 Johannes Westman
Johannes Westman Sergio Grinstein
Sergio Grinstein Pedro Elias Marques
Pedro Elias Marques- 1Program in Cell Biology, Hospital for Sick Children, Toronto, ON, Canada
- 2Department of Biochemistry, University of Toronto, Toronto, ON, Canada
- 3Keenan Research Centre of the Li Ka Shing Knowledge Institute, St. Michael's Hospital, Toronto, ON, Canada
- 4Laboratory of Molecular Immunology, Department of Microbiology, Immunology and Transplantation, Rega Institute for Medical Research, KU Leuven, Leuven, Belgium
Clearance of cellular debris is required to maintain the homeostasis of multicellular organisms. It is intrinsic to processes such as tissue growth and remodeling, regeneration and resolution of injury and inflammation. Most of the removal of effete and damaged cells is performed by macrophages and neutrophils through phagocytosis, a complex phenomenon involving ingestion and degradation of the disposable particles. The study of the clearance of cellular debris has been strongly biased toward the removal of apoptotic bodies; as a result, the mechanisms underlying the removal of necrotic cells have remained relatively unexplored. Here, we will review the incipient but growing knowledge of the phagocytosis of necrotic debris, from their recognition and engagement to their internalization and disposal. Critical insights into these events were gained recently through the development of new in vitro and in vivo models, along with advances in live-cell and intravital microscopy. This review addresses the classes of “find-me” and “eat-me” signals presented by necrotic cells and their cognate receptors in phagocytes, which in most cases differ from the extensively characterized counterparts in apoptotic cell engulfment. The roles of damage-associated molecular patterns, chemokines, lipid mediators, and complement components in recruiting and activating phagocytes are reviewed. Lastly, the physiological importance of necrotic cell removal is emphasized, highlighting the key role of impaired debris clearance in autoimmunity.
Introduction
Cell death is inherent to living multicellular organisms. It is a key regulator of homeostasis, being required during development, growth and maintenance of tissues; it is also a turning point in the immune response. Healthy humans lose billions of cells per day constitutively via the process of apoptotic cell death. Apoptosis, the prototypical form of programmed cell death, was described morphologically in the early seventies (1) as involving cell shrinkage and chromatin condensation, followed by fragmentation of the entire cell into smaller, sealed apoptotic bodies. These apoptotic bodies are promptly cleared by neighboring phagocytes and parenchymal cells through phagocytosis, in this case termed efferocytosis (meaning “carrying to the grave”), without initiating an inflammatory response or disturbing tissue homeostasis.
While apoptosis has been studied most extensively, there are many other ways for cells to experience death. The intrinsic activity of organisms often puts them in contact with extreme temperatures, strong mechanical forces and harmful chemical agents. These situations frequently culminate in a catastrophic form of cell death with loss of plasma membrane integrity and pro-inflammatory properties named necrosis (2). Necrotic cell death can either be accidental or programmed (e.g., pyroptosis and necroptosis), leading to the release of intracellular contents into the extracellular environment. Necrosis differs qualitatively from apoptosis, which is clearly demonstrated by the lack of conversion of necrotic cells into apoptotic bodies, a process that requires enzymatic activity and energy. Importantly, these differences also predict that the means of clearance of the cell debris generated by necrosis vs. apoptosis may be drastically different.
Efferocytosis has received a great deal of attention in the past decades, and is by now a well-understood process involving dozens of described receptors and molecular effectors (Figure 1). Because of the profusion of studies, a casual reader may be left with the mistaken impression that efferocytosis is the only means of clearance of cell debris in the body. This is certainly not the case, as is most graphically shown by the existence of apoptosis-defective organisms, such as mice deficient in the initiator caspases 2 (3) and 9 (4), and effector caspases 3 (5), 6 (6), and 7 (7), that nevertheless develop and survive rather normally! Clearly, other mechanisms of cell death and debris clearance must exist. The main purpose of this chapter is to review the clearance of cell debris of necrotic origin. Parallels will be drawn between apoptosis and necrosis, stressing how each mode of cell death may produce different “find-me” and “eat-me” signals that will ultimately lead to clearance of debris by different cell types and phagocytic receptors. In addition, the immunological consequences of defective clearance of cell debris will be discussed: this can take the form of delayed tissue regeneration upon injury or even severe autoimmunity in the long-term. In collating the available information on necrotic cell clearance, this review aims to shed new light on diseases in which necrotic debris are central, such as in atherosclerosis, liver injury, arthritis, severe trauma, lupus, and many others.
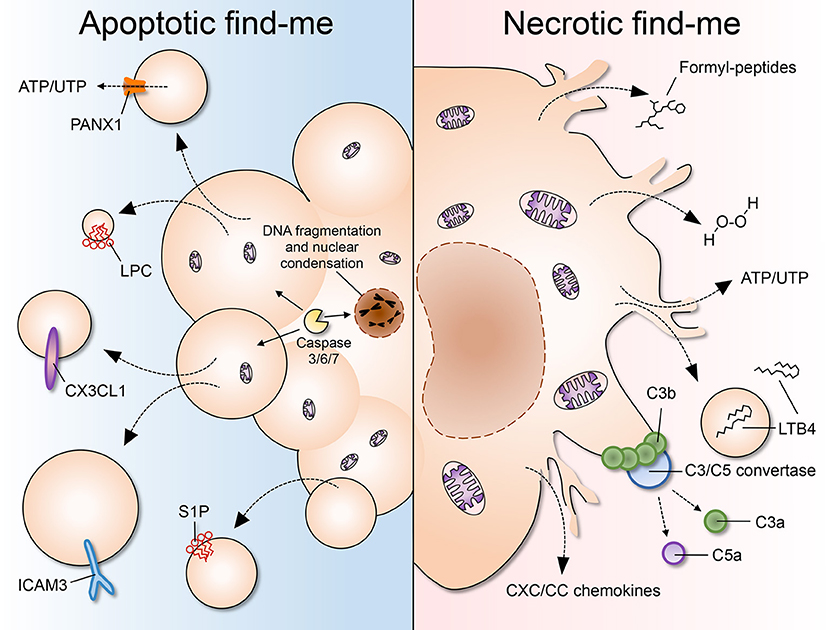
Figure 1. A comparison of apoptotic and necrotic “find-me” signals. (Left) Apoptosis is characterized by cell shrinkage, membrane blebbing, DNA fragmentation and nuclear condensation. As cells undergo apoptosis, “find-me” signals such as lysophoshatidylcholine (LPC), CX3CL1, ICAM3, and sphingosine 1-phosphate (S1P) are secreted, exposed on the outer leaflet of the plasma membrane, and/or released via apoptotic bodies or exosomes. Pannexin 1 (PANX1) is an important membrane channel involved in formation of membrane protrusions and ATP/UTP release during apoptosis. LPC, lysophosphatidylcholine; S1P, sphingosine-1-phosphate. (Right) Necrosis is considered to be an uncontrolled form of cell death characterized by nuclear and organellar swelling, plasma membrane rupture and leakage of intracellular contents, which many fall into the category of damage-associated molecular patterns (DAMPs or danger signals). “Find-me” signals released by necrotic cells include mitochondria-derived formylated peptides, as well as molecules released from the cytosol such as H2O2, ATP/UTP, leukotriene B4 (LTB4), and CXC/CC chemokines. LTB4 can also be released via sealed extracellular vesicles. The chemotactic complement components C3a and C5a are generated after complement activation on the surface of necrotic cells.
Apoptosis and Efferocytosis
Approximately 200 billion cells undergo turnover (ostensibly by apoptosis) every day in the human body (8). Yet, few apoptotic cells are found in the steady state in healthy humans, suggesting that these cells are rapidly cleared. In order to orchestrate efferocytosis, three main signaling programs are required. First, chemotactic “find-me” signals are produced to attract professional phagocytes toward the dying cell. Second, “eat-me” signals appear on the surface of the apoptotic cell, which will help phagocytes recognize and engulf it. Lastly, the internalized apoptotic body is degraded in the phagolysosomal compartment by proteases, DNAses and lipases.
During apoptosis, cellular components are modified by the activity of caspases and packaged into sealed vesicles—the so-called apoptotic bodies—that expose phosphatidylserine (PS) (9). The activation of initiator caspases (2, 8–10) leads to the cleavage-dependent activation of the effector caspases (3, 6, 7), which, being promiscuous proteases, cause the widespread cleavage of proteins in the cell (10). This, in turn, promotes the degradation of nuclear and cytoskeletal proteins and the activation of accessory enzymes, such as the caspase-activated DNAse (CAD), that degrades chromosomal DNA (11). The concomitant cleavage of nuclear scaffold proteins such as lamins leads to nuclear fragmentation (12), while proteolysis of actin, fodrin, and gelsolin (13) causes cell shrinkage and membrane blebbing. In addition, caspase activation is central to drive PS exposure on the outer leaflet of the plasma membrane, a key event in apoptotic cell recognition and clearance (14). Thus, caspase activity is largely accountable for the morphological and biochemical hallmarks of apoptosis, including the auto-digestion of cellular components and the generation of “find-me” and “eat-me” signals. Caspases can be activated when proteases that are normally secreted are released into the cytosol. For example, neutrophil elastase induces the unfolded protein response in vascular endothelial cells, promoting apoptosis via caspase-3/7 activation (15). The best-characterized apoptotic “find-me” signals are the nucleotides ATP and UTP (16), the chemokine CX3CL1 (17), ICAM3 (18), and the lipids lysophosphatidylcholine (LPC) (19) and sphingosine 1-phosphate (S1P) (20). Interestingly, these signals can be released as soluble mediators or become exposed on the surface of apoptotic microparticles, which detach from the main apoptotic body and are capable of diffusing in the extracellular environment (21).
Apoptotic bodies are easily engulfed by leukocytes (professional phagocytes) and are cleared from the tissue without any inflammatory impact. This process depends largely on the exposure of PS on the outer leaflet of the membrane, an evolutionary conserved “eat-me” signal for apoptotic cells. PS is recognized by a plethora of receptors, including TIM-1, TIM-3, TIM-4, BAI1, MerTK, and the stabilins 1 and 2, which will cause internalization of the apoptotic bodies by phagocytes (2, 22–25). Also, scavenger receptors such as CD36, might be able to interact directly with exofacial PS due to its negative charge (26). In addition to direct receptor-binding to PS, several soluble molecules were described to bridge phagocyte receptors to the phospholipid. They may originate from the phagocyte, the dying cell or the interstitial fluid. Examples of phagocyte-derived bridging molecules include milk fat globule EGF factor 8 (MFG-E8), developmental endothelial locus 1 (Del-1), growth arrest-specific 6 (Gas6), protein S and the complement factor C1q. Bridging proteins interact with PS via different PS-binding domains. For example, MFG-E8 secreted by macrophages and immature dendritic cells binds to PS on apoptotic cells via its Ca2+-independent discoidin-like C2 domain, while interacting with ανβ3/5 integrins on the phagocyte membrane, resulting in cell engulfment (27, 28). In contrast, Gas6 and protein S bind PS via their γ-carboxyglutamic acid (Gla) domain. Unlike the discoidin-like C2 domain, binding of the Gla domain to PS requires Ca2+, in this way promoting apoptotic cell internalization (29, 30). C1q binds to apoptotic cells via its cationic globular head, and interacts with calreticulin-CD91 on phagocytes to promote efferocytosis (31, 32). Another phagocyte-derived protein, Annexin A1, can be translocated to the plasma membrane to interact with PS exposed on the apoptotic cell target, and this may contribute significantly to the anti-inflammatory effects of apoptotic cell clearance (33).
Complement C1q and additional bridging molecules such as IgM and collectins were proven to bind to “defects” in the plasma membrane of the apoptotic cell, including the presence of phosphatidylcholine, phosphatidylethanolamine, lyso-phospholipids, carbohydrates, and DNA (34). Collectins, such as mannose-binding lectin (MBL) and complement C1q, bind late apoptotic cells and also drive engulfment via interaction with CD91 and calreticulin on the macrophage in vitro. Calreticulin is an endoplasmic reticulum (ER)-localized chaperone that normally facilitates folding and quality control of N-glycosylated proteins. As cells undergo apoptosis, calreticulin escapes and translocates to the plasma membrane, where it acts as an “eat-me” signal that is recognized by CD91 on phagocytes (35). In addition, a variety of other receptors and adaptor molecules have been reported to contribute to efferocytosis. These include Fcγ receptors, β2-glycoprotein I receptor, lectins, CD14, ABC transporters, scavenger receptors, and complement components [reviewed in (31, 36–42)]. Together this indicates that there is marked redundancy in receptors and ligands for the engulfment of apoptotic cells.
Interestingly, although PS exposure is a hallmark of apoptosis, forced PS exposure on viable cells does not trigger internalization (43). This is due to the presence of “don't eat-me” signals on viable cells, including CD31, CD46, CD47, and CD61, which disable target cell engulfment. The downregulation of “don't eat-me” signals, such as CD47 and its binding partner SIRPα, contributes to internalization of apoptotic bodies, indicating that a coordinated effort between the dying cell and the phagocyte likely exists (44).
Non-Apoptotic Cell Death
Necrosis is generally considered to be a drastic and uncontrolled form of cell death, characterized morphologically by nuclear and organellar swelling (oncosis) and plasma membrane rupture (Figure 1) (10). Due to the loss of membrane integrity, the intracellular contents are spilled out by the dying cell. The exposure of necrotic cell content (or debris) is abrupt and lacking in processing, causing it to be released in a disorderly fashion into the tissue, without the specific cues of its apoptotic counterpart. This causes necrotic debris to be potent inducers of inflammation, through activation of pattern-recognition receptors such as toll-like receptors (TLRs), NOD-like receptors (NLRs), and C-type lectins (CLECs), among others. Generally, necrotic cell death is problematic to tissues, prompting the need for immediate removal of debris, delaying the regeneration required after injury and sustaining collateral inflammatory damage. Interestingly, the recent identification of signaling pathways that are activated before and during necrosis have prompted reconsideration of this type of cell death as multiple, distinct types of events, at least some of which are tightly regulated and not always accidental. Recent subclassifications of necrosis include pyroptosis, necroptosis, parthanatos, ferroptosis, oxytosis, ETosis, and secondary necrosis (45).
