 Justin Jacobse
Justin Jacobse Jing Li2
Jing Li2 Janneke N. Samsom
Janneke N. Samsom Jeremy A. Goettel
Jeremy A. Goettel- 1Department of Pediatrics, Willem-Alexander Children’s Hospital, Leiden University Medical Center, Leiden, Netherlands
- 2Department of Pathology, Microbiology, and Immunology, Vanderbilt University, Nashville, TN, United States
- 3Department of Medicine, Division of Gastroenterology, Hepatology and Nutrition, Vanderbilt University Medical Center, Nashville, TN, United States
- 4Department of Pediatrics, Sophia Children’s Hospital, Erasmus University, Erasmus University Medical Center, Rotterdam, Netherlands
- 5Laboratory of Pediatrics, Division of Gastroenterology and Nutrition, Erasmus University Medical Center, Rotterdam, Netherlands
- 6Program in Cancer Biology, Vanderbilt University School of Medicine, Nashville, TN, United States
- 7Vanderbilt Institute for Infection, Immunology, and Inflammation, Vanderbilt University Medical Center, Nashville, TN, United States
- 8Center for Mucosal Inflammation and Cancer, Vanderbilt University Medical Center, Nashville, TN, United States
FOXP3+ regulatory T cells (Treg cells) are a specialized population of CD4+ T cells that restrict immune activation and are essential to prevent systemic autoimmunity. In the intestine, the major function of Treg cells is to regulate inflammation as shown by a wide array of mechanistic studies in mice. While Treg cells originating from the thymus can home to the intestine, the majority of Treg cells residing in the intestine are induced from FOXP3neg conventional CD4+ T cells to elicit tolerogenic responses to microbiota and food antigens. This process largely takes place in the gut draining lymph nodes via interaction with antigen-presenting cells that convert circulating naïve T cells into Treg cells. Notably, dysregulation of Treg cells leads to a number of chronic inflammatory disorders, including inflammatory bowel disease. Thus, understanding intestinal Treg cell biology in settings of inflammation and homeostasis has the potential to improve therapeutic options for patients with inflammatory bowel disease. Here, the induction, maintenance, trafficking, and function of intestinal Treg cells is reviewed in the context of intestinal inflammation and inflammatory bowel disease. In this review we propose intestinal Treg cells do not compose fixed Treg cell subsets, but rather (like T helper cells), are plastic and can adopt different programs depending on microenvironmental cues.
Introduction
The immune system provides host protection against pathogenic microorganisms as well as anti-tumor immunity. Importantly, regulatory mechanisms prevent immune reactivity against self- and harmless foreign antigens. However, in some individuals this form of tolerance is compromised leading to various autoimmune conditions. Key observations made in the 1960’s and 1970’s began to unveil the mediators of autoimmunity and the concept of immunological tolerance. In the early 60’s, Jacques Miller showed that thymectomy of mice at birth results in a lack of peripheral T cell responses to skin grafts, identifying the thymus as the essential organ of T cell development (1, 2). Yet, it was the seminal finding in 1970 by Richard Gershon that the thymus also gives rise to cells essential for immune tolerance (3). Then in 1976, the developmental timing of tolerance was first reported by Kojima and colleagues who showed that thymectomy in mice between 2-4 days after birth leads to autoimmune thyroiditis, whereas no lesions are observed if the thymectomy is performed at birth or after day 5 (4). This autoimmunity following thymectomy can be prevented by the adoptive transfer of thymocytes or splenocytes from adult euthymic mice (4–6). Intriguingly, in the thymus a limited number of autoreactive CD4+ T cells differentiate into highly suppressive T cells, termed regulatory T cells (Treg cells), in a process known as agonist selection to ensure central tolerance to self-antigens, and thereby prevent autoimmunity (7–9). Despite the recognition that T cells could play a “suppressor” role in the control of effector T cell responses, it would take nearly two decades to define this population. Seminal work by Fiona Powrie in 1993 described two populations of T cells that can induce or protect immunodeficient mice from chronic colitis following adoptive transfer of T cells based on the expression level of CD45RB (naïve T cells are CD45Bhigh) (10). Shortly after, Sakaguchi et al. reported that CD4+ T cells that immunosuppress T helper cells express the cell surface marker CD25 (11) and that these cells reside within the CD45RBlow fraction of T cells, the same fraction that prevents colitis mediated by adoptive transfer of CD45RBhigh T cells (12).
Treg cells are essential for maintaining both central as well as peripheral immunological tolerance (13) and are found in many tissues and mucosal surfaces including the intestine. The intestinal mucosa is lined by a single cell layer of columnar epithelial cells with the lamina propria (LP) below the epithelial cells and muscularis mucosa at the base. The LP serves as an effector site, whereas adaptive immune cells are primed within inductive sites including lymph nodes that drain anatomically distinct regions of the intestine and/or mucosa-associated lymphoid tissue such as Peyer’s patches. For example, the duodenum drains into a lymph node embedded in pancreatic tissue, while the jejunum, ileum, cecum, and ascending colon drain into the mesenteric lymph nodes (mLNs). Two small lymph nodes in the pancreatic tissue also drain the transverse colon and descending colon while the distal colon and rectum primarily drain to caudal lymph nodes (14–19).
Just as different anatomical regions of the intestine have specific functions, immune cells residing in, or trafficking to, distinct regions of the gut carry out specialized functions. Naïve T cells circulate from blood into lymph nodes via afferent lymphatic vessels where they interact with antigen-presenting cells (APCs) such as dendritic cells (DCs) that have acquired peptides in the intestinal lumen or LP and migrated to the draining lymph nodes. Together, the local microenvironment in the lymph nodes draining these specific intestinal segments and the function of the resident DCs contribute to mounting either tolerogenic or pro-inflammatory adaptive immune responses. Besides the draining lymph node compartment, the intestinal microenvironment in the various segments of the GI-tract is also markedly different. In the small intestine (SI), the relative amount of soluble, luminal food antigens decreases from the duodenum to the terminal ileum and colon. In stark contrast, the large intestinal micro-environment is characterized by high abundance and diversity of commensal bacteria.
One of the major functions of the intestinal immune system is to maintain immune homeostasis among the non-pathogenic gut microbiome, harmless exogenous food antigens, pathogenic microbes, and damage-associated self-antigens. Microenvironmental cues by both immune and non-immune cells alike help define the phenotype and function of intestinal Treg cells. Suppression of effector T cells by Treg cells requires a T-cell receptor (TCR)-dependent signal. Under physiological conditions, Treg cells regulate immune responses to self-antigens, microbes, and food antigens. While Treg cell activation requires a TCR-specific signal, Treg cells can also suppress effector T cells with different antigen specificity through a process called bystander suppression that contributes to overall immune homeostasis. In addition, Treg cells can secrete immunosuppressive cytokines such as interleukin (IL)-10. Consequently, loss of suppressive function caused by defective Treg cells results in intestinal inflammation in mice and contributes to inflammatory bowel disease (IBD) in humans.
This review will provide the reader with the current evidence regarding the role Treg cells play in maintaining immune homeostasis in the colon and SI, both in mouse models and in human disease.
Intestinal Treg Cells Are a Diverse Class of Immunoregulatory Immune Cells
The most commonly studied Treg cell populations are defined by expression of CD4, CD25, and Forkhead box P3 (FOXP3). Expression of the transcription factor FOXP3 is necessary to bestow suppressive capacity on Treg cells and maintain the Treg cell phenotype [Reviewed in (20)]. Unfortunately, classification of human FOXP3+ cells is not as definitive as in mice as both FOXP3 and CD25 can be observed in activated conventional T cells (21). To identify human Treg cells by flow cytometry, a minimum set of cell surface markers consisting of CD3, CD4, CD25, CD127, and FOXP3 are used with Ki67 and CD45RA providing additional information about Treg cell proliferation and activation status (21, 22). Studying human Treg cells is further complicated by FOXP3 being an intranuclear protein, and as such requires fixation and permeabilization that limits the ability to purify cells using cell sorting for functional characterization.
Other regulatory CD4+ T cells have been described that do not express FOXP3 but nevertheless regulate intestinal immune responses, possessing both distinct and similar features of FOXP3+ Treg cells. Type 1 regulatory T cells (Tr1) were initially described by Roncarolo et al. (23, 24) as CD4+FOXP3neg T cells that co-express CD49b, LAG-3, CD226, CCR5, and PD1 (25, 26). In contrast to FOXP3+ Treg cells, Tr1 cells are more abundant in the SI and Peyer’s patches (PP) and rapidly secrete IL-10 and TGFβ upon stimulation in the absence of IL-4 (27). Functionally, Tr1 cells suppress antigen-specific T-cell proliferation in an IL-10-dependent manner and are protective in the adoptive naïve T cell transfer model of colitis (23, 24, 28). Whereas both FOXP3+ Treg cells and Tr1 cells produce IL-10, Tr1 cells seem to be especially important for maintaining tolerance to commensal bacteria. Nevertheless, FOXP3+ Treg cells are still necessary for initial tolerance induction (29–31). For the remainder of this review we will focus on FOXP3+ Treg cells.
According to the recommended nomenclature, Treg cells can be subdivided into tTreg cells when they originate from the thymus and pTreg cells when they are locally induced. In addition, naïve T cells can be cultured in vitro under conditions that promote Treg cell induction; these are commonly referred to as iTreg cells (32). In vitro induced Treg cells differ from ex vivo Treg cells (33) and this should be taken into consideration when interpreting studies performed with iTreg cells. Regardless of their site of origin or ontogeny, Treg cells must exhibit suppressive properties to be considered bona fide. Assessing Treg cell function is generally carried out using in vitro suppression assays. Despite some limitations (34), this assay quantifies the proliferation of conventional T cells when activated in the presence of Treg cells. Altogether, Treg cells are defined by a collection of markers and confirmation of suppressive function commonly performed via in vitro suppression assay.
The peripheral Treg cell pool can be further classified into three groups based on location and function: central, effector, and tissue-resident. The main properties of these populations, as reviewed in detail by Liston and Gray (35), will be summarized here. Central Treg cells (mouse- CD62LhighCCR7+ or CD45RAhighCD25low) are considered to be naïve and represent the majority of Treg cells in circulation and in secondary lymphoid organs. Effector memory, or activated Treg cells, (mouse- CD62LlowCCR7lowCD44hiKLRG1+CD103+ or CD25RAlowCD25hi) possess characteristics of conventional activated CD4+ T cells with recent antigen exposure and are less abundant. In contrast to the central and effector Treg cells, tissue Treg cells reside in non-lymphoid tissue such as the colon. In absence of inflammation, the majority of Treg cells in the gut are tissue-resident pTreg cells.
Like conventional T cells, Treg cells are governed by transcription factors (e.g., T-BET, STAT3, and IRF4) that also regulate the function of effector T cells during Th1-, Th2-, and Th17-mediated inflammation respectively (36–38). Thus, it is not surprising that the transcriptome of tissue Treg cells varies according to their location which regulates expression of these transcription factors [Reviewed in (39)]. For many Treg cell genes regulated by the local environment, expression tends to reflect a gradient rather than binary, suggesting plasticity and adaptation to their microenvironment (40). Although not entirely definitive, some markers used to discriminate intestinal Treg cells from each other are RORγt (microbiome, highly suppressive), IL-33R (tissue protection during inflammation), IL-10 (suppression of autoimmunity), GATA3 (suppression of Th2), HELIOS (probable thymic origin), and NRP1 (probable thymic origin) (Table 1).
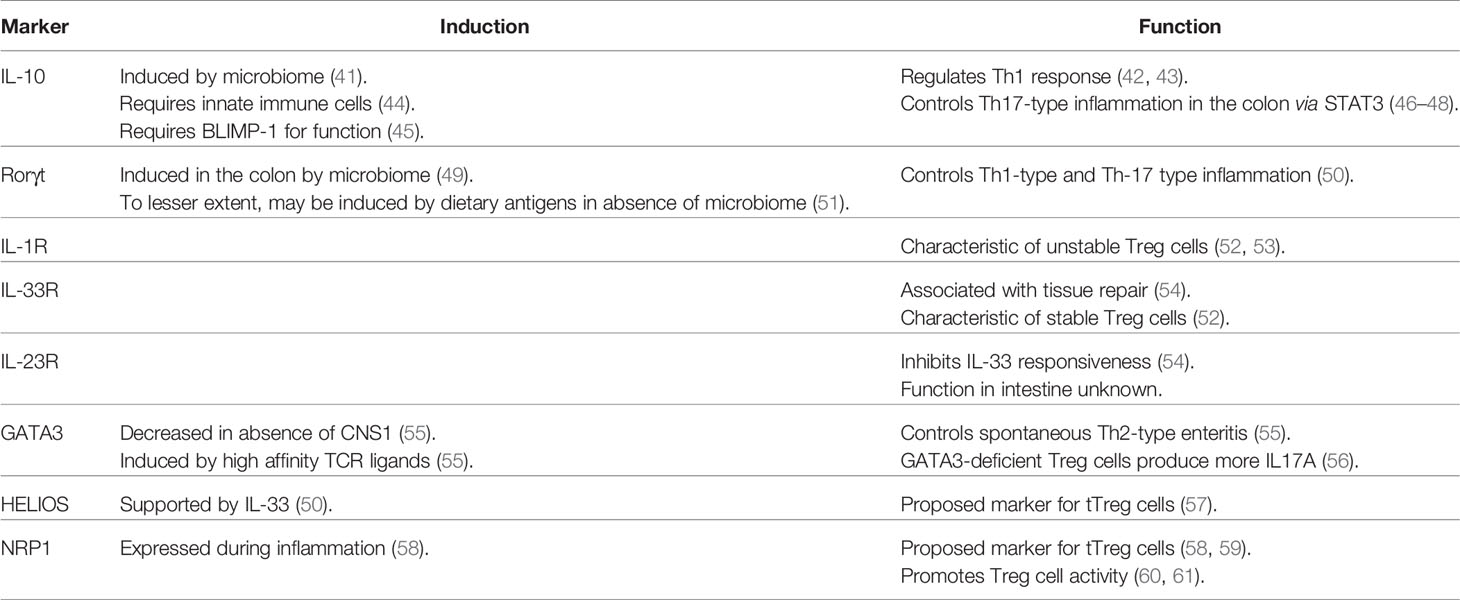
Table 1 Markers of intestinal Treg cells and the gist of their phenotype translated to intestinal immune homeostasis.
The T Cell Receptor Repertoire of Intestinal Treg Cells Is Influenced by Luminal Antigens
In the intestine, Treg cells have a distinct TCR repertoire (repertoire) from both non-Treg cells as well as Treg cells residing in other organs, suggesting that tissue-specific factors shape the Treg cell repertoire (62). Germ-free (GF) mice have fewer intestinal Treg cells than conventionally housed mice. When CD4+CD25+ Treg cells are isolated from conventionally housed mice or GF mice and co-injected with naïve T cells in the adoptive T cell transfer model of colitis, recipients of GF Treg cells exhibit increased inflammation (63). It has been suggested that these effects may be due to the limited repertoire of GF Treg cells as compared to the larger repertoire of Treg cells isolated from conventionally housed mice (64). This would be consistent with locally induced intestinal Treg cells having TCRs specific for microbial antigens and small quantities of antigens being sufficient for efficient pTreg induction (65). Indeed, in vitro data show that some TCRs of colonic Treg cells are specific for food or microbial antigens in stool from conventional mice but not from GF mice (62). While a limited Treg cell repertoire is one possible explanation, it has also been shown that CD4+CD25+ cells from GF mice express less Foxp3 and exhibit reduced functionality, which could also account for the increased inflammation (66). Interestingly, a study from the Rudensky laboratory showed that activated CD25+ cells in Foxp3-/-mice have a similar repertoire as Treg cells in wild-type mice, which implies a cell-intrinsic mechanism directing Treg cell TCR development but likely restricted to those of thymic origin (67). Furthermore, mice that develop spontaneous colitis due to defects in IL-2, IL-10, or TGFβ have many effector and memory T cells with TCRs that in wild-type mice are found on colonic FOXP3+ cells (62). These studies suggest that Treg cells in physiological conditions exert TCR-specific suppression of inflammation mediated by other T cell subsets. The importance of TCR diversity in Treg cells comes from a study showing that Treg cells isolated from mice with a limited repertoire are not able to suppress the development of spontaneous, microbiome-dependent, Th17-type intestinal inflammation (68).
It is clear that a proportion of colonic Treg cells exhibit specificity for colonic luminal antigens, mostly microbial (62, 69). However, the ontogeny of intestinal Treg cells with TCR specificity for the gut microbiota is not entirely known. The TCR specificity of a Treg in itself does not necessarily pre-determine the location in which the cell will reside. This follows from the observation that a T cell expressing a TCR from a pTreg becomes either a pTreg cell or an intraepithelial lymphocyte, dependent on the location (70). T cells in this model were generated by transferring the nucleus of an Nrp1lowFoxp3+ cell from the mLN to the donor cell, i.e. generating a transnuclear mouse. In the SI these cells are poised to become intraepithelial lymphocytes (in a FOXP3-independent manner), whereas in the mLN they are poised to become pTreg cells, as evidenced by accumulation of these cells in these respective locations (70).
Although colonic Treg cell TCRs retrovirally expressed in thymocytes were reported to be unable to facilitate thymic Treg cell differentiation (62), a fraction of thymic Treg cells share TCRs with pTreg cells indicating that a portion of intestinal Treg cells originate from the thymus (69). Surprisingly, a recent landmark paper from the Diehl laboratory shows that microbial antigens are presented to T cells in the thymus, suggesting that a part of the intestinal Treg cells can originate from the thymus (71). Additional evidence highlighting the influence of the microbiome on colonic Treg cell TCR comes from Cebula et al. who reported that the repertoire of colonic Treg cells changes following antibiotic treatment (69). In this study, antibiotic treatment decreased some TCR clones and expanded other TCR clones. However, overall, antibiotic treatment did not alter the repertoire diversity of colonic Treg cells as compared to other Treg cells. Furthermore, the TCR clones that did change with antibiotic treatment were also found on thymic Treg cells. Finally, colonic Treg cells TCRs cloned into hybridomas were reactive against fecal extracts were also found on thymic Treg cells. These findings can be interpreted to mean at least some intestinal Treg cells originate from the thymus (69).
Collectively, these highly informative studies raise new questions that need to be reviewed with a few considerations. Several of the studies listed above employ fixed TCR-β chains. When using a transgenic mouse with fixed TCR-β, the TCR diversity and antigen specificity is determined by the TCR-α chain, allowing for analysis of the diversity of the repertoire in different T cell subsets with high certainty but not reflecting the true diversity of the human repertoire (72). Nevertheless, as a whole the data strongly argue that a large proportion of intestinal Treg cells have TCRs specific to microbial antigens.
Mucosal CD103+ Dendritic Cells and Retinoic Acid Are Pivotal Players in the Induction of Small Intestinal Treg Cells
Oral tolerance develops upon first encounter of the antigen ovalbumin (OVA), which is a harmless exogenous chicken egg white protein and the most widely used model food antigen. Oral tolerance to OVA is critically dependent on resident intestinal DCs carrying the antigen to gut-associated lymphoid tissue (GALT) and mLN (73). In contrast to the spleen, OVA-specific FOXP3+ Treg cells are preferentially induced in these two sites, emphasizing the compartmentalization of this response (Figure 1) (74–76). Within 24-48 hours after OVA oral gavage, CD25+ Treg cells differentiate in PP and mLN and exhibit functional suppressive capacity, conferring oral tolerance when adoptively transferred to OVA-naïve recipient mice (75). Subsequent studies demonstrated that a large proportion of these mucosally-induced pTreg cells express FOXP3 (74, 76). Since Treg cell induction in response to a harmless dietary antigen occurs with higher frequency in mucosal versus non-mucosal draining lymphoid tissue, it was questioned whether particular subsets of DCs in the PP and mLN are required. As compared to splenic DCs, SI DCs are better able to convert naïve CD4+ cells to stable suppressive FOXP3+ cells when stimulated with anti-CD3 in the presence of TGFβ in vitro. Furthermore, DCs in the SI and cLP, as well as their respective draining lymph nodes, can be divided into four subsets based on CD11b and CD103 expression (19). CD103+ DCs are generally CD11c+MHCII+78 and represent a significant population of SI DCs, with a smaller population of cLP DCs being CD11c+CD103+. SI LP CD103+ DCs are more efficient in inducing Treg cells as compared to LP CD103neg DCs, which is attributed to retinoic acid (RA) being a co-factor for TGFβ-dependent Treg cell induction, with TGFβ thought to be the rate-limiting factor (74, 76, 77).
