 Gabrielle Keb
Gabrielle Keb Kenneth A. Fields*
Kenneth A. Fields*- Department of Microbiology, Immunology & Molecular Genetics, University of Kentucky College of Medicine, Lexington, KY, United States
Dynamic interactions that govern the balance between host and pathogen determine the outcome of infection and are shaped by evolutionary pressures. Eukaryotic hosts have evolved elaborate and formidable defense mechanisms that provide the basis for innate and adaptive immunity. Proteins containing a membrane attack complex/Perforin (MACPF) domain represent an important class of immune effectors. These pore-forming proteins induce cell killing by targeting microbial or host membranes. Intracellular bacteria can be shielded from MACPF-mediated killing, and Chlamydia spp. represent a successful paradigm of obligate intracellular parasitism. Ancestors of present-day Chlamydia likely originated at evolutionary times that correlated with or preceded many host defense pathways. We discuss the current knowledge regarding how chlamydiae interact with the MACPF proteins Complement C9, Perforin-1, and Perforin-2. Current evidence indicates a degree of resistance by Chlamydia to MACPF effector mechanisms. In fact, chlamydiae have acquired and adapted their own MACPF-domain protein to facilitate infection.
Introduction
Obligate intracellular bacteria depend on survival within eukaryotic host cells. The family Chlamydiaceae contains at least nine designated species of obligate intracellular pathogens exhibiting a diverse host range in higher eukaryotes. C. trachomatis and C. pneumoniae represent species commonly impacting human health. C. pneumoniae infects the upper respiratory tract and is associated with 10–20% of adult community-acquired pneumonia (1). C. trachomatis urogenital infection (serovars D-K) continues to be the most common sexually transmitted bacterial infection in the US (2) and ocular infection (serovars A-C) is the leading cause of infectious blindness in developing countries (3). Interestingly, anecdotal evidence suggests that chlamydial ocular infections have affected humans for millennia (4). Regardless of species, all Chlamydia share a biphasic developmental cycle that alternates between infectious elementary bodies (EBs) and non-infectious reticulate bodies (RBs). EBs have minimal metabolic activity and are often referred to as “spore-like” due to a durable cell wall that is resistant to mechanical and osmotic pressures [reviewed in (5)]. EB envelopes are comprised of an “atypical” Gram-negative lipid bilayer that is stabilized through disulfide bonds among cysteine-rich outer membrane proteins [reviewed in (6)]. During invasion, EBs traverse the host-cell plasma membrane and establish an intracellular niche within a membrane bound vesicle termed the inclusion. Once the inclusion is established, EBs differentiate into non-infectious RBs which are capable of robust protein synthesis and replication. This stage of the developmental cycle occurs entirely within the protection of the infected host cell. The cycle is completed by asynchronous differentiation of RBs back into EBs capable of infecting neighboring cells after release. Escape of EBs from the host cell is accomplished by either lysis of the host cell or extrusion of intact inclusions (7).
The Chlamydiales order also contains Chlamydia-related bacteria often referred to as environmental Chlamydia due to their obligate intracellular parasitism of amoeba (8). The Chlamydiaceae family diverged from Chlamydia-related bacteria an estimated 700 million years ago at a time when all eukaryotes were single cell [reviewed in (9)]. Due to a dependence on intracellular survival and long evolution with eukaryotic hosts, it is not surprising that Chlamydia may have adapted multiple immune evasion strategies. During C. trachomatis genital infection, both innate and adaptive immune responses are clearly elicited with innate immunity functioning to limit ascension of infection and a Th1-, IFNγ-dependent adaptive response being required for control and resolution [reviewed in (10)]. Reinfection is common, and the majority of infections are asymptomatic, particularly in women (11). Progress in developing an efficacious vaccine has been challenged by poor protective immunity and increased pathology (12). Detrimental patient outcomes, such as tubal factor infertility and pelvic inflammatory disease, are associated with severe immunopathology which is initiated by the infected epithelium (13). Taken together, it is evident that Chlamydia maintain a finely tuned relationship with their host to interfere with productive immune recognition and clearance.
Formation of targeted membrane spanning pores using membrane attack complex/Perforin (MACPF)-domain proteins represents one mechanism used by both innate and adaptive arms of immunity. The MACPF-containing host proteins Perforin-2 and Complement C9 represent innate immune effectors whereas Perforin-1 functions during adaptive immunity (14). Both C9 and Perforin-2 are evolutionarily ancient whereas Perforin-1 likely arose by gene duplication of Perforin-2 during evolution of adaptive immunity in multi-cellular organisms (15, 16). All three function by killing microbes (C9 and Perforin-2) or host cells (Perforin-1) via polymerization and pore formation in target membranes (17). Interestingly, chlamydial genomes also contain a gene encoding a MACPF domain protein. It is hypothesized that this domain was acquired through horizontal gene transfer with a mammalian host (18, 19). A recent metagenomic study found only a small number of protein families that were taxonomically restricted within Chlamydiaceae (20). MACPF-containing proteins were among factors related to specific host interactions, providing further evidence that Chlamydia likely acquired this domain through co-evolution with a mammalian host.
Given the apparent long co-evolution of the host-pathogen interaction exemplified by Chlamydia, this review will summarize current evidence of chlamydial resistance and susceptibility to MACPF domain-mediated attack strategies while highlighting immune evasion mechanisms adapted through co-evolution. The discussion will focus on the more thoroughly characterized human pathogen C. trachomatis and corresponding immune modeling in mice using C. muridarum. We will also discuss the implications of the endogenous chlamydial MACPF domain protein in infection biology.
The C9 MACPF Domain
Complement is a central defense mechanism of the innate immune system that evolved to inactivate extracellularly localized pathogens (16). The complement system consists of more than 30 soluble serum proteins culminating in formation of the membrane attack complex (MAC) for complement-mediated cell lysis. The pore forming complex targets outer membranes of Gram-negative bacteria, enveloped viruses, and parasites. Activation of the complement cascade can occur via lectin, classical, or alternative pathways. Each pathway differs in the early mechanism used to recognize pathogens, but all converge through the covalent attachment of C3b to the target cell which then recruit downstream factors leading to MAC formation. Complement activation clearly occurs during chlamydial infection with the antibody-independent alternative pathway playing a major role (21). Multiple studies using a tissue-culture infection model have demonstrated that Chlamydia inclusion formation is significantly inhibited when EBs are pre-incubated with normal human sera (21–25) indicating complement factors may be important for controlling infection.