Pyroptosis is a type of necrotic cell death caused by extensive inflammasome activation. It occurs in cell types that express inflammasome components, such as macrophages, upon infection (e.g., with the intracellular pathogen Salmonella typhimurium) or LPS treatment (46). Pyroptosis is caused by the formation of gasdermin D pores, which assemble at the plasma membrane after proteolytic processing of their precursor by inflammasomes containing activated caspase-1 or -11 (47, 48). Gasdermin pores create a path for the release of IL-1β, but secondarily cause cell lysis by excessive permeabilization of the plasma membrane. Necroptosis differs from other modalities of necrosis by the involvement of receptor-interacting protein kinase 1 (RIPK1) and RIPK3, which recruit and phosphorylate the mixed lineage kinase domain-like protein MLKL (45). Subsequently, MLKL oligomerizes, translocates to the inner leaflet of the plasma membrane and promotes membrane permeabilization and cell death (49, 50). Curiously, necroptosis requires the inhibition of caspase-8, which otherwise causes the cells to die by apoptosis. This may restrict the relevance of this death pathway in vivo, since caspase-8 inhibition may only occur in some viral infections (51). Parthanatos is a necrotic mode of cell death that depends on poly(ADP-ribose) polymerase proteins (PARPs). PARPs are typically activated by DNA breaks from ultraviolet light and by alkylating agents (52). By causing poly (ADP-ribosyl)ation of target proteins, PARPs may deplete cells of NAD+ and consequently of ATP, causing necrotic cell death.
Ferroptosis is the necrotic cell death induced by iron-dependent oxidative stress (53). It was postulated that iron catalyzes the lipid peroxidation triggered by the ferroptosis-inducing molecules erastin and RSL3, or by inhibiting the glutamate/cystine antiporter. It was later found that these pathways converge on the reduction of intracellular glutathione (GSH) levels and impaired GSH peroxidase 4 (GPX4) activity, leading to the accumulation of lipid-based reactive oxygen species and cell death (54). As expected, iron-chelators and lipophilic antioxidants were found to be potent inhibitors of ferroptosis (55). A related oxidative stress-dependent necrotic cell death, oxytosis, involves GSH depletion, 12-lipoxygenase activation and opening of cGMP-gated channels on the plasma membrane (56). This leads to calcium influx and activation of the calpain-cathepsin cascade, causing lysosome membrane permeabilization and necrosis (57).
In contrast to the “passive” nature of classical necrosis, one of the necrotic death pathways involves purposeful intracellular content exposure. Since its description in 2004 by Zychlinsky and collaborators (58) (neutrophil) extracellular traps (ETs), which consist primarily of extruded DNA, have been studied extensively. Subsequent work determined that cells may die during ET production, a process dubbed NETosis (59, 60). NETosis was later shown to occur in several cell types other than neutrophils, such as monocytes (61), mast cells (62), and eosinophils (63), making the name ETosis more appropriate. Generally, ETosis requires NADPH oxidase-dependent reactive oxygen species production, leading to chromatin decondensation, nuclear disruption and release of chromatin complexed with granular/cytoplasmic proteins (59), although the mechanisms underlying the process may vary between cell types.
Necrosis may also take place even after apoptosis has occurred. If apoptotic cells are not cleared in a timely fashion, the apoptotic bodies may decay and lose plasma membrane integrity, leaking their contents in a similar manner as a primary necrotic cell would (64). This event is named secondary necrosis and it shares common features with both apoptotic and necrotic cell death. The intracellular debris produced by secondary necrosis undergo apoptotic caspase processing, causing it to be qualitatively different from primary necrotic debris (65). For instance, secondary necrotic debris are considerably smaller, contain digested chromatin, prostaglandin E2 and high levels of uric acid, but very low ATP levels (65). These change drastically the manner by which the organism deals with the debris, as exemplified by the higher efficiency of complement C1q and DNAse I in degrading chromatin from secondary necrotic cells (66) and the potent anti-inflammatory polarization of macrophages elicited by C1q-covered late apoptotic debris (67).
Necrotic “Find-me” Signals and Their Receptors
In contrast to apoptotic “find-me” signals, necrotic cells may not have enough time or energy to process their own signals. A myriad of molecules has been shown to be released by dying cells, many of which fall into the category of damage-associated molecular patterns (DAMPs): bona fide cellular components that are normally concealed inside the cell, but that become exposed to the extracellular environment upon cell damage or death. Some well-established DAMPs include mitochondria-derived N-formylated peptides, DNA and RNA, the nuclear protein HMGB1, histones, actin, calcium-binding S100 proteins, heat-shock proteins (HSPs), ATP and uric acid, among many others (68, 69). Necrotic cells may also release pre-stored inflammatory mediators, such as IL-1α, IL-33, and chemokines, which may directly or indirectly recruit phagocytes to the vicinity. In addition, the occurrence of necrosis and the consequent exposure of “unusual” molecules promptly activates the proteolytic cascades of complement and coagulation. The activation of complement on necrotic debris can itself generate several “find-me” signals, including the powerful chemoattractant C5a. Below, we discuss established necrotic “find-me” signals. After reading this section the reader may agree that neutrophils and monocytes respond to a “complex pool of exogenous signals, of which no single cue is absolutely required for migration” (70).
Formyl-Peptides
Formyl-peptides (or N-formylated peptides) are classically generated in the course of bacterial protein synthesis, which is initiated by N-formyl-methionine residues. Mitochondria, sharing the bacterial ancestry, initiate protein synthesis similarly, thus creating a formylated protein reservoir inside the eukaryotic cell. Upon necrosis, the release and cleavage of mitochondrial proteins produces formyl-peptides, causing massive leukocyte activation and recruitment in a variety of necrotic states (71, 72).
Formyl-peptides are powerful chemoattractants. The most used analog, fMLP, activates neutrophils in the picomolar range (73, 74). It is considered an end-target chemoattractant, which bypasses the signaling of intermediate chemotactic molecules such as CXCL8 (IL-8) and LTB4 (75). Formyl-peptides bind FPR1, FPR2, and FPR3 receptors, although the classical chemotactic effects are mainly mediated by FPR1 activation. Whereas, FPR1 and FPR2 are expressed in several cell types, especially neutrophils and macrophages, FPR3 is much less understood (76). Upon ligand binding, FPR1, induces multiple intracellular signaling pathways: Gα activation signals via the MAPK pathway and the small GTPases CDC42 and RAC to stimulate migration and phagocytosis; Gβγ transduce signals via PI3Kγ and PLCβ to stimulate superoxide production and transcriptional regulation in phagocytes (77).
The role of formyl-peptides as a necrotic “find-me” signal is firmly established in the literature. In a seminal paper where McDonald et al. (71) used focal thermal injury of the liver, FPR1 activation of neutrophils was the key step required for migration into the necrotic area. The neutrophils initially traveled to the liver stimulated by an intravascular gradient of CXC chemokines. Upon reaching the edge of the necrotic site, the neutrophils switched to a FPR1-dependent migration mode, presumably chasing formyl-peptides emanating from the mitochondria of necrotic cells. Furthermore, the dependence of neutrophils on formyl-peptide gradients for recruitment to necrotic sites was confirmed in clinically-relevant disease models of drug-induced liver injury (78) and liver ischemia-reperfusion (79). Interestingly, when the necrotic injury is extensive enough, as in severe trauma (crushes, fractures, burns) or acute liver failure, the formyl peptides can be released in such a significant amount that they cause systemic inflammation, affecting lung function in mice and humans (72, 78).
Though most studies have focused on neutrophil chemotaxis and activation by formyl-peptides, macrophages also express FPR1 and are sensitive to formyl-peptide stimulation. Human PBMCs produce significant amounts of CXCL8 in the presence of mitochondrial extracts containing formyl-peptides (80). Interestingly, the response is stronger when formyl-peptides are applied in conjunction with other stimulatory DAMPs such as HMGB1. This suggests that formyl-peptides may indirectly recruit phagocytes to necrotic sites by inducing the production of additional chemoattractants (CXCL8) by resident macrophages. Formyl-peptides induce monocyte recruitment in vitro (81, 82), however, the relevance of FRP1 signaling in monocyte recruitment to necrotic sites in vivo remains elusive.
Importantly, questions about formyl-peptides as necrotic “find-me” signals remain. For example, the mechanism by which peptides are retained in necrotic areas in order to signal to leukocytes is unclear. Chemokines interact with glycosaminoglycans in order to form a gradient in the vasculature (83), but no mechanism has been proposed for the gradient formation of formyl-peptides. One should also consider that necrotic formyl-peptides may be very heterogeneous, varying in peptide length from a few to several amino acids. This may also impact the localization and agonistic activity of the formyl-peptides in vivo.
Chemokines
Chemokines are chemotactic cytokines that dictate the localization and mobilization of leukocyte populations in the organism (84). Chemokines are produced in a constitutive fashion and/or in response to stimuli such as those that activate pattern-recognition receptor (85). In the context of necrosis, chemokines play the roles of primary and secondary “find-me” signals, meaning that they can originate from both the dying cells and from healthy bystander cells. However, the multitude and promiscuity of chemokines and chemokine receptors adds a significant layer of complexity to the study of these mediators in vivo (84). CC and CXC chemokines can be released essentially by any cell type, including resident leukocytes (85). For instance, the chemokine CXCL1 can be produced by endothelial cells, pericytes, hepatocytes, macrophages, and fibroblasts (86–89). In addition, leukocytes can produce chemokines in an autocrine fashion, such as when neutrophils secrete CXCL2 during transendothelial migration (87) and Kupffer cells that release CCL2 during necrotic injury (90). Thus, there is an abundance of chemokine sources that can direct the migration of phagocytes to necrotic sites, reflecting the importance of chemokines as necrotic “find-me” signals.
The chemokines CXCL1 and CXCL2 (in mice) or CXCL8 (in humans), among others, have been known as powerful chemoattractants for neutrophils for decades (91). They activate CXCR1 and CXCR2 receptors to induce neutrophil polarization and migration, an effect strongly dependent on PI3Kγ signaling (92). CXC chemokines were shown to be the first signal guiding neutrophils to sites of focal necrosis in the liver (71) and suffice to induce neutrophil accumulation in zebrafish in vivo (93, 94). In contrast to formyl-peptides, the CXC chemokines were actually shown to form an intravascular gradient in the vicinity of necrotic areas, which is required for proper neutrophil recruitment to the injury site. These chemokine gradients are built on heparan sulfate proteoglycans expressed by the endothelium; they are long lasting and extend hundreds of microns from the site of injury (71, 94). This promotes the recruitment of patrolling neutrophils from the vasculature far from the original insult area. Moreover, CXCL1/CXCL2 signaling via CXCR1/CXCR2 receptors act in conjunction with formyl-peptides in the case of widespread hepatic necrosis (e.g., drug-induced liver injury), in which both pathways are required for maximal neutrophil recruitment to the interior of necrotic areas (95). Despite being considered redundant, evidence shows that CXCL1 and CXCL2 act on neutrophils sequentially to promote successful diapedesis and recruitment to inflamed muscle (87). Conversely, there is mounting evidence that neutrophils expressing lower levels of CXCR1 may transmigrate in reverse into the bloodstream (96). This has been corroborated by observations of neutrophil reverse migration (e.g., away from the site of necrosis and back into the bloodstream) in both zebrafish (97) and mice (98). This suggests that the role of CXC chemokines as “find-me” signals may be much more complex than anticipated, regulating initially the recruitment of neutrophils to necrotic areas and subsequently directing their egress back to the vasculature.
CC chemokines such as CCL2, CCL3, and CCL5 are notably active in cells of the monocytic lineage. Even though they are present in a variety of parenchymal and non-parenchymal cells, the CC chemokine receptors, especially CCR2, are highly expressed in monocytes and macrophages (85). Using transgenic mice, Dal-Secco et al. identified two different monocyte subsets that are recruited to necrotic sites: a classical pro-inflammatory CCR2hi-CX3CR1low population and an alternative patrolling CCR2low-CX3CR1hi population (99). They showed that CCR2hi monocytes migrate to the edge of the necrotic area after the initial wave of neutrophil recruitment, and this was dependent on CCR2 expression. The monocytes persisted in the necrotic area for days, where they transitioned in situ into a CX3CR1hi population. The reprogramming of the monocyte population was dependent at least partially on the cytokines IL-4 and IL-10, and was required for the timely resolution of the necrotic injury. Interestingly, CCL2 and CCL3 were found to be significantly increased in the necrotic liver of humans, correlating to a CCR2-dependent recruitment of CD68-positive monocytes to the necrotic areas (100).
Of note, chemokine receptors other than the ones mentioned above may control different leukocyte populations, playing roles that are still unclear in the context of necrotic debris. CXCR3 and its ligands CXCL9 and CXCL10 seem to control the population of NK and NKT cells, such that deficiency of CXCR3 causes a significant reduction of both cell populations in necrotic liver (101). Similarly, the chemokine CXCL12, known to control neutrophil egress from the bone marrow via CXCR4 (102) may control later events in tissue necrosis, such as re-vascularization (103).
Leukotriene B4 (LTB4)
LTB4 is a mediator derived from membrane phospholipids. The activation of cytosolic phospholipase 2 (cPLA2) initiates the cleavage of phospholipids to generate arachidonic acid. This fatty acid is used as substrate by the lipoxygenase 5-LOX to produce LTB4, among other intermediate eicosanoids. LTB4 is a powerful chemoattractant to neutrophils. It activates the BLT1/ LTB4R1 receptor, which, coupled to Gαi, stimulates neutrophil migration via Src-family kinases and Rho GTPases (104). Since LTB4 synthesis demands several enzymatic steps, it is unlikely to be released by necrotic cells as a DAMP. Instead, it can be produced in a matter of minutes by leukocytes such as neutrophils, macrophages, and mast cells upon demand (104). However, it has been recently demonstrated that the enzymatic machinery for LTB4 synthesis can be localized to multivesicular bodies and secreted as exosomes in vitro (105). In this way, LTB4 may also be produced independently of the cell and travel in the aqueous environment concealed in exosomes, increasing its diffusion range and persistence in the tissue.
LTB4 is considered an “intermediate target” chemoattractant. Nevertheless, it is required for the rapid migration and concentration of neutrophils in focal necrotic sites, a phenomenon dubbed “neutrophil swarming” (106). LTB4 is produced by neutrophils recruited to necrotic foci in order to further amplify neutrophil recruitment to the area, forming the typical densely-populated clusters that are associated to neutrophil swarming. Neutrophil-derived LTB4 can act as a signal relay molecule (107) that is necessary for cell-cell communication to produce optimal aggregation of neutrophils at the injury site (106). Moreover, LTB4 was shown to act in conjunction with other necrotic “find-me” signals such as formyl-peptides and chemokines (106, 107), supporting the idea that there is no absolute necrotic “find-me” signal, but instead, a synergistic pool of signals that vary in chemotactic potency and range to mediate an integrated response.