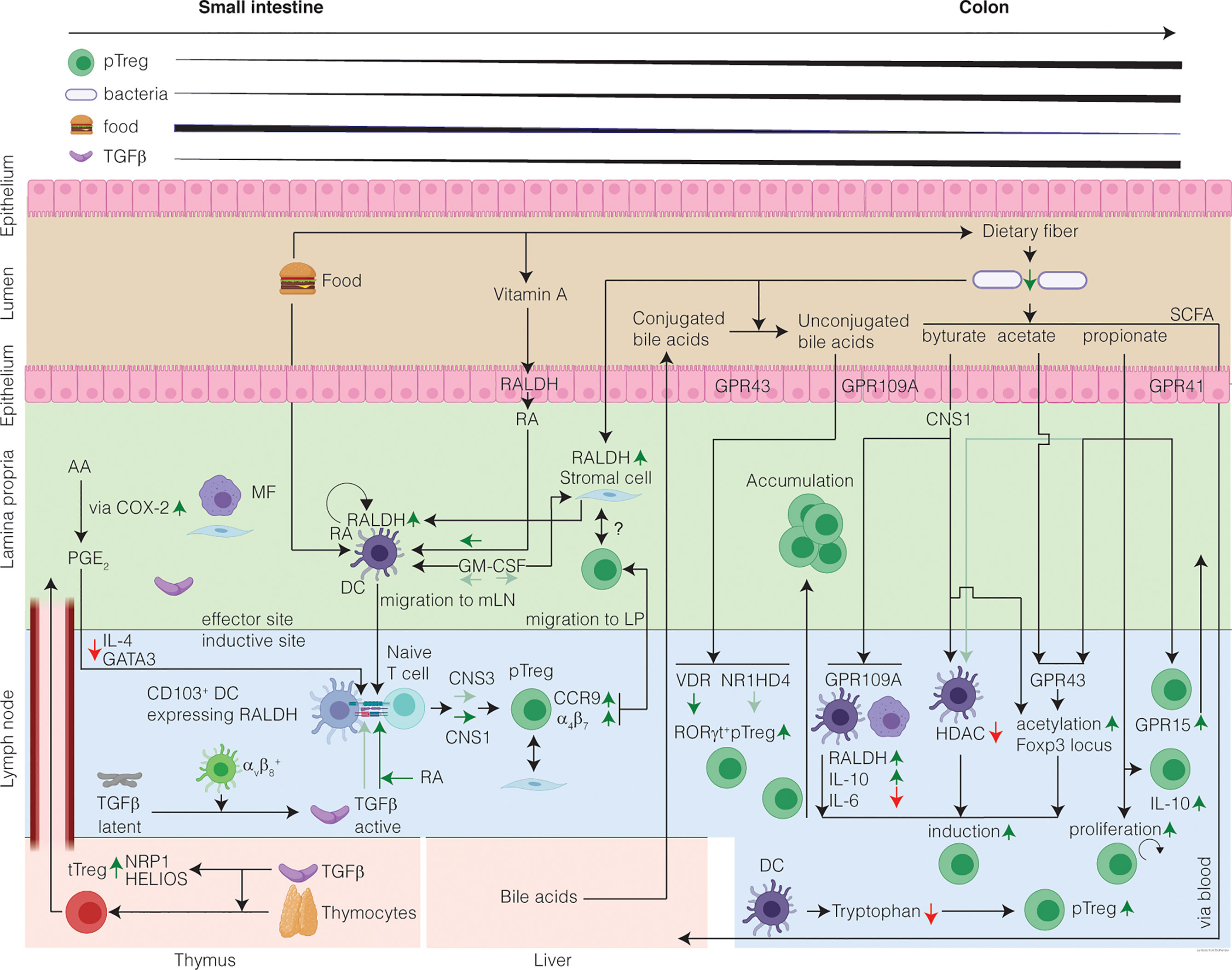
Figure 1 The healthy intestine favors Treg-cell induction in response to harmless antigen and local microenvironmental conditioning maintains the Treg cell phenotype. The mechanisms of intestinal Treg cell induction depend on the structure of the encountered antigen and on the inductive site: in the SI encounter of harmless food antigens predominates eliciting DC-mediated Treg cell induction from naïve T cells. In the colon, commensal microbiota produce SCFA which in presence of TGFβ induce RORγt+ Treg cells. In the SI inductive sites, DC-derived TGFβ induction converts circulating naïve T cells into pTreg cells, which is further potentiated by diet-derived RA. Treg cell induction in the SI is CNS1-dependent with a minor contribution from CNS3. In the colon inductive sites, the SCFA induce Treg cells. SCFA induce acetylation of the Foxp3 locus and alter the phenotype of the pTreg cells by increasing RORγt. Moreover, bile acids unconjugated by the microbiome contribute to the induction of colonic RORγt+ Treg cells. After induction, pTreg cells migrate from the inductive sites to the LP, where they accumulate and maintain immune homeostasis. In the LP, Treg cells might interact with stromal cells, which receive signals from the microbiome, through a cell-contact dependent mechanism. A portion of the intestinal Treg cells is tTreg cells, induced in the thymus in presence of TGFβ, and that have migrated to the intestine and are highly expressing putative markers for tTreg cells. Finally, a gradient of Treg cells exists increasing from SI to colon. Question marks indicate research areas that remain relatively unexplored.
The SI microenvironment is high in RA as enterocytes metabolize retinol (vitamin A) to RA via retinal aldehyde dehydrogenases (RALDH) (78). CD103+ DCs in the mLN also highly express RALDH (encoded by Aldh1a2) and produce RA, whereas CD103neg DCs in the mLN do not (76). In addition, TGFβ induces CD103+ expression on DCs (77) and selectively potentiates the ability of CD103+ but not CD103neg DCs to induce Treg cells in a dose-dependent manner. Moreover, RA enhances active TGFβ to induce FOXP3 in T cells in vitro by both GALT and splenic DCs, but this is specific to the CD103+ DC subset (74, 76). Furthermore, IL-2 is required for this induction as anti-IL-2 abolishes in vitro induction of FOXP3+ cells in the presence of RA and TGFβ. Another contributing factor to DC-mediated Treg-cell induction is the interaction of mucosal DCs with stromal cells, which has been shown to imprint RA production on DCs. Stromal cell production of RA is independent of vitamin A, but rather depends on the microbiome (79). Importantly, these DC-mediated induced Treg cells are functional in vivo as demonstrated by their ability to suppress colitis in the adoptive T cell transfer setting (77). Altogether, these studies show that a specialized subset of CD103+ DCs present in GALT favors the Treg cell induction in vitro and in vivo, requires TGFβ and IL-2, and is potentiated by RA. The exact mechanism by which RA potentiates TGFβ to induce Treg cells remains to be elucidated.
Like Treg cells, the distribution of DCs along the GI-tract is not random. The fraction of CD11b+CD103+ DCs in the colon is lower than in the SI, whereas the CD11bnegCD103+ DC subset comprises a larger fraction of the cLP DCs, with a similar pattern in their respective draining lymph nodes (19). Consistent with this gradient and the above described function of CD103, RALDH activity in the SI is highest as compared to other compartments such as the mLN and colon (80). Additionally, CD103 expression by DCs in the mucosa is higher than DCs in the gut draining lymph nodes (19). Interestingly, only SI DCs, but not cLP DCs, can present OVA in the mLN (19). Despite this gradient of CD11b+CD103b+ DCs, Treg cell induction to soluble protein antigen delivered via rectal enema can be established in the colon draining iliac/caudal lymph nodes (ILN) independent of CD103+ DCs (18). Of note, it has recently been shown that induction of CD11b+CD103+ DCs is dependent on TGFβ signaling (81), and it is suspected that TGFβ, which is equally present at the mRNA level in the ILNs and mLN, may strongly contribute to DC-mediated FOXP3+ Treg cell induction in both ILN and mLN. In keeping with this pivotal role of TGFβ, SI CD103+ DCs can induce FOXP3+ Treg cells independently of RA via integrin αvβ8-mediated activation of TGFβ (82, 83). Collectively, these data highlight the interplay between resident mucosal DCs, RA, and TGFβ, and the local microenvironment in the mucosa draining lymphoid tissues are essential for the induction of SI Treg cells to promote immunological tolerance.
Stromal- and Epithelial-Dependent Induction of Intestinal Treg Cells
The contribution of non-immune cells to intestinal immune regulation has not been well-characterized. In humans, direct interactions between intestinal epithelial cells and the immune system contribute to intestinal Treg cell development via DCs. This is especially relevant in patients with Crohn’s disease, where epithelial cells have been shown to be functionally altered (84). Epithelial cells can induce tolerogenic DCs directed by TGFβ and RA (84) as well as produce thymic stromal lymphopoietin, which is also known to contribute to Treg cell induction (84, 85). Thus, intestinal epithelial cells are important players in Treg cell induction and function.
Directly underlying the intestinal epithelium, stromal cells interact with both immune and epithelial cells and have been proposed to regulate immune responses (86, 87). Small intestinal stromal cells induce DCs to produce RA under influence of the microbiome and increase iTreg cell differentiation (79). Additionally, stromal cells from the mLN are essential to induce CCR9 and α4β7 on T cells in a RALDH-dependent manner (88), and neonatally imprinted mLN stromal cells are also required for the Treg cell induction (89, 90). These stromal cells from the mLN can also produce vesicles that carry TGFβ and can induce Treg cells (91). Kashiwakura et al. showed that stromal cells from the spleen and lymph nodes have cell-to-cell contact with Treg cells from these same tissues to decrease apoptosis in a CD2-dependent manner (92). An elegant study showed that human Treg cells can be induced from peripheral blood naïve T cells in the presence of colonic stromal cells in a PGE2-dependent manner. When these stromal cells are derived from patients with IBD, their capacity to induce Treg cells is slightly reduced (93). Thus, stromal cells play a role in Treg cell induction that may be similar to the role of professional APCs and use a variety of functions to alter/induce intestinal Treg cells.
Cofactors That Contribute to Mucosal pTreg-Cell Induction
Cyclooxygenase 2 (COX-2) is an enzyme involved in the conversion of arachidonic acid (AA) to prostaglandins (PG). Prostaglandins are typically induced upon inflammation; however, in the SI COX-2 is present during homeostasis and expressed by SI stromal cells and macrophages (94, 95). The predominant COX-2-dependent metabolite of AA in the intestine is PGE2, and mice lacking COX-2 have undetectable intestinal PGE2 that leads to increased inflammation upon challenge with dextran sulphate sodium (DSS) (96). Moreover, COX-2 inhibition during dietary antigen feeding in mice elecits loss of oral tolerance with pathological changes in the proximal SI including increased epithelial crypt elongation, villus blunting, and increased LP mononuclear cell proliferation (95). These histological features highly resemble the histological changes seen in patients with Celiac disease caused by aberrant oral tolerance to the dietary protein gluten (97). In keeping with these findings, COX-2-derived PGs enhance SI Treg cell induction in the mLN after OVA ingestion via suppression of IL-4 production and GATA3 expression (98). This mechanism is dependent on mLN DCs which are high COX-2 expressers and dependent on PGE2. Altogether, these studies show that local COX-2 contributes to SI Treg cell induction and subsequent oral tolerance. For an elaborate discussion about the role of COX-2 in IBD and colorectal cancer the reader is referred to a review by Wang and Dubois (99).
Finally, an additional mechanism by which DCs have been shown to induce Treg cells is through indoleamine 2,3-dioxygenase (IDO). IDO is expressed by CD11c+CD103+ DCs, reduces local tryptophan concentration, and produces tryptophan metabolites to promote a tolerogenic environment, possibly through Treg cell induction via aryl hydrocarbon receptor signaling (100–102). Together, these data emphasize that mucosally induced pTreg differentiation during oral tolerance is highly regulated and dependent on numerous local, tissue-specific, interactions.
Additional Cues From the Microenvironment Influence Intestinal Treg Cells
Factors that regulate FOXP3 expression include TCR signaling, co-stimulation, cytokine-mediated signals, and the epigenetic status of the FOXP3 locus (103). In the thymus, Foxp3 expression is induced following three signals: (1) TCR; (2) co-stimulation by CD28; and (3) IL-2R (64). In contrast, the induction of peripheral Treg cells from naïve CD4+ T cells is dependent on a TCR signal and the cytokine TGFβ. In the thymus TGFβ-signaling is not required for FOXP3 induction but rather is required for survival of newly generated Treg cells (104, 105).
Studies examining the regulation of Foxp3 expression showed that the Foxp3 promotor has several enhancer elements, found in conserved non-coding sequences (CNS) 1 through 3 (106, 107). Histone methylation of these enhancer elements regulates transcription of Foxp3, as shown for the first time by Kim and Leonard (107). The CNS regulating Foxp3 expression each have different functions. TGFβ-dependent induction of pTreg cells relies on CNS1 located within the Foxp3 locus and deficiency of this region selectively reduces intestinal pTreg cells without affecting tTreg cells (55, 108). Thus, selective targeting of CNS1 enables the study of extrathymically generated Treg cells, with the caveat that CNS3 also contributes to pTreg cell induction, albeit to a lesser extent (55).
Distinguishing tTreg cells from pTreg cells remains challenging. The transcription factor HELIOS was initially described as a marker to discriminate tTreg cells and pTreg cells, where tTreg cells express HELIOS and pTreg cells do not (57). However, there is conflicting evidence and this topic remains controversial (109). Another marker implicated in the distinction between tTreg and pTreg cells is NEUROPILIN-1 (NRP1). Expression of NRP1 on murine Treg cells is controlled by TGFβ both in vitro and in vivo (58). In the absence of inflammation, NRP1 appears to predominate in tTreg cells and exhibits low expression in pTreg cells (58, 59). However, Treg cells at sites of inflammation can also be NRP1high, confounding this distinction (58). Moreover, TCR sequencing of Treg cells isolated from different compartments (thymus, colon, mLN) show that Treg cells cluster according to their location rather than NRP1 expression, indicating that the TCR repertoire is more similar between compartments than between Treg cells with similar NRP1 expression (110). Altogether, the immunophenotype and functional differences of tTreg cells versus pTreg cells remain active areas of investigation.
Under homeostatic conditions, Treg cell numbers are generally associated with the function of the intestinal site in which they are found (15). In the SI, the major function is the uptake of soluble, luminal, food antigens. Conceptually it makes sense that the SI microenvironment is well-suited to promote Treg cell induction to promote tolerance to food antigens (74). Tolerance to an antigen is actively acquired and results in local and systemic immune unresponsiveness specific to the ingested antigen. As food deprivation studies are considerably more difficult to perform given their inherent ethical considerations, most of our knowledge about Treg cell induction in the SI is limited to supplemented model dietary antigens. Oral tolerance to OVA is induced in naïve mice upon by direct intragastric administration of OVA or providing OVA in the drinking water. Both local and systemic unresponsiveness are dependent on de novo induction of pTreg cells, while tTreg cells are dispensable for oral tolerance to soluble food antigens (111).
In contrast to the SI, Treg cell induction in the colon is largely associated with gut resident bacteria. During the resolution of adoptive transfer colitis with Treg cells, almost all Treg cells in the spleen, mLN, and colon originate from transferred Treg cells. Thus, the majority of cLP Treg cells in the transfer model is not induced from donor naïve T cells but most likely reflects proliferation of transferred Treg cell population in a lymphopenic host, typically referred to as homeostatic proliferation (112). However, these observations do not yield information pertaining to the homeostatic induction of intestinal Treg cells.
Germ-free mice lacking all microbes exhibit an immature mucosal immune system with considerably lower Treg cell frequencies as compared to mice housed under SPF conditions (113–115). Similarly, administering antibiotics to SPF mice reduces the frequency of colonic Treg cells in conventionally housed mice, though neither GF mice nor antibiotic treated SPF mice are entirely devoid of Treg cells (113). In the SI of both GF and antibiotic treated SPF mice, Treg cell frequency and numbers resemble those of conventionally housed mice, supporting the notion the Treg cells in this anatomic location are primarily responsive to dietary antigens (113, 116). In a landmark paper, Atarashi and colleagues showed that colonic Treg cell induction can be directly modulated by Clostridia species (113). The authors inoculated GF mice with a cocktail of 46 strains of Clostridia or 16 strains of Bacteroides, which increases colonic Treg cells as compared to colonization with SPF stool, with the Clostridia cocktail yielding the highest level of Treg cell induction (113). Expansion of Clostridia species is associated with a TGFβ-rich environment and, in support of microbial antigens driving colonic Treg induction, there is no increase in SI Treg cell frequency following colonization with Clostridia species. These findings also hold true for human Clostridia isolates, suggesting that these bacteria may also be relevant for colonic Treg cell induction in humans (117). Thus, the intimate relationship between commensal bacteria and the immune system drives colonic Treg-cell induction. Studying changes in the Treg cell population following manipulation of dietary or microbial antigens can elucidate factors that modulate intestinal Treg cells (92).
Similar to mucosally induced pTreg induction to dietary antigen in the SI, pTreg cells can be induced to microbial protein antigens as well. Using a commensal microbe-derived (Cbir) flagellin expressed by a subset of the Clostridium XIVa cluster of bacteria bound to the tolerogenic, non-toxic portion of cholera toxin, it was demonstrated that APCs induce antigen-specific intestinal Treg cells in an endogenous TGFβ−dependent manner (118). This observation of microbial influence on tolerogenic intestinal Treg cells once more emphasizes the critical role of the gut microbiome in intestinal immune homeostasis.
Elegant studies by Sefik et al. show that colon FOXP3+ Treg cells have a markedly distinct transcriptome expressing high levels of Rorc, the gene encoding RAR-related orphan receptor gamma-t (RORγt), as compared to Treg cells residing in other locations (50). This was surprising at the time given that RORγt is a known marker for IL-17 producing Th17 cells. These RORγt+ Treg cells are NRP1lowHELIOSlow, indicating that they are likely pTreg cells. Importantly, RORγt+ Treg cells are induced by a diverse group of microbes; moreover, this study suggests short-chain fatty acid (SCFA)-dependent mechanisms make only a minor contribution to colonic Treg cell induction. In addition to the mouse intestine, RORγt+ Treg cells have been detected in human colon tissue (50). These RORγt+ Treg cells differ from Treg cells expressing IL-33R or GATA3, as these two latter populations are HELIOS+ whereas most RORγt+ Treg cells are HELIOSneg (54, 119). Interestingly, despite being RORγt+, the gene signature of this Treg cell population only partially overlaps with Th17 cells (50). These studies show that Treg cells with different properties are present throughout the intestine.
Mechanisms of Intestinal Treg Cell Induction Mediated by Intestinal Microbial Metabolites
SCFA are a fermentation byproduct of fiber digestion, which primarily occurs in the colon. Consequently, the concentrations of SCFA are highest in this part of the intestine (120). As expected, GF mice exhibit lower concentrations of SCFA in the intestine, and supplementation of SCFA in the drinking water elevates intestinal SCFA and increases colonic Treg cells (121, 122). In addition to the gut lumen, SCFA are also present in the vena porta blood. The concentration of SCFA in the vena porta is higher than in peripheral blood, indicating that SCFA are transported from the intestine to the liver and from there are taken up and dispersed throughout the periphery (120, 123). SCFA are often considered only present in the colon and not in the SI, but this is an oversimplification. Although concentrations of SCFA are higher in the colon, SCFA are indeed present and absorbed in mouse and human SI (124–126). Since luminal content (e.g., fiber) resides in the SI for a shorter duration than in the colon, SCFA-producing bacteria in the SI have to be more efficient (126).
The three SCFA produced in the gut and shown to be relevant for Treg cell induction are propionate, butyrate, and acetate. Interestingly, these SCFA each have different mechanisms of action. Mechanistically, SCFA signal through three G-protein-coupled receptors (GPCRs): GPR41, GPR43, and GPR109A, and inhibit histone deacetylase (HDAC) (127, 128). HDAC inhibitors increase Treg cell production and promote their suppressive function (129). These GPCRs are expressed by both immune and epithelial cells and their function is diverse (128, 130). GRP109A is only activated by butyrate, but not by acetate or propionate, and GPR109A deficiency was shown to specifically reduce colonic Treg cells (131, 132). Butyrate binding to GPR109A on DCs and macrophages also induces anti-inflammatory molecules, including IL-10 and RALDH to promote pTreg induction in a CNS1-dependent manner and can act directly on T cells to induce FOXP3 by inhibiting HDAC (132, 133). Conversely, all three SCFA are able to activate GPR43. Intestinal Treg cells express relatively high amounts of GPR43, but only in the presence of microbiota. The mechanism by which propionate increases intestinal Treg cell accumulation, by both increasing induction/homing as well as proliferation, likely occurs through GPR43. Smith et al. demonstrated treatment of wild-type mice with propionate increases colonic Treg cell numbers as compared to GPR43-deficient mice also treated with propionate (122). The authors also demonstrate that propionate increases histone acetylation, which was not observed in GPR43-deficient mice. Furthermore, when Rag-/- mice co-injected with naïve T cells and GPR43-sufficient or -deficient Treg cells are treated with propionate, mice receiving GPR43-sufficient Treg cells exhibited greater protection from intestinal inflammation as compared to mice co-injected with GPR43-deficient Treg cells. Not only does GPR43 induce colonic Treg cells, it also promotes proliferation of Treg cells already present. In contrast to propionate, butyrate only increases pTreg-cell induction (not proliferation), whereas acetate does not drive induction but rather Treg cell accumulation, independent of CNS1 (133, 134). The mechanism by which butyrate induces Treg cells is through direct acetylation of the Foxp3 locus (133). Finally, butyrate and propionate have also been shown to prime DCs to drive Treg cell induction in vitro (133). To summarize, SCFA induce intestinal Treg cells through various mechanisms.
