 Afef Rais1,2,3†
Afef Rais1,2,3† Najla Mekki1,2,3†
Najla Mekki1,2,3† Faten Fedhila2,3,4
Faten Fedhila2,3,4 Mohammed Faraj Alosaimi5
Mohammed Faraj Alosaimi5 Monia Ben Khaled2,3,6
Monia Ben Khaled2,3,6 Amal Zameli1,3
Amal Zameli1,3 Nourhen Agrebi1,3Maryam Kallel Sellami2,3,7
Nourhen Agrebi1,3Maryam Kallel Sellami2,3,7 Raif Geha8
Raif Geha8 Imen Ben-Mustapha1,2,3*‡
Imen Ben-Mustapha1,2,3*‡ Mohamed-Ridha Barbouche1,2,3‡
Mohamed-Ridha Barbouche1,2,3‡- 1Laboratory of Transmission, Control and Immunobiology of Infections (LR11IPT02), Institut Pasteur de Tunis, Tunis, Tunisia
- 2Faculty of Medicine, Université de Tunis El Manar, Tunis, Tunisia
- 3Université de Tunis El Manar, Tunis, Tunisia
- 4Department of Pediatrics A, Children’s Hospital, Tunis, Tunisia
- 5Department of Pediatrics, College of Medicine, King Saud University, Riyadh, Saudi Arabia
- 6Pediatric Immuno-Hematology unit, Bone Marrow Transplantation Center Tunis, Tunis, Tunisia
- 7Department of Immunology, La Rabta University Hospital, Tunis, Tunisia
- 8Division of Immunology, Boston Children’s Hospital, Harvard Medical School, Boston, MA, United States
ALPS and IPEX are two well-characterized inborn errors of immunity with immune dysregulation, considered as two master models of monogenic auto-immune diseases. Thus, with autoimmunity as their primary clinical manifestation, these two entities may show clinical overlap. Traditionally, immunological biomarkers are used to establish an accurate differential diagnosis. Herein, we describe a patient who presented with clinical features and biomarkers fulfilling the diagnostic criteria of ALPS. Severe apoptotic defect was also shown in the patient’s cell lines and PHA-activated peripheral blood lymphocytes. Sanger sequencing of the FAS gene did not reveal any causal mutation. NGS screening revealed a novel deleterious variant located in the N terminal repressor domain of FOXP3 but no mutations in the FAS pathway-related genes. TEMRA cells (terminally differentiated effector memory cells re-expressing CD45RA) and PD1 expression were increased arguing in favor of T-cell exhaustion, which could be induced by unrestrained activation of T effector cells because of Treg deficiency. Moreover, defective FOXP3 observed in the patient could intrinsically induce increased proliferation and resistance to apoptosis in T effector cells. This observation expands the spectrum of FOXP3 deficiency and underscores the role of NGS in detecting mutations that induce overlapping phenotypes among inborn errors of immunity with immune dysregulation. In addition, these findings suggest a potential link between FOXP3 and FAS pathways.
Introduction
Primary immune dysregulation diseases are a group of monogenic disorders characterized by defective immune regulatory pathways (1–3). Their clinical presentation mainly comprises organ-specific autoimmunity, hyperinflammation, and nonmalignant lymphoproliferation. New sequencing technologies have improved the understanding of this ever-expanding group of disorders but they still often pose a diagnostic challenge because of their variable and complex phenotypic expressions and the overlap of symptoms between different entities (1, 4). Immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) and autoimmune lymphoproliferative syndrome (ALPS) are archetypes of inborn errors of immunity (IEI) with immune dysregulation and have gained attention as models of monogenic autoimmunity.
IPEX syndrome is secondary to mutations in the Forkhead Box Protein 3 (FOXP3) gene, a transcription factor essential for regulatory T cell differentiation and function. Its clinical expression encompasses various combinations of autoimmune manifestations appearing early in life, with a characteristic triad comprising enteropathy, dermatitis, and autoimmune endocrinopathy, usually, type 1 diabetes (5).
ALPS represents a distinct pathologic mechanism for loss of immune tolerance caused by a defect in the Fas-FasL pathway. Noninfectious and nonmalignant lymphoproliferation associated with autoimmune cytopenias and a greatly increased lifetime risk of lymphoma constitute the major features of ALPS. The marking immunological phenotype remains an expansion of autoreactive double-negative T (DNT) cells (6), a subset of CD3+ cells bearing α/β T cell receptor (TCR), and negative for both CD4 and CD8 co-receptors. They massively accumulate in patients with ALPS but are also reported in other inflammatory and autoimmune conditions (7, 8). A chronic non-malignant lymphadenopathy, an elevation of peripheral DNT cells, and a defective apoptosis assay were first proposed as a required triad of criteria to establish the diagnosis of ALPS (9). Later on, serum sFasL, IL-10, and vitamin B12 were shown to be elevated in ALPS because of FAS mutations (ALPS-FAS), and their association to high DNT cells rose as an accurate predictor for the presence of germline or somatic FAS mutation (10, 11). Consequently, these biomarkers were included in the revised diagnostic criteria of ALPS to facilitate the patients’ identification particularly in settings where no access to advanced genetic analysis or functional testing are available (12).
The functional apoptosis test allows to detect a defective response upon Fas stimulation which results in abnormal cell survival and is sufficient for definitive ALPS diagnosis provided that required criteria are fulfilled (12). Therefore, defective Fas driven apoptosis is considered specific to ALPS and prompts molecular testing when a patient has suggestive clinical and laboratory features.
Despite the establishment of well-characterized ALPS diagnostic guidelines, phenotypic overlap with other primary immune dysregulation conditions still poses a diagnostic challenge (13). Indeed, previous reports have shown that LRBA deficiency, STAT3 GOF mutations and ITK deficiency may be misdiagnosed as ALPS and may share with it some clinical and immunological features (13–17).
Herein, we describe a patient bearing a novel FOXP3 mutation in the N terminal repressor domain of the protein who fulfills the current criteria for definitive diagnosis for ALPS (12, 18).