After typical C3b deposition on a bacterial surface, subsequently recruitmented components C5b-C8 then recruit soluble C9 monomers and facilitate C9 polymerization and the assembly of an 88-strand β-barrel membrane spanning pore (26). CryoEM studies have revealed that active pores contain 6 poly peptide chains, C5b, C6, C7, C8a, C8β, and C8γ, with 18 C9 monomers (27, 28). In experiments using C3 deficient mice, C. muridarum infectivity was not impacted during genital infections (29), however, C. psittaci pneumonia was significantly exacerbated when chlamydiae were introduced via a respiratory route (30). These data raise the possibility that C5b-C8 recruitment of C9 and pore formation may lead to fatal disruption of the chlamydial envelope; yet, formation of the MAC appears to be dispensable as a primary control mechanism for Chlamydia infections. Depletion of factors C5 and C8 from serum had no effect on in vitro anti-chlamydial activity (24). Additionally, C. muridarum shedding and ascension into the upper genital tract was not impacted in C5-deficient mice (29). Together these data indicate that late complement factors do not play a significant role in directly inactivating Chlamydia. The anaphylatoxin activities of C5a and C3a have been proposed as mediators of complement-dependent effects on infectivity and pathogenesis in mice (29–31). The antibody-independent inhibitory activity of complement in human serum could be mediated by opsonization and inactivation of chlamydial surface proteins required for cellular attachment and invasion (21), or deposition of other components such as properdin leading to targeting of Chlamydia to the lysosomal pathway for degradation (32). It remains undefined, however, how findings in cell culture are related to those observed in vivo. Clearly, further investigation of this interesting area is warranted.
Given that the complement system primarily targets extracellular invaders, the obligate intracellular lifestyle of all Chlamydia spp. represents the most obvious defense mechanism. Beyond that, the biphasic developmental cycle represents an additional layer of protection. The extracellular, infectious EB possess a rigid and highly disulfide crosslinked outer envelope. Assembly of the MAC requires a fluid membrane capable of allowing lateral diffusion of MAC components and dramatic structural changes associated with pore formation (33). We therefore speculate that the EB envelope would be impervious to the MAC. A highly conserved chlamydial protease, CPAF, cleaves C3, and factor B in vitro and may inhibit activation of the alternate complement pathway (25). Host cell escape through extrusion, one of two chlamydial exit strategies, may be another defense mechanism. During extrusion, the inclusion pinches off from the host cell in an exocytosis-like mechanism. Both the host cell and inclusion remain intact, and the now double-membrane-bound inclusion is released into the extracellular space where it is stable up to 4 h in vitro (7, 34–36). We predict that it is unlikely that complement factors could gain access through the multiple layers of membrane comprising this barrier. Chlamydia-mediated recruitment of CD59 to the inclusion membrane (37) would represent an additional layer of defense. CD59 regulates formation of the MAC to prevent uncontrolled complement-mediated cell lysis (38) and would prevent any lytic pore formation should barrier integrity be compromised.
Perforin
The cytotoxic functions of natural killer cells (NKs) and cytotoxic T lymphocytes (CTLs) represent an adaptive defense mechanism against viral and intracellular pathogens. NKs and CTLs release cytoplasmic granules containing Perforin and proteolytic granzymes onto the surface of infected cells. In the presence of Ca2+, Perforin binds to the target membrane and forms a transmembrane β-barrel pore. The N-terminal domain of Perforin contains a MACPF domain that allows insertion into lipid bilayers (39). Once assembled, the Perforin pore functions to deliver the proteolytic granzymes to the cytosol of the targeted cell. Two models of Perforin mediated granzyme delivery exist. The original model proposes that the Perforin pore provides direct delivery of granzymes to the cytosol, and a second model proposes that both Perforin and granzymes are endocytosed into the cell with subsequent delivery of granzymes by Perforin (40, 41). This model is supported by data demonstrating Perforin alters membrane curvature and stimulates the formation of endocytic vesicles (42).
During respiratory infection with C. muridarum, NKs infiltrate the lungs and become activated (43). Multiple studies have shown that NKs contribute modestly to clearance of chlamydial infection, however, this effect may be driven by IFN-γ expression and independent of Perforin targeting (43–45). Chlamydia infected cells are highly resistant to induction of apoptosis which is predicted to be due to chlamydial proteins that interrupt events such as cytochrome C release from mitochondria (46). NK cells extracted from C. trachomatis infected patients have also be shown to have decreased lytic capability (47). In two studies, Perforin knockout mice were not compromised in their ability to clear C. muridarum genital infection (48, 49), indicating Perforin-mediated cytotoxicity is not required for clearance of primary chlamydial infection. A third study, using lower infectious doses, did note a delayed clearance of infection in Perforin -/- mice (50); however, the authors concluded from their additional data that the phenotype occurred independently of direct interaction of cytotoxic cells with infected epithelia. Finally, the IFN-γ dependent/Perforin independent clearance of Chlamydia is supported by the finding that NK cells have a differential effect during infection where IFN-γ production is increased, yet cytolytic function is decreased (43).
Although some studies indicated Chlamydia-mediated interference with inducible expression of class I MHC on infected cells (51), primary chlamydial defense against Perforin may be more passive. Ex vivo studies indicated that Chlamydia-infected epithelial cells can be lysed by cytotoxic cells (52–54). Whether host cell lysis would directly contribute to control of chlamydial infection, however, is unclear since the disrupted cells would merely release any infectious EBs that had formed. Indeed, in vivo work noted above is consistent with Perforin-independent control mechanisms. In addition, the female genital tract represents one location where tolerance to foreign antigens must be greater for sustainment of the natural microbial flora. In both the gastrointestinal and female genital tracts, CD8+ T-cells have decreased expression of Perforin, thus comparatively limited cytotoxic activity (8, 55). In endocervical samples from both non-infected and C. trachomatis infected patients, effector memory T cell subsets showed decreased Perforin expression as compared to paired blood controls (55). Therefore, infection of the genital tract likely provides an advantageous niche for chlamydial infection.
Perforin-2
In contrast to the relative lack of susceptibility of Chlamydia to Perforin-1 and Complement C9, there does appear to be a role for the most recently described MACPF protein, Perforin-2. Perforin-2, encoded by the intronless MPEG1, represents perhaps the most evolutionary ancient and conserved member of the MACPF family of proteins (16, 56, 57). Originally shown to have anti-bacterial properties in sponges (58) and zebrafish (59), it is now established that Perforin-2 is capable of killing a range of cell-associated bacteria including Gram positive, Gram negative, and acid fast bacteria (60). A model has emerged where Perforin-2 is trafficked to bacteria-containing vacuoles and disrupts the integrity of bacterial envelopes by polymerizing into multi-subunit pores (14, 57). To date, the susceptibility of Chlamydia spp. to Perforin-2 represents the sole indication for how an obligate intracellular bacterium might respond to this novel innate immune mechanism (61).
