 Wenbin Liu1,2
Wenbin Liu1,2 Chang Gao1,3Haoran Dai4
Chang Gao1,3Haoran Dai4 Yang Zheng1Zhaocheng Dong1,3Yu Gao1,2Fei Liu3Zihan Zhang3Zhiyuan Liu5Weijing Liu6Baoli Liu1*†
Yang Zheng1Zhaocheng Dong1,3Yu Gao1,2Fei Liu3Zihan Zhang3Zhiyuan Liu5Weijing Liu6Baoli Liu1*† Qingquan Liu1*†Jialan Shi7*†
Qingquan Liu1*†Jialan Shi7*†- 1Beijing Hospital of Traditional Chinese Medicine, Capital Medical University, Beijing, China
- 2Capital Medical University, Beijing, China
- 3Beijing University of Chinese Medicine, Beijing, China
- 4Shunyi Branch, Beijing Hospital of Traditional Chinese Medicine, Beijing, China
- 5Taishan Medical University, Shandong, China
- 6Key Laboratory of Chinese Internal Medicine of Ministry of Education, Beijing, China
- 7Departments of Medicine, VA Boston Healthcare System, Brigham and Women's Hospital, Harvard Medical School, Boston, MA, United States
Membranous nephropathy (MN) is the major cause of nephrotic syndrome with special pathological features, caused by the formation of immune complexes in the space between podocytes and the glomerular basement membrane. In idiopathic membranous nephropathy (IMN) the immune complexes are formed by circulating antibodies binding mainly to one of two naturally-expressed podocyte antigens: the M-type receptor for secretory phospholipase A2 (PLA2R1) and the Thrombospondin type-1 domain-containing 7A (THSD7A). Formation of antibodies against PLA2R1 is much more common, accounting for 70–80% of IMN. However, the mechanism of anti-podocyte antibody production in IMN is still unclear. In this review, we emphasize that the exposure of PLA2R1 is critical for triggering the pathogenesis of PLA2R1-associated MN, and propose the potential association between inflammation, pollution and PLA2R1. Our review aims to clarify the current research of these precipitating factors in a way that may suggest future directions for discovering the pathogenesis of MN, leading to additional therapeutic targets and strategies for the prevention and early treatment of MN.
Introduction
Membranous nephropathy (MN) is an immune-mediated glomerular disease that affects 12 new cases out of every million adults every year globally (1, 2). MN is characterized by a pathological change in the glomerular basement membrane (GBM) caused by the accumulation of immune complexes which appear as granular deposits of immunoglobulin (Ig)G when imaged with immunofluorescence and as electron-dense deposits under electron microscopy. These immune deposits in the space between podocytes and GBM contain the complement membrane attack complex (C5b-9). MN can be either idiopathic (idiopathic membranous nephropathy or IMN) or caused partially by clinical disease such as hepatitis B, systemic lupus erythematosus, cancer, or drug side-effect (secondary membranous nephropathy or SMN) (1). However, it is not clear whether these clinical diseases are direct causes of MN or merely triggers, such as the potential association between cancer and MN (3–5). In addition to the typical GBM changes, SMN exhibits mesangial deposits, “full-house” immunostaining (IgG, IgA, IgM, C3, and C1q, mesangial deposits, and reticular aggregates) in membranous lupus nephritis, and amyloid or fibrillary glomerulonephritis which are seen as fibrils under electron microscopy (Table 1) (6). The deposition of immune complexes in SMN may be caused by exogenous antigens “pre-planted” between podocytes and the GBM which bind to circulating antibodies, such as in hepatitis B virus-associated MN (7). The formation of glomeruli subepithelial immune complex deposits in IMN is now believed to be mediated by specific intrinsic podocyte antigens and their corresponding autoantibodies in humans, such as neutral endopeptidase (NEP), M-type receptor for secretory phospholipase A2 (PLA2R1), and Thrombospondin type-1 domain-containing 7A (THSD7A) (8–10). PLA2R1 (70–80% of IMN) and THSD7A (3–5% of IMN), the two major podocyte antigens identified in adult IMN, can be detected through both direct immunofluorescence staining of renal tissue and detection of their autoantibodies in serum for diagnosis and prognosis (1).
