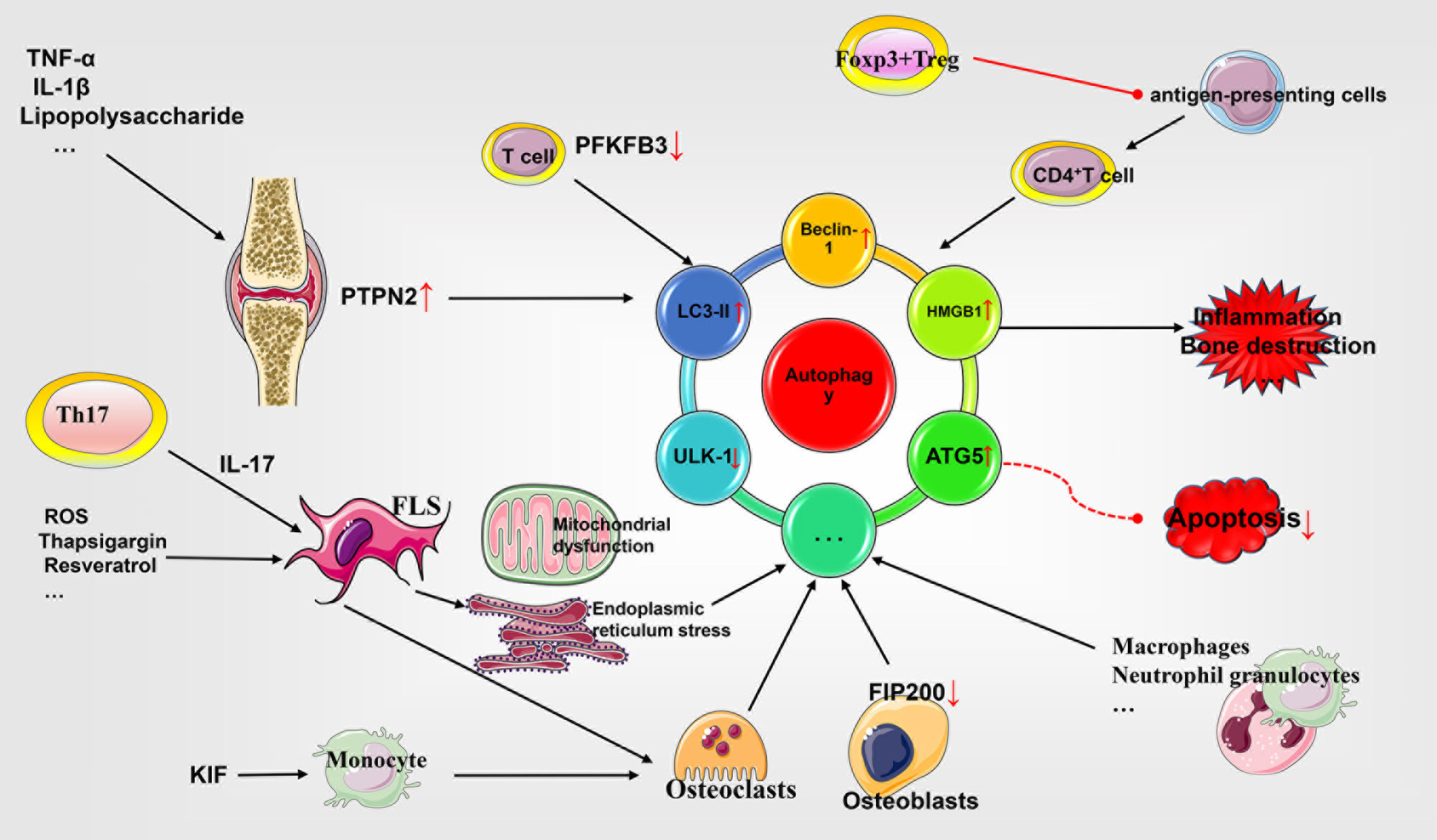
Figure 2 Role of autophagy in the pathophysiology of rheumatoid arthritis. Autophagy and apoptosis appear to be antagonistic to each other. FLS enhances autophagy by expressing different molecular patterns and antagonizes apoptosis to promote synovial cell proliferation. In addition, insufficient autophagy flux in T cells causes premature senescence and apoptosis. Autophagy of antigen-presenting cells is involved in inflammation. Autophagy of osteoblasts and osteoclasts affects the process of bone destruction. Other cells affect RA through different mechanisms. TNF, tumor necrosis factor; IL, interleukin; PTPN2, phosphatase nonreceptor type 2; ROS, reactive oxygen species; LC3, light chain 3; ULK-1, UNC51-like kinase 1; FLS, fibroblast-like synoviocytes; PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3; FIP200, focal adhesion kinase family interacting protein of 200 Kd; HMGB1, high mobility group box 1; ATG5, autophagy related 5.
Autophagy Inhibits the FLS Apoptosis and Enhances Synoviocytes Proliferation in RA
Autophagy in RA has a close association with apoptosis, wherein autophagy exhibits an antagonistic effect on apoptosis. Assessment of synovial tissues from patients with RA and osteoarthritis (OA) has revealed that while the apoptosis of RA synovial tissues was reduced, the expression of autophagy related proteins, Beclin-1 and LC3, were highly augmented. These findings, thus suggest a significant negative correlation between apoptosis and autophagy (97).Furthermore, studies have shown that TNF-α activates the NF-KB signaling in FLS and enhances autophagy, which in turn enhances the resistance of FLS to anti-TNF-α therapy for RA (98, 99). Similarly, FLS induces the formation of autophagosomes and resistance to methotrexate in RA by enhancing the expression of HMGB1 and Beclin-1 (100). Thus, various mechanisms promote the autophagy of FLS and antagonize cell apoptosis while also enhance the proliferation ability of FLS. The expression of protein tyrosine phosphatase non-receptor type 2 (PTPN2) in the synovial tissue of RA patients is higher than that in OA patients. Moreover, TNF-α, IL-1β or lipopolysaccharide have been reported to stimulate the upregulation of PTPN2, which aids in the prevention of apoptosis and increasing the level of autophagy in FLS. However, silencing of PTPN2 aggravates the production of IL-6, the mechanism underlying which requires further experimental elucidation (101). The upregulation of dynamin 1-like protein (DNM1L) has been shown to promote the survival of FLS. It also stimulates inflammation by increasing the expression of autophagy protein LC3B, ROS production, inhibiting cell apoptosis, and regulating AKT/IKK/NFKBIA/NF-Kb signaling (102). Furthermore, the IL-17 secreted by the Th17 cells have been reported to induce mitochondrial dysfunction and autophagy in FLS by activating STAT3, thereby antagonizing the apoptosis of FLS in RA (103, 104). Resveratrol has been demonstrated to reduces autophagy related proteins such as Beclin-1 and LC3A/B, thereby augmenting mitochondrial dysfunction as well as FLS apoptosis, and alleviating the symptoms of adjuvant-arthritis (AA) in rats (105). Knockdown of PADI4, a genetic susceptibility gene for RA, has been shown to inhibit FLS autophagy while promoting apoptosis (106). Exogenous administration of ROS to stimulate RA FLS expressing TLR4 receptors induces a vital state of oxidative stress state and promotes the release of high mobility group box 1(HMGB1) and autophagy, which has an antagonistic effect on apoptosis (107).The endoplasmic reticulum (ER) stress inducer thapsigargin reportedly results in lower expression of the ER stress protein CHOP in FLS and induces autophagy, through the formation of autophagosomes, Beclin-1 expression, and LC3-II transformation, to increase the resistance of FLS to cell death caused by ER stress, such as apoptosis in RA (108). In addition, thapsigargin promotes FLS cell death in a caspase-3 independent manner, accompanied by remarkable aggregation of p62-positive polyubiquitinated protein and reduced expression of autophagy-linked FYVE protein (AFLY); suggesting that autophagy may have a dual role in regulating FLS survival and proliferation in RA (109).
Autophagy Regulates T Cell Subsets in RA
The increased autophagy of FLS has been shown to antagonize apoptosis, while the decreased autophagy of T cells results in augmented apoptosis, leading to chronic T cell loss and lymphocytopenia, which are risk factors for RA. The expression of 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase 3 (PFKFB3) has been found to be reduced in RA associated T cells. Consequently, it is easily reprogrammed by metabolism and lack of energy and ROS and autophagy, leading to senescence and apoptosis. Overexpression of PFKFB3 has been reported to promote glycolytic flux and regulate autophagy, thereby preventing excessive T cell apoptosis (110–112). In addition, autophagy may be attributed to the conference of memory to the inflammatory CD4+ T cells, which leads to continuous recognition and activation of autoantigens that promotes inflammation in RA. Furthermore, it has been shown that inhibition of MYC may serve as a putative mechanism for the augmentation of autophagy in RA (113). Simultaneously, the increase in the level of autophagy has been demonstrated to inhibit the apoptosis of inflammatory CD4+ T cells and prolong cell survival time, further facilitating RA progression (114).
Autophagy Increases Auto-Antigen Presentation by Antigen-Presenting Cells
Various studies support the notion that autophagy is involved in the process of autoantigen presentation by APC cells to CD4+ T cells. For instance, starvation of B cells results in the production of citrullinated peptides, which is also accompanied by an increase in the expression of autophagy protein ATG5. Furthermore, the treatment of B cells by autophagy inhibitor abrogates the presentation of citrullinated peptides. Nevertheless, it does not affect other unmodified peptides (115). Another study has shown that macrophages, dendritic cells, and B cells undergo autophagy to produce citrullinated peptides after the antigen contacts the autophagic vesicle (116). Further, it has been reported that induction of FLS autophagy in vitro is accompanied by peptidyl arginine deiminase 4 activation and protein citrullination. This was further corroborated by a significant positive correlation between autophagy and anti-CCP levels in RA patients (117). Additionally, Foxp3+ Treg cells have been reported to inhibit the autophagy as well as the autoimmune response of dendritic cells in a cytotoxic T-lymphocyte-associated protein 4-dependent (CTLA4) dependent manner. The underlying mechanism for this phenomena possibly involves the interaction between CTLA4 and PI3K/AKT/mTOR axis, which leads to reduced autophagy levels (118). In addition, studies have shown that autophagy may be involved in the carbamylation of FLS proteins, a non-enzymatic post-translational modification mechanism, which results in their accumulation and subsequent participation in the pathogenesis of RA. However, the specific mechanism underlying this phenomenon is not yet clear (119).
Regulation of Autophagy in Osteoblasts and Osteoclasts in RA
The autophagy defect of osteoblasts leads to suppression of bone formation. It has been reported that defective focal adhesion kinase family interacting protein of 200 kDa (FIP200) in osteoblasts results in insufficient autophagy, accompanied by abnormal expression of p62, defects in autophagy flux, and ultimately lower bone mass (120). Similarly, defects in ATG7, the key autophagy protein in osteoblasts, have been documented to decrease osteoblast formation, matrix mineralization, and secretion of osteoprotegerin TNFRSF11B/OPG; and augment osteoclast population, secretion of TNFSF11/RANKL, and endoplasmic reticulum stress, to promote apoptosis (121). The increased autophagy in osteoclasts leads to enhanced production, which mediates bone destruction. Remarkably, the defects of autophagy-related proteins have been shown to facilitate prevention of bones and joints destruction. For instance, TNF-α has been reported to stimulate the expression of osteoclast autophagy proteins Beclin1, and ATG7; activate autophagy; and regulate osteoclast differentiation, and bone resorption in RA (122, 123). The expression of the autophagy receptor optineurin in FLS has been documented to be decreased under the action of a variety of inflammatory factors (e.g., TNF-α). Subsequently, it reportedly stimulates the expression of RANKL and the differentiation of osteoclasts (124). Kruppel-like factor 2 (KIF) negatively regulates the expression of the autophagy protein Beclin1 in monocytes and induces osteoclast differentiation and autophagy levels to enhance joint inflammation and bone destruction (125). Deficiency of the autophagy protein Afly leads to the up-regulation of TRAF6 in osteoclasts and regulates RANKL-induced osteoclastogenesis (126).
Autophagy Regulates Other Cells Associated With RA and Targeting Autophagy Significantly Alleviates RA
RA associated macrophages highly express SNAPIN, mainly to maintain autophagy and healthy lysosome function and prevent proton leakage from the lysosomes (127). A variety of cell surface proteins such as CD244 bind to autophagy proteins such as Vps34 and Beclin-1, to facilitate inhibition of autophagy. The down-regulation of CD244 has been shown to augment autophagy, which is associated with the severity and prognosis of RA (128). The concentrations of IL-6, IL-8, IL-10, and MCP-1 in RA synovial fluid are reportedly elevated and mediate the autophagy of neutrophils through their corresponding cytokine receptors (129). The autophagy level in the peripheral blood mononuclear cells has been reported to be elevated in RA with concomitant decrease in apoptosis. Moreover, anti-TNF-α therapy has been documented to decrease the levels of autophagy protein LC3-II, which also correlates with the DAS28 score, thereby resulting in augmented apoptosis (130).
Many studies have shown that targeting autophagy can significantly improve the prognosis of RA through a variety of mechanisms. For instance, celastrol has been shown to inhibit sarcoplasmic/endoplasmic reticulum calcium ATPase pump (SERCA) induced autophagy by regulating the Ca2+/calmodulin-dependent kinase kinase-β (CaMKKβ) -AMP-activated protein kinase (AMPK) -mTOR pathway. Moreover, such induction of autophagy is accompanied by the alleviation of arthritis symptoms in rats with adjuvant-induced arthritis (AIA) (131). The autophagy-associated protein, microtubule-associated protein light chain 3b (LC3b) has been reported to be upregulated, and while UNC51-like kinase 1 (ULK-1) is downregulated in experimental arthritis mice. In contrast, the extract of tomorou, an indigenous herb of Hunza-Nagar Valley, Pakistan has been reported to inhibit LC3B protein by up-regulating caspase-3. Furthermore, its water and ethyl acetate extracts also result in the normalization of abnormal ULK-1 protein levels, thereby alleviating arthritis symptoms (132).
It is worth noting that targeting AKT-related pathways to regulate autophagy is also a potential direction for the treatment of RA. For instance, dexamethasone has been reported to induce autophagy by increasing the ROS levels in human chondrocytes by regulating the AKT/FOXO3 signal pathway to inhibit apoptosis (133). Furthermore, tangeretin and 5-hydroxy-6,7,8,3’,4’-pentamethoxyflavone have also been shown to inhibit autophagy by regulating ROS levels in synovial cells and AKT/mTOR signaling, thereby inhibiting the pathological changes associated with bovine type II collagen-induced arthritis in animals (134). Moreover, astragalus polysaccharides have been shown to regulate FLS autophagy and enhance the expression of pro-apoptotic proteins such as Bax and Caspase-3, through PI3K/AKT/mTOR signalling to promote apoptosis of FLS and improve RA prognosis (135). Remarkably, the combination therapy of pterostilbene and physical exercise has been documented to inhibit the proliferation and IL-1 stimulated apoptosis of rat synovial cells through the PI3K/AKT/NF-KB signaling pathway and enhance the level of autophagy to alleviate the symptoms of experimental arthritis in rats (136). Similarly, autophagy inhibitors have also been demonstrated to directly reduce the inflammatory response of experimental arthritis rats, inhibit the proliferation of FLS through the PI3K/AKT pathway, and promote their apoptosis (137).