Case Description
Born to a non-consanguineous marriage, the patient had a family history of infant deaths without clearly identified causes. His clinical manifestations started at the age of 2 months and were marked by generalized squamous dermatitis and multiple adenopathies without hepatosplenomegaly. Langerhans cell histiocytosis diagnosis was initially proposed based on the presence of multiple CD1a+PS100+ cells in a retroauricular lymph node biopsy, then ruled out because of the atypical cutaneous manifestation and the absence of histiocytic proliferation. The dermatitis significantly improved after the use of topical corticosteroids and evolved to skin xerosis. At the age of 10 months, the clinical course was marked by the occurrence of a severe prolonged diarrhea episode with edema and hypoproteinemia requiring hospitalization. Etiological investigations, including coeliac disease autoantibodies screening, sweat test, and RAST test to cow milk proteins were all negative. The diarrhea resolved under symptomatic treatment with exclusion of cow milk and did not relapse. Interestingly, during this episode, biological investigations revealed hemolytic anemia with positive direct Coombs test (AIHA). The patient received oral prednisone at 2 mg/kg/day during 1 month followed by a progressive decrease during 3 months with a stabilization of the hemoglobin rate. At the age of 14 months, he presented with an Evans syndrome as revealed by epistaxis and purpura. The patient’s platelet level was very low with a normal platelet size, and his anemia was critical, requiring transfusion. He underwent corticosteroid treatment (prednisone, 2 mg/kg/day) and received intravenous immunoglobulins (1 g/kg) with a poor response. During hospitalization, he also experienced a resolutive episode of pneumonia. The clinical picture was further worsened by multiple relapses of Evans syndrome, which required full-dose corticosteroid (prednisone, 2 mg/kg) and two hospitalizations at age 18 months and 3 years (Figure 1). Following the initial period of recurrent corticosteroid-dependent cytopenias, he continued to receive full dose of corticosteroids, allowing full remission at the age of 4 years. Interestingly, corticosteroid has been decreased then stopped for 3 years. His follow-up at age 7 years showed a favorable outcome. His condition is now characterized by the persistence of chronic adenopathies with no histological signs of malignancy and skin xerosis. During this follow-up, no gastro-intestinal manifestations, no endocrinopathies, or other auto-immune manifestations were recorded with the exception of a recent episode of thrombocytopenia. His infectious course was characterized by the occurrence of recurrent episodes of otitis media.
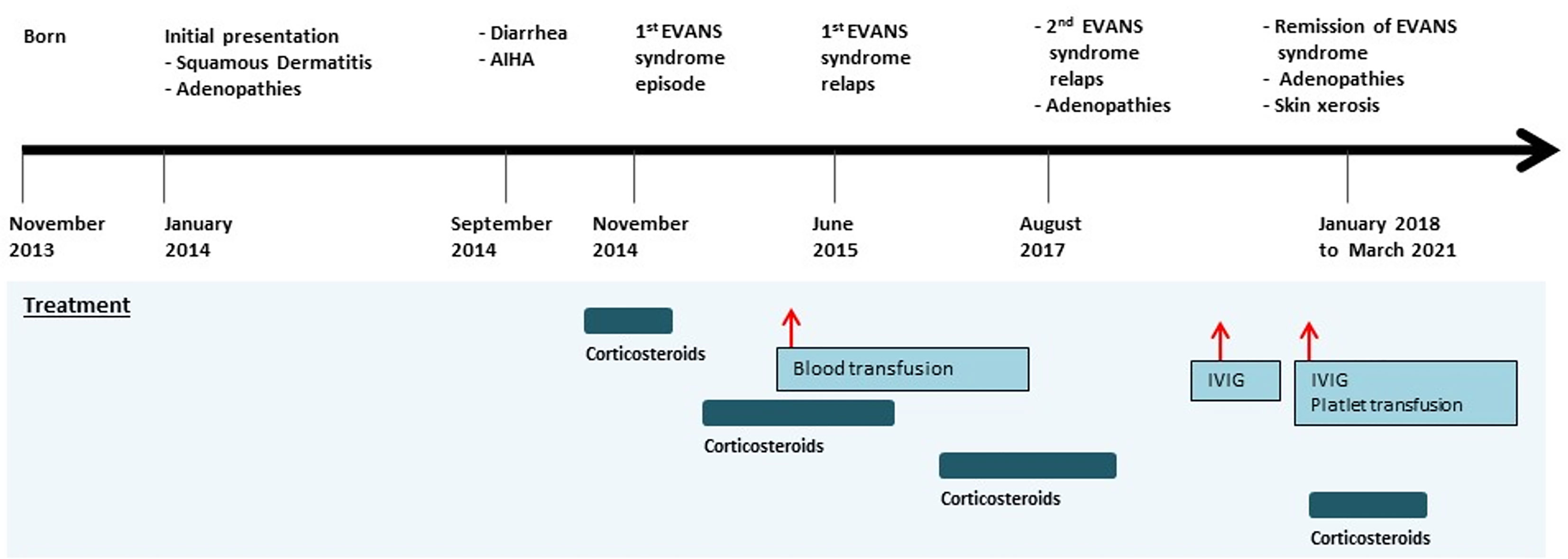
Figure 1 Timeline of the clinical events and treatment strategies. AIHA, autoimmune hemolytic anemia; IVIG, intravenous immunoglobulin.
Timeline
The timeline is presented in Figure 1.
Diagnostic Assessment
The diagnosis of IEI was suspected given the patient’s presentation. Preliminary immunological investigations showed polyclonal hypergammaglobulinemia (IgG: 15.51 g/L), elevated IgE, and hypereosinophilia. The standard immunophenotyping of lymphocyte subpopulations (T, B, and NK cells) assessed by flow cytometry was within the normal range. Lymphoproliferative responses to mitogens and antigens were normal. Recurrent Evans syndrome associated with chronic lymphoproliferations prompted the evaluation of ALPS criteria. Double negative T cells CD3+TCRα/β+CD4−CD8− percentage was increased, reaching 5.1%. Soluble FasL and IL-10 plasma levels were high. Apoptosis functional assay was performed on the patient’s EBV-transformed B-cells and showed severe resistance to Fas-mediated cell death in comparison to a healthy control, as confirmed in two separate experiments (Figure 2A). It has also been performed on the patient’s PHA-activated peripheral blood lymphocytes and confirmed the resistance to apoptosis as compared with healthy control (Figure 2A). This defect of apoptosis is persistent at age 7 years. All biological and immunological investigations are summarized in Table 1.
