 Lan-Hui Li1,2Tzu-Ling Chen1,2Hsiao-Wen Chiu3Chung-Hua Hsu1,4Chien-Chun Wang5Tzu-Ting Tai3
Lan-Hui Li1,2Tzu-Ling Chen1,2Hsiao-Wen Chiu3Chung-Hua Hsu1,4Chien-Chun Wang5Tzu-Ting Tai3 Tz-Chuen Ju6
Tz-Chuen Ju6 Fang-Hsin Chen7
Fang-Hsin Chen7 Oleg V. Chernikov8
Oleg V. Chernikov8 Wen-Chiuan Tsai2
Wen-Chiuan Tsai2 Kuo-Feng Hua2,3,9*
Kuo-Feng Hua2,3,9*- 1Department of Laboratory Medicine, Linsen, Chinese Medicine and Kunming Branch, Taipei City Hospital, Taipei, Taiwan
- 2Department of Pathology, Tri-Service General Hospital, National Defense Medical Center, Taipei, Taiwan
- 3Department of Biotechnology and Animal Science, National Ilan University, Ilan, Taiwan
- 4Institute of Traditional Medicine, School of Medicine, National Yang-Ming University, Taipei, Taiwan
- 5Infectious Disease Division, Linsen, Chinese Medicine and Kunming Branch, Taipei City Hospital, Taipei, Taiwan
- 6Department of Animal Science and Biotechnology, Tunghai University, Taichung, Taiwan
- 7Department of Medical Imaging and Radiological Sciences, Chang Gung University, Taoyuan, Taiwan
- 8G.B. Elyakov Pacific Institute of Bioorganic Chemistry FEB RAS, Vladivostok, Russia
- 9Department of Medical Research, China Medical University Hospital, China Medical University, Taichung, Taiwan
Shigella is one of the leading bacterial causes of diarrhea worldwide, affecting more than 165 million people annually. Among the serotypes of Shigella, Shigella sonnei is physiologically unique and endemic in human immunodeficiency virus-infected men who have sex with men. The NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) inflammasome, a protein complex composed of NLRP3, apoptosis-associated speck-like protein, and caspase-1, recognizes, and responds to pathogen infection and diverse sterile host-derived or environmental danger signals to induce IL-1β and IL-18 production. Although the Shigella flexneri-mediated activation of the NLRP3 inflammasome has been reported, the effect of S. sonnei on NLRP3 inflammasome activation remains unclear. We found that S. sonnei induced IL-1β production through NLRP3-dependent pathways in lipopolysaccharide-primed macrophages. A mechanistic study revealed that S. sonnei induced IL-1β production through P2X7 receptor-mediated potassium efflux, reactive oxygen species generation, lysosomal acidification, and mitochondrial damage. In addition, the phagocytosis of viable S. sonnei was important for IL-1β production. Furthermore, we demonstrated that NLRP3 negatively regulated phagocytosis and the bactericidal activity of macrophages against S. sonnei. These findings provide mechanistic insight into the activation of the NLRP3 inflammasome by S. sonnei in macrophages.
Introduction
Shigellosis is a bacillary dysentery caused by Gram-negative, non-motile rod-shaped Shigella species. Shigella infects the intestines of humans and higher primates, resulting in acute diarrhea that may contain blood and mucus (1). Globally, there were at least 26 million cases of shigellosis from 1990 to 2016 and 212,438 deaths in 2016 (2). Each year, ~500,000 cases of diarrhea and 40 deaths caused by Shigella are reported in the United States (3). In Taiwan, there were 172 notifiable cases in 2018 according to a report by the Center of Disease Control (Taiwan CDC). Shigella belongs to the Enterobacteriacae family, which comprises four Shigella species, namely, Shigella dysenteriae, Shigella flexneri, Shigella boydii, and Shigella sonnei; these species are distributed worldwide, and their resistance to ciprofloxacin, ceftriaxone, and azithromycin is emerging (4, 5). Particularly in men who have sex with men (MSM) and HIV-positive populations, azithromycin-resistant Shigella is spreading globally (6). Outbreaks of S. flexneri and S. sonnei among MSM have been reported more frequently in the US, Canada, England, and Spain in recent years (1, 5, 7). In addition, from 2015 to 2016 in Taiwan, an outbreak of shigellosis was reported in MSM living with HIV (8). Taipei City Hospital isolated S. sonnei from several clinical shigellosis cases in MSM with HIV. Watery or bloody diarrhea caused by S. sonnei is usually relatively mild illness; however, its spread between MSM by sexual transmission is a public health concern.
Shigella invades and destroys the lining of the colon and the rectum mucosa and then enters resident macrophages and dendritic cells (9). Once these cells are infected, Shigella induces vacuole lysis, intracellular replication, and inflammatory cell death. It eventually disseminates and triggers a severe inflammatory response and cause acute bloody diarrhea (10). During shigellosis, the formation of micro-ulcers and inflammatory exudates of the colonic epithelium lead to polymorphonuclear leucocytes, and blood appears in the feces. Inflammation is a protective process that restricts microbial infection. Nucleotide-binding and oligomerization domain (NOD)-like receptor (NLR) is an intracellular innate immune receptor that recognizes and triggers inflammation against bacterial infection (11). Inflammasomes are multiprotein complexes comprised of members of the NLR family and/or apoptosis-associated speck-like protein (ASC) in response to intracellular pathogen- or damage-associated molecular patterns (12). Of the discovered inflammasomes, the NLRP3 inflammasome is the most well-investigated because it is highly relevant to human diseases (13–15).
Infection with Gram-negative bacteria from the Enterobacteriaceae family, such as Salmonella typhimurium, Escherichia coli, and Citrobacter rodentium, activate the NLRP3 inflammasome (16–19). In 2007, Suzuki et al., and Willingham et al., demonstrated that S. flexneri infection induces interleukin (IL)-1β production through Ipaf/ASC- and NLRP3/ASC-dependent pathways in macrophages (20, 21). In 2014, Suzuki et al. further determine that the S. flexneri type III secreted protein invasion plasmid antigen H7.8 enzyme 3 ubiquitin ligase plays pivotal role in NLRP3 inflammasome activation in macrophages (22). Although the effect of S. flexneri on the NLRP3 inflammasome has been well-studied, the effect of S. sonnei, a physiologically unique serotype of Shigella, on the NLRP3 inflammasome has not yet been addressed. In this study, we demonstrated the importance of the NLRP3 inflammasome in S. sonnei-mediated IL-1β and IL-18 production in macrophages. The roles of phagocytosis, P2X7 receptor-mediated potassium efflux, reactive oxygen species generation, lysosomal acidification, and mitochondrial damage in S. sonnei-mediated NLRP3 inflammasome activation were further investigated. Furthermore, the effect of NLRP3 knockout on the bactericidal activity of macrophages against S. sonnei was studied. This study provides evidence for the NLRP3 inflammasome as a promising drug target for S. sonnei infection.
Materials and Methods
Reagents and Chemicals
YVAD-CHO, ammonium chloride (NH4Cl), chloroquine diphosphate (CQ), N-acetyl cysteine (NAC), potassium chloride (KCl), glibenclamide, probenecid, carbenoxolone, LPS (Escherichia coli O111:B4), cyclosporine A, CA-074-Me, nordihydroguaiaretic acid (NDGA) were purchased from Sigma-Aldrich (St. Louis, MO). TLR2 shRNA lentiviral particles (sc-40257-V), control shRNA lentiviral particles (sc-108080), P2X7 shRNA plasmids (sc-42576-SH), control shRNA plasmids (sc-108060), Cryopyrin CRISPR/Cas9 KO plasmids (sc-432122), Manganese (III) tetrakis (4-benzoic acid) porphyrin chloride (MnTBAP), and antibodies against ASC (SC-22514-R, polyclonal antibody), IL-18 (SC-6177, polyclonal antibody), P2X7 (SC-514962, monoclonal antibody), and actin (SC-47778, monoclonal antibody) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against NLRP3 (AG-20B-0014, monoclonal antibody) and mouse caspase-1 (AG-20B-0044, monoclonal antibody) were purchased from Adipogen International (San Diego, CA). Antibodies against IL-1β (AB-401-NA, polyclonal antibody) were purchased from R&D Systems (Minneapolis, MN). MCC950 was purchased from TargetMol (Wellesley Hills, MA). DiOC2(3) and ELISA kits for IL-1β and tumor necrosis factor-α (TNF-α) were purchased from Thermo Fisher Scientific (Waltham, MA). Phorbol 12-myristate 13-acetate was purchased from Merck Millipore (Bedford, MA). Macrophage Colony Stimulating Factor (M-CSF) was purchased from Peprotech (London, UK).
Cell Lines and Culture
Mouse J774A.1 macrophages and human THP-1 monocytes were purchased from the American Type Culture Collection (Rockville, MD). THP-1 macrophages were differentiated from THP-1 monocytes by treatment with 50 nM PMA for 48 h. Human peripheral blood mononuclear cells (PBMCs) were separated from whole blood from healthy volunteers by density gradient centrifugation using Histopaque-1077 (23), and all experimental protocols were performed in accordance with the guidelines and regulations provided and accepted by the Institutional Review Board of the Tri-Service General Hospital, National Defense Medical Center and the volunteers' informed consent (TSGH-IRB-2-106-05-190 and TSGH-IRB-2-106-05-009). Mouse primary bone marrow derived macrophages (BMDM) were prepared from bone marrow collected from C57BL/6 mouse femur and tibia by differentiating in the M-CSF containing medium for 7 days. Animal experiments were performed with the approval of the Institutional Animal Care and Use Committee of the National Ilan University (approval number: No. 106-13). TLR2-knockdown and scramble control J774A.1 macrophages were generated by stably infection of TLR2 shRNA lentiviral particles and control shRNA lentiviral particles, respectively. P2X7-knockdown and scramble control J774A.1 macrophages were generated by stably transfection of P2X7 shRNA plasmids and control shRNA plasmids, respectively. NLRP3-knockout J774A.1 macrophages were generated by transfection of Cryopyrin CRISPR/Cas9 KO plasmids, and the clone with significantly reduced NLRP3 protein expression was selected for further studies. All cells were cultured in RPMI-1640 medium containing 10% fetal bovine serum in a 37°C CO2 incubator.