In subsequent sections, we predominantly discuss three types of PCD: necroptosis, pyroptosis and NETosis. Notably, the research related to these three types of PCD in the context of RA is not as advanced as in case of apoptosis and autophagy as discussed in previous sections. Hence, further sophisticated clinical pre-experiments and clinical trials are essential to understand the in-depth role of PCD in RA prognosis.
NETosis as a Source of Citrullinated Autoantigens and an Inflammatory Driver in Rheumatoid Arthritis
Neutrophils are white blood cells that mature in the bone marrow and are released into the circulation. They are characterized by a granular cytoplasm and lobulated nuclei. Neutrophils in healthy humans have a short lifespan. Instigation with a variety of anti-inflammatory or pro-inflammatory signals triggers the chemotaxis and accumulation of neutrophils at the inflammation site under the influence of a variety of cytokines. This ultimately results in the occurrence of apoptosis that participates in the process of inflammation subsidence (138, 139). An important physiological defense mechanism of neutrophils is the production of neutrophil extracellular traps (NETs), which are extracellular network-like structures that contain DNA, histones, neutrophil proteins (alarmins), myeloperoxidase (MPO), neutrophil elastase (NE), and cathepsin G and are designed to eliminate harmful pathogens (138, 140, 141). For instance, neutrophils themselves participate in the enhanced citrullination of histones by expressing the peptidylarginine deiminase (PAD)2/PAD4 enzymes, whereas NADPH oxidase (NOX) is involved in the generation of ROS, and a variety of serines contribute to chromatin remodeling. The process also involves the participation of proteases, including neutrophil elastase and cathepsin G, thereby resulting in NETosis which releases an array of inflammatory mediators to drive RA progression (142–144).
The susceptibility factors in patients with RA include microorganisms associated with periodontitis and smoking (145). Among them, P. gingivalis has been reported to express PAD enzyme homologues to induce the production of citrullinated antigens in patients with periodontitis, which enhance NETosis, further promoting RA. A remarkable reduction in the serum levels of carbamylated protein (CarP) and NETs post periodontal treatment, further substantiates the possible association between RA and periodontitis (146–149). In addition, studies have shown that in the lungs of smokers, PAD2 and PAD4 expressed by neutrophils can citrullinate the cathelicidin human cationic antimicrobial protein-18 (chcap-18) that can be potentially externalized by NETosis to promote RA (146, 148, 150, 151). Some studies have identified that the production of some extracellular citrullinated antigens in RA may be associated with the inflammatory process of NETosis in neutrophils. The active PAD2/4 enzyme is evidently released into the synovial fluid of RA patients to promote the generation of autoantibodies, which results in inflammation. However, the phenomenon has no association with the apoptosis process of neutrophils (152, 153). Activated neutrophils are significantly elevated in synovial fluid of RA patients. Studies suggest that they might be involved in responding to the synergistic effect of the immune complex and complement system; resulting in excessive NETosis formation and release of a variety of inflammatory mediators to mediate inflammation and joint destruction (148, 154, 155). The spontaneous increase of neutrophil population in RA patients is associated with increased ROS production, MPO expression, and citrullination mediated by PAD4 activity. The NETosis derivatives including plasma cell-free nucleosomes, MPO, neutrophil elastase, and cathepsin G are closely related to the severity of RA. Studies have shown that anti-TNF-α and anti-IL-6R treatments result in decreased formation of NETs and are accompanied by reduction of inflammation and serum markers, endothelial dysfunction, and suppression of immune cell activation (156, 157). The MPO-DNA complex in the serum of patients with RA is reported to be markedly higher than that in the healthy control group. Its levels are associated with the levels of anti-citrullinated protein antibodies (ACPA) and RF, and thus may prove be useful as a supplementary biomarker for clinical assessment of RA (158). The enhanced NETosis observed in the circulation and RA synovial fluid is related to ACPA and systemic inflammation markers. The serum and immunoglobulin components of RA patients with high levels of ACPA and RF evidently demonstrate enhanced NETosis. Furthermore, IL-8, IL-17A, and TNF-α have been shown to induce NETosis. In turn, NETosis containing citrullinated peptides is internalized and taken up by FLS through the RAGE-TLR9 pathway thereby upregulating the expression of MHC class II (MHCII). The MHCII molecule is then presented to CD4+ T cells, which strengthens the autoimmune B cell response and the production of ACPA. These events ultimately result in the induction of IL-6, IL-8, in addition to a variety of other chemokines and adhesion factors, to promote the formation of a vicious circle of inflammation (148, 156, 159, 160). Wright et al. have shown that low-density granulocytes (LDG) in RA exhibit lower TNFR1/2 mRNA and protein expression, indicating the presence of an immature neutrophil population in the peripheral blood, which may ultimately result in poor response to anti-TNF-α therapy (161). In addition, the level of calprotectin in RA has been shown to be elevated in the systemic and synovial fluid. However, their levels are markedly decreased with effective treatment. As a result, calprotectin can serve as a significant predictor for the progression of joint destruction and drug efficacy in RA (162). Anti-CCP, IgA, IgM, and RF-positive RA patients exhibit higher calprotectin levels as compared to the control groups that correlates with the baseline levels of inflammation markers, CRP, ESR, and anti-CCP (163). Consistently, a study by Bach et al. has demonstrated that the levels of calprotectin and NETs of neutrophils are markedly increased in RA patients, which represents transitional activation and cell death, respectively. The study further revealed that the calprotectin levels were significantly related to disease severity, and the levels of ACPA cooperatively enhanced the prediction of bone erosion and disability in patients with RA. In addition, NETosis is significantly associated with inflammatory disease markers and thus, can be used as a predictor of nodule development in patients with RA (164). Zhu et al. have shown that emodin, a natural anthraquinone derivative, can (1) reduce neutrophil infiltration and the release of pro-inflammatory factors, such as IL-6, IFN-γ, and TNF-α; (2) promote the expression of pro-apoptotic proteins, such as caspase-3 and Bax; and (3) inhibit the expression of anti-apoptotic protein Bcl-2 for promoting neutrophil apoptosis and inhibiting Atg5, LC3B, Beclin-1, and other autophagy protein levels by restraining autophagy and NETosis, which alleviates the symptoms RA in the mouse model of adjuvant-induced arthritis (AA) (165). Felty syndrome, a severe form of RA, is characterized by the presence of a large amount of anti-histone autoantibodies, which may be derived from neutrophil NETosis. Moreover, studies have shown that some circulating autoantibodies in Felty syndrome preferentially target PAD4-deaminated histones, such as H3, H4, H2A, and bind to activated neutrophils and NETs, further clarifying the role of NETosis in RA (166) (See Figure 3).
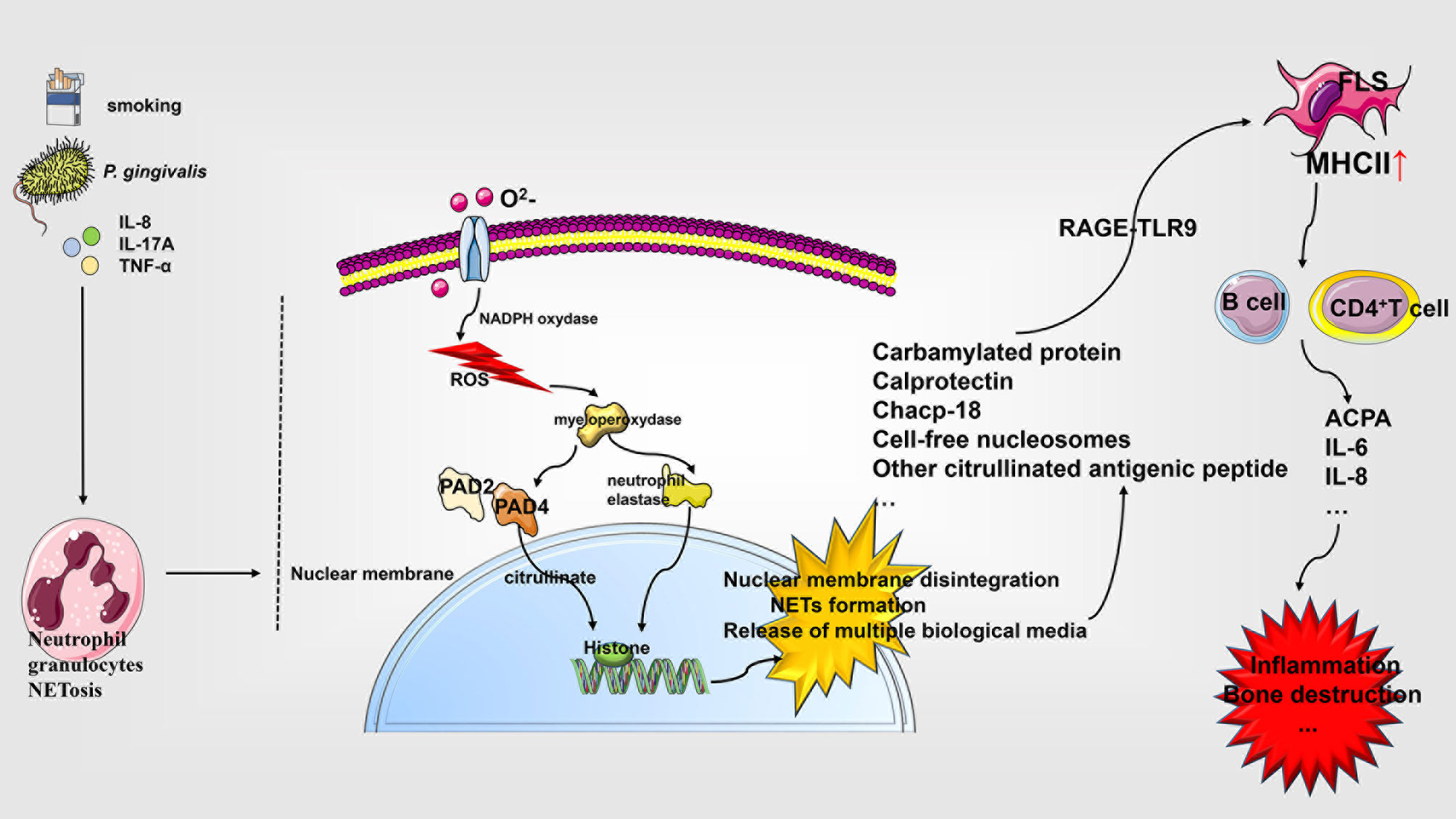
Figure 3 Role of NETosis in the pathophysiology of rheumatoid arthritis. Neutrophils respond to diverse stimuli for developing NETosis, which starts with the NADPH oxidase complex and promotes ROS production. ROS activate the hydrolytic activity of PAD4 and neutrophil elastase in an MPO-dependent manner, cleaving the histones over-citrullinated by PAD4. This triggers a series of events, including the rupture of the nuclear membrane, the formation of NETs, and the release of various biological agents, which interact with a variety of cells affecting RA progression. IL, interleukin; PAD, peptidylarginine deiminase; chcap-18, cathelicidin human cationic antimicrobial protein-18; NETs, neutrophil extracellular traps; NADPH, nicotinamide adenine dinucleotide phosphate; TLR, Toll-like receptor; MHCII, MHC class II; ACPA, anti-citrullinated protein antibodies; FLS, fibroblast-like synoviocytes; ROS, reactive oxygen species.
Necroptosis in the Pathophysiology of Rheumatoid Arthritis
Necroptosis is essentially a PCD mediated by receptor-interacting protein kinase-1 (RIPK1), RIPK3, mixed lineage kinase-like (MLKL). The interaction between RIPK1 and RIPK3 is related to the crosstalk between necroptosis and apoptosis. Its morphological characteristics have been widely reviewed, and involve the release of cellular contents, ultimately mediating inflammation in RA (167–170). Neutrophils in the joints of RA patients activate RIPK1, RIPK3, and MLKL under the influence of CD44 and granulocyte-macrophage colony-stimulating factor (GM-CSF) to cause necroptosis, which can be blocked through the application of fibroblast activation protein-α (FAP-α) suggesting its importance as an potent drug for RA treatment (171). The expression of 14-3-3η can be detected in the synovium and serum of RA patients and is closely related to disease severity and the level of anti-cyclic citrullinated peptide antibodies. Macrophages in the RA synovium respond to TNF-α stimulation, and p-MLKL significantly increases the release of 14-3-3η in the induction of necroptosis (172). A spontaneous RA study involving a non-human primate (NHP) model showed that RIPK1 binds to voltage-dependent anion-selective channel 1 (VDAC1) in the heart causing its enhanced oligomerization. This ultimately increases the death and impairment of cardiomyocytes thereby resulting in aggravated RA-related heart function abnormalities (173). In addition, studies with articular cartilage in rats with AA and in vitro chondrocytes have shown an acid-sensitive ion channel (ASIC)-1a-mediated increase in the expression of RIPK1, RIPK3, and P-MLKL. Moreover, treatment with necrostatin (NST)-1s, a RIPK1 inhibitor, has been shown to reduce joint damage and inflammation in AA rats (174). Similarly, NST-1s can inhibit the expression of necroptosis-related molecules (RIPK1, RIPK3, and MLKL) in mice with experimental CIA arthritis, thereby reducing the population of osteoclasts, Th1, and Th17 cells; and augmenting the population of Th2 and Treg cells (175). Furthermore, the lack of interferon γ (IFN- γ) has also been reported to promote the expression of RIPK1, RIPK3 and MLKL in CIA mice, increase the number of Th17 cells, promote the release of IL17 and TNF-α, and activate STAT3, thereby aggravating inflammation and joint damage in RA (176).