Professional phagocytes, including macrophages and neutrophils, represent a functionally important arm of innate immunity, and Perforin-2 expression is constitutive in these cells (60). Cumulative data using murine-specific C. muridarum in a well-established mouse model of genital tract infection indicate robust recruitment of professional phagocytes to infected tissues. Although innate immunity is not required for resolution of infection, it has been proposed to function in opposing ascension of chlamydial infection into the upper genital tract (10). Although some degree of chlamydial growth can be detected in macrophage cell lines, Chlamydia spp. do not productively infect primary cells (62). RNAi knock-down of MPEG1 message was used to provide direct evidence for Perforin-2-mediated eradication of Chlamydia in infected macrophages (61). Transmission electron microscopy revealed that mock-treated murine BV2 macrophages contained vacuoles harboring debris and few intact chlamydiae, whereas Perforin-2-deficient cells contained intact inclusions that yielded 103 more progeny 24 h post infection. Knock-down of Perforin-2 resulted in levels of progeny C. trachomatis L2 EB production equivalent to similarly infected HeLa cells. Comparable results were seen for C. trachomatis serovars B and D indicating that Perforin-2 is capable of inhibiting a range of C. trachomatis serovars. In addition, C. muridarum was also susceptible to Perforin-2 activity. The BV2 line was initially tested due to the comparatively high level of constitutive Perforin-2 expression, yet similar results were obtained using the murine RAW 264.7 cell line (61). To date, the potential role of Perforin-2-dependent inhibition of Chlamydia has not been tested in human cell lines such as THP-1 or HL-60; however, Perforin-2 has been shown in these cells to limit growth of other bacteria such as Salmonella, S. aureus, and Mycobacterium (60) raising the probability that the observed anti-Chlamydia potential of Perforin-2 also occurs in humans cells.
These data are consistent with macrophage-produced Perforin-2 having a significant role in controlling chlamydial infection. How Perforin-2 inactivates intra-inclusion chlamydiae remains an open question. Chlamydia are rapidly targeted to lysosomal compartments in macrophages (61), yet develop normally in Perforin-2 deficient macrophages. Hence, Perforin-2 plays an active role in clearance at the cellular level. Based on a proposed working model for susceptibility of intracellular bacteria (14), deployment of Perforin-2 to the luminal face of subsequent Chlamydia-containing vacuolar membranes would culminate in insertion and polymerization of the Perforin-2 pore in chlamydial membranes. This would be predicted to disrupt the integrity of the RB envelope and lead to lysis. This model is supported by electron micrographs of Perforin-2 sufficient and deficient cells were clearly lysed chlamydial material is detected within an apparently intact vacuole (61). The Chlamydia highly cross-linked envelope of the EB developmental form (6) is likely resistant to Perforin-2 insertion. However, Perforin-2-mediated killing of Mycobacteria spp., which possess a highly impermeable, mycolic acid-containing envelope, suggest that the atypical RB envelope may be susceptible to Perforin-2 attack. Perforation of bacterial envelopes has also been shown to promote access of antimicrobial substances like reactive oxygen species (63). This mechanism may contribute to observed Chlamydia clearance since RAW 264.7 macrophages generate ROS and iNOS in response to infection, and pharmacologic inhibitors or scavengers of reactive species benefit chlamydial survival (64). Finally, macrophage-mediated killing of C. trachomatis has also been linked to autophagy (65–67). It is possible that Perforin-2 and autophagy mechanisms are linked, yet Perforin-2 has been proposed to function upstream of autophagy based on the greater impact on chlamydial survival (61).
Interestingly, chlamydial exit from infected epithelial cells via extrusion may subvert Perforin-2-mediated killing. Chlamydia-containing extrusionsare phagocytosed by bone marrow-derived macrophages and retain infectivity compared to non-encapsulated Chlamydia (36). In this scenario, the extra lipid bilayer could shield Chlamydia from detection and Perforin-2-mediated killing since machinery necessary for targeting Perforin-2 to membranes would be absent from the extrusion membrane (68) Finally, C. pneumoniae appears to be capable of replication in a subset of primary phagocytes (62), raising the possibility of additional, species-specific protective mechanisms. This would be consistent with observations that other pathogens such as Salmonella and enteropathogenic E. coli have evolved Perforin-2-specific mitigation mechanisms (68).
Columnar epithelial cells lining target mucosa represent the productive replication niche for all Chlamydia spp. Therefore, Chlamydia would be predicted to possess effective Perforin-2 protective mechanisms in this cell type. MPEG1 is inducible in non-myeloid cells, and Perforin-2-specific signal is below detection in multiple epithelial lines (60). Indeed, Perforin-2 was absent in both mock-treated and Chlamydia-infected cells in a HeLa-cell infection model (61). In contrast, treatment of cells with heat-killed Chlamydia resulted in significant up-regulation of Perforin-2, indicating that stealthy subversion of signals leading to MPEG1 expression as one protective mechanism. MPEG1 is inducible with IFNγ and type I interferons (60), both of which play important roles in limiting chlamydial infection in vivo (10). Indeed, MPEG1 upregulation was evident in early microarray analyses of IFNγ-treated oviduct epithelial cells (69). Interestingly, cells containing established inclusions prevent IFNγ-mediated induction of Perforin-2 in epithelial cells. This effect required viable Chlamydia. The role of IFNβ-mediated induction of MPEG1 during chlamydial infection has not been tested, yet a recent report indicated a requirement of Perforin-2 in transducing activation signals through the cognate receptor, IFNAR (70). Hence, Perforin-2 mediated killing is counter-indicated in infected epithelial cells. Importantly, ex vivo treatment of murine genital tract-derived epithelial cells with Chlamydia-conditioned media resulted in upregulation of MPEG1 (50). Proinflammatory signals are certainly capable of acting on uninfected, bystander cells which could then be more resistant to chlamydial infection in a Perforin-2-dependent manner. In support of this notion, ectopic overexpression of RFP-tagged Perforin-2 in HeLa cells resulted in efficient killing of Chlamydia (61). It is important to note that polymerization of Perforin-2 for crystallographic studies required an acidic environment and is postulated to reflect delivery of Perforin-2 to acidified phagolysosomes (71, 72). The chlamydial inclusion, however, does not acidify and remains segregated from the lysosomal pathway (73), raising the possibility of alternative polymerization mechanisms. It is also possible that non-physiologic levels due to ectopic expression favor polymerization in vivo. Regardless, it seems apparent that Chlamydia avoid Perforin-2 killing in epithelial cells by avoiding and suppressing MPEG1 expression. Although Perforin-2 proteins levels were not shown, inhibition of C. muridarum in IFNγ-primed murine embryonic fibroblasts was unchanged after Perforin-2 knockdown using siRNAs (74). These data may, once again, elude to alternative, species-specific susceptiblity of Chlamydia to Perforin-2.