The importance of LTB4 as a necrotic “find-me” signal has been confirmed in several models other than laser-induced focal skin injury. In spinal cord injury, LTB4-BLT1 signaling was required for the recruitment of neutrophils to the injury site (108). Interestingly, BLT1 knockout or pharmacological blockage of the receptor reduced neutrophil recruitment significantly, but did not alter monocyte recruitment to the injured spinal cord area. In the K/BxN model of inflammatory arthritis, inhibition of 5-LO led to a significant reduction of neutrophil migration to arthritic joints and amelioration of the disease (109). There, LTB4 was also produced locally by infiltrating neutrophils. In drug-induced liver injury, deficiency of 5-LO prevented mortality associated with acetaminophen overdose, which was correlated with reduced recruitment of phagocytes to the necrotic liver (110).
It is clear that LTB4 is an essential necrotic “find-me” signal to neutrophils. Yet, many questions pertaining its production and release still remain. For example, the transport of lipid mediators across membranes is still poorly defined, as is its mode of release from the nanoscopic exosomes. It would be interesting to assess whether exosomes are able to bind to the vasculature, perhaps stimulating leukocyte recruitment at long range. In addition, based on the role of LTB4 in the skin, one could wonder if it is especially relevant in tissues with abundant extracellular matrix, where it would be better retained and less prone to degradation.
Hydrogen Peroxide (H2O2)
H2O2 is a reactive oxygen species commonly generated in organelles such as mitochondria and phagosomes (111). The signaling capabilities of H2O2 are not limited to mammalian cells: it also serves as a major chemotactic signal in other species, such as zebrafish (Danio rerio) and Drosophila. Niethammer et al. showed formation of a H2O2 gradient minutes after wounding zebrafish, which extended up to 200 μm from the site of injury (112). The H2O2 was created by the activity of the enzyme Duox, a NADPH oxidase expressed in epithelial cells, and was necessary for rapid leukocyte recruitment to the injury site. In Drosophila, hemocytes also respond to H2O2 emanating from the wound (113). In this species, H2O2 was also derived from Duox and inhibition of the enzyme by siRNA knockdown or using diphenylene iodonium blocked the recruitment of hemocytes to the wound. Interestingly, it was later shown that wounding of tissues in zebrafish and flies causes a calcium wave across the tissue, which precedes and is responsible for the activation of Duox via its EF-hand motif, initiating the production of the H2O2 gradient (114).
Leukocytes must have a mechanism to sense this transient H2O2 gradient emanating from the injured cells. The redox sensor is seemingly the Src family kinase Lyn, which is activated by wound-derived H2O2 and mediates recruitment of neutrophils to injury sites in zebrafish (115). Oxidation of cysteine C466 by H2O2 activates Lyn, which in turn contributes to neutrophil migration toward the wound. Of interest, H2O2 is also chemotactic in murine (116) and human neutrophils (115), and Lyn is expressed in all mammalian leukocytes, with the exception of T cells (which nevertheless express related Src-family kinases). Thus, the role of H2O2 as a necrotic “find-me” signal spans several species and leukocyte types. Beyond the direct effects that H2O2 has on phagocyte recruitment to injury sites, it can also regulate the chemotactic responses to other “find-me” signals, such as fMLP, LTB4, and CXCL8 (117). Indeed, generation of reactive oxygen species by the NADPH oxidase at the leading edge of neutrophils is important to oxidize and inhibit the phosphoinositide phosphatase PTEN, maintaining high levels of PI(3,4,5)P3 at the leading edge and supporting the directional migration of neutrophils (118).
Purines
Nucleotides are among the earliest molecules released by damaged and dying cells (119). Nucleotide sensing occurs via P2Y and P2X receptor families, which are G protein-coupled receptors and nucleotide-gated ion channels, respectively. These receptors are numerous and vary in sensitivity to different nucleotides (e.g., ATP, ADP, UTP), but the majority of the studies have focused on the role of ATP and its degradation products. ATP is very abundant in the cytoplasm, ranging from 3 to 10 mM (120). When released actively or passively by cells, ATP is rapidly hydrolyzed by ectonucleotidases into ADP, AMP, and adenosine (119). Yet, despite its very short half-life outside the cell, ATP nevertheless has pivotal effects in leukocyte activation and migration.
Chen and collaborators demonstrated that neutrophils exposed to a gradient of fMLP release ATP at the leading edge of the cell, amplifying the chemotactic response to the formyl-peptide. This effect was mediated by ATP signaling via P2Y2 receptors and subsequently by activation of A3 receptors by adenosine derived from ATP hydrolysis (121). P2Y2 activation by ATP was also required for chemotaxis of human neutrophils toward CXCL8 (122), but in this case adenosine signaling did not play a role. Moreover, it was demonstrated that macrophages utilize the same autocrine ATP amplification loop to migrate toward C5a. Blockage of P2Y2 also impaired macrophage chemotaxis in vitro and in vivo (123). Clearly, purinergic signaling is involved in phagocyte migration to several stimuli, but this is not sufficient to characterize it as a chemotactic agent. Indeed, it was demonstrated that ATP itself is not directly chemotactic to macrophages. Instead, it induces lamellipodial extensions and chemokinesis (increased random displacement) (124), without directing the migration. These studies suggest an indirect effect of ATP, that though not acting as a chemoattractant, acts in an autocrine capacity in phagocytes to maximize the response to other chemoattractants, including fMLP and chemokines.
ATP was initially implicated as a “find-me” signal of apoptotic cells (16). The authors showed that ATP and UTP released during apoptosis were required for monocyte migration toward supernatant of apoptotic cells, in a P2Y2-dependent manner. Also, the migration of monocytes toward apoptotic cells in vivo was impaired in the absence of P2Y2. In necrotic injuries, the role of purinergic signaling is even more interesting. Applying focal necrotic injury to the liver, it was shown that ATP is required for invasion of peritoneal macrophages into the necrotic area (125). Curiously, the peritoneal macrophages, which can be found floating in the peritoneal fluid, took this avascular route to the necrotic site by recognizing ATP released from the dead cells, which prompted the macrophages to arrest at that site. The use of apyrase (to degrade ATP and ADP) and P2X7 blockage reduced significantly the infiltration of macrophages from the peritoneum to the injury site. In the case of focal injury of the brain, the extension of microglial processes to the area of injury was also found to be mediated by ATP (126). The rapid convergence of microglial extensions to the necrotic site took place without displacement of the main cell body and was dependent on ATP and P2Y receptors. Uderhardt and collaborators found a similar response of peritoneal macrophages to a focal necrotic injury. Sessile resident macrophages extended membrane processes toward dead cells in order to cloak the debris from patrolling neutrophils, thereby minimizing inflammation (127). The extension of the macrophage processes could be blocked by apyrase or joint inhibition of P2X and P2Y receptors, indicating an elevated redundancy in purinergic signaling. In zebrafish, wounding causes ATP release and P2Y receptor activation, which in turn activates Duox to produce H2O2, recruiting phagocytes to the injury site (128). In this species, the effects of the nucleotide are not limited to phagocytes, as ATP is also involved in rapid wound closure by stimulating epithelial cell motility (129).
It is important to highlight the differences in the function of P2Y and P2X receptors. As mentioned, P2Y receptors, especially P2Y2, have been implicated in regulating the migration of phagocytes to diverse necrotic “find-me” stimuli. P2Y receptors are metabotropic, transducing signals via RhoA, Rac and PLCβ, leading to cytoskeletal rearrangement and increased intracellular calcium (130). P2X receptors, on the other hand, being ion channels activated by nucleotides, signal in a fundamentally different way. A classic example is the role of P2X7 in inflammasome activation in macrophages. In this instance, P2X7 mediates K+ efflux from cells stimulated by ATP, a major step in the activation of the inflammasome complex and caspase-1 that eventually culminates in the release of interleukin-1β (IL-1β). IL-1β is able to prime the production of several other chemotactic agents such as chemokines and lipid mediators, but like P2X7, lacks intrinsic chemotactic activity.
Complement
The complement system comprises an evolutionary ancient set of fluid-phase proteins and receptors, present in vertebrates and invertebrates. It is at the core of the immune system and mediates a cross-talk between innate and adaptive responses (131–134). The complement cascade is activated by a myriad of self and non-self molecules, which initiate the proteolytic cleavage of complement proteins into fragments that deposit onto the target or are released into the extracellular fluid to signal to neighboring cells and leukocytes.
Necrotic debris can initiate complement activation through all 3 pathways: classical, alternative and lectin (133). Exposure of intracellular components such as DNA or mitochondria activate complement directly (135, 136), and natural IgM and IgG autoantibodies can bind necrotic debris to initiate complement by the classical pathway (137). In addition, there are numerous adaptors and pattern-recognition receptors that detect necrotic debris and initiate the complement cascade by themselves, including mannose-binding lectin (MBL), pentraxins, ficolins, and histidine-rich glycoprotein (34). In the specific context of this section, the production of complement C3a and C5a fragments (anaphylatoxins) is central, since these stimulate chemotaxis of leukocytes (138). Their respective receptors, C3aR and C5aR, that are expressed primarily in myeloid cells, are G protein-coupled receptors signaling via PI3K activation, MAPK activation and intracellular calcium mobilization (139).
The activation of complement on dying or necrotic cells, measured by the deposition of C3b/iC3b, has been demonstrated in several tissues, including the liver (140–142), muscle (143, 144), brain (145, 146), joint (147, 148), and intestines (149, 150). The presence of complement deposited on damaged tissues was already strong indication that C3a and C5a were being generated, but subsequent studies focused on the exact role of each fragment in disease. In muscle injury induced by cardiotoxin, it was shown that complement is activated via the alternative pathway (spontaneously), and that C3a-C3aR signaling was required for monocyte migration to the tissue (143). Deficiency in C3aR reduced the recruitment of monocytes to the injured muscle significantly, although neutrophil migration was unaffected. Moreover, C5aR was not required for the migration of either monocytes or neutrophils to the muscle, indicating specificity of C3a activity in this setting. In liver injury by ischemia/reperfusion, neutrophil migration requires complement activity. C5a is produced early during injury and formation of the complement membrane attack complex (MAC) plays an additional role in amplifying neutrophil recruitment, likely via release of IL-1β and additional DAMPs (140).
Complement inhibition in intestinal ischemia/reperfusion injury, a severe model of intestinal damage, also minimizes neutrophil recruitment and disease severity (149, 150). Interestingly, whilst complement inhibition presumably inhibited the generation of C3a and C5a in the injured intestine, it also inhibited the production of another chemoattractant, LTB4. This shows again a synergistic relationship between different classes of “find-me” signals, acting simultaneously or sequentially to guide leukocyte recruitment to necrotic debris. In the joints, the synovium is a site of both synthesis and deposition of complement (131). The alternative complement pathway plays a major role in the pathogenesis of arthritis from the initiation phase (when synoviocytes can be damaged directly by complement) to the chronic inflammatory stage (147, 148). Importantly, both C3aR and C5aR are required for the recruitment of neutrophils and macrophages to the damaged joint (148), showing yet again a degree of redundancy in the role of anaphylatoxins in phagocyte recruitment to the joint. Altogether, there is abundant evidence that complement by-products are released during necrosis and that they play a role in attracting phagocytes to injury sites.
Necrotic “eat-me” Signals and Their Receptors
Apoptotic cells have to be cleared quickly and efficiently to prevent secondary necrosis, which would lead to the release of intracellular components and inflammation. Similarly, cells dying from primary necrosis need to be removed efficiently, as they could be a detrimental source of autoantigens and may trigger excessive inflammation (Figure 2). In the case of necrotic debris, the mechanism of recognition by professional phagocytes is not fully understood. As the necrotic cell can be disintegrated into small debris, it has been suggested that engulfment of necrotic cells resembles macropinocytosis, in which macrophages develop membrane ruffles which protrude around the target material (151–153). As reviewed briefly above, multiple receptors implicated in the clearance of apoptotic debris have been described. By contrast, much less is known about the receptors and ligands involved in the uptake of necrotic debris. Remarkably, as the evidence emerges, it is becoming apparent that some necrotic “eat-me” ligands overlap with the equivalent apoptotic signals. For example, necrotic cells also expose PS, although the mechanism underlying such exposure differs drastically. In line with this, many of the molecules that bridge PS for efferocytosis (complement, collectins and pentraxins) have also been shown to bind necrotic cells. Nevertheless, there are “eat-me” signals that apply uniquely to necrotic debris. For instance, complement C1q deposition represents a hallmark of necrotic debris, but it is absent on apoptotic debris (154). Another distinguishing “eat-me” signal is annexin A1, which is translocated to the plasma membrane of necrotic cells to promote phagocytic uptake (155). Below, we will summarize and discuss necrotic “eat-me” signals, comparing and contrasting them to apoptotic “eat-me” signals.
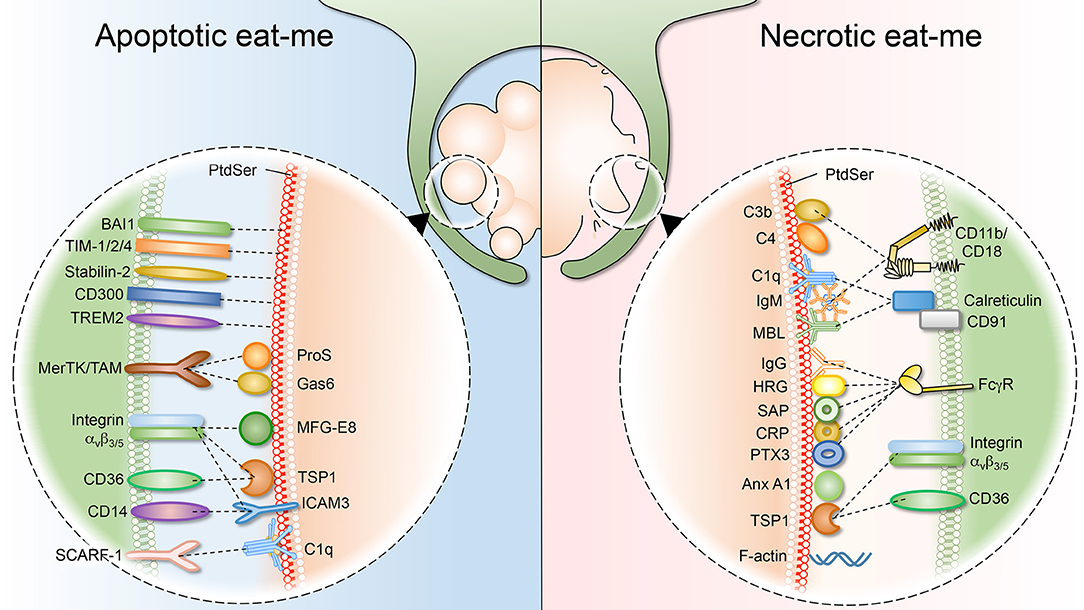
Figure 2. Apoptotic and necrotic “eat-me” signals, and their respective phagocytic receptors. (Left) As cells undergo apoptosis, they expose “eat-me” signals on their surface. The best studied eat-me signals for apoptotic cells is phosphatidylserine (PS). PS can either be bound directly by macrophage receptors such as BAI1, TIM 1/2/4, Stabilin-2, CD300, and TREM2. Alternatively, bridging molecules such as ProS, Gas6, MFG-E8, TSP1, and ICAM function to connect macrophage receptors (MerTK/TAM, integrin avβ3/5, CD36, and CD14) to the apoptotic surface. (Right) Necrotic cells also expose “eat-me” signals on their surface to engage professional phagocytes. Necrotic cells share some exposed “eat-me” signals, such as PS, with apoptotic cells, although the means of exposure likely differ. Other “eat-me” signals are unique to necrotic cells, such as deposition of C1q, MBL (Mannose-binding lectin), C3b, and C4 as well as IgG/IgM opsonization, and the subsequent involvement of integrin CD11b/CD18 and Fcγ receptors.
