 Haoyu Sun
Haoyu Sun Cheng Sun
Cheng Sun- 1The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, China
- 2Division of Molecular Medicine, Hefei National Laboratory for Physical Sciences at Microscale, The CAS Key Laboratory of Innate Immunity and Chronic Disease, School of Life Sciences, University of Science and Technology of China, Hefei, China
- 3Institute of Immunology, University of Science and Technology of China, Hefei, China
Checkpoint immunotherapy that targets inhibitory receptors of T cells, thereby reversing the functional exhaustion of T cells, marks a breakthrough in anticancer therapy. The success of T cell-directed checkpoint inhibitors of CTLA-4 and PD-1/PD-L1 has opened a new approach for cancer immunotherapy and resulted in extensive research on immune checkpoints. However, it is only in recent years that research on NK cell exhaustion and potential checkpoints impacting NK cells has become popular. NK cells, as the major player in innate immunity, are critical for immune surveillance, particularly the control of metastasis and hematological cancers. The balance between activating and inhibitory signals fine tunes the activation and effector functions of NK cells, and transformed cells modulate NK cells by upregulating negative signaling that “exhausts” NK cells. Exhausted NK cells with excessive expression of inhibitory receptors (checkpoint molecules) are impaired in the recognition of tumor cells as well as antitumor cytotoxicity and cytokine secretion. Therefore, an understanding of the potential checkpoint molecules involved in NK cell exhaustion is particularly important in terms of NK cell-targeted cancer immunotherapy. In this review, we summarize recent advances in NK cell checkpoint inhibitors and their progress in clinical trials. Moreover, we highlight some of the latest findings in fundamental NK cell receptor biology and propose potential NK cell checkpoint molecules for future immunotherapeutic applications.
Introduction
Excessive negative regulation of immune cells by inhibitory receptors (checkpoint molecules) results in functional exhaustion of these cells, which is one of the major reasons for tumor escape. The activation and function of immune cells are regulated through activating and inhibitory receptors on these cells, and establishing an equilibrium between activating and inhibitory signaling is critical because it assures effective control against pathogenic factors (such as tumors, viruses, and bacteria) meanwhile helping to avoid self-directed attacks (such as autoimmune disease) (1, 2). The negative feedback provided by inhibitory receptors is the key to immune regulation; however, unfortunately, tumor cells can take advantage of this negative feedback system, as they upregulate the surface expression of corresponding ligands to ingratiate excessive expression of inhibitory receptors on immune cells that automatically leads to reduced activation and functional exhaustion of these cells (3, 4). Antibodies that specifically target these inhibitory checkpoints can effectively block the interaction between the checkpoint molecule and its ligands, thereby reversing the functional exhaustion of immune cells and restoring antitumor immunity.
Checkpoint immunotherapy targeting checkpoint molecules that reverses functional exhaustion in immune cells marks a major breakthrough in anticancer therapy. Blocking inhibitory receptors on T cells to reverse functional exhaustion in these cells has made great progress. T cells from cancer patients highly express inhibitory receptors including cytotoxicity T-lymphocyte-associated protein 4 (CTLA-4), programmed cell death protein 1 (PD-1), T cell immunoglobulin- and mucin-domain-containing molecule 3 (TIM-3), lymphocyte activation gene 3 (LAG-3), T cell immunoreceptor with Ig and ITIM domains (TIGIT), etc., which contribute to T cell functional exhaustion (5, 6). Blocking these checkpoint molecules can effectively reverse T cell exhaustion and restore the antitumor capacity of T cells. During the past 10 years, the efficacy and feasibility of checkpoint immunotherapy have been verified in clinical settings, and antibodies targeting CTLA-4, PD-1/programmed death-ligand 1 (PD-L1), TIM-3, LAG-3, or TIGIT have entered clinical trials (7, 8). Monoclonal antibodies (mAbs) targeting CTLA-4 and PD-1/PD-L1 were approved by the U.S. Food and Drug Administration (FDA) in 2011 and 2014, respectively. The combined use of an anti-CTLA-4 mAb with an anti-PD-1 mAb showed better efficacy than either antibody used as a monotherapy (9–11).
However, although remarkable clinical benefits derived from anti-CTLA-4 and anti-PD-1/PD-L1 antibody therapies have been noted in some patients, there are still many patients who do not respond to these treatments. Currently, researchers are still trying to understand these “non-responders;” additional unknown inhibitory pathways that suppress immune responses could be an explanation for nonresponse. On the other hand, tumors escape T cell-mediated immunity by downregulating the expression of major histocompatibility complex class I (MHC-I) molecules; these MHC-I-null tumor cells are not attacked by T cells, but they are still targets of natural killer (NK) cells (12). Moreover, Zhang et al. demonstrated the significance of NK cells in cancer immunotherapy and noted that mAb targeting checkpoint molecule TIGIT, which is expressed by both T cells and NK cells, could improve the antitumor immunity of both T and NK cells. Furthermore, they found that TIGIT blockade was effective even in the absence of T cells and B cells, highlighting the importance of NK cells in checkpoint-targeted immunotherapy (13, 14).
NK cells are innate lymphocytes that play a critical role in the early defense against transformed cells, and they are particularly important in the control of cancer metastasis and hematological cancers (1, 15–17). NK cells can directly kill tumor cells, secrete various cytokines such as interferon (IFN)-γ and tumor necrosis factor (TNF)-α to initiate antitumor responses, and recruit other immune cells into the antitumor response (1, 18, 19). Alterations in NK cells, for example, excessive expression of inhibitory receptors or reduced expression of activating receptors, can result in impaired cytotoxicity against tumor cells and a decreased ability to recruit other immune cells (20–22). Some checkpoint molecules in cytotoxic T lymphocytes (CTLs), such as PD-1, LAG-3, TIGIT, and TIM-3, are shared with NK cells. Blockade with checkpoint inhibitors that reverses the functional exhaustion of NK cells opens a new strategy for cancer immunotherapy that may complement the limitations of T cell-based immunotherapy. This review summarizes recent advances in NK cell checkpoint molecules in humans (Figure 1) and the corresponding antibodies being studied in clinical trials (Tables 1, 2). In addition, we highlight some of the latest findings in fundamental NK cell receptor biology that may provide a fundamental basis for future NK cell-based immunotherapeutic applications.
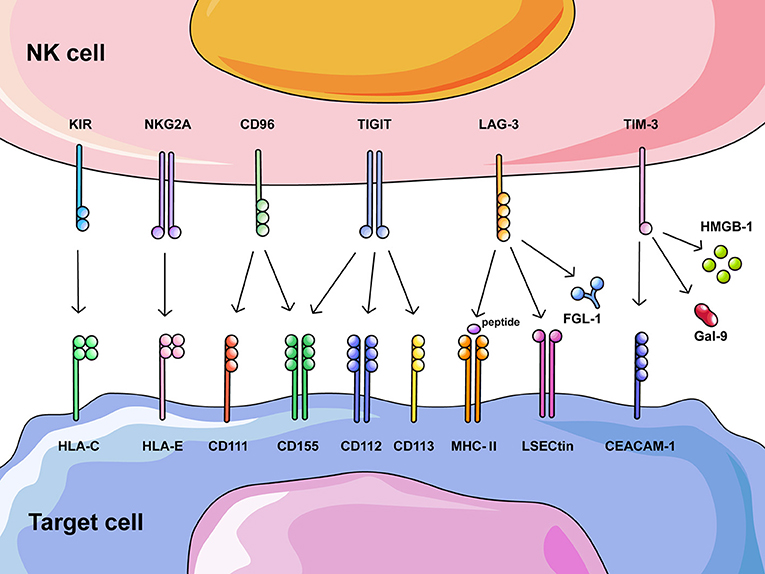
Figure 1. Overview of potential NK cell checkpoint molecules and their corresponding ligands. Recognition and clearance of tumor cells by NK cells are regulated through activating and inhibitory receptors on NK cells that bind their corresponding ligands on tumor cells. Increased expression of ligands on tumor cells induces altered expression of inhibitory receptors on NK cells, excessive negative regulation results in functional exhaustion of NK cells. This figure summarizes inhibitory receptors on NK cells that could also act as checkpoints in cancer immunotherapy, including HLA class I-specific receptors (KIR and NKG2A) and those recognizing ligands other than HLA class I molecules (CD96, TIGIT, LAG-3, and TIM-3).

Table 1. Potential NK cell checkpoint molecules in cancer.