Mice deficient in MPEG1 have subsequently been employed to test the anti-bacterial role of Perforin-2 in an animal model. Cells derived from MPEG1 -/- mice are deficient in ex vivo killing of a range of bacteria including L. monocytogenes (75), Mycobacteria spp., S. aureus, S. enterica Typhimurium, and enteropathogenic E. coli (60, 68). MPEG1 -/- mice are more susceptible to systemic listeriosis (75) and succumb to sublethal doses of S. aureus and Salmonella in cutaneous and orogastric infection models, respectively (60). Collectively, these data indicate the fundamental importance of Perforin-2 in controlling a diverse array of bacterial infections. It remained unclear how the impact of Perforin-2 would manifest during a mammalian model of chlamydial infection. To that end, the well-established intravaginal infection model was employed were Chlamydia are deposited at the cervical vault and bacterial shedding is enumerated by swabbing over time. We infected groups of 5 wild type or MPEG1 -/- mice with 5 × 104 infectious particles of C. muridarum and monitored chlamydial shedding and mouse body weight over time (Figure 1). Time to resolution was not extended in the absence of Perforin-2. However, MPEG -/- mice shed statistically fewer IFUs overall with a ca. log10 decrease observed on days 11 and 15. Interestingly, the MPEG1 -/- mice did appear ill with ruffled fur (Fields, unpublished) and decreased body weight (Figure 1B). These symptoms persisted until times corresponding to resolution. We speculate that these data could indicate systemic dissemination of chlamydiae beyond the genital tract. Infiltration of macrophages and neutrophils to the sites of active infection functions to contain chlamydiae (10). Less efficient killing by professional phagocytes could allow rapid ascension and seeding of peripheral sites. The role of these myeloid cells could be easily tested using bone-marrow chimeric mice expressing or lacking MPEG1. A functional adaptive response is likely intact given that infections in MPEG1 -/- mice were resolved comparable to wild type. As is the case with the molecular mechanisms of Perforin-2 function in cell culture, many provocative questions remain to be resolved.
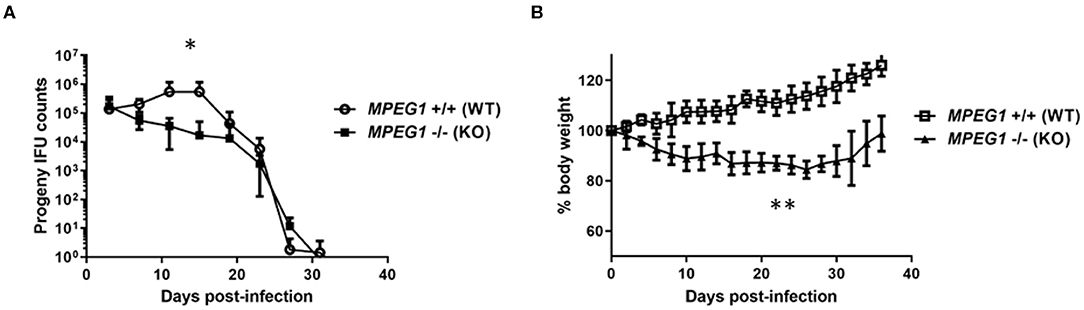
Figure 1. C. muridarum infection of Perforin-2 KO mice. Groups of 5, 6–7 week-old female wild type (MPEG1+/+) or KO (MPEG1 -/-) C57BL/6 × 129 × 1/SJV mice (60) were infected intravaginally with 5 × 104 C. muridarum 5 days after synchronization with medroxyprogesterone. Shed IFUs (A) and body weights (B) were measured over time (days). (A) Shed IFUs were enumerated every 4 days according to standard protocols and averages within groups are shown with standard deviations. Curves were different (*p < 0.01) by two-way ANOVA. (B) Average body weights (+/– standard deviation) within groups are shown. KO weights were significantly different (**p < 0.0001) from WT via two-way ANOVA. Data represent one of two replicate experiments (n = 2).
The Chlamydial MACPF
Whole-genome sequencing of Chlamydia spp. originally identified a gene encoding a MACPF domain protein (18). In the reference strain C. trachomatis serovar D, this protein is designated CT153 (76). Gene orthologs were conserved in all C. trachomatis genomes sampled and located within the highly variable plasticity zone, immediately upstream pzPLD genes encoding putative lipid-modifying proteins (77, 78). Although similarity varies, orthologs are also present in other Chlamydia spp (Figure 2). Consistent with acquisition through horizontal gene transfer during co-evolution, the MACPF gene sequence of C. pneumoniae can differentiate bacteria isolated from indigenous and non-indigenous human sources from varying geographical regions (79). C-terminal amino acid residues 427–621 share homology with the MACPF domain found in C9 and Perforin. CT153 appears to undergo some proteolytic processing (78). The full length p91 was observed as the dominant polypeptide in EBs and is rapidly cleaved (15 min) into p57 and p41 fragments independent of de novo chlamydial protein synthesis. Processing may occur through host-mediated proteolytic cleavage or auto-proteolysis. This suggests distinct functions for full-length and processed peptides, yet it should be noted that the possibility of post-lysis degradation has not been ruled out (80). Currently, there is limited understanding regarding the function and cytolytic activity of this protein. It was originally postulated that the MACPF domain protein may be essential for Chlamydia since a saturating screen for chemically-induced mutations in C. muridarum failed to reveal nonsense mutations in tc0431 (81). However, inactivating transposon insertions were subsequently observed in both C. muridarum and C. trachomatis (82, 83). None of the mutations abolished intracellular survival, indicating that this protein is not essential for cultivation in tissue culture. These data are in line with the apparent lack of a MACPF-encoding gene in C. abortus which can infect the same cells (84). CT153 is expressed during mid-cycle (85) and has been shown to localize with both RBs near the inclusion membrane and within the inclusion membrane (78). These data suggest that CT153 may permeabilize the inclusion membrane. Complete lysis of the inclusion membrane is likely retarded by structural integrity conferred by chlamydial inclusion membrane proteins (86). For example, loss of specific Incs results in premature lysis of the parasitophorous vacuole (87, 88). The embedded pore could instead facilitate diffusion of small molecules into or out of the inclusion or be involved in fusion events during exocytic exit from cells. Clearly, this is a rich area needing further investigation.