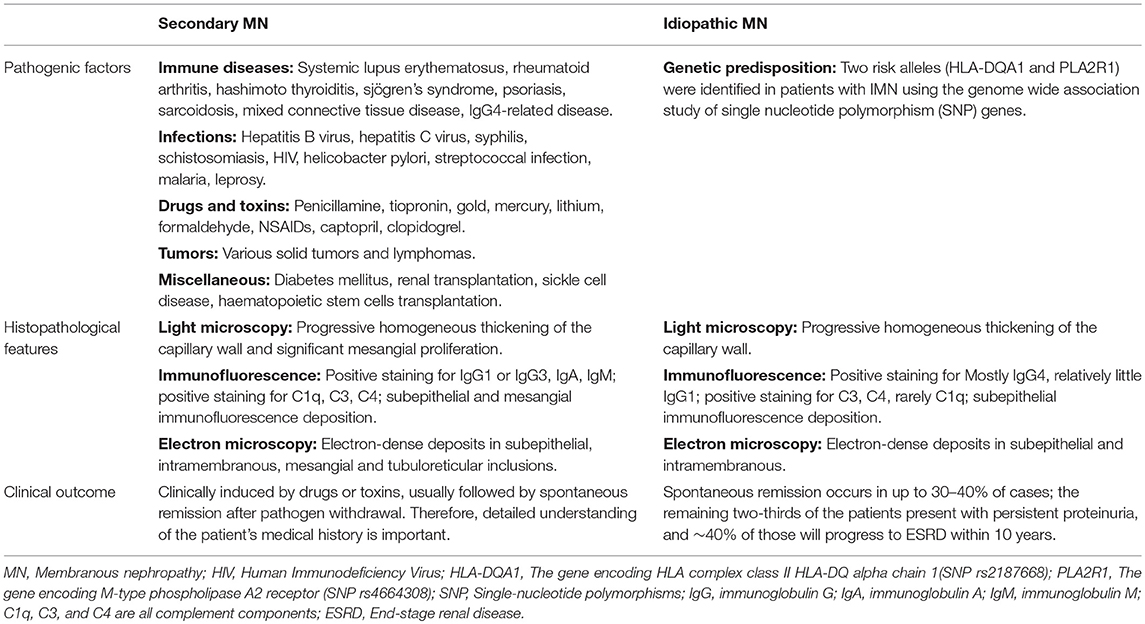
Table 1. Pathogenic factors, histopathological features, and clinical outcomes of secondary and idiopathic membranous nephropathy.
In recent years in China, the morbidity of MN has been gradually increasing possibly due to long-term exposure to air pollution, mainly fine particulate matter of <2.5 μm (PM2.5). Each 10 mg/m3 increase in PM2.5 concentration over 70 mg/m3 is associated with 14% higher odds for MN (11). However, the association between the air pollution and the pathogenesis of MN remains unclear. It is worth noting that PLA2R1 and THSD7A are naturally expressed in other parts of the human body, such as the lungs (3). In this review, we have focused on the possible mechanisms of anti-podocyte antibody production, particularly in PLA2R1-associated MN, and proposed several hypotheses, which may be beneficial to further exploration of the pathogenesis of IMN.
The Definition and Pathology of Membranous Nephropathy
Membranous nephropathy, originally called membranous glomerulonephritis (MGN), was first described in 1946 by Bell as a type of glomerular disease, which manifests pathologically as a thickening of the GBM and is clinically characterized by marked proteinuria and edema (12). In the later half of the 1950s, understanding of the pathology of MN developed rapidly, driven by several outstanding pioneers. In 1956, Mellors and Orgeta discovered immunoglobulins containing in the glomeruli deposits using immunofluorescence. In 1957, Jones demonstrated the presence of silver-positive rods projecting from the GBM using periodic acid silver methenamine stain. And in 1959, Movat defined the causal relationship between thickening of the GBM and protein deposition between the GBM and the podocyte using electron microscopy (12). In this way, these pioneers identified the basic features of glomerular lesions in MN, consisting of changes in GBM structure caused by subepithelial electron-dense deposits (Figure 1A). Moreover, in 1968, Ehrenreich used repeated renal biopsy to describe the four stages of glomerular lesions in MN: Stage I consists of only a few small subepithelial deposits such that the GBM may appear normal or be slightly thickened under light microscopy; in stage II spikes protruding from the GBM can be observed using appropriate staining; in stage III the deposits are incorporated within the GBM; in stage IV the GBM appears to be irregularly thickened by reabsorbed deposits, and in complete clinical remission the deposits may disappear and leave some areas lucent or the GBM may return to normal (13) (Figure 1A). These pathological discoveries in the middle of the last century not only defined MN as a unique type of renal pathology, but also guided the development of clinical practices that are still in use today (6).