Activation of NLRP3 Inflammasome by S. sonnei Infection
S. sonnei (strain 25,931) was purchased from American Type Culture Collection. Generally, the bacteria were grown and subcultured twice a week on chocolate agar purchased from Creative Lifesciences (Taipei, Taiwan) at 37°C in 5% CO2. S. sonnei was subcultured again 1 day before infection. J774A.1 macrophages, THP-1 macrophages, or BMDM were primed for 4 h with 1 μg/ml LPS and then infected with S. sonnei at different multiplicities of infections (MOIs) for 1 h at 37°C. The extracellular bacteria were washed with sterile PBS and cultured in fresh medium for an additional 20 h with 3 μg/ml gentamicin (to kill the residual extracellular bacteria and avoid excessive bacterial growth in the medium). The intracellular penetration of gentamicin is low and did not have significant effect on intracellular bacteria (24). If the inhibitor (YVAD-CHO, MCC950, NH4Cl, CQ, NAC, KCl, glibenclamide, probenecid, carbenoxolone, cyclosporine A, MnTBAP, CA-074-Me, and NDGA) was used, it was added to the medium after LPS priming and 30 min before S. sonnei infection. The expression levels of IL-1β and TNF-α in the culture medium were measured by ELISA as described previously (25). In addition, to detect the expression levels of proIL-1β/IL-1β, proIL-18/IL-18, p45/p10, NLRP3, and ASC in the culture medium, the medium was concentrated with methanol/chloroform as described previously (26) and then analyzed by Western blotting. To detect the expression levels of proIL-1β, NLRP3, and actin in the cells, the cell lysates were analyzed by Western blotting. Heat-killed S. sonnei were prepared by incubating the bacteria in 80°C hot-plate for 30 min. Freeze/thaw-killed S. sonnei were prepared by repeatedly freezing the bacteria at −80°C for 1 h and thawing at room temperature five times. The loss of bacterial viability was confirmed by plating on a chocolate agar plate.
Detection of Intracellular ROS
Intracellular ROS levels were measured by staining the cells with the general oxidative stress indicator CM-H2DCFDA (Thermo Fisher Scientific). J774A.1 macrophages were primed with 1 μg/ml LPS for 4 h and then infected with 50 MOI S. sonnei for an additional 20 h. Then, the cells were stained with 2 μM CM-H2DCFDA for 15 min, and the intracellular fluorescence intensity was detected by flow cytometry (Cytomics FC500 Flow Cytometry CXP, Beckman Coulter Life Sciences).
Detection of Mitochondrial ROS and Membrane Potential
To detect mitochondrial ROS production, J774A.1 macrophages were primed with 1 μg/ml LPS for 4 h and then incubated with or without 20 μM MnTBAP for 30 min. The cells were infected with S. sonnei at 50 MOI for an additional 20 h. Then, the cells were stained with 5 nM MitoSOX for 15 min, and the fluorescence signal was acquired by flow cytometry. To detect the mitochondrial membrane potential, J774A.1 macrophages were primed with 1 μg/ml LPS for 4 h and then infected with 50 MOI S. sonnei for an additional 20 h. Then, the cells were stained with 50 nM DiOC2(3) for 15 min. The fluorescence signals were detected by flow cytometry.
Phagocytosis and Bactericidal Activity Assay
For short-term treatment, wild-type and NLRP3 knockout J774A.1 macrophages were infected with S. sonnei at 50 MOI for 15 min at 37°C in a CO2 incubator. The extracellular bacteria were washed out with sterile PBS and incubated in PBS containing 300 μg/ml gentamicin for an additional 1 h at 37°C in a CO2 incubator to completely kill the extracellular bacteria. The cells were washed with PBS and lysed with 300 μl distilled water for 40 min at 37°C in a CO2 incubator. The lysate was diluted 300 times with PBS, and 200 μl diluted lysate was then inoculated on chocolate agar plates and incubated at 37°C in a CO2 incubator overnight. The number of colony-forming units (CFUs) was counted and calculated. For long-term treatment, wild-type and NLRP3 knockout J774A.1 macrophages were infected with S. sonnei at 50 MOI for 15 min at 37°C in a CO2 incubator. The extracellular bacteria were washed out with sterile PBS and cultured in medium containing 100 μg/ml gentamicin for an additional 20 h at 37°C in a CO2 incubator. The cells were washed with PBS and lysed in 300 μl distilled water for 40 min at 37°C in a CO2 incubator. The lysate was diluted 50 times with PBS, and then 200 μl diluted lysate was inoculated on chocolate agar plates and incubated at 37°C in a CO2 incubator overnight. The number of CFUs was counted and calculated. Bactericidal activity was presented as the number of killed bacteria, which was calculated by subtracting the long-term CFU assay result from the short-term CFU assay result.
Statistical Analysis
Statistical significance was determined by GraphPad Prism 7.0 software. Two-tailed t-tests were used for two groups, and ANOVA with Dunnett's multiple comparisons test was used for three or more groups. *, **, and *** indicate a significant difference at the levels of p < 0.05, p < 0.01 and p < 0.001, respectively.
Results
S. sonnei Induces the Secretion of IL-1β, IL-18, NLRP3, ASC, and Active Caspase-1 in Macrophages
To investigate whether S. sonnei infection induces IL-1β secretion, untreated or LPS-primed mouse J774A.1 macrophages were infected with S. sonnei at 25, 50, or 100 MOI for 24 h. We found that S. sonnei induced IL-1β secretion in LPS-primed J774A.1 macrophages but not in untreated J774A.1 macrophages (Figure 1A). These results indicated that S. sonnei provided the activation signal but not the priming signal of inflammasome in macrophages. S. sonnei infection also induced IL-1β secretion in LPS-primed human THP-1 macrophages, human PBMCs, and mouse BMDM (Figure 1A). The S. sonnei-mediated induction of IL-1β was confirmed by detecting IL-1β expression in the culture medium of J774A.1 and THP-1 macrophages by Western blotting (Figure 1B). In addition, S. sonnei infection increased the expression of another inflammasome product, IL-18, in the culture medium of J774A.1 and THP-1 macrophages, as analyzed by Western blotting (Figure 1C). LPS- and ATP-activated J774A.1 macrophages were a positive control for IL-1β and IL-18 secretion, because ATP strongly activates the NLRP3 inflammasome in mouse macrophages, but only slightly activates the NLRP3 inflammasome in THP-1 macrophages. As the maturation and secretion of IL-1β and IL-18 are regulated by caspase-1, we asked whether caspase-1 is activated by S. sonnei infection. We found that the level of active caspase-1 (p10) in the culture medium of J774A.1 macrophages was increased by S. sonnei infection, as analyzed by Western blotting (Figure 1D). It has been demonstrated that upon the activation of inflammasome, inflammasome components are released from macrophages, act as extracellular danger signals and amplify the inflammatory response (27). We found that S. sonnei infection induced NLRP3 and ASC release into the culture medium of J774A.1 macrophages (Figure 1E).
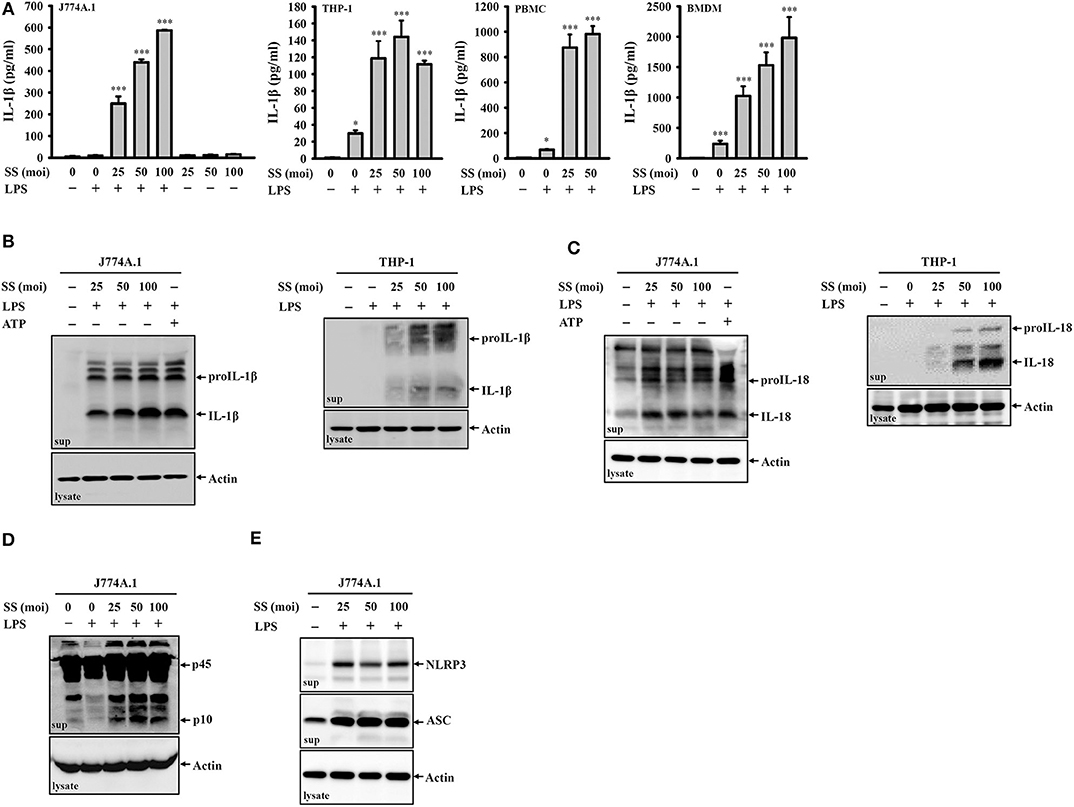
Figure 1. S. sonnei induces the secretion of IL-1β, IL-18, NLRP3, ASC, and active caspase-1 in macrophages. (A) J774A.1 macrophages, THP-1 macrophages, PBMCs or BMDM were primed with 1 μg/ml LPS for 4 h and then infected with S. sonnei for an additional 20 h. The levels of IL-1β in the supernatants were measured by ELISA. (B–E) J774A.1 macrophages or THP-1 macrophages were primed with 1 μg/ml LPS for 4 h followed and then infected with S. sonnei for an additional 20 h or stimulated with 5 mM ATP for an additional 0.5 h. The levels of IL-1β (B), IL-18 (C), caspase-1 (D), NLRP3, and ASC (E) in the supernatants were measured by Western blotting. The ELISA data are expressed as the mean ± SD of four separate experiments. The Western blotting results are representative of three different experiments. * and *** indicate significant differences at the levels of p < 0.05 and p < 0.001, respectively, compared to untreated control cells (one-way ANOVA with Dunnett's multiple comparisons test).