Apoptosis and necroptosis are associated with each other in the progression of RA. Studies have shown that second mitochondria-derived activator of caspases (SMAC) mimetics (SMs) can inhibit the anti-apoptotic effect of Smac, which is a member of the IAP family protein, by competing with CIAP1/2 protein to induce the activation of caspase-3, caspase-7, caspase-8, caspase-9, and Fas; thereby facilitating the release of TNF-α; and in vitro activation of RIPK1, RIPK3, and MLKL in pro-inflammatory M1 macrophages to activate apoptosis and necroptosis. Blockade of the IAP and caspase pathways, facilitates M2c phenotype induction by SMs, resulting in necroptosis of M0 macrophages (177). Compared with etanercept, a TNF-α inhibitor, geldanamycin inhibits the RIPK1 and NF-KB activity while augmenting the activation of caspase-8 to promote MH7A cell apoptosis, and inhibit TNF-α-induced necroptosis. Thus, geldanamycin demonstrably results in better clinical outcomes and lowered synovial hyperplasia in CIA mouse model (178). A multicenter, randomized, double-blind, placebo-controlled clinical trial has evaluated the effect of the RIPK1 inhibitor GSK2982772 in RA. The study revealed that as compared with the placebo group, the inhibiter administered group demonstrated no significant differences in certain common clinical indicators and disease severity, such as DAS28-CRP and ACR20/50/70, (NCT02858492) (179) (See Figure 4).
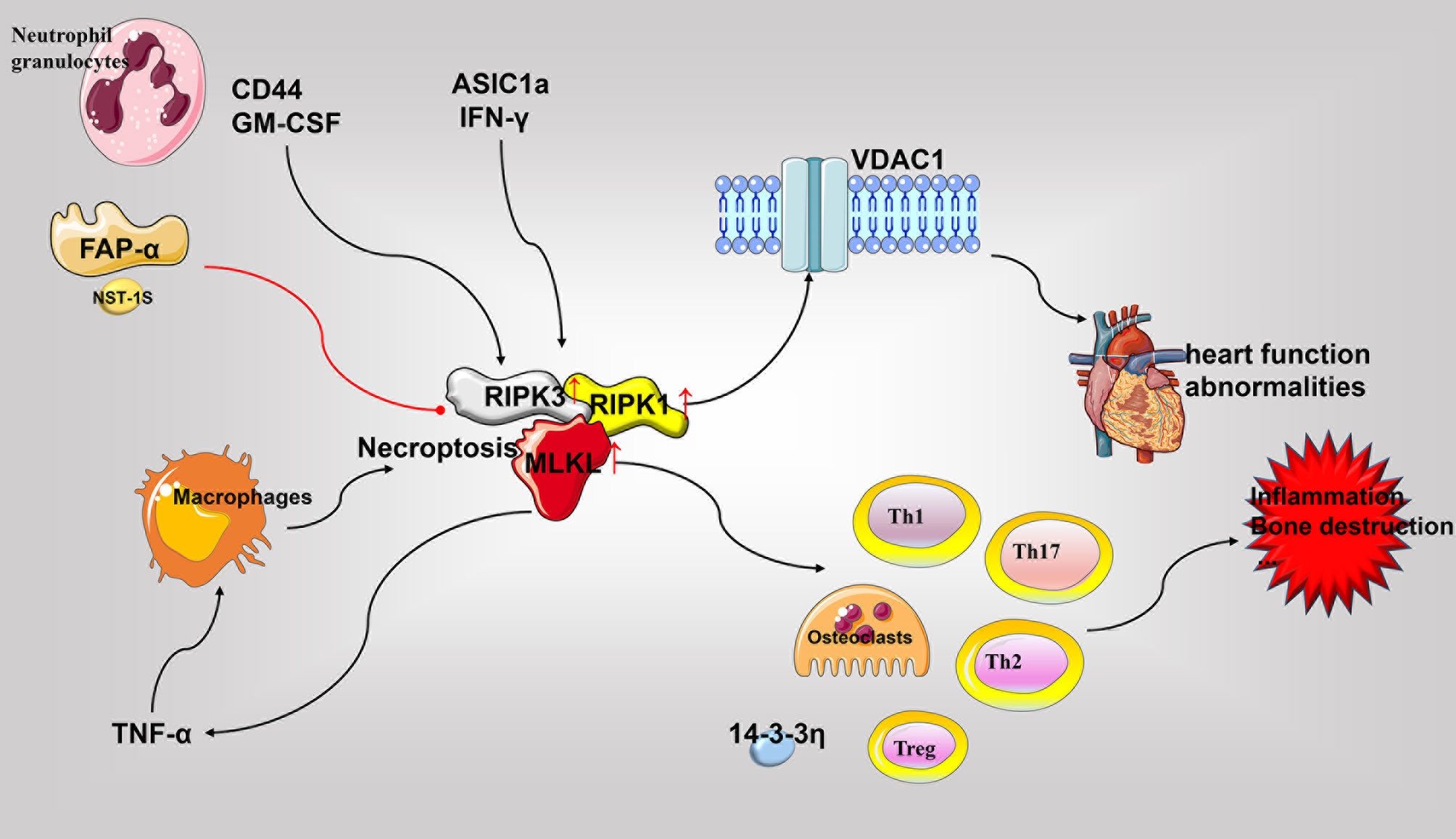
Figure 4 Role of necroptosis in the pathophysiology of rheumatoid arthritis. The critical molecules of necroptosis, RIPK1, RIPK3, and MLKL, are elevated in RA. Various cell types undergo necroptosis in response to diverse stimuli and release various pro-inflammatory mediators to promote inflammation, bone destruction, and other RA associated pathological processes. Necroptosis in cardiomyocytes may cause RA-related heart damage. Accordingly, inhibition of RIPK1 and other necroptotic molecules may be beneficial for disease treatment. GM-CSF, granulocyte-macrophage colony-stimulating factor; RIPK, receptor-interacting protein kinase; MLKL, pseudokinase mixed lineage kinase domain-like; TNF, tumor necrosis factor; VDAC1, voltage-dependent anion-selective channel 1.
Pyroptosis in the Pathophysiology of Rheumatoid Arthritis
Pyroptosis is a type of pro-inflammatory PCD. Its morphological characteristics include membrane rupture and the release of cellular contents. Pyroptosis is mainly composed of ASC, caspase-1, and other proteins of the NLRP3 inflammasome, GSDMD, IL-1β, and IL-18. The characteristic purpose of inflammasome assembly is to resist pathogens that are harmful to the body and maintain homeostasis. However, when NLRP3 inflammasomes and pyroptosis are over-activated, the released inflammatory mediators can cause inflammation and damaging reactions. The role of inflammasomes in a variety of autoimmune inflammatory conditions has been widely reviewed. Here, we essentially discuss the connection of inflammasomes and pyroptosis with respect to prominent cell types known to play significant roles in the pathogenesis of RA including monocytes-macrophages, chondrocytes, CD4+ T cells, and FLS (180–183).
Elevated IL-1β and IL-18 levels are detected in the serum and synovial fluid of patients with RA (184). In RA synovium, transforming growth factor (TGF) β-1 reverses the activation of succinate dehydrogenase to increase succinate accumulation. This facilitates the assembly of NLRP3 inflammasomes via HIF-1α to activate pyroptosis for releasing IL-1β and IL-18 (185). Further, IL-18 reportedly induces inflammation by activating IFN-γ. Moreover, the expression of IL-18 is closely associated with the inflammation of synovial tissue in RA patients, and involved in the promotion of monocyte chemotaxis in addition to angiogenesis, thereby playing an important role in the induction of RA in experimental arthritis mouse models (183). Notably, TLR3 and TLR4 stimulation of patients with active RA has been documented to increase the levels of NLRP3, pyroptosis, and IL-1β released from whole blood cells (186), This is consistent with previous reports demonstrating that RA associated monocytes can release IL-1β through NLRP3-mediated pyroptosis (187). Furthermore, the expression of ASC, NLRP3-FL, NLRP-SL, and CASP1 in the monocytes derived from RA patients as well as the serum levels of caspase-1 and IL-18 have been found to be elevated in RA patients as compared to the control group. Moreover, the SNPs of NLRP3 was also identified to be involved in determining the susceptibility to RA and resistance to TNF-α therapy (188). C1q in the serum of RA patients has been shown to promote the binding of pentaxin 3 (PTX3) to CD14 monocytes, resulting in the lysis of gasdermin D (GSDMD), activation of NLRP3 inflammasomes, swelling of cells into bullae, and ultimate release of caspase-1, TNF-α, IL -6, and IL-1β concurrently to promote pyroptosis (189). In addition, studies have found that M1 macrophages and pyroptosis in the CIA model are the main sources of inflammatory factors. Punicalagin (PUN) is an active substance extracted from pomegranate peel. Its treatment has been documented to transform macrophages into anti-inflammatory phenotypes to reduce the release of inducible nitric oxide synthase (iNOS), increase the levels of IL-10 and other anti-inflammatory mediators for inhibiting pyroptosis, and facilitate the NLRP3 -mediated release of IL-1β and IL-18 in addition to caspase-1, ultimately resulting in the inhibition of inflammation (190). The A20/TNFAIP3 mouse model has been reported to develop spontaneous erosive polyarthritis, in which A20-deficient macrophages exhibit enhanced NLRP3, caspase-1, IL-1β, and pyroptosis. Furthermore, the absence of NLRP3, caspase-1, and IL-1R notably inhibits the inflammation and joint destruction in A20/TNFAIP3 mice (191). Similarly, IL-1β gene-deficient mice are completely protected from chronic inflammation-mediated joint destruction and damage (192). The frequency of caspase-1 activation by CD4+ T cells in patients with RA is higher than that in healthy controls. Defects in MRE11A, the DNA repair nuclease, has been reported to cause CD4+ T cell mitochondrial dysfunction and the leakage of mitochondrial DNA (mtDNA), leading to the assembly of NLRP3 inflammasomes, activation of caspase-1, and pyroptosis. Moreover, the overexpression of MRE11A reverses these effects associated with defective MRE11A. Thus, MRE11A participates in RA associated inflammation by mediating mitochondrial homeostasis and pyroptosis (193). ASIC1a not only participates in the regulation of necroptosis in RA but also mediates pyroptosis in AA model of chondrocytes by promoting the assembly of Ca2+-related NLRP3 inflammasomes, the expression of caspase-1, and the release of IL-1β and IL-18 (194). The primary rat articular chondrocytes acidified in vitro demonstrate increased ASIC1a, calpain-2, and calcineurin, NLRP3, caspase-1 expression, and pyroptosis. In contrast, the inhibition of ASIC1a, calpain-2, and calcineurin reduces pyroptosis, IL-1β, and other inflammatory mediators (195). Furthermore, the overexpression of miR-20a has been documented to inhibit FLS pyroptosis in the AA model by downregulating the expression of TXNIP, resulting in reduced levels of NLRP3, ASC, caspase-1, and IL-1β (196) (See Figure 5).
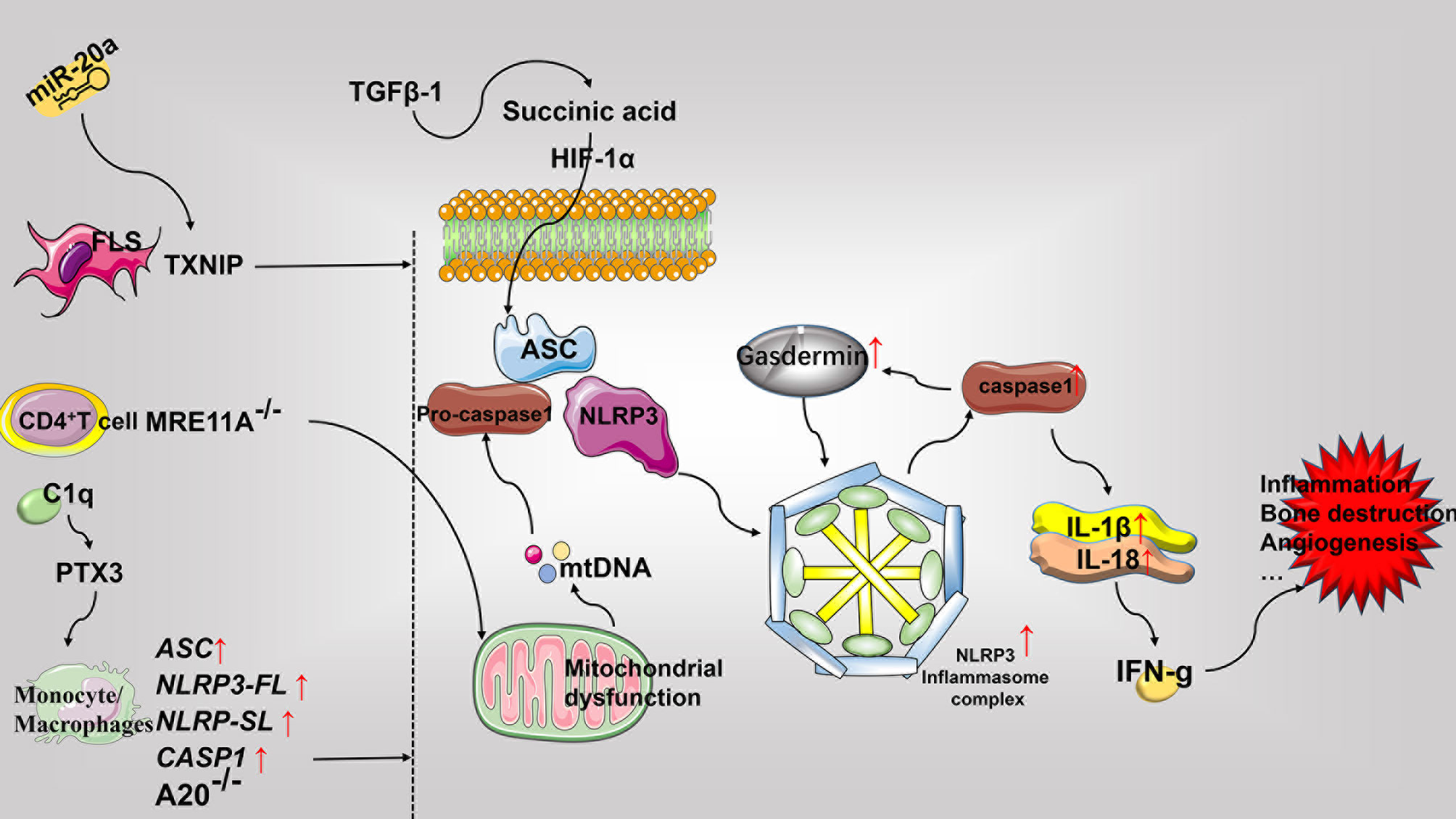
Figure 5 Role of pyroptosis in the pathophysiology of rheumatoid arthritis. Pyroptosis mainly involves the assembly of the NLRP3 inflammasome and the release of the pro-inflammatory mediators including IL-1β and IL-18. Most pyroptosis-related molecules are elevated in RA. Predominantly, the FLS, CD4+T cells, and monocytes-macrophages express different molecular patterns and promote pyroptosis, inflammation, bone destruction, and angiogenesis, that ultimately worsens exacerbates RA. FLS, fibroblast-like synoviocytes; PTX3, pentaxin 3; GSDMD, gasdermin D; mtDNA, mitochondrial DNA; TGF, transforming growth factor; HIF-1α, hypoxia-inducible factor-1α.