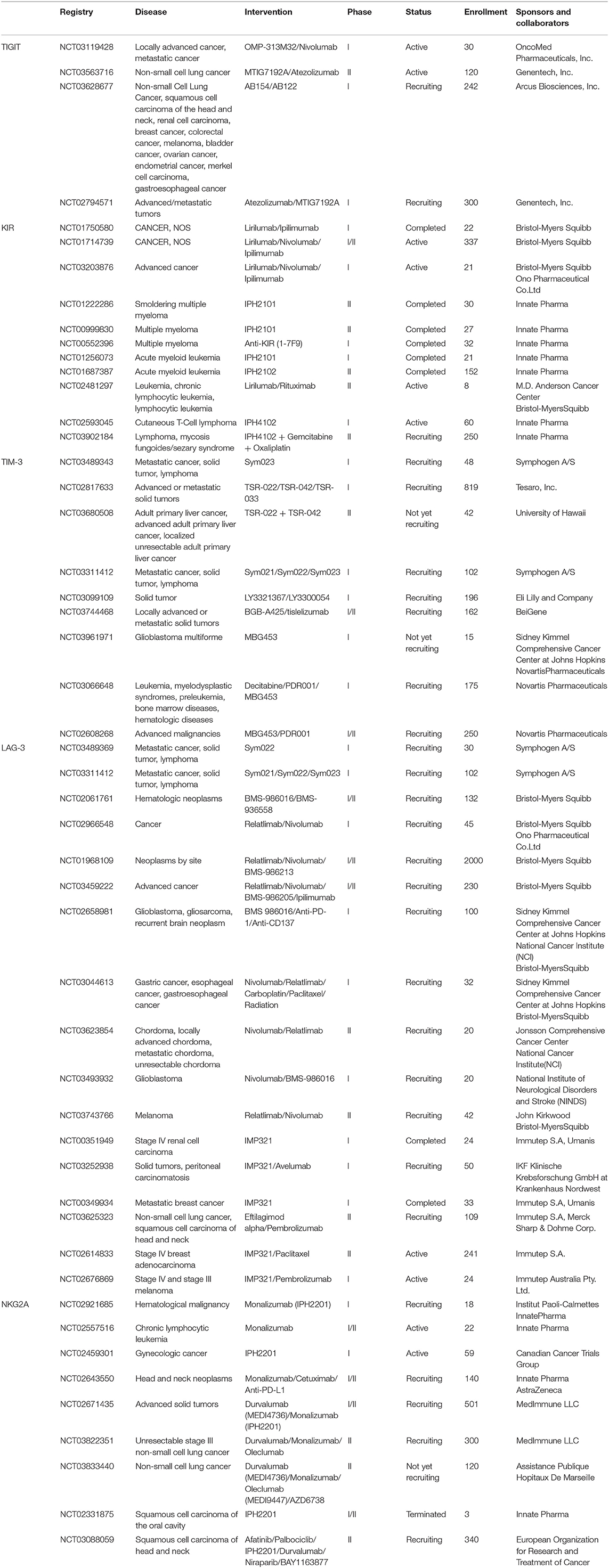
Table 2. Clinical trials based on potential NK cell checkpoint inhibitors in cancer.
Killer Cell Lectin-Like Receptor Family
NKG2A
NK group 2 member A (NKG2A) is a type II membrane receptor that forms a heterodimer with CD94 (23). NKG2A binds a human leukocyte antigen (HLA) class I molecule (HLA-E) (24) and transduces inhibitory signaling that suppresses the cytokine secretion and cytotoxicity of NK cells (25–27). NKG2A+ NK cells infiltrate the tumor microenvironment, and increased expression of NKG2A in NK cells has been observed in patients with non-small cell lung cancer (28, 29), breast cancer (30), colorectal cancer (31, 32), acute myeloid leukemia (33, 34), hepatocellular carcinoma (35), breast cancer (36), cervical cancer (32), etc. A large proportion of NK cells with high NKG2A expression has also been found in tumor-draining lymph nodes (36). NK cells with elevated NKG2A expression are functionally exhausted and associated with a poor prognosis in human hepatocellular carcinoma (35). NKG2A expression predominantly increases on CD56dim NK cells compared to CD56bright NK cells, and these NKG2A+ CD56dim NK cells are functionally exhausted and highly correlated with massive tumor size in human hepatocellular carcinoma (35).
Due to the strong capability of NKG2A to suppress NK cells, blockade of NKG2A is effective in restoring functions of NK cells. A mAb targeting NKG2A, namely, monalizumab (formerly IPH2201), has been tested in both phase I and phase II clinical trials (Table 2). NKG2A is overexpressed in NK cells from chronic lymphocytic leukemia patients, and blocking NKG2A with monalizumab is sufficient to restore the direct cytotoxicity of NK cells against HLA-E-expressing tumor cells (37). Treatment with IPH2201 has been shown to trigger NKG2A+ NK cell-mediated lysis of HLA-E+ target cells in vitro and abolish HLA-E+ leukemia and lymphoma tumors in xenograft mouse models of human neoplastic disease (NOD-SCID mice injected with HLA-E+ Epstein-Barr virus-positive cells or acute myeloid leukemia cells) (38). Interestingly, although NKG2A is predominantly expressed by NK cells, a study by the Vivier group showed that blockade of NKG2A enhanced the effector functions of both NK cells and CD8+ T cells in mice and humans (32). The use of monalizumab not only promoted human NK cell antibody-dependent cell-mediated cytotoxicity (ADCC) against various tumor cells but also rescued the function of CD8+ T cells when combined with PD-1 blockade (32). This group also reported impressive clinical outcomes: the use of monalizumab combined with cetuximab (an anti-EGFR antibody) in previously treated patients with squamous cell carcinoma of the head and neck showed a 30% response rate with limited side effects [fatigue (17%), pyrexia (13%), and headache (10%)] (32). Interestingly, a study by Kamiya et al. showed that NKG2Anull NK cells, which were generated through transduction of anti-NKG2A protein expression blockers (PEBLs), exhibited relatively high cytotoxicity against HLA-E+ tumor cells; moreover, this method generated more potent cytotoxicity than blockade with an anti-NKG2A mAb (39), suggesting a new method for developing NKG2A-targeted cancer immunotherapy.
Killer Cell Immunoglobulin-Like Receptor Family
KIRs
The killer-cell immunoglobulin-like receptors (KIRs) on human NK cells include both activating and inhibitory receptors, among which the inhibitory KIRs exhibit an inhibitory signaling motif and are named with the convention KIRxDL (40). KIR2DL and KIR3DL specifically bind to HLA-C and HLA-A/B allotypes, respectively (41, 42). KIR2DL includes KIR2DL1 and KIR2DL2/3, which bind distinct HLA-C allotypes to suppress the activation and effector functions of NK cells (41). Tumor cells induce the upregulated expression of KIRs on NK cells; for example, the expression of KIR2DL2 and HLA-C1 is significantly elevated in breast cancer patients (43); KIR2D (L1, L3, L4, and S4) and KIR3DL1 are expressed on tumor cells and TILs from non-small cell lung cancer patients, and patients without expression of KIR2D (L1, L3, L4, and S4) or KIR3DL1 on their tumor cells or TILs exhibit extended overall survival (44). KIR centromeric B haplotype is associated with significant risks of multiple basal cell carcinoma and squamous cell carcinoma, suggesting that interactions between KIRs and HLA molecules may modify the risks of basal cell carcinoma and squamous cell carcinoma (45). Interestingly, patients with bile duct cancer show multiple alterations at KIR gene loci (46), and genetic variations in KIRs are also present in non-small cell lung cancer patients who are resistant to anti-PD-1 monotherapy (47).
Due to their impressive suppressive effect on NK cells, human mAbs targeting KIRs have shown some clinical benefits. Lirilumab (1-7F9, IPH2101) targeting KIR2DL1, KIR2DL2, and KIR2DL3 increases NK cell cytotoxicity against autologous acute myeloid leukemia blasts and mediates the lysis of HLA-C-expressing tumor cells both in vitro and in vivo (48). Lirilumab also enhances NK cell activity against autologous multiple myeloma cells by preventing inhibitory KIR-ligand interactions (49). Phase I studies of lirilumab in patients with acute myeloid leukemia, hematological malignancies or solid tumors have shown that lirilumab can effectively block KIRs with mild adverse events (50, 51). However, a study by Carlsten et al. demonstrated that lirilumab not only reduced KIR2D expression on NK cells but also rapidly reduced NK cell functions, resulting in significantly diminished overall responses (52). On the other hand, IPH4102 targeting KIR3DL2 shows encouraging clinical activity in patients with relapsed or refractory cutaneous T-cell lymphoma, particularly those with Sézary syndrome (53).
An in vitro study found that stimulation with IL-12/IL-15/IL-18 also downregulated the expression of KIR2DL2/3, KIR2DL1, and KIR3DL1 on peripheral blood NK cells, resulting in reduced inhibitory KIR signaling and elevated CD16-dependent cytotoxicity (54). Furthermore, these IL-12/IL-15/IL-18-stimulated NK cells showed increased cytotoxicity against tumor cells (54).
Immunoglobulin Superfamily
TIGIT
TIGIT is an immunoglobulin protein that belongs to the CD28 family (55, 56). It was discovered as a surface receptor on T cells that recognizes CD155 in 2009 (57); however, TIGIT is also expressed on NK cells and interacts with other ligands, such as CD112 and CD113 (56). Together with CD226 and CD96, TIGIT participates in a balanced system to control the activation and function of T cells and NK cells. Unlike CD96, which inhibits only IFN-γ production in NK cells and has no effects on cytotoxicity, TIGIT can inhibit NK cell cytotoxicity directly through its ITIM domain in both humans and mice (58, 59). A study showed that the cytotoxicity of YTS NK cells (human NK cell line) transfected with TIGIT was strongly inhibited by CD155-transfected 721.221 cells and this inhibition could be blocked with an anti-TIGIT mAb in vitro (58). Furthermore, blockade of TIGIT has also been shown to significantly increase mouse NK cell-mediated killing of CD155-expressing B12 cells and enhance the secretion of IFN-γ (59). In an in vivo study, NK cells isolated from TIGIT-transgenic mice produced reduced amounts of IFN-γ after incubation with Yac-1 cells (murine T cell lymphoma cell line), while NK cells isolated from TIGIT−/− mice produced increased amounts of IFN-γ. The suppression of IFN-γ production was mediated by TIGIT-CD155 ligation through the NF-κB pathway (60). In humans, NK cells with lower levels of TIGIT isolated from healthy individuals were shown to have a higher ability to secrete cytokines, degranulate, and kill target cells than those with higher TIGIT expression (61), suggesting the ability of TIGIT to regulate immune responses.