S. sonnei Induces IL-1β Secretion Through the NLRP3 Inflammasome
To investigate whether S. sonnei-induced IL-1β secretion requires the activation of the NLRP3 inflammasome, LPS-primed J774A.1 macrophages were incubated for 0.5 h with the caspase-1 inhibitor YVAD-CHO before S. sonnei infection. We found that YVAD-CHO reduced IL-1β secretion in a dose-dependent manner (Figure 2A). In addition, the NLRP3-specific inhibitor MCC950 reduced S. sonnei-induced IL-1β secretion in LPS-primed J774A.1 macrophages and BMDM, indicating the important role of NLRP3 in IL-1β secretion (Figure 2B). The role of NLRP3 in S. sonnei-mediated IL-1β secretion was confirmed in J774A.1 macrophages by CRISPR/Cas9-mediated NLRP3 knockout, as S. sonnei and ATP failed to induce IL-1β secretion (Figure 2C) and caspase-1 activation in LPS-primed NLRP3 knockout cells (Figure 2D). These results demonstrate that S. sonnei induces IL-1β secretion through the NLRP3 inflammasome.
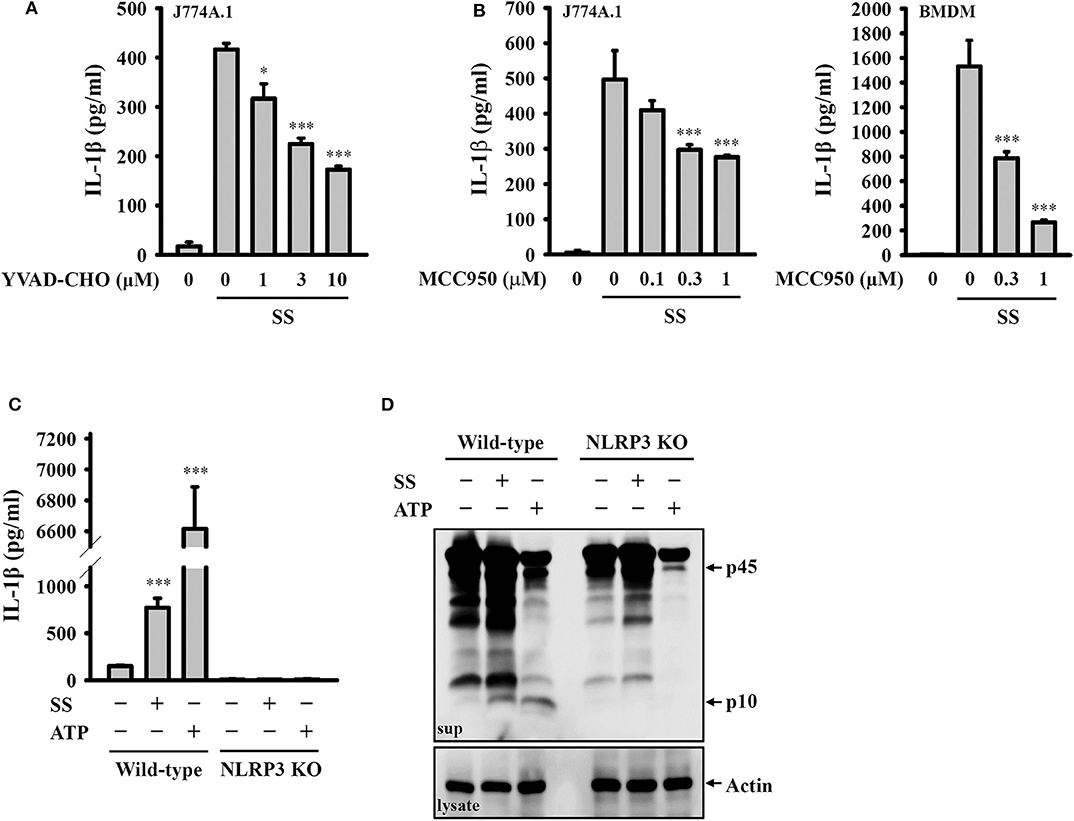
Figure 2. S. sonnei induces IL-1β secretion through the NLRP3 inflammasome. (A,B) J774A.1 macrophages or BMDM were primed with 1 μg/ml LPS for 4 h and then treated with YVAD-CHO (A) or MCC950 (B) for 0.5 h. The cells were then infected with 50 MOI S. sonnei for an additional 20 h. The levels of IL-1β in the supernatants were measured by ELISA. (C,D) Wild-type or NLRP3 knockout J774A.1 macrophages were primed with 1 μg/ml LPS for 4 h and then infected with 50 MOI S. sonnei for an additional 20 h. The levels of IL-1β (C) and caspase-1 (D) in the supernatants were measured by ELISA and Western blotting, respectively. The ELISA data are expressed as the mean ± SD of four separate experiments. The Western blotting results are representative of three different experiments. * and *** indicate significant differences at the levels of p < 0.05 and p < 0.001, respectively, compared to S. sonnei-infected cells (A,B) or untreated control cells (C) (one-way ANOVA with Dunnett's multiple comparisons test).
S. sonnei Activates the NLRP3 Inflammasome Through P2X7 Receptor-Mediated Potassium Efflux
Potassium efflux plays important roles in NLRP3 inflammasome activation in response to various NLRP3 stimulators (28). To investigate whether S. sonnei mediates NLRP3 inflammasome activation through potassium efflux, high extracellular potassium concentrations (12.5 and 25 mM KCl in the culture medium) were used to block potassium efflux. We found that S. sonnei-mediated IL-1β secretion and caspase-1 activation in J774A.1 macrophages were significantly inhibited by extracellular KCl (Figure 3A). To confirm the role of potassium efflux in S. sonnei-mediated NLRP3 inflammasome activation, potassium efflux was blocked by the adenosine triphosphate-sensitive potassium channel blocker glibenclamide. We found that S. sonnei-mediated IL-1β secretion and caspase-1 activation in J774A.1 macrophages were significantly inhibited by glibenclamide in a dose-dependent manner (Figure 3B). To further determine the receptor involved in S. sonnei-mediated potassium efflux, the effects of probenecid (a P2X7 receptor inhibitor) and carbenoxolone (a pannexin-1 inhibitor) on S. sonnei-induced NLRP3 inflammasome activation were investigated. We found that the P2X7 receptor inhibitor probenecid, but not the pannexin-1 inhibitor carbenoxolone, reduced IL-1β secretion in S. sonnei-infected J774A.1 macrophages (Figure 3C). To provide direct evidence of the important role of the P2X7 receptor in S. sonnei-mediated NLRP3 inflammasome activation, we knocked down the P2X7 receptor in J774A.1 macrophages by shRNA technology (26). Although the cell surface expression of P2X7 receptor was partially reduced by shRNA, S. sonnei-, and ATP-mediated IL-1β secretion was significantly reduced in P2X7 receptor knockdown cells compared to scramble shRNA-treated control cells (Figure 3D). Notably, although ATP-mediated caspase-1 activation was significantly reduced in P2X7 receptor knockdown cells, S. sonnei-mediated caspase-1 activation was not reduced in P2X7 receptor knockdown cells compared to control cells (Figure 3E). To understand why P2X7 receptor knockdown reduced IL-1β secretion without affecting caspase-1 activation, we investigated the expression levels of NLRP3 and proIL-1β in LPS-activated control and P2X7 receptor knockdown J774A.1 macrophages. We found that LPS-induced NLRP3 and proIL-1β expression was reduced in P2X7 receptor knockdown cells (Figure 3F). These results indicate that the P2X7 receptor participates in the priming of the NLRP3 inflammasome.