Complement
Opsonization of targets by complement components C1q, C3b, and C4 alerts phagocytes bearing complement receptors such as CR1, CR3, and CR4. Moreover, it is clear that a functioning complement system is required for efficient handling of dying and dead cells. Recent evidence points to a role of complement deposition in the clearance of late apoptotic/necrotic cells, rather than early apoptotic cells (154). In fact, most apoptotic cells are cleared while at an early apoptosis stage, when complement plays a minor role. It is only when apoptotic cells persist into late apoptotic/secondary necrotic stages that complement opsonization enhances recognition by phagocytes (156).
Phagocytosis of late apoptotic/necrotic Jurkat cells is impaired in individuals with deficiencies in C1q, C2, C3, or C4. In contrast, the MBL and alternative pathway did not participate in phagocytosis of debris, suggesting that opsonization by C3 fragments and the involvement of the classical pathway are mostly responsible for the clearance of necrotic cells (157). Similarly, complement components C3 and C4 bind immediately to necrotic peripheral blood lymphocytes. In contrast, irradiated lymphocytes undergoing apoptosis only displayed a weak binding of complement components for up to 2 days. At day 3, when secondary necrosis had ensued, C1q, C3b, and C4 all bound with higher affinity (154). Also, the clearance of necrotic cells is increased in presence of serum, and adding C1q to C1q-depleted serum markedly increased uptake of primary necrotic cells (158). Another study investigated complement deposition on viable, early apoptotic and late apoptotic (secondary necrotic) Jurkat cells (159). In this study, binding of C3 and C4 to early apoptotic cells was similar to that of viable cells, while secondary necrotic cells had a substantial binding of C3, C4, and at some extent C1q. The necrotic cells also bound IgM, and depletion of plasma IgM abolished most of the complement binding, supporting a role for the classical pathway of complement activation on late apoptotic/necrotic cell clearance (159).
Macrophages were shown to engulf apoptotic cells after C1q and MBL opsonization. Calreticulin released from dying cells bound macrophages via CD91/LDL receptor related protein 1, and was shown to recognize the collagen tails of C1q and MBL attached to the surface of the dying cell (31). Besides IgM, C1q binding to necrotic cells can be initiated via molecules of the acute-phase protein pentraxin family. The classical (short) pentraxins—including serum amyloid protein (SAP) and C-reactive protein (CRP)—are produced in the liver in response to IL-6 and play a role as opsonins by binding to cellular debris and late apoptotic cells (160, 161). The long pentraxin PTX3 is produced by hematopoietic and stromal cells as a response to a primary pro-inflammatory signal such as LPS, IL-1β, and TNF-α. PTX3 has multiple functions including complement activation on necrotic cells that results in cell clearance and reduced tissue damage (162). However, PTX3 can also limit excessive complement activation by promoting deposition of complement Factor H, a major inhibitor of the alternative pathway of the complement system. Under normal conditions, Factor H binds to self-surfaces, where it inactivates accidental C3b deposition on healthy cells. By directing Factor H to the surface of dying cells, PTX3 limits tissue damage while still increasing phagocytic clearance (163). Moreover, both human and mouse pentraxins recognize FcγRI and FcγRII, and binding of pentraxins to cellular surfaces results in phagocyte activation (164). In line with data suggesting complement deposition on late apoptotic/necrotic cells, the collectin MBL was found to bind to both late apoptotic and necrotic cells, but not early apoptotic cells. MBL binding initiated C4 deposition onto the necrotic cells and addition of C1q inhibited MBL opsonization of cells (165). Taken together, these observations imply that complement deposition and recognition function as a mechanism for the clearance of necrotic cells and as a backup for clearance of late apoptotic material undergoing secondary necrosis.
Phosphatidylserine
Although PS was long thought to be an exclusive marker of apoptosis, evidence is gathering that exposure of PS is also a hallmark of necrosis. In a study comparing internalization of apoptotic and necrotic cells, macrophages were shown to selectively engulf apoptotic and necrotic cells, while leaving living cells untouched. The engulfment of both apoptotic and necrotic cells was PS-dependent, suggesting that externalization of PS is a common trigger for the clearance of both types of cell debris (152). Annexin A5 (or Annexin V), commonly used as a marker of apoptosis due to its PS binding capacity, also binds to necrotic cells, supporting the occurrence of PS exposure during necrosis. Moreover, treatment with recombinant Annexin V inhibited phagocytosis of both apoptotic and necrotic cells by mouse macrophages, suggesting that PS exposure is required in both instances (152).
The difference in morphology between apoptotic and necrotic cells suggests that the mechanisms of PS exposure differ. PS exposure was observed in necrotic neurons of the nematode Caenorhabditis elegans, where it was facilitated by the homolog of the calcium-dependent scramblase TMEM16F and by CED-7, a member of the ATP-binding cassette (ABC) transporter family. However, rupture of the necrotic cells into particles can also readily account for exposure of PS without invoking specific externalization mechanisms. Zargarian et al. demonstrated that necroptotic cells also expose PS as an “eat-me” signal as phosphorylated MLKL translocates to the plasma membrane. The externalization of PS by necroptotic cells induced recognition and phagocytosis; they stained positive for Annexin A5 and exposed PS prior to overt permeabilization. The dying cells also released PS-exposing extracellular vesicles, thereby alerting neighboring cells of the impending cell death (166).
Although both apoptotic and necrotic cells expose PS, the efficiency of their clearance differs drastically: the engulfment of necrotic cells is considerably less effective, both quantitatively and kinetically. The mechanisms underlying this difference remain obscure, but down-regulation of “don't eat-me” signals in apoptotic, but not necrotic cells is a distinct possibility. Importantly, clearance of necrotic cells is carried out not only by phagocytes like macrophages, but also by non-professional phagocytes. In comparison to macrophages, engulfment by non-professional cells is slow and engulfment events were only detectable after 2.5 h. But, by taking up neighboring necrotic cells, non-professional cells remove a portion of the billions of cells that die daily during normal turnover (167).
Annexin A1
Annexin A1 was first believed to translocate to the surface of apoptotic cells, where it was proposed to function as a bridging protein that facilitates their phagocytic uptake (168, 169). However, this interpretation was recently revised, as it was demonstrated that annexin A1 rarely translocates in apoptotic cells; instead, its translocation to the cell surface is rather a hallmark of secondary necrosis (155). As proposed earlier for apoptosis, in necrotic cells annexin A1 is believed to function by bridging PS to the phagocyte surface to promote uptake. This interaction also dampens the secretion of pro-inflammatory cytokines by the macrophages that ingested the necrotic cell. This implies that clearance of necrotic debris can have anti-inflammatory effects. After translocation, annexin A1 is proteolytically cleaved at the cell surface by ADAM10, which generates a small peptide with chemotactic activity toward monocytes, thus generating a monocytic “find-me” signal for the necrotic debris (170).
Histidine-Rich Glycoprotein (HRG)
HRG is an abundant 75 kDa plasma glycoprotein that has a multi-domain structure known to interact with many ligands including Zn2+, heparin, heparan sulfate and plasminogen. HRG has been shown to function as an adaptor molecule that tethers plasminogen to glycosaminoglycan-bearing surfaces to regulate plasminogen activation (171). HRG was also demonstrated to distinguish between apoptotic cells and necrotic cells by binding to cytoplasmic ligands exposed by necrotic cells. This interaction, mediated by the amino-terminal domain of HRG, results in an opsonic function, encouraging the phagocytosis of the necrotic cell. In contrast, HRG does not opsonize apoptotic cells and thus, may play an important physiological role in the selective clearance of necrotic debris (34).
CD14
Initially, it was thought that the macrophage plasmalemmal glycoprotein CD14 was specific for recognition and clearance of apoptotic cells, as treatment with an anti-CD14 antibody reduced the phagocyte interaction with apoptotic but not necrotic cells (172). CD14 also recognizes LPS and it was initially thought its interaction with apoptotic cells occurs also via its LPS-binding domain, but this view was subsequently revised (173). Indeed, unlike LPS, binding of macrophages to apoptotic cells does not generate pro-inflammatory signaling. Later studies found a significant role for CD14 also in the clearance of necrotic cells. (158). ICAM-3 on the surface of dying cells may serve as the ligand recognized by CD14 (174).
Scavenger Receptors
Scavenger receptors were originally discovered by their capacity to recognize and remove modified lipoproteins. They are structurally diverse and recognize a variety of ligands, including DAMPs, oxidized PS and phosphatidylcholine (175). As PS is exposed on necrotic cells, this raises the possibility that PS-binding scavenger receptors may function as receptors for necrotic cells. SR-B1 and CD36 are class B scavenger receptors, and were the first cell surface receptors appreciated to recognize anionic phospholipids such as PS (26). CD36 for instance, which is highly expressed in macrophages, is involved in the phagocytosis of necrotic lymphocytes in vitro (158). Antibody blockage of CD36 caused a significant, yet partial, reduction in the macrophages ability to bind and internalize necrotic cells. Macrophages, dendritic cells and endothelial cells also express the scavenger receptor class F (SCARF1), that can recognize and engulf apoptotic cells via C1q (176). Given the earlier findings that C1q binds to late apoptotic/secondary necrotic cells, SCARF1 can potentially operate as a receptor for necrotic cells. In fact, loss of SCARF1 impaired uptake of dying cells, and SCARF1-deficient mice had accumulation of dying cells in tissues, leading to generation of autoantibodies to DNA-containing antigens and development of lupus-like disease (176).
Phagocyte-Independent Clearance of Necrotic Debris
During pregnancy, a large number of multinucleated fragments of dying syncytiothrophoblasts are shed daily into the maternal circulation. These trophoblasts, shed by the placenta, are rapidly cleared from the circulation by endothelial cells during normal pregnancy in order to prevent clogging of the maternal pulmonary circulation. Indeed, failure to clear such fragments often results in pre-eclampsia. Endothelial cells can internalize dying trophoblasts regardless of whether they are apoptotic or necrotic. However, while engulfment of apoptotic trophoblasts does not induce endothelial cell activation, phagocytosis of necrotic trophoblasts causes endothelial activation and ICAM-I expression (177).
The organism also counts with acellular routes for degradation of necrotic debris. It has been shown that serum components such as the nuclease DNAse I, the complement protein C1q and the protease plasmin act in synergy to degrade chromatin even in the absence of leukocytes. For instance, the binding of C1q to necrotic chromatin strongly enhances the activity of DNAse I, even though C1q lacks nuclease activity (178). It was postulated that C1q was able to enhance the access of DNAse I to necrotic DNA, improving the degradation of debris. Moreover, plasminogen was shown to penetrate necrotic cells, where it was activated into plasmin (179). The proteolytic activity of plasmin caused the cleavage of histone H1, which in turn facilitated the cleavage of DNA by DNAse I. The synergy between these enzymes is required for the fast and effective breakdown of necrotic chromatin.
“Don't Eat-me” Signals
Effective engulfment of dead cells entails not only the exposure of “eat-me” determinants, but requires a reduction of surface “don't eat-me” signals. Eukaryotic cells display CD47, a surface protein that is recognized by SIRPα, expressed by myeloid cells (44). CD47 functions by directly binding SIRPα on macrophages and monocytes, signaling inhibition of phagocytosis that is partly due to impaired myosin assembly at the phagocytic synapse (180).
Aging and the subsequent elimination of erythrocytes by efferocytosis correlates with a decrease in their surface CD47 (181). The importance of CD47 as a “don't eat-me” signal was demonstrated by Kojima and colleagues, who showed that dysregulation of CD47 signaling contributes to the development or atherosclerotic plaques. In this setting, instead of downregulating CD47, dying cells upregulated it, making apoptotic cells resistant to phagocytic clearance and thereby driving plaque formation. Interestingly, administration of a blocking CD47 antibody reversed this effect, stimulating efferocytosis and reducing atherosclerosis, making CD47 a potential drug target for the clinic (182).
CD47 can alter also the phagocytosis of necrotic debris. One explanation why necrotic debris are engulfed at a slower rate than apoptotic cells is that they have, comparatively, an increased surface expression of CD47. Moreover, CD47 was found to be clustered on necrotic cells, and these clusters stimulated RhoA-pMLC signaling in macrophages that promoted “nibbling” of the necrotic cells, rather than whole-cell internalization (183). This process—commonly known as trogocytosis—is shared by amoeba, lymphocytes, neutrophils and macrophages, and polarization of CD47 might explain the preferential nibbling over whole-cell engulfment.
CD46 is a widely expressed complement regulatory protein. It inhibits complement by binding C3b and C4b and acting as a cofactor for their proteolytic cleavage (184). CD46 is a “don't eat-me” signal that is lost during apoptosis and necrosis. In both types of dying cells, CD46 is clustered and shed in microparticles alongside nucleic acids and PS (185). The loss of CD46 correlates with an increase in deposition of C1q and C3b on the dying cells. However, only necrotic cells proceed to form membrane attack complexes, because they also undergo significant reduction in the expression of the complement regulators CD55 and CD59. This study indicated that the dying cells specifically lose “don't eat-me” signals that block complement activation in healthy cells, allowing them to be opsonized by complement and engulfed (185).