Given the abundance of SCFA along the GI tract as discussed above, SCFA are surprisingly important for maintenance of oral tolerance. As compared to mice given a low-fiber diet, the CD11c+CD103+ DCs in mice fed a high fiber diet display more RALDH activity and induce more antigen-specific Treg cells in vitro and in vivo (135). In vivo, the increase in Treg cells was observed both in mLN as well in the SI LP. By using a vitamin A-deficient diet, the authors showed that Treg cells might also be induced by the high-fiber diet in a CD103+-independent manner. However, Treg cells induced in the absence of vitamin A have impaired suppressive capacity, indicating that both fiber and vitamin A are essential for induced Treg cell function in the SI. Lastly, the authors showed that epithelial GRP43 and immune cell GPR109A are essential for induction of tolerance.
Colonic Treg cells can also be induced independent of bacterial-derived SCFA. One reported mechanism that involves the symbiont Bacteroides fragilis that produces polysaccharide A (PSA), which induces IL-10 producing FOXP3+ Treg cells in a toll-like receptor 2 and plasmacytoid DC-dependent manner (136, 137). PSA does not appear to alter the frequency of colonic Treg cells, but rather enhances their function. Another recently discovered mechanism of colonic Treg induction is through bile acids and bacteria-derived bile acid derivatives. These have been shown to selectively induce colonic RORγt+ Treg cells, once more emphasizing the importance of interactions between the gut microbiome and immune system for intestinal Treg cells (138).
These observations show that although there are distinct and similar drivers of Treg cell induction in the SI and colon, the local microenvironment governs the dominant mechanism of Treg cell induction. Whereas Treg cell induction in the SI is driven by DC-presented food antigens in the presence of TGFβ and diet-derived RA, Treg cell induction in the colon largely occurs TCR stimulation in the presence of fermentation products, microbial metabolites, and TGFβ, the latter resulting in tolerance to the resident commensal microbes populating the colon.
Trafficking of Treg Cells to the Intestine Under Homeostatic Conditions
Migration of conventional T cells occurs via expression of adhesion molecules and chemokine receptors and is essential for a physiological immune response (139, 140). T cells migrate to non-lymphoid tissue in the absence of inflammation, and also in response to stimuli such as commensal bacteria and dietary antigens (116, 141, 142). To reach the intestine, Treg cells express homing receptors that partially overlap with those of conventional T cells (143, 144). Lee et al. elegantly showed that tTreg cells first undergo changes in homing receptors to egress the thymus, and migrate to secondary lymphoid tissue where they undergo changes following antigen priming and migration to non-lymphoid tissue (143). GALT and mLN, but not splenic, DCs can induce α4β7 expression on Treg cells, which enables MADCAM-1-dependent extravasation from circulation into the intestine (144–146). Additionally, they induce expression of the chemokine receptor CCR9, enabling trafficking to the SI (146, 147). Interestingly, in vivo induction of these molecules is preferential after oral administration of OVA as compared to i.p. administration, which is attributed to the transport of OVA by DCs to the mLN (145). Induction of α4β7 on Treg cells is more efficient by mLN DCs as compared to DCs from peripheral lymph nodes or spleen and is also attributed to RA (74, 144, 148). These findings can be translated from mice to human as Bakdash et al. showed that human monocyte-derived DCs cultured in presence of RA resemble LP DCs in terms of CD103 expression and RA production (149). When cultured together with T cells, these DCs induce a suppressive FOXP3neg T cell population, as well an IL-10+ T cell population, although suppression of naïve T-cell proliferation in vitro was independent of IL-10. The RA-conditioned human DCs also induced CCR9 and α4β7 on T cells. Moreover, IL-10 production was only observed in the CCR9+ T cells and not in the CCR9neg T cells. In contrast to RA, TGFβ induces FOXP3+ cells rather than IL-10 producing T cells in these assays. This human in vitro data shows RA induces CCR9 and α4β7 on an IL-10 producing suppressive T cell population with improved intestinal homing capacity, in contrast to TGFβ, which induces FOXP3 expressing T cells. Altogether, these studies demonstrate that RA from CD103+ DCs induces CCR9 and α4β7 on Treg cells, which licenses them with improved homing to the SI. In the context of antigen presentation in the lymph nodes, it makes sense that DCs license activated Treg cells to migrate to the site of antigen exposure.
The induction of chemokine receptors in Treg cells can also be independent of RA production by DCs (150). Treg cells can specifically acquire capacity to home to the colon. For instance, the microbiome can modulate expression of GPR15 (150) and GPR15-deficient mice exhibit fewer FOXP3+ Treg cells in the colon but not the SI. In addition to GPR15 and α4β7, colonic Treg cell homing, and homing of other leukocyte subsets, is also dependent on CCR6 (151, 152). Together with the observation that OVA-specific T cells preferentially differentiate into CCR6+FOXP3+ cells (151) in mucosal tissue upon antigen exposure, the induction of CCR6 expression and subsequent intestinal homing and suppression is likely antigen driven. Further insights in Treg cell homing comes from a study by Nakanishi et al., who utilized a photoconvertible mouse, showing for the first time that Treg cells can move bidirectionally between colon and lymph nodes (153). The movement of Treg cells from the colon to the lymph nodes depends largely on Sphingosine-1-phosphate receptor 1, as blocking this pathway with FTY720 (a S1PR1 agonist that downregulates S1PR1), substantially decreases the number of photoconverted cells in the lymph nodes.
In conclusion, Treg cells utilize a variety of trafficking molecules to migrate bidirectionally between lymph nodes and mucosal tissues in an antigen-dependent manner.
The Sustenance of Intestinal Treg Cells Is a Delicate Balance Between Apoptosis and Proliferation
In contrast to early viewpoints that Treg cells are quiescent, Treg cells are now known to have substantial proliferative capacity. Liston and Gray reviewed the turnover of the central Treg cell population in circulation and in lymphoid organs (35). Nearly half of the circulating Treg cells in both humans and mice undergo division every eight to ten days (154, 155). Under homeostatic conditions, a high rate of proliferation is balanced out by a high rate of apoptosis with the latter occurring in Treg cells through FOXP3-dependent phosphorylation of the pro-apoptotic protein BIM (156). BIM is antagonized via MCL1 through IL-2 signaling (154) and under steady-state conditions, Treg cells repress paracrine and autocrine IL-2 production. The high affinity IL-2R is a trimeric protein, consisting of an IL-2Rα (CD25), IL-2Rβ (CD122), and IL-2Rγ (CD132) subunit with Treg cells constitutively expressing CD25. In the absence of CD25, the IL-2Rβ and IL-2Rγ exhibit intermediate-affinity for IL-2 (157). Thus, Treg cells exhibit increased sensitivity to IL-2 as compared to conventional T cells.
Following partial depletion of Treg cells, apoptosis decreases, and proliferation increases in an IL-2-dependent manner, likely through the “de-repression” of IL-2 production. This balance of proliferation and apoptosis of central Treg cells is thought to drive a homeostatic network of rapid self-correction and IL-2-dependent competition effects (35). In the SI, the IL-2 for intestinal Treg cells is in part supplied by group 3 innate lymphoid cells (ILC3s), whereas in the cLP conventional T cells are a significant source of IL-2. Furthermore, the lifespan of Treg cells also differs in various compartments. In contrast to the half-life of circulatory Treg cells, the half-life of tissue resident Treg cells in the intestine appears longer. For example, the estimated half-life of pTreg cells in the SI LP has been is 4-6 weeks, which is based on the sharp decline of SI Treg cells in mice after deprivation of Treg inducing food antigens (51).
The mechanisms that maintain Treg cells in non-lymphoid tissues differs from central Treg cells. Rather, the transcription factor BLIMP-1 (encoded by Prdm1) is expressed by effector Treg cells and together with IRF4, is required for IL-10 production (45). In T cells, BLIMP-1 is induced by IL-2 and has a negative feedback loop for IL-2 production (158, 159). In BLIMP-1-deficient mice, activated T cells accumulate in the liver, lung and intestine, leading to inflammation in these organs (160, 161). Treg cells express high levels of BLIMP-1, suggesting that the autoimmune pathology in BLIMP-1-deficient mice may be due to defects in Treg cells (160, 161). BLIMP-1+ Treg cells produce IL-10 and resemble an effector Treg cell phenotype, whereas IL-10 is not produced by BLIMP-1-deficient Treg cells (45). In Treg cells specifically, BLIMP-1 represses the pro-survival protein BCL-2, and the chemokine receptor CCR6 that mediates trafficking of T cells to the intestine. Thus, BLIMP-1 may antagonize the accumulation of effector Treg cells in the intestine (45). The similarities between Il10-/- and BLIMP-1-deficient mice suggests that the pathology in BLIMP-1-deficient mice may be attributed to a lack of IL-10 production by effector Treg cells (45). Altogether these studies identify BLIMP-1 as an important factor in intestinal Treg cell homeostasis, preventing auto-immune disease through modulation of sustenance and function of Treg cells.
Interestingly, modulation of food antigens reduces the frequency of RORγtneg Treg cells in the intestine, whereas manipulation of the microbiome reduces the RORγt+ Treg cells (51). Manipulation of the microbiome does not reduce GATA3+/Helios+ RORγtneg Treg cells, indicating that these Treg cell subsets are not sustained by microbial signals but rather by other factors such as IL-33 (54, 162). Meanwhile, in GF or antibiotic treated mice, the proportion of Treg cells that are IL-10+ decreases specifically in the cLP (90). Thus, the gut microbiome is essential to sustain both the intestinal RORγt+ Treg and IL-10+ cLP Treg cell compartment.
To summarize, IL-2 is a key cytokine involved in Treg cell maintenance and regulates the balance between apoptosis and proliferation via redundant mechanisms. The central Treg cell turnover is higher as compared to intestinal Treg cells with intestinal Treg cells having different mechanisms of sustenance.
Intestinal Treg Cells Reside in Lymphoid Follicles and Are Interspersed Throughout the Lamina Propria
Under homeostatic conditions, Treg cell frequency increases from duodenum to jejunum and from ileum to colon (163). In mice, colonic Treg cells reside in the LP and under homeostatic conditions are mostly observed in lymphoid clusters (112). In a model where mice reconstituted with OVA-specific T cells are challenged with OVA-producing bacteria, cLP Treg cells are preferentially found in lymphoid follicles and colocalize with DCs while macrophages reside outside the follicles (164). The Treg cells actively suppress effector T cell cytokine production, as indicated by lower IFNγ production in stimulated T cells from mice that receive OVA-specific Treg cells (together with conventional T cells) as compared to mice that receive only OVA-specific conventional T cells.
Similar to mice, most Treg cells in the human colon are found in lymphoid follicles, with a smaller fraction scattered throughout the LP (112). Interestingly, decreased intestinal Treg cells have not been observed in patients with IBD, nor do these Treg cells exhibit reduced suppressive capacity in vitro as compared to healthy controls (165–170). Rather, during colonic inflammation the number of colonic Treg cells increases, whereas the number of colonic T cells remains rather unchanged (112, 171). Although the localization of Treg cells in IBD is often not detailed, the FOXP3+ T cells in patients with IBD appear to colocalize with lymphocytes, which begs the question: why are they not able to suppress colitogenic T cell responses? (172) Several possibilities have been put forward. For example, it has been reported that LP T cells might be resistant to Treg cell mediated suppression in a SMAD7-dependent manner (173). Furthermore, the local inflammatory microenvironment may attenuate Treg cell function during active inflammation, effector Treg cells may transiently upregulate FOXP3, or the FOXP3+ cells represent an uncommitted, i.e. non-suppressive, population of FOXP3+ cells that proliferate within an inflammatory micro-environment (174, 175). Recent work using single-cell RNA-seq uncovered marked changed in T cell expression profiles in patients with ileal Crohn’s disease depending on whether the cells are from inflammatory lesions or adjacent non-inflamed tissue (176).
In contrast to the LP and its lymphoid follicles, murine Treg cells are sparse in the intestinal epithelium. Rather, amongst lineage labelled Treg cells that have migrated into the epithelium, 50% and 10% of the SI and cLP Treg cells, respectively, lose FOXP3 expression (177). Interestingly, when mice are treated with broad-spectrum antibiotics the number of Treg cells in the SI epithelium increases. Thus, this study shows that in the SI, the microbiome destabilizes epithelial Treg cells.
Intestinal Treg Cell Maintain Intestinal Immune Homeostasis via Contact-Dependent and -Independent Mechanisms
Most evidence for the role of intestinal Treg cells in maintaining intestinal immune homeostasis comes from studies showing dysfunction of Treg cells in mice. An often-used model is the adoptive T cell transfer model of colitis. In this model, immunodeficient mice lacking mature T and B cells (e.g., Rag1 -/-) develop colitis a few weeks following the transfer of naïve CD4+ T cells (CD25negCD45RBhigh) unless a fraction containing Treg cells (e.g., CD45RBlow or CD4+CD25+FOXP3+) are co-injected (10, 178). The intestinal inflammation in the adoptive T cell transfer model of colitis is mediated by inflammatory cytokines produced by effector T cells in response to gut microbial antigens and requires the presence of Helicobacter spp (179–182). Although inflammation is generally restricted to the large intestine, there can be limited small bowel involvement as well (178). The adoptive T cell transfer colitis model illustrates how Treg cells are instrumental in controlling effector T cell responses to prevent T cell-mediated inflammation in the gut. Thus, immune homeostasis in the intestine requires tolerance to commensal intestinal microbiota.
The adoptive transfer model of colitis really allows to tease apart Treg cell function. For example, Denning et al. performed adoptive transfer experiments using CD4+CD25+β7-/- Treg cells and found that β7-/- Treg cells are impaired in intestinal homing, but still have comparable suppressive function as wild-type Treg cells in vitro as well as in vivo (183). Thus, migration and suppressive function of Treg cells may be independent processes.
The presence of tTreg cells without intestinal pTreg cells is insufficient to maintain intestinal immune homeostasis, as CNS1-deficient mice develop Th2 skewed intestinal inflammation (55). The Th2-type intestinal inflammation, rather than Th1 or Th17, is attributed to GATA3-specific Treg cell function (55). Evidence for synergistic roles between pTreg cells and tTreg cells further comes from studies examining the treatment of established adoptive transfer colitis (12) with Treg cells. Haribhai et al. generated in situ induced Treg cells (pTreg cells) by transferring CD45RBhigh naïve T cells into Rag1-/-mice and found that these pTreg cells exhibit comparable suppressive function in vitro as compared to ex vivo Treg cells isolated from the spleen and mLN (184). Induced mLN Treg cells have a slightly different phenotype than mLN Treg cells from a wildtype mouse, in terms of CD25 (higher during inflammation), CD62 (lower during inflammation), and CD103 (lower during inflammation) expression. This finding supports that the pTreg cells are indeed suppressive and also shows that pTreg cells in the mLN during inflammation are dissimilar from Treg cells residing in the mLN under homeostatic conditions. The authors then switched to showing the function of pTreg cells during intestinal inflammation but first examined the function of in vitro induced Treg cells (iTreg cells) with ex vivo Treg cells. They used the iTreg cells as a model for in situ pTreg cells – it is not feasible to get enough in situ pTreg Treg cells from the cLP in this model to treat other mice with. Mice transferred with ex vivo Treg cells had significantly less colitis as compared to mice transferred with iTreg cells, which shows that iTreg cells have dissimilar suppressive capacity in vitro as compared to ex vivo Treg cells, which, as we now know and as discussed above, are a group of both pTreg cells and tTreg cells. Subsequently, the rescue of adoptive transfer colitis was examined in absence of pTreg cells by transferring in naïve T cells from mice lacking functional FOXP3 (Foxp3ΔEGFP). Interestingly, a combination of ex vivo Treg cells and iTreg cells ameliorated established disease whereas ex vivo Treg cells alone or iTreg cells could not. Altogether, this study shows that resolution of intestinal inflammation it is beneficial to have a combination of in vitro induced Treg cells and ex vivo Treg cells.
Treg cells can exert their suppressive effects using different modes of action (Figure 2). These suppressive mechanisms include production of tolerogenic cytokines, cytolysis, metabolic disruption, and the modulation of APC function (185). Colonic Treg cells have a transcriptome that resembles highly suppressive Treg cells (50). Recently, a group performed single-cell RNA-seq on Treg cells and memory T cells isolated from the colon, skin, and respective draining lymph nodes under homeostatic conditions (40). Treg cells from the colon differ from Treg cells in the draining lymph nodes by differential gene expression related to the TNFR-NF-κB pathway, and several functional genes, e.g. Il10, granzyme b (Gzmb), and cytotoxic lymphocyte antigen 4 (Ctla4). In the lymphoid tissue specifically, a small population of Treg cells expresses genes overlapping with effector Treg cells and non-lymphoid tissue T cells, suggesting functional migration to non-lymphoid tissue. In the absence of intestinal inflammation, colon Treg cells travelling to lymph nodes express higher levels of immunosuppressive molecules (ICOS and LAG3) than lymph node Treg cells that have not trafficked to the colon (153). The same observation was made during DSS-induced intestinal inflammation. That is, expression of ICOS, PD-1, CTLA-4, and IL-10 is higher in cLP Treg cells then in lymph node Treg cells. An in vitro suppression assay showed that colonic Treg cells are superior suppressors of T-cell proliferation as compared to lymph node Treg cells, suggesting that highly suppressive Treg cells migrate back to the lymph nodes during inflammation.
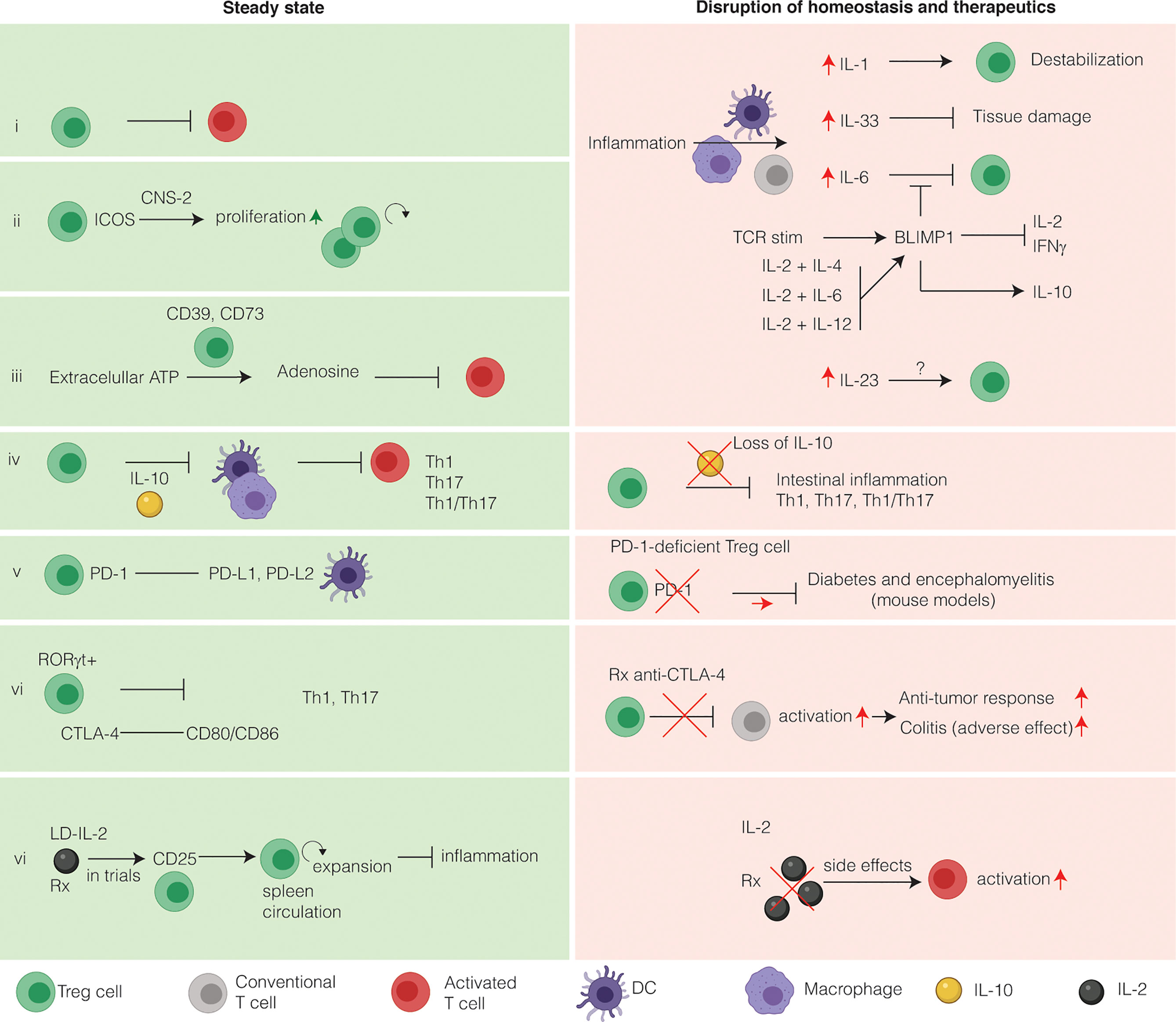
Figure 2 Intestinal Treg cells tightly maintain intestinal immune homeostasis and relevance for immunotherapy. Intestinal Treg cells have several markers that discriminate them from other Treg cells. Under homeostatic conditions, Treg cells suppress effector T cells to prevent inflammation (i). During inflammation, several inflammatory cytokines alter the phenotype of intestinal Treg cells. (ii) Inducible co-stimulator (ICOS) stabilizes intestinal Treg cells in a CNS2-dependent manner. (iii) Extracellular ATP is converted to the immunosuppressive adenosine by Treg cells expressing CD39 and CD73. CD73 is induced by TGFβ. (iv) The main immunosuppressive cytokine secreted by intestinal Treg cells is IL-10. IL-10 functions to inhibit T-helper 1 (Th1), Th17 and Th1/Th17 inflammation in a STAT3-dependent manner via IL-10R signaling on APCs, limiting inflammasome activation. On the other hand, loss of IL-10 both in mice and in humans leads to intestinal inflammation and IBD, respectively. The main co-stimulatory receptors expressed on Treg cells are programmed death receptor (PD-1) (v) and cytotoxic T-lymphocyte associated protein 4 (CTLA-4) (vi). Both these co-stimulatory receptors are exploited therapeutically to increase the anti-cancer immune response. Anti-CTLA-4 therapy has colitis as a frequent side-effect, whereas colitis is not associated with blockade of the PD-1 pathway. (vi) Because Treg cells express the high affinity receptor CD25, they respond to low doses of IL-2 by expansion, thus limiting inflammation. LD-IL-2 is investigated in a clinical trial for its use in patients with IBD. In contract to LD-IL-2, a high dose of IL-2 will activate T cells and lead to deleterious side effects and therefore is not used in clinical practice.