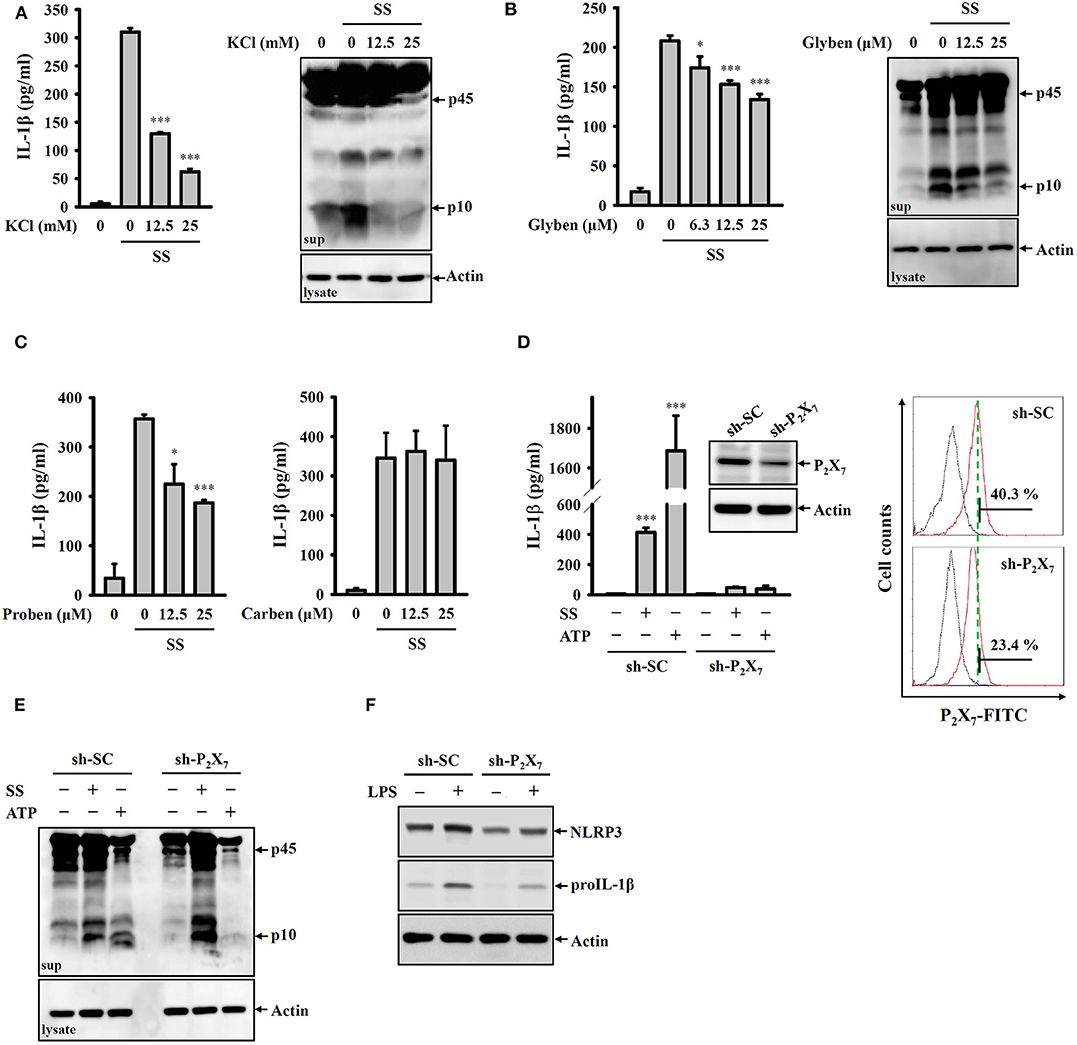
Figure 3. S. sonnei activates the NLRP3 inflammasome through P2X7 receptor-mediated potassium efflux. (A–C) J774A.1 macrophages were primed with 1 μg/ml LPS for 4 h and then treated with KCl (A), glibenclamide (B), probenecid or carbenoxolone (C) for 0.5 h. The cells were then infected with 50 MOI S. sonnei for an additional 20 h. The levels of IL-1β and caspase-1 in the supernatants were measured by ELISA and Western blotting, respectively. (D,E) Mock or P2X7 knockdown J774A.1 macrophages were primed with 1 μg/ml LPS for 4 h and then infected with 50 MOI S. sonnei for an additional 20 h or stimulated with 5 mM ATP for an additional 0.5 h. The levels of IL-1β (D) and caspase-1 (E) in the supernatants were measured by ELISA and Western blotting, respectively. The levels of P2X7 in mock or P2X7 knockdown J774A.1 macrophages were measured by Western blotting and flow cytometry (D). (F) Mock or P2X7 knockdown J774A.1 macrophages were stimulated with 1 μg/ml LPS for 6 h. The levels of NLRP3 and proIL-1β in the cell lysates were measured by Western blotting. The ELISA data are expressed as the mean ± SD of four separate experiments. The Western blotting results are representative of three different experiments. * and *** indicate significant differences at the levels of p < 0.05 and p < 0.001, respectively, compared to S. sonnei-infected cells (A–C) or untreated control cells (D) (one-way ANOVA with Dunnett's multiple comparisons test).
S. sonnei Activates the NLRP3 Inflammasome Through H2O2 Production and Lysosomal Damage
It has been proposed that ROS are crucial elements for NLRP3 inflammasome activation (29). We investigated whether S. sonnei infection activates the NLRP3 inflammasome through the generation of ROS. We found that S. sonnei infection increased H2O2 production in LPS-primed J774A.1 macrophages, as analyzed by H2DCFDA staining (Figure 4A). The inhibition of H2O2 by the antioxidant NAC reduced IL-1β secretion and caspase-1 activation in S. sonnei-infected J774A.1 macrophages (Figure 4B). NAC also reduced IL-1β secretion in S. sonnei-infected BMDM (Figure 4B). In addition, the inhibition of ROS generation enzyme lipoxygenase by NDGA reduced IL-1β secretion in S. sonnei-infected J774A.1 macrophages and BMDM (Figure 4C), confirming the importance of ROS in S. sonnei-mediated IL-1β secretion. Furthermore, the inhibition of the lysosomal cysteine protease cathepsin B by CA-074-me attenuates NLRP3 inflammasome activation in Mycobacterium tuberculosis- and Neisseria gonorrhoeae-infected cells (30, 31), suggesting an important role for lysosomes in bacterial infection-mediated NLRP3 inflammasome activation. As cathepsin B can be released from damaged lysosomes to drive NLRP3 inflammasome activation through binding to NLRP3 (32), NH4Cl, and CQ, which both inhibit endosomal/lysosomal acidification, were used to block lysosomal damage. We found that both NH4Cl and CQ significantly reduced IL-1β secretion in S. sonnei-infected J774A.1 macrophages (Figure 4D), confirming the role of lysosomes in NLRP3 inflammasome activation in response to S. sonnei infection. Furthermore, the cathepsin B inhibitor CA-074-me reduced IL-1β secretion in S. sonnei-infected J774A.1 macrophages (Figure 4E). These results indicate that S. sonnei activates the NLRP3 inflammasome through H2O2 production and lysosomal damage.
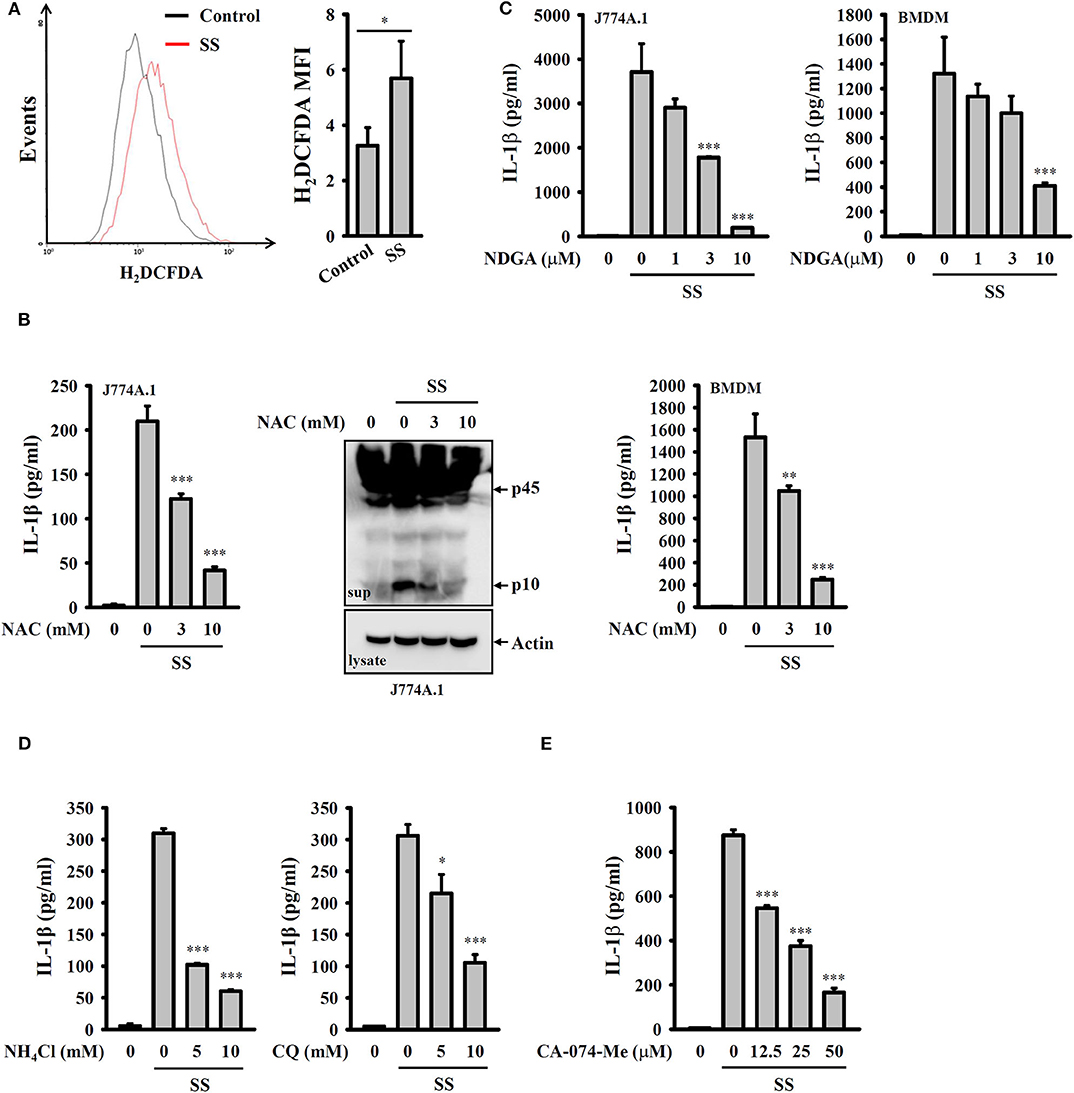
Figure 4. S. sonnei activates the NLRP3 inflammasome through H2O2 production and lysosomal damage. (A) J774A.1 macrophages were primed with 1 μg/ml LPS for 4 h and then infected with 50 MOI S. sonnei for an additional 20 h. The levels of intracellular ROS were measured by CM-H2DCFDA staining, and the data were acquired by flow cytometry. (B) J774A.1 macrophages or BMDM were primed with 1 μg/ml LPS for 4 h and then treated with NAC for 0.5 h. The cells were then infected with 50 MOI S. sonnei for an additional 20 h. The levels of IL-1β and caspase-1 in the supernatants were measured by ELISA and Western blotting, respectively. (C) J774A.1 macrophages or BMDM were primed with 1 μg/ml LPS for 4 h and then treated with NDGA for 0.5 h. The cells were then infected with 50 MOI S. sonnei for an additional 20 h. The levels of IL-1β in the supernatants were measured by ELISA. (D,E) J774A.1 macrophages were primed with 1 μg/ml LPS for 4 h and then treated with NH4Cl and CQ (D) or CA-074-Me (E) for 0.5 h. The cells were then infected with 50 MOI S. sonnei for an additional 20 h. The levels of IL-1β in the supernatants were measured by ELISA. The ELISA data are expressed as the mean ± SD of four separate experiments. The flow cytometry and Western blotting results are representative of three different experiments. *, **, and *** indicate significant differences at the levels of p < 0.05, p < 0.01 and p < 0.001, respectively, compared to untreated control cells (A) or S. sonnei-infected cells (B–E) [two-tailed t-test in panel (A); one-way ANOVA with Dunnett's multiple comparisons test in panels (B–E)].