Conclusion
In a healthy organism, cell death is important for the proper maintenance of the balance between the body and tissue cells. When any part of the pathway becomes defective, the balance between survival and death of various cells is disrupted, which results in manifestations of abnormal patho-physiological conditions. Several studies have shown that the imbalance of PCD in promoting the chronic inflammation of the synovial membrane, joint erosion, and destruction, as well as angiogenesis plays an important role in promoting RA. In this review, we predominantly discuss the relationship between a variety of PCD and RA pathogenesis. We have first discussed upon how the insufficient apoptosis of FLS leads to abnormal proliferation of synovium, autoimmune T cells, B cells, inflammatory monocytes, and inflammation. Further, we have deliberated over the irregular apoptotic pattern of autoimmune T cells, B cells, inflammatory monocytes, inflammatory macrophages, and osteoclasts, which results in prolonged cell survival, thereby facilitating continuous presentation of autoantigens, the release of inflammatory factors, and the destruction of bones and joints. The excessive apoptosis of osteoblasts leads to a decrease in bone formation. Secondly, we reviewed the role of autophagy in the progression of RA in the context of various cell types involved in the process. The autophagy of FLS antagonizes apoptosis and contributes to the abnormal proliferation of synovium. Furthermore, the autophagy of a variety of APC cells enhances the presentation of autoantigens and the activation of CD4+ T cells. Moreover, the abnormal autophagy patterns of osteoblasts and osteoclasts are attributable to the low bone mass and increased bone resorption in RA, respectively. Additionally, the autophagy of other cells has different degrees of contribution to the pathological process of RA. Finally, we have described the role of the three forms of PCD, namely, NETosis, Necroptosis, and Pyroptosis in the pathogenesis of RA. NETosis of neutrophils is mainly involved in the formation of citrullinated proteins in RA. Necroptosis of a variety of cells primarily leads to the release of cell contents, which further promotes chronic inflammation. In addition, pyroptosis of various cells types mainly releases pro-inflammatory cytokines (IL-18 and IL-1β) and leading to the formation of inflammasomes that mediate RA associated chronic inflammation. Exhaustive literature mining reveals that multiple PCDs involved in the pathogenesis of RA communicate with each other and cooperatively promote the multiple pathological processes associated with RA. Rapid progress in the field of cell death research in the last decade, has led to the development of various experimental methods and technologies that can be helpful in detecting the link between PCD-related molecules and RA. The various studies in the field have achieved certain degree of progress, which has resulted in the identification of some biomarkers associated with the pathogenesis of RA (Table 1).In conclusion, in this review, we have summarized a variety of abnormal PCD and their mechanisms in RA in addition to provided corresponding clinical strategies for their application (Table 2). With the development and application of various PCD inhibitors and agonists, researchers can conduct an in-depth examination of the role of various PCDs in RA progression. Thus, future research directions will be based on the efficacy of preclinical experiments, which should essentially involve comprehensive experimental programs devoted to the application of multiple agonists and inhibitors for validating the effects of various PCD-related molecules on different cell types associated with RA. The immense potential attributed to this line of research is based on the complexity of RA and PCD mechanisms which can facilitate the expansion of existing repertoire of promising drug candidates for the development of effective RA drugs. The ultimate purpose of designing such treatment strategies is to rebalance the abnormal cell survival and death to combat the consequences of excessive or insufficient PCD in the various cell types involved in RA progression.

Table 1 Rheumatoid arthritis related biomarkers of programmed cell death.
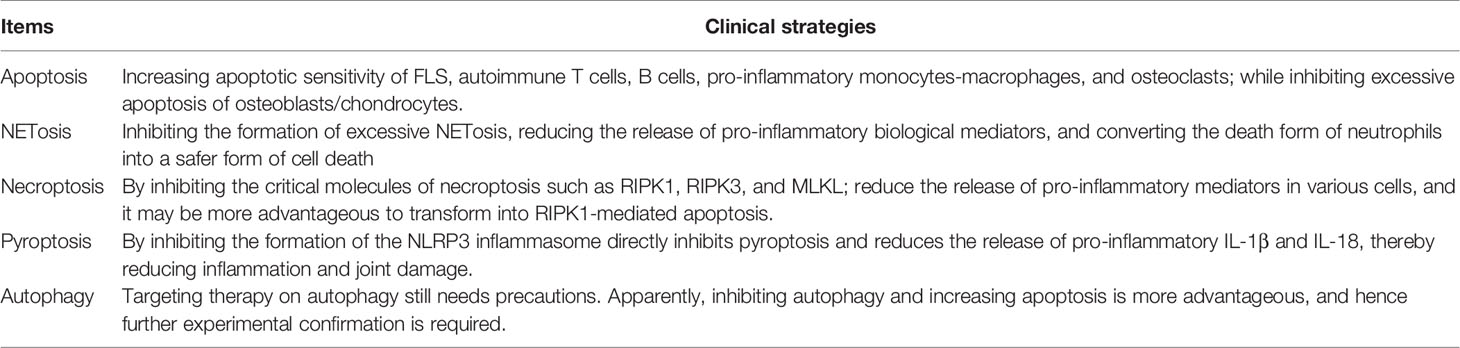
Table 2 Rheumatoid arthritis related clinical strategies for targeting programmed cell death.
Author Contributions
JZ is responsible for the collection of data, collation, and writing of the original manuscript. PJ is responsible for the organization of the original manuscript. SG, SS, and DH are responsible for the concept development, revision, and review of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the National Natural Science Funds of China (81774114), Shanghai Chinese Medicine Development Office, Shanghai Chinese and Western Medicine Clinical Pilot Project (ZY(2018-2020)-FWTX-1010), Shanghai Chinese Medicine Development Office, Shanghai Traditional Chinese Medicine Specialty Alliance Project (ZY(2018-2020)-FWTX-4017), National Administration of Traditional Chinese Medicine, and Regional Chinese Medicine (Specialist) Diagnosis and Treatment Center Construction Project-Rheumatology.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
RA, rheumatoid arthritis; PCD, programmed cell death; AICD, activation-induced cell death; FLS, fibroblasts like synoviocytes; Bcl-2, B cell lymphoma/leukemia-2; Bax, accumulation of Bcl-2–associated X protein; Bak, Bcl-2 homologous antagonist/killer; Cyt c, cytochrome c; TNF, tumor necrosis factor; TNFR1, tumor necrosis factor receptor 1; STAT3, activators of transcription 3; HSP70, heat-shock-protein-70; PBL, peripheral blood lymphocyte; PADI4, peptidylarginine deiminase IV; NSAIDs, non-steroidal anti-inflammatory drugs; PPAR gamma, peroxisome proliferator-activated receptor gamma; IκB, the intracellular calcium, phosphorylated I-kappa-B; IKK, the intracellular calcium, phosphorylated I-kappa-B (IκB) kinase; NF-κB, nuclear factor kappa-light-chain enhancer of activated B cells; AA, adjuvant arthritis; TIPE2, TNF-alpha-induced protein-8-like-2; DR5, Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) receptor 2; MCL-1, myelogenous cell leukemia-1; Bim, Bcl-2 interacting mediator of cell death; SYVN1, the E3 ubiquitin ligase synoviolin; IRE1, inositol-requiring enzyme 1; CIA, collagen-induced arthritis; sFas, soluble Fas; mFas, membrane-bound Fas; ERK, extracellular signal-regulated kinase; PI3K, phosphatidylinositol 3-kinase; MMP, matrix metalloproteinase; IPT, imperatorin; TIMP-3, tissue inhibitor of metalloproteinases-3; CIP2A, cancerous inhibitor of protein phosphatase 2A; PDGF, platelet-derived growth factor; HIF-1α, hypoxia-inducible factor-1α; BAFF, B-cell activating factor; ASIC3, acid-sensing ion channel 3; TRPV1, transient receptor potential vanilloid 1; ROS, reactive oxygen species; NO, nitric oxide; TRPM8, transient receptor potential melastatin subtype 8; POGel, prolonged O2/Ca2+-supporting phototherapy hydrogel; CHOP, EBP-homologous protein; GA, Geldanamycin; PDCD5, programmed cell death 5; ALLN, calpain inhibitor 1; CBP, CREB binding protein; IAPs, inhibitors of apoptosis proteins; DC, dendritic cells; IL, Interleukin; RANKL, receptor activator of nuclear factor (NF)-κB ligand; TRAIL-R2, TNF-related apoptosis-inducing ligand-receptor 2; NADPH, nicotinamide adenine dinucleotide phosphate; PARP-1, poly (ADP-ribose) polymerase-1; CARD6, caspase recruitment domain protein 6; TRAF2, tumor necrosis factor receptor -associated factor-2; HOCl, hypochlorous acid; MPCs, mesenchymal progenitor cells; NETs, neutrophil extracellular traps; MPO, myeloperoxidase; NE, neutrophil elastase; CarP, carbamylated protein; chcap-18, cathelicidin human cationic antimicrobial protein-18; PAD, peptidylarginine deiminase; MHCII, MHC class II; LDG, low-density granulocytes; RF, rheumatoid factor; anti-CCP, anti-cyclic citrullinated peptide; ACPA, anti-citrullinated protein antibodies; AA, adjuvant-induced arthritis; RIPK, receptor-interacting protein kinase; MLKL, pseudokinase mixed lineage kinase domain-like; FAP-α, fibroblast activation protein-α; NHP, non-human primate; VDAC1, voltage-dependent anion-selective channel 1; ASICs, acid-sensitive ion channels; IFN-γ, interferon γ; SMAC, second mitochondria-derived activator of caspases; SMs, SMAC mimetics; PTX3, pentaxin 3; GSDMD, gasdermin D; PUN, punicalagin; mtDNA, mitochondrial DNA; PTPN2, phosphatase nonreceptor type 2; OA, osteoarthritis; PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3; SERCA, sarcoplasmic/endoplasmic reticulum Ca ATPase pump; AIA, adjuvant-induced arthritis; LC3b, light chain 3b; ULK-1, UNC51-like kinase 1; TLR, Toll-like receptor; GM-CSF, granulocyte-macrophage colony-stimulating factor; TGF, transforming growth factor; BID, the BH3 interacting-domain death agonist.
Glossary

References
1. Aletaha D, Neogi T, Silman A, Funovits J, Felson D, Bingham C, et al. 2010 Rheumatoid Arthritis Classification Criteria: An American College of Rheumatology/European League Against Rheumatism Collaborative Initiative. Arthritis Rheum-us (2010) 62(9):2569–81. doi: 10.1002/art.27584
2. Alamanos Y, Voulgari P, Drosos A. Incidence and Prevalence of Rheumatoid Arthritis, Based on the 1987 American College of Rheumatology Criteria: A Systematic Review. Semin Arthritis Rheum (2006) 36(3):182–8. doi: 10.1016/j.semarthrit.2006.08.006
3. Firestein G, McInnes I. Immunopathogenesis of Rheumatoid Arthritis. Immunity (2017) 46(2):183–96. doi: 10.1016/j.immuni.2017.02.006
4. Guo S, Jin Y, Zhou J, Zhu Q, Jiang T, Bian Y, et al. MicroRNA Variants and HLA-miRNA Interactionsare Novel Rheumatoid Arthritis Susceptibility Factors. Front Genet (2021) 12:747274(747274). doi: 10.3389/fgene.2021.747274
5. Zhao J, Guo S, Schrodi SJ, D H. Molecular and Cellular Heterogeneity in Rheumatoid Arthritis:Mechanisms and Clinical Implications. Front Immunol (2021) 12:790122(790122). doi: 10.3389/fimmu.2021.790122
6. McInnes I, Schett G. Pathogenetic Insights From the Treatment of Rheumatoid Arthritis. Lancet (2017) 389(10086):2328–37. doi: 10.1016/s0140-6736(17)31472-1
7. Nakano K, Saito K, Mine S, Matsushita S, Tanaka Y. Engagement of CD44 Up-Regulates Fas Ligand Expression on T Cells Leading to Activation-Induced Cell Death. Apoptosis (2007) 12(1):45–54. doi: 10.1007/s10495-006-0488-8
8. Zhao J, Hu Y, Peng J. Targeting Programmed Cell Death in Metabolic Dysfunction-Associated Fatty Liver Disease (MAFLD): A Promising New Therapy. Cell Mol Biol Lett (2021) 26(1):17. doi: 10.1186/s11658-021-00254-z
9. Wakisaka S, Suzuki N, Takeba Y, Shimoyama Y, Nagafuchi H, Takeno M, et al. Modulation by Proinflammatory Cytokines of Fas/Fas Ligand-Mediated Apoptotic Cell Death of Synovial Cells in Patients With Rheumatoid Arthritis (RA). Clin Exp Immunol (1998) 114(1):119–28. doi: 10.1046/j.1365-2249.1998.00701.x
10. Krause A, Scaletta N, Ji J, Ivashkiv L. Rheumatoid Arthritis Synoviocyte Survival Is Dependent on Stat3. J Immunol (2002) 169(11):6610–6. doi: 10.4049/jimmunol.169.11.6610
11. Moodley D, Mody G, Chuturgoon A. Initiation But No Execution - Modulation of Peripheral Blood Lymphocyte Apoptosis in Rheumatoid Arthritis - A Potential Role for Heat Shock Protein 70. J Inflammation (Lond) (2011) 8(1):30. doi: 10.1186/1476-9255-8-30
12. Hung H, Lin C, Liao Y, Hsu P, Tsay G, Liu G. The Functional Haplotype of Peptidylarginine Deiminase IV (S55G, A82V and A112G) Associated With Susceptibility to Rheumatoid Arthritis Dominates Apoptosis of Acute T Leukemia Jurkat Cells. Apoptosis (2007) 12(3):475–87. doi: 10.1007/s10495-006-0005-0
13. Ohshima S, Mima T, Sasai M, Nishioka K, Shimizu M, Murata N, et al. Tumour Necrosis Factor Alpha (TNF-Alpha) Interferes With Fas-Mediated Apoptotic Cell Death on Rheumatoid Arthritis (RA) Synovial Cells: A Possible Mechanism of Rheumatoid Synovial Hyperplasia and a Clinical Benefit of Anti-TNF-Alpha Therapy for RA. Cytokine (2000) 12(3):281–8. doi: 10.1006/cyto.1999.0552
14. Koshiba M, Kosaka H, Nakazawa T, Hayashi N, Saura R, Kitamura N, et al. 2-Chloroadenosine But Not Adenosine Induces Apoptosis in Rheumatoid Fibroblasts Independently of Cell Surface Adenosine Receptor Signalling. Br J Pharmacol (2002) 135(6):1477–86. doi: 10.1038/sj.bjp.0704612
15. Yamazaki R, Kusunoki N, Matsuzaki T, Hashimoto S, Kawai S. Nonsteroidal Anti-Inflammatory Drugs Induce Apoptosis in Association With Activation of Peroxisome Proliferator-Activated Receptor Gamma in Rheumatoid Synovial Cells. J Pharmacol Exp Ther (2002) 302(1):18–25. doi: 10.1124/jpet.302.1.18
16. Shen C, Maerten P, Geboes K, Van Assche G, Rutgeerts P, Ceuppens J. Infliximab Induces Apoptosis of Monocytes and T Lymphocytes in a Human-Mouse Chimeric Model. Clin Immunol (2005) 115(3):250–9. doi: 10.1016/j.clim.2005.01.007
17. Borker A, Choudhary N. Rituximab. Indian Pediatr (2011) 48(8):627–32. doi: 10.1007/s13312-011-0098-6
18. Emori T, Kasahara M, Sugahara S, Hashimoto M, Ito H, Narumiya S, et al. Role of JAK-STAT Signaling in the Pathogenic Behavior of Fibroblast-Like Synoviocytes in Rheumatoid Arthritis: Effect of the Novel JAK Inhibitor Peficitinib. Eur J Pharmacol (2020) 882:173238. doi: 10.1016/j.ejphar.2020.173238
19. Zhai Y, Wu B, Li J, Yao X, Zhu P, Chen Z. CD147 Promotes IKK/IκB/NF-κB Pathway to Resist TNF-Induced Apoptosis in Rheumatoid Arthritis Synovial Fibroblasts. J Mol Med (Berl) (2016) 94(1):71–82. doi: 10.1007/s00109-015-1334-7
20. Shi C, Zhang S, Hong S, Pang J, Yesibulati Y, Yin P, et al. The Pro-Apoptotic Effects of TIPE2 on AA Rat Fibroblast-Like Synoviocytes via Regulation of the DR5-Caspase-NF-κB Pathway In Vitro. Onco Targets Ther (2016) 9:993–1000. doi: 10.2147/ott.S92907
21. Liu H, Eksarko P, Temkin V, Haines G, Perlman H, Koch A, et al. Mcl-1 Is Essential for the Survival of Synovial Fibroblasts in Rheumatoid Arthritis. J Immunol (2005) 175(12):8337–45. doi: 10.4049/jimmunol.175.12.8337
22. Kabala P, Angiolilli C, Yeremenko N, Grabiec A, Giovannone B, Pots D, et al. Endoplasmic Reticulum Stress Cooperates With Toll-Like Receptor Ligation in Driving Activation of Rheumatoid Arthritis Fibroblast-Like Synoviocytes. Arthritis Res Ther (2017) 19(1):207. doi: 10.1186/s13075-017-1386-x
23. Gao B, Lee S, Chen A, Zhang J, Zhang D, Kannan K, et al. Synoviolin Promotes IRE1 Ubiquitination and Degradation in Synovial Fibroblasts From Mice With Collagen-Induced Arthritis. EMBO Rep (2008) 9(5):480–5. doi: 10.1038/embor.2008.37
24. Matsumoto S, Müller-Ladner U, Gay R, Nishioka K, Gay S. Ultrastructural Demonstration of Apoptosis, Fas and Bcl-2 Expression of Rheumatoid Synovial Fibroblasts. J Rheumatol (1996) 23(8):1345–52.