The suppressive phenotype of intestinal Treg cells is related to their regulatory function. CTLA-4 functions as a co-inhibitory molecule on T cells by binding CD80/CD86 on activated APCs (186), is critical for Treg-cell function to prevent lethal auto-immunity in mice, and is highly expressed by intestinal Rorγt+ Treg cells (50, 162, 187). This is also true in humans as patients with CTLA-4 haploinsufficiency have impaired Treg cell function and are highly susceptible to very-early-onset Crohn’s-like intestinal inflammation (188–190). In agreement with this, colitis is one of the most common side-effects of immune checkpoint blockade with anti-CTLA-4 in cancer (191).
Another co-inhibitory receptor expressed on Treg cells is programmed death receptor 1 (PD-1), although the role of this receptor has not been fully dissected (192). PD-1 functions by binding to PD ligand 1 (PD-L1) and PD-L2 and signaling via this receptor-ligand complex results in inhibition of CD28, thus inhibiting co-stimulation of T cells in the early phase after antigen encounter (193). PD-L1 synergizes with TGF-β to promote pTreg cell induction via PD-1 on differentiating Treg cells (192, 194, 195). Moreover, several studies argue that PD-1 is involved in Treg cell maintenance through maintaining the suppressive phenotype (196), preventing conversion of Treg cells into pro-inflammatory effector memory T cells (197) and rendering them less sensitive to apoptosis (198). Initially, PD-1 signaling was suggested to may have less profound effects on suppressive function as IL-2-induced Treg cells isolated from PD-1 deficient mice are not altered in their suppressive capacity (198). However, the recent generation of mice that selectively lack PD-1 in Treg cells allowed to establish that PD-1 deficient Treg have an activated phenotype and exert enhanced immunosuppressive function, as compared to wild-type Treg cells, in experimental diabetes and autoimmune encephalomyelitis (199). These studies would argue that blockade of PD-1 on Treg cells should enhance their regulatory capacity rather than diminish it. In keeping with this hypothesis the prevalence of colitis as adverse effect in cancer patients receiving PD-1 blockade is much lower than in patients receiving anti-CTLA-4 therapy (200). Of note, the role of PD-1 in Treg cells in IBD has mostly focused on IL-10 producing Tr1 cells (201). PD-1 expression on intestinal CD4+Foxp3neg Tr1 cells enriches for IL-10 secreting cells. In mice, co-transfer of IL-10-producing CCR5+PD-1+ Tr1 cells strongly inhibits colitis induced by transfer of Th17 cells, whereas IL-10-producing control T cells lacking CCR5 and PD-1 are less efficient (202).
Besides the enrichment in co-inhibitory receptors Treg cells also carry co-stimulatory receptors. In particular, inducible co-stimulator (ICOS) is involved in Treg cell maintenance. ICOS-deficient mice have reduced frequencies of Treg cells in secondary lymphoid tissues but do not develop spontaneous disease when housed under SPF conditions (203). In mice with specific deletion of ICOS in Treg cells, thymic output of Treg cells and the numbers of intestinal IL-10+ cells are increased, perhaps as a compensatory mechanism (203). However, together with the fact that ICOS-deficient Treg cells are equally suppressive as wildtype Treg cells in adoptive transfer colitis it appears that ICOS plays a key role in the stability of intestinal pTreg cells rather than directly regulating suppressive capacity. In line with this, ICOS increases antigen-specific Treg cell expansion in a CNS2-dependent manner (204). In the intestinal LP ICOS-L is expressed by CD11c+CD103+ DCs, suggesting these APCs can provide a signal to sustain the intestinal ICOS+ Treg cell population (203). This ICOS mediated sustenance is important as, although ICOS-deficient Treg cells are equally suppressive as wildtype Treg cells in adoptive transfer colitis they eventually cannot prevent mortality in Foxp3 deficient mice (203, 204). Moreover, ICOS-deficient Treg cells cannot reverse established adoptive transfer colitis (203). Altogether, these results indicate that ICOS is required for the stability of intestinal Treg cells to maintain suppressive function during intestinal inflammation.
Treg cells also exert suppressive function by metabolic disruption. Treg cells express both CD39 and CD73 (205, 206), and SI Treg cells in particular express high levels of CD39 and CD73 in comparison to Treg cells from other compartments (207). The expression of CD73, but not CD39, is regulated by TGFβ yet both CD39 and CD73 convert pro-inflammatory extracellular ATP into immunosuppressive adenosine (208, 209). In T cells, adenosine signals via the A2A adenosine receptor and is required for the suppression of intestinal inflammation independent of TGFβ and IL-10 (210–212). However, a surplus of adenosine in adenosine deaminase deficiency results in severe combined immunodeficiency (SCID) [for the function of Treg cells in relation to autoimmunity in ADA-SCID see Sauer et al. (213)]. Altogether, these studies identify a role for metabolic disruption of intestinal Treg cells during intestinal inflammation.
A cell-contact dependent mechanism intestinal Treg cells employ is through latent activation gene 3 (LAG-3) (214). The mechanism of LAG-3 mediated suppression occurs via inhibition of production of the inflammatory cytokine IL-23 by CX3CR1+ macrophages, which promotes IL-22 production by group 3 innate lymphoid cells (ILC3s) during anti-CD40 colitis (215). Another mechanism Treg cells may use to suppress effector T cells is GZMB that functions to lyse target cells (e.g., effector T cells) but to date the role for GZMB in intestinal Treg cells is lacking.
Intestinal Treg cells also maintain intestinal immune homeostasis through secretion of immunosuppressive cytokines including IL-10, TGFβ, and IL-35. The complex role of TGFβ in Treg cells has been extensively reviewed by Sakaguchi et al. (9). The importance of IL-10 and the intestinal microbiome as a driver of inflammation was initially described by Kuhn and colleagues who discovered spontaneous enterocolitis development in IL-10-deficient mice housed under non-specific-pathogen free (SPF) conditions (216). Later studies showed that colitis in SPF housed Il10 -/- mice required the presence of Helicobacter species (217). Although IL-10 can be secreted by multiple types of immune cells, IL-10 produced by FOXP3+ Treg cells plays an especially critical role in maintaining colonic immune homeostasis (218). IL-10 production by FOXP3+ Treg cells is found more frequently in the colon as compared to SI and secondary lymphoid tissue and is regulated by the microbiome (113, 219). In humans, deficiency in IL-10 production or IL-10 receptor signaling causes infantile colitis (220–222). The IL-10 receptor is more abundantly expressed on APCs than T cells (223), and cell-specific deletion of the IL-10R alpha chain in APCs is sufficient to induce colitis (44, 224, 225). IL-10R signaling activates the transcription factor STAT3 and perhaps not surprising, global or DC-specific deficiency for STAT3 largely phenocopies the pathology of Il10-/- mice (36, 226). In macrophages, autocrine IL-10 signaling impairs inflammasome activation (227). Via caspase, activation of the inflammasome results in increased levels of active IL-1β (228). Fitting to this pathway, inhibition of IL-1β with anakinra in IL-10R deficient patients decreases intestinal inflammation (229, 230). The consequence of IL-10-mediated regulation of intestinal APCs is largely though inhibition of inflammatory Th17 (CD4+IL-17A+) cells, Th1 (CD4+IFNγ+) cells (42, 43) as well as IL-17A+IFNγ+CD4+ double positive T cells (47). In summary, IL-10 is a central, non-redundant, effector cytokine used by intestinal Treg cells to suppress inflammation in the murine GI-tract.
The versatile role of intestinal Treg cells during inflammation is exemplified by a study by Schiering et al., who investigated the role of tissue specificity of Treg cells in a setting of inflammation (54). They found that signaling in colonic Treg cells by the alarmin IL-33 may limit tissue damage, and that the balance between IL-23 and IL-33 might determine the outcome of an inflammatory response in the intestine. Consistent with this, expression of IL-33R (ST2) was found to be increased on colonic Treg cells (54). ST2 expression is generally limited to GATA3+ Treg cells and IL-33 increases TGFβ-dependent Treg induction in vitro (54). In addition, IL-33 signaling exerts positive feedback on the expression of IL-33R in vitro (54) and increases intestinal Treg cell accumulation, proliferation, and stability under inflammatory conditions by directly binding to the Foxp3 promotor (54). In an IL-23-dependent model of intestinal inflammation, soluble ST2 was found to be released by stromal cells (54) and has been suggested to act as a decoy receptor to antagonize IL-33 activity. Finally, IL-33 also acts on epithelial cells to produce retinoic acid and thymic stromal lymphopoietin, independent of DCs (231). Together, these studies indicate that the IL-33 pathway regulates intestinal Treg cells to yield a tissue protective response.
Another key cytokine driving IBD pathogenesis is IL-23. IL-23 is a pro-inflammatory cytokine maintaining the stability of Th17 cells. To date, surprisingly little is known about the cell-specific contribution of IL-23R signaling in driving inflammation. It has been reported that Il23r is highly expressed on RORγt+ colonic Treg cells (50). Moreover, IL-23 may inhibit intestinal Treg cell induction while other studies have failed to show a role of IL-23R signaling in Treg cells (46, 232). In vitro Treg-cell induction studies have demonstrated that IL-23 impairs the responsiveness of Treg cells to IL-33 (54). Furthermore, ST2-deficient T cells were impaired in pTreg induction as compared to wild-type T cells in a Il23a-/- host. Finally, ST2+ Treg cells from the colon express Il23r and IL-33 signaling induces genes co-regulated by FOXP3 and GATA3, which is inhibited by IL-23 in a STAT3-dependent manner (54). Thus, IL-33 functions to increase intestinal TGFβ pTreg induction and IL-23 inhibits IL-33 action on Treg cells that may contribute to intestinal inflammation. These studies indicate that cytokines present during inflammation modulate intestinal Treg cells in various, yet relatively unexplored, ways.
Intestinal Treg Cells in Inflammatory Bowel Disease
IBD is a group of chronic inflammatory diseases of the intestine whose incidence is increasing in both the Westernized world and in developing countries (233). Ulcerative colitis (UC) and Crohn’s disease (CD), the most common clinical entities of IBD, are chronic conditions lacking a permanent cure resulting in significant long-term morbidity. Although the exact etiology remains unknown, IBD primarily results from dysregulated immune responses to commensal gut microbiota in genetically predisposed hosts that might be triggered by unknown environmental factors (234). In absence of inflammation, Treg cells mediate tolerance though the secretion of soluble mediators such as IL-10, as well as through direct interaction with other immune cells (9). In the case of IBD, the local microenvironment and the microbiome greatly influences Treg cell function and specificity (235). To date, there is no conclusive evidence that polygenic IBD arises due to numerical insufficiency of Treg cells or FOXP3 dysfunction. Nevertheless, there is ample evidence that functional changes in T cells in polygenic IBD can more severely impact Treg cells (e.g., deficiency in CTLA-4 or IL-10).
In patients with IBD, tolerance to commensal bacteria appears to be compromised, as circulating IgG antibodies to Cbir flagellin are associated with Crohn’s disease (236). Murine models have been employed to address whether such loss of tolerance could occur upon acute gastrointestinal infection eliciting transient bacterial translocation. Indeed, during a gastrointestinal infection with Toxoplasmosis gondii, tolerance to commensal bacteria is lost and microbiota-specific Cbir1 TCR-transgenic T cells, which are normally quiescent, become activated and differentiate to inflammatory effector and memory T cells (237, 238). Furthermore, in mice infected with Toxoplasmosis gondii Cbir-specific T cells upregulate T-BET and mostly secrete IFNγ, whereas infected mice treated with DSS results in Cbir-specific T cells upregulating RORγt (237). Together, these findings argue that during infection or acute inflammation transient commensal microbiota translocation elicits distinct commensal-specific pathogenic memory T cell responses.
In humans, many studies investigating Treg cells rely on changes in peripheral blood Treg cell frequencies as an outcome parameter as blood draws are less invasive than obtaining biopsies through endoscopy and can be better quantitated. However, with the evidence discussed so far, it is clear that peripheral blood Treg cells do not necessarily reflect the complex dynamics of intestinal Treg cells. Therefore, we will limit our discussion to studies examining intestinal Treg cells. During inflammation in patients with IBD, the number of Treg cells in the inflamed tissue typically increases (239). Expression of CCR6 is increased in both inflamed colon as well as in Th17 cells (240, 241). Adoptive transfer studies in mice previously have shown that CCR6-deficient Treg cells are defective in homing capacity and as a consequence, their suppressive function impaired exacerbating colitis as compared to wild-type Treg cells (151). This suppressive defect may be due to a decrease in local IL-10 levels as CCR6 is preferentially expressed by IL-10 producing Treg cells.
IL-10 signaling is highly relevant for a subset of patients with IBD. Rare loss-of-function mutations in the IL-10 pathway are sufficient to cause very-early-onset IBD presenting in children less than 6 years of age (222, 242–245). Polymorphisms of IL-10, IL-10RA, and IL-10RB resulting in functional hypomorphs are also associated with adult onset IBD (246, 247). Conflicting data has been reported regarding the relationship between IL-10 and maintenance of FOXP3 cells in mice and it has been suggested the maintenance of intestinal FOXP3+ is dependent on IL-10 only in an inflammatory microenvironment (218, 248–250).
Monogenic Disorders Driven by Treg Cell Dysfunction
Humans with a spontaneous X-linked mutation in Foxp3 develop a rare X-linked recessive disorder defined by systemic autoimmunity termed immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome (251). Patients presenting with IPEX syndrome lack functional Treg cells due to loss of protein expression or function (252, 253). To date, over 70 mutations are associated with IPEX syndrome, with patients having varying degrees of autoimmunity (254). IPEX syndrome typically presents in male infants with a triad of enteropathy, autoimmune endocrinopathy, and dermatitis (255). In the absence of functional Treg cells, the balance of T helper cells is perturbed, resulting in T cell skewing towards Th2, IL-17 producing T cells, and expansion of autoreactive B cells (256–258). For this reason, inflammation in patients with IPEX syndrome is marked by a Th2 skewed immune response with the patients often having food allergies, eosinophilia, eczema, and elevated levels of serum immunoglobulin E (255, 259). The intestinal pathology in patients with IPEX syndrome mostly involves enteritis and it is atypical for patients to develop large bowel inflammation. Patients with IPEX syndrome commonly have antibodies directed against enterocytes targeting a 75 kDa autoimmune enteropathy related antigen and VILLIN (260–262). Treatment options for patients with IPEX syndrome generally consist of immunosuppression to control autoimmunity, nutritional interventions, and/or allogeneic hematopoietic stem cell transplantation, which can be curative but is not without risk (254).
Other primary immunodeficiencies (PID) have also been described that are caused by diminished Treg cell function independent of FOXP3 (263–266). These PID can occur in both males and females and present with IPEX-like symptoms but due to mutations in IL2RA, the gene encoding CD25 which is a component of the interleukin (IL)-2 receptor (267–269). These mutations may have differential effects on Treg cells versus effector T cells as CD25 deficiency results in defective IL-10 expression in CD4+ T cells (267) and lack of IL-10 expression by Treg cells in mice contributes to intestinal inflammation (46). Recently, a microduplication of the IL2RA locus associated with very-early-onset IBD which is attributed to increased IL-2 signaling (270). This increased IL-2 signaling potentiates the IFNγ response after TCR activation (270, 271).
Low-Dose IL-2 Expands Treg Cells
In recent years, the sensitivity of Treg cells for IL-2 has been exploited for therapeutic purposes to treat chronic inflammatory diseases. Initially therapeutic applications employed high-dose IL-2 to promote effector T cell expansion to treat metastatic melanoma but is rarely used clinically due to deleterious side effects. Conversely, low-dose IL-2 (LD IL-2), selectively promotes the generation, expansion, survival, and functional activity of Treg cells (157). LD IL-2 has been shown to be an effective strategy to expand autologous Treg cells to ameliorate various inflammatory diseases in humans (e.g., GvHD, hepatitis C-induced vasculitis, WAS, and SLE) (157, 272–275). To date it is unknown whether LD IL-2 would be an effective therapeutic used to treat IBD. Using a translational humanized mouse model, LD IL-2 expands human Treg cells and is protective against TNBS-induced colitis (276) that is independent of CD4+ HLA restriction (277). Of note, expansion of Treg cells in this model was not observed in the intestine but rather in peripheral sites including blood and spleen. A phase 1b/2a clinical trial investigating the use of LD IL-2 to treat moderate to several ulcerative colitis is ongoing (NCT02200445).
Enteral Graft Versus Host Disease
Patients who undergo allogenic hematopoietic stem cell transplantation frequently develop graft versus host disease (GvHD). GvHD can be either acute or chronic, and in most cases the gut is affected. A high frequency of circulatory Treg cells primed to home to the intestine is, perhaps not surprisingly, associated with better outcomes. However, knowledge regarding the role intestinal Treg cells play in GvHD is rather limited (278). One study showed that IL-33R expression in intestinal Treg cells improves their function and since RORγt-deficient Treg cells express higher levels of IL-33R, absence of RORγt results in less severe GvHD (250, 279).
Outstanding Questions
Additional focus on intestinal Treg cells might yield further insights into Treg cell targeted approaches to mitigate inflammation in patients with IBD and/or autoimmunity. The mechanisms underlying microbial specificity of intestinal Treg cells remain to be determined. Moreover, the extent to which SCFA induce new Treg cells from naïve T cells versus increase the proliferation of intestinal Treg cells already present has not yet been fully elucidated (20, 280). A definitive marker to discriminate tTreg cells and pTreg cells is needed to get to the origin of the intestinal Treg cells. Related to the origin, the processes ongoing in lymphoid follicles with regards to intestinal Treg cells remain unclear (281). Together the evidence shows that colonic Treg preferentially localize in lymphoid follicles during homeostasis and inflammation, where they colocalize with DCs and alter the migration and phenotype of conventional T cells. It is tempting to speculate that Treg cells in the lymphoid follicles and those in the LP are phenotypically heterogenous. Much remains to be learned about interaction between epithelial cells, for example whether epithelial cells have differential interactions with Treg cells based on location (i.e., SI versus large intestine) remains to be determined. More insight in the interaction between intestinal Treg cells and stromal cells both during homeostasis and disease is especially warranted since mesenchymal stromal cells (mesenchymal stem cells) are now actively being investigated for a range of diseases including Crohn’s disease (282, 283). Last, how Treg cell expression profiles are altered in settings of chronic intestinal inflammation remains to be elucidated.