The expression of TIGIT is highly variable among different cancer types. The highest expressions of TIGIT in lymphocytes are found in Hodgkin's lymphoma, Warthin's tumors, medullary breast cancer, intestinal stomach cancer, and seminoma, while the lowest expressions of TIGIT in lymphocytes are found in renal oncocytoma, papillary renal cell cancer, desmoid tumors, pancreatic neuroendocrine cancer, chromophobic renal cell cancer, and adrenocortical cancer (62). Indeed, TIGIT−/− mice show no resistance to lung metastasis in three different experimental lung metastasis models (B16F10, murine melanoma cell line; RM-1, murine prostate cancer cell line; E0771, murine breast cancer cell line) (63); moreover, TIGIT expression on NK cells is not significantly different between pancreatic cancer patients and healthy controls (64). Furthermore, a reduced proportion of TIGIT+ NK cells has been observed in the intratumoral region of hepatocellular carcinoma compared to the peritumoral region (65).
In contrast, TIGIT is overexpressed on CD8+ tumor-infiltrating lymphocytes (TILs) and tumor antigen-specific CD8+ T cells from melanoma patients and often coexpressed with the inhibitory receptor PD-1 (66). Coblockade of TIGIT and PD-1 can reverse dysfunctions in CD8+ TILs and antigen-specific CD8+ T cells by increasing their proliferation and effector functions (66). TIGIT expression was also found to be significantly increased in CD4+ T cells from chronic lymphocytic leukemia patients, and an increased number of TIGIT+ CD4+ T cells was found in patients with advanced disease stage (67). Moreover, TIGIT−/− was shown to significantly inhibit tumorigenicity in both CT26 tumor-bearing BALB/c mice and MC38 tumor-bearing C57BL/6 mice, whereas an anti-TIGIT mAb significantly inhibited tumor growth in both of these colorectal tumor models (68). Furthermore, a study showed that TIGIT−/− mice intravenously injected with B16 melanoma cells had relatively few lung metastases and improved overall survival (13). It is also important to note that TIGIT inhibits IFN-γ secretion of both CD8+ T cells and NK cells in the above mentioned colorectal tumor models; interestingly, CT26 tumor-bearing TIGIT knockout (KO) mice develop tumors early after NK cells are depleted, suggesting that NK cells and T cells collaborate to eliminate tumors (68).
The Tian group has demonstrated that TIGIT is highly expressed on tumor-infiltrating NK cells and associated with NK cell exhaustion in different tumor models [CT26 colon cancer, 4T1 breast cancer, B16 melanoma, and fibrosarcoma induced by methylcholanthrene (MCA)] and patients with colon cancer (13). NK cell-specific TIGIT KO in mice results in significantly prolonged survival, while TIGIT blockade inhibits NK cell exhaustion in colon tumors, breast tumors, and MCA-induced fibrosarcomas (13). Surprisingly, they showed that anti-TIGIT mAb could reduce tumor mass and slow tumor growth in T cell-deficient mice; in addition, NK cell deficiency resulted in an increased metastasis and number of exhausted CD8+ T cells, and abolished the effect of TIGIT blockade even in the presence of TIGIT-expressing CD8+ T cells (13). Notably, the therapeutic effects of anti-TIGIT mAbs, anti-PD-L1 mAbs or anti-TIGIT mAbs combined with anti-PD-L1 mAbs all depended on the presence of NK cells (13), indicating the importance of NK cells in checkpoint-targeted immunotherapy. Other studies have also indicated the importance of TIGIT+ NK cells in the tumor microenvironment. For example, blocking TIGIT could increase cytokine production by NK cells after an incubation with trastuzumab-coated breast cancer cells (69); the proportion of TIGIT+ NK cells was significantly increased in the peripheral blood mononuclear cell (PBMC) population of non-muscle invasive bladder cancer patients compared to that of healthy controls (70); endometrial tumor-resident CD103+ NK cells expressed higher levels of TIGIT than circulating CD103− NK cells, and tumor-resident NK cells from patients with lymph node invasion showed significantly higher expressions of TIGIT than those from patients with no lymph node invasion (71); and TIGIT+ NK cells showed increased susceptibility to functional suppression by CD155-expressing myeloid-derived suppressor cells (MDSCs) (72). Currently, several clinical trials (phase I and phase II) focused on testing the feasibility of targeting this new pathway and improving therapeutic effects through combination with existing immunotherapies are either active or recruiting (Table 2).
CD96
CD96 is a transmembrane glycoprotein that belongs to the immunoglobulin superfamily (73, 74). It was identified as a key receptor on NK cells that recognizes the ligand CD155 in 2004 (75) and was initially identified as a possible costimulatory receptor. However, 10 years later, its inhibitory characteristics were revealed by the Smyth group (76). Their study showed that CD96 competes with CD226 for CD155 binding and negatively regulates IFN-γ secretion in NK cells (77); however, it does not affect the direct killing of tumor cells by NK cells. Furthermore, CD96−/− mice are resistant to MCA-induced fibrosarcoma and experimental lung metastasis modeled by injecting B16F10 melanoma cells (77), and blocking CD96 with a mAb inhibits experimental metastases in three different models (B16F10 melanoma, 3LL lung carcinoma, and RM-1 prostate cancer) (63). Blockade of the CD96-CD155 interaction was also shown to be effective in controlling lung metastases in NCR2-transgenic mice injected with B16-PDGFD cells (78). An anti-CD96 mAb was shown to be superior to other well-characterized checkpoint inhibitors, such as anti-CTLA-4 and anti-PD-1 antibodies, and the combination of an anti-CD96 mAb with an anti-CTLA-4 or anti-PD-1/PD-L1 mAb could further inhibit experimental lung metastases (63). Notably, although CD96 was also expressed by T cells, the control of metastases by an anti-CD96 mAb appeared to be dependent on NK cells, CD226 and IFN-γ production (63), suggesting a non-negligible role for NK cells in cancer immunotherapy. Further, blocking CD96 reduced the number of B16F10 metastases in Tigit−/− mice compared to wildtype mice, indicating the synergistic potential of blocking CD96 and TIGIT in treating cancer (63).
The structural basis for the CD96-CD155 interaction involves the “ancillary key” motif that is critical for CD155 recognition; moreover, CD96 and CD155 interact via the “lock-and-key” docking mode (79). However, surprisingly, a comparison between three anti-CD96 mAbs, including two that block the CD96-CD155 interaction (3.3 and 6A6) and one that does not block this interaction (8B10), revealed that although the two blocking mAbs showed higher potency than the non-blocking mAb in the control of metastases, it was not necessary to block the CD96-CD155 interaction to promote NK cell antimetastatic functions (80, 81). In contrast, another study using a transgenic mouse model of resectable pancreatic ductal adenocarcinoma showed that a mAb targeting the CD96-CD155 interaction (6A6) significantly reduced distant metastases, while a mAb that did not target the CD96-CD155 interaction (8B10) showed no effect on the frequency of metastases (82). One possible explanation for the contradictions occurred involves various microenvironmental cues in in vitro vs. in vivo settings, given that the microenvironment in an in vivo experiment is much more complicated and involves various cell-cell interactions and consequences (for example, cytokine secretions, etc.) following these interactions, which may contribute to the differences raised between the studies.
In humans, a significantly decreased percentage of CD96+ NK cells in pancreatic cancer patients and associations of this decreased percentage with lymph node metastasis and tumor histological grade were observed (64), suggesting a possible protective role for CD96+ NK cells in pancreatic cancer. Contradictorily, another study showed increased serum levels of soluble CD96 in NK cells from late-stage melanoma patients (83). In addition, a study noted an elevated proportion and number of CD96+CD56dim NK cells in hepatocellular carcinoma tissues, and these NK cells were functionally exhausted with impaired IFN-γ and TNF-α productions (65). Furthermore, patients with higher CD96+ NK cell infiltration within tumors have been shown to exhibit relatively short disease-free survival times (65). These studies suggest a protumor role for CD96+ NK cells in melanoma and hepatocellular carcinoma.
LAG-3
LAG-3 is a negative coinhibitory receptor expressed on T cells and NK cells that binds MHC class II (MHC-II) molecules on target cells (84, 85). LAG-3 also interacts with LSECtin, a cell surface lectin that belongs to the C-type lectin receptor superfamily, to inhibit IFN-γ production by effector T cells (86). Recently, Chen and colleagues identified fibrinogen-like protein 1 (FGL1), a liver-secreted protein, as an MHC-II-independent ligand for LAG-3 in both humans and mice (87). Previous studies have shown that LAG-3 negatively regulates the proliferation and activation of T cells (88, 89) and that it also interacts with FGL1 to inhibit antigen-mediated T cell responses both in vitro and in vivo (87). LAG-3 and PD-L1 coregulate the exhaustion of CD8+ T cells, and compared to anti-PD-L1 mAb or anti-LAG-3 mAb monotherapy, dual blockade of LAG-3 and PD-L1 increases the number and effector functions of functional virus-specific CD8+ T cells (90), suggesting that the combination of anti-LAG-3 and anti-PD-L1 antibodies results in an improved reversal of exhaustion.