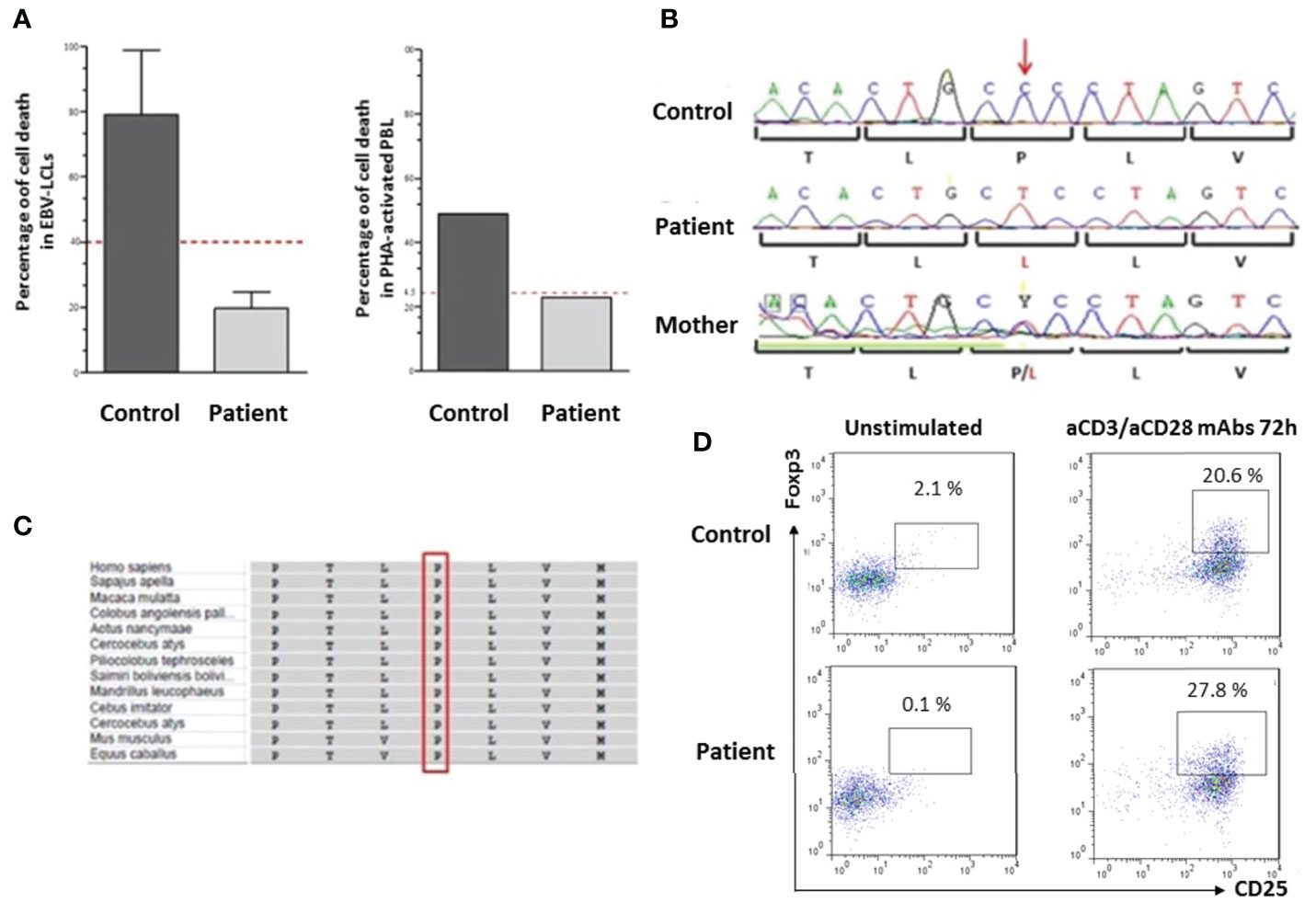
Figure 2 Genetic and functional investigations (A) Apoptosis assay: The IPEX patient exhibits a significantly altered apoptosis. Fas-induced apoptosis in EBV-lymphoblastoid cell lines (EBV-LCL) or PHA-activated peripheral blood lymphocytes of the patient and a healthy control were stimulated for 24 h with or without APO-1-3 in the presence of protein A. Subsequently apoptosis was measured by flow cytometric detection of Annexin V-PE and 7AAD. For apoptosis in EBV-LCL, data represent two separate experiments and the T bars indicate the standard deviation. The dotted red line represents 50% of the cell death observed in the control. (B) Genomic DNA sequence analysis of FOXP3 gene showing a missens hemizygous mutation p.Pro75Leu in the patient, the mother was heterozygous for the mutation. (C) Multiple protein alignment showing the position of the conserved proline 75 residue in different species using the NCBI Multiple Sequence Alignment Viewer, Version 1.19.1. (D) FOXP3 expression in PBMCs after staining with anti-CD4/APC, anti-CD25/BB-515 and anti-Foxp3/PE as compared to healthy control. Treg cells were absent in patient’s unstimulated cells and induced after activation with anti-CD3 (aCD3)/anti-CD28 (aCD28) mAb during 72 h.