Conclusions
In summary, intestinal Treg cells are characterized by a unique transcriptome that is regulated in part by cues from location, site of induction, proximity to the gut microbiome, and interaction with other immune and non-immune cells. Thus, intestinal Treg cells cannot be easily categorized into a distinct and limited number of subsets. Rather, we suggest that the complex local microenvironment, specificity, cell-cell interactions, and signaling receptors contribute to the distinct populations and phenotypes that exhibit functional diversity representing a spectrum of regulators of intestinal immune homeostasis.
Author Contributions
JJ wrote the manuscript with editing by JS and JG, with input of ER. JL summarized Tr1 cells. The authors approved the final version of the manuscript.
Funding
The work was supported by an Academy Ter Meulen Grant from the Royal Netherlands Academy of Arts and Sciences and the Cultural Foundation Grant from Prince Bernhard Cultural Foundation (to JJ) and by grants from the National Institute of Diabetes and Digestive and Kidney Diseases DK123489 (to JG).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AA, Arachidonic acid; CD, Crohn’s disease; CNS1, Conserved noncoding sequence 1; CLTA-4, Cytotoxic lymphocyte antigen-4; DC, Dendritic cell; DSS, dextran sulphate sodium; Foxp3, Forkhead box P3; GALT, Gut associated lymphoid tissue; GF, Germ-free; GPCR, G-protein-coupled receptor; GI-tract, Gastrointestinal tract; GvHD, Graft versus Host Disease; GZMB, Granzyme B; IBD, Inflammatory bowel disease; IL, Interleukin; ICOS, Inducible T-cell costimulator; iTreg, in vitro induced Treg cell; IPEX, Immune dysregulation, polyendocrinopathy, enteropathy, X-linked; LP, Lamina propria; mLN, Mesenteric lymph node; MHC, Major histocompatibility complex; NRP1, Neuropilin-1; PD-1, Programmed cell death protein 1; PG, Prostaglandin; pTreg, Peripherally derived Treg cell; PID, Primary immunodeficiency; RA, Retinoic acid; Repertoire, TCR repertoire; RORγt, RAR-related orphan receptor gamma-t; SCFA, Short-chain fatty acids; SI, Small intestine; SPF, Specific pathogen-free; SLE, Systemic lupus erythematosus; tTreg, Thymus-derived Treg cell; Th, T helper; TCR, T cell receptor; Tr1, Type 1 regulatory T cells; Treg cell, Regulatory T cell; TSDR, Treg cell specific demethylation region; UC, Ulcerative Colitis; Treg cell, Regulatory T cell; WAS, Wiskott-Aldrich Syndrome.
References
1. Miller JFAP. Immunological Function of the Thymus. Lancet (1961) 278:748–9. doi: 10.1016/S0140-6736(61)90693-6
2. Miller JFAP. Role of the Thymus in Transplantation Immunity*. Ann N Y Acad Sci (1962) 99:340–54. doi: 10.1111/j.1749-6632.1962.tb45319.x
3. Gershon RK, Kondo K. Cell Interactions in the Induction of Tolerance: The Role of Thymic Lymphocytes. Immunology (1970) 18:723–37.
4. Kojima A, Tanaka-Kojima Y, Sakakura T, Nishizuka Y. Prevention of Postthymectomy Autoimmune Thyroiditis in Mice. Lab Invest (1976) 34:601–5.
5. Nishizuka Y, Sakakura T. Thymus and Reproduction: Sex-Linked Dysgenesia of the Gonad After Neonatal Thymectomy in Mice. Science (1969) 166:753–5. doi: 10.1126/science.166.3906.753
6. Sakakura T, Nishizuka Y, Tanaka Y. Thymic Control Mechanism in Ovarian Development: Reconstitution of Ovarian Dysgenesis in Thymectomized Mice by Replacement With Thymic and Other Lymphoid Tissues. Endocrinology (1972) 90:431–7. doi: 10.1210/endo-90-2-431
7. Takaba H, Takayanagi H. The Mechanisms of T Cell Selection in the Thymus. Trends Immunol (2017) 38:805–16. doi: 10.1016/j.it.2017.07.010
8. Josefowicz SZ, Rudensky A. Control of Regulatory T Cell Lineage Commitment and Maintenance. Immunity (2009) 30:616–25. doi: 10.1016/j.immuni.2009.04.009
9. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T Cells and Immune Tolerance. Cell (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
10. Powrie F, Leach MW, Mauze S, Caddle LB, Coffman RL. Phenotypically Distinct Subsets of CD4+ T Cells Induce or Protect From Chronic Instestinal Inflammation. Int Immunol (1993) 5:1461–71. doi: 10.1093/intimm/5.11.1461
11. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic Self-Tolerance Maintained by Activated T Cells Expressing IL-2 Receptor Alpha-Chains (CD25). Breakdown of a Single Mechanism of Self-Tolerance Causes Various Autoimmune Diseases. J Immunol (1995) 155:1151–64.
12. Mottet C, Uhlig HH, Powrie F. Cutting Edge: Cure of Colitis by CD4 + CD25 + Regulatory T Cells. J Immunol (2003) 170:3939–43. doi: 10.4049/jimmunol.170.8.3939
13. Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3 + Regulatory T Cells in the Human Immune System. Nat Rev Immunol (2010) 10:490–500. doi: 10.1038/nri2785
14. Carter PB, Collins FM. The Route of Enteric Infection in Normal Mice. J Exp Med (1974) 139:1189–203. doi: 10.1084/jem.139.5.1189
15. Mowat AM, Agace WW. Regional Specialization Within the Intestinal Immune System. Nat Rev Immunol (2014) 14:667–85. doi: 10.1038/nri3738
16. Tilney NL. Patterns of Lymphatic Drainage in the Adult Laboratory Rat. J Anat (1971) 109:369–83.
17. Broeck W, Van den Derore A, Simoens P. Anatomy and Nomenclature of Murine Lymph Nodes: Descriptive Study and Nomenclatory Standardization in BALB/cAnNCrl Mice. J Immunol Methods (2006) 312:12–9. doi: 10.1016/j.jim.2006.01.022
18. Veenbergen S, Van Berkel LA, Du Pré MF, He J, Karrich JJ, Costes LMM, et al. Colonic Tolerance Develops in the Iliac Lymph Nodes and Can Be Established Independent of CD103 + Dendritic Cells. Mucosal Immunol (2016) 9:894–906. doi: 10.1038/mi.2015.118
19. Houston SA, Cerovic V, Thomson C, Brewer J, Mowat AM, Milling S, et al. The Lymph Nodes Draining the Small Intestine and Colon Are Anatomically Separate and Immunologically Distinct. Mucosal Immunol (2016) 9:468–78. doi: 10.1038/mi.2015.77
20. Li X, Zheng Y. Regulatory T Cell Identity: Formation and Maintenance. Trends Immunol (2015) 36:344–53. doi: 10.1016/j.it.2015.04.006
21. Wang J, Ioan-Facsinay A, Voort EIH, van der Huizinga TWJ, Toes REM. Transient Expression of FOXP3 in Human Activated Nonregulatory CD4+ T Cells. Eur J Immunol (2007) 37:129–38. doi: 10.1002/eji.200636435
22. Santegoets SJAM, Dijkgraaf EM, Battaglia A, Beckhove P, Britten CM, Gallimore A, et al. Monitoring Regulatory T Cells in Clinical Samples: Consensus on an Essential Marker Set and Gating Strategy for Regulatory T Cell Analysis by Flow Cytometry. Cancer Immunol Immunother (2015) 64:1271–86. doi: 10.1007/s00262-015-1729-x
23. Roncarolo MG, Yssel H, Touraine JL, Betuel H, De Vries JE, Spits H, et al. Autoreactive T Cell Clones Specific for Class I and Class II HLA Antigens Isolated From a Human Chimera. J Exp Med (1988) 167:1523–34. doi: 10.1084/jem.167.5.1523
24. Groux H, O'Garra A, Bigler M, Rouleau M, GAntonenkoroux S, De Vries JE, et al. A CD4+ T-Cell Subset Inhibits Antigen-Specific T-Cell Responses and Prevents Colitis. Nature (1997) 389:737–42. doi: 10.1038/39614
25. Gagliani N, Magnani CF, Huber S, Gianolini ME, Pala M, Licona-Limon P, et al. Coexpression of CD49b and LAG-3 Identifies Human and Mouse T Regulatory Type 1 Cells. Nat Med (2013) 19:739–46. doi: 10.1038/nm.3179
26. Magnani CF, Alberigo G, Bacchetta R, Serafini G, Andreani M, Roncarolo MG, et al. Killing of Myeloid APCs via HLA Class I, CD2 and CD226 Defines a Novel Mechanism of Suppression by Human Tr1 Cells. Eur J Immunol (2011) 41:1652–62. doi: 10.1002/eji.201041120
27. Akdis M, Verhagen J, Taylor A, Karamloo F, Karagiannidis C, Crameri R, et al. Immune Responses in Healthy and Allergic Individuals Are Characterized by a Fine Balance Between Allergen-Specific T Regulatory 1 and T Helper 2 Cells. J Exp Med (2004) 199:1567–75. doi: 10.1084/jem.20032058
28. Bacchetta R, Bigler M, Touraine JL, Parkman R, Tovo PA, Abrams J, et al. High Levels of Interleukin 10 Production In Vivo Are Associated With Tolerance in SCID Patients Transplanted With HLA Mismatched Hematopoietic Stem Cells. J Exp Med (1994) 179:493–502. doi: 10.1084/jem.179.2.493
29. Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, Roncarolo M-G, et al. Rapamycin Promotes Expansion of Functional CD4 + CD25 + FOXP3 + Regulatory T Cells of Both Healthy Subjects and Type 1 Diabetic Patients. J Immunol (2006) 177:8338–47. doi: 10.4049/jimmunol.177.12.8338
30. Gagliani N, Gregori S, Jofra T, Valle A, Stabilini A, Rothstein DM, et al. Rapamycin Combined With Anti-CD45RB mAB and IL-10 or With G-CSF Induces Tolerance in a Stringent Mouse Model of Islet Transplantation. PloS One (2011) 6:e28434. doi: 10.1371/journal.pone.0028434
31. Roncarolo MG, Gregori S, Bacchetta R, Battaglia M, Gagliani N. The Biology of T Regulatory Type 1 Cells and Their Therapeutic Application in Immune-Mediated Diseases. Immunity (2018) 49:1004–19. doi: 10.1016/j.immuni.2018.12.001
32. Abbas AK, Benoist C, Bluestone JA, Campbell D, Ghosh J, Hori S, et al. Regulatory T Cells: Recommendations to Simplify the Nomenclature. Nat Immunol (2013) 14:307–8. doi: 10.1038/ni.2554
33. Kanamori M, Nakatsukasa H, Okada M, Lu Q, Yoshimura A. Induced Regulatory T Cells: Their Development, Stability, and Applications. Trends Immunol (2016) 37:803–11. doi: 10.1016/j.it.2016.08.012
34. Shevach EM. Foxp3+T Regulatory Cells: Still Many Unanswered Questions-A Perspective After 20 Years of Study. Front Immunol (2018) 9:1–9. doi: 10.3389/fimmu.2018.01048
35. Liston A, Gray DHD. Homeostatic Control of Regulatory T Cell Diversity. Nat Rev Immunol (2014) 14:154–65. doi: 10.1038/nri3605
36. Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, Kas A, et al. CD4+ Regulatory T Cells Control TH17 Responses in a Stat3-Dependent Manner. Science (2009) 326:986–91. doi: 10.1126/science.1172702
37. Koch MA, Tucker-Heard G, Perdue NR, Killebrew J, Urdahl R, Campbell KB, et al. The Transcription Factor T-Bet Controls Regulatory T Cell Homeostasis and Function During Type 1 Inflammation. Nat Immunol (2009) 10:595–602. doi: 10.1038/ni.1731
38. Zheng Y, Chaudhry A, Kas A, DeRoos P, Kim JM, Chu TT, et al. Regulatory T-Cell Suppressor Program Co-Opts Transcription Factor IRF4 to Control TH2 Responses. Nature (2009) 458:351–6. doi: 10.1038/nature07674
39. Burzyn D, Benoist C, Mathis D. Regulatory T Cells in Nonlymphoid Tissues. Nat Immunol (2013) 14:1007–13. doi: 10.1038/ni.2683
40. Miragaia RJ, Gomes T, Chomka A, Jardine L, Riedel A, Hegazy AN, et al. Single-Cell Transcriptomics of Regulatory T Cells Reveals Trajectories of Tissue Adaptation. Immunity (2019) 50:493–504.e7. doi: 10.1016/j.immuni.2019.01.001
41. Round JL, Lee SM, Li J, Tran G, Jabri BA, Chatila T, et al. The Toll-Like Receptor Pathway Establishes Commensal Gut Colonization. Science (2011) 332:974–7. doi: 10.1126/science.1206095
42. Fiorentino DF, Zlotnik A, Vieira P, Mosmann TR, Howard M, Moore KW, et al. IL-10 Acts on the Antigen-Presenting Cell to Inhibit Cytokine Production by Th1 Cells. J Immunol (1991) 146(10):3444–51.
43. Kullberg MC, Jankovic D, Feng CG, Hue S, Gorelick PL, McKenzie BS, et al. IL-23 Plays a Key Role in Helicobacter Hepaticus –Induced T Cell–Dependent Colitis. J Exp Med (2006) 203:2485–94. doi: 10.1084/jem.20061082
44. Shouval DS, Biswas A, Goettel DJA, McCann K, Conaway E, Redhu NS, et al. Interleukin-10 Receptor Signaling in Innate Immune Cells Regulates Mucosal Immune Tolerance and Anti-Inflammatory Macrophage Function. Immunity (2014) 40:706–19. doi: 10.1016/j.immuni.2014.03.011
45. Cretney E, Xin A, Shi W, Minnich M, Masson F, Miasari M, et al. The Transcription Factors Blimp-1 and IRF4 Jointly Control the Differentiation and Function of Effector Regulatory T Cells. Nat Immunol (2011) 12:304–12. doi: 10.1038/ni.2006
46. Chaudhry A, Samstein RM, Treuting P, Liang Y, Pils MC, Heinrich JM, et al. Interleukin-10 Signaling in Regulatory T Cells Is Required for Suppression of Th17 Cell-Mediated Inflammation. Immunity (2011) 34:566–78. doi: 10.1016/j.immuni.2011.03.018
47. Huber S, Gagliani N, Esplugues E, O’Connor W, Huber FJ, Chaudhry A, et al. Th17 Cells Express Interleukin-10 Receptor and Are Controlled by Foxp3– and Foxp3+ Regulatory CD4+ T Cells in an Interleukin-10-Dependent Manner. Immunity (2011) 34:554–65. doi: 10.1016/j.immuni.2011.01.020
48. Asseman C, Mauze S, Leach MW, Coffman RL, Powrie F. An Essential Role for Interleukin 10 in the Function of Regulatory T Cells That Inhibit Intestinal Inflammation. J Exp Med (1999) 190:995–1003. doi: 10.1084/jem.190.7.995
49. Lochner M, Bérard M, Sawa S, Hauer S, Gaboriau-Routhiau V, Fernandez TD, et al. Restricted Microbiota and Absence of Cognate TCR Antigen Leads to an Unbalanced Generation of Th17 Cells. J Immunol (2011) 186:1531–7. doi: 10.4049/jimmunol.1001723
50. Sefik E, Geva-Zatorsky N, Oh S, Konnikova L, Zemmour D, McGuire AM, et al. Individual Intestinal Symbionts Induce a Distinct Population of Rorγ+ Regulatory T Cells. Science (2015) 349:993–7. doi: 10.1126/science.aaa9420
51. Kim KS, Hong SW, Han D, Yi J, Jung J, Yang B, et al. Dietary Antigens Limit Mucosal Immunity by Inducing Regulatory T Cells in the Small Intestine. Science (2016) 351:858–63. doi: 10.1126/science.aac5560
52. Alvarez F, Istomine F, Shourian F, Pavey F, Al-Aubodah F, Qureshi F, et al. The Alarmins IL-1 and IL-33 Differentially Regulate the Functional Specialisation of Foxp3 + Regulatory T Cells During Mucosal Inflammation. Mucosal Immunol (2019) 12:746–60. doi: 10.1038/s41385-019-0153-5
53. Basu R, Whitley SK, Bhaumik S, Zindl CL, Schoeb TR, Benveniste EN, et al. IL-1 Signaling Modulates Activation of STAT Transcription Factors to Antagonize Retinoic Acid Signaling and Control the TH17 cell-iT Reg Cell Balance. Nat Immunol (2015) 16:286–95. doi: 10.1038/ni.3099
54. Schiering C, Krausgruber T, Chomka A, Fröhlich A, Adelmann K, Wohlfert EA, et al. The Alarmin IL-33 Promotes Regulatory T-Cell Function in the Intestine. Nature (2014) 513:564–8. doi: 10.1038/nature13577
55. Josefowicz SZ, Niec RE, Kim HY, Treuting P, Chinen T, Zheng Y, et al. Extrathymically Generated Regulatory T Cells Control Mucosal TH2 Inflammation. Nature (2012) 482:395–9. doi: 10.1038/nature10772
56. Wang Y, Su MA, Wan YY. An Essential Role of the Transcription Factor GATA-3 for the Function of Regulatory T Cells. Immunity (2011) 35:337–48. doi: 10.1016/j.immuni.2011.08.012
57. Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, et al. Expression of Helios, an Ikaros Transcription Factor Family Member, Differentiates Thymic-Derived From Peripherally Induced Foxp3 + T Regulatory Cells. J Immunol (2010) 184:3433–41. doi: 10.4049/jimmunol.0904028
58. Weiss JM, Bilate AM, Gobert M, Ding Y, Curotto de Lafaille MA, Parkhurst CN, et al. Neuropilin 1 Is Expressed on Thymus-Derived Natural Regulatory T Cells, But Not Mucosa-Generated Induced Foxp3 + T Reg Cells. J Exp Med (2012) 209:1723–42. doi: 10.1084/jem.20120914
59. Yadav M, Louvet C, Davini D, Gardner JM, Martinez-Llordella M, Bailey-Bucktrout S, et al. Neuropilin-1 Distinguishes Natural and Inducible Regulatory T Cells Among Regulatory T Cell Subsets. Vivo J Exp Med (2012) 209:1713–22. doi: 10.1084/jem.20120822
60. Glinka Y, Prud’homme GJ. Neuropilin-1 Is a Receptor for Transforming Growth Factor β-1, Activates Its Latent Form, and Promotes Regulatory T Cell Activity. J Leukoc Biol (2008) 84:302–10. doi: 10.1189/jlb.0208090
61. Sarris M, Andersen KG, Randow F, Mayr L, Betz AG. Neuropilin-1 Expression on Regulatory T Cells Enhances Their Interactionsac With Dendritic Cells During Antigen Recognition. Immunity (2008) 28:402–13. doi: 10.1016/j.immuni.2008.01.012
62. Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio CW, Santacruz N, et al. Peripheral Education of the Immune System by Colonic Commensal Microbiota. Nature (2011) 478:250–4. doi: 10.1038/nature10434
63. Singh B, Read S, Asseman C, Malmström V, Mottet C, Stephens LA, et al. Control of Intestinal Inflammation by Regulatory T Cells. Immunol Rev (2001) 182:190–200. doi: 10.1034/j.1600-065X.2001.1820115.x
64. Ai TL, Solomon BD, Hsieh CS. T-Cell Selection and Intestinal Homeostasis. Immunol Rev (2014) 259:60–74. doi: 10.1111/imr.12171
65. Kretschmer K, Apostolou I, Hawiger D, Khazaie K, Nussenzweig MC, von Boehmer H, et al. Inducing and Expanding Regulatory T Cell Populations by Foreign Antigen. Nat Immunol (2005) 6:1219–27. doi: 10.1038/ni1265
66. Östman S, Rask C, Wold AE, Hultkrantz S, Telemo E. Impaired Regulatory T Cell Function in Germ-Free Mice. Eur J Immunol (2006) 36:2336–46. doi: 10.1002/eji.200535244