LAG-3 has been shown to suppress immune responses in several tumors, including Hodgkin's lymphoma, gastric cancer, breast cancer, and other solid tumors (91). In studies of squamous cell carcinoma mouse models, both CD8+ and CD4+ TILs coexpressed LAG-3 and PD-1, and dual blockade of LAG-3 and PD-1 significantly suppressed tumor growth (92). LAG-3 has been detected in TILs from 41.5% of non-small cell lung cancer patients and associated with the checkpoint molecules PD-1 and TIM-3 (93). Moreover, elevated LAG-3 expression has been associated with reduced progression-free survival in patients with advanced non-small cell lung cancer treated with PD-1 blockade (93). Elevated expression of LAG-3 has also been observed in patients with peripheral T cell lymphoma or NK/T cell lymphoma (94). LAG-3+ TIL numbers are increased in MHC-II+ tumors (lung cancer, melanoma, and breast cancer), and MHC-II+ tumors acquire immunosuppressive signals through LAG-3; thus, combined PD-1/PD-L1 and LAG-3 blockade can provide a particular advantage against MHC-II+ tumors (95). Interestingly, expression of the newly identified ligand FGL1 has also been shown to be upregulated in cancer, and blockade of the FGL1-LAG-3 interaction stimulates immune responses and exhibits therapeutic effects on mouse tumor models (MC38 colon cancer and Hepa1-6 liver cancer) (87). Furthermore, compared to monotherapy, an anti-FGL1 mAb or anti-LAG-3 mAb in combination with an anti-B7-H1 mAb significantly reduces tumor burden and prolongs survival (87).
An early study in 1996 indicated that mice lacking the lag3 gene exhibited reduced lysis of Yac-1 cells; in addition, polyclonal antibodies against LAG-3 could reduce NK-mediated lysis of Yac-1 cells but leave MHC-II-deficient target cells intact, suggesting the existence of an independent mode of natural killing through LAG-3 (96). However, this independent mode of natural killing has not been observed with human NK cells, and blockade of LAG-3 has no effect on the natural killing of various target cells (97). Notably, although studies on LAG-3+ NK cells are limited, the role of these cells in antitumor immunity should not be neglected. A study showed that using IL-12 to boost the cytotoxicity of NK cells in a lung cancer model (BALB/c mice injected with 4T1 cells) increased the NK cell population expressing high levels of coinhibitory molecules, including LAG-3, which limited NK cell-mediated antimetastatic activity (98). The combination of an anti-LAG-3 mAb with IL-12 significantly reduces lung metastasis, whereas monotherapy fails to achieve this effect (98). Furthermore, synergy between an anti-LAG-3 mAb and IL-12 contributes to the increased efficacy of IL-12 immunotherapy in breast cancer, which is solely dependent on NK cells, suggesting that LAG-3 is applicable in not only T cell-mediated immunotherapies but also NK cell-mediated antimetastatic immunotherapies (98).
TIM-3
TIM-3 is a type I glycoprotein that binds galectin-9 (Gal-9), high mobility group box 1 protein (HMGB1), and carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM-1) on target cells to act as an NK cell coreceptor (91, 99, 100). An early in vitro study showed that an NK92 cell line overexpressing TIM-3 secreted an increased amount of IFN-γ, while TIM-3 blockade resulted in reduced IFN-γ production (101). However, although human TIM-3+ NK cells are functional in terms of cytokine production and cytotoxicity, they become suppressed when TIM-3 is cross-linked with antibodies (102), suggesting that an interaction between TIM-3 and its ligand can result in NK cell dysfunction.
TIM-3 can mediate cell exhaustion and suppress immune responses under both chronic viral and cancerous conditions. For example, TIM-3 mediates suppression of NK cells in chronic hepatitis B patients, while TIM-3 blockade results in increased NK cell cytotoxicity both in vitro and ex vivo (103). TIM-3 is highly expressed in various tumor types, including gastrointestinal stromal tumor (104), lung adenocarcinoma (105), perineural squamous cell carcinoma (106), melanoma (107), gastric cancer (108), acute myeloid leukemia (109), colon cancer (110), bladder cancer (70), renal cell carcinoma (111), pancreatic cancer (112), glioma (113), anaplastic thyroid cancer (114), peripheral T cell lymphoma, NK/T cell lymphoma (94), etc. Significant overexpression of TIM-3 has been observed in peripheral NK cells from non-muscle invasive bladder cancer patients (70); moreover, TIM-3 is expressed in TILs from 25.3% of non-small cell lung cancer patients and associated with the expression of PD-1 and LAG-3 (93). TIM-3 expression was found to be higher on peripheral NK cells from glioma patients than on those from healthy controls, and these TIM-3+ NK cells showed a reduced capability for IFN-γ production and correlated with the proportion of Ki-67+ tumor cells (113). Furthermore, TIM-3 expression is upregulated on NK cells in late-stage melanoma patients, and blockade of TIM-3 reverses NK cell exhaustion in these patients (107).
TIM-3 functions as a potential prognostic marker in several tumor types. Upregulated expression of TIM-3 in peripheral NK cells from lung adenocarcinoma patients correlates with decreased overall survival, while blockade of TIM-3 enhances cytotoxicity and IFN-γ production in peripheral NK cells (105). Overexpression of TIM-3 in NK cells from gastric cancer patients has been associated with an advanced tumor stage (108). In addition, a study found that endometrial tumor-resident CD103+ NK cells expressed higher levels of TIM-3 than circulating CD103− NK cells, and tumor NK cells from patients with lymph node invasion showed significantly higher expression of TIM-3 than those from patients with no lymph node invasion (71). Bladder cancer patients have high levels of TIM-3+ NK cells and Gal-9+ tumor cells, and patients with relatively low levels of TIM-3+ NK cells and Gal-9+ tumor cells have an improved prognosis (115). Furthermore, TIM-3+ NK cells are defective in esophageal cancer, and relatively high TIM-3 expression on NK cells correlates with a poor prognosis in esophageal carcinoma (116).
However, contradictorily, studies have also reported stimulatory functions of TIM-3 (117). For example, after short-term stimulation with anti-CD3/CD28 antibodies, TIM-3 can enhance the secretion of IL-2 and signaling pathways that lead to T-cell activation (118, 119). TIM-3 engagement during antigen stimulation directly promoted CD8 T cell differentiation through mTORC1 (120). Furthermore, activation of human T cells was not affected by the presence of Gal-9 or antibodies to TIM-3 (121), which also reported a contradictory role of Gal-9 as a ligand for TIM-3. These studies suggest that the use of anti-TIM-3 should be particularly careful because TIM-3 also plays an activating role under certain circumstances, whether its antibodies act as agonist or antagonist remains to be questioned. Both anti-murine and anti-human TIM-3 antibodies bind to TIM-3 in a manner that interfere with the binding of TIM-3 to both phosphatidylserine and CEACAM1, the understanding of the interaction between TIM-3 and its ligands plays an important role in the screening of anti-TIM-3 antibody candidates (122).
Some studies have explored the reasons underlying the upregulation of TIM-3 expression on NK cells in the tumor microenvironment. One study reported that the LPHN1/PKC/mTOR-TIM-3-Gal-9 pathway in human acute myeloid leukemia induced high levels of Gal-9 secretion and the release of soluble TIM-3 (109). Gal-9 impaired the killing of tumor cells by NK cells, whereas soluble TIM-3 impaired the ability of T cells to produce IL-2, contributing to the breakdown of immune surveillance and thus to the progression of tumors (109). Another proposed mechanism is that MHC-I-deficient tumors induce coexpression of TIM-3 and PD-1 on NK cells, resulting in functional NK cell exhaustion in both tumor-bearing mice and cancer patients; functional recovery in these exhausted NK cells induced by vaccination requires IL-21 produced by NKT cells (110). Furthermore, carcinoma-associated fibroblasts (CAFs) have been shown to promote the expression of TIM-3 in pancreatic cancer patients (112). Sustained IL-15 stimulation upregulates TIM-3 expression on both T and NK cells (123). TIM-3 expression can also be induced by TNF-α through the NF-κB signaling pathway (116). In addition, IL-27/NFIL3 signaling axis has been identified crucial for the induction of Tim-3, IL-10 and T-cell dysfunction (124).
Perspective
The success of mAbs targeting CTLA-4 and PD-1 has shed light on cancer immunotherapy, and restoring exhausted T cells has shown promising clinical outcomes in some patients. However, there were still many patients who are nonresponsive to these treatments. However, recent findings indicate that improved survival highly correlates with the frequency of DNAM-1+CD56dim NK and NKp46+CD56dim NK cells after treatment with anti-CTLA-4 in patients with malignant mesothelioma (125). Hsu and Hodgins et al. demonstrated in multiple tumor models that PD-1 is upregulated on the most activated and functionally responsive intratumoral NK cells, suggesting that the efficacy of PD-1 blockade depends in part on inducing an NK cell-based antitumor response (126). Thereof, we propose that NK cell-targeted immunotherapy may provide an alternative or complementary approach to overcome the limitations of T cell immunotherapy and combination with NK cell immunotherapy could increase the response rate of T cell-targeted treatments. NK cells are critical for immunosurveillance, particularly in the control of metastasis and hematological cancers. A study by the Tian group indicated the importance of NK cells in checkpoint immunotherapy; TIGIT blockade prevented NK cell exhaustion in the absence of T cells and B cells, and an anti-TIGIT mAb improved T cell responses in an NK cell-dependent manner (13). These findings suggest that certain checkpoint molecules expressed by both T cells and NK cells may exert a greater effect on NK cells than on T cells and that NK cells could be essential for the T cell-mediated antitumor response in such a scenario.