25. Matsumura T, Saito Y, Suzuki T, Teramoto A, Ozasa Y, Yamashita T, et al. Phosphorylated Platelet-Derived Growth Factor Receptor-Positive Cells With Anti-Apoptotic Properties Accumulate in the Synovium of Patients With Rheumatoid Arthritis. Front Immunol (2019) 10:241. doi: 10.3389/fimmu.2019.00241
26. Perlman H, Georganas C, Pagliari L, Koch A, Haines K, Pope R. Bcl-2 Expression in Synovial Fibroblasts Is Essential for Maintaining Mitochondrial Homeostasis and Cell Viability. J Immunol (2000) 164(10):5227–35. doi: 10.4049/jimmunol.164.10.5227
27. Audo R, Calmon-Hamaty F, Papon L, Combe B, Morel J, Hahne M. Distinct Effects of Soluble and Membrane-Bound Fas Ligand on Fibroblast-Like Synoviocytes From Rheumatoid Arthritis Patients. Arthritis Rheumatol (2014) 66(12):3289–99. doi: 10.1002/art.38806
28. Tauzin S, Chaigne-Delalande B, Selva E, Khadra N, Daburon S, Contin-Bordes C, et al. The Naturally Processed CD95L Elicits a C-Yes/Calcium/PI3K-Driven Cell Migration Pathway. PloS Biol (2011) 9(6):e1001090. doi: 10.1371/journal.pbio.1001090
29. Hasunuma T, Kayagaki N, Asahara H, Motokawa S, Kobata T, Yagita H, et al. Accumulation of Soluble Fas in Inflamed Joints of Patients With Rheumatoid Arthritis. Arthritis Rheum-us (1997) 40(1):80–6. doi: 10.1002/art.1780400112
30. Kim W, Kwok S, Hong K, Yoo S, Kong J, Choe J, et al. Soluble Fas Ligand Inhibits Angiogenesis in Rheumatoid Arthritis. Arthritis Res Ther (2007) 9(2):R42. doi: 10.1186/ar2181
31. Yamaguchi A, Nozawa K, Fujishiro M, Kawasaki M, Takamori K, Ogawa H, et al. Estrogen Inhibits Apoptosis and Promotes CC Motif Chemokine Ligand 13 Expression on Synovial Fibroblasts in Rheumatoid Arthritis. Immunopharmacol Immunotoxicol (2012) 34(5):852–7. doi: 10.3109/08923973.2012.664149
32. Zhai K, Duan H, Chen Y, Khan G, Cao W, Gao G, et al. Apoptosis Effects of Imperatorin on Synoviocytes in Rheumatoid Arthritis Through Mitochondrial/Caspase-Mediated Pathways. Food Funct (2018) 9(4):2070–9. doi: 10.1039/c7fo01748k
33. Drynda A, Quax P, Neumann M, van der Laan W, Pap G, Drynda S, et al. Gene Transfer of Tissue Inhibitor of Metalloproteinases-3 Reverses the Inhibitory Effects of TNF-Alpha on Fas-Induced Apoptosis in Rheumatoid Arthritis Synovial Fibroblasts. J Immunol (2005) 174(10):6524–31. doi: 10.4049/jimmunol.174.10.6524
34. Lee J, Jeong H, Park E, Hwang J, Huang B, Bae E, et al. CIP2A Facilitates Apoptotic Resistance of Fibroblast-Like Synoviocytes in Rheumatoid Arthritis Independent of C-Myc Expression. Rheumatol Int (2013) 33(9):2241–8. doi: 10.1007/s00296-013-2711-6
35. Migita K, Honda S, Yamasaki S, Hirai Y, Fukuda T, Aoyagi T, et al. Regulation of Rheumatoid Synovial Cell Growth by Ceramide. Biochem Biophys Res Commun (2000) 269(1):70–5. doi: 10.1006/bbrc.2000.2239
36. Lee J, Lee J, Um S, Moon E. Synovial Cell Death Is Regulated by TNF-α-Induced Expression of B-Cell Activating Factor Through an ERK-Dependent Increase in Hypoxia-Inducible Factor-1α. Cell Death Dis (2017) 8(4):e2727. doi: 10.1038/cddis.2017.26
37. Gong W, Kolker S, Usachev Y, Walder R, Boyle D, Firestein G, et al. Acid-Sensing Ion Channel 3 Decreases Phosphorylation of Extracellular Signal-Regulated Kinases and Induces Synoviocyte Cell Death by Increasing Intracellular Calcium. Arthritis Res Ther (2014) 16(3):R121. doi: 10.1186/ar4577
38. Hu F, Sun W, Zhao X, Cui Z, Yang W. TRPV1 Mediates Cell Death in Rat Synovial Fibroblasts Through Calcium Entry-Dependent ROS Production and Mitochondrial Depolarization. Biochem Biophys Res Commun (2008) 369(4):989–93. doi: 10.1016/j.bbrc.2008.02.155
39. Hu F, Yang S, Zhao D, Zhu S, Wang Y, Li J. Moderate Extracellular Acidification Inhibits Capsaicin-Induced Cell Death Through Regulating Calcium Mobilization, NF-κB Translocation and ROS Production in Synoviocytes. Biochem Biophys Res Commun (2012) 424(1):196–200. doi: 10.1016/j.bbrc.2012.06.115
40. Migita K, Yamasaki S, Kita M, Ida H, Shibatomi K, Kawakami A, et al. Nitric Oxide Protects Cultured Rheumatoid Synovial Cells From Fas-Induced Apoptosis by Inhibiting Caspase-3. Immunology (2001) 103(3):362–7. doi: 10.1046/j.1365-2567.2001.01252.x
41. Whiteman M, Armstrong J, Cheung N, Siau J, Rose P, Schantz J, et al. Peroxynitrite Mediates Calcium-Dependent Mitochondrial Dysfunction and Cell Death via Activation of Calpains. FASEB J (2004) 18(12):1395–7. doi: 10.1096/fj.03-1096fje
42. Zhu S, Wang Y, Pan L, Yang S, Sun Y, Wang X, et al. Involvement of Transient Receptor Potential Melastatin-8 (TRPM8) in Menthol-Induced Calcium Entry, Reactive Oxygen Species Production and Cell Death in Rheumatoid Arthritis Rat Synovial Fibroblasts. Eur J Pharmacol (2014) 725:1–9. doi: 10.1016/j.ejphar.2014.01.001
43. Rui X, Yang Y, Chen Q, Wu J, Chen J, Zhang Q, et al. Imperative and Effective Reversion of Synovial Hyperplasia and Cartilage Destruction in Rheumatoid Arthritis Through Multiple Synergistic Effects of O and Ca. Mater Sci Eng C Mater Biol Appl (2020) 114:111058. doi: 10.1016/j.msec.2020.111058
44. Yan C, Kong D, Ge D, Zhang Y, Zhang X, Su C, et al. Mitomycin C Induces Apoptosis in Rheumatoid Arthritis Fibroblast-Like Synoviocytes via a Mitochondrial-Mediated Pathway. Cell Physiol Biochem (2015) 35(3):1125–36. doi: 10.1159/000373938
45. Shin G, Kim C, Lee J, Cho W, Lee S, Jeong M, et al. Apigenin-Induced Apoptosis is Mediated by Reactive Oxygen Species and Activation of ERK1/2 in Rheumatoid Fibroblast-Like Synoviocytes. Chem Biol Interact (2009) 182(1):29–36. doi: 10.1016/j.cbi.2009.07.016
46. Jeong M, Cho J, Shin J, Jeon Y, Kim J, Lee S, et al. Hempseed Oil Induces Reactive Oxygen Species- and C/EBP Homologous Protein-Mediated Apoptosis in MH7A Human Rheumatoid Arthritis Fibroblast-Like Synovial Cells. J Ethnopharmacol (2014) 154(3):745–52. doi: 10.1016/j.jep.2014.04.052
47. Nakayama H, Yaguchi T, Yoshiya S, Nishizaki T. Resveratrol Induces Apoptosis MH7A Human Rheumatoid Arthritis Synovial Cells in a Sirtuin 1-Dependent Manner. Rheumatol Int (2012) 32(1):151–7. doi: 10.1007/s00296-010-1598-8
48. Ma C, Chen J, Li P. Geldanamycin Induces Apoptosis and Inhibits Inflammation in Fibroblast-Like Synoviocytes Isolated From Rheumatoid Arthritis Patients. J Cell Biochem (2019) 120(9):16254–63. doi: 10.1002/jcb.28906
49. Wang J, Guan Z, Zhang S, Pei Z, Chen Y, Pan H. Programmed Cell Death 5 Correlates With Disease Activity and Interleukin-17 in Serum and Synovial Fluid of Rheumatoid Arthritis Patients. Chin Med J (2013) 126(2):296–9.
50. Wang J, Guan Z, Ge Z. Plasma and Synovial Fluid Programmed Cell Death 5 (PDCD5) Levels Are Inversely Associated With TNF-α and Disease Activity in Patients With Rheumatoid Arthritis. Biomarkers (2013) 18(2):155–9. doi: 10.3109/1354750x.2012.759277
51. Wang N, Lu H, Guan Z, Sun T, Chen Y, Ruan G, et al. Involvement of PDCD5 in the Regulation of Apoptosis in Fibroblast-Like Synoviocytes of Rheumatoid Arthritis. Apoptosis (2007) 12(8):1433–41. doi: 10.1007/s10495-007-0070-z
52. Shu K, Kuang N, Zhang Z, Hu Z, Zhang Y, Fu Y, et al. Therapeutic Effect of Daphnetin on the Autoimmune Arthritis Through Demethylation of Proapoptotic Genes in Synovial Cells. J Transl Med (2014) 12:287. doi: 10.1186/s12967-014-0287-x
53. You X, Boyle D, Hammaker D, Firestein G. PUMA-Mediated Apoptosis in Fibroblast-Like Synoviocytes Does Not Require P53. Arthritis Res Ther (2006) 8(6):R157. doi: 10.1186/ar2052
54. Aupperle K, Boyle D, Hendrix M, Seftor E, Zvaifler N, Barbosa M, et al. Regulation of Synoviocyte Proliferation, Apoptosis, and Invasion by the P53 Tumor Suppressor Gene. Am J Pathol (1998) 152(4):1091–8.
55. Akaike A, Banno Y, Osawa Y, Oshita H, Fushimi K, Kodama H, et al. Synergistic Induction of Apoptosis of Rheumatoid Arthritis Synovial Cells by H(2)O(2) and N-Acetyl-Leucyl-Leucyl-Norleucinal. J Orthop Sci (2003) 8(3):346–51. doi: 10.1007/s10776-003-0645-8
56. Nakazawa M, Aratani S, Hatta M, Araya N, Daitoku H, Kawahara K, et al. TNFalpha Induces Acetylation of P53 But Attenuates its Transcriptional Activation in Rheumatoid Synoviocytes. Int J Mol Med (2002) 10(3):269–75.