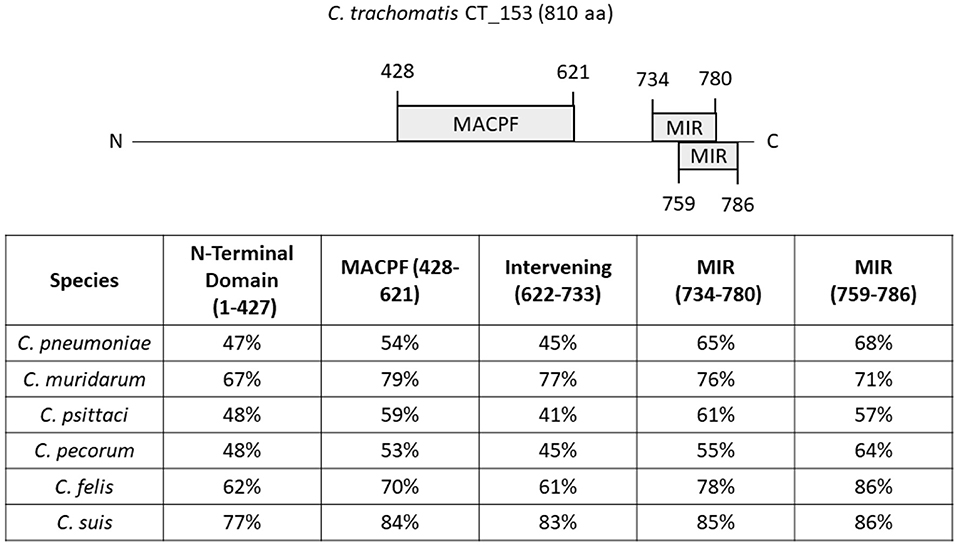
Figure 2. Similarity of C. trachomatis CT153 domains to orthologs in other Chlamydia spp. The 810 residue CT153 of C. trachomatis D/UW-3 is shown schematically with predicted functional domains highlighted. These include the MACPF and Mannosyltransferase, Inositol 1,4,5-trisphosphate receptor and Ryanodine receptor (MIR) domains. Residues corresponding to the N-Terminal domain (aa 1-427), MACPF (aa 428-621), Intervening domain (aa 622-733), or MIR (aa 734-780 and aa 759-786) were used as query sequences in NCBI BLAST searches. Searches were performed using C. pneumoniae TW-183, C. psittaci 6 BC, C. muridarum Nigg, C. suis S45, C. felis DSM:26967, and C. pecorum Bo/E58 data bases. Calculated percent identity in respective species is shown for each domain.
Concluding Remarks
Chlamydia are ancient bacteria that have evolved with mammalian hosts for of millions of years. To sustain a privileged niche and intracellular survival, Chlamydia have adapted resistance mechanisms to major host defenses that include formation of the MAC and Perforin mediated cytotoxicity. Chlamydia are not completely resistant to MACPF attack strategies since these pathogens are susceptible to Perforin-2 activities of professional phagocytes. Finally, acquisition of the MACPF domain from hosts may have given the bacteria an edge for survival, however it is unclear whether this domain is responsible for resistance to host immunity or has other functions.
Author Contributions
Article preparation and writing was performed by GK and KF. KF performed work associated with Figure 1. All authors contributed to the article and approved the submitted version.
Funding
KF and GK were supported by Public Health Service grants from the National Institutes of Health/NIAD including AI117876 to KF and AI147417 to GK.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. K. Wolf, R. Hayman, and M. Clouse for critical reading of the manuscript.
References
1. Grayston JT, Campbell LA, Kuo C-C, Mordhorst CH, Saikku P, Thorn DH, et al. A new respiratory tract pathogen: Chlamydia pneumoniae strain TWAR. J Infect Dis. (1990) 161:618–25. doi: 10.1093/infdis/161.4.618
2. National Center for HIV/AIDS Viral Hepatitis STD and TB Prevention (U.S.). Division of STD Prevention. Sexually Transmitted Disease Surveillance 2018. Atlanta, GA: CDC (2019).
3. Mariotti SP, Pascolini D, Rose-Nussbaumer J. Trachoma: global magnitude of a preventable cause of blindness. Br J Ophthalmol. (2009) 93:563–8. doi: 10.1136/bjo.2008.148494
4. Halberstädter L, Prowazek SV. Über zelleinschlüsse parasitärer natur beim trachom. Arb Aus Dem Kais Gesundheitsamte. (1907) 26:44–7.
5. Cossé MM, Hayward RD, Subtil A. One face of Chlamydia trachomatis: the infectious elementary body. Curr Top Microbiol Immunol. (2018) 412:35–58. doi: 10.1007/82_2016_12
6. Christensen S, McMahon RM, Martin JL, Huston WM. Life inside and out: making and breaking protein disulfide bonds in Chlamydia. Crit Rev Microbiol. (2019) 45:33–50. doi: 10.1080/1040841X.2018.1538933
7. Hybiske K, Stephens RS. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc Natl Acad Sci. USA. (2007) 104:11430–5. doi: 10.1073/pnas.0703218104
8. Bayramova F, Jacquier N, Greub G. Insight in the biology of Chlamydia-related bacteria. Microbes Infect. (2018) 20:432–40. doi: 10.1016/j.micinf.2017.11.008
9. Nunes A, Gomes JP. Evolution, phylogeny, and molecular epidemiology of Chlamydia. Infect Genet Evol. (2014) 23:49–64. doi: 10.1016/j.meegid.2014.01.029
10. Darville T, Hiltke TJ. Pathogenesis of genital tract disease due to Chlamydia trachomatis. J Infect Dis. (2010) 201(Suppl. 2):S114–25. doi: 10.1086/652397
11. Hosenfeld CB, Workowski KA, Berman S, Zaidi A, Dyson J, Mosure D, et al. Repeat infection with Chlamydia and gonorrhea among females: a systematic review of the literature. Sex Transm Dis. (2009) 36:478–89. doi: 10.1097/OLQ.0b013e3181a2a933
12. Schachter J. Overview of Chlamydia trachomatis infection and the requirements for a vaccine. Rev Infect Dis. (1985) 7:713–6. doi: 10.1093/clinids/7.6.713
13. Stephens RS. The cellular paradigm of chlamydial pathogenesis. Trends Microbiol. (2003) 11:44–51. doi: 10.1016/S0966-842X(02)00011-2
14. McCormack R, de Armas L, Shiratsuchi M, Podack ER. Killing machines: three pore-forming proteins of the immune system. Immunol Res. (2013) 57:268–78. doi: 10.1007/s12026-013-8469-9
15. Sunyer JO, Boshra H, Lorenzo G, Parra D, Freedman B, Bosch N. Evolution of complement as an effector system in innate and adaptive immunity. Immunol Res. (2003) 27:549–64. doi: 10.1385/IR:27:2-3:549
16. McCormack R, Podack ER. Perforin-2/Mpeg1 and other pore-forming proteins throughout evolution. J Leukoc Biol. (2015) 98:761–8. doi: 10.1189/jlb.4MR1114-523RR
17. Podack ER, Munson GP. Killing of microbes and cancer by the immune system with three mammalian pore-forming killer proteins. Front Immunol. (2016) 7:464. doi: 10.3389/fimmu.2016.00464
18. Ponting CP. Chlamydial homologues of the MACPF (MAC/perforin) domain. Curr Biol. (1999) 9:R911–3. doi: 10.1016/S0960-9822(00)80102-5
19. Wolf YI, Aravind L, Koonin EV. Rickettsiae and Chlamydiae: evidence of horizontal gene transfer and gene exchange. Trends Genet. (1999) 15:173–5. doi: 10.1016/S0168-9525(99)01704-7
20. Dharamshi JE, Tamarit D, Eme L, Stairs CW, Martijn J, Homa F, et al. Marine sediments illuminate Chlamydiae diversity and evolution. Curr Biol. (2020) 30:1032–48.e7. doi: 10.1016/j.cub.2020.02.016