The recent success of anti-NKG2A mAb on clinical trials unleashes its role as a promising checkpoint inhibitor in treating cancers with minimum side effects, and the success of NKG2A blockade also points out the importance of NK cells in anti-tumor immunity and advances the idea that combined reversal of both T and NK cell exhaustion is truly important in anti-tumor immunotherapy. Anti-NKG2A could be the third potential checkpoint inhibitor approved by the FDA following anti-CTLA-4 and anti-PD-1/PD-L1. Another promising checkpoint molecule targeting NK cells is TIGIT based on the results observed by the Tian group (13). However, due to its constitutive expression on peripheral human NK cells, more studies are required to fulfill its role as a checkpoint in treating cancer. The balance between CD96, TIGIT, and CD226 is critical for proper immune responses by NK cells. The accumulation of CD96 in NK cells in hepatocellular carcinoma patients disrupts the balance between these three receptors, which subsequently results in NK cell dysfunction and exhaustion (65); therefore, careful examination of the CD96-TIGIT-CD226 system should be involved in developing immunotherapies targeting these receptors. In addition to CD155, CD112, and CD113, human TIGIT can also bind to the Fap2 protein of Fusobacterium nucleatum, and the interaction between TIGIT and Fap2 inhibits NK cell cytotoxicity (127). This finding unleashes a new mechanism of tumor immune evasion that depends on the bacterium and identifies new possibilities for NK cell immunotherapy. Alterations in KIR and HLA gene loci affect NK cell functions, which should be considered when developing immunotherapies against KIRs (46). The interaction between LAG-3 and the newly defined ligand FGL1 suppresses the functions of T cells, and whether FGL1 also interacts with LAG-3 on NK cells merits further research (87).
Other checkpoint molecules on NK cells have been proven to be potential targets, and further experiments are needed to prove these novel targets for NK-based immunotherapy (22, 128). There are ways to improve the efficacy of NK cell immunotherapy. For example, NKG2Anull NK cells are more effective than NKG2A+ NK cells treated with an anti-NKG2A mAb, suggesting a new immunotherapeutic approach using NKG2Anull NK cells (39, 129). In addition, cytokines can enhance the efficacy of mAbs (130), and a combination of cytokine treatment with checkpoint immunotherapy may boost the effects of mAbs. Furthermore, we believe that combined blockade of checkpoint molecules expressed by T cells and NK cells could unleash antitumor immunity mediated by innate and adaptive populations, which not only improve overall antitumor immune responses but also allow the two approaches to complement each other; this strategy might be the solution for the “non-responders.” Accumulating evidence suggests that NK cell-targeted immunotherapy is highly feasible; however, our knowledge of the inhibitory mechanisms in NK cells is still inadequate, and more fundamental research is required to identify the best inhibitory pathways to be targeted for future clinical applications.
Author Contributions
HS and CS were involved in the search and analysis of the literature, design and writing of the manuscript, and revision of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (#31670908, #81701631), and Fundamental Research Funds for the Central Universities (#WK2070000107).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
NK, Natural killer; CTL, Cytotoxic T lymphocyte; PBMC, Peripheral blood mononuclear cell; MDSC, Myeloid-derived suppressor cell; mAb, Monoclonal antibody; IFN, Interferon; TNF, Tumor necrosis factor; IL, Interleukin; KO, Knockout; HLA, Human leukocyte antigen; ADCC, Antibody-dependent cell-mediated cytotoxicity; MHC, Major histocompatibility complex; FDA, Food and Drug Administration; MCA, Methylcholanthrene; PD-1, Programmed death-1; CTLA-4, Cytotoxic T-lymphocyte-associated protein 4; TIM-3, T cell immunoglobulin and mucin domain 3; TIGIT, T cell immunoreceptor with Ig and ITIM domains; KIR, Killer cell immunoglobulin-like receptor; LAG-3, Lymphocyte activating gene 3; FGL1, Fibrinogen-like protein 1; NKG2A, NK group 2 member A; Gal-9, Galectin-9; CAF, Carcinoma-associated fibroblast; PEBL, Protein expression blocker; HMGB1, High mobility group box 1 protein; CEACAM-1, Carcinoembryonic antigen-related cell adhesion molecule 1.
References
1. Souza-Fonseca-Guimaraes F, Cursons J, Huntington ND. The emergence of natural killer cells as a major target in cancer immunotherapy. Trends Immunol. (2019) 40:142–58. doi: 10.1016/j.it.2018.12.003
2. Zou W. Mechanistic insights into cancer immunity and immunotherapy. Cell Mol Immunol. (2018) 15:419–20. doi: 10.1038/s41423-018-0011-5
3. Crespo J, Sun H, Welling TH, Tian Z, Zou W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol. (2013) 25:214–21. doi: 10.1016/j.coi.2012.12.003
4. Sun H, Xu J, Huang M, Huang Q, Sun R, Xiao W, et al. CD200R, a co-inhibitory receptor on immune cells, predicts the prognosis of human hepatocellular carcinoma. Immunol Lett. (2016) 178:105–13. doi: 10.1016/j.imlet.2016.08.009
5. Xu W, Hieu T, Malarkannan S, Wang L. The structure, expression, and multifaceted role of immune-checkpoint protein VISTA as a critical regulator of anti-tumor immunity, autoimmunity, and inflammation. Cell Mol Immunol. (2018) 15:438–46. doi: 10.1038/cmi.2017.148
6. Zhai L, Ladomersky E, Lenzen A, Nguyen B, Patel R, Lauing KL, et al. IDO1 in cancer: a Gemini of immune checkpoints. Cell Mol Immunol. (2018) 15:447–57. doi: 10.1038/cmi.2017.143
7. Burugu S, Dancsok AR, Nielsen TO. Emerging targets in cancer immunotherapy. Semin Cancer Biol. (2018) 52:39–52. doi: 10.1016/j.semcancer.2017.10.001
8. Darvin P, Toor SM, Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med. (2018) 50:165. doi: 10.1038/s12276-018-0191-1
9. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. (2013) 369:122–33. doi: 10.1056/NEJMoa1302369
10. Hodi FS, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Cowey CL, et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. (2018) 19:1480–92. doi: 10.1016/S1470-2045(18)30700-9
11. Turajlic S, Gore M, Larkin J. First report of overall survival for ipilimumab plus nivolumab from the phase III Checkmate 067 study in advanced melanoma. Ann Oncol. (2018) 29:542–3. doi: 10.1093/annonc/mdy020
12. Stojanovic A, Cerwenka A. Checkpoint inhibition: NK cells enter the scene. Nat Immunol. (2018) 19:650–2. doi: 10.1038/s41590-018-0142-y
13. Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. (2018) 19:723–32. doi: 10.1038/s41590-018-0132-0
14. Liu Y, Zheng J, Liu Y, Wen L, Huang L, Xiang Z, et al. Uncompromised NK cell activation is essential for virus-specific CTL activity during acute influenza virus infection. Cell Mol Immunol. (2018) 15:827–37. doi: 10.1038/cmi.2017.10
15. Poznanski SM, Ashkar AA. Shining light on the significance of NK cell CD56 brightness. Cell Mol Immunol. (2018) 15:1071–3. doi: 10.1038/s41423-018-0163-3
16. Wu LS, Wang JY. Warm up, cool down, and tearing apart in NK cell memory. Cell Mol Immunol. (2018) 15:1095–7. doi: 10.1038/s41423-018-0188-7
17. Chen Y, Chen B, Yang T, Xiao W, Qian L, Ding Y, et al. Human fused NKG2D-IL-15 protein controls xenografted human gastric cancer through the recruitment and activation of NK cells. Cell Mol Immunol. (2017) 14:293–307. doi: 10.1038/cmi.2015.81
18. Kwon B. IFN-gamma in tissue-immune homeostasis and antitumor immunity. Cell Mol Immunol. (2018) 15:531–2. doi: 10.1038/cmi.2017.95
19. Chyuan IT, Hsu PN. Tumor necrosis factor: the key to hepatitis B viral clearance. Cell Mol Immunol. (2018) 15:731–3. doi: 10.1038/cmi.2017.139
20. Zhang QF, Yin WW, Xia Y, Yi YY, He QF, Wang X, et al. Liver-infiltrating CD11b(-)CD27(-) NK subsets account for NK-cell dysfunction in patients with hepatocellular carcinoma and are associated with tumor progression. Cell Mol Immunol. (2017) 14:819–29. doi: 10.1038/cmi.2016.28
21. Sun C, Sun HY, Xiao WH, Zhang C, Tian ZG. Natural killer cell dysfunction in hepatocellular carcinoma and NK cell-based immunotherapy. Acta Pharmacol Sin. (2015) 36:1191–9. doi: 10.1038/aps.2015.41
22. Sun H, Xu J, Huang Q, Huang M, Li K, Qu K, et al. Reduced CD160 expression contributes to impaired NK-cell function and poor clinical outcomes in patients with HCC. Cancer Res. (2018) 78:6581–93. doi: 10.1158/0008-5472.CAN-18-1049
23. Haanen JB, Cerundolo V. NKG2A, a new kid on the immune checkpoint block. Cell. (2018) 175:1720–2. doi: 10.1016/j.cell.2018.11.048
24. Lazetic S, Chang C, Houchins JP, Lanier LL, Phillips JH. Human natural killer cell receptors involved in MHC class I recognition are disulfide-linked heterodimers of CD94 and NKG2 subunits. J Immunol. (1996) 157:4741–5.
25. Braud VM, Allan DS, O'Callaghan CA, Soderstrom K, D'Andrea A, Ogg GS, et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature. (1998) 391:795–9. doi: 10.1038/35869
26. Lee N, Llano M, Carretero M, Ishitani A, Navarro F, Lopez-Botet M, et al. HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc Natl Acad Sci USA. (1998) 95:5199–204. doi: 10.1073/pnas.95.9.5199
27. Houchins JP, Lanier LL, Niemi EC, Phillips JH, Ryan JC. Natural killer cell cytolytic activity is inhibited by NKG2-A and activated by NKG2-C. J Immunol. (1997) 158:3603–9.