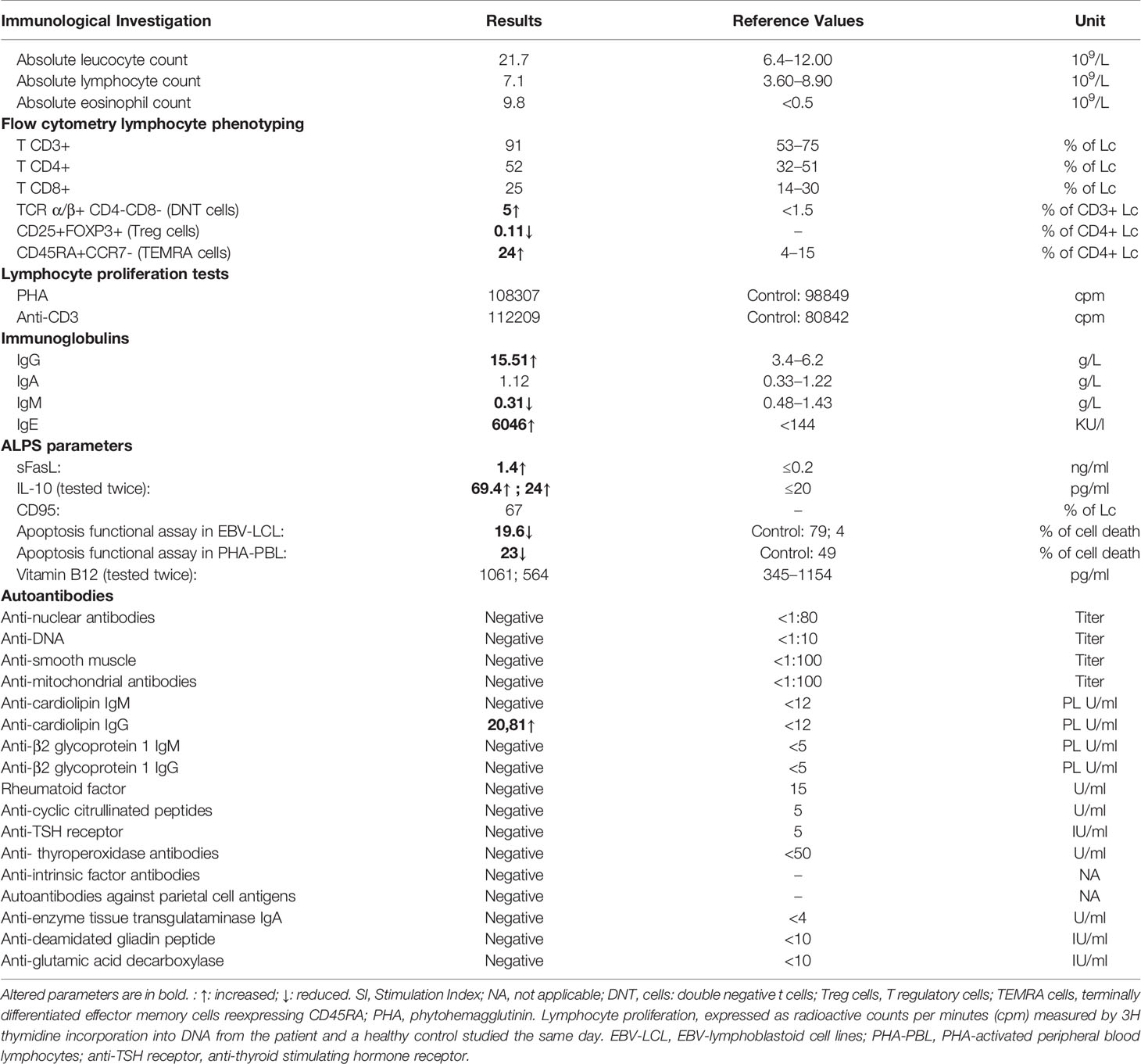
Table 1 Results of the immunological investigations.
The criteria for definitive ALPS diagnosis were fulfilled. Indeed, the two required criteria (i.e., expanded double negative T cells and chronic lymphadenopathy) were present together with one accessory primary criterion, namely, defective lymphocyte apoptosis in two separate assays and two accessory secondary criteria, namely, autoimmune cytopenia with elevated polyclonal IgG and elevated plasma sFasL levels (>200 pg/ml) and IL-10 levels (>20 pg/ml). The resistance to Fas-driven apoptosis argued in favor of a defect in the Fas pathway. The FAS gene was screened by Sanger sequencing for mutations in all exons and intron-exon junctions, because FAS deficiency accounts for approximately 65% of all ALPS cases. Surprisingly, no FAS germline mutation was found. Somatic FAS mutations were excluded because their presence does not fit with the defective apoptosis assay.
Targeted NGS, using the PID v2 gene panel (including all known ALPS genes) and an Ion Torrent S5 sequencer identified a novel missense mutation c.224C>T (p.Pro75Leu) located in FOXP3. No variants in other genes were found, which might have contributed to the patient’s phenotype. Sanger sequencing confirmed the mutation using forward and reverse specific primers (5′CCATGAGCCTCAGTTTCCAT and 5′ CACCTTTGACCCCCAGAGTA). The patient’s mother was heterozygous for the same mutation (Figure 2B). This mutation is located in the repressor domain of FOXP3 and is not reported in 1000G or ExAC databases. The affected residue is conserved among vertebrates (Figure 2C), and the mutation is predicted to be deleterious in multiple in silico prediction tools (probably damaging in Polyphen2, damaging in SIFT and CADD score of 24).
To assess the consequences of the FOXP3 mutation, FOXP3 expression was performed in patient’s PBMCs as compared to healthy control, after staining with anti-CD4/APC, anti-CD25/BB-515 and anti-Foxp3/PE with or without activation for 72 h using anti-CD3/anti-CD28 monoclonal antibodies. FOXP3+ CD25+ cells were absent in patient’s unstimulated cells (0.1% of CD4+) but were induced after activation (27.8%) (Figure 2D).
The search for the presence of autoantibodies using an extensive set of antigens displayed negative results except for anti-cardiolipin IgG (Table 1).
To decipher the potential link between FOXP3 defect and the ALPS phenotype, and based on the hypothesis that Treg deficiency would induce an unrestrained activation of T effector cells that are resistant to Fas driven apoptosis (19), we performed an immunophenotyping of TEMRA cells. These cells have been defined as the latest stage of T-cell differentiation which accumulate through successive rounds of antigen encounters (20, 21) and could be induced by persistent re-stimulation of individual clones (22). We found increased percentage of CD4+ TEMRA cells in the patient reaching 24% (normal range for the age, 4–15%) (Table 1 and Figure 3A). Moreover, the patient showed high PD1 expression by 10.9% on CD4+T cells, one of the hallmarks of T cell exhaustion (23), in comparison to healthy controls (Figure 3B).