67. Hsieh CS, Zheng Y, Liang Y, Fontenot JD, Rudensky AY. An Intersection Between the Self-Reactive Regulatory and Nonregulatory T Cell Receptor Repertoires. Nat Immunol (2006) 7:401–10. doi: 10.1038/ni1318
68. Nishio J, Baba M, Atarashi K, Tanoue T, Negishi H, Yanai H, et al. Requirement of Full TCR Repertoire for Regulatory T Cells to Maintain Intestinal Homeostasis. Proc Natl Acad Sci USA (2015) 112:12770–5. doi: 10.1073/pnas.1516617112
69. Cebula A, Seweryn M, Rempala GA, Pabla SS, McIndoe RA, Denning TL, et al. Thymus-Derived Regulatory T Cells Contribute to Tolerance to Commensal Microbiota. Nature (2013) 497:258–62. doi: 10.1038/nature12079
70. Bilate AM, Bousbaine D, Mesin L, Agudelo M, Leube J, Kratzert A, et al. Tissue-Specific Emergence of Regulatory and Intraepithelial T Cells From a Clonal T Cell Precursor. Sci Immunol (2016) 1:eaaf7471–eaaf7471. doi: 10.1126/sciimmunol.aaf7471
71. Zegarra-ruiz DF, Kim DV, Norwood K, Kim M, Wu WH, Saldana-Morales FB, et al. Thymic Development of Gut-Microbiota-Specific T Cells. Nature (2021) 594(7863):413–7. doi: 10.1038/s41586-021-03531-1
72. Izraelson M, Nakonechnaya TO, Moltedo B, Egorov E, Kasatskaya S, Putintseva SA, et al. Comparative Analysis of Murine T-Cell Receptor Repertoires. Immunology (2018) 153:133–44. doi: 10.1111/imm.12857
73. Hoffmann MW, Hintzen G, Bernhardt G, Förster R, Pabst O, Bode U, et al. Oral Tolerance Originates in the Intestinal Immune System and Relies on Antigen Carriage by Dendritic Cells. J Exp Med (2006) 203:519–27. doi: 10.1084/jem.20052016
74. Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, et al. Small Intestine Lamina Propria Dendritic Cells Promote De Novo Generation of Foxp3 T Reg Cells via Retinoic Acid. J Exp Med (2007) 204:1775–85. doi: 10.1084/jem.20070602
75. Hauet-Broere F, Unger WWJJ, Garssen J, Hoijer MA, Kraal G, Samsom JN, et al. Functional CD25- and CD25+ Mucosal Regulatory T Cells Are Induced in Gut-Draining Lymphoid Tissue Within 48 H After Oral Antigen Application. Eur J Immunol (2003) 33:2801–10. doi: 10.1002/eji.200324115
76. Coombes JL, Siddiqui KRR, Arancibia-Cárcamo CV, Hall J, Sun CM, Belkaid Y, et al. A Functionally Specialized Population of Mucosal CD103+ DCs Induces Foxp3+ Regulatory T Cells via a TGF-β -and Retinoic Acid-Dependent Mechanism. J Exp Med (2007) 204:1757–64. doi: 10.1084/jem.20070590
77. Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, et al. Reciprocal TH17 and Regulatory T Cell Differentiation Mediated by Retinoic Acid. Science (2007) 317:256–60. doi: 10.1126/science.1145697
78. Manicassamy S, Pulendran B. Retinoic Acid-Dependent Regulation of Immune Responses by Dendritic Cells and Macrophages. Semin Immunol (2009) 21:22–7. doi: 10.1016/j.smim.2008.07.007
79. Vicente-Suarez I, Larange A, Reardon C, Matho M, Feau S, Chodaczek G, et al. Unique Lamina Propria Stromal Cells Imprint the Functional Phenotype of Mucosal Dendritic Cells. Mucosal Immunol (2015) 8:141–51. doi: 10.1038/mi.2014.51
80. McDonald KG, Leach MR, Brooke KWM, Wang C, Wheeler LW, Hanly EK, et al. Epithelial Expression of the Cytosolic Retinoid Chaperone Cellular Retinol Binding Protein II Is Essential for In Vivo Imprinting of Local Gut Dendritic Cells by Lumenal Retinoids. Am J Pathol (2012) 180:984–97. doi: 10.1016/j.ajpath.2011.11.009
81. Bain CC, Montgomery J, Scott CL, Kel JM, Girard-Madoux MJH, Martens L, et al. Tgfβr Signalling Controls CD103+CD11b+ Dendritic Cell Development in the Intestine. Nat Commun (2017) 8:620. doi: 10.1038/s41467-017-00658-6
82. Worthington JJ, Czajkowska BI, Melton AC, Travis MA. Intestinal Dendritic Cells Specialize to Activate Transforming Growth Factor-β and Induce Foxp3+ Regulatory T Cells via Integrin αvβ8. Gastroenterology (2011) 141:1802–12. doi: 10.1053/j.gastro.2011.06.057
83. Païdassi H, Acharya M, Zhang A, Mukhopadhyay S, Kwon M, Chow C, et al. Preferential Expression of Integrin αvβ8 Promotes Generation of Regulatory T Cells by Mouse CD103+ Dendritic Cells. Gastroenterology (2011) 141:1813–20. doi: 10.1053/j.gastro.2011.06.076
84. Iliev ID, Spadoni I, Mileti E, Matteoli G, Sonzogni A, Sampietro GM, et al. Human Intestinal Epithelial Cells Promote the Differentiation of Tolerogenic Dendritic Cells. Gut (2009) 58:1481–9. doi: 10.1136/gut.2008.175166
85. Spadoni I, Iliev ID, Rossi G, Rescigno M. Dendritic Cells Produce TSLP That Limits the Differentiation of Th17 Cells, Fosters Treg Development, and Protects Against Colitis. Mucosal Immunol (2012) 5:184–93. doi: 10.1038/mi.2011.64
86. Owens BMJ, Simmons A. Intestinal Stromal Cells in Mucosal Immunity and Homeostasis. Mucosal Immunol (2013) 6:224–34. doi: 10.1038/mi.2012.125
87. Pinchuk IV, Powell DW. Immunosuppression by Intestinal Stromal Cells. Adv Exp Med Biol (2018) 1060:115–29. doi: 10.1007/978-3-319-78127-3_7
88. Hammerschmidt SI, Ahrendt M, Bode U, Wahl B, Kremmer E, Förster R, et al. Stromal Mesenteric Lymph Node Cells Are Essential for the Generation of Gut- Homing T Cells In Vivo. J Exp Med (2008) 205:2483–90. doi: 10.1084/jem.20080039
89. Cording S, Wahl B, Kulkarni D, Chopra H, Pezoldt J, Buettner M, et al. The Intestinal Micro-Environment Imprints Stromal Cells to Promote Efficient Treg Induction in Gut-Draining Lymph Nodes. Mucosal Immunol (2014) 7:359–68. doi: 10.1038/mi.2013.54
90. Pezoldt J, Pasztoi M, Zou M, Wiechers C, Beckstette M, Thierry GR, et al. Neonatally Imprinted Stromal Cell Subsets Induce Tolerogenic Dendritic Cells in Mesenteric Lymph Nodes. Nat Commun (2018) 9:3903. doi: 10.1038/s41467-018-06423-7
91. Pasztoi M, Pezoldt J, Beckstette M, Lipps C, Wirth D, Rohde M, et al. Mesenteric Lymph Node Stromal Cell-Derived Extracellular Vesicles Contribute to Peripheral De Novo Induction of Foxp3+ Regulatory T Cells. Eur J Immunol (2017) 47:2142–52. doi: 10.1002/eji.201746960
92. Kashiwakura Y, Sakurai D, Kanno Y, Hashiguchi M, Kobayashi A, Kurosu A, et al. CD2-Mediated Regulation of Peripheral CD4+ CD25+ Regulatory T-Cell Apoptosis Accompanied by Down-Regulation of Bim. Immunology (2013) 139:48–60. doi: 10.1111/imm.12054
93. Pinchuk IV, Beswick EJ, Saada JI, Boya G, Schmitt D, Raju GS, et al. Human Colonic Myofibroblasts Promote Expansion of CD4+ CD25high Foxp3+ Regulatory T Cells. Gastroenterology (2011) 140:2019–30. doi: 10.1053/j.gastro.2011.02.059
94. Newberry RD, McDonough JS, Stenson WF, Lorenz RG. Spontaneous and Continuous Cyclooxygenase-2-Dependent Prostaglandin E 2 Production by Stromal Cells in the Murine Small Intestine Lamina Propria: Directing the Tone of the Intestinal Immune Response. J Immunol (2001) 166:4465–72. doi: 10.4049/jimmunol.166.7.4465
95. Newberry RD, Stenson WF, Lorenz RG. Cyclooxygenase-2-Dependent Arachidonic Acid Metabolites Are Essential Modulators of the Intestinal Immune Response to Dietary Antigen. Nat Med (1999) 5:900–6. doi: 10.1038/11341
96. Morteau O, Morham SG, Sellon R, Dieleman LA, Langenbach R, Smithies O, et al. Impaired Mucosal Defense to Acute Colonic Injury in Mice Lacking Cyclooxygenase-1 or Cyclooxygenase-2. J Clin Invest (2000) 105:469–78. doi: 10.1172/JCI6899
97. Marsh MN, Crowe PT. 5 Morphology of the Mucosal Lesion in Gluten Sensitivity. Baillieres Clin Gastroenterol (1995) 9:273–93. doi: 10.1016/0950-3528(95)90032-2
98. Broere F, du Pré MF, van Berkel LA, Garssen J, Schmidt-Weber CB, Lambrecht BN, et al. Cyclooxygenase-2 in Mucosal DC Mediates Induction of Regulatory T Cells in the Intestine Through Suppression of IL-4. Mucosal Immunol (2009) 2:254–64. doi: 10.1038/mi.2009.2
99. Wang D, Dubois RN. The Role of COX-2 in Intestinal Inflammation and Colorectal Cancer. Oncogene (2010) 29:781–8. doi: 10.1038/onc.2009.421
100. Matteoli G, Mazzini E, Iliev ID, Mileti E, Fallarino F, Puccetti P, et al. Gut CD103+ Dendritic Cells Express Indoleamine 2,3-Dioxygenase Which Influences T Regulatory/T Effector Cell Balance and Oral Tolerance Induction. Gut (2010) 59:595–604. doi: 10.1136/gut.2009.185108
101. Munn DH, Mellor AL. IDO and Tumor-Induced Tolerance. J Clin Invest (2007) 117:1147–54. doi: 10.1172/JCI31178
102. Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, et al. The Combined Effects of Tryptophan Starvation and Tryptophan Catabolites Down-Regulate T Cell Receptor ζ-Chain and Induce a Regulatory Phenotype in Naive T Cells. J Immunol (2006) 176:6752–61. doi: 10.4049/jimmunol.176.11.6752
103. Huehn J, Polansky JK, Hamann A. Epigenetic Control of FOXP3 Expression: The Key to a Stable Regulatory T-Cell Lineage? Nat Rev Immunol (2009) 9:83–9. doi: 10.1038/nri2474
104. Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen WJ, et al. A Critical Function for TGF-β Signaling in the Development of Natural CD4+CD25+Foxp3+ Regulatory T Cells. Nat Immunol (2008) 9:632–40. doi: 10.1038/ni.1607
105. Ouyang W, Beckett O, Ma Q, Li MO. Transforming Growth Factor-β Signaling Curbs Thymic Negative Selection Promoting Regulatory T Cell Development. Immunity (2010) 32:642–53. doi: 10.1016/j.immuni.2010.04.012
106. Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M, et al. Smad3 and NFAT Cooperate to Induce Foxp3 Expression Through Its Enhancer. Nat Immunol (2008) 9:194–202. doi: 10.1038/ni1549
107. Kim HP, Leonard WJ. CREB/ATF-Dependent T Cell Receptor-Induced FoxP3 Gene Expression: A Role for DNA Methylation. J Exp Med (2007) 204:1543–51. doi: 10.1084/jem.20070109
108. Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY, et al. Role of Conserved non-Coding DNA Elements in the Foxp3 Gene in Regulatory T-Cell Fate. Nature (2010) 463:808–12. doi: 10.1038/nature08750
109. Thornton AM, Shevach EM. Helios: Still Behind the Clouds. Immunology (2019) 158:161–70. doi: 10.1111/imm.13115
110. Szurek E, Cebula A, Wojciech L, Pietrzak M, Rempala G, Kisielow P, et al. Differences in Expression Level of Helios and Neuropilin-1 do Not Distinguish Thymus-Derived From Extrathymically-Induced CD4+Foxp3+ Regulatory T Cells. PloS One (2015) 10:1–16. doi: 10.1371/journal.pone.0141161
111. Mucida D, Kutchukhidze N, Erazo A, Russo M, Lafaille JJ, Curotto De Lafaille MA, et al. Oral Tolerance in the Absence of Naturally Occurring Tregs. J Clin Invest (2005) 115:1923–33. doi: 10.1172/JCI24487
112. Uhlig HH, Coombes J, Mottet C, Izcue A, Thompson C, Fanger A, et al. Characterization of Foxp3 + CD4 + CD25 + and IL-10-Secreting CD4 + CD25 + T Cells During Cure of Colitis. J Immunol (2006) 177:5852–60. doi: 10.4049/jimmunol.177.9.5852
113. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, et al. Induction of Colonic Regulatory T Cells by Indigenous Clostridium Species. Science (2011) 331:337–41. doi: 10.1126/science.1198469
114. Geuking MB, Cahenzli J, Lawson MAE, Ng DCK, Slack E, Hapfelmeier S, et al. Intestinal Bacterial Colonization Induces Mutualistic Regulatory T Cell Responses. Immunity (2011) 34:794–806. doi: 10.1016/j.immuni.2011.03.021
115. Chung H, Pamp SJ, Hill JA, Surana NK, Edelman SM, Troy EB, et al. Gut Immune Maturation Depends on Colonization With a Host-Specific Microbiota. Cell (2012) 149:1578–93. doi: 10.1016/j.cell.2012.04.037
116. Ivanov II, Frutos R de L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, et al. Specific Microbiota Direct the Differentiation of IL-17-Producing T-Helper Cells in the Mucosa of the Small Intestine. Cell Host Microbe (2008) 4:337–49. doi: 10.1016/j.chom.2008.09.009
117. Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, et al. Treg Induction by a Rationally Selected Mixture of Clostridia Strains From the Human Microbiota. Nature (2013) 500:232–6. doi: 10.1038/nature12331
118. Alexander KL, Katz J, Elson CO. CBirTox Is a Selective Antigen-Specific Agonist of the Treg-IgA-Microbiota Homeostatic Pathway. PloS One (2017) 12:1–20. doi: 10.1371/journal.pone.0181866
119. Wohlfert EA, Grainger JR, Bouladoux N, Konkel JE, Oldenhove G, Ribeiro CH, et al. GATA3 Controls Foxp3 + Regulatory T Cell Fate During Inflammation in Mice. J Clin Invest (2011) 121:4503–15. doi: 10.1172/JCI57456
120. Cummings JH, Pomare EW, Branch HWJ, Naylor CPE, MacFarlane GT. Short Chain Fatty Acids in Human Large Intestine, Portal, Hepatic and Venous Blood. Gut (1987) 28:1221–7. doi: 10.1136/gut.28.10.1221
121. Høverstad T, Midtvedt T. Short-Chain Fatty Acids in Germfree Mice and Rats. J Nutr (1986) 116:1772–6. doi: 10.1093/jn/116.9.1772
122. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, et al. The Microbial Metabolites, Short-Chain Fatty Acids, Regulate Colonic Treg Cell Homeostasis. Science (2013) 341:569–73. doi: 10.1126/science.1241165
123. Dankert J, Zijlstra JB, Wolthers BG. Volatile Fatty Acids in Human Peripheral and Portal Blood: Quantitative Determination by Vacuum Distillation and Gas Chromatography. Clin Chim Acta (1981) 110:301–7. doi: 10.1016/0009-8981(81)90359-4
124. Schmitt MG, Soergel KH, Wood CM. Absorption of Short Chain Fatty Acids From the Human Jejunum. Gastroenterology (1976) 70:211–5. doi: 10.1016/S0016-5085(76)80011-X
125. Pearce SC, Weber GJ, Van Sambeek DM, Soares J, Racicot W, Breault K, et al. Intestinal Enteroids Recapitulate the Effects of Short-Chain Fatty Acids on the Intestinal Epithelium. PloS One (2020) 15:1–23. doi: 10.1371/journal.pone.0230231
126. Zoetendal EG, Raes J, Van Den Bogert B, Arumugam M, Booijink CC, Troost FJ, et al. The Human Small Intestinal Microbiota Is Driven by Rapid Uptake and Conversion of Simple Carbohydrates. ISME J (2012) 6:1415–26. doi: 10.1038/ismej.2011.212
127. Venegas DP, De La Fuente MK, Landskron G, González MJ, Quera R, Dijkstra G, et al. Short Chain Fatty Acids (SCFAs)mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front Immunol (2019) 10:277. doi: 10.3389/fimmu.2019.00277
128. Bilotta AJ, Cong Y. Gut Microbiota Metabolite Regulation of Host Defenses at Mucosal Surfaces: Implication in Precision Medicine. Precis Clin Med (2019) 2:110–9. doi: 10.1093/pcmedi/pbz008
129. Tao R, De Zoeten EF, Özkaynak E, Chen C, Wang L, Porrett PM, et al. Deacetylase Inhibition Promotes the Generation and Function of Regulatory T Cells. Nat Med (2007) 13:1299–307. doi: 10.1038/nm1652
130. Vinolo MAR, Rodrigues HG, Nachbar RT, Curi R. Regulation of Inflammation by Short Chain Fatty Acids. Nutrients (2011) 3:858–76. doi: 10.3390/nu3100858
131. Thangaraju M, Cresci GA, Liu K, Ananth S, Gnanaprakasam JP, Browning DD, et al. GPFM 09A Is a G-Protein-Coupled Receptor for the Bacterial Fermentation Product Butyrate and Functions as a Tumor Suppressor in Colon. Cancer Res (2009) 69:2826–32. doi: 10.1158/0008-5472.CAN-08-4466
132. Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, et al. Activation of Gpr109a, Receptor for Niacin and the Commensal Metabolite Butyrate, Suppresses Colonic Inflammation and Carcinogenesis. Immunity (2014) 40:128–39. doi: 10.1016/j.immuni.2013.12.007
133. Arpaia N, Campbell C, Fan X, Dikiy S, Van Der Veeken J, Deroos P, et al. Metabolites Produced by Commensal Bacteria Promote Peripheral Regulatory T-Cell Generation. Nature (2013) 504:451–5. doi: 10.1038/nature12726
134. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal Microbe-Derived Butyrate Induces the Differentiation of Colonic Regulatory T Cells. Nature (2013) 504:446–50. doi: 10.1038/nature12721
135. Tan J, McKenzie C, Vuillermin PJ, Goverse G, Vinuesa CG, Mebius RE, et al. Dietary Fiber and Bacterial SCFA Enhance Oral Tolerance and Protect Against Food Allergy Through Diverse Cellular Pathways. Cell Rep (2016) 15:2809–24. doi: 10.1016/j.celrep.2016.05.047
136. Dasgupta S, Erturk-Hasdemir D, Ochoa-Reparaz J, Reinecker H-C, Kasper DL. Plasmacytoid Dendritic Cells Mediate Anti-Inflammatory Responses to a Gut Commensal Molecule via Both Innate and Adaptive Mechanisms. Cell Host Microbe (2014) 15:413–23. doi: 10.1016/j.chom.2014.03.006
137. Round JL, Mazmanian SK. Inducible Foxp3+ Regulatory T-Cell Development by a Commensal Bacterium of the Intestinal Microbiota. Proc Natl Acad Sci USA (2010) 107:12204–9. doi: 10.1073/pnas.0909122107
138. Song X, Song X, Oh SF, Wu M, Zhang Y, Zheng W, et al. Microbial Bile Acid Metabolites Modulate Gut Rorγ+ Regulatory T Cell Homeostasis. Nature (2020) 577:410–5. doi: 10.1038/s41586-019-1865-0
139. Krummel MF, Bartumeus F, Gérard A. T Cell Migration, Search Strategies and Mechanisms. Nat Rev Immunol (2016) 16:193–201. doi: 10.1038/nri.2015.16
140. Marsal J, Agace WW. Targeting T-Cell Migration in Inflammatory Bowel Disease. J Intern Med (2012) 272:411–29. doi: 10.1111/j.1365-2796.2012.02588.x
141. Kimpton WG, Washington EA, Cahill RNP. Virgin αβ and γδ T Cells Recirculate Extensively Through Peripheral Tissues and Skin During Normal Development of the Fetal Immune System. Int Immunol (1995) 7:1567–77. doi: 10.1093/intimm/7.10.1567