28. Platonova S, Cherfils-Vicini J, Damotte D, Crozet L, Vieillard V, Validire P, et al. Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res. (2011) 71:5412–22. doi: 10.1158/0008-5472.CAN-10-4179
29. Gillard-Bocquet M, Caer C, Cagnard N, Crozet L, Perez M, Fridman WH, et al. Lung tumor microenvironment induces specific gene expression signature in intratumoral NK cells. Front Immunol. (2013) 4:19. doi: 10.3389/fimmu.2013.00019
30. Mamessier E, Sylvain A, Thibult ML, Houvenaeghel G, Jacquemier J, Castellano R, et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J Clin Invest. (2011) 121:3609–22. doi: 10.1172/JCI45816
31. Meckawy GR, Mohamed AM, Zaki WK, Khattab MA, Amin MM, ElDeeb MA, et al. Natural killer NKG2A and NKG2D in patients with colorectal cancer. J Gastrointest Oncol. (2019) 10:218–25. doi: 10.21037/jgo.2018.12.13
32. Andre P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell. (2018) 175:1731–43 e13. doi: 10.1016/j.cell.2018.10.014
33. Stringaris K, Sekine T, Khoder A, Alsuliman A, Razzaghi B, Sargeant R, et al. Leukemia-induced phenotypic and functional defects in natural killer cells predict failure to achieve remission in acute myeloid leukemia. Haematologica. (2014) 99:836–47. doi: 10.3324/haematol.2013.087536
34. Sandoval-Borrego D, Moreno-Lafont MC, Vazquez-Sanchez EA, Gutierrez-Hoya A, Lopez-Santiago R, Montiel-Cervantes LA, et al. Overexpression of CD158 and NKG2A inhibitory receptors and underexpression of NKG2D and NKp46 activating receptors on NK cells in acute myeloid leukemia. Arch Med Res. (2016) 47:55–64. doi: 10.1016/j.arcmed.2016.02.001
35. Sun C, Xu J, Huang Q, Huang M, Wen H, Zhang C, et al. High NKG2A expression contributes to NK cell exhaustion and predicts a poor prognosis of patients with liver cancer. Oncoimmunology. (2017) 6:e1264562. doi: 10.1080/2162402X.2016.1264562
36. Frazao A, Messaoudene M, Nunez N, Dulphy N, Roussin F, Sedlik C, et al. CD16(+)NKG2A(high) natural killer cells infiltrate breast cancer-draining lymph nodes. Cancer Immunol Res. (2019) 7:208–18. doi: 10.1158/2326-6066.CIR-18-0085
37. McWilliams EM, Mele JM, Cheney C, Timmerman EA, Fiazuddin F, Strattan EJ, et al. Therapeutic CD94/NKG2A blockade improves natural killer cell dysfunction in chronic lymphocytic leukemia. Oncoimmunology. (2016) 5:e1226720. doi: 10.1080/2162402X.2016.1226720
38. Ruggeri L, Urbani E, Andre P, Mancusi A, Tosti A, Topini F, et al. Effects of anti-NKG2A antibody administration on leukemia and normal hematopoietic cells. Haematologica. (2016) 101:626–33. doi: 10.3324/haematol.2015.135301
39. Kamiya T, Seow SV, Wong D, Robinson M, Campana D. Blocking expression of inhibitory receptor NKG2A overcomes tumor resistance to NK cells. J Clin Invest. (2019) 130:2094–106. doi: 10.1172/JCI123955
40. Mahaweni NM, Ehlers FAI, Bos GMJ, Wieten L. Tuning natural killer cell anti-multiple myeloma reactivity by targeting inhibitory signaling via KIR and NKG2A. Front Immunol. (2018) 9:2848. doi: 10.3389/fimmu.2018.02848
41. Colonna M, Spies T, Strominger JL, Ciccone E, Moretta A, Moretta L, et al. Alloantigen recognition by two human natural killer cell clones is associated with HLA-C or a closely linked gene. Proc Natl Acad Sci USA. (1992) 89:7983–5. doi: 10.1073/pnas.89.17.7983
42. He Y, Tian Z. NK cell education via nonclassical MHC and non-MHC ligands. Cell Mol Immunol. (2017) 14:321–30. doi: 10.1038/cmi.2016.26
43. Jobim MR, Jobim M, Salim PH, Portela P, Jobim LF, Leistner-Segal S, et al. Analysis of KIR gene frequencies and HLA class I genotypes in breast cancer and control group. Hum Immunol. (2013) 74:1130–3. doi: 10.1016/j.humimm.2013.06.021
44. He Y, Bunn PA, Zhou C, Chan D. KIR 2D (L1, L3, L4, S4) and KIR 3DL1 protein expression in non-small cell lung cancer. Oncotarget. (2016) 7:82104–11. doi: 10.18632/oncotarget.13486
45. Vineretsky KA, Karagas MR, Christensen BC, Kuriger-Laber JK, Perry AE, Storm CA, et al. Skin cancer risk is modified by KIR/HLA interactions that influence the activation of natural killer immune cells. Cancer Res. (2016) 76:370–6. doi: 10.1158/0008-5472.CAN-15-0547
46. Cornillet M, Jansson H, Schaffer M, Hertwig L, Berglin L, Zimmer CL, et al. Imbalance of genes encoding natural killer immunoglobulin-like receptors and human leukocyte antigen in patients with biliary cancer. Gastroenterology. (2019) 157:1067–80 e9. doi: 10.1053/j.gastro.2019.06.023
47. Trefny MP, Rothschild SI, Uhlenbrock F, Rieder D, Kasenda B, Stanczak MA, et al. A variant of a killer cell immunoglobulin-like receptor is associated with resistance to PD-1 blockade in lung cancer. Clin Cancer Res. (2019) 25:3026–34. doi: 10.1158/1078-0432.CCR-18-3041
48. Romagne F, Andre P, Spee P, Zahn S, Anfossi N, Gauthier L, et al. Preclinical characterization of 1-7F9, a novel human anti-KIR receptor therapeutic antibody that augments natural killer-mediated killing of tumor cells. Blood. (2009) 114:2667–77. doi: 10.1182/blood-2009-02-206532
49. Benson DM Jr, Bakan CE, Zhang S, Collins SM, Liang J, Srivastava S, et al. IPH2101, a novel anti-inhibitory KIR antibody, and lenalidomide combine to enhance the natural killer cell versus multiple myeloma effect. Blood. (2011) 118:6387–91. doi: 10.1182/blood-2011-06-360255
50. Vey N, Bourhis JH, Boissel N, Bordessoule D, Prebet T, Charbonnier A, et al. A phase 1 trial of the anti-inhibitory KIR mAb IPH2101 for AML in complete remission. Blood. (2012) 120:4317–23. doi: 10.1182/blood-2012-06-437558
51. Vey N, Karlin L, Sadot-Lebouvier S, Broussais F, Berton-Rigaud D, Rey J, et al. A phase 1 study of lirilumab (antibody against killer immunoglobulin-like receptor antibody KIR2D; IPH2102) in patients with solid tumors and hematologic malignancies. Oncotarget. (2018) 9:17675–88. doi: 10.18632/oncotarget.24832
52. Carlsten M, Korde N, Kotecha R, Reger R, Bor S, Kazandjian D, et al. Checkpoint inhibition of KIR2D with the monoclonal antibody IPH2101 induces contraction and hyporesponsiveness of NK cells in patients with myeloma. Clin Cancer Res. (2016) 22:5211–22. doi: 10.1158/1078-0432.CCR-16-1108
53. Bagot M, Porcu P, Marie-Cardine A, Battistella M, William BM, Vermeer M, et al. IPH4102, a first-in-class anti-KIR3DL2 monoclonal antibody, in patients with relapsed or refractory cutaneous T-cell lymphoma: an international, first-in-human, open-label, phase 1 trial. Lancet Oncol. (2019) 20:1160–70. doi: 10.1016/S1470-2045(19)30320-1
54. Ewen EM, Pahl JHW, Miller M, Watzl C, Cerwenka A. KIR downregulation by IL-12/15/18 unleashes human NK cells from KIR/HLA-I inhibition and enhances killing of tumor cells. Eur J Immunol. (2018) 48:355–65. doi: 10.1002/eji.201747128
55. Solomon BL, Garrido-Laguna I. TIGIT: a novel immunotherapy target moving from bench to bedside. Cancer Immunol Immunother. (2018) 67:1659–67. doi: 10.1007/s00262-018-2246-5
56. Dougall WC, Kurtulus S, Smyth MJ, Anderson AC. TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol Rev. (2017) 276:112–20. doi: 10.1111/imr.12518
57. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. (2009) 10:48–57. doi: 10.1038/ni.1674
58. Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci USA. (2009) 106:17858–63. doi: 10.1073/pnas.0903474106
59. Stanietsky N, Rovis TL, Glasner A, Seidel E, Tsukerman P, Yamin R, et al. Mouse TIGIT inhibits NK-cell cytotoxicity upon interaction with PVR. Eur J Immunol. (2013) 43:2138–50. doi: 10.1002/eji.201243072
60. Li M, Xia P, Du Y, Liu S, Huang G, Chen J, et al. T-cell immunoglobulin and ITIM domain (TIGIT) receptor/poliovirus receptor (PVR) ligand engagement suppresses interferon-gamma production of natural killer cells via beta-arrestin 2-mediated negative signaling. J Biol Chem. (2014) 289:17647–57. doi: 10.1074/jbc.M114.572420