21. Hall RT, Strugnell T, Wu X, Devine DV, Stiver HG. Characterization of kinetics and target proteins for binding of human complement component C3 to the surface-exposed outer membrane of Chlamydia trachomatis serovar L2. Infect Immun. (1993) 61:1829–34. doi: 10.1128/IAI.61.5.1829-1834.1993
22. Johnson AP, Osborn MF, Rowntree S, Thomas BJ, Taylor-Robinson D. A study of inactivation of Chlamydia trachomatis by normal human serum. Br J Vener Dis. (1983) 59:369–72. doi: 10.1136/sti.59.6.369
23. Osborn MF, Johnson AP, Taylor-Robinson D. Susceptibility of different serovars of Chlamydia trachomatis to inactivation by normal human serum. Genitourin Med. (1985) 61:244–6. doi: 10.1136/sti.61.4.244
24. Lin JS, Yan LL, Ho Y, Rice PA. Early complement components enhance neutralization of Chlamydia trachomatis infectivity by human sera. Infect Immun. (1992) 60:2547–50. doi: 10.1128/IAI.60.6.2547-2550.1992
25. Yang Z, Tang L, Zhou Z, Zhong G. Neutralizing antichlamydial activity of complement by chlamydia-secreted protease CPAF. Microbes Infect. (2016) 18:669–74. doi: 10.1016/j.micinf.2016.07.002
26. Dudkina NV, Spicer BA, Reboul CF, Conroy PJ, Lukoyanova N, Elmlund H, et al. Structure of the poly-C9 component of the complement membrane attack complex. Nat Commun. (2016) 7:10588. doi: 10.1038/ncomms10588
27. Sharp TH, Koster AJ, Gros P. Heterogeneous MAC initiator and pore structures in a lipid bilayer by phase-plate cryo-electron tomography. Cell Rep. (2016) 15:1–8. doi: 10.1016/j.celrep.2016.03.002
28. Menny A, Serna M, Boyd CM, Gardner S, Joseph AP, Morgan BP, et al. CryoEM reveals how the complement membrane attack complex ruptures lipid bilayers. Nat Commun. (2018) 9:5316. doi: 10.1038/s41467-018-07653-5
29. Yang Z, Conrad T, Zhou Z, Chen J, Dutow P, Klos A, et al. Complement factor C5 but not C3 contributes significantly to hydrosalpinx development in mice infected with Chlamydia muridarum. Infect Immun. (2014) 82:3154–63. doi: 10.1128/IAI.01833-14
30. Bode J, Dutow P, Sommer K, Janik K, Glage S, Tümmler B, et al. A new role of the complement system: C3 provides protection in a mouse model of lung infection with intracellular Chlamydia psittaci. PLoS ONE. (2012) 7:e50327. doi: 10.1371/journal.pone.0050327
31. Dutow P, Fehlhaber B, Bode J, Laudeley R, Rheinheimer C, Glage S, et al. The complement C3a receptor is critical in defense against Chlamydia psittaci in mouse lung infection and required for antibody and optimal T cell response. J Infect Dis. (2014) 209:1269–78. doi: 10.1093/infdis/jit640
32. Cortes C, Ferreira VP, Pangburn MK. Native properdin binds to Chlamydia pneumoniae and promotes complement activation. Infect Immun. (2011) 79:724–31. doi: 10.1128/IAI.00980-10
33. Bayly-Jones C, Bubeck D, Dunstone M. The mystery behind membrane insertion: a review of the complement membrane attack complex. Philos Trans R Soc Lond Biol Sci. (2017) 372:20160221. doi: 10.1098/rstb.2016.0221
34. Doughri AM, Storz J, Altera KP. Mode of entry and release of Chlamydiae in infections of intestinal epithelial cells. J Infect Dis. (1972) 126:652–7. doi: 10.1093/infdis/126.6.652
35. Chin E, Kirker K, Zuck M, James G, Hybiske K. Actin recruitment to the Chlamydia inclusion is spatiotemporally regulated by a mechanism that requires host and bacterial factors. PLoS ONE. (2012) 7:e46949. doi: 10.1371/journal.pone.0046949
36. Zuck M, Ellis T, Venida A, Hybiske K. Extrusions are phagocytosed and promote Chlamydia survival within macrophages: extrusions are phagocytosed and promote Chlamydia survival within macrophages. Cell Microbiol. (2017) 19:e12683. doi: 10.1111/cmi.12683
37. Hasegawa A, Sogo LF, Tan M, Sutterlin C. Host complement regulatory protein CD59 is transported to the chlamydial inclusion by a golgi apparatus-independent pathway. Infect Immun. (2009) 77:1285–92. doi: 10.1128/IAI.01062-08
38. Kim DD, Song W-C. Membrane complement regulatory proteins. Clin Immunol. (2006) 118:127–36. doi: 10.1016/j.clim.2005.10.014
39. Law RHP, Lukoyanova N, Voskoboinik I, Caradoc-Davies TT, Baran K, Dunstone MA, et al. The structural basis for membrane binding and pore formation by lymphocyte perforin. Nature. (2010) 468:447–51. doi: 10.1038/nature09518
40. Froelich CJ, Orth K, Turbov J, Seth P, Gottlieb R, Babior B, et al. New paradigm for lymphocyte granule-mediated cytotoxicity: target cells bind and internalize granzyme b, but an endosomolytic agent is necessary for cytosolic delivery and subsequent apoptosis. J Biol Chem. (1996) 271:29073–9. doi: 10.1074/jbc.271.46.29073
41. Pipkin ME, Lieberman J. Delivering the kiss of death: progress on understanding how perforin works. Curr Opin Immunol. (2007) 19:301–8. doi: 10.1016/j.coi.2007.04.011
42. Praper T, Sonnen AF-P, Kladnik A, Andrighetti AO, Viero G, Morris KJ, et al. Perforin activity at membranes leads to invaginations and vesicle formation. Proc Natl Acad Sci. USA. (2011) 108:21016–21. doi: 10.1073/pnas.1107473108
43. Zhao L, Gao X, Peng Y, Joyee AG, Bai H, Wang S, et al. Differential modulating effect of natural killer (NK) T cells on interferon-γ production and cytotoxic function of NK cells and its relationship with NK subsets in Chlamydia muridarum infection: differential modulation of NKT on NK function. Immunology. (2011) 134:172–84. doi: 10.1111/j.1365-2567.2011.03477.x