142. Thome JJC, Bickham KL, Ohmura Y, Kubota M, Matsuoka N, Gordon C, et al. Early-Life Compartmentalization of Human T Cell Differentiation and Regulatory Function in Mucosal and Lymphoid Tissues. Nat Med (2016) 22:72–7. doi: 10.1038/nm.4008
143. Lee JH, Kang SG, Kim CH. FoxP3 + T Cells Undergo Conventional First Switch to Lymphoid Tissue Homing Receptors in Thymus But Accelerated Second Switch to Nonlymphoid Tissue Homing Receptors in Secondary Lymphoid Tissues. J Immunol (2007) 178:301–11. doi: 10.4049/jimmunol.178.1.301
144. Siewert C, Menning A, Dudda J, Siegmund K, Lauer U, Floess S, et al. Induction of Organ-Selective CD4+ Regulatory T Cell Homing. Eur J Immunol (2007) 37:978–89. doi: 10.1002/eji.200636575
145. Johansson-Lindbom B, Svensson M, Pabst O, Palmqvist C, Marquez G, Förster R, et al. Functional Specialization of Gut CD103+ Dendritic Cells in the Regulation of Tissue-Selective T Cell Homing. J Exp Med (2005) 202:1063–73. doi: 10.1084/jem.20051100
146. Johansson-Lindbom B, Svensson M, Wurbel MA, Malissen B, Márquez G, Agace W, et al. Selective Generation of Gut Tropic T Cells in Gut-Associated Lymphoid Tissue (GALT): Requirement for GALT Dendritic Cells and Adjuvant. J Exp Med (2003) 198:963–9. doi: 10.1084/jem.20031244
147. Mora JR, Bono MR, Manjunath N, Weninger W, Cavanagh LL, Rosemblatt M, et al. Selective Imprinting of Gut-Homing T Cells by Peyer’s Patch Dendritic Cells. Nature (2003) 424:88–93. doi: 10.1038/nature01726
148. Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY, et al. Retinoic Acid Imprints Gut-Homing Specificity on T Cells. Immunity (2004) 21:527–38. doi: 10.1016/j.immuni.2004.08.011
149. Bakdash G, Vogelpoel LTC, Capel TMM, Van Kapsenberg ML, de Jong EC. Retinoic Acid Primes Human Dendritic Cells to Induce Gut-Homing, IL-10-Producing Regulatory T Cells. Mucosal Immunol (2015) 8:265–78. doi: 10.1038/mi.2014.64
150. Balzola F, Cullen G, Ho GT, Russell RK. GPR15-Mediated Homing Controls Immune Homeostasis in the Large Intestine Mucosa. Inflamm Bowel Dis Monit (2013) 14:29. doi: 10.1126/science.1237013
151. Kitamura K, Farber JM, Kelsall BL. CCR6 Marks Regulatory T Cells as a Colon-Tropic, IL-10–Producing Phenotype. J Immunol (2010) 185:3295–304. doi: 10.4049/jimmunol.1001156
152. Schutyser E, Struyf S, Damme J. Van The CC Chemokine CCL20 and Its Receptor CCR6. Cytokine Growth Factor Rev (2003) 14:409–26. doi: 10.1016/S1359-6101(03)00049-2
153. Nakanishi Y, Ikebuchi R, Chtanova T, Kusumoto Y, Okuyama H, Moriya T, et al. Regulatory T Cells With Superior Immunosuppressive Capacity Emigrate From the Inflamed Colon to Draining Lymph Nodes. Mucosal Immunol (2018) 11:437–48. doi: 10.1038/mi.2017.64
154. Pierson W, Cauwe B, Policheni A, Schlenner SM, Franckaert D, Berges J, et al. Antiapoptotic McI-1 Is Critical for the Survival and Niche-Filling Capacity of Foxp3+ Regulatory T Cells. Nat Immunol (2013) 14:959–65. doi: 10.1038/ni.2649
155. Vukmanovic-Stejic M, Zhang Y, Cook JE, Fletcher JM, McQuaid A, Masters JE, et al. Human CD4+CD25hiFoxp3+ Regulatory T Cells Are Derived by Rapid Turnover of Memory Populations. Vivo J Clin Invest (2006) 116:2423–33. doi: 10.1172/JCI28941
156. Tai X, Erman B, Alag A, Mu J, Kimura M, Katz G, et al. Foxp3 Transcription Factor Is Proapoptotic and Lethal to Developing Regulatory T Cells Unless Counterbalanced by Cytokine Survival Signals. Immunity (2013) 38:1116–28. doi: 10.1016/j.immuni.2013.02.022
157. Abbas AK, Trotta E, Simeonov DR, Marson A, Bluestone JA. Revisiting IL-2: Biology and Therapeutic Prospects. Sci Immunol (2018) 3:1–8. doi: 10.1126/sciimmunol.aat1482
158. Gong D, Malek TR. Cytokine-Dependent Blimp-1 Expression in Activated T Cells Inhibits IL-2 Production. J Immunol (2007) 178:242–52. doi: 10.4049/jimmunol.178.1.242
159. Santner-Nanan B, Berberich-Siebelt F, Xiao Z, Poser N, Sennefelder H, Rauthe S, et al. Blimp-1 Is Expressed in Human and Mouse T Cell Subsets and Leads to Loss of IL-2 Production and to Defective Proliferation. Signal Transduction (2006) 6:268–79. doi: 10.1002/sita.200500062
160. Kallies A, Hawkins ED, Belz GT, Metcalf D, Hommel M, Corcoran LM, et al. Transcriptional Repressor Blimp-1 Is Essential for T Cell Homeostasis and Self-Tolerance. Nat Immunol (2006) 7:466–74. doi: 10.1038/ni1321
161. Martins GA, Cimmino L, Shapiro-Shelef M, Szabolcs M, Herron A, Magnusdottir E, et al. Transcriptional Repressor Blimp-1 Regulates T Cell Homeostasis and Function. Nat Immunol (2006) 7:457–65. doi: 10.1038/ni1320
162. Ohnmacht C, Park JH, Cording S, Wing JB, Atarashi K, Obata Y, et al. The Microbiota Regulates Type 2 Immunity Through Rorγt+ T Cells. Science (2015) 349:989–93. doi: 10.1126/science.aac4263
163. Denning TL, Norris BA, Medina-Contreras O, Manicassamy S, Geem D, Madan R, et al. Functional Specializations of Intestinal Dendritic Cell and Macrophage Subsets That Control Th17 and Regulatory T Cell Responses Are Dependent on the T Cell/APC Ratio, Source of Mouse Strain, and Regional Localization. J Immunol (2011) 187:733–47. doi: 10.4049/jimmunol.1002701
164. Watanabe T, Yamori M, Kita T. CD4 + CD25 + T Cells Regulate Colonic Localization of CD4 T Cells Reactive to a Microbial Antigen. Inflamm Bowel Dis (2005) 11:541–50. doi: 10.1097/01.MIB.0000163696.26969.e4
165. Reikvam DH, Perminow G, Lyckander LG, Gran JM, Brandtzaeg P, Vatn M, et al. Increase of Regulatory T Cells in Ileal Mucosa of Untreated Pediatric Crohn’s Disease Patients. Scand J Gastroenterol (2011) 46:550–60. doi: 10.3109/00365521.2011.551887
166. Kelsen J, Agnholt J, Hoffmann J, Rømer HJ, Hvas JL, Dahlerup CL, et al. FoxP3+CD4+CD25+ T Cells With Regulatory Properties can be Cultured From Colonic Mucosa of Patients With Crohn’s Disease. Clin Exp Immunol (2005) 141:549–57. doi: 10.1111/j.1365-2249.2005.02876.x
167. Makita S, Kanai T, Oshima S, Uraushihara K, Totsuka T, Sawada T, et al. CD4 + CD25 Bright T Cells in Human Intestinal Lamina Propria as Regulatory Cells. J Immunol (2004) 173:3119–30. doi: 10.4049/jimmunol.173.5.3119
168. Canavan JB, Scottà C, Vossenkämper A, Goldberg R, Elder MJ, Shoval I, et al. Developing In Vitro Expanded CD45RA+ Regulatory T Cells as an Adoptive Cell Therapy for Crohn’s Disease. Gut (2016) 65:584–94. doi: 10.1136/gutjnl-2014-306919
169. Holmén N, Lundgren A, Lundin S, Bergin AM, Rudin A, Sjövall H, et al. Functional CD4+CD25high Regulatory T Cells Are Enriched in the Colonic Mucosa of Patients With Active Ulcerative Colitis and Increase With Disease Activity. Inflamm Bowel Dis (2006) 12:447–56. doi: 10.1097/00054725-200606000-00003
170. Ricciardelli I, Lindley KJ, Londei M, Quaratino S. Anti Tumour Necrosis-α Therapy Increases the Number of FOXP3 + Regulatory T Cells in Children Affected by Crohn’s Disease. Immunology (2008) 125:178–83. doi: 10.1111/j.1365-2567.2008.02839.x
171. Maul J, Loddenkemper C, Mundt P, Berg E, Giese T, Stallmach A, et al. Peripheral and Intestinal Regulatory CD4+CD25high T Cells in Inflammatory Bowel Disease. Gastroenterology (2005) 128:1868–78. doi: 10.1053/j.gastro.2005.03.043
172. Saruta M, Yu QT, Fleshner PR, Mantel PY, Schmidt-Weber CB, Banham AH, et al. Characterization of FOXP3+CD4+ Regulatory T Cells in Crohn’s Disease. Clin Immunol (2007) 125:281–90. doi: 10.1016/j.clim.2007.08.003
173. Fantini MC, Rizzo A, Fina D, Caruso R, Sarra M, Stolfi C, et al. Smad7 Controls Resistance of Colitogenic T Cells to Regulatory T Cell-Mediated Suppression. Gastroenterology (2009) 136:1308–16.e3. doi: 10.1053/j.gastro.2008.12.053
174. Miyao T, Floess S, Setoguchi R, Luche H, Fehling H, Waldmann J, et al. Plasticity of Foxp3 + T Cells Reflects Promiscuous Foxp3 Expression in Conventional T Cells But Not Reprogramming of Regulatory T Cells. Immunity (2012) 36:262–75. doi: 10.1016/j.immuni.2011.12.012
175. Hori S. Developmental Plasticity of Foxp3+ Regulatory T Cells. Curr Opin Immunol (2010) 22:575–82. doi: 10.1016/j.coi.2010.08.004
176. Martin JC, Chang C, Boschetti G, Ungaro R, Giri M, Grout JA, et al. Single-Cell Analysis of Crohn’s Disease Lesions Identifies a Pathogenic Cellular Module Associated With Resistance to Anti-TNF Therapy. Cell (2019) 178:1493–508.e20. doi: 10.1016/j.cell.2019.08.008
177. Sujino T, London M, Hoytema van Konijnenburg DP, Rendon T, Buch T, Silva HM, et al. Tissue Adaptation of Regulatory and Intraepithelial CD4+ T Cells Controls Gut Inflammation. Science (2016) 352:1581–6. doi: 10.1126/science.aaf3892
178. Ostanin DV, Bao J, Koboziev I, Gray L, Robinson-Jackson SA, Kosloski-Davidson M, et al. T Cell Transfer Model of Chronic Colitis: Concepts, Considerations, and Tricks of the Trade. AJP Gastrointest Liver Physiol (2008) 296:G135–46. doi: 10.1152/ajpgi.90462.2008
179. Eden K. Adoptive Transfer Colitis. In: AC Irving, editor. Mouse Models of Innate Immunity: Methods and Protocols, Methods in Molecular Biology Springer Science+Business Media, LLC, part of Springer Nature (2019), 1960, 207–14. doi: 10.1007/978-1-4939-9167-9_18
180. Feng T, Wang L, Schoeb TR, Elson CO, Cong Y. Microbiota Innate Stimulation Is a Prerequisite for T Cell Spontaneous Proliferation and Induction of Experimental Colitis. J Exp Med (2010) 207:1321–32. doi: 10.1084/jem.20092253
181. Kieper WC, Troy A, Burghardt JT, Ramsey C, Lee JY, Jiang H-Q, et al. Cutting Edge: Recent Immune Status Determines the Source of Antigens That Drive Homeostatic T Cell Expansion. J Immunol (2005) 174:3158–63. doi: 10.4049/jimmunol.174.6.3158
182. Cahill RJ, Foltz CJ, Fox JG, Dangler CA, Powrie D, Schauer DB, et al. Inflammatory Bowel Disease: An Immunity-Mediated Condition Triggered by Bacterial Infection With Helicobacter Hepaticus. Infect Immun (1997) 65:3126–31. doi: 10.1128/iai.65.8.3126-3131.1997
183. Denning TL, Kim G, Kronenberg M. Cutting Edge: CD4 + CD25 + Regulatory T Cells Impaired for Intestinal Homing Can Prevent Colitis. J Immunol (2005) 174:7487–91. doi: 10.4049/jimmunol.174.12.7487
184. Haribhai D, Lin W, Edwards B, Ziegelbauer J, Salzman NH, Carlson MR, et al. A Central Role for Induced Regulatory T Cells in Tolerance Induction in Experimental Colitis. J Immunol (2009) 182:3461–8. doi: 10.4049/jimmunol.0802535
185. Vignali DAA, Collison LW, Workman CJ. How Regulatory T Cells Work. Nat Rev Immunol (2008) 8:523–32. doi: 10.1038/nri2343
186. Wing K, Yamaguchi T, Sakaguchi S. Cell-Autonomous and -non-Autonomous Roles of CTLA-4 in Immune Regulation. Trends Immunol (2011) 32:428–33. doi: 10.1016/j.it.2011.06.002
187. Knickelbein JE, Khanna KM, Yee MB, Baty CJ, Kinchington PR, Hendricks RL, et al. CTLA-4 Control Over Foxp3+ Regulatory T Cell Function. Science (2008) 322:268–71. doi: 10.1126/science.1160062
188. Zeissig S, Petersen B-S, Tomczak M, Melum E, Huc-Claustre E, Dougan SK, et al. Early-Onset Crohn’s Disease and Autoimmunity Associated With a Variant in CTLA-4. Gut (2015) 64:1889–97. doi: 10.1136/gutjnl-2014-308541
189. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal Dominant Immune Dysregulation Syndrome in Humans With CTLA4 Mutations. Nat Med (2014) 20:1410–6. doi: 10.1038/nm.3746
190. Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune Dysregulation in Human Subjects With Heterozygous Germline Mutations in CTLA4. Science (2014) 345:1623–7. doi: 10.1126/science.1255904
191. Gupta A, Felice KM, De Loftus EV, Khanna S. Systematic Review: Colitis Associated With Anti-CTLA-4 Therapy. Aliment Pharmacol Ther (2015) 42:406–17. doi: 10.1111/apt.13281
192. Francisco LM, Salinas VH, Brown KE, Vanguri V, Freeman K, Kuchroo GJ, et al. PD-L1 Regulates the Development, Maintenance, and Function of Induced Regulatory T Cells. J Exp Med (2009) 206:3015–29. doi: 10.1084/jem.20090847
193. Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front Oncol (2018) 8:1–14. doi: 10.3389/fonc.2018.00086
194. Amarnath S, Mangus CW, Wang JCM, Wei F, He A, Kapoor V, et al. The PDL1-PD1 Axis Converts Human T H1 Cells Into Regulatory T Cells. Sci Transl Med (2011) 3:1–14. doi: 10.1126/scitranslmed.3003130
195. Wang L, Pino-Lagos K, De Vrie VC, Guleria I, Sayegh MH, Noelle RJ, et al. Programmed Death 1 Ligand Signaling Regulates the Generation of Adaptive Foxp3+CD4+ Regulatory T Cells. Proc Natl Acad Sci USA (2008) 105:9331–6. doi: 10.1073/pnas.0710441105
196. Sharma M, Shinde R, McGaha T, Huang L, Holmgaard R, Wolchok J, et al. The PTEN Pathway in Tregs Functions as a Critical Driver of the Immunosuppressive Tumor Microenvironment and Tolerance to Apoptotic Cells. J Immunother Cancer (2015) 3:1–16. doi: 10.1186/2051-1426-3-S2-O19
197. Zhang B, Chikuma S, Hori S, Fagarasan S, Honjo T. Nonoverlapping Roles of PD-1 and FoxP3 in Maintaining Immune Tolerance in a Novel Autoimmune Pancreatitis Mouse Model. Proc Natl Acad Sci USA (2016) 113:8490–5. doi: 10.1073/pnas.1608873113
198. Asano T, Meguri Y, Yoshioka T, Kishi Y, Iwamoto M, Nakamura M, et al. PD-1 Modulates Regulatory T-Cell Homeostasis During Low-Dose Interleukin-2 Therapy. Blood (2017) 129:2186–97. doi: 10.1182/blood-2016-09-741629
199. Tan CL, Kuchroo JR, Sage PT, Liang D, Francisco LM, Buck J, et al. PD-1 Restraint of Regulatory T Cell Suppressive Activity Is Critical for Immune Tolerance. J Exp Med (2021) 218(1):e20182232. doi: 10.1084/jem.20182232
200. Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, et al. Survival, Durable Tumor Remission, and Long-Term Safety in Patients With Advanced Melanoma Receiving Nivolumab. J Clin Oncol (2014) 32:1020–30. doi: 10.1200/JCO.2013.53.0105
201. Gianchecchi E, Fierabracci A. Inhibitory Receptors and Pathways of Lymphocytes: The Role of PD-1 in Treg Development and Their Involvement in Autoimmunity Onset and Cancer Progression. Front Immunol (2018) 9:1–12. doi: 10.3389/fimmu.2018.02374
202. Alfen JS, Larghi P, Facciotti F, Gagliani N, Bosotti R, Paroni M, et al. Intestinal IFN-γ–Producing Type 1 Regulatory T Cells Coexpress CCR5 and Programmed Cell Death Protein 1 and Downregulate IL-10 in the Inflamed Guts of Patients With Inflammatory Bowel Disease. J Allergy Clin Immunol (2018) 142:1537–47.e8. doi: 10.1016/j.jaci.2017.12.984
203. Landuyt AE, Klocke BJ, Colvin TB, Schoeb TR, Maynard CL. Cutting Edge: ICOS-Deficient Regulatory T Cells Display Normal Induction of Il10 But Readily Downregulate Expression of Foxp3. J Immunol (2019) 202:1039–44. doi: 10.4049/jimmunol.1801266
204. Burmeister Y, Lischke T, Dahler AC, Mages HW, Lam K-P, Coyle AJ, et al. ICOS Controls the Pool Size of Effector-Memory and Regulatory T Cells. J Immunol (2008) 180:774–82. doi: 10.4049/jimmunol.180.2.774
205. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine Generation Catalyzed by CD39 and CD73 Expressed on Regulatory T Cells Mediates Immune Suppression. J Exp Med (2007) 204:1257–65. doi: 10.1084/jem.20062512
206. Kobie JJ, Shah PR, Yang L, Jonathan A, Fowell DJ, Mosmann TR, et al. T Regulatory and Primed Uncommitted CD4 T Cells Express CD73, Which Suppresses Effector CD4 T Cells by Converting 5 ′ -Adenosine Monophosphate to Adenosine. J Immunol (2006) 177:6780–6. doi: 10.4049/jimmunol.177.10.6780
207. Francois V, Shehade H, Acolty V, Preyat N, Delrée P, Moser M, et al. Intestinal Immunopathology Is Associated With Decreased CD73-Generated Adenosine During Lethal Infection. Mucosal Immunol (2015) 8:773–84. doi: 10.1038/mi.2014.108
208. Resta R, Yamashite Y, Thompson LF. Ecto-Enzyme and Signaling Functions of Lymphocyte CD73. Immunol Rev (1998) 161:95–110. doi: 10.1111/j.1600-065X.1998.tb01574.x
209. Israel B, Medical D. CD39 Is the Dominant Langerhans Cell–Associated Ecto- NTPDase: Modulatory Roles in Inflammation and Immune Responsiveness. Nat Med (2002) 8:358–65. doi: 10.1038/nm0402-358
210. Naganuma M, Wiznerowicz EB, Courtney M, Linden J, Worthington MT, Peter B, et al. Cutting Edge: Critical Role for A 2a Adenosine Receptors in the T Cell-Mediated Regulation of Colitis. J Immunol (2021) 177:2765–9. doi: 10.4049/jimmunol.177.5.2765
211. Huang S, Apasov S, Koshiba M, Sitkovsky M. Adenosine-Mediated Inhibition of T-Cell Activation and Expansion. Blood (1997) 90:1600–10. doi: 10.1182/blood.V90.4.1600.1600_1600_1610
212. Odashima M, Bamias G, Rivera-Nieves J, Linden J, Nast CC, Moskaluk CA, et al. Activation of A2A Adenosine Receptor Attenuates Intestinal Inflammation in Animal Models of Inflammatory Bowel Disease. Gastroenterology (2005) 129:26–33. doi: 10.1053/j.gastro.2005.05.032
213. Scid A, Sauer AV, Brigida I, Carriglio N, Hernandez RJ, Scaramuzza S, et al. Alterations in the Adenosine Metabolism and CD39/CD73 Adenosinergic Machinery Cause Loss of Treg Cell Function and Autoimmunity. in ADA-Deficient SCID. Blood (2012) 119:1428–39. doi: 10.1182/blood-2011-07-366781