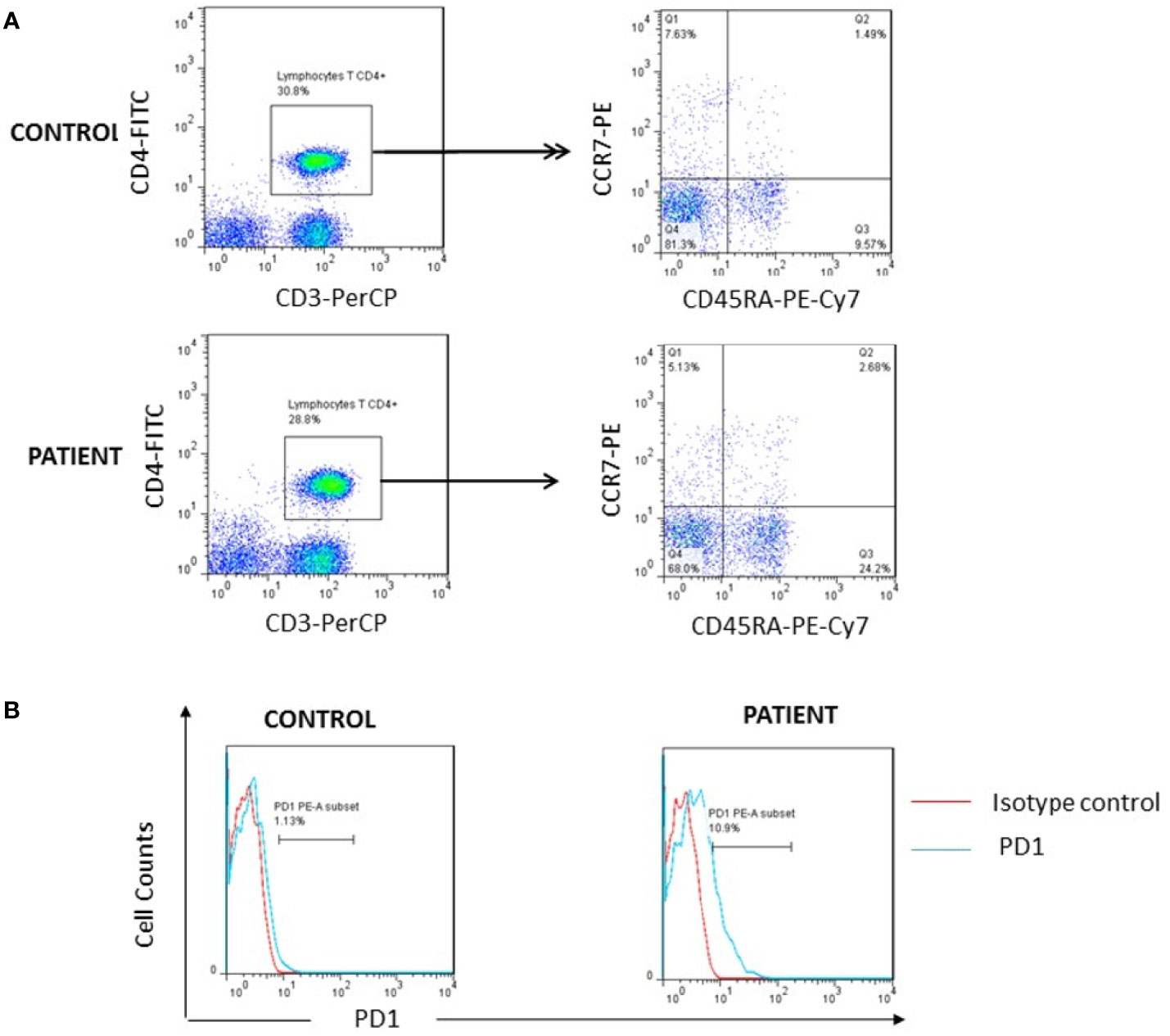
Figure 3 Investigation of T cell exhaustion. (A) High percentage of TEMRA cells (Q3) was found in patient after whole blood staining with anti-CD3/PerCP, anti-CD4/FITC, anti-CD45RA/PE-Cy7 and anti-CCR7/PE as compared to one representative healthy control. (B) Increased surface expression of PD1 on the patient’s CD4+ lymphocytes as compared to one representative control..
Discussion
The classical IPEX presentation consists of a triad of symptoms, comprising diarrhea, autoimmune endocrinopathies and dermatitis (5, 24, 25). The most consistent clinical finding, often inaugural in this disease, is the exudative enteropathy (24). It classically begins very early, during the first few months of life, characterized by its persistence, and usually complicates with malabsorption and failure to thrive (5, 24, 25). The patient we report did first present with dermatitis and multiple adenopathies. Unlike in the IPEX classical presentation, this patient had a single limited episode of severe diarrhea at age ten months. Shortly after, he developed Evans syndrome associated with bleeding complications. Autoimmune cytopenias are a characteristic feature of ALPS. However, they are also observed in up to 26% of IPEX patients (25, 26). Moreover, the patient did not develop autoimmune endocrinopathies, which are frequently reported in IPEX patients and consist most commonly in type 1 diabetes mellitus occurring in the first year of life (5, 24, 25). It has been reported that in the absence of clinical disease, anti-glutamic acid decarboxylase antibodies or anti-islet cell antibodies may be positive (5). The screening of this patient’s serum for autoantibodies associated with endocrinopathies was negative, and no endocrine disorders were recorded during the follow-up. The patient’s clinical presentation was also marked by the occurrence of infectious manifestations consisting of recurrent media otitis and one episode of resolving pneumonia. This contrasts with the classical severe infections observed in IPEX patients, which may be life-threatening sepsis (5, 24, 25). Thus, the IPEX clinical presentation in this patient is unusual with mild or absent features of the characteristic IPEX triad and the predominance of an Evans syndrome. In this context, the persistence of benign adenopathies and hypergamma- globulinemia shaped a clinical picture more reminiscent of ALPS, the most typical IEI associated with autoimmune cytopenias (27). Moreover, the immunohistological pattern of sinus histiocytosis with S100 positive cells observed in the patient’s lymph node biopsy has not only been reported in ALPS (6) but also in a single description of IPEX patient (28).