S. sonnei Activates the NLRP3 Inflammasome Through Mitochondrial Damage
It has been demonstrated that mitochondrial damage drives downstream signaling that leads to NLRP3 inflammasome activation (33). Mitochondrial ROS are one downstream signals of damaged mitochondria that activate the NLRP3 inflammasome. Using the mitochondrial ROS indicator MitoSOX, we demonstrated that S. sonnei infection significantly induced mitochondrial ROS production in J774A.1 macrophages and that this effect was inhibited by MnTBAP, a mimic of superoxide dismutase (Figure 5A). We also found that the inhibition of mitochondrial ROS production by MnTBAP inhibited IL-1β secretion in S. sonnei-infected J774A.1 macrophages (Figure 5B). In addition, we found that S. sonnei infection caused mitochondrial integrity loss in J774A.1 macrophages, as analyzed by the mitochondrial membrane-potential-sensitive probe DiOC2(3) (Figure 5C). Furthermore, preserving mitochondrial integrity with cyclosporine A, an inhibitor of mitochondrial membrane permeability transition (33), reduced IL-1β secretion in S. sonnei-infected J774A.1 macrophages (Figure 5D). These results suggest that S. sonnei activates the NLRP3 inflammasome through mitochondrial damage.
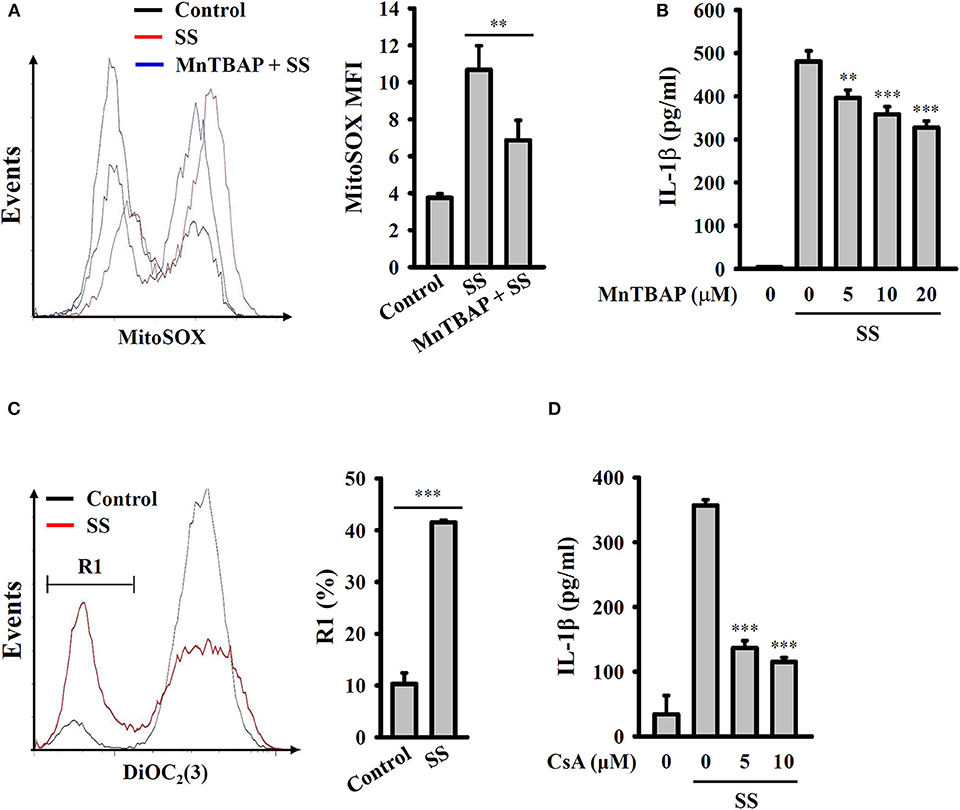
Figure 5. S. sonnei activates the NLRP3 inflammasome through mitochondrial damage. (A) J774A.1 macrophages were primed with 1 μg/ml LPS for 4 h and then treated with 20 μM MnTBAP for 0.5 h. The cells were then infected with 50 MOI S. sonnei for an additional 20 h. The levels of mitochondrial ROS were measured by MitoSOX staining and flow cytometry. (B) J774A.1 macrophages were primed with 1 μg/ml LPS for 4 h and then treated with MnTBAP for 0.5 h. The cells were then infected with 50 MOI S. sonnei for an additional 20 h. The levels of IL-1β in the supernatants were measured by ELISA. (C) J774A.1 macrophages were primed with 1 μg/ml LPS for 4 h and then infected with 50 MOI S. sonnei for an additional 20 h. The mitochondrial membrane potential was measured by DiOC2(3) staining and flow cytometry. (D) J774A.1 macrophages were primed with 1 μg/ml LPS for 4 h and then treated with cyclosporine A for 0.5 h. The cells were then infected with 50 MOI S. sonnei for an additional 20 h. The levels of IL-1β in the supernatants were measured by ELISA. The ELISA data are expressed as the mean ± SD of four separate experiments. The flow cytometry results are representative of three different experiments. ** and *** indicate significant differences at the levels of p < 0.01 and p < 0.001, respectively, compared to S. sonnei-infected cells (B,D) or as indicated (A,C) [two-tailed t-test in panels (A,C); one-way ANOVA with Dunnett's multiple comparisons test in panels (B,D)].
Phagocytosis of Live S. sonnei Is Required for the Full Activation of the NLRP3 Inflammasome
To investigate whether the activation of the NLRP3 inflammasome requires phagocytosis of S. sonnei, J774A.1 macrophages were incubated with cytochalasin D, a cell-permeable actin polymerization inhibitor, which blocks the phagocytosis of macrophages. We found that cytochalasin D significantly reduced IL-1β secretion but only slightly reduced TNF-α secretion in S. sonnei-infected J774A.1 macrophages (Figure 6A). These results indicate that phagocytosis of S. sonnei is required for the full activation of the NLRP3 inflammasome but plays a smaller role in NLRP3-independent TNF-α secretion. In addition, our previous study indicated that TLR2 and phagocytosis of live Neisseria gonorrhoeae are important for the NLRP3 inflammasome activation (26). To investigate the role of TLR2 and live S. sonnei in NLRP3 inflammasome activation, we generated TLR2 knockdown J774A.1 macrophages by shRNA technology (26). We found that TLR2 mRNA expression was significantly reduced in sh-TLR2 cells compared to the scramble control (sh-SC) cells, and TLR2 ligand Pam3CSK4-mediated TNF-α secretion was significantly reduced in sh-TLR2 cells compared to sh-SC cells, indicating the functional knockdown of TLR2 in the cells (Figure 6B). We investigated the effect of live, heat-killed, or freeze/thaw-killed S. sonnei on IL-1β and TNF-α in sh-TLR2 and sh-SC cells. We found that heat-killed and freeze/thaw-killed S. sonnei induced less IL-1β and TNF-α secretion than that induced by live S. sonnei (Figure 6C). These results indicate that the NLRP3 inflammasome activation and TNF-α expression requires phagocytosis of live S. sonnei. In addition, we found that live, heat-killed, and freeze/thaw-killed S. sonnei induced less IL-1β and TNF-α secretion in sh-TLR2 cells than that in sh-SC cells (Figure 6C). These results indicate that TLR2 plays an important role in S. sonnei-mediated IL-1β and TNF-α secretion in macrophages.