Several “don't eat-me” signals that have been implicated in apoptosis have not yet been investigated in the context of necrosis yet. CD31 (also known as platelet-endothelial cell adhesion molecule 1, PECAM-1) is an important “don't eat-me” signal, acting as a repulsive signal through homotypic CD31-CD31 interactions between cells. The ligation of CD31 on viable leukocytes promotes cell detachment. Apoptotic cells that lack CD31 bind tightly to leukocytes and are subsequently engulfed (186). Plasminogen activator inhibitor (PAI)-1, is a member of the serpin family of serine protease inhibitors. It appears to co-localize with calreticulin on viable neutrophils, where it is thought to impair signaling to macrophages. This impairment is lost during neutrophil apoptosis, suggesting that PAI-1 is a “don't eat-me” signal (187). Also, the urokinase receptor (uPAR), which normally plays a role in fibrinolysis, cell migration and adhesion, was shown to modulate efferocytosis (188). Macrophages from uPAR-deficient mice demonstrated enhanced ability to engulf viable neutrophils in vitro and in vivo. In line with this, expression of uPAR was reduced in apoptotic neutrophils compared to viable neutrophils, suggesting that uPAR is also a bona fide “don't eat-me” signal that is downregulated in apoptotic cells (189). Proteinase 3 (PR3) is a neutrophil granular protein that is co-externalized with PS during neutrophil apoptosis (190). PR3 impairs phagocytosis of apoptotic neutrophils by macrophages via inhibition of calreticulin function, a powerful “eat-me” signal. Another regulator is CD24, a heavily glycosylated GPI-anchored surface protein. It interacts with Siglec-10 in leukocytes to dampen inflammation in a variety of diseases (191). Recently, CD24 was described as a major “don't eat-me” signal exploited by tumor cells to evade the immune response (192). CD24 is overexpressed by a variety of tumor cells, inhibiting phagocytosis by neighboring tumor-associated macrophages, which express high levels of Siglec-10. Blockage of CD24 by a monoclonal antibody reduced tumor growth in vivo, suggesting that inhibition of this “don't eat-me” signal suffices to enable phagocytosis of live cancer cells.
Immune Consequences of Defective debris clearance
The generation of necrotic debris is a severe occurrence; yet, it is immediately met by a barrage of fluid-phase proteins, mediators and cells, which cause it to be essentially uneventful. Tissue inflammation resolves in a timely manner and immune responses against self-develop very rarely. However, if the organism fails to contain or clear the necrotic debris appropriately, tissue inflammation is prolonged and autoimmunity can ensue. Mutations that impair the ability of leukocytes to recognize or eliminate debris have been connected to defects in tissue regeneration and to diseases such as systemic lupus erythematosus (SLE). Below, we highlight a series of studies describing the catastrophic consequences of tampering with the response to necrotic cell death.
Inhibition of phagocyte recruitment or function at necrotic sites results in a clear defect in recovery from injury. Depletion of neutrophils prevents the clearance of debris from necrotic sites, leading to an impairment of regeneration and revascularization of the focal injury (98). Moreover, inhibition of monocyte recruitment to necrotic foci, whether due to CCR2 deficiency or to interference with their transition into CX3CR1+ cells delays the regeneration of the necrotic injury (99). Similar observations were made in complement-deficient models, reinforcing the notion that the removal of necrotic debris by phagocytes is paramount to tissue repair.
As described above, complement contributes “find-me” and “eat-me” signals to necrotic cells, and several studies have shown its major role in tissue regeneration (132, 140, 193–195). In the long term, defects in the complement cascade have been strongly associated to the development of SLE. Although a multi-factorial disease, SLE and related lupus-like syndromes are clearly connected to mutations or deficiency in C1q, C2, C3, and C4 complement factors (196). In addition, the disease has been associated to decreased expression of complement receptors CR1 and CR2 (133). Defects in complement activation, such as in the classical pathway, also yield organisms susceptible to autoimmunity, as is the case of IgM-deficient mice (197). Interestingly, a mutation of CD11b (ITGAM) has been correlated to the development of SLE as well (198). Phagocytes express high levels of CD11b, which is used as both complement receptor (CR3) and as adhesion molecule (Mac-1). Whether the polymorphism affects the phagocytic or adhesive functions of CD11b is still unclear, but the findings nevertheless provide further indication that impairment of the ability of phagocytes to clear debris causes immediate and long-term disadvantages to the host.
The exposure of extracellular DNA is a key factor in SLE development. The accumulation of DNA in tissues and bloodstream has to be rapidly counteracted by the activity of DNAses to minimize inflammation and autoimmunity. An abundant source of DNAse activity in the organism is serum, which contains two major nucleases, DNAse I and DNAse IL3 (199, 200). The two enzymes have non-redundant roles in DNA/chromatin degradation; DNAse I acts preferentially against internucleosomal “naked” DNA, whereas DNAse IL3 cleaves chromatin (protein-bound DNA) with high activity (201). DNAse I deficiency causes mice to develop anti-nuclear antibodies and SLE (200). DNAse IL3 deficiency is also sufficient to cause autoimmunity and SLE in mice (202, 203). In humans, mutations of DNAse I (204), DNAse IL3 (205) and DNAse III (TREX1) (206) have already been implicated in the incidence of SLE or lupus-like disease. Global deficiency in DNAse II, an isoform found in lysosomes, is embryonically lethal due to the accumulation of undigested DNA from red cell nuclei inside macrophages, which mount a type I interferon response that leads to embryonic demise (207). Interestingly, the lethality can be abrogated by simultaneously knocking out STING, a central adaptor of cytoplasmic DNA immunity (208), suggesting that leakage of undegraded debris from the phagosome to the cytosol fuels the deleterious response. The deficiency in DNAse II also correlates with worsening of heart failure (209) and development of polyarthritis in mice (210). It is clear that removal of necrotic debris is a multi-layered response: the phagocytes must be able not only to reach and ingest the debris, but also to effectively degrade it in order to avoid overt inflammation and autoimmunity.
Like DNA, actin is a very abundant component of cells and a DAMP conserved across species (211, 212). Necrotic cells expose actin after the plasma membrane integrity is breached, and the released actin is recognized by Clec9A (DNGR-1), a C-type lectin receptor expressed primarily in dendritic cells (211, 213). Clec9A specifically recognizes filamentous (F)-actin, which—remarkably—persists in necrotic cells even after their death. F-actin is able to bind Clec9A even when forming complexes with actin-binding proteins such as spectrin, α-actinin, and myosin II (213, 214). Importantly, the recognition of actin-rich necrotic debris by dendritic cell Clec9A prompted the cross presentation of self-antigens to CD8 T cells, a mechanism that explains how autoimmunity is initiated by the exposure of necrotic debris. Exposure of F-actin can be further regulated, as it is depolymerized by circulating DNase I. Full F-actin depolymerization requires ATP, which could be present as necrotic cells release ATP (described in section Purines) (215).
Concluding Remarks
Consideration of necrotic cells as an important, ongoing contributor to overall cell death provides a different vantage point of how debris are sensed and cleared and their contribution to inflammation and autoimmunity. Clearly, inhibiting the host's ability to eliminate and process necrotic debris has harmful effects. Strikingly, therapies for acute inflammation are largely confined to the use of anti-inflammatory drugs. In the short term, this approach reduces tissue inflammation and the associated symptoms (swelling, pain), but it comes at the cost of delayed resolution of injury. Prevention of inflammation retards debris clearance, re-growth of parenchymal cells and tissue regeneration. Thus, an alternative approach would be to stimulate debris clearance in addition to minimizing the uncomfortable symptoms. This can be accomplished by the application of pro-resolving mediators such as resolvins, lipoxins, hydrogen sulfide, IL-10, and annexin A1, which can stimulate clearance mechanisms without the damaging effects of excessive inflammation (216, 217).
Author Contributions
JW and PM prepared the figures. JW, SG, and PM wrote the manuscript.
Funding
JW was supported by The Swedish Society of Medicine and The Foundation Blanceflor Boncompagni Ludovisi, née Bildt. SG was supported by Canadian Institutes of Health Research grant FDN-143202. PM was supported by the Rega Foundation (Belgium), by Fonds Wetenschappelijk Onderzoek (FWO—Belgium), and by a Marie Sklodowska-Curie Individual Fellowship.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. (1972) 26:239–57. doi: 10.1038/bjc.1972.33
2. Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nat Rev Immunol. (2009) 9:353–63. doi: 10.1038/nri2545
3. Bergeron L, Perez GI, Macdonald G, Shi L, Sun Y, Jurisicova A, et al. Defects in regulation of apoptosis in caspase-2-deficient mice. Genes Dev. (1998) 12:1304–14. doi: 10.1101/gad.12.9.1304
4. Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, et al. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. (1998) 94:325–37. doi: 10.1016/S0092-8674(00)81476-2
5. Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, et al. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. (1996) 384:368–72. doi: 10.1038/384368a0
6. Zandy AJ, Lakhani S, Zheng T, Flavell RA, Bassnett S. Role of the executioner caspases during lens development. J Biol Chem. (2005) 280:30263–72. doi: 10.1074/jbc.M504007200
7. Lakhani SA, Masud A, Kuida K, Porter GA Jr, Booth CJ, Mehal WZ, et al. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science. (2006) 311:847–51. doi: 10.1126/science.1115035
8. Morioka S, Maueroder C, Ravichandran KS. Living on the edge: efferocytosis at the interface of homeostasis and pathology. Immunity. (2019) 50:1149–62. doi: 10.1016/j.immuni.2019.04.018
9. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. (2007) 35:495–516. doi: 10.1080/01926230701320337
10. Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. (2005) 73:1907–16. doi: 10.1128/IAI.73.4.1907-1916.2005
11. Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. (1998) 391:43–50. doi: 10.1038/34112
12. Rao L, Perez D, White E. Lamin proteolysis facilitates nuclear events during apoptosis. J Cell Biol. (1996) 135:1441–55. doi: 10.1083/jcb.135.6.1441
13. Kothakota S, Azuma T, Reinhard C, Klippel A, Tang J, Chu K, et al. Caspase-3-generated fragment of gelsolin: effector of morphological change in apoptosis. Science. (1997) 278:294–8. doi: 10.1126/science.278.5336.294
14. Segawa K, Nagata S. An apoptotic ‘eat me' signal: phosphatidylserine exposure. Trends Cell Biol. (2015) 25:639–50. doi: 10.1016/j.tcb.2015.08.003
15. Grechowa I, Horke S, Wallrath A, Vahl CF, Dorweiler B. Human neutrophil elastase induces endothelial cell apoptosis by activating the PERK-CHOP branch of the unfolded protein response. FASEB J. (2017) 31:3868–81. doi: 10.1096/fj.201700012R
16. Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. (2009) 461:282–6. doi: 10.1038/nature08296
17. Truman LA, Ford CA, Pasikowska M, Pound JD, Wilkinson SJ, Dumitriu IE, et al. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood. (2008) 112:5026–36. doi: 10.1182/blood-2008-06-162404
18. Torr EE, Gardner DH, Thomas L, Goodall DM, Bielemeier A, Willetts R, et al. Apoptotic cell-derived ICAM-3 promotes both macrophage chemoattraction to and tethering of apoptotic cells. Cell Death Differ. (2012) 19:671–9. doi: 10.1038/cdd.2011.167
19. Lauber K, Bohn E, Krober SM, Xiao YJ, Blumenthal SG, Lindemann RK, et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell. (2003) 113:717–30. doi: 10.1016/S0092-8674(03)00422-7
20. Gude DR, Alvarez SE, Paugh SW, Mitra P, Yu J, Griffiths R, et al. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J. (2008) 22:2629–38. doi: 10.1096/fj.08-107169
21. Poon IK, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol. (2014) 14:166–80. doi: 10.1038/nri3607
22. Kobayashi N, Karisola P, Pena-Cruz V, Dorfman DM, Jinushi M, Umetsu SE, et al. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity. (2007) 27:927–40. doi: 10.1016/j.immuni.2007.11.011
23. Nakayama M, Akiba H, Takeda K, Kojima Y, Hashiguchi M, Azuma M, et al. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood. (2009) 113:3821–30. doi: 10.1182/blood-2008-10-185884
24. Park SY, Jung MY, Lee SJ, Kang KB, Gratchev A, Riabov V, et al. Stabilin-1 mediates phosphatidylserine-dependent clearance of cell corpses in alternatively activated macrophages. J Cell Sci. (2009) 122:3365–73. doi: 10.1242/jcs.049569
25. Park SY, Jung MY, Kim HJ, Lee SJ, Kim SY, Lee BH, et al. Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Differ. (2008) 15:192–201. doi: 10.1038/sj.cdd.4402242
26. Rigotti A, Acton SL, Krieger M. The class B scavenger receptors SR-BI and CD36 are receptors for anionic phospholipids. J Biol Chem. (1995) 270:16221–4. doi: 10.1074/jbc.270.27.16221
27. Akakura S, Singh S, Spataro M, Akakura R, Kim JI, Albert ML, et al. The opsonin MFG-E8 is a ligand for the alphavbeta5 integrin and triggers DOCK180-dependent Rac1 activation for the phagocytosis of apoptotic cells. Exp Cell Res. (2004) 292:403–16. doi: 10.1016/j.yexcr.2003.09.011
28. Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a factor that links apoptotic cells to phagocytes. Nature. (2002) 417:182–7. doi: 10.1038/417182a
29. Anderson HA, Maylock CA, Williams JA, Paweletz CP, Shu H, Shacter E. Serum-derived protein S binds to phosphatidylserine and stimulates the phagocytosis of apoptotic cells. Nat Immunol. (2003) 4:87–91. doi: 10.1038/ni871
30. Ishimoto Y, Ohashi K, Mizuno K, Nakano T. Promotion of the uptake of PS liposomes and apoptotic cells by a product of growth arrest-specific gene, gas6. J Biochem. (2000) 127:411–7. doi: 10.1093/oxfordjournals.jbchem.a022622
31. Ogden CA, deCathelineau A, Hoffmann PR, Bratton D, Ghebrehiwet B, Fadok VA, et al. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp Med. (2001) 194:781–95. doi: 10.1084/jem.194.6.781
32. Paidassi H, Tacnet-Delorme P, Garlatti V, Darnault C, Ghebrehiwet B, Gaboriaud C, et al. C1q binds phosphatidylserine and likely acts as a multiligand-bridging molecule in apoptotic cell recognition. J Immunol. (2008) 180:2329–38. doi: 10.4049/jimmunol.180.4.2329
33. Scannell M, Flanagan MB, deStefani A, Wynne KJ, Cagney G, Godson C, et al. Annexin-1 and peptide derivatives are released by apoptotic cells and stimulate phagocytosis of apoptotic neutrophils by macrophages. J Immunol. (2007) 178:4595–605. doi: 10.4049/jimmunol.178.7.4595
34. Poon IK, Hulett MD, Parish CR. Molecular mechanisms of late apoptotic/necrotic cell clearance. Cell Death Differ. (2010) 17:381–97. doi: 10.1038/cdd.2009.195
35. Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. (2005) 123:321–34. doi: 10.1016/j.cell.2005.08.032
36. Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. (2002) 2:965–75. doi: 10.1038/nri957
37. Hart SP, Smith JR, Dransfield I. Phagocytosis of opsonized apoptotic cells: roles for ‘old-fashioned' receptors for antibody and complement. Clin Exp Immunol. (2004) 135:181–5. doi: 10.1111/j.1365-2249.2003.02330.x
38. Manfredi AA, Rovere P, Heltai S, Galati G, Nebbia G, Tincani A, et al. Apoptotic cell clearance in systemic lupus erythematosus. II. Role of beta2-glycoprotein I. Arthritis Rheum. (1998) 41:215–23.