The patient’s laboratory investigations showed hyper- eosinophilia and elevated IgE. The increase of these two parameters is common in IPEX patients. Although they are not usually characteristic of ALPS classical presentation, their increase was reported in patients with ALPS (29–31). Besides these overlapping features, the most striking finding in this observation is the presence of almost all ALPS biomarkers.
To our best knowledge, there are no published reports of IPEX associated with elevated ALPS biomarkers and a defective Fas apoptosis assay. Elevated DNT cells were initially considered as a hallmark of ALPS (9) and are still a required criterion for the diagnosis (12). This biomarker specificity has been challenged because other IEIs have displayed elevated DNT cells, such as LRBA deficiency, STAT3 GOF disease and CVID (13, 16). Elevation of DNT cells has also been reported in patients with autoimmune diseases (8, 32). However, when they exceed 4%, they are considered as highly predictive of FAS mutations (11). Nevertheless, high percentages of this subset, exceeding this threshold, have been recorded in LRBA deficiency, reaching 10% and in STAT3 GOF disease reaching 20% (13, 16). In our patient, DNT cells reached 5% and were associated with other ALPS biomarkers, including elevation of IL-10 and sFasL and a defective Fas-induced apoptosis. Interestingly, in a recent review of key diagnostic markers of ALPS, the combination of elevated DNTs and an abnormal in vitro apoptosis functional test was considered the most useful in identifying all types of ALPS patients (33). The combination of an abnormal in vitro apoptosis functional test and elevated sFasL was a predictive marker for ALPS-FAS group classification (33).
The FOXP3 mutation identified changed the diagnosis from ALPS to IPEX, clearly showing that NGS use is critical to unmask atypical presentations of IPEX and is reshaping the clinical and immunological spectrum of this rare and probably under-diagnosed disease (34). No less than 70 pathological variants have been identified in the FOXP3 gene (5, 35). Interestingly, some mutations were more frequently associated with a mild or atypical presentation (34), such as reported here with currently a favorable outcome. This contrasts with the classical outcome of IPEX syndrome, generally associated with a poor prognosis (24). The mutation we report resides in the N terminal proline-rich domain, thought to mediate the repressive activity of FOXP3 (36). On another hand, the mutation is located in exon 3, which is the second coding exon (the first being a non-coding one) of a total of 12 exons. It is worth mentioning that some of these exons could be alternatively spliced leading to naturally occurring FOXP3 isoforms and may be associated with slower kinetics of FOXP3 export (37, 38). The mutation we identified was associated with the absence of CD4+CD25+FOXP3+ cells. Since steroid administration may reduce FOXP3 expression, the latter was assessed after activation and found to be restored. This finding is in accordance with the study of Gambineri et al. (39), showing that some FOXP3 mutations may not hinder protein expression after activation despite initial very low levels of Treg. The assessment of the correlation between restored FOXP3 expression and residual Treg function must be further investigated and may explain the moderate clinical phenotype of the patient.
The defect of apoptosis assay observed in the patient is somewhat intriguing. An altered functional apoptosis assay was previously observed in other immune regulation disorders, such as STAT3 GOF disease and LRBA deficiency (14, 16). Resistance to apoptosis in STAT3 GOF disease appears to be secondary to a disturbance in the balance of BCL2 family proteins, which are critical intrinsic regulators of the apoptotic pathway (16). The compromised Fas triggered apoptosis described in LRBA deficiency is suggested to be due to an altered autophagy. Indeed, autophagy is significantly reduced in LRBA deficient cells and has been shown to facilitate Fas-mediated apoptosis (14).
The Treg deficiency in the patient could result in unrestrained activation of T effector cells. The latter would become exhausted and thus resistant to apoptosis (19). Consistent with this hypothesis, we found increased CD4+TEMRA cells and PD1 expression in patient as compared with healthy controls.
On the other hand, defective FOXP3 observed in the patient could intrinsically induce increased proliferation and resistance to apoptosis in T effector cells, as herein reported. There is evidence in the literature that the amount of FOXP3 expressed within a cell controls the balance between life and death. Indeed, it has been shown that T cells from mice and humans that lack functional FOXP3 are hyper-responsive to TCR stimulation, and those from mice that overexpress FOXP3 are hypo-responsive to TCR-mediated stimulation as manifested by the reduction in proliferative capacity. Consequently, defect in cell cycle progression after TCR engagement may result in altered apoptosis (40).
In summary, this case expands the clinical spectrum of Foxp3 deficiency and suggests a potential link between FOXP3 and the FAS apoptotic pathway that requires further investigation.
Patient Perspective
The patient did adhere to the treatment proposed. He and his family were satisfied by the relative improvement of his clinical condition.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/clinvar/variation/1098427/.