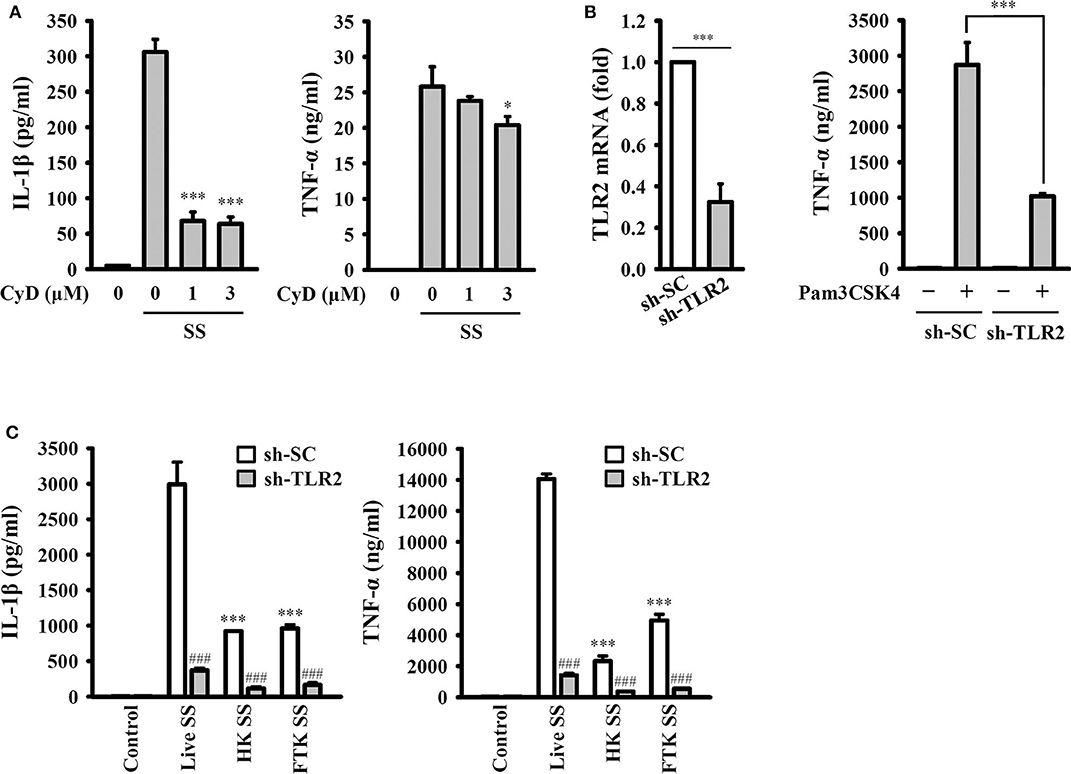
Figure 6. Phagocytosis of live S. sonnei is required for the full activation of the NLRP3 inflammasome. (A) J774A.1 macrophages were primed with 1 μg/ml LPS for 4 h and then treated with cytochalasin D for 0.5 h. The cells were then infected with 50 MOI S. sonnei for an additional 20 h. The levels of IL-1β and TNF-α in the supernatants were measured by ELISA. (B) The TLR2 mRNA expression of mock or TLR2 knockdown J774A.1 macrophages was measured by RT-qPCR. Mock or TLR2 knockdown J774A.1 macrophages were stimulated with 1 μg/ml Pam3CSK4 for 6 h. The levels of TNF-α in the supernatants were measured by ELISA. (C) Mock or TLR2 knockdown J774A.1 macrophages were primed with 1 μg/ml LPS for 4 h and then infected with 50 MOI live, heat-killed or freeze/thaw-killed S. sonnei for an additional 20 h. The levels of IL-1β and TNF-α in the supernatants were measured by ELISA. The ELISA data are expressed as the mean ± SD of four separate experiments. * and *** indicate significant differences at the levels of p < 0.05 and p < 0.001, respectively, compared to S. sonnei-infected cells (A) or live S. sonnei-infected cells (C) or as indicated (B). ###indicates significant differences at the levels of p < 0.001 compared to sh-SC cells [one-way ANOVA with Dunnett's multiple comparisons test in panels (A,C); two-tailed t-test in panel (B)].
NLRP3 Knockout Increases the Bactericidal Activity of Macrophages Against S. sonnei
Our previous study demonstrated that the knockout of NLRP3 by CRISPR/Cas9 technology enhances phagocytosis of pHrodo Green E. coli BioParticles Conjugate and Neisseria gonorrhoeae by macrophages (26). In this study, we assessed the functional consequence of NLRP3 knockout on phagocytosis of S. sonnei by macrophages. The number of engulfed bacteria in wild-type and NLRP3 knockout J774A.1 macrophages was determined by the CFU assay after 15 min of infection. The extracellular bacteria were washed out and completely killed by gentamicin. We found that the number of engulfed bacteria in NLRP3 knockout cells (17,400 ± 1,665 CFUs) was higher than that in wild-type cells (3,240 ± 332 CFUs; Figures 7A,B). These results indicated that NLRP3-knockout increased phagocytosis of S. sonnei by macrophages. To further investigate the functional consequence of NLRP3 knockout on the bactericidal activity of macrophages against S. sonnei, the number of intracellular live S. sonnei cells after 20 h of infection was determined by the CFU assay. We found that the number of intracellular live S. sonnei cells in NLRP3 knockout cells and wild-type cells was 7,370 ± 451 and 760 ± 57, respectively (Figures 7A,B). The number of killed S. sonnei cells was calculated by subtracting the 20 h CFU assay result from the 15 min CFU assay result. We found that NLRP3 knockout significantly increased the bactericidal activity of macrophages against S. sonnei (10,030 S. sonnei cells were killed) compared to wild-type cells (2,480 S. sonnei cells were killed; Figure 7B). These results indicate that NLRP3 knockout increases the phagocytic and bactericidal activity of macrophages against S. sonnei.
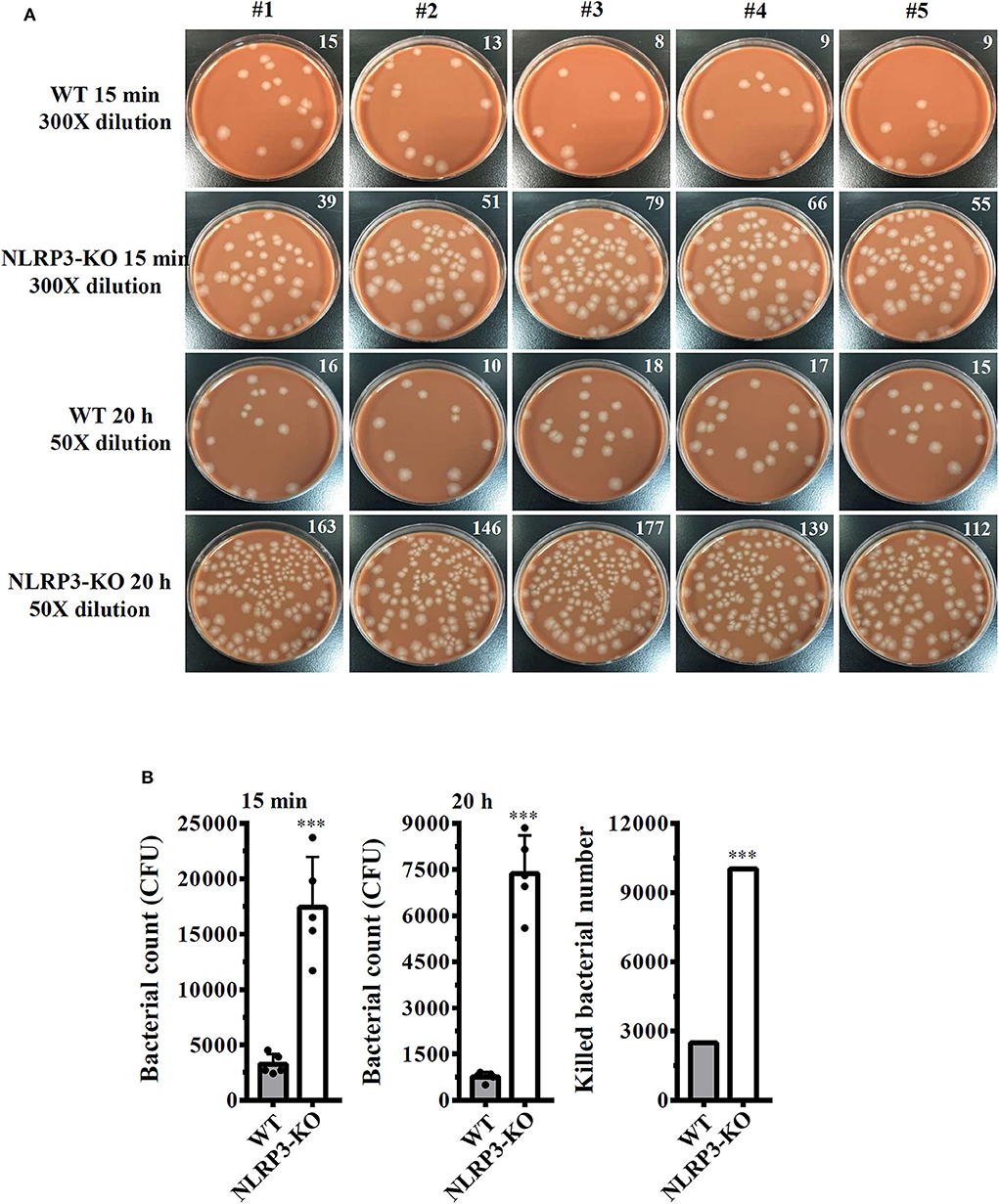
Figure 7. NLRP3 knockout increases the bactericidal activity of macrophages against S. sonnei. (A,B) Wild-type or NLRP3-knockout J774A.1 macrophages were infected with 50 MOI S. sonnei for 15 min or 20 h. The cells were lysed, and the number of engulfed live S. sonnei cells was determined by the CFU assay and is indicated in the upper right corner (A). The mean CFUs of cells infected with S. sonnei for 15 min or 20 h and the number of killed bacteria were calculated by subtracting the 20 h CFU assay result from the 15 min CFU assay result and are shown in panel (B). The data are expressed as the mean ± SD of five separate experiments. ***indicates a significant difference at the level of p < 0.001 compared to wild-type cells (two-tailed t-test).
Discussion
Inflammation is a process of the immune system that regulates the host defense machinery and controls microbial invasion; however, over-reactive or prolonged inflammation increases the risk for the development of inflammatory diseases. Precise control of inflammatory responses is important for limiting pathogen infection without causing host damage. The NLRP3 inflammasome controls the maturation and secretion of the proinflammatory cytokines IL-1β and IL-18 and is important for the innate immunity against pathogen infection (12, 26). The dysregulation of NLRP3 inflammasome activation has been demonstrated to participate in the pathogenesis of metabolic disorders and neurodegenerative diseases (15, 34). The findings of this study clearly showed that S. sonnei infection caused IL-1β and IL-18 production through the NLRP3 inflammasome in macrophages, suggesting that S. sonnei infection may increase the risk for NLRP3-associated inflammatory diseases, including diabetes, atherosclerosis, inflammatory bowel disease, chronic kidney disease, gout, and Alzheimer's disease (13–15).