57. Lattuada D, Casnici C, Crotta K, Seneci P, Corradini C, Truzzi M, et al. Proapoptotic Activity of a Monomeric Smac Mimetic on Human Fibroblast-Like Synoviocytes From Patients With Rheumatoid Arthritis. Inflammation (2015) 38(1):102–9. doi: 10.1007/s10753-014-0012-1
58. Jüngel A, Baresova V, Ospelt C, Simmen B, Michel B, Gay R, et al. Trichostatin A Sensitises Rheumatoid Arthritis Synovial Fibroblasts for TRAIL-Induced Apoptosis. Ann Rheum Dis (2006) 65(7):910–2. doi: 10.1136/ard.2005.044065
59. Pundt N, Peters M, Wunrau C, Strietholt S, Fehrmann C, Neugebauer K, et al. Susceptibility of Rheumatoid Arthritis Synovial Fibroblasts to FasL- and TRAIL-Induced Apoptosis is Cell Cycle-Dependent. Arthritis Res Ther (2009) 11(1):R16. doi: 10.1186/ar2607
60. Pericolini E, Alunno A, Gabrielli E, Bartoloni E, Cenci E, Chow S, et al. The Microbial Capsular Polysaccharide Galactoxylomannan Inhibits IL-17A Production in Circulating T Cells From Rheumatoid Arthritis Patients. PloS One (2013) 8(1):e53336. doi: 10.1371/journal.pone.0053336
61. Zhang X, Burch E, Cai L, So E, Hubbard F, Matteson E, et al. CD40 Mediates Downregulation of CD32B on Specific Memory B Cell Populations in Rheumatoid Arthritis. J Immunol (2013) 190(12):6015–22. doi: 10.4049/jimmunol.1203366
62. Dong H, Strome S, Matteson E, Moder K, Flies D, Zhu G, et al. Costimulating Aberrant T Cell Responses by B7-H1 Autoantibodies in Rheumatoid Arthritis. J Clin Invest (2003) 111(3):363–70. doi: 10.1172/jci16015
63. Rapetti L, Chavele K, Evans C, Ehrenstein M. B Cell Resistance to Fas-Mediated Apoptosis Contributes to Their Ineffective Control by Regulatory T Cells in Rheumatoid Arthritis. Ann Rheum Dis (2015) 74(1):294–302. doi: 10.1136/annrheumdis-2013-204049
64. Izawa T, Kondo T, Kurosawa M, Oura R, Matsumoto K, Tanaka E, et al. Fas-Independent T-Cell Apoptosis by Dendritic Cells Controls Autoimmune Arthritis in MRL/lpr Mice. PloS One (2012) 7(12):e48798. doi: 10.1371/journal.pone.0048798
65. Wang C, Shiau A, Chen S, Cheng Z, Li Y, Lee C, et al. Intra-Articular Lentivirus-Mediated Delivery of Galectin-3 shRNA and Galectin-1 Gene Ameliorates Collagen-Induced Arthritis. Gene Ther (2010) 17(10):1225–33. doi: 10.1038/gt.2010.78
66. Felix K, Teng F, Bates N, Ma H, Jaimez I, Sleiman K, et al. P2RX7 Deletion in T Cells Promotes Autoimmune Arthritis by Unleashing the Tfh Cell Response. Front Immunol (2019) 10:411. doi: 10.3389/fimmu.2019.00411
67. Taneja V, Behrens M, Basal E, Sparks J, Griffiths M, Luthra H, et al. Delineating the Role of the HLA-DR4 "Shared Epitope" in Susceptibility Versus Resistance to Develop Arthritis. J Immunol (2008) 181(4):2869–77. doi: 10.4049/jimmunol.181.4.2869
68. Taneja V, Taneja N, Paisansinsup T, Behrens M, Griffiths M, Luthra H, et al. CD4 and CD8 T Cells in Susceptibility/Protection to Collagen-Induced Arthritis in HLA-DQ8-Transgenic Mice: Implications for Rheumatoid Arthritis. J Immunol (2002) 168(11):5867–75. doi: 10.4049/jimmunol.168.11.5867
69. Rodríguez-Frade J, Guedán A, Lucas P, Martínez-Muñoz L, Villares R, Criado G, et al. In VivoUse of Lentiviral Particles As a Cell Membrane-Based Mfasl Delivery System for Treatment of Inflammatory Arthritis. Front Immunol (2017) 8:460. doi: 10.3389/fimmu.2017.00460
70. Bremer E, Abdulahad W, de Bruyn M, Samplonius D, Kallenberg C, Armbrust W, et al. Selective Elimination of Pathogenic Synovial Fluid T-Cells From Rheumatoid Arthritis and Juvenile Idiopathic Arthritis by Targeted Activation of Fas-Apoptotic Signaling. Immunol Lett (2011) 138(2):161–8. doi: 10.1016/j.imlet.2011.04.004
71. Röhner E, Matziolis G, Perka C, Füchtmeier B, Gaber T, Burmester G, et al. Inflammatory Synovial Fluid Microenvironment Drives Primary Human Chondrocytes to Actively Take Part in Inflammatory Joint Diseases. Immunol Res (2012) 52(3):169–75. doi: 10.1007/s12026-011-8247-5
72. Kawakami A, Eguchi K, Matsuoka N, Tsuboi M, Koji T, Urayama S, et al. Fas and Fas Ligand Interaction Is Necessary for Human Osteoblast Apoptosis. J Bone Miner Res (1997) 12(10):1637–46. doi: 10.1359/jbmr.1997.12.10.1637
73. Yumoto K, Nifuji A, Rittling S, Tsuchiya Y, Kon S, Uede T, et al. Osteopontin Deficiency Suppresses Tumor Necrosis Factor-α-Induced Apoptosis in Chondrocytes. Cartilage (2012) 3(1):79–85. doi: 10.1177/1947603511421502
74. Ye W, Zhong Z, Zhu S, Zheng S, Xiao J, Song S, et al. Advanced Oxidation Protein Products Induce Chondrocyte Death Through a Redox-Dependent, Poly (ADP-Ribose) Polymerase-1-Mediated Pathway. Apoptosis (2017) 22(1):86–97. doi: 10.1007/s10495-016-1314-6
75. Relić B, Bentires-Alj M, Ribbens C, Franchimont N, Guerne P, Benoît V, et al. TNF-Alpha Protects Human Primary Articular Chondrocytes From Nitric Oxide-Induced Apoptosis via Nuclear Factor-Kappab. Lab Invest (2002) 82(12):1661–72. doi: 10.1097/01.lab.0000041714.05322.c0
76. Chaea H, Kim H, Kang Y, Hyun K, Kim H, Seo H, et al. Heme Oxygenase-1 Induction by (S)-Enantiomer of YS-51 (YS-51S), a Synthetic Isoquinoline Alkaloid, Inhibits Nitric Oxide Production and Nuclear factor-kappaB Translocation in ROS 17/2.8 Cells Activated With Inflammatory Stimulants. Int Immunopharmacol (2007) 7(12):1559–68. doi: 10.1016/j.intimp.2007.07.023
77. Penolazzi L, Lampronti I, Borgatti M, Khan M, Zennaro M, Piva R, et al. Induction of Apoptosis of Human Primary Osteoclasts Treated With Extracts From the Medicinal Plant Emblica Officinalis. BMC Complement Altern Med (2008) 8:59. doi: 10.1186/1472-6882-8-59
78. Kuo P, Huang Y, Chang C, Chang J. Fraxetin Inhibits the Induction of Anti-Fas IgM, Tumor Necrosis Factor-Alpha and Interleukin-1beta-Mediated Apoptosis by Fas Pathway Inhibition in Human Osteoblastic Cell Line MG-63. Int Immunopharmacol (2006) 6(7):1167–75. doi: 10.1016/j.intimp.2006.02.010
79. Rajasekhar M, Olsson A, Steel K, Georgouli M, Ranasinghe U, Brender Read C, et al. MicroRNA-155 Contributes to Enhanced Resistance to Apoptosis in Monocytes From Patients With Rheumatoid Arthritis. J Autoimmun (2017) 79:53–62. doi: 10.1016/j.jaut.2017.01.002
80. Perlman H, Pagliari L, Georganas C, Mano T, Walsh K, Pope R. FLICE-Inhibitory Protein Expression During Macrophage Differentiation Confers Resistance to Fas-Mediated Apoptosis. J Exp Med (1999) 190(11):1679–88. doi: 10.1084/jem.190.11.1679
81. Gockel H, Lügering A, Heidemann J, Schmidt M, Domschke W, Kucharzik T, et al. Thalidomide Induces Apoptosis in Human Monocytes by Using a Cytochrome C-Dependent Pathway. J Immunol (2004) 172(8):5103–9. doi: 10.4049/jimmunol.172.8.5103
82. Cutolo M, Sulli A, Craviotto C, Felli L, Pizzorni C, Seriolo B, et al. Modulation of Cell Growth and Apoptosis by Sex Hormones in Cultured Monocytic THP-1 Cells. Ann N Y Acad Sci (2002) 966:204–10. doi: 10.1111/j.1749-6632.2002.tb04216.x
83. Cutolo M, Capellino S, Montagna P, Ghiorzo P, Sulli A, Villaggio B. Sex Hormone Modulation of Cell Growth and Apoptosis of the Human Monocytic/Macrophage Cell Line. Arthritis Res Ther (2005) 7(5):R1124–32. doi: 10.1186/ar1791
84. Cutolo M, Barone A, Accardo S, Setti M, Villaggio B. Effect of Cyclosporin on Apoptosis in Human Cultured Monocytic THP-1 Cells and Synovial Macrophages. Clin Exp Rheumatol (1998) 16(4):417–22.
85. Zhao M, He H, Yin J. CARD6 Protects Against Collagen-Induced Rheumatoid Arthritis in Mice Through Attenuating the Inflammatory Response and Joint Destruction via Suppression of TNFR1/TRAF2 Signaling. Biochem Biophys Res Commun (2020) 526(4):1092–9. doi: 10.1016/j.bbrc.2020.04.006
86. Tsuda M, Kawaida R, Kobayashi K, Shinagawa A, Sawada T, Yamada R, et al. POSH Promotes Cell Survival in Drosophila and in Human RASF Cells. FEBS Lett (2010) 584(22):4689–94. doi: 10.1016/j.febslet.2010.10.048
87. Whiteman M, Chu S, Siau J, Rose P, Sabapathy K, Schantz J, et al. The Pro-Inflammatory Oxidant Hypochlorous Acid Induces Bax-Dependent Mitochondrial Permeabilisation and Cell Death Through AIF-/EndoG-Dependent Pathways. Cell Signal (2007) 19(4):705–14. doi: 10.1016/j.cellsig.2006.08.019
88. Krenn V, Molitoris R, Berek C, Sack U, König A, Müller-Deubert S, et al. A Novel Monospecific IgG2/lambda-Autoantibody With Specificity for a Mitochondrial Antigen: Evidence for an Antigen-Driven Pathogenetic B-Cell Response in Rheumatoid Synovial Tissue, Induced by Tissue Alteration. Lab Invest (1998) 78(4):485–96.
89. Wahle M, Pierer M, Krause A, Kolker S, Baerwald C. Decreased Catecholamine-Induced Cell Death in B Lymphocytes From Patients With Rheumatoid Arthritis. Ann N Y Acad Sci (2002) 966:425–8. doi: 10.1111/j.1749-6632.2002.tb04243.x
90. Tan S, Xiao L, Pi X, Holoshitz J. Aberrant Gi Protein Coupled Receptor-Mediated Cell Survival Signaling in Rheumatoid Arthritis B Cell Lines. Front Biosci (2007) 12:1651–60. doi: 10.2741/2177
91. Badadani M. Autophagy Mechanism, Regulation, Functions, and Disorders. Int Sch Res Notices (2012) 2012:11. doi: 10.5402/2012/927064
92. Walker S, Ktistakis N. Autophagosome Biogenesis Machinery. J Mol Bio (2020) 432(8):2449–61. doi: 10.1016/j.jmb.2019.10.027
93. Feng Y, He D, Yao Z, Klionsky D. The Machinery of Macroautophagy. Cell Res (2014) 24(1):24–41. doi: 10.1038/cr.2013.168
94. Chen Y, Chang C, Chen H, Hsieh C, Tang K, Yang M, et al. Association Between Autophagy and Inflammation in Patients With Rheumatoid Arthritis Receiving Biologic Therapy. Arthritis Res Ther (2018) 20(1):268. doi: 10.1186/s13075-018-1763-0
95. Zhu L, Wang H, Wu Y, He Z, Qin Y, Shen Q. The Autophagy Level Is Increased in the Synovial Tissues of Patients With Active Rheumatoid Arthritis and Is Correlated With Disease Severity. Mediators Inflamm (2017) 2017:7623145. doi: 10.1155/2017/7623145
96. Schrezenmeier E, Dörner T. Mechanisms of Action of Hydroxychloroquine and Chloroquine: Implications for Rheumatology. Nat Rev Rheumatol (2020) 16(3):155–66. doi: 10.1038/s41584-020-0372-x
97. Xu K, Xu P, Yao J, Zhang Y, Hou W, Lu S. Reduced Apoptosis Correlates With Enhanced Autophagy in Synovial Tissues of Rheumatoid Arthritis. Inflammation Res (2013) 62(2):229–37. doi: 10.1007/s00011-012-0572-1
98. Wang Y, Gao W. Effects of TNF-α on Autophagy of Rheumatoid Arthritis Fibroblast-Like Synoviocytes and Regulation of the NF-κB Signaling Pathway. Immunobiology (2021) 226(2):152059. doi: 10.1016/j.imbio.2021.152059
99. Dai Y, Ding J, Yin W, He Y, Yu F, Ye C, et al. Increased Autophagy Enhances the Resistance to Tumor Necrosis Factor-Alpha Treatment in Rheumatoid Arthritis Human Fibroblast-Like Synovial Cell. BioMed Res Int (2018) 2018:4941027. doi: 10.1155/2018/4941027
100. Xu K, Cai Y, Lu S, Li X, Liu L, Li Z, et al. Autophagy Induction Contributes to the Resistance to Methotrexate Treatment in Rheumatoid Arthritis Fibroblast-Like Synovial Cells Through High Mobility Group Box Chromosomal Protein 1. Arthritis Res Ther (2015) 17:374. doi: 10.1186/s13075-015-0892-y
101. Aradi B, Kato M, Filkova M, Karouzakis E, Klein K, Scharl M, et al. Protein Tyrosine Phosphatase Nonreceptor Type 2: An Important Regulator of Lnterleukin-6 Production in Rheumatoid Arthritis Synovial Fibroblasts. Arthritis Rheumatol (2015) 67(10):2624–33. doi: 10.1002/art.39256
102. Wang X, Chen Z, Fan X, Li W, Qu J, Dong C, et al. Inhibition of DNM1L and Mitochondrial Fission Attenuates Inflammatory Response in Fibroblast-Like Synoviocytes of Rheumatoid Arthritis. J Cell Mol Med (2020) 24(2):1516–28. doi: 10.1111/jcmm.14837
103. Kim E, Kwon J, Lee S, Lee E, Kim D, Moon S, et al. IL-17-Mediated Mitochondrial Dysfunction Impairs Apoptosis in Rheumatoid Arthritis Synovial Fibroblasts Through Activation of Autophagy. Cell Death Dis (2017) 8(1):e2565. doi: 10.1038/cddis.2016.490