Ethics Statement
The studies involving human participants were reviewed and approved by Comité d’éthique biomédicale de l’Institut Pasteur de Tunis. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
AR, NM, NA and IB-M performed immunological investigations and interpretation of results. MS performed the screening of autoantibodies and interpretation of results. FF and MB were the clinicians in charge of patient care and management. MFA and RG performed targeted NGS and interpretation of results. IB-M, M-RB, and RG contributed to conception and design of the study. AR and IB-M wrote the initial manuscript draft. AR, NM, MA, RG, IB-M and M-RB reviewed the manuscript and contributed to the final draft. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Tunisian ministry of higher education and research.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Chandrakasan S, Chandra S, Davila Saldana BJ, Torgerson TR, Buchbinder D. Primary Immune Regulatory Disorders for the Pediatric Hematologist and Oncologist: A Case-Based Review. Pediatr Blood Cancer (2019) 66(5):e27619. doi: 10.1002/pbc.27619
2. Cepika AM, Sato Y, Liu JM, Uyeda MJ, Bacchetta R, Roncarolo MG. Tregopathies: Monogenic Diseases Resulting in Regulatory T-Cell Deficiency. J Allergy Clin Immunol (2018) 142(6):1679–95. doi: 10.1016/j.jaci.2018.10.026
3. Bousfiha A, Jeddane L, Picard C, Ailal F, Bobby Gaspar H, Al-Herz W, et al. The 2017 IUIS Phenotypic Classification for Primary Immunodeficiencies. J Clin Immunol (2018) 38(1):129–43. doi: 10.1007/s10875-017-0465-8
4. Takagi M, Hoshino A, Yoshida K, Ueno H, Imai K, Piao J, et al. Genetic Heterogeneity of Uncharacterized Childhood Autoimmune Diseases With Lymphoproliferation. Pediatr Blood Cancer (2018) 65(2). doi: 10.1002/pbc.26831
5. Gambineri E, Ciullini Mannurita S, Hagin D, Vignoli M, Anover-Sombke S, DeBoer S, et al. Clinical, Immunological, and Molecular Heterogeneity of 173 Patients With the Phenotype of Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked (IPEX) Syndrome. Front Immunol (2018) 9:2411. doi: 10.3389/fimmu.2018.02411
6. Matson DR, Yang DT. Autoimmune Lymphoproliferative Syndrome: An Overview. Arch Pathol Lab Med (2020) 144(2):245–51. doi: 10.5858/arpa.2018-0190-RS
7. Crispín JC, Oukka M, Bayliss G, Cohen RA, Van Beek CA, Stillman IE, et al. Expanded Double Negative T Cells in Patients With Systemic Lupus Erythematosus Produce IL-17 and Infiltrate the Kidneys. J Immunol (Baltimore Md: 1950) (2008) 181(12):8761–6. doi: 10.4049/jimmunol.181.12.8761
8. Tarbox JA, Keppel MP, Topcagic N, Mackin C, Ben Abdallah M, Baszis KW, et al. Elevated Double Negative T Cells in Pediatric Autoimmunity. J Clin Immunol (2014) 34(5):594–9. doi: 10.1007/s10875-014-0038-z
9. Straus SE, Sneller M, Lenardo MJ, Puck JM, Strober W. An Inherited Disorder of Lymphocyte Apoptosis: The Autoimmune Lymphoproliferative Syndrome. Ann Internal Med (1999) 130(7):591–601. doi: 10.7326/0003-4819-130-7-199904060-00020
10. Magerus-Chatinet A, Stolzenberg MC, Loffredo MS, Neven B, Schaffner C, Ducrot N, et al. FAS-L, IL-10, and Double-Negative CD4- CD8- TCR Alpha/Beta+ T Cells Are Reliable Markers of Autoimmune Lymphoproliferative Syndrome (ALPS) Associated With FAS Loss of Function. Blood (2009) 113(13):3027–30. doi: 10.1182/blood-2008-09-179630
11. Caminha I, Fleisher TA, Hornung RL, Dale JK, Niemela JE, Price S, et al. Using Biomarkers to Predict the Presence of FAS Mutations in Patients With Features of the Autoimmune Lymphoproliferative Syndrome. J Allergy Clin Immunol (2010) 125(4):946–9.e6. doi: 10.1016/j.jaci.2009.12.983
12. Oliveira JB, Bleesing JJ, Dianzani U, Fleisher TA, Jaffe ES, Lenardo MJ, et al. Revised Diagnostic Criteria and Classification for the Autoimmune Lymphoproliferative Syndrome (ALPS): Report From the 2009 NIH International Workshop. Blood (2010) 116(14):e35–40. doi: 10.1182/blood-2010-04-280347
13. Cagdas D, Halaçlı SO, Tan Ç, Lo B, Çetinkaya PG, Esenboğa S, et al. A Spectrum of Clinical Findings From ALPS to CVID: Several Novel LRBA Defects. J Clin Immunol (2019) 39(7):726–38. doi: 10.1007/s10875-019-00677-6
14. Revel-Vilk S, Fischer U, Keller B, Nabhani S, Gámez-Díaz L, Rensing-Ehl A, et al. Autoimmune Lymphoproliferative Syndrome-Like Disease in Patients With LRBA Mutation. Clin Immunol (Orlando Fla) (2015) 159(1):84–92. doi: 10.1016/j.clim.2015.04.007
15. Oliveira JB. The Expanding Spectrum of the Autoimmune Lymphoproliferative Syndromes. Curr Opin Pediatr (2013) 25(6):722–9. doi: 10.1097/MOP.0000000000000032
16. Nabhani S, Schipp C, Miskin H, Levin C, Postovsky S, Dujovny T, et al. STAT3 Gain-of-Function Mutations Associated With Autoimmune Lymphoproliferative Syndrome Like Disease Deregulate Lymphocyte Apoptosis and can be Targeted by BH3 Mimetic Compounds. Clin Immunol (Orlando Fla) (2017) 181:32–42. doi: 10.1016/j.clim.2017.05.021
17. Wallace JG, Alosaimi MF, Khayat CD, Jaber F, Almutairi A, Beaussant-Cohen S, et al. ITK Deficiency Presenting as Autoimmune Lymphoproliferative Syndrome. J Allergy Clin Immunol (2021) 147(2):743–5.e1. doi: 10.1016/j.jaci.2020.06.019
18. Seidel MG, Kindle G, Gathmann B, Quinti I, Buckland M, van Montfrans J, et al. The European Society for Immunodeficiencies (ESID) Registry Working Definitions for the Clinical Diagnosis of Inborn Errors of Immunity. J Allergy Clin Immunol In Pract (2019) 7(6):1763–70. doi: 10.1016/j.jaip.2019.02.004
19. Gupta S, Gollapudi S. CD95-Mediated Apoptosis in Naïve, Central and Effector Memory Subsets of CD4+ and CD8+ T Cells in Aged Humans. Exp Gerontol (2008) 43(4):266–74. doi: 10.1016/j.exger.2007.12.006
20. Weekes MP, Wills MR, Mynard K, Hicks R, Sissons JG, Carmichael AJ. Large Clonal Expansions of Human Virus-Specific Memory Cytotoxic T Lymphocytes Within the CD57+ CD28- CD8+ T-Cell Population. Immunology (1999) 98(3):443–9. doi: 10.1046/j.1365-2567.1999.00901.x
21. Pangrazzi L, Naismith E, Meryk A, Keller M, Jenewein B, Trieb K, et al. Increased IL-15 Production and Accumulation of Highly Differentiated CD8(+) Effector/Memory T Cells in the Bone Marrow of Persons With Cytomegalovirus. Front Immunol (2017) 8:715. doi: 10.3389/fimmu.2017.00715
22. Larbi A, Fulop T. From “Truly Naïve” to “Exhausted Senescent” T Cells: When Markers Predict Functionality. Cytomet Part A: J Int Soc Anal Cytol (2014) 85(1):25–35. doi: 10.1002/cyto.a.22351
24. Tan QKG, Louie R, Sleasman JW. IPEX Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2021 (2004). Available at: https://www.ncbi.nlm.nih.gov/books/NBK1118/.