44. Williams DM, Grubbs BG, Schachter J, Magee DM. Gamma interferon levels during Chlamydia trachomatis pneumonia in mice. Infect Immun. (1993) 61:3556–8. doi: 10.1128/IAI.61.8.3556-3558.1993
45. Tseng C-TK, Rank RG. Role of NK cells in early host response to chlamydial genital infection. Infect Immun. (1998) 66:5867–75. doi: 10.1128/IAI.66.12.5867-5875.1998
46. Fan T, Lu H, Hu H, Shi L, McClarty GA, Nance DM, et al. Inhibition of apoptosis in Chlamydia-infected cells: blockade of mitochondrial cytochrome c release and caspase activation. J Exp Med. (1998) 187:487–96. doi: 10.1084/jem.187.4.487
47. Mavoungou E, Poaty-Mavoungou V, Touré FS, Sall A, Delicat A, Yaba P, et al. Impairment of natural killer cell activity in Chlamydia trachomatis infected individuals. Trop Med Int Health. (1999) 4:719–27. doi: 10.1046/j.1365-3156.1999.00479.x
48. Perry LL, Feilzer K, Hughes S, Caldwell HD. Clearance of Chlamydia trachomatis from the murine genital mucosa does not require perforin-mediated cytolysis or fas-mediated apoptosis. Infect Immun. (1999) 67:1379–85. doi: 10.1128/IAI.67.3.1379-1385.1999
49. Murthy AK, Li W, Chaganty BKR, Kamalakaran S, Guentzel MN, Seshu J, et al. Tumor necrosis factor alpha production from CD8+ T cells mediates oviduct pathological sequelae following primary genital chlamydia muridarum infection. Infect Immun. (2011) 79:2928–35. doi: 10.1128/IAI.05022-11
50. Johnson RM, Kerr MS, Slaven JE. Perforin is detrimental to controllinγ C. muridarum replication in vitro, but not in vivo. PLoS ONE. (2013) 8:e63340. doi: 10.1371/journal.pone.0063340
51. Zhong G, Liu L, Fan T, Fan P, Ji H. Degradation of transcription factor RFX5 during the inhibition of both constitutive and interferon gamma-inducible major histocompatibility complex class I expression in chlamydia-infected cells. J Exp Med. (2000) 191:1525–34. doi: 10.1084/jem.191.9.1525
52. Beatty PR, Stephens RS. CD8+ T lymphocyte-mediated lysis of Chlamydia-infected L cells using an endogenous antigen pathway. J Immunol Baltim Md. (1994) 153:4588–95.
53. Starnbach MN, Bevan MJ, Lampe MF. Protective cytotoxic T lymphocytes are induced during murine infection with Chlamydia trachomatis. J Immunol Baltim Md. (1994) 153:5183–9.
54. Starnbach MN, Bevan MJ, Lampe MF. Murine cytotoxic T lymphocytes induced following Chlamydia trachomatis intraperitoneal or genital tract infection respond to cells infected with multiple serovars. Infect Immun. (1995) 63:3527–30. doi: 10.1128/IAI.63.9.3527-3530.1995
55. Ibana JA, Myers L, Porretta C, Lewis M, Taylor SN, Martin DH, et al. The major CD8 T cell effector memory subset in the normal and Chlamydia trachomatis-infected human endocervix is low in perforin. BMC Immunol. (2012) 13:66. doi: 10.1186/1471-2172-13-66
56. McCormack R, de Armas LR, Shiratsuchi M, Ramos JE, Podack ER. Inhibition of intracellular bacterial replication in fibroblasts is dependent on the perforin-like protein (Perforin-2) encoded by macrophage-expressed gene 1. J Innate Immun. (2013) 5:185–94. doi: 10.1159/000345249
57. Ni T, Gilbert RJC. Repurposing a pore: highly conserved perforin-like proteins with alternative mechanisms. Philos Trans R Soc B Biol Sci. (2017) 372:20160212. doi: 10.1098/rstb.2016.0212
58. Wiens M, Korzhev M, Krasko A, Thakur NL, Perović-Ottstadt S, Breter HJ, et al. Innate immune defense of the sponge Suberites domuncula against bacteria involves a MyD88-dependent signaling pathway: induction of a perforin-like molecule. J Biol Chem. (2005) 280:27949–59. doi: 10.1074/jbc.M504049200
59. Benard EL, Racz PI, Rougeot J, Nezhinsky AE, Verbeek FJ, Spaink HP, et al. Macrophage-expressed perforins Mpeg1 and Mpeg1.2 have an anti-bacterial function in zebrafish. J Innate Immun. (2015) 7:136–52. doi: 10.1159/000366103
60. McCormack RM, de Armas LR, Shiratsuchi M, Fiorentino DG, Olsson ML, Lichtenheld MG, et al. Perforin-2 is essential for intracellular defense of parenchymal cells and phagocytes against pathogenic bacteria. eLife. (2015) 4:e06508. doi: 10.7554/eLife.06508
61. Fields KA, McCormack R, de Armas LR, Podack ER. Perforin-2 restricts growth of Chlamydia trachomatis in macrophages. Infect Immun. (2013) 81:3045–54. doi: 10.1128/IAI.00497-13
62. Herweg J-A, Rudel T. Interaction of Chlamydiae with human macrophages. FEBS J. (2016) 283:608–18. doi: 10.1111/febs.13609
63. Bai F, McCormack RM, Hower S, Plano GV, Lichtenheld MG, Munson GP. Perforin-2 breaches the envelope of phagocytosed bacteria allowing antimicrobial effectors access to intracellular targets. J Immunol Baltim Md. (2018) 201:2710–20. doi: 10.4049/jimmunol.1800365
64. Rajaram K, Nelson DE. Chlamydia muridarum infection of macrophages elicits bactericidal nitric oxide production via reactive oxygen species and cathepsin B. Infect Immun. (2015) 83:3164–75. doi: 10.1128/IAI.00382-15
65. Yasir M, Pachikara ND, Bao X, Pan Z, Fan H. Regulation of chlamydial infection by host autophagy and vacuolar ATPase-bearing organelles. Infect Immun. (2011) 79:4019–28. doi: 10.1128/IAI.05308-11
66. Sun HS, Eng EWY, Jeganathan S, Sin AT-W, Patel PC, Gracey E, et al. Chlamydia trachomatis vacuole maturation in infected macrophages. J Leukoc Biol. (2012) 92:815–27. doi: 10.1189/jlb.0711336
67. Al-Zeer MA, Al-Younes HM, Lauster D, Abu Lubad M, Meyer TF. Autophagy restricts Chlamydia trachomatis growth in human macrophages via IFNG-inducible guanylate binding proteins. Autophagy. (2013) 9:50–62. doi: 10.4161/auto.22482