104. Chang L, Feng X, Gao W. Proliferation of Rheumatoid Arthritis Fibroblast-Like Synoviocytes Is Enhanced by IL-17-Mediated Autophagy Through STAT3 Activation. Connective Tissue Res (2019) 60(4):358–66. doi: 10.1080/03008207.2018.1552266
105. Zhang J, Song X, Cao W, Lu J, Wang X, Wang G, et al. Autophagy and Mitochondrial Dysfunction in Adjuvant-Arthritis Rats Treatment With Resveratrol. Sci Rep-UK (2016) 6:32928. doi: 10.1038/srep32928
106. Fan T, Zhang C, Zong M, Fan L. Hypoxia−induced Autophagy Is Inhibited by PADI4 Knockdown, Which Promotes Apoptosis of Fibroblast−Like Synoviocytes in Rheumatoid Arthritis. Mol Med Rep (2018) 17(4):5116–24. doi: 10.3892/mmr.2018.8501
107. Alsousi A, Igwe O. Autophagy Protects Against Redox-Active Trace Metal-Induced Cell Death in Rabbit Synovial Fibroblasts Through Toll-Like Receptor 4 Activation. Exp Cell Res (2019) 374(1):19–28. doi: 10.1016/j.yexcr.2018.11.003
108. Shin Y, Han S, Kim D, Lee G, Yoo W, Kang Y, et al. Autophagy Induction and CHOP Under-Expression Promotes Survival of Fibroblasts From Rheumatoid Arthritis Patients Under Endoplasmic Reticulum Stress. Arthritis Res Ther (2010) 12(1):R19. doi: 10.1186/ar2921
109. Kato M, Ospelt C, Gay R, Gay S, Klein K. Dual Role of Autophagy in Stress-Induced Cell Death in Rheumatoid Arthritis Synovial Fibroblasts. Arthritis Rheumatol (2014) 66(1):40–8. doi: 10.1002/art.38190
110. Yang Z, Goronzy J, Weyand C. Autophagy in Autoimmune Disease. J Mol Med (Berl) (2015) 93(7):707–17. doi: 10.1007/s00109-015-1297-8
111. Yang Z, Fujii H, Mohan S, Goronzy J, Weyand C. Phosphofructokinase Deficiency Impairs ATP Generation, Autophagy, and Redox Balance in Rheumatoid Arthritis T Cells. J Exp Med (2013) 210(10):2119–34. doi: 10.1084/jem.20130252
112. Yang Z, Goronzy J, Weyand C. The Glycolytic Enzyme PFKFB3/phosphofructokinase Regulates Autophagy. Autophagy (2014) 10(2):382–3. doi: 10.4161/auto.27345
113. Kumar P, Yao L, Saidin S, Paleja B, van Loosdregt J, Chua C, et al. Molecular Mechanisms of Autophagic Memory in Pathogenic T Cells in Human Arthritis. J Autoimmun (2018) 94:90–8. doi: 10.1016/j.jaut.2018.07.014
114. van Loosdregt J, Rossetti M, Spreafico R, Moshref M, Olmer M, Williams G, et al. Increased Autophagy in CD4 T Cells of Rheumatoid Arthritis Patients Results in T-Cell Hyperactivation and Apoptosis Resistance. Eur J Immunol (2016) 46(12):2862–70. doi: 10.1002/eji.201646375
115. Ireland J, Unanue E. Autophagy in Antigen-Presenting Cells Results in Presentation of Citrullinated Peptides to CD4 T Cells. J Exp Med (2011) 208(13):2625–32. doi: 10.1084/jem.20110640
116. Ireland J, Unanue E. Processing of Proteins in Autophagy Vesicles of Antigen-Presenting Cells Generates Citrullinated Peptides Recognized by the Immune System. Autophagy (2012) 8(3):429–30. doi: 10.4161/auto.19261
117. Sorice M, Iannuccelli C, Manganelli V, Capozzi A, Alessandri C, Lococo E, et al. Autophagy Generates Citrullinated Peptides in Human Synoviocytes: A Possible Trigger for Anti-Citrullinated Peptide Antibodies. Rheumatology (Oxford) (2016) 55(8):1374–85. doi: 10.1093/rheumatology/kew178
118. Alissafi T, Banos A, Boon L, Sparwasser T, Ghigo A, Wing K, et al. Tregs Restrain Dendritic Cell Autophagy to Ameliorate Autoimmunity. J Clin Invest (2017) 127(7):2789–804. doi: 10.1172/jci92079
119. Manganelli V, Recalchi S, Capozzi A, Riitano G, Mattei V, Longo A, et al. Autophagy Induces Protein Carbamylation in Fibroblast-Like Synoviocytes From Patients With Rheumatoid Arthritis. Rheumatology (Oxford) (2018) 57(11):2032–41. doi: 10.1093/rheumatology/key174
120. Liu F, Fang F, Yuan H, Yang D, Chen Y, Williams L, et al. Suppression of Autophagy by FIP200 Deletion Leads to Osteopenia in Mice Through the Inhibition of Osteoblast Terminal Differentiation. J Bone Miner Res (2013) 28(11):2414–30. doi: 10.1002/jbmr.1971
121. Li H, Li D, Ma Z, Qian Z, Kang X, Jin X, et al. Defective Autophagy in Osteoblasts Induces Endoplasmic Reticulum Stress and Causes Remarkable Bone Loss. Autophagy (2018) 14(10):1726–41. doi: 10.1080/15548627.2018.1483807
122. Lin N, Beyer C, Giessl A, Kireva T, Scholtysek C, Uderhardt S, et al. Autophagy Regulates Tnfα-Mediated Joint Destruction in Experimental Arthritis. Ann Rheum Dis (2013) 72(5):761–8. doi: 10.1136/annrheumdis-2012-201671
123. Lin N, Stefanica A, Distler J. Autophagy: A Key Pathway of TNF-Induced Inflammatory Bone Loss. Autophagy (2013) 9(8):1253–5. doi: 10.4161/auto.25467
124. Lee W, Kato M, Sugawara E, Kono M, Kudo Y, Kono M, et al. Protective Role of Optineurin Against Joint Destruction in Rheumatoid Arthritis Synovial Fibroblasts. Arthritis Rheumatol (2020) 72(9):1493–504. doi: 10.1002/art.41290
125. Laha D, Deb M, Das H. KLF2 (Kruppel-Like Factor 2 [Lung]) Regulates Osteoclastogenesis by Modulating Autophagy. Autophagy (2019) 15(12):2063–75. doi: 10.1080/15548627.2019.1596491
126. Wu D, Gu R, Sarin R, Zavodovskaya R, Chen C, Christiansen B, et al. Autophagy-Linked FYVE Containing Protein WDFY3 Interacts With TRAF6 and Modulates RANKL-Induced Osteoclastogenesis. J Autoimmun (2016) 73:73–84. doi: 10.1016/j.jaut.2016.06.004
127. Shi B, Huang Q, Birkett R, Doyle R, Dorfleutner A, Stehlik C, et al. SNAPIN Is Critical for Lysosomal Acidification and Autophagosome Maturation in Macrophages. Autophagy (2017) 13(2):285–301. doi: 10.1080/15548627.2016.1261238
128. Chaudhary A, Leite M, Kulasekara B, Altura M, Ogahara C, Weiss E, et al. Human Diversity in a Cell Surface Receptor That Inhibits Autophagy. Curr Biol (2016) 26(14):1791–801. doi: 10.1016/j.cub.2016.05.003
129. An Q, Yan W, Zhao Y, Yu K. Enhanced Neutrophil Autophagy and Increased Concentrations of IL-6, IL-8, IL-10 and MCP-1 in Rheumatoid Arthritis. Int Immunopharmacol (2018) 65:119–28. doi: 10.1016/j.intimp.2018.09.011
130. Vomero M, Manganelli V, Barbati C, Colasanti T, Capozzi A, Finucci A, et al. Reduction of Autophagy and Increase in Apoptosis Correlates With a Favorable Clinical Outcome in Patients With Rheumatoid Arthritis Treated With Anti-TNF Drugs. Arthritis Res Ther (2019) 21(1):39. doi: 10.1186/s13075-019-1818-x
131. Wong V, Qiu C, Xu S, Law B, Zeng W, Wang H, et al. Ca Signalling Plays a Role in Celastrol-Mediated Suppression of Synovial Fibroblasts of Rheumatoid Arthritis Patients and Experimental Arthritis in Rats. Br J Pharmacol (2019) 176(16):2922–44. doi: 10.1111/bph.14718
132. Jannat A, John P, Bhatti A, Hayat M. Tomorou Attenuates Progression of Rheumatoid Arthritis Through Alteration in ULK-1 Independent Autophagy Pathway in Collagen Induced Arthritis Mice Model. Cell Death Dis (2019) 5:142. doi: 10.1038/s41420-019-0222-2
133. Shen C, Cai G, Peng J, Chen X. Autophagy Protects Chondrocytes From Glucocorticoids-Induced Apoptosis via ROS/Akt/FOXO3 Signaling. Osteoarthr Cartil (2015) 23(12):2279–87. doi: 10.1016/j.joca.2015.06.020
134. Yang G, Xia X, Zhong H, Shen J, Li S. Protective Effect of Tangeretin and 5-Hydroxy-6,7,8,3',4'-Pentamethoxyflavone on Collagen-Induced Arthritis by Inhibiting Autophagy via Activation of the ROS-AKT/mTOR Signaling Pathway. J Agric Food Chem (2021) 69(1):259–66. doi: 10.1021/acs.jafc.0c06801
135. Meng Q, Du X, Wang H, Gu H, Zhan J, Zhou Z. Astragalus Polysaccharides Inhibits Cell Growth and Pro-Inflammatory Response in IL-1β-Stimulated Fibroblast-Like Synoviocytes by Enhancement of Autophagy via PI3K/AKT/mTOR Inhibition. Apoptosis (2017) 22(9):1138–46. doi: 10.1007/s10495-017-1387-x
136. Yang G, Sun J, Lu K, Shan S, Li S, Sun C. Pterostilbene Coupled With Physical Exercise Effectively Mitigates Collagen-Induced Articular Synovial by Correcting the PI3K/Akt/NF-κB Signal Pathway. J Agric Food Chem (2021) 69(46):13821–30. doi: 10.1021/acs.jafc.1c05819
137. Li S, Chen J, Xie X, Tian J, Deng C, Wang J, et al. Autophagy Inhibitor Regulates Apoptosis and Proliferation of Synovial Fibroblasts Through the Inhibition of PI3K/AKT Pathway in Collagen-Induced Arthritis Rat Model. Am J Trans Res (2017) 9(5):2065–76. doi: 10.1136/annrheumdis-2017-eular.2044
138. Apel F, Zychlinsky A, Kenny E. The Role of Neutrophil Extracellular Traps in Rheumatic Diseases. Nat Rev Rheumatol (2018) 14(8):467–75. doi: 10.1038/s41584-018-0039-z
139. Kolaczkowska E, Kubes P. Neutrophil Recruitment and Function in Health and Inflammation. Nat Rev Immunol (2013) 13(3):159–75. doi: 10.1038/nri3399
140. Kaplan M. Role of Neutrophils in Systemic Autoimmune Diseases. Arthritis Res Ther (2013) 15(5):219. doi: 10.1186/ar4325
141. Yang D, de la Rosa G, Tewary P, Oppenheim J. Alarmins Link Neutrophils and Dendritic Cells. Trends Immunol (2009) 30(11):531–7. doi: 10.1016/j.it.2009.07.004
142. Mantovani A, Cassatella M, Costantini C, Jaillon S. Neutrophils in the Activation and Regulation of Innate and Adaptive Immunity. Nat Rev Immunol (2011) 11(8):519–31. doi: 10.1038/nri3024
143. Kenny EF, Herzig A, Krüger R, Muth A, Mondal S, Thompson PR, et al. Diverse Stimuli Engage Different Neutrophil Extracellular Trap Pathways. eLife (2017) 6:e24437. doi: 10.7554/eLife.24437
144. Zhou Y, Chen B, Mittereder N, Chaerkady R, Strain M, An L, et al. Spontaneous Secretion of the Citrullination Enzyme PAD2 and Cell Surface Exposure of PAD4 by Neutrophils. Front Immunol (2017) 8:1200. doi: 10.3389/fimmu.2017.01200
145. Yin J, He D, Jiang L, Cheng F, Guo Q, Huang S, et al. Influence of Cigarette Smoking on Rheumatoid Arthritis Risk in the Han Chinese Population. Front Med (Lausanne) (2017) 4:76. doi: 10.3389/fmed.2017.00076
146. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils Activate Plasmacytoid Dendritic Cells by Releasing Self-DNA-Peptide Complexes in Systemic Lupus Erythematosus. Sci Transl Med (2011) 3(73):73ra19. doi: 10.1126/scitranslmed.3001180
147. Delbosc S, Alsac J, Journe C, Louedec L, Castier Y, Bonnaure-Mallet M, et al. Porphyromonas Gingivalis Participates in Pathogenesis of Human Abdominal Aortic Aneurysm by Neutrophil Activation. Proof of Concept in Rats. PloS One (2011) 6(4):e18679. doi: 10.1371/journal.pone.0018679
148. Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight J, et al. NETs Are a Source of Citrullinated Autoantigens and Stimulate Inflammatory Responses in Rheumatoid Arthritis. Sci Transl Med (2013) 5(178):178ra40. doi: 10.1126/scitranslmed.3005580
149. Kaneko C, Kobayashi T, Ito S, Sugita N, Murasawa A, Nakazono K, et al. Circulating Levels of Carbamylated Protein and Neutrophil Extracellular Traps Are Associated With Periodontitis Severity in Patients With Rheumatoid Arthritis: A Pilot Case-Control Study. PloS One (2018) 13(2):e0192365. doi: 10.1371/journal.pone.0192365
150. Kilsgård O, Andersson P, Malmsten M, Nordin S, Linge H, Eliasson M, et al. Peptidylarginine Deiminases Present in the Airways During Tobacco Smoking and Inflammation can Citrullinate the Host Defense Peptide LL-37, Resulting in Altered Activities. Am J Respir Cell Mol Biol (2012) 46(2):240–8. doi: 10.1165/rcmb.2010-0500OC
151. Villanueva E, Yalavarthi S, Berthier C, Hodgin J, Khandpur R, Lin A, et al. Netting Neutrophils Induce Endothelial Damage, Infiltrate Tissues, and Expose Immunostimulatory Molecules in Systemic Lupus Erythematosus. J Immunol (2011) 187(1):538–52. doi: 10.4049/jimmunol.1100450
152. Blachère N, Parveen S, Fak J, Frank M, Orange D. Inflammatory But Not Apoptotic Death of Granulocytes Citrullinates Fibrinogen. Arthritis Res Ther (2015) 17:369. doi: 10.1186/s13075-015-0890-0