25. Jamee M, Zaki-Dizaji M, Lo B, Abolhassani H, Aghamahdi F, Mosavian M, et al. Clinical, Immunological, and Genetic Features in Patients With Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked (IPEX) and IPEX-Like Syndrome. J Allergy Clin Immunol In Pract (2020) 8(8):2747–60.e7. doi: 10.1016/j.jaip.2020.04.070
26. Barzaghi F, Amaya Hernandez LC, Neven B, Ricci S, Kucuk ZY, Bleesing JJ, et al. Long-Term Follow-Up of IPEX Syndrome Patients After Different Therapeutic Strategies: An International Multicenter Retrospective Study. J Allergy Clin Immunol (2018) 141(3):1036–49.e5. doi: 10.1016/j.jaci.2017.10.041
27. Seidel MG. Autoimmune and Other Cytopenias in Primary Immunodeficiencies: Pathomechanisms, Novel Differential Diagnoses, and Treatment. Blood (2014) 124(15):2337–44. doi: 10.1182/blood-2014-06-583260
28. Sayar E, Uygun DF, Islek A, Hazar-Sayar E, Akkaya B, Vignoli M, et al. Langerhans Cell Histiocytosis in IPEX Syndrome: Possible Role for Natural Regulatory T Cells? Pediatr Allergy Immunol: Off Publ Eur Soc Pediatr Allergy Immunol (2014) 25(6):601–3. doi: 10.1111/pai.12219
29. Butt D, Chan TD, Bourne K, Hermes JR, Nguyen A, Statham A, et al. FAS Inactivation Releases Unconventional Germinal Center B Cells That Escape Antigen Control and Drive IgE and Autoantibody Production. Immunity (2015) 42(5):890–902. doi: 10.1016/j.immuni.2015.04.010
30. Kim YJ, Dale JK, Noel P, Brown MR, Nutman TB, Straus SE, et al. Eosinophilia Is Associated With a Higher Mortality Rate Among Patients With Autoimmune Lymphoproliferative Syndrome. Am J Hematol (2007) 82(7):615–24. doi: 10.1002/ajh.20851
31. Price S, Shaw PA, Seitz A, Joshi G, Davis J, Niemela JE, et al. Natural History of Autoimmune Lymphoproliferative Syndrome Associated With FAS Gene Mutations. Blood (2014) 123(13):1989–99. doi: 10.1182/blood-2013-10-535393
32. El-Sayed ZA, El-Owaidy RH. Alpha Beta Double Negative T Cells in Children With Systemic Lupus Erythematosus: The Relation to Disease Activity and Characteristics. Mod Rheumatol (2018) 28:(4):654–60. doi: 10.1080/14397595.2017.1377146
33. Molnár E, Radwan N, Kovács G, Andrikovics H, Henriquez F, Zarafov A, et al. Key Diagnostic Markers for Autoimmune Lymphoproliferative Syndrome With Molecular Genetic Diagnosis. Blood (2020) 136(17):1933–45. doi: 10.1182/blood.2020005486
34. Consonni F, Ciullini Mannurita S, Gambineri E. Atypical Presentations of IPEX: Expect the Unexpected. Front Pediatr (2021) 9:643094. doi: 10.3389/fped.2021.643094
35. Bacchetta R, Barzaghi F, Roncarolo MG. From IPEX Syndrome to FOXP3 Mutation: A Lesson on Immune Dysregulation. Ann New York Acad Sci (2018) 1417(1):5–22. doi: 10.1111/nyas.13011
36. Deng G, Xiao Y, Zhou Z, Nagai Y, Zhang H, Li B, et al. Molecular and Biological Role of the FOXP3 N-Terminal Domain in Immune Regulation by T Regulatory/Suppressor Cells. Exp Mol Pathol (2012) 93(3):334–8. doi: 10.1016/j.yexmp.2012.09.013
37. Magg T, Mannert J, Ellwart JW, Schmid I, Albert MH. Subcellular Localization of FOXP3 in Human Regulatory and Nonregulatory T Cells. Eur J Immunol (2012) 42(6):1627–38. doi: 10.1002/eji.201141838
38. Mailer RKW. Alternative Splicing of FOXP3-Virtue and Vice. Front Immunol (2018) 9:530. doi: 10.3389/fimmu.2018.00530
39. Gambineri E, Perroni L, Passerini L, Bianchi L, Doglioni C, Meschi F, et al. Clinical and Molecular Profile of a New Series of Patients With Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked Syndrome: Inconsistent Correlation Between Forkhead Box Protein 3 Expression and Disease Severity. J Allergy Clin Immunol (2008) 122(6):1105–12 e1. doi: 10.1016/j.jaci.2008.09.027
Keywords: FOXP3, IPEX, ALPS, NGS, inborn errors of immunity
Citation: Rais A, Mekki N, Fedhila F, Alosaimi MF, Ben Khaled M, Zameli A, Agrebi N, Sellami MK, Geha R, Ben-Mustapha I and Barbouche MR (2021) Case Report: FOXP3 Mutation in a Patient Presenting With ALPS. Front. Immunol. 12:692107. doi: 10.3389/fimmu.2021.692107
Received: 07 April 2021; Accepted: 19 July 2021;
Published: 31 August 2021.
Edited by:
Antonio Condino-Neto, University of São Paulo, BrazilReviewed by:
Rosa Bacchetta, Stanford University, United StatesClaudio Pignata, University of Naples Federico II, Italy
Copyright © 2021 Rais, Mekki, Fedhila, Alosaimi, Ben Khaled, Zameli, Agrebi, Sellami, Geha, Ben-Mustapha and Barbouche. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Imen Ben-Mustapha, imen.benmustapha@pasteur.rns.tn
†These authors share first authorship
‡These authors share last authorship