The activation of the NLRP3 inflammasome is initiated by 2-step priming and activating signals. The priming step involves pathogen-associated molecular patterns (e.g., LPS) through toll-like receptors to induce the protein expression of NLRP3 and the IL-1β precursor (25). The activating step involves a broad range medically relevant stimuli, including saturated fatty acids (type II diabetes), cholesterol crystals (atherosclerosis), uric acid crystals (gouty inflammation), and amyloid-β (Alzheimer's disease) (15). In this study, we demonstrated that S. sonnei infection did not provide the priming signal of the NLRP3 inflammasome because S. sonnei infection induced IL-1β secretion in LPS-primed macrophages but not in macrophages without LPS priming (Figure 1A). However, other studies have shown that S. flexneri (strain YSH6000) infection induces IL-1β secretion in bone marrow-derived macrophages without LPS priming (20, 22). These differences may come from the different serotypes of Shigella or cell types. The activating signals of the NLRP3 inflammasome include extracellular ATP, bacterial pore-forming toxins, and crystal substances that induce ROS production, ion efflux, lysosomal damage, and mitochondria stress (15). Finally, the NLRP3 protein recruits adaptor protein ASC to form oligomers, activate caspase-1, and induce the maturation of IL-1β and IL-18 (34, 35). It has been demonstrated that invasion plasmid antigen H7.8 enzyme 3 ubiquitin ligase is important for S. flexneri-mediated NLRP3 and NLR family CARD domain containing 4 (NLRC4) inflammasome activation, as it digests the NLRP3 and NLRC4 inflammasome inhibitory proteins glomulin/flagellar-associated protein 68 (22, 36). Furthermore, needle- or rod-shapes proteins secreted by the S. flexneri type III secretion system are recognized by NAIP1 and NAIP2 to induce robust NLRC4 inflammasome activation (37–40). RAW264.7 macrophages infected with S. flexneri induce NLRP1B inflammasome activation (41). This study was limited because it did not identify the virulence factor of S. sonnei for NLRP3 inflammasome activation. We demonstrated that, compared to live S. sonnei, killed S. sonnei significantly reduced the IL-1β induction activity (Figure 6C). In addition, a phagocytosis inhibitor also significantly reduced IL-1β induction in S. sonnei-infected macrophages (Figure 6A). These results indicate that the intracellular active delivery of the virulence factor by S. sonnei is important for NLRP3 inflammasome activation. Recent study demonstrated that S. sonnei infection caused caspase-1 activation, ASC speck formation, and IL-18 expression in THP-1 macrophages. However, S. sonnei infection caused less caspase-1 dependent pyroptosis of macrophages than S. flexneri infection. In a mechanistic study, the O-antigen on the surface of S. sonnei reduced bacterial uptake and cytosolic escape, which results in reduced activation of caspase-1 (42).
The P2X7 receptor is a cation-specific ion channel that recognizes and responds to extracellular ATP and induces potassium efflux. the activation of the P2X7 receptor is associated with the immune response and regulates pathogen infection (43). In this study, we demonstrated that the P2X7 receptor participated in IL-1β release in S. sonnei-infected macrophages (Figure 3D). Notably, although P2X7 receptor knockdown significantly reduced caspase-1 activation in ATP-activated macrophages, caspase-1 activation was not affected in S. sonnei-infected macrophages (Figure 3E). One explanation for is P2X7 receptor knockdown-mediated IL-1β inhibition in S. sonnei-infected macrophages is the reduced expression of NLRP3 and proIL-1β (Figure 3F). These results indicated that P2X7 receptor knockdown reduced NLRP3 inflammasome activation by inhibiting the priming signal but not by affecting the activation signal in S. sonnei-infected macrophages. It should be noted that only partial inhibition of P2X7 receptor with shRNA, but total inhibition of IL-1β in S. sonnei-infected macrophages (Figure 3D). It has been demonstrated that stimulation of P2X7 receptor activates caspase-1 dependent pyroptosis and induces the formation of a non-selective pore, allowing the release of intracellular components including IL-1β (44). We suggested that P2X7 receptor knockdown not only reduced the proIL-1β expression but also inhibited the IL-1β release by reducing pyroptosis. A previous study demonstrated that the P2X7 receptor positively regulates LPS-induced TNF-α secretion by increasing the extracellular activity of the TNF-α converting enzyme (45), indicating that the P2X7 receptor can regulate LPS-mediated proinflammatory signaling. The inhibition of the P2X7 receptor not only reduces IL-1β production by S. sonnei-infected macrophages but also reduces IL-1β production by Neisseria gonorrhoeae- and Group A Streptococcus-infected macrophages (26, 46). These results suggest that the P2X7 receptor is a potential therapeutic target for pathogen infection (47, 48) or inflammatory diseases (49).
In this study, we demonstrated that S. sonnei infection induces inflammatory responses by activating the NLRP3 inflammasome. We also found that NLRP3 knockout enhanced the phagocytic and bactericidal activity of macrophages against S. sonnei (Figure 7). This study provides a rational strategy for protecting against S. sonnei infection and reducing inflammatory damage by targeting the NLRP3 inflammasome. In addition, the in vitro S. sonnei infection model can be used to screen and develop potential compounds or ingredients to ameliorate S. sonnei-induced inflammation. However, limitation of this study is the lack of an animal model of S. sonnei infection. Because of the complications of Shigella pathogenesis, there is no ideal model animal model that faithfully recapitulates Shigella infection in humans. Recently, Koestler et al. (50) cultured human intestinal stem cell-derived intestinal enteroids ex vivo and mimicked the human intestinal environment. The effect of S. sonnei infection on the NLRP3 inflammasome can be confirmed in the human intestinal enteroid model in the future.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Author Contributions
K-FH was the guarantor of the article. L-HL and K-FH conceived and designed the study, wrote, and finished the manuscript. L-HL, T-LC, H-WC, and T-TT performed the experiments and analyzed the data. C-HH, C-CW, and W-CT contributed to critical revision of the manuscript. T-CJ, F-HC, and OC assisted with some experiments.
Funding
This research work was supported by the funding from the Ministry of Science and Technology of Taiwan (MOST 108-2628-B-197-001; MOST 108-2923-B-197-001-MY3; MOST 108-2321-B-197-001; MOST 107-2635-B-532-001).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Toro C, Arroyo A, Sarria A, Iglesias N, Enríquez A, Baquero M, et al. Shigellosis in subjects with traveler's diarrhea versus domestically acquired diarrhea: implications for antimicrobial therapy and human immunodeficiency virus surveillance. Am J Trop Med Hyg. (2015) 93:491–6. doi: 10.4269/ajtmh.14-0804
2. Khalil IA, Troeger C, Blacker BF, Rao PC, Brown A, Atherly DE, et al. Morbidity and mortality due to shigella and enterotoxigenic Escherichia coli diarrhoea: the global burden of disease study 1990-2016. Lancet Infect Dis. (2018) 18:1229–40. doi: 10.1016/S1473-3099(18)30475-4
3. McCrickard LS, Crim SM, Kim S, Bowen A. Disparities in severe shigellosis among adults - foodborne diseases active surveillance network, 2002-2014. BMC Public Health. (2018) 18:221. doi: 10.1186/s12889-018-5115-4
4. Bowen A, Grass J, Bicknese A, Campbell D, Hurd J, Kirkcaldy RD. Elevated risk for antimicrobial drug-resistant shigella infection among men who have sex with men, United States, 2011-2015. Emerg Infect Dis. (2016) 22:1613–6. doi: 10.3201/eid2209.160624
5. Chiou CS, Izumiya H, Kawamura M, Liao YS, Su YS, Wu HH, et al. The worldwide spread of ciprofloxacin-resistant Shigella sonnei among HIV-infected men who have sex with men, Taiwan. Clin Microbiol Infect. (2016) 22:383.e11–e6. doi: 10.1016/j.cmi.2015.12.021
6. Kotloff KL, Riddle MS, Platts-Mills JA, Pavlinac P, Zaidi AKM. Shigellosis. Lancet. (2018) 391:801–12. doi: 10.1016/S0140-6736(17)33296-8
7. Ratnayake R, Allard R, Pilon PA. Shifting dominance of Shigella species in men who have sex with men. Epidemiol Infect. (2012) 140:2082–6. doi: 10.1017/S0950268812000738
8. Wu HH, Shen YT, Chiou CS, Fang CT, Lo YC. Shigellosis outbreak among MSM living with HIV: a case-control study in Taiwan, 2015-2016. Sex Transm Infect. (2019) 95:67–70. doi: 10.1136/sextrans-2017-053410
9. Cunha LD, Zamboni DS. Subversion of inflammasome activation and pyroptosis by pathogenic bacteria. Front Cell Infect Microbiol. (2013) 3:76. doi: 10.3389/fcimb.2013.00076
10. Paciello I, Silipo A, Lembo-Fazio L, Curcurù L, Zumsteg A, Noël G, et al. Intracellular shigella remodels its LPS to dampen the innate immune recognition and evade inflammasome activation. Proc Natl Acad Sci U S A. (2013) 110:E4345–54. doi: 10.1073/pnas.1303641110