153. Spengler J, Lugonja B, Ytterberg A, Zubarev R, Creese A, Pearson M, et al. Release of Active Peptidyl Arginine Deiminases by Neutrophils Can Explain Production of Extracellular Citrullinated Autoantigens in Rheumatoid Arthritis Synovial Fluid. Arthritis Rheumatol (2015) 67(12):3135–45. doi: 10.1002/art.39313
154. Paoliello-Paschoalato A, Marchi L, de Andrade M, Kabeya L, Donadi E, Lucisano-Valim Y. Fcγ and Complement Receptors and Complement Proteins in Neutrophil Activation in Rheumatoid Arthritis: Contribution to Pathogenesis and Progression and Modulation by Natural Products. Evid Based Complement Alternat Med (2015) 2015:429878. doi: 10.1155/2015/429878
155. Pillinger M, Abramson S. The Neutrophil in Rheumatoid Arthritis. Rheum Dis Clin North Am (1995) 21(3):691–714. doi: 10.1016/S0889-857X(21)00463-4
156. Sur Chowdhury C, Giaglis S, Walker U, Buser A, Hahn S, Hasler P. Enhanced Neutrophil Extracellular Trap Generation in Rheumatoid Arthritis: Analysis of Underlying Signal Transduction Pathways and Potential Diagnostic Utility. Arthritis Res Ther (2014) 16(3):R122. doi: 10.1186/ar4579
157. Pérez-Sánchez C, Ruiz-Limón P, Aguirre M, Jiménez-Gómez Y, Arias-de la Rosa I, Ábalos-Aguilera M, et al. Diagnostic Potential of NETosis-Derived Products for Disease Activity, Atherosclerosis and Therapeutic Effectiveness in Rheumatoid Arthritis Patients. J Autoimmun (2017) 82:31–40. doi: 10.1016/j.jaut.2017.04.007
158. Wang W, Peng W, Ning X. Increased Levels of Neutrophil Extracellular Trap Remnants in the Serum of Patients With Rheumatoid Arthritis. Int J Rheum Dis (2018) 21(2):415–21. doi: 10.1111/1756-185x.13226
159. Gupta A, Hasler P, Holzgreve W, Gebhardt S, Hahn S. Induction of Neutrophil Extracellular DNA Lattices by Placental Microparticles and IL-8 and Their Presence in Preeclampsia. Hum Immunol (2005) 66(11):1146–54. doi: 10.1016/j.humimm.2005.11.003
160. Carmona-Rivera C, Carlucci PM, Moore E, Lingampalli N, Uchtenhagen H, James E, et al. Synovial Fibroblast-Neutrophil Interactions Promote Pathogenic Adaptive Immunity in Rheumatoid Arthritis. Sci Immunol (2017) 2(10):eaag3358. doi: 10.1126/sciimmunol.aag3358
161. Wright H, Makki F, Moots R, Edwards S. Low-Density Granulocytes: Functionally Distinct, Immature Neutrophils in Rheumatoid Arthritis With Altered Properties and Defective TNF Signalling. J Leukoc Biol (2017) 101(2):599–611. doi: 10.1189/jlb.5A0116-022R
162. Abildtrup M, Kingsley G, Scott D. Calprotectin as a Biomarker for Rheumatoid Arthritis: A Systematic Review. J Rheumatol (2015) 42(5):760–70. doi: 10.3899/jrheum.140628
163. Hammer HB, Ødegård S, Syversen SW, Landewé R, van der Heijde D, Uhlig T, et al. Calprotectin (a Major S100 Leucocyte Protein) Predicts 10-Year Radiographic Progression in Patients With Rheumatoid Arthritis. Ann Rheum Dis (2010) 69(1):150–4. doi: 10.1136/ard.2008.103739
164. Bach M, Moon J, Moore R, Pan T, Nelson J, Lood C. A Neutrophil Activation Biomarker Panel in Prognosis and Monitoring of Patients With Rheumatoid Arthritis. Arthritis Rheumatol (2020) 72(1):47–56. doi: 10.1002/art.41062
165. Zhu M, Yuan K, Lu Q, Zhu Q, Zhang S, Li X, et al. Emodin Ameliorates Rheumatoid Arthritis by Promoting Neutrophil Apoptosis and Inhibiting Neutrophil Extracellular Trap Formation. Mol Immunol (2019) 112:188–97. doi: 10.1016/j.molimm.2019.05.010
166. Dwivedi N, Upadhyay J, Neeli I, Khan S, Pattanaik D, Myers L, et al. Felty's Syndrome Autoantibodies Bind to Deiminated Histones and Neutrophil Extracellular Chromatin Traps. Arthritis Rheum-US (2012) 64(4):982–92. doi: 10.1002/art.33432
167. Cho Y, Challa S, Moquin D, Genga R, Ray T, Guildford M, et al. Phosphorylation-Driven Assembly of the RIP1-RIP3 Complex Regulates Programmed Necrosis and Virus-Induced Inflammation. Cell (2009) 137(6):1112–23. doi: 10.1016/j.cell.2009.05.037
168. He S, Wang L, Miao L, Wang T, Du F, Zhao L, et al. Receptor Interacting Protein Kinase-3 Determines Cellular Necrotic Response to TNF-Alpha. Cell (2009) 137(6):1100–11. doi: 10.1016/j.cell.2009.05.021
169. Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, et al. Fas Triggers an Alternative, Caspase-8-Independent Cell Death Pathway Using the Kinase RIP as Effector Molecule. Nat Immunol (2000) 1(6):489–95. doi: 10.1038/82732
170. Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, et al. RIP3, an Energy Metabolism Regulator That Switches TNF-Induced Cell Death From Apoptosis to Necrosis. Science (2009) 325(5938):332–6. doi: 10.1126/science.1172308
171. Wang X, Gessier F, Perozzo R, Stojkov D, Hosseini A, Amirshahrokhi K, et al. RIPK3-MLKL-Mediated Neutrophil Death Requires Concurrent Activation of Fibroblast Activation Protein-α. J Immunol (2020) 205(6):1653–63. doi: 10.4049/jimmunol.2000113
172. Trimova G, Yamagata K, Iwata S, Hirata S, Zhang T, Uemura F, et al. Tumour Necrosis Factor Alpha Promotes Secretion of 14-3-3η by Inducing Necroptosis in Macrophages. Arthritis Res Ther (2020) 22(1):24. doi: 10.1186/s13075-020-2110-9
173. Zeng F, Wen W, Cui W, Zheng W, Liu Y, Sun X, et al. Central Role of RIPK1-VDAC1 Pathway on Cardiac Impairment in a non-Human Primate Model of Rheumatoid Arthritis. J Mol Cell Cardiol (2018) 125:50–60. doi: 10.1016/j.yjmcc.2018.10.015
174. Chen Y, Zhu C, Zhu F, Dai B, Song S, Wang Z, et al. Necrostatin-1 Ameliorates Adjuvant Arthritis Rat Articular Chondrocyte Injury via Inhibiting ASIC1a-Mediated Necroptosis. Biochem Biophys Res Commun (2018) 504(4):843–50. doi: 10.1016/j.bbrc.2018.09.031
175. Jhun J, Lee S, Kim S, Ryu J, Kwon J, Na H, et al. RIPK1 Inhibition Attenuates Experimental Autoimmune Arthritis via Suppression of Osteoclastogenesis. J Transl Med (2019) 17(1):84. doi: 10.1186/s12967-019-1809-3
176. Lee S, Kwon J, Kim S, Jung K, Cho M. Interferon-Gamma Regulates Inflammatory Cell Death by Targeting Necroptosis in Experimental Autoimmune Arthritis. Sci Rep-UK (2017) 7(1):10133. doi: 10.1038/s41598-017-09767-0
177. Ali H, Caballero R, Dong SXM, Gajnayaka N, Vranjkovic A, Ahmed D, et al. Selective Killing of Human M1 Macrophages by Smac Mimetics Alone and M2 Macrophages by Smac Mimetics and Caspase Inhibition. J Leukoc Biol (2021) 110(4):693–710. doi: 10.1002/jlb.4a0220-114rr
178. Saeki Y, Okita Y, Igashira-Oguro E, Udagawa C, Murata A, Tanaka T, et al. Modulation of TNFR 1-Triggered Two Opposing Signals for Inflammation and Apoptosis via RIPK 1 Disruption by Geldanamycin in Rheumatoid Arthritis. Clin Rheumatol (2021) 40(6):2395–405. doi: 10.1007/s10067-021-05579-w
179. Weisel K, Berger S, Thorn K, Taylor P, Peterfy C, Siddall H, et al. A Randomized, Placebo-Controlled Experimental Medicine Study of RIPK1 Inhibitor GSK2982772 in Patients With Moderate to Severe Rheumatoid Arthritis. Arthritis Res Ther (2021) 23(1):85. doi: 10.1186/s13075-021-02468-0
180. Spel L, Martinon F. Inflammasomes Contributing to Inflammation in Arthritis. Immunol Rev (2020) 294(1):48–62. doi: 10.1111/imr.12839
181. Shen H, Yang Y, Meng X, Luo X, Li X, Shuai Z, et al. NLRP3: A Promising Therapeutic Target for Autoimmune Diseases. Autoimmun Rev (2018) 17(7):694–702. doi: 10.1016/j.autrev.2018.01.020
182. Saavedra P, Demon D, Van Gorp H, Lamkanfi M. Protective and Detrimental Roles of Inflammasomes in Disease. Semin Immunopathol (2015) 37(4):313–22. doi: 10.1007/s00281-015-0485-5
183. Chadha S, Behl T, Bungau S, Kumar A, Arora R, Gupta A, et al. Mechanistic Insights Into the Role of Pyroptosis in Rheumatoid Arthritis. Curr Res Transl Med (2020) 68(4):151–8. doi: 10.1016/j.retram.2020.07.003
184. Burska A, Boissinot M, Ponchel F. Cytokines as Biomarkers in Rheumatoid Arthritis. Mediators Inflamm (2014) 2014:545493. doi: 10.1155/2014/545493
185. Li Y, Zheng J, Liu J, Yang J, Liu Y, Wang C, et al. Succinate/NLRP3 Inflammasome Induces Synovial Fibroblast Activation: Therapeutical Effects of Clematichinenoside AR on Arthritis. Front Immunol (2016) 7:532. doi: 10.3389/fimmu.2016.00532
186. Choulaki C, Papadaki G, Repa A, Kampouraki E, Kambas K, Ritis K, et al. Enhanced Activity of NLRP3 Inflammasome in Peripheral Blood Cells of Patients With Active Rheumatoid Arthritis. Arthritis Res Ther (2015) 17:257. doi: 10.1186/s13075-015-0775-2
187. Ruscitti P, Cipriani P, Di Benedetto P, Liakouli V, Berardicurti O, Carubbi F, et al. Monocytes From Patients With Rheumatoid Arthritis and Type 2 Diabetes Mellitus Display an Increased Production of Interleukin (IL)-1β via the Nucleotide-Binding Domain and Leucine-Rich Repeat Containing Family Pyrin 3(NLRP3)-Inflammasome Activation: A Possible Implication for Therapeutic Decision in These Patients. Clin Exp Immunol (2015) 182(1):35–44. doi: 10.1111/cei.12667
188. Mathews R, Robinson J, Battellino M, Wong C, Taylor J, Eyre S, et al. Evidence of NLRP3-Inflammasome Activation in Rheumatoid Arthritis (RA); Genetic Variants Within the NLRP3-Inflammasome Complex in Relation to Susceptibility to RA and Response to Anti-TNF Treatment. Ann Rheum Dis (2014) 73(6):1202–10. doi: 10.1136/annrheumdis-2013-203276
189. Wu X, Li K, Yang H, Yang B, Lu X, Zhao L, et al. Complement C1q Synergizes With PTX3 in Promoting NLRP3 Inflammasome Over-Activation and Pyroptosis in Rheumatoid Arthritis. J Autoimmun (2020) 106:102336. doi: 10.1016/j.jaut.2019.102336
190. Ge G, Bai J, Wang Q, Liang X, Tao H, Chen H, et al. Punicalagin Ameliorates Collagen-Induced Arthritis by Downregulating M1 Macrophage and Pyroptosis via NF-κB Signaling Pathway. Sci China Life Sci (2021). doi: 10.1007/s11427-020-1939-1
191. Vande Walle L, Van Opdenbosch N, Jacques P, Fossoul A, Verheugen E, Vogel P, et al. Negative Regulation of the NLRP3 Inflammasome by A20 Protects Against Arthritis. Nature (2014) 512(7512):69–73. doi: 10.1038/nature13322
192. Joosten L, Abdollahi-Roodsaz S, Heuvelmans-Jacobs M, Helsen M, van den Bersselaar L, Oppers-Walgreen B, et al. T Cell Dependence of Chronic Destructive Murine Arthritis Induced by Repeated Local Activation of Toll-Like Receptor-Driven Pathways: Crucial Role of Both Interleukin-1beta and Interleukin-17. Arthritis Rheum-US (2008) 58(1):98–108. doi: 10.1002/art.23152
193. Li Y, Shen Y, Jin K, Wen Z, Cao W, Wu B, et al. The DNA Repair Nuclease MRE11A Functions as a Mitochondrial Protector and Prevents T Cell Pyroptosis and Tissue Inflammation. Cell Metab (2019) 30(3):477–92.e6. doi: 10.1016/j.cmet.2019.06.016
194. Wu X, Ren G, Zhou R, Ge J, Chen F. The Role of Ca in Acid-Sensing Ion Channel 1a-Mediated Chondrocyte Pyroptosis in Rat Adjuvant Arthritis. Lab Invest (2019) 99(4):499–513. doi: 10.1038/s41374-018-0135-3
195. Zu S, Feng Y, Zhu C, Wu X, Zhou R, Li G, et al. Acid-Sensing Ion Channel 1a Mediates Acid-Induced Pyroptosis Through Calpain-2/Calcineurin Pathway in Rat Articular Chondrocytes. Cell Biol Int (2020) 44(10):2140–52. doi: 10.1002/cbin.11422
Keywords: rheumatoid arthritis, programmed cell death, apoptosis, NETosis, necroptosis, pyroptosis, autophagy
Citation: Zhao J, Jiang P, Guo S, Schrodi SJ and He D (2021) Apoptosis, Autophagy, NETosis, Necroptosis, and Pyroptosis Mediated Programmed Cell Death as Targets for Innovative Therapy in Rheumatoid Arthritis. Front. Immunol. 12:809806. doi: 10.3389/fimmu.2021.809806
Received: 05 November 2021; Accepted: 13 December 2021;
Published: 24 December 2021.
Edited by:
Haitao Wang, National Cancer Institute, United StatesReviewed by:
Malvina Hoxha, Catholic University Our Lady of Good Counsel, AlbaniaAntonella Capozzi, Sapienza University of Rome, Italy
Cristiana Barbati, Sapienza University of Rome, Italy
Copyright © 2021 Zhao, Jiang, Guo, Schrodi and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Steven J. Schrodi, Schrodi@wisc.edu; Shicheng Guo, Shicheng.Guo@wisc.edu; Dongyi He, hedongyi1967@shutcm.edu.cn