68. McCormack RM, Lyapichev K, Olsson ML, Podack ER, Munson GP. Enteric pathogens deploy cell cycle inhibiting factors to block the bactericidal activity of Perforin-2. eLife. (2015) 4:e06505. doi: 10.7554/eLife.06505
69. Nelson DE, Virok D, Wood H, Roshick C, Johnson RM, Whitmire WM, et al. Chlamydial IFN-gamma immune evasion is linked to host infection tropism. Proc Natl Acad Sci USA. (2005) 26:10658–63. doi: 10.1073/pnas.0504198102
70. McCormack R, Hunte R, Podack ER, Plano GV, Shembade N. An essential role for Perforin-2 in type I IFN signaling. J Immunol Baltim Md. (2020) 204:2242–56. doi: 10.4049/jimmunol.1901013
71. Pang SS, Bayly-Jones C, Radjainia M, Spicer BA, Law RHP, Hodel AW, et al. The cryo-EM structure of the acid activatable pore-forming immune effector macrophage-expressed gene 1. Nat Commun. (2019) 10:4288. doi: 10.1038/s41467-019-12279-2
72. Ni T, Jiao F, Yu X, Aden S, Ginger L, Williams SI, et al. Structure and mechanism of bactericidal mammalian Perforin-2, an ancient agent of innate immunity. Sci Adv. (2020) 6:eaax8286. doi: 10.1126/sciadv.aax8286
73. Heinzen RA, Scidmore MA, Rockey DD, Hackstadt T. Differential interaction with endocytic and exocytic pathways distinguish parasitophorous vacuoles of Coxiella burnetii and Chlamydia trachomatis. Infect Immun. (1996) 64:796–809. doi: 10.1128/IAI.64.3.796-809.1996
74. Giebel AM, Hu S, Rajaram K, Finethy R, Toh E, Brothwell JA, et al. Genetic screen in Chlamydia muridarum reveals role for an interferon-induced host cell death program in antimicrobial inclusion rupture. MBio. (2019) 10:e00385–19. doi: 10.1128/mBio.00385-19
75. McCormack R, Bahnan W, Shrestha N, Boucher J, Barreto M, Barrera CM, et al. Perforin-2 protects host cells and mice by restricting the vacuole to cytosol transitioning of a bacterial pathogen. Infect Immun. (2016) 84:1083–91. doi: 10.1128/IAI.01434-15
76. Stephens RS, Kalman S, Lammel C, Fan J, Marathe R, Aravind L, et al. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science. (1998) 282:754–9. doi: 10.1126/science.282.5389.754
77. Read TD. Genome sequences of Chlamydia trachomatis MoPn and Chlamydia pneumoniae AR39. Nucleic Acids Res. (2000) 28:1397–406. doi: 10.1093/nar/28.6.1397
78. Taylor LD, Nelson DE, Dorward DW, Whitmire WM, Caldwell HD. Biological characterization of Chlamydia trachomatis plasticity zone MACPF domain family protein CT153. Infect Immun. (2010) 78:2691–9. doi: 10.1128/IAI.01455-09
79. Mitchell CM, Hutton S, Myers GSA, Brunham R, Timms P. Chlamydia pneumoniae is genetically diverse in animals and appears to have crossed the host barrier to humans on (at least) two occasions. PLoS Pathog. (2010) 6:e1000903. doi: 10.1371/journal.ppat.1000903
80. Chen AL, Johnson KA, Lee JK, Sutterlin C, Tan M. CPAF: a chlamydial protease in search of an authentic substrate. PLoS Path. (2012) 8:e1002842. doi: 10.1371/Journal.ppat.1002842
81. Rajaram K, Giebel AM, Toh E, Hu S, Newman JH, Morrison SG, et al. Mutational analysis of the Chlamydia muridarum plasticity zone. Infect Immun. (2015) 83:2870–81. doi: 10.1128/IAI.00106-15
82. LaBrie SD, Dimond ZE, Harrison KS, Baid S, Wickstrum J, Suchland RJ, et al. Transposon mutagenesis in Chlamydia trachomatis identifies CT339 as a ComEC homolog important for DNA uptake and lateral gene transfer. MBio. (2019) 10:e01343–19. doi: 10.1128/mBio.01343-19
83. Wang Y, LaBrie SD, Carrell SJ, Suchland RJ, Dimond ZE, Kwong F, et al. Development of transposon mutagenesis for Chlamydia muridarum. J Bacteriol. (2019) 201:e00366–19. doi: 10.1128/JB.00366-19
84. Thomson NR, Yeats C, Bell K, Holden MTG, Bentley SD, Livingstone M, et al. The Chlamydophila abortus genome sequence reveals an array of variable proteins that contribute to interspecies variation. Genome Res. (2005) 15:629–40. doi: 10.1101/gr.3684805
85. Belland RJ, Zhong G, Crane DD, Hogan D, Sturdevant D, Sharma J, et al. Genomic transcriptional profiling of the developmental cycle of Chlamydia trachomatis. Proc Natl Acad Sci USA. (2003) 100:8478–83. doi: 10.1073/pnas.1331135100
86. Moore ER, Ouellette SP. Reconceptualizing the chlamydial inclusion as a pathogen-specified parasitic organelle: an expanded role for Inc proteins. Front Cell Infect Microbiol. (2014) 4:157. doi: 10.3389/fcimb.2014.00157
87. Sixt BS, Bastidas RJ, Finethy R, Baxter RM, Carpenter VK, Kroemer G, et al. The Chlamydia trachomatis inclusion membrane protein CpoS counteracts STING-mediated cellular surveillance and suicide programs. Cell Host Microbe. (2017) 21:113–21. doi: 10.1016/j.chom.2016.12.002
Keywords: immunity, evolution, pathogenesis, pore-forming, obligate intracellular
Citation: Keb G and Fields KA (2020) An Ancient Molecular Arms Race: Chlamydia vs. Membrane Attack Complex/Perforin (MACPF) Domain Proteins. Front. Immunol. 11:1490. doi: 10.3389/fimmu.2020.01490
Received: 20 April 2020; Accepted: 08 June 2020;
Published: 14 July 2020.
Edited by:
George P. Munson, The University of Miami Leonard M. Miller School of Medicine, United StatesReviewed by:
Mary M. Weber, University of Iowa, United StatesErika Ildiko Lutter, Oklahoma State University, United States
David E. Nelson, Indiana University School of Medicine, United States
Kevin Hybiske, University of Washington, United States
Copyright © 2020 Keb and Fields. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kenneth A. Fields, Ken.fields@uky.edu