11. Liao KC, Sandall CF, Carlson DA, Ulke-Lemée A, Platnich JM, Hughes PF, et al. Application of immobilized ATP to the study of NLRP inflammasomes. Arch Biochem Biophys. (2019) 670:104–15. doi: 10.1016/j.abb.2018.12.031
12. Verma V, Dhanda RS, Møller NF, Yadav M. Inflammasomes and their role in innate immunity of sexually transmitted infections. Front Immunol. (2016) 7:540. doi: 10.3389/fimmu.2016.00540
13. Kanneganti TD. Inflammatory bowel disease and the NLRP3 inflammasome. N Engl J Med. (2017) 377:694–6. doi: 10.1056/NEJMcibr1706536
14. Tsai YL, Hua KF, Chen A, Wei CW, Chen WS, Wu CY, et al. NLRP3 inflammasome: Pathogenic role and potential therapeutic target for IgA nephropathy. Sci Rep. (2017) 7:41123. doi: 10.1038/srep41123
15. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. (2015) 21:677–87. doi: 10.1038/nm.3893
16. Bierschenk D, Monteleone M, Moghaddas F, Baker PJ, Masters SL, Boucher D, et al. The Salmonella pathogenicity island-2 subverts human NLRP3 and NLRC4 inflammasome responses. J Leukoc Biol. (2019) 105:401–10. doi: 10.1002/JLB.MA0318-112RR
17. Loss H, Aschenbach JR, Ebner F, Tedin K, Lodemann U. Effects of a pathogenic ETEC strain and a probiotic Enterococcus faecium strain on the inflammasome response in porcine dendritic cells. Vet Immunol Immunopathol. (2018) 203:78–87. doi: 10.1016/j.vetimm.2018.08.004
18. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. (2015) 526:666–71. doi: 10.1038/nature15541
19. Bording-Jorgensen M, Alipour M, Danesh G, Wine E. Inflammasome activation by ATP enhances Citrobacter rodentium clearance through ROS generation. Cell Physiol Biochem. (2017) 41:193–204. doi: 10.1159/000455988
20. Suzuki T, Franchi L, Toma C, Ashida H, Ogawa M, Yoshikawa Y, et al. Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog. (2007) 3:e111. doi: 10.1371/journal.ppat.0030111
21. Willingham SB, Bergstralh DT, O'Connor W, Morrison AC, Taxman DJ, Duncan JA, et al. Microbial pathogen-induced necrotic cell death mediated by the inflammasome components CIAS1/cryopyrin/NLRP3 and ASC. Cell Host Microbe. (2007) 2:147–59. doi: 10.1016/j.chom.2007.07.009
22. Suzuki S, Mimuro H, Kim M, Ogawa M, Ashida H, Toyotome T, et al. Shigella IpaH7.8 E3 ubiquitin ligase targets glomulin and activates inflammasomes to demolish macrophages. Proc Natl Acad Sci U S A. (2014) 111:E4254–63. doi: 10.1073/pnas.1324021111
23. Chernikov OV, Wong WT, Li LH, Chikalovets IV, Molchanova VI, Wu SH, et al. A GalNAc/Gal-specific lectin from the sea mussel Crenomytilus grayanus modulates immune response in macrophages and in mice. Sci Rep. (2017) 7:6315. doi: 10.1038/s41598-017-06647-5
24. Bongers S, Hellebrekers P, Leenen LPH, Koenderman L, Hietbrink F. Intracellular penetration and effects of antibiotics on Staphylococcus aureus inside human neutrophils: a comprehensive review. Antibiotics (Basel). (2019) 8:E54. doi: 10.3390/antibiotics8020054
25. Liao PC, Chao LK, Chou JC, Dong WC, Lin CN, Lin CY, et al. Lipopolysaccharide/adenosine triphosphate-mediated signal transduction in the regulation of NLRP3 protein expression and caspase-1-mediated interleukin-1β secretion. Inflamm Res. (2013) 62:89–96. doi: 10.1007/s00011-012-0555-2
26. Li LH, Lin JS, Chiu HW, Lin WY, Ju TC, Chen FH, et al. Mechanistic insight into the activation of the NLRP3 inflammasome by Neisseria gonorrhoeae in macrophages. Front Immunol. (2019) 10:1815. doi: 10.3389/fimmu.2019.01815
27. Baroja-Mazo A, Martín-Sánchez F, Gomez AI, Martínez CM, Amores-Iniesta J, Compan V, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol. (2014) 15:738–48. doi: 10.1038/ni.2919
28. Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. (2013) 38:1142–53. doi: 10.1016/j.immuni.2013.05.016
29. Tschopp J, Schroder K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. (2010) 10:210–5. doi: 10.1038/nri2725
30. Amaral EP, Riteau N, Moayeri M, Maier N, Mayer-Barber KD, Pereira RM, et al. lysosomal cathepsin release is required for NLRP3-inflammasome activation by Mycobacterium tuberculosis in infected macrophages. Front Immunol. (2018) 9:1427. doi: 10.3389/fimmu.2018.01427
31. Duncan JA, Gao X, Huang MT, O'Connor BP, Thomas CE, Willingham SB, et al. Neisseria gonorrhoeae activates the proteinase cathepsin B to mediate the signaling activities of the NLRP3 and ASC-containing inflammasome. J Immunol. (2009) 182:6460–9. doi: 10.4049/jimmunol.0802696
32. Bruchard M, Mignot G, Derangère V, Chalmin F, Chevriaux A, Végran F, et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med. (2013) 19:57–64. doi: 10.1038/nm.2999
33. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. (2011) 12:222–30. doi: 10.1038/ni.1980
34. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. (2019) 20:E3328. doi: 10.3390/ijms20133328
35. Stutz A, Golenbock DT, Latz E. Inflammasomes: too big to miss. J Clin Invest. (2009) 119:3502–11. doi: 10.1172/JCI40599
36. Suzuki S, Franchi L, He Y, Muñoz-Planillo R, Mimuro H, Suzuki T, et al. Shigella type III secretion protein MxiI is recognized by Naip2 to induce Nlrc4 inflammasome activation independently of Pkcδ. PLoS Pathog. (2014) 10:e1003926. doi: 10.1371/journal.ppat.1003926
37. Suzuki S, Suzuki T, Mimuro H, Mizushima T, Sasakawa C. Shigella hijacks the glomulin-cIAPs-inflammasome axis to promote inflammation. EMBO Rep. (2018) 19:89–101. doi: 10.15252/embr.201643841
38. Zhao Y, Shi J, Shi X, Wang Y, Wang F, Shao F. Genetic functions of the NAIP family of inflammasome receptors for bacterial ligands in mice. J Exp Med. (2016) 213:647–56. doi: 10.1084/jem.20160006
39. Jessen DL, Osei-Owusu P, Toosky M, Roughead W, Bradley DS, Nilles ML. Type III secretion needle proteins induce cell signaling and cytokine secretion via Toll-like receptors. Infect Immun. (2014) 82:2300–9. doi: 10.1128/IAI.01705-14
40. Yang J, Zhao Y, Shi J, Shao F. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc Natl Acad Sci U S A. (2013) 110:14408–13. doi: 10.1073/pnas.1306376110
41. Neiman-Zenevich J, Stuart S, Abdel-Nour M, Girardin SE, Mogridge J. Listeria monocytogenes and Shigella flexneri activate the NLRP1B inflammasome. Infect Immun. (2017) 85:e00338–17. doi: 10.1128/IAI.00338-17
42. Watson JL, Sanchez-Garrido J, Goddard PJ, Torraca V, Mostowy S, Shenoy AR, et al. Shigella sonnei O-antigen inhibits internalization, vacuole escape, and inflammasome activation. mBio. (2019) 10:e02654–19. doi: 10.1128/mBio.02654-19
43. Chaves MM, Sinflorio DA, Thorstenberg ML, Martins MDA, Moreira-Souza ACA, Rangel TP, et al. Non-canonical NLRP3 inflammasome activation and IL-1β signaling are necessary to L. amazonensis control mediated by P2X7 receptor and leukotriene B4. PLoS Pathog. (2019) 15:e1007887. doi: 10.1371/journal.ppat.1007887
44. Zhao H, Chen Y, Feng H. P2X7 Receptor-associated programmed cell death in the pathophysiology of hemorrhagic stroke. Curr Neuropharmacol. (2018) 16:1282–95. doi: 10.2174/1570159X16666180516094500
45. Barberà-Cremades M, Gómez AI, Baroja-Mazo A, Martínez-Alarcón L, Martínez CM, de Torre-Minguela C, et al. P2X7 receptor induces tumor necrosis factor-α converting enzyme activation and release to boost TNF-α production. Front Immunol. (2017) 8:862. doi: 10.3389/fimmu.2017.00862
46. Westerlund E, Valfridsson C, Yi DX, Persson JJ. The secreted virulence factor NADase of group A Streptococcus INHIBITS P2X7 receptor-mediated release of IL-1β. Front Immunol. (2019) 10:1385. doi: 10.3389/fimmu.2019.01385
47. Rosli S, Kirby FJ, Lawlor KE, Rainczuk K, Drummond GR, Mansell A, et al. Repurposing drugs targeting the P2X7 receptor to limit hyperinflammation and disease during influenza virus infection. Br J Pharmacol. (2019) 176:3834–44. doi: 10.1111/bph.14787
48. Martínez-García JJ, Martínez-Banaclocha H, Angosto-Bazarra D, de Torre-Minguela C, Baroja-Mazo A, Alarcón-Vila C, et al. P2X7 receptor induces mitochondrial failure in monocytes and compromises NLRP3 inflammasome activation during sepsis. Nat Commun. (2019) 10:2711. doi: 10.1038/s41467-019-10626-x
49. Cao F, Hu LQ, Yao SR, Hu Y, Wang DG, Fan YG, et al. P2X7 receptor: a potential therapeutic target for autoimmune diseases. Autoimmun Rev. (2019) 18:767–77. doi: 10.1016/j.autrev.2019.06.009
Keywords: shigellosis, NLRP3 inflammasome, macrophages, P2X7 receptor, mitochondria
Citation: Li L-H, Chen T-L, Chiu H-W, Hsu C-H, Wang C-C, Tai T-T, Ju T-C, Chen F-H, Chernikov OV, Tsai W-C and Hua K-F (2020) Critical Role for the NLRP3 Inflammasome in Mediating IL-1β Production in Shigella sonnei-Infected Macrophages. Front. Immunol. 11:1115. doi: 10.3389/fimmu.2020.01115
Received: 29 December 2019; Accepted: 07 May 2020;
Published: 03 June 2020.
Edited by:
Eric Pearlman, University of California, Irvine, United StatesReviewed by:
Sheryl Coutermarsh-Ott, Virginia Tech, United StatesRoberta Olmo Pinheiro, Oswaldo Cruz Foundation, Brazil
Copyright © 2020 Li, Chen, Chiu, Hsu, Wang, Tai, Ju, Chen, Chernikov, Tsai and Hua. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kuo-Feng Hua, kuofenghua@gmail.com