39. Manfredi AA, Rovere P, Galati G, Heltai S, Bozzolo E, Soldini L, et al. Apoptotic cell clearance in systemic lupus erythematosus. I. Opsonization by antiphospholipid antibodies. Arthritis Rheum. (1998) 41:205–14.
40. Botto M, Dell'Agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, et al. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet. (1998) 19:56–9. doi: 10.1038/ng0598-56
41. Mevorach D, Mascarenhas JO, Gershov D, Elkon KB. Complement-dependent clearance of apoptotic cells by human macrophages. J Exp Med. (1998) 188:2313–20. doi: 10.1084/jem.188.12.2313
42. Savill J, Hogg N, Ren Y, Haslett C. Thrombospondin cooperates with CD36 and the vitronectin receptor in macrophage recognition of neutrophils undergoing apoptosis. J Clin Invest. (1992) 90:1513–22. doi: 10.1172/JCI116019
43. van den Eijnde SM, van den Hoff MJ, Reutelingsperger CP, van Heerde WL, Henfling ME, Vermeij-Keers C, et al. Transient expression of phosphatidylserine at cell-cell contact areas is required for myotube formation. J Cell Sci. (2001) 114:3631–42.
44. Hochreiter-Hufford A, Ravichandran KS. Clearing the dead: apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harb Perspect Biol. (2013) 5:a008748. doi: 10.1101/cshperspect.a008748
45. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. (2015) 517:311–20. doi: 10.1038/nature14191
46. Lieberman J, Wu H, Kagan JC. Gasdermin D activity in inflammation and host defense. Sci Immunol. (2019) 4:eaav1447. doi: 10.1126/sciimmunol.aav1447
47. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. (2015) 526:660–5. doi: 10.1038/nature15514
48. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. (2015) 526:666–71. doi: 10.1038/nature15541
49. Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. (2014) 16:55–65. doi: 10.1038/ncb2883
50. Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. (2014) 7:971–81. doi: 10.1016/j.celrep.2014.04.026
51. Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. (2014) 15:135–47. doi: 10.1038/nrm3737
52. Lonskaya I, Potaman VN, Shlyakhtenko LS, Oussatcheva EA, Lyubchenko YL, Soldatenkov VA. Regulation of poly(ADP-ribose) polymerase-1 by DNA structure-specific binding. J Biol Chem. (2005) 280:17076–83. doi: 10.1074/jbc.M413483200
53. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
54. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. (2014) 156:317–31. doi: 10.1016/j.cell.2013.12.010
55. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA. (2016) 113:E4966–75. doi: 10.1073/pnas.1603244113
56. Tan S, Schubert D, Maher P. Oxytosis: a novel form of programmed cell death. Curr Top Med Chem. (2001) 1:497–506. doi: 10.2174/1568026013394741
57. Yamashima T. Ca2+-dependent proteases in ischemic neuronal death: a conserved ‘calpain-cathepsin cascade' from nematodes to primates. Cell Calcium. (2004) 36:285–93. doi: 10.1016/j.ceca.2004.03.001
58. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
59. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. (2007) 176:231–41. doi: 10.1083/jcb.200606027
60. Steinberg BE, Grinstein S. Unconventional roles of the NADPH oxidase: signaling, ion homeostasis, and cell death. Sci STKE. (2007) 2007:pe11. doi: 10.1126/stke.3792007pe11
61. Webster SJ, Daigneault M, Bewley MA, Preston JA, Marriott HM, Walmsley SR, et al. Distinct cell death programs in monocytes regulate innate responses following challenge with common causes of invasive bacterial disease. J Immunol. (2010) 185:2968–79. doi: 10.4049/jimmunol.1000805
62. von Kockritz-Blickwede M, Goldmann O, Thulin P, Heinemann K, Norrby-Teglund A, Rohde M, et al. Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood. (2008) 111:3070–80. doi: 10.1182/blood-2007-07-104018
63. Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E, et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med. (2008) 14:949–53. doi: 10.1038/nm.1855
64. Silva MT. Secondary necrosis: the natural outcome of the complete apoptotic program. FEBS Lett. (2010) 584:4491–9. doi: 10.1016/j.febslet.2010.10.046
65. Sachet M, Liang YY, Oehler R. The immune response to secondary necrotic cells. Apoptosis. (2017) 22:1189–204. doi: 10.1007/s10495-017-1413-z
66. Liang YY, Arnold T, Michlmayr A, Rainprecht D, Perticevic B, Spittler A, et al. Serum-dependent processing of late apoptotic cells for enhanced efferocytosis. Cell Death Dis. (2014) 5:e1264. doi: 10.1038/cddis.2014.210
67. Fraser DA, Laust AK, Nelson EL, Tenner AJ. C1q differentially modulates phagocytosis and cytokine responses during ingestion of apoptotic cells by human monocytes, macrophages, and dendritic cells. J Immunol. (2009) 183:6175–85. doi: 10.4049/jimmunol.0902232
68. Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. (2010) 10:826–37. doi: 10.1038/nri2873
69. Westman J, Papareddy P, Dahlgren MW, Chakrakodi B, Norrby-Teglund A, Smeds E, et al. Extracellular histones induce chemokine production in whole blood ex vivo and leukocyte recruitment in vivo. PLoS Pathog. (2015) 11:e1005319. doi: 10.1371/journal.ppat.1005319
70. de Oliveira S, Rosowski EE, Huttenlocher A. Neutrophil migration in infection and wound repair: going forward in reverse. Nat Rev Immunol. (2016) 16:378–91. doi: 10.1038/nri.2016.49
71. McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. (2010) 330:362–6. doi: 10.1126/science.1195491
72. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. (2010) 464:104–7. doi: 10.1038/nature08780
73. Yuo A, Kitagawa S, Kasahara T, Matsushima K, Saito M, Takaku F. Stimulation and priming of human neutrophils by interleukin-8: cooperation with tumor necrosis factor and colony-stimulating factors. Blood. (1991) 78:2708–14. doi: 10.1182/blood.V78.10.2708.2708
74. Heit B, Tavener S, Raharjo E, Kubes P. An intracellular signaling hierarchy determines direction of migration in opposing chemotactic gradients. J Cell Biol. (2002) 159:91–102. doi: 10.1083/jcb.200202114
75. Heit B, Robbins SM, Downey CM, Guan Z, Colarusso P, Miller BJ, et al. PTEN functions to ‘prioritize' chemotactic cues and prevent ‘distraction' in migrating neutrophils. Nat Immunol. (2008) 9:743–52. doi: 10.1038/ni.1623
76. Weiss E, Kretschmer D. Formyl-peptide receptors in infection, inflammation, and cancer. Trends Immunol. (2018) 39:815–29. doi: 10.1016/j.it.2018.08.005
77. Dorward DA, Lucas CD, Chapman GB, Haslett C, Dhaliwal K, Rossi AG. The role of formylated peptides and formyl peptide receptor 1 in governing neutrophil function during acute inflammation. Am J Pathol. (2015) 185:1172–84. doi: 10.1016/j.ajpath.2015.01.020
78. Marques PE, Amaral SS, Pires DA, Nogueira LL, Soriani FM, Lima BH, et al. Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology. (2012) 56:1971–82. doi: 10.1002/hep.25801
79. Honda M, Takeichi T, Hashimoto S, Yoshii D, Isono K, Hayashida S, et al. Intravital imaging of neutrophil recruitment reveals the efficacy of FPR1 blockade in hepatic ischemia-reperfusion injury. J Immunol. (2017) 198:1718–28. doi: 10.4049/jimmunol.1601773
80. Crouser ED, Shao G, Julian MW, Macre JE, Shadel GS, Tridandapani S, et al. Monocyte activation by necrotic cells is promoted by mitochondrial proteins and formyl peptide receptors. Crit Care Med. (2009) 37:2000–9. doi: 10.1097/CCM.0b013e3181a001ae
81. Cockx M, Gouwy M, Ruytinx P, Lodewijckx I, Van Hout A, Knoops S, et al. Monocytes from patients with Primary Ciliary Dyskinesia show enhanced inflammatory properties and produce higher levels of pro-inflammatory cytokines. Sci Rep. (2017) 7:14657. doi: 10.1038/s41598-017-15027-y
82. Obrist R, Reilly R, Leavitt T, Knapp RC, Bast RC Jr. Monocyte chemotaxis mediated by formyl-methionyl-leucyl-phenylalanine conjugated with monoclonal antibodies against human ovarian carcinoma. Int J Immunopharmacol. (1983) 5:307–14. doi: 10.1016/0192-0561(83)90033-4
83. Kuschert GS, Coulin F, Power CA, Proudfoot AE, Hubbard RE, Hoogewerf AJ, et al. Glycosaminoglycans interact selectively with chemokines and modulate receptor binding and cellular responses. Biochemistry. (1999) 38:12959–68. doi: 10.1021/bi990711d
84. Zlotnik A, Yoshie O. The chemokine superfamily revisited. Immunity. (2012) 36:705–16. doi: 10.1016/j.immuni.2012.05.008
85. Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. (2014) 32:659–702. doi: 10.1146/annurev-immunol-032713-120145
86. Hol J, Wilhelmsen L, Haraldsen G. The murine IL-8 homologues KC, MIP-2, and LIX are found in endothelial cytoplasmic granules but not in Weibel-Palade bodies. J Leukoc Biol. (2010) 87:501–8. doi: 10.1189/jlb.0809532
87. Girbl T, Lenn T, Perez L, Rolas L, Barkaway A, Thiriot A, et al. Distinct compartmentalization of the chemokines CXCL1 and CXCL2 and the atypical receptor ACKR1 determine discrete stages of neutrophil diapedesis. Immunity. (2018) 49:1062–76 e6. doi: 10.1016/j.immuni.2018.09.018
88. Su L, Li N, Tang H, Lou Z, Chong X, Zhang C, et al. Kupffer cell-derived TNF-alpha promotes hepatocytes to produce CXCL1 and mobilize neutrophils in response to necrotic cells. Cell Death Dis. (2018) 9:323. doi: 10.1038/s41419-018-0377-4
89. Bigorgne AE, John B, Ebrahimkhani MR, Shimizu-Albergine M, Campbell JS, Crispe IN. TLR4-dependent secretion by hepatic stellate cells of the neutrophil-chemoattractant CXCL1 mediates liver response to gut microbiota. PLoS ONE. (2016) 11:e0151063. doi: 10.1371/journal.pone.0151063
90. Leifeld L, Dumoulin FL, Purr I, Janberg K, Trautwein C, Wolff M, et al. Early up-regulation of chemokine expression in fulminant hepatic failure. J Pathol. (2003) 199:335–44. doi: 10.1002/path.1298
91. Lindley I, Aschauer H, Seifert JM, Lam C, Brunowsky W, Kownatzki E, et al. Synthesis and expression in Escherichia coli of the gene encoding monocyte-derived neutrophil-activating factor: biological equivalence between natural and recombinant neutrophil-activating factor. Proc Natl Acad Sci USA. (1988) 85:9199–203. doi: 10.1073/pnas.85.23.9199
92. Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, et al. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science. (2000) 287:1049–53. doi: 10.1126/science.287.5455.1049
93. de Oliveira S, Reyes-Aldasoro CC, Candel S, Renshaw SA, Mulero V, Calado A. Cxcl8 (IL-8) mediates neutrophil recruitment and behavior in the zebrafish inflammatory response. J Immunol. (2013) 190:4349–59. doi: 10.4049/jimmunol.1203266
94. Sarris M, Masson JB, Maurin D, Van der Aa LM, Boudinot P, Lortat-Jacob H, et al. Inflammatory chemokines direct and restrict leukocyte migration within live tissues as glycan-bound gradients. Curr Biol. (2012) 22:2375–82. doi: 10.1016/j.cub.2012.11.018
95. Marques PE, Oliveira AG, Pereira RV, David BA, Gomides LF, Saraiva AM, et al. Hepatic DNA deposition drives drug-induced liver injury and inflammation in mice. Hepatology. (2015) 61:348–60. doi: 10.1002/hep.27216
96. Buckley CD, Ross EA, McGettrick HM, Osborne CE, Haworth O, Schmutz C, et al. Identification of a phenotypically and functionally distinct population of long-lived neutrophils in a model of reverse endothelial migration. J Leukoc Biol. (2006) 79:303–11. doi: 10.1189/jlb.0905496
97. Mathias JR, Perrin BJ, Liu TX, Kanki J, Look AT, Huttenlocher A. Resolution of inflammation by retrograde chemotaxis of neutrophils in transgenic zebrafish. J Leukoc Biol. (2006) 80:1281–8. doi: 10.1189/jlb.0506346
98. Wang J, Hossain M, Thanabalasuriar A, Gunzer M, Meininger C, Kubes P. Visualizing the function and fate of neutrophils in sterile injury and repair. Science. (2017) 358:111–6. doi: 10.1126/science.aam9690
99. Dal-Secco D, Wang J, Zeng Z, Kolaczkowska E, Wong CH, Petri B, et al. A dynamic spectrum of monocytes arising from the in situ reprogramming of CCR2+ monocytes at a site of sterile injury. J Exp Med. (2015) 212:447–56. doi: 10.1084/jem.20141539
100. Antoniades CG, Quaglia A, Taams LS, Mitry RR, Hussain M, Abeles R, et al. Source and characterization of hepatic macrophages in acetaminophen-induced acute liver failure in humans. Hepatology. (2012) 56:735–46. doi: 10.1002/hep.25657
101. Zaldivar MM, Berres ML, Sahin H, Nellen A, Heinrichs D, Schmitz P, et al. The chemokine receptor CXCR3 limits injury after acute toxic liver damage. Lab Invest. (2012) 92:724–34. doi: 10.1038/labinvest.2012.48
102. Martin C, Burdon PC, Bridger G, Gutierrez-Ramos JC, Williams TJ, Rankin SM. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity. (2003) 19:583–93. doi: 10.1016/S1074-7613(03)00263-2
103. Christoffersson G, Lomei J, O'Callaghan P, Kreuger J, Engblom S, Phillipson M. Vascular sprouts induce local attraction of proangiogenic neutrophils. J Leukoc Biol. (2017) 102:741–51. doi: 10.1189/jlb.1MA0117-018R
104. Kienle K, Lammermann T. Neutrophil swarming: an essential process of the neutrophil tissue response. Immunol Rev. (2016) 273:76–93. doi: 10.1111/imr.12458
105. Majumdar R, Tavakoli Tameh A, Parent CA. Exosomes mediate LTB4 release during neutrophil chemotaxis. PLoS Biol. (2016) 14:e1002336. doi: 10.1371/journal.pbio.1002336
106. Lammermann T, Afonso PV, Angermann BR, Wang JM, Kastenmuller W, Parent CA, et al. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature. (2013) 498:371–5. doi: 10.1038/nature12175
107. Afonso PV, Janka-Junttila M, Lee YJ, McCann CP, Oliver CM, Aamer KA, et al. LTB4 is a signal-relay molecule during neutrophil chemotaxis. Dev Cell. (2012) 22:1079–91. doi: 10.1016/j.devcel.2012.02.003
108. Saiwai H, Ohkawa Y, Yamada H, Kumamaru H, Harada A, Okano H, et al. The LTB4-BLT1 axis mediates neutrophil infiltration and secondary injury in experimental spinal cord injury. Am J Pathol. (2010) 176:2352–66. doi: 10.2353/ajpath.2010.090839
109. Chen M, Lam BK, Kanaoka Y, Nigrovic PA, Audoly LP, Austen KF, et al. Neutrophil-derived leukotriene B4 is required for inflammatory arthritis. J Exp Med. (2006) 203:837–42. doi: 10.1084/jem.20052371
110. Hohmann MS, Cardoso RD, Pinho-Ribeiro FA, Crespigio J, Cunha TM, Alves-Filho JC, et al. 5-lipoxygenase deficiency reduces acetaminophen-induced hepatotoxicity and lethality. Biomed Res Int. (2013) 2013:627046. doi: 10.1155/2013/627046
111. Nathan C, Cunningham-Bussel A. Beyond oxidative stress: an immunologist's guide to reactive oxygen species. Nat Rev Immunol. (2013) 13:349–61. doi: 10.1038/nri3423
112. Niethammer P, Grabher C, Look AT, Mitchison TJ. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature. (2009) 459:996–9. doi: 10.1038/nature08119
113. Moreira S, Stramer B, Evans I, Wood W, Martin P. Prioritization of competing damage and developmental signals by migrating macrophages in the Drosophila embryo. Curr Biol. (2010) 20:464–70. doi: 10.1016/j.cub.2010.01.047
114. Razzell W, Evans IR, Martin P, Wood W. Calcium flashes orchestrate the wound inflammatory response through DUOX activation and hydrogen peroxide release. Curr Biol. (2013) 23:424–9. doi: 10.1016/j.cub.2013.01.058
115. Yoo SK, Starnes TW, Deng Q, Huttenlocher A. Lyn is a redox sensor that mediates leukocyte wound attraction in vivo. Nature. (2011) 480:109–12. doi: 10.1038/nature10632
116. Klyubin IV, Kirpichnikova KM, Gamaley IA. Hydrogen peroxide-induced chemotaxis of mouse peritoneal neutrophils. Eur J Cell Biol. (1996) 70:347–51.