61. Wang F, Hou H, Wu S, Tang Q, Liu W, Huang M, et al. TIGIT expression levels on human NK cells correlate with functional heterogeneity among healthy individuals. Eur J Immunol. (2015) 45:2886–97. doi: 10.1002/eji.201545480
62. Blessin NC, Simon R, Kluth M, Fischer K, Hube-Magg C, Li W, et al. Patterns of TIGIT expression in lymphatic tissue, inflammation, and cancer. Dis Markers. (2019) 2019:5160565. doi: 10.1155/2019/5160565
63. Blake SJ, Stannard K, Liu J, Allen S, Yong MC, Mittal D, et al. Suppression of metastases using a new lymphocyte checkpoint target for cancer immunotherapy. Cancer Discov. (2016) 6:446–59. doi: 10.1158/2159-8290.CD-15-0944
64. Peng YP, Xi CH, Zhu Y, Yin LD, Wei JS, Zhang JJ, et al. Altered expression of CD226 and CD96 on natural killer cells in patients with pancreatic cancer. Oncotarget. (2016) 7:66586–94. doi: 10.18632/oncotarget.11953
65. Sun H, Huang Q, Huang M, Wen H, Lin R, Zheng M, et al. Human CD96 correlates to natural killer cell exhaustion and predicts the prognosis of human hepatocellular carcinoma. Hepatology. (2018) 70:168–83. doi: 10.1002/hep.30347
66. Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, et al. TIGIT and PD-1 impair tumor antigen-specific CD8(+) T cells in melanoma patients. J Clin Invest. (2015) 125:2046–58. doi: 10.1172/JCI80445
67. Catakovic K, Gassner FJ, Ratswohl C, Zaborsky N, Rebhandl S, Schubert M, et al. TIGIT expressing CD4+T cells represent a tumor-supportive T cell subset in chronic lymphocytic leukemia. Oncoimmunology. (2017) 7:e1371399. doi: 10.1080/2162402X.2017.1371399
68. Zhou XM, Li WQ, Wu YH, Han L, Cao XG, Yang XM, et al. Intrinsic expression of immune checkpoint molecule TIGIT could help tumor growth in vivo by suppressing the function of NK and CD8(+) T cells. Front Immunol. (2018) 9:2821. doi: 10.3389/fimmu.2018.02821
69. Xu F, Sunderland A, Zhou Y, Schulick RD, Edil BH, Zhu Y. Blockade of CD112R and TIGIT signaling sensitizes human natural killer cell functions. Cancer Immunol Immunother. (2017) 66:1367–75. doi: 10.1007/s00262-017-2031-x
70. Audenet F, Farkas AM, Anastos H, Galsky MD, Bhardwaj N, Sfakianos JP. Immune phenotype of peripheral blood mononuclear cells in patients with high-risk non-muscle invasive bladder cancer. World J Urol. (2018) 36:1741–8. doi: 10.1007/s00345-018-2359-7
71. Degos C, Heinemann M, Barrou J, Boucherit N, Lambaudie E, Savina A, et al. Endometrial tumor microenvironment alters human NK cell recruitment, and resident NK cell phenotype and function. Front Immunol. (2019) 10:877. doi: 10.3389/fimmu.2019.00877
72. Sarhan D, Cichocki F, Zhang B, Yingst A, Spellman SR, Cooley S, et al. Adaptive NK cells with low TIGIT expression are inherently resistant to myeloid-derived suppressor cells. Cancer Res. (2016) 76:5696–706. doi: 10.1158/0008-5472.CAN-16-0839
73. Georgiev H, Ravens I, Papadogianni G, Bernhardt G. Coming of age: CD96 emerges as modulator of immune responses. Front Immunol. (2018) 9:1072. doi: 10.3389/fimmu.2018.01072
74. Minton K. Natural killer cells: a TACTILE restraint. Nat Rev Immunol. (2014) 14:285. doi: 10.1038/nri3664
75. Fuchs A, Cella M, Giurisato E, Shaw AS, Colonna M. Cutting edge: CD96 (tactile) promotes NK cell-target cell adhesion by interacting with the poliovirus receptor (CD155). J Immunol. (2004) 172:3994–8. doi: 10.4049/jimmunol.172.7.3994
76. Bernhardt G. TACTILE becomes tangible: CD96 discloses its inhibitory peculiarities. Nat Immunol. (2014) 15:406–8. doi: 10.1038/ni.2855
77. Chan CJ, Martinet L, Gilfillan S, Souza-Fonseca-Guimaraes F, Chow MT, Town L, et al. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat Immunol. (2014) 15:431–8. doi: 10.1038/ni.2850
78. Barrow AD, Edeling MA, Trifonov V, Luo J, Goyal P, Bohl B, et al. Natural killer cells control tumor growth by sensing a growth factor. Cell. (2018) 172:534–48 e19. doi: 10.1016/j.cell.2017.11.037
79. Deuss FA, Watson GM, Fu Z, Rossjohn J, Berry R. Structural basis for CD96 immune receptor recognition of nectin-like protein-5, CD155. Structure. (2019) 27:219–28 e3. doi: 10.1016/j.str.2018.10.023
80. Roman Aguilera A, Lutzky VP, Mittal D, Li XY, Stannard K, Takeda K, et al. CD96 targeted antibodies need not block CD96-CD155 interactions to promote NK cell anti-metastatic activity. Oncoimmunology. (2018) 7:e1424677. doi: 10.1080/2162402X.2018.1424677
81. Karade SS, Mariuzza RA. How natural killer cell receptors stick to cell-cell adhesion proteins. Structure. (2019) 27:209–10. doi: 10.1016/j.str.2019.01.007
82. Brooks J, Fleischmann-Mundt B, Woller N, Niemann J, Ribback S, Peters K, et al. Perioperative, spatiotemporally coordinated activation of T and NK cells prevents recurrence of pancreatic cancer. Cancer Res. (2018) 78:475–88. doi: 10.1158/0008-5472.CAN-17-2415
83. Vujanovic L, Chuckran C, Lin Y, Ding F, Sander CA, Santos PM, et al. CD56(dim) CD16(-) natural killer cell profiling in melanoma patients receiving a cancer vaccine and interferon-alpha. Front Immunol. (2019) 10:14. doi: 10.3389/fimmu.2019.00014
84. Baixeras E, Huard B, Miossec C, Jitsukawa S, Martin M, Hercend T, et al. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J Exp Med. (1992) 176:327–37. doi: 10.1084/jem.176.2.327
85. Sun H, Sun C, Xiao W. Expression regulation of co-inhibitory molecules on human natural killer cells in response to cytokine stimulations. Cytokine. (2014) 65:33–41. doi: 10.1016/j.cyto.2013.09.016
86. Xu F, Liu J, Liu D, Liu B, Wang M, Hu Z, et al. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer Res. (2014) 74:3418–28. doi: 10.1158/0008-5472.CAN-13-2690
87. Wang J, Sanmamed MF, Datar I, Su TT, Ji L, Sun J, et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell. (2019) 176:334–47 e12. doi: 10.1016/j.cell.2018.11.010
88. Workman CJ, Vignali DA. The CD4-related molecule, LAG-3 (CD223), regulates the expansion of activated T cells. Eur J Immunol. (2003) 33:970–9. doi: 10.1002/eji.200323382
89. Workman CJ, Cauley LS, Kim IJ, Blackman MA, Woodland DL, Vignali DA. Lymphocyte activation gene-3 (CD223) regulates the size of the expanding T cell population following antigen activation in vivo. J Immunol. (2004) 172:5450–5. doi: 10.4049/jimmunol.172.9.5450
90. Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. (2009) 10:29–37. doi: 10.1038/ni.1679
91. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity. (2016) 44:989–1004. doi: 10.1016/j.immuni.2016.05.001
92. Mishra AK, Kadoishi T, Wang X, Driver E, Chen Z, Wang XJ, et al. Squamous cell carcinomas escape immune surveillance via inducing chronic activation and exhaustion of CD8+ T cells co-expressing PD-1 and LAG-3 inhibitory receptors. Oncotarget. (2016) 7:81341–56. doi: 10.18632/oncotarget.13228
93. Datar I, Sanmamed MF, Wang J, Henick BS, Choi J, Badri T, et al. Expression analysis and significance of PD-1, LAG-3, and TIM-3 in human non-small cell lung cancer using spatially resolved and multiparametric single-cell analysis. Clin Cancer Res. (2019) 25:4663–73. doi: 10.1158/1078-0432.CCR-18-4142
94. Liao Z, Lv X, Liu S, He Z, Chen S, Wang L, et al. Different aberrant expression pattern of immune checkpoint receptors in patients with PTCL and NK/T-CL. Asia Pac J Clin Oncol. (2018) 14:e252–8. doi: 10.1111/ajco.12850
95. Johnson DB, Nixon MJ, Wang Y, Wang DY, Castellanos E, Estrada MV, et al. Tumor-specific MHC-II expression drives a unique pattern of resistance to immunotherapy via LAG-3/FCRL6 engagement. JCI Insight. (2018) 3:120360. doi: 10.1172/jci.insight.120360
96. Miyazaki T, Dierich A, Benoist C, Mathis D. Independent modes of natural killing distinguished in mice lacking Lag3. Science. (1996) 272:405–8. doi: 10.1126/science.272.5260.405
97. Huard B, Tournier M, Triebel F. LAG-3 does not define a specific mode of natural killing in human. Immunol Lett. (1998) 61:109–12. doi: 10.1016/S0165-2478(97)00170-3