214. Huang C, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in Regulatory T Cells. Immunity (2004) 21:503–13. doi: 10.1016/j.immuni.2004.08.010
215. Bauché D, Joyce-Shaikh B, Jain R, Grein J, Ku K, Blumenschein S, et al. LAG3 + Regulatory T Cells Restrain Interleukin-23-Producing CX3CR1 + Gut-Resident Macrophages During Group 3 Innate Lymphoid Cell-Driven Colitis. Immunity (2018) 49:342–52.e5. doi: 10.1016/j.immuni.2018.07.007
216. Kühn R, Löhler J, Rennick D, Rajewsky K, Müller W. Interleukin-10-Deficient Mice Develop Chronic Enterocolitis. Cell (1993) 75:263–74. doi: 10.1016/0092-8674(93)80068-P
217. Kullberg MC, Ward JM, Gorelick PL, Caspar P, Hieny S, Cheever A, et al. Helicobacter Hepaticus Triggers Colitis in Specific-Pathogen-Free Interleukin-10 (IL-10)-Deficient Mice Through an IL-12-and Gamma Interferon- Dependent Mechanism. Infect Immun (1998) 66:5157–66. doi: 10.1128/IAI.66.11.5157-5166.1998
218. Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, et al. Regulatory T Cell-Derived Interleukin-10 Limits Inflammation at Environmental Interfaces. Immunity (2008) 28:546–58. doi: 10.1016/j.immuni.2008.02.017
219. Kamanaka M, Kim ST, Wan YY, Sutterwala FS, Lara-Tejero M, Galán JE, et al. Expression of Interleukin-10 in Intestinal Lymphocytes Detected by an Interleukin-10 Reporter Knockin Tiger Mouse. Immunity (2006) 25:941–52. doi: 10.1016/j.immuni.2006.09.013
220. Veenbergen S, Van Leeuwen MA, Driessen GJ, Kersseboom R, De Ruiter LF, Raatgeep RHC, et al. Development and Function of Immune Cells in an Adolescent Patient With a Deficiency in the Interleukin-10 Receptor. J Pediatr Gastroenterol Nutr (2017) 65:e5–15. doi: 10.1097/MPG.0000000000001559
221. Kotlarz D, Beier R, Murugan D, Diestelhorst J, Jensen O, Boztug K, et al. Loss of Interleukin-10 Signaling and Infantile Inflammatory Bowel Disease: Implications for Diagnosis and Therapy. Gastroenterology (2012) 143:347–55. doi: 10.1053/j.gastro.2012.04.045
222. Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schaffer AA, Noyan F, et al. Inflammatory Bowel Disease and Mutations Affecting the Interleukin-10 Receptor. N Engl J Med (2009) 361:2033–45. doi: 10.1056/NEJMoa0907206
223. Veenbergen S, Li P, Raatgeep HC, Lindenbergh-Kortleve DJ, Simons-Oosterhuis Y, Farrel A, et al. IL-10 Signaling in Dendritic Cells Controls IL-1β-Mediated Ifnγ Secretion by Human CD4+ T Cells: Relevance to Inflammatory Bowel Disease. Mucosal Immunol (2019) 12:1201–11. doi: 10.1038/s41385-019-0194-9
224. Girard-Madoux MJH, Ober-Blöbaum JL, Costes LMM, Kel JM, Lindenbergh-Kortleve DJ, Brouwers-Haspels I, et al. IL-10 Control of CD11c+ Myeloid Cells Is Essential to Maintain Immune Homeostasis in the Small and Large Intestine. Oncotarget (2016) 7:32015–30. doi: 10.18632/oncotarget.8337
225. Zigmond E, Bernshtein B, Friedlander G, Walker CR, Yona S, Kim KW, et al. Macrophage-Restricted Interleukin-10 Receptor Deficiency, But Not IL-10 Deficiency, Causes Severe Spontaneous Colitis. Immunity (2014) 40:720–33. doi: 10.1016/j.immuni.2014.03.012
226. Melillo JA, Song L, Bhagat G, Blazquez AB, Plumlee CR, Lee C, et al. Dendritic Cell (DC)-Specific Targeting Reveals Stat3 as a Negative Regulator of DC Function. J Immunol (2010) 184:2638–45. doi: 10.4049/jimmunol.0902960
227. Gurung P, Li B, Subbarao Malireddi RK, Lamkanfi M, Geiger TL, Kanneganti TD, et al. Chronic TLR Stimulation Controls NLRP3 Inflammasome Activation Through IL-10 Mediated Regulation of NLRP3 Expression and Caspase-8 Activation. Sci Rep (2015) 5:1–10. doi: 10.1038/srep14488
228. Chan AH, Schroder K. Inflammasome Signaling and Regulation of Interleukin-1 Family Cytokines. J Exp Med (2020) 217:1–10. doi: 10.1084/jem.20190314
229. Li J, Shouval DS, Doty AL, Snapper SB, Glover SC. Increased Mucosal IL-22 Production of an IL-10ra Mutation Patient Following Anakinra Treatment Suggests Further Mechanism for Mucosal Healing. J Clin Immunol (2017) 37:104–7. doi: 10.1007/s10875-016-0365-3
230. Shouval DS, Biswas A, Kang YH, Griffith AE, Konnikova L, Mascanfroni ID, et al. Interleukin 1β Mediates Intestinal Inflammation in Mice and Patients With Interleukin 10 Receptor Deficiency. Gastroenterology (2016) 151:1100–4. doi: 10.1053/j.gastro.2016.08.055
231. Duan L, Chen J, Zhang H, Yang H, Zhu P, Xiong A, et al. Interleukin-33 Ameliorates Experimental Colitis Through Promoting Th2/Foxp3+ Regulatory T-Cell Responses in Mice. Mol Med (2012) 18:753–61. doi: 10.2119/molmed.2011.00428
232. Izcue A, Hue S, Buonocore S, Arancibia-Cárcamo CV, Ahern PP, Iwakura Y, et al. Interleukin-23 Restrains Regulatory T Cell Activity To Drive T Cell-Dependent Colitis. Immunity (2008) 28:559–70. doi: 10.1016/j.immuni.2008.02.019
233. M’Koma AE. Inflammatory Bowel Disease: An Expanding Global Health Problem. Clin Med Insights Gastroenterol (2013) 6:33–47. doi: 10.4137/CGast.S12731
234. Souza HSP. De & Fiocchi, C. Immunopathogenesis of IBD: Current State of the Art. Nat Rev Gastroenterol Hepatol (2016) 13:13–27. doi: 10.1038/nrgastro.2015.186
235. Nutsch K, Chai JN, Ai TL, Russler-Germain E, Feehley T, Nagler CR, et al. Rapid and Efficient Generation of Regulatory T Cells to Commensal Antigens in the Periphery. Cell Rep (2016) 17:206–20. doi: 10.1016/j.celrep.2016.08.092
236. Alexander KL, Zhao Q, Reif M, Rosenberg AF, Mannon PJ, Duck LW, et al. Human Microbiota Flagellins Drive Adaptive Immune Responses in Crohn’s Disease. Gastroenterology (2021) 1–14. doi: 10.1053/j.gastro.2021.03.064
237. Hand TW, Dos Santos LM, Bouladoux N, Molloy M, Pagán J, Pepper AJ, et al. Acute Gastrointestinal Infection Induces Long-Lived Microbiota-Specific T Cell Responses. Science (2012) 337:1553–6. doi: 10.1126/science.1220961
238. Cong Y, Feng T, Fujihashi K, Schoeb TR, Elson CO. A Dominant, Coordinated T Regulatory Cell-IgA Response to the Intestinal Microbiota. Proc Natl Acad Sci USA (2009) 106:19256–61. doi: 10.1073/pnas.0812681106
239. Tang Q, Bluestone JA, Kang SM. CD4 +Foxp3 + Regulatory T Cell Therapy in Transplantation. J Mol Cell Biol (2012) 4:11–21. doi: 10.1093/jmcb/mjr047
240. Puleston J, Cooper M, Murch S, Bid K, Makh S, Ashwood P, et al. A Distinct Subset of Chemokines Dominates the Mucosal Chemokine Response in Inflammatory Bowel Disease. Aliment Pharmacol Ther (2005) 21:109–20. doi: 10.1111/j.1365-2036.2004.02262.x
241. Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, et al. Surface Phenotype and Antigenic Specificity of Human Interleukin 17-Producing T Helper Memory Cells. Nat Immunol (2007) 8:639–46. doi: 10.1038/ni1467
242. Engelhardt K.R GB. IL-10 in Humans: Lessons From the Gut, IL-10/IL-10 Receptor Deficiencies, and IL-10 Polymorphisms. Curr Top Microbiol Immunol (2014) 380:1–8. doi: 10.1007/978-3-662-43492-5_1
243. Glocker EO, Frede N, Perro M, Sebire N, Elawad M, Shah N, et al. Infant Colitis-its in the Genes. Lancet (2010) 376:1272. doi: 10.1016/S0140-6736(10)61008-2
244. Glocker EO, Kotlarz D, Klein C, Shah N, Grimbacher B. IL-10 and IL-10 Receptor Defects in Humans. Ann N Y Acad Sci (2011) 1246:102–7. doi: 10.1111/j.1749-6632.2011.06339.x
245. Moran CJ, Walters TD, Guo C-HH, Kugathasan S, Klein C, Turner D, et al. IL-10r Polymorphisms Are Associated With Very-Early-Onset Ulcerative Colitis. Inflamm Bowel Dis (2013) 19:115–23. doi: 10.1002/ibd.22974
246. Ellinghaus D, Jostins L, Spain SL, Cortes A, Bethune J, Han B, et al. Analysis of Five Chronic Inflammatory Diseases Identifies 27 New Associations and Highlights Disease-Specific Patterns at Shared Loci. Nat Genet (2016) 48:510–8. doi: 10.1038/ng.3528
247. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-Microbe Interactions Have Shaped the Genetic Architecture of Inflammatory Bowel Disease. Nature (2012) 491:119–24. doi: 10.1038/nature11582
248. Turovskaya O, Kim G, Cheroutre H, Kronenberg M, Madan R. Interleukin 10 Acts on Regulatory T Cells to Maintain Expression of the Transcription Factor Foxp3 and Suppressive Function in Mice With Colitis. Nat Immunol (2009) 10:1178–84. doi: 10.1038/ni.1791
249. Ouyang W, O’Garra A. IL-10 Family Cytokines IL-10 and IL-22: From Basic Science to Clinical Translation. Immunity (2019) 50:871–91. doi: 10.1016/j.immuni.2019.03.020
250. Fulton LM, Carlson MJ, Coghill JM, Ott LE, West ML, Panoskaltsis-Mortari A, et al. Attenuation of Acute Graft-Versus-Host Disease in the Absence of the Transcription Factor Rorγt. J Immunol (2012) 189:1765–72. doi: 10.4049/jimmunol.1200858
251. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a New Forkhead/Winged-Helix Protein, Scurfin, Results in the Fatal Lymphoproliferative Disorder of the Scurfy Mouse. Nat Genet (2001) 27:68–73. doi: 10.1038/83784
252. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Mazzella M, et al. The Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked Syndrome (IPEX) Is Caused by Mutations of FOXP3. Nat Genet (2001) 27:20–1. doi: 10.1038/83713
253. Powell BR, Buist NRM, Stenzel P. An X-Linked Syndrome of Diarrhea, Polyendocrinopathy, and Fatal Infection in Infancy. J Pediatr (1982) 100:731–7. doi: 10.1016/S0022-3476(82)80573-8
254. Bacchetta R, Barzaghi F, Roncarolo MG. From IPEX Syndrome to FOXP3 Mutation: A Lesson on Immune Dysregulation. Ann N Y Acad Sci (2016) 1417:5–22. doi: 10.1111/nyas.13011
255. Braskett MJ, Chatila T. IPEX: Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked (2020). Available at: https://www.uptodate.com/contents/ipex-immune-dysregulation-polyendocrinopathy-enteropathy-x-linked.
256. Bacchetta R, Passerini L, Gambineri E, Dai M, Allan SE, Perroni L, et al. Defective Regulatory and Effector T Cell Functions in Patients With FOXP3 Mutations. J Clin Invest (2006) 116:1713–22. doi: 10.1172/JCI25112
257. Kinnunen T, Chamberlain N, Morbach H, Choi J, Kim S, Craft J, et al. Accumulation of Peripheral Autoreactive B Cells in the Absence of Functional Human Regulatory T Cells. Blood (2012) 121:1595–603. doi: 10.1182/blood-2012-09-457465
258. Passerini L, Olek S, Di Nunzio S, Barzaghi F, Hambleton S, Abinun M, et al. Forkhead Box Protein 3 (FOXP3) Mutations Lead to Increased T H 17 Cell Numbers and Regulatory T-Cell Instability. J Allergy Clin Immunol (2011) 128:1376–80. doi: 10.1016/j.jaci.2011.09.010
259. Hansmann L, Schmidl C, Kett J, Steger L, Andreesen R, Hoffmann P, et al. Dominant Th2 Differentiation of Human Regulatory T Cells Upon Loss of FOXP3 Expression. J Immunol (2012) 188:1275–82. doi: 10.4049/jimmunol.1102288
260. Kobayashi I, Imamura K, Yamada M, Okano M, Yara A, Ikema S, et al. A 75-kD Autoantigen Recognized by Sera From Patients With X-Linked Autoimmune Enteropathy Associated With Nephropathy. Clin Exp Immunol (1998) 111:527–31. doi: 10.1046/j.1365-2249.1998.00523.x
261. Kobayashi I, Kubota M, Yamada M, Tanaka H, Itoh S, Sasahara Y, et al. Autoantibodies to Villin Occur Frequently in IPEX, a Severe Immune Dysregulation, Syndrome Caused by Mutation of FOXP3. Clin Immunol (2011) 141:83–9. doi: 10.1016/j.clim.2011.05.010
262. Patey-Mariaud De Serre N, Canioni D, Ganousse S, Rieux-Laucat F, Goulet O, Ruemmele F, et al. Digestive Histopathological Presentation of IPEX Syndrome. Mod Pathol (2009) 22:95–102. doi: 10.1038/modpathol.2008.161
263. Alroqi FJ, Chatila TA. T Regulatory Cell Biology in Health and Disease. Curr Allergy Asthma Rep (2016) 16:27. doi: 10.1007/s11882-016-0606-9
264. Demeyer A, Skordos I, Driege Y, Kreike M, Hochepied T, Baens M, et al. MALT1 Proteolytic Activity Suppresses Autoimmunity in a T Cell Intrinsic Manner. Front Immunol (2019) 10:1–13. doi: 10.3389/fimmu.2019.01898
265. McKinnon ML, Rozmus J, Fung S-Y, Hirschfeld AF, Del Bel KL, Thomas L, et al. Combined Immunodeficiency Associated With Homozygous MALT1 Mutations. J Allergy Clin Immunol (2014) 133:1458–62.e7. doi: 10.1016/j.jaci.2013.10.045
266. Verbsky JW, Chatila TA. T-Regulatory Cells in Primary Immune Deficiencies. Curr Opin Allergy Clin Immunol (2011) 11:539–44. doi: 10.1097/ACI.0b013e32834cb8fa
267. Caudy AA, Reddy ST, Chatila T, Atkinson JP, Verbsky JW. CD25 Deficiency Causes an Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked-Like Syndrome, And Defective IL-10 Expression From CD4 lymphocytes. J Allergy Clin Immunol (2007) 119:482–7. doi: 10.1016/j.jaci.2006.10.007
268. Roifman CM. Human IL-2 Receptor α Chain Deficiency. Pediatr Res (2000) 48:6–11. doi: 10.1203/00006450-200007000-00004
269. Sharfe N, Dadi HK, Shahar M, Roifman CM. Human Immune Disorder Arising From Mutation of the Chain of the Interleukin-2 Receptor (CD25CD1bcl-2thymocytelymphocytic Infiltration). Immunology (1997) 94:3168–71. doi: 10.1073/pnas.94.7.3168
270. Joosse ME, Charbit-henrion F, Boisgard R, Raatgeep RHC, Lindenbergh-kortleve DJ, Costes LMM, et al. Duplication of the IL2RA Locus Causes Excessive IL-2 Signaling and may Predispose to Very Early Onset Colitis. Mucosal Immunol (2021). doi: 10.1038/s41385-021-00423-5
271. Shatrova AN, Mityushova EV, Vassilieva IO, Aksenov ND, Zenin VV, Nikolsky NN, et al. Time-Dependent Regulation of IL-2r α-Chain (CD25) Expression by TCR Signal Strength and IL-2-Induced STAT5 Signaling in Activated Human Blood T Lymphocytes. PloS One (2016) 11:1–19. doi: 10.1371/journal.pone.0167215
272. He J, Zhang X, Wei Y, Sun X, Chen Y, Deng J, et al. Low-Dose Interleukin-2 Treatment Selectively Modulates CD4+ T Cell Subsets in Patients With Systemic Lupus Erythematosus. Nat Med (2016) 22:991. doi: 10.1038/nm.4148
273. Jyonouchi S, Gwafila B, Gwalani LA, Ahmad M, Moertel C, Holbert C, et al. Phase I Trial of Low-Dose Interleukin 2 Therapy in Patients With Wiskott-Aldrich Syndrome. Clin Immunol (2017) 179:47–53. doi: 10.1016/j.clim.2017.02.001
274. Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP, et al. Interleukin-2 and Regulatory T Cells in Graft-Versus-Host Disease. N Engl J Med (2011) 365:2055–66. doi: 10.1056/NEJMoa1108188
275. Saadoun D, Rosenzwajg M, Joly F, Six A, Carrat F, Thibault V, et al. Regulatory T-Cell Responses to Low-Dose Interleukin-2 in HCV-Induced Vasculitis. N Engl J Med (2011) 365:2067–77. doi: 10.1056/NEJMoa1105143
276. Goettel JA, Kotlarz D, Emani R, Canavan JB, Konnikova L, Illig D, et al. Low-Dose Interleukin-2 Ameliorates Colitis in a Preclinical Humanized Mouse Model. Cell Mol Gastroenterol Hepatol (2019) 8:193–5. doi: 10.1016/j.jcmgh.2019.05.001
277. Tyagi RK, Jacobse J, Li J, Allaman MM, Otipoby KL, Sampson ER, et al. HLA-Restriction of Human Treg Cells Is Not Required for Therapeutic Efficacy of Low-Dose IL-2 in Humanized Mice. Front Immunol (2021) 12:1–8. doi: 10.3389/fimmu.2021.630204
278. Akpek G, Chinratanalab W, Lee LA, Torbenson M, Hallick JP, Anders V, et al. Gastrointestinal Involvement in Chronic Graft-Versus-Host Disease: A Clinicopathologic Study. Biol Blood Marrow Transpl (2003) 9:46–51. doi: 10.1053/bbmt.2003.49999
279. Yang J, Ramadan A, Reichenbach DK, Loschi M, Zhang J, Griesenauer B, et al. Rorc Restrains the Potency of ST2+ Regulatory T Cells in Ameliorating Intestinal Graft-Versus-Host Disease. JCI Insight (2019) 4:1–14. doi: 10.1172/jci.insight.122014
280. Russler-Germain EV, Rengarajan S, Hsieh C-S. Antigen-Specific Regulatory T-Cell Responses to Intestinal Microbiota. Mucosal Immunol (2017) 10:1375–86. doi: 10.1038/mi.2017.65
281. Sipos F, Muzes G. Isolated Lymphoid Follicles in Colon: Switch Points Between Inflammation and Colorectal Cancer? World J Gastroenterol (2011) 17:1666–73. doi: 10.3748/wjg.v17.i13.1666
282. Negi N, Griffin MD. Effects of Mesenchymal Stromal Cells on Regulatory T Cells: Current Understanding and Clinical Relevance. Stem Cells (2020) 38:596–605. doi: 10.1002/stem.3151
Keywords: intestine, regulatory T (Treg) cells, homeostasis, IBD, intestinal inflammation
Citation: Jacobse J, Li J, Rings EHHM, Samsom JN and Goettel JA (2021) Intestinal Regulatory T Cells as Specialized Tissue-Restricted Immune Cells in Intestinal Immune Homeostasis and Disease. Front. Immunol. 12:716499. doi: 10.3389/fimmu.2021.716499
Received: 28 May 2021; Accepted: 16 July 2021;
Published: 04 August 2021.
Edited by:
Femke Van Wijk, University Medical Center Utrecht, NetherlandsReviewed by:
David Hoytema Van Konijnenburg, Boston Children’s Hospital and Harvard Medical School, United StatesCraig L. Maynard, University of Alabama at Birmingham, United States
Copyright © 2021 Jacobse, Li, Rings, Samsom and Goettel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeremy A. Goettel, jeremy.goettel@vumc.org
†These authors share last authorship