117. Hattori H, Subramanian KK, Sakai J, Jia Y, Li Y, Porter TF, et al. Small-molecule screen identifies reactive oxygen species as key regulators of neutrophil chemotaxis. Proc Natl Acad Sci USA. (2010) 107:3546–51. doi: 10.1073/pnas.0914351107
118. Kuiper JW, Sun C, Magalhaes MA, Glogauer M. Rac regulates PtdInsP(3) signaling and the chemotactic compass through a redox-mediated feedback loop. Blood. (2011) 118:6164–71. doi: 10.1182/blood-2010-09-310383
119. Burnstock G, Boeynaems JM. Purinergic signalling and immune cells. Purinergic Signal. (2014) 10:529–64. doi: 10.1007/s11302-014-9427-2
120. Junger WG. Purinergic regulation of neutrophil chemotaxis. Cell Mol Life Sci. (2008) 65:2528–40. doi: 10.1007/s00018-008-8095-1
121. Chen Y, Corriden R, Inoue Y, Yip L, Hashiguchi N, Zinkernagel A, et al. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science. (2006) 314:1792–5. doi: 10.1126/science.1132559
122. Kukulski F, Ben Yebdri F, Lecka J, Kauffenstein G, Levesque SA, Martin-Satue M, et al. Extracellular ATP and P2 receptors are required for IL-8 to induce neutrophil migration. Cytokine. (2009) 46:166–70. doi: 10.1016/j.cyto.2009.02.011
123. Kronlage M, Song J, Sorokin L, Isfort K, Schwerdtle T, Leipziger J, et al. Autocrine purinergic receptor signaling is essential for macrophage chemotaxis. Sci Signal. (2010) 3:ra55. doi: 10.1126/scisignal.2000588
124. Isfort K, Ebert F, Bornhorst J, Sargin S, Kardakaris R, Pasparakis M, et al. Real-time imaging reveals that P2Y2 and P2Y12 receptor agonists are not chemoattractants and macrophage chemotaxis to complement C5a is phosphatidylinositol 3-kinase (PI3K)- and p38 mitogen-activated protein kinase (MAPK)-independent. J Biol Chem. (2011) 286:44776–87. doi: 10.1074/jbc.M111.289793
125. Wang J, Kubes P. A reservoir of mature cavity macrophages that can rapidly invade visceral organs to affect tissue repair. Cell. (2016) 165:668–78. doi: 10.1016/j.cell.2016.03.009
126. Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. (2005) 8:752–8. doi: 10.1038/nn1472
127. Uderhardt S, Martins AJ, Tsang JS, Lammermann T, Germain RN. Resident macrophages cloak tissue microlesions to prevent neutrophil-driven inflammatory damage. Cell. (2019) 177:541–55 e17. doi: 10.1016/j.cell.2019.02.028
128. de Oliveira S, Lopez-Munoz A, Candel S, Pelegrin P, Calado A, Mulero V. ATP modulates acute inflammation in vivo through dual oxidase 1-derived H2O2 production and NF-kappaB activation. J Immunol. (2014) 192:5710–9. doi: 10.4049/jimmunol.1302902
129. Gault WJ, Enyedi B, Niethammer P. Osmotic surveillance mediates rapid wound closure through nucleotide release. J Cell Biol. (2014) 207:767–82. doi: 10.1083/jcb.201408049
130. Erb L, Weisman GA. Coupling of P2Y receptors to G proteins and other signaling pathways. Wiley Interdiscip Rev Membr Transp Signal. (2012) 1:789–803. doi: 10.1002/wmts.62
131. Holers VM. Complement and its receptors: new insights into human disease. Annu Rev Immunol. (2014) 32:433–59. doi: 10.1146/annurev-immunol-032713-120154
132. Thorgersen EB, Barratt-Due A, Haugaa H, Harboe M, Pischke SE, Nilsson PH, et al. The role of complement in liver injury, regeneration, and transplantation. Hepatology. (2019) 70:725–36. doi: 10.1002/hep.30508
133. Tsokos GC, Fleming SD. Autoimmunity, complement activation, tissue injury and reciprocal effects. Curr Dir Autoimmun. (2004) 7:149–64. doi: 10.1159/000075691
134. Zhang S, Cui P. Complement system in zebrafish. Dev Comp Immunol. (2014) 46:3–10. doi: 10.1016/j.dci.2014.01.010
135. Brinkmann CR, Jensen L, Dagnaes-Hansen F, Holm IE, Endo Y, Fujita T, et al. Mitochondria and the lectin pathway of complement. J Biol Chem. (2013) 288:8016–27. doi: 10.1074/jbc.M112.430249
136. Jiang H, Cooper B, Robey FA, Gewurz H. DNA binds and activates complement via residues 14-26 of the human C1q A chain. J Biol Chem. (1992) 267:25597–601.
137. Holers VM. Complement receptors and the shaping of the natural antibody repertoire. Springer Semin Immunopathol. (2005) 26:405–23. doi: 10.1007/s00281-004-0186-y
138. Ehrengruber MU, Geiser T, Deranleau DA. Activation of human neutrophils by C3a and C5A. Comparison of the effects on shape changes, chemotaxis, secretion, and respiratory burst. FEBS Lett. (1994) 346:181–4. doi: 10.1016/0014-5793(94)00463-3
139. Karsten CM, Laumonnier Y, Kohl J. Functional analysis of C5a effector responses in vitro and in vivo. Methods Mol Biol. (2014) 1100:291–304. doi: 10.1007/978-1-62703-724-2_23
140. Marshall KM, He S, Zhong Z, Atkinson C, Tomlinson S. Dissecting the complement pathway in hepatic injury and regeneration with a novel protective strategy. J Exp Med. (2014) 211:1793–805. doi: 10.1084/jem.20131902
141. Singhal R, Ganey PE, Roth RA. Complement activation in acetaminophen-induced liver injury in mice. J Pharmacol Exp Ther. (2012) 341:377–85. doi: 10.1124/jpet.111.189837
142. Roychowdhury S, McMullen MR, Pritchard MT, Hise AG, van Rooijen N, Medof ME, et al. An early complement-dependent and TLR-4-independent phase in the pathogenesis of ethanol-induced liver injury in mice. Hepatology. (2009) 49:1326–34. doi: 10.1002/hep.22776
143. Zhang C, Wang C, Li Y, Miwa T, Liu C, Cui W, et al. Complement C3a signaling facilitates skeletal muscle regeneration by regulating monocyte function and trafficking. Nat Commun. (2017) 8:2078. doi: 10.1038/s41467-017-01526-z
144. Weiser MR, Williams JP, Moore FD Jr, Kobzik L, Ma M, Hechtman HB, et al. Reperfusion injury of ischemic skeletal muscle is mediated by natural antibody and complement. J Exp Med. (1996) 183:2343–8. doi: 10.1084/jem.183.5.2343
145. Pedersen ED, Loberg EM, Vege E, Daha MR, Maehlen J, Mollnes TE. In situ deposition of complement in human acute brain ischaemia. Scand J Immunol. (2009) 69:555–62. doi: 10.1111/j.1365-3083.2009.02253.x
146. Van Beek J, Bernaudin M, Petit E, Gasque P, Nouvelot A, MacKenzie ET, et al. Expression of receptors for complement anaphylatoxins C3a and C5a following permanent focal cerebral ischemia in the mouse. Exp Neurol. (2000) 161:373–82. doi: 10.1006/exnr.1999.7273
147. Ji H, Ohmura K, Mahmood U, Lee DM, Hofhuis FM, Boackle SA, et al. Arthritis critically dependent on innate immune system players. Immunity. (2002) 16:157–68. doi: 10.1016/S1074-7613(02)00275-3
148. Banda NK, Thurman JM, Kraus D, Wood A, Carroll MC, Arend WP, et al. Alternative complement pathway activation is essential for inflammation and joint destruction in the passive transfer model of collagen-induced arthritis. J Immunol. (2006) 177:1904–12. doi: 10.4049/jimmunol.177.3.1904
149. Eror AT, Stojadinovic A, Starnes BW, Makrides SC, Tsokos GC, Shea-Donohue T. Antiinflammatory effects of soluble complement receptor type 1 promote rapid recovery of ischemia/reperfusion injury in rat small intestine. Clin Immunol. (1999) 90:266–75. doi: 10.1006/clim.1998.4635
150. Rehrig S, Fleming SD, Anderson J, Guthridge JM, Rakstang J, McQueen CE, et al. Complement inhibitor, complement receptor 1-related gene/protein y-Ig attenuates intestinal damage after the onset of mesenteric ischemia/reperfusion injury in mice. J Immunol. (2001) 167:5921–7. doi: 10.4049/jimmunol.167.10.5921
151. Krysko DV, Brouckaert G, Kalai M, Vandenabeele P, D'Herde K. Mechanisms of internalization of apoptotic and necrotic L929 cells by a macrophage cell line studied by electron microscopy. J Morphol. (2003) 258:336–45. doi: 10.1002/jmor.10161
152. Brouckaert G, Kalai M, Krysko DV, Saelens X, Vercammen D, Ndlovu MN, et al. Phagocytosis of necrotic cells by macrophages is phosphatidylserine dependent and does not induce inflammatory cytokine production. Mol Biol Cell. (2004) 15:1089–100. doi: 10.1091/mbc.e03-09-0668
153. Krysko DV, Denecker G, Festjens N, Gabriels S, Parthoens E, D'Herde K, et al. Macrophages use different internalization mechanisms to clear apoptotic and necrotic cells. Cell Death Differ. (2006) 13:2011–22. doi: 10.1038/sj.cdd.4401900
154. Gaipl US, Kuenkele S, Voll RE, Beyer TD, Kolowos W, Heyder P, et al. Complement binding is an early feature of necrotic and a rather late event during apoptotic cell death. Cell Death Differ. (2001) 8:327–34. doi: 10.1038/sj.cdd.4400826
155. Blume KE, Soeroes S, Waibel M, Keppeler H, Wesselborg S, Herrmann M, et al. Cell surface externalization of annexin A1 as a failsafe mechanism preventing inflammatory responses during secondary necrosis. J Immunol. (2009) 183:8138–47. doi: 10.4049/jimmunol.0902250
156. Trouw LA, Blom AM, Gasque P. Role of complement and complement regulators in the removal of apoptotic cells. Mol Immunol. (2008) 45:1199–207. doi: 10.1016/j.molimm.2007.09.008
157. Gullstrand B, Martensson U, Sturfelt G, Bengtsson AA, Truedsson L. Complement classical pathway components are all important in clearance of apoptotic and secondary necrotic cells. Clin Exp Immunol. (2009) 156:303–11. doi: 10.1111/j.1365-2249.2009.03896.x
158. Bottcher A, Gaipl US, Furnrohr BG, Herrmann M, Girkontaite I, Kalden JR, et al. Involvement of phosphatidylserine, alphavbeta3, CD14, CD36, and complement C1q in the phagocytosis of primary necrotic lymphocytes by macrophages. Arthritis Rheum. (2006) 54:927–38. doi: 10.1002/art.21660
159. Zwart B, Ciurana C, Rensink I, Manoe R, Hack CE, Aarden LA. Complement activation by apoptotic cells occurs predominantly via IgM and is limited to late apoptotic (secondary necrotic) cells. Autoimmunity. (2004) 37:95–102. doi: 10.1080/0891693042000196183
160. Bijl M, Horst G, Bijzet J, Bootsma H, Limburg PC, Kallenberg CG. Serum amyloid P component binds to late apoptotic cells and mediates their uptake by monocyte-derived macrophages. Arthritis Rheum. (2003) 48:248–54. doi: 10.1002/art.10737
161. Hart SP, Alexander KM, MacCall SM, Dransfield I. C-reactive protein does not opsonize early apoptotic human neutrophils, but binds only membrane-permeable late apoptotic cells and has no effect on their phagocytosis by macrophages. J Inflamm. (2005) 2:5. doi: 10.1186/1476-9255-2-5
162. Bottazzi B, Inforzato A, Messa M, Barbagallo M, Magrini E, Garlanda C, et al. The pentraxins PTX3 and SAP in innate immunity, regulation of inflammation and tissue remodelling. J Hepatol. (2016) 64:1416–27. doi: 10.1016/j.jhep.2016.02.029
163. Deban L, Jarva H, Lehtinen MJ, Bottazzi B, Bastone A, Doni A, et al. Binding of the long pentraxin PTX3 to factor H: interacting domains and function in the regulation of complement activation. J Immunol. (2008) 181:8433–40. doi: 10.4049/jimmunol.181.12.8433
164. Lu J, Marjon KD, Mold C, Du Clos TW, Sun PD. Pentraxins and Fc receptors. Immunol Rev. (2012) 250:230–8. doi: 10.1111/j.1600-065X.2012.01162.x
165. Nauta AJ, Raaschou-Jensen N, Roos A, Daha MR, Madsen HO, Borrias-Essers MC, et al. Mannose-binding lectin engagement with late apoptotic and necrotic cells. Eur J Immunol. (2003) 33:2853–63. doi: 10.1002/eji.200323888
166. Zargarian S, Shlomovitz I, Erlich Z, Hourizadeh A, Ofir-Birin Y, Croker BA, et al. Phosphatidylserine externalization, “necroptotic bodies” release, and phagocytosis during necroptosis. PLoS Biol. (2017) 15:e2002711. doi: 10.1371/journal.pbio.2002711
167. Schwegler M, Wirsing AM, Dollinger AJ, Abendroth B, Putz F, Fietkau R, et al. Clearance of primary necrotic cells by non-professional phagocytes. Biol Cell. (2015) 107:372–87. doi: 10.1111/boc.201400090
168. Arur S, Uche UE, Rezaul K, Fong M, Scranton V, Cowan AE, et al. Annexin I is an endogenous ligand that mediates apoptotic cell engulfment. Dev Cell. (2003) 4:587–98. doi: 10.1016/S1534-5807(03)00090-X
169. Fan X, Krahling S, Smith D, Williamson P, Schlegel RA. Macrophage surface expression of annexins I and II in the phagocytosis of apoptotic lymphocytes. Mol Biol Cell. (2004) 15:2863–72. doi: 10.1091/mbc.e03-09-0670
170. Blume KE, Soeroes S, Keppeler H, Stevanovic S, Kretschmer D, Rautenberg M, et al. Cleavage of annexin A1 by ADAM10 during secondary necrosis generates a monocytic “find-me” signal. J Immunol. (2012) 188:135–45. doi: 10.4049/jimmunol.1004073
171. Poon IK, Olsson AK, Hulett MD, Parish CR. Regulation of histidine-rich glycoprotein (HRG) function via plasmin-mediated proteolytic cleavage. Biochem J. (2009) 424:27–37. doi: 10.1042/BJ20090794
172. Devitt A, Moffatt OD, Raykundalia C, Capra JD, Simmons DL, Gregory CD. Human CD14 mediates recognition and phagocytosis of apoptotic cells. Nature. (1998) 392:505–9. doi: 10.1038/33169
173. Thomas L, Bielemeier A, Lambert PA, Darveau RP, Marshall LJ, Devitt A. The N-terminus of CD14 acts to bind apoptotic cells and confers rapid-tethering capabilities on non-myeloid cells. PLoS ONE. (2013) 8:e70691. doi: 10.1371/journal.pone.0070691
174. Moffatt OD, Devitt A, Bell ED, Simmons DL, Gregory CD. Macrophage recognition of ICAM-3 on apoptotic leukocytes. J Immunol. (1999) 162:6800–10.