98. Ohs I, Ducimetiere L, Marinho J, Kulig P, Becher B, Tugues S. Restoration of natural killer cell antimetastatic activity by IL12 and checkpoint blockade. Cancer Res. (2017) 77:7059–71. doi: 10.1158/0008-5472.CAN-17-1032
99. Li Y, Li D, Du M. TIM-3: a crucial regulator of NK cells in pregnancy. Cell Mol Immunol. (2017) 14:948–50. doi: 10.1038/cmi.2017.85
100. Bertheloot D, Latz E. HMGB1, IL-1alpha, IL-33 and S100 proteins: dual-function alarmins. Cell Mol Immunol. (2017) 14:43–64. doi: 10.1038/cmi.2016.34
101. Gleason MK, Lenvik TR, McCullar V, Felices M, O'Brien MS, Cooley SA, et al. Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood. (2012) 119:3064–72. doi: 10.1182/blood-2011-06-360321
102. Ndhlovu LC, Lopez-Verges S, Barbour JD, Jones RB, Jha AR, Long BR, et al. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood. (2012) 119:3734–43. doi: 10.1182/blood-2011-11-392951
103. Ju Y, Hou N, Meng J, Wang X, Zhang X, Zhao D, et al. T cell immunoglobulin- and mucin-domain-containing molecule-3 (Tim-3) mediates natural killer cell suppression in chronic hepatitis B. J Hepatol. (2010) 52:322–9. doi: 10.1016/j.jhep.2009.12.005
104. Komita H, Koido S, Hayashi K, Kan S, Ito M, Kamata Y, et al. Expression of immune checkpoint molecules of T cell immunoglobulin and mucin protein 3/galectin-9 for NK cell suppression in human gastrointestinal stromal tumors. Oncol Rep. (2015) 34:2099–105. doi: 10.3892/or.2015.4149
105. Xu L, Huang Y, Tan L, Yu W, Chen D, Lu C, et al. Increased Tim-3 expression in peripheral NK cells predicts a poorer prognosis and Tim-3 blockade improves NK cell-mediated cytotoxicity in human lung adenocarcinoma. Int Immunopharmacol. (2015) 29:635–41. doi: 10.1016/j.intimp.2015.09.017
106. Linedale R, Schmidt C, King BT, Ganko AG, Simpson F, Panizza BJ, et al. Elevated frequencies of CD8 T cells expressing PD-1, CTLA-4 and Tim-3 within tumour from perineural squamous cell carcinoma patients. PLoS ONE. (2017) 12:e0175755. doi: 10.1371/journal.pone.0175755
107. da Silva IP, Gallois A, Jimenez-Baranda S, Khan S, Anderson AC, Kuchroo VK, et al. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol Res. (2014) 2:410–22. doi: 10.1158/2326-6066.CIR-13-0171
108. Wang Z, Zhu J, Gu H, Yuan Y, Zhang B, Zhu D, et al. The clinical significance of abnormal Tim-3 expression on NK cells from patients with gastric cancer. Immunol Invest. (2015) 44:578–89. doi: 10.3109/08820139.2015.1052145
109. Goncalves Silva I, Yasinska IM, Sakhnevych SS, Fiedler W, Wellbrock J, Bardelli M, et al. The Tim-3-galectin-9 secretory pathway is involved in the immune escape of human acute myeloid leukemia cells. EBioMedicine. (2017) 22:44–57. doi: 10.1016/j.ebiom.2017.07.018
110. Seo H, Jeon I, Kim BS, Park M, Bae EA, Song B, et al. IL-21-mediated reversal of NK cell exhaustion facilitates anti-tumour immunity in MHC class I-deficient tumours. Nat Commun. (2017) 8:15776. doi: 10.1038/ncomms15776
111. Granier C, Dariane C, Combe P, Verkarre V, Urien S, Badoual C, et al. Tim-3 expression on tumor-infiltrating PD-1(+)CD8(+) T cells correlates with poor clinical outcome in renal cell carcinoma. Cancer Res. (2017) 77:1075–82. doi: 10.1158/0008-5472.CAN-16-0274
112. Gorchs L, Fernandez Moro C, Bankhead P, Kern KP, Sadeak I, Meng Q, et al. Human pancreatic carcinoma-associated fibroblasts promote expression of co-inhibitory markers on CD4(+) and CD8(+) T-cells. Front Immunol. (2019) 10:847. doi: 10.3389/fimmu.2019.00847
113. Li X, Wang B, Gu L, Zhang J, Li X, Gao L, et al. Tim-3 expression predicts the abnormal innate immune status and poor prognosis of glioma patients. Clin Chim Acta. (2018) 476:178–84. doi: 10.1016/j.cca.2017.11.022
114. Yin M, Di G, Bian M. Dysfunction of natural killer cells mediated by PD-1 and Tim-3 pathway in anaplastic thyroid cancer. Int Immunopharmacol. (2018) 64:333–9. doi: 10.1016/j.intimp.2018.09.016
115. Zhang W, Feng H, Chen Q, Lu X, Ge J. The functional potency of natural killer cells in response to IL-2/IL-15/IL-21 stimulation is limited by a concurrent upregulation of Tim-3 in bladder cancer. Exp Cell Res. (2018) 372:92–8. doi: 10.1016/j.yexcr.2018.09.013
116. Zheng Y, Li Y, Lian J, Yang H, Li F, Zhao S, et al. TNF-alpha-induced Tim-3 expression marks the dysfunction of infiltrating natural killer cells in human esophageal cancer. J Transl Med. (2019) 17:165. doi: 10.1186/s12967-019-1917-0
117. De Sousa Linhares A, Leitner J, Grabmeier-Pfistershammer K, Steinberger P. Not all immune checkpoints are created equal. Front Immunol. (2018) 9:1909. doi: 10.3389/fimmu.2018.01909
118. Lee J, Su EW, Zhu C, Hainline S, Phuah J, Moroco JA, et al. Phosphotyrosine-dependent coupling of Tim-3 to T-cell receptor signaling pathways. Mol Cell Biol. (2011) 31:3963–74. doi: 10.1128/MCB.05297-11
119. Tomkowicz B, Walsh E, Cotty A, Verona R, Sabins N, Kaplan F, et al. TIM-3 Suppresses anti-CD3/CD28-induced TCR activation and IL-2 expression through the NFAT signaling pathway. PLoS ONE. (2015) 10:e0140694. doi: 10.1371/journal.pone.0140694
120. Sabins NC, Chornoguz O, Leander K, Kaplan F, Carter R, Kinder M, et al. TIM-3 engagement promotes effector memory T cell differentiation of human antigen-specific CD8 T cells by activating mTORC1. J Immunol. (2017) 199:4091–102. doi: 10.4049/jimmunol.1701030
121. Leitner J, Rieger A, Pickl WF, Zlabinger G, Grabmeier-Pfistershammer K, Steinberger P. TIM-3 does not act as a receptor for galectin-9. PLoS Pathog. (2013) 9:e1003253. doi: 10.1371/journal.ppat.1003253
122. Sabatos-Peyton CA, Nevin J, Brock A, Venable JD, Tan DJ, Kassam N, et al. Blockade of Tim-3 binding to phosphatidylserine and CEACAM1 is a shared feature of anti-Tim-3 antibodies that have functional efficacy. Oncoimmunology. (2018) 7:e1385690. doi: 10.1080/2162402X.2017.1385690
123. Tallerico R, Cristiani CM, Staaf E, Garofalo C, Sottile R, Capone M, et al. IL-15, TIM-3 and NK cells subsets predict responsiveness to anti-CTLA-4 treatment in melanoma patients. Oncoimmunology. (2017) 6:e1261242. doi: 10.1080/2162402X.2016.1261242
124. Zhu C, Sakuishi K, Xiao S, Sun Z, Zaghouani S, Gu G, et al. An IL-27/NFIL3 signalling axis drives Tim-3 and IL-10 expression and T-cell dysfunction. Nat Commun. (2015) 6:6072. doi: 10.1038/ncomms8657
125. Sottile R, Tannazi M, Johansson MH, Cristiani CM, Calabro L, Ventura V, et al. NK- and T-cell subsets in malignant mesothelioma patients: Baseline pattern and changes in the context of anti-CTLA-4 therapy. Int J Cancer. (2019) 145:2238–48. doi: 10.1002/ijc.32363
126. Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault MC, Trevino TN, et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J Clin Invest. (2018) 128:4654–68. doi: 10.1172/JCI99317
127. Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity. (2015) 42:344–55. doi: 10.1016/j.immuni.2015.01.010
128. Sedy JR, Bjordahl RL, Bekiaris V, Macauley MG, Ware BC, Norris PS, et al. CD160 activation by herpesvirus entry mediator augments inflammatory cytokine production and cytolytic function by NK cells. J Immunol. (2013) 191:828–36. doi: 10.4049/jimmunol.1300894
129. Cichocki F, Miller JS. Setting traps for NKG2A gives NK cell immunotherapy a fighting chance. J Clin Invest. (2019) 130:1839–41. doi: 10.1172/JCI128480
Keywords: NK cell, NK cell exhaustion, inhibitory receptor, checkpoint inhibitor, cancer immunotherapy
Citation: Sun H and Sun C (2019) The Rise of NK Cell Checkpoints as Promising Therapeutic Targets in Cancer Immunotherapy. Front. Immunol. 10:2354. doi: 10.3389/fimmu.2019.02354
Received: 14 July 2019; Accepted: 18 September 2019;
Published: 17 October 2019.
Edited by:
Daniel Olive, Aix Marseille Université, FranceReviewed by:
Eric Tartour, Hôpital Européen Georges-Pompidou (HEGP), FrancePeter Steinberger, Medical University of Vienna, Austria
Copyright © 2019 Sun and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haoyu Sun, haoyusun@ustc.edu.cn; Cheng Sun, charless@ustc.edu.cn