175. Rigotti A, Miettinen HE, Krieger M. The role of the high-density lipoprotein receptor SR-BI in the lipid metabolism of endocrine and other tissues. Endocr Rev. (2003) 24:357–87. doi: 10.1210/er.2001-0037
176. Ramirez-Ortiz ZG, Pendergraft WF III, Prasad A, Byrne MH, Iram T, Blanchette CJ, et al. The scavenger receptor SCARF1 mediates the clearance of apoptotic cells and prevents autoimmunity. Nat Immunol. (2013) 14:917–26. doi: 10.1038/ni.2670
177. Chen Q, Stone PR, McCowan LM, Chamley LW. Phagocytosis of necrotic but not apoptotic trophoblasts induces endothelial cell activation. Hypertension. (2006) 47:116–21. doi: 10.1161/01.HYP.0000196731.56062.7c
178. Gaipl US, Beyer TD, Heyder P, Kuenkele S, Bottcher A, Voll RE, et al. Cooperation between C1q and DNase I in the clearance of necrotic cell-derived chromatin. Arthritis Rheum. (2004) 50:640–9. doi: 10.1002/art.20034
179. Napirei M, Wulf S, Mannherz HG. Chromatin breakdown during necrosis by serum Dnase1 and the plasminogen system. Arthritis Rheum. (2004) 50:1873–83. doi: 10.1002/art.20267
180. Tsai RK, Discher DE. Inhibition of “self” engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J Cell Biol. (2008) 180:989–1003. doi: 10.1083/jcb.200708043
181. Khandelwal S, van Rooijen N, Saxena RK. Reduced expression of CD47 during murine red blood cell (RBC) senescence and its role in RBC clearance from the circulation. Transfusion. (2007) 47:1725–32. doi: 10.1111/j.1537-2995.2007.01348.x
182. Kojima Y, Volkmer JP, McKenna K, Civelek M, Lusis AJ, Miller CL, et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature. (2016) 536:86–90. doi: 10.1038/nature18935
183. Gerlach BD, Marinello M, Heinz J, Rymut N, Sansbury BE, Riley CO, et al. Resolvin D1 promotes the targeting and clearance of necroptotic cells. Cell Death Differ. (2019). doi: 10.1038/s41418-019-0370-1. [Epub ahead of print].
184. Astier AL. T-cell regulation by CD46 and its relevance in multiple sclerosis. Immunology. (2008) 124:149–54. doi: 10.1111/j.1365-2567.2008.02821.x
185. Elward K, Griffiths M, Mizuno M, Harris CL, Neal JW, Morgan BP, et al. CD46 plays a key role in tailoring innate immune recognition of apoptotic and necrotic cells. J Biol Chem. (2005) 280:36342–54. doi: 10.1074/jbc.M506579200
186. Brown S, Heinisch I, Ross E, Shaw K, Buckley CD, Savill J. Apoptosis disables CD31-mediated cell detachment from phagocytes promoting binding and engulfment. Nature. (2002) 418:200–3. doi: 10.1038/nature00811
187. Park YJ, Liu G, Lorne EF, Zhao X, Wang J, Tsuruta Y, et al. PAI-1 inhibits neutrophil efferocytosis. Proc Natl Acad Sci USA. (2008) 105:11784–9. doi: 10.1073/pnas.0801394105
188. Park YJ, Liu G, Tsuruta Y, Lorne E, Abraham E. Participation of the urokinase receptor in neutrophil efferocytosis. Blood. (2009) 114:860–70. doi: 10.1182/blood-2008-12-193524
189. Blasi F, Sidenius N. Efferocytosis: another function of uPAR. Blood. (2009) 114:752–3. doi: 10.1182/blood-2009-05-220657
190. Gabillet J, Millet A, Pederzoli-Ribeil M, Tacnet-Delorme P, Guillevin L, Mouthon L, et al. Proteinase 3, the autoantigen in granulomatosis with polyangiitis, associates with calreticulin on apoptotic neutrophils, impairs macrophage phagocytosis, and promotes inflammation. J Immunol. (2012) 189:2574–83. doi: 10.4049/jimmunol.1200600
191. Chen GY, Tang J, Zheng P, Liu Y. CD24 and Siglec-10 selectively repress tissue damage-induced immune responses. Science. (2009) 323:1722–5. doi: 10.1126/science.1168988
192. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature. (2019) 572:392–6. doi: 10.1038/s41586-019-1456-0
193. Strey CW, Markiewski M, Mastellos D, Tudoran R, Spruce LA, Greenbaum LE, et al. The proinflammatory mediators C3a and C5a are essential for liver regeneration. J Exp Med. (2003) 198:913–23. doi: 10.1084/jem.20030374
194. Markiewski MM, Mastellos D, Tudoran R, DeAngelis RA, Strey CW, Franchini S, et al. C3a and C3b activation products of the third component of complement (C3) are critical for normal liver recovery after toxic injury. J Immunol. (2004) 173:747–54. doi: 10.4049/jimmunol.173.2.747
195. Daveau M, Benard M, Scotte M, Schouft MT, Hiron M, Francois A, et al. Expression of a functional C5a receptor in regenerating hepatocytes and its involvement in a proliferative signaling pathway in rat. J Immunol. (2004) 173:3418–24. doi: 10.4049/jimmunol.173.5.3418
196. Tsokos GC, Lo MS, Costa Reis P, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol. (2016) 12:716–30. doi: 10.1038/nrrheum.2016.186
197. Boes M, Schmidt T, Linkemann K, Beaudette BC, Marshak-Rothstein A, Chen J. Accelerated development of IgG autoantibodies and autoimmune disease in the absence of secreted IgM. Proc Natl Acad Sci USA. (2000) 97:1184–9. doi: 10.1073/pnas.97.3.1184
198. Nath SK, Han S, Kim-Howard X, Kelly JA, Viswanathan P, Gilkeson GS, et al. A nonsynonymous functional variant in integrin-alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat Genet. (2008) 40:152–4. doi: 10.1038/ng.71
199. Napirei M, Wulf S, Eulitz D, Mannherz HG, Kloeckl T. Comparative characterization of rat deoxyribonuclease 1 (Dnase1) and murine deoxyribonuclease 1-like 3 (Dnase1l3). Biochem J. (2005) 389:355–64. doi: 10.1042/BJ20042124
200. Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Moroy T. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet. (2000) 25:177–81. doi: 10.1038/76032
201. Napirei M, Gultekin A, Kloeckl T, Moroy T, Frostegard J, Mannherz HG. Systemic lupus-erythematosus: deoxyribonuclease 1 in necrotic chromatin disposal. Int J Biochem Cell Biol. (2006) 38:297–306. doi: 10.1016/j.biocel.2005.10.023
202. Keyel PA. Dnases in health and disease. Dev Biol. (2017) 429:1–11. doi: 10.1016/j.ydbio.2017.06.028
203. Sisirak V, Sally B, D'Agati V, Martinez-Ortiz W, Ozcakar ZB, David J, et al. Digestion of chromatin in apoptotic cell microparticles prevents autoimmunity. Cell. (2016) 166:88–101. doi: 10.1016/j.cell.2016.05.034
204. Yasutomo K, Horiuchi T, Kagami S, Tsukamoto H, Hashimura C, Urushihara M, et al. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat Genet. (2001) 28:313–4. doi: 10.1038/91070
205. Al-Mayouf SM, Sunker A, Abdwani R, Abrawi SA, Almurshedi F, Alhashmi N, et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet. (2011) 43:1186–8. doi: 10.1038/ng.975
206. Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K, et al. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am J Hum Genet. (2007) 80:811–5. doi: 10.1086/513443
207. Kawane K, Fukuyama H, Kondoh G, Takeda J, Ohsawa Y, Uchiyama Y, et al. Requirement of DNase II for definitive erythropoiesis in the mouse fetal liver. Science. (2001) 292:1546–9. doi: 10.1126/science.292.5521.1546
208. Ahn J, Gutman D, Saijo S, Barber GN. STING manifests self DNA-dependent inflammatory disease. Proc Natl Acad Sci USA. (2012) 109:19386–91. doi: 10.1073/pnas.1215006109
209. Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. (2012) 485:251–5. doi: 10.1038/nature10992
210. Kawane K, Ohtani M, Miwa K, Kizawa T, Kanbara Y, Yoshioka Y, et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature. (2006) 443:998–1002. doi: 10.1038/nature05245
211. Ahrens S, Zelenay S, Sancho D, Hanc P, Kjaer S, Feest C, et al. F-actin is an evolutionarily conserved damage-associated molecular pattern recognized by DNGR-1, a receptor for dead cells. Immunity. (2012) 36:635–45. doi: 10.1016/j.immuni.2012.03.008
212. Srinivasan N, Gordon O, Ahrens S, Franz A, Deddouche S, Chakravarty P, et al. Actin is an evolutionarily-conserved damage-associated molecular pattern that signals tissue injury in Drosophila melanogaster. Elife. (2016) 5:e19662. doi: 10.7554/eLife.19662
213. Zhang JG, Czabotar PE, Policheni AN, Caminschi I, Wan SS, Kitsoulis S, et al. The dendritic cell receptor Clec9A binds damaged cells via exposed actin filaments. Immunity. (2012) 36:646–57. doi: 10.1016/j.immuni.2012.03.009
214. Schulz O, Hanc P, Bottcher JP, Hoogeboom R, Diebold SS, Tolar P, et al. Myosin II synergizes with F-actin to promote DNGR-1-dependent cross-presentation of dead cell-associated antigens. Cell Rep. (2018) 24:419–28. doi: 10.1016/j.celrep.2018.06.038
215. Hitchcock SE, Carisson L, Lindberg U. Depolymerization of F-actin by deoxyribonuclease I. Cell. (1976) 7:531–42. doi: 10.1016/0092-8674(76)90203-8
216. Serhan CN. Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu Rev Immunol. (2007) 25:101–37. doi: 10.1146/annurev.immunol.25.022106.141647
Keywords: cell death, necrosis, apoptosis, phagocytosis, inflammation, cell debris, “find-me,” “eat-me”
Citation: Westman J, Grinstein S and Marques PE (2020) Phagocytosis of Necrotic Debris at Sites of Injury and Inflammation. Front. Immunol. 10:3030. doi: 10.3389/fimmu.2019.03030
Received: 14 October 2019; Accepted: 10 December 2019;
Published: 09 January 2020.
Edited by:
Florence Niedergang, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Ian Dransfield, University of Edinburgh, United KingdomBarbara Bottazzi, Humanitas Clinical and Research Center, Milan University, Italy
Copyright © 2020 Westman, Grinstein and Marques. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sergio Grinstein, sergio.grinstein@sickkids.ca