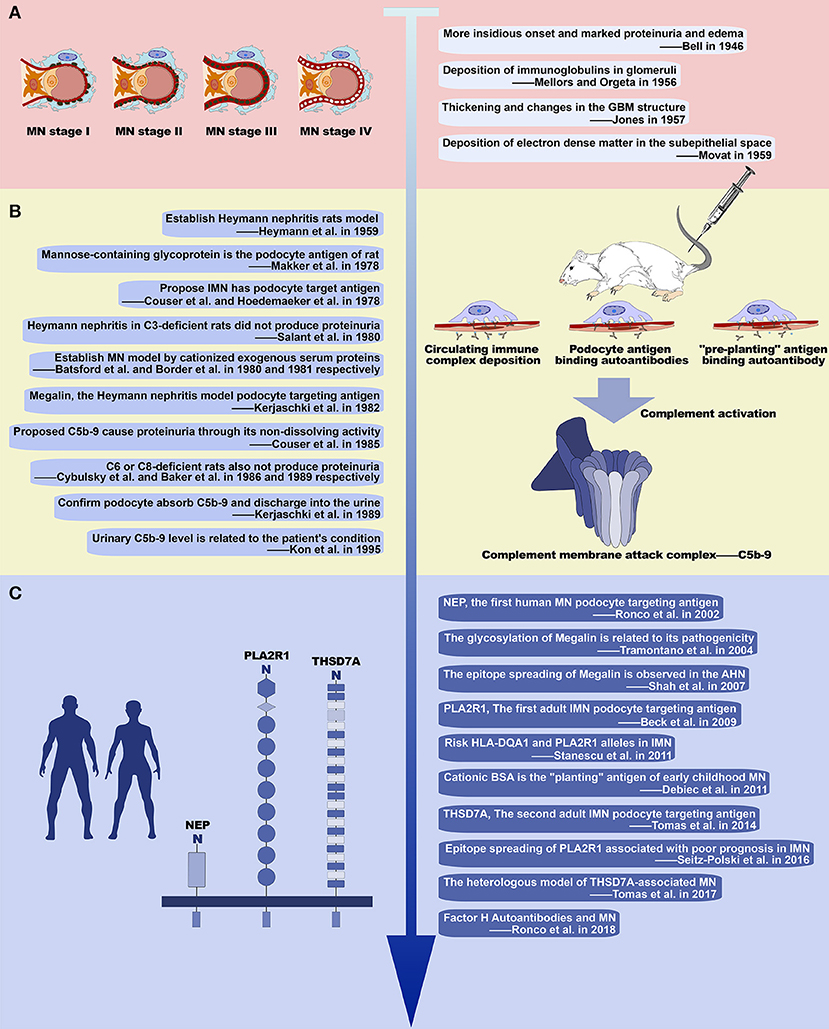
Figure 1. History of the study of membranous nephropathy. (A) Basic pathological features of MN. Illustration of the progression of glomerular lesions in MN (left) with four early studies describing its basic pathological features (right). (B) Studies of experimental MN and important results. Ten seminal studies of experimental MN (left) and the major discoveries relating to development of MN, including immune complex deposition and complement activation (right). (C) Recent advances in our understanding of MN. Three major podocyte antigens found in human MN (left), and other major work on MN since 2000 (right).
Enlightenment Brought by Heymann Nephritis
Megalin, the Rat Podocyte Antigen
In 1959, Heymann et al. established an animal model that develops pathological manifestations similar to human MN by immunizing rats with tissue components of the proximal tubular brush border (Heymann nephritis or HN) (Figure 1B) (14). This animal model relies on the active immune response of the rat and therefore is often referred to as active Heymann nephritis (AHN). In passive Heymann nephritis (PHN), an analogous lesion can be induced more rapidly by administering heterologous antibody to tubular brush border (anti-Fx1A antibody) (15). At the time, it was thought that the deposition of subepithelial immune complex was caused by circulating immune complexes because Fx1A and its antibody are simultaneously present in glomerular deposits and circulation in HN (16, 17), and the development of immune complexes in circulation was found to be coincident with the glomerular deposits (18). However, in 1978, Couser et al. and Hoedemaeker et al. simultaneously induced HN pathological changes in isolated rat kidneys using anti-Fx1A antibodies suggesting that IMN is not caused by the deposition of circulating immune complexes, but in instead by in situ immune complex formed by the autoantibodies binding to antigens located on podocytes (19, 20). In the same year, Makker et al. further confirmed that the podocyte antigen in HN is a mannose-containing glycoprotein (21). And then in 1982, Kerjaschki et al. identified megalin as an antigen on the rat podocyte membrane and tubular brush border (22) (Table 2). Consistent with previous studies, the pathogenicity of megalin is related to glycosylation (23), and anti-megalin autoantibodies fail to react with megalin in Western blots run under reducing conditions, indicating that autoantibodies recognize the spatial conformation of their epitopes (24). As the rat-specific podocyte target antigen in HN, megalin contains multiple epitopes (25–27), and demonstrates intramolecular epitope spreading during the progression of HN proteinuria (28). Intermolecular epitope spreading has also been observed with the development of autoantibodies to plasminogen, a known ligand of megalin (24). Although megalin is not the pathogenic antigen in human MN, this series of animal studies reveals the possible pathogenesis of human MN, suggesting that autoantibodies bind to podocyte intrinsic antigens to form in situ immune complexes in the glomerular subepithelial. Thus, the search for human podocyte-specific target antigens has become the focus of further studies (29).

Table 2. Morphological and serological connections between animal and human membranous nephropathy.
Human Podocyte Antigens
The results from animal model studies have since been validated in humans (Figure 1C). In 2002, Ronco et al. found that the pathogenic antigen in three families of neonatal MN was neutral endopeptidase (NEP) located on the foot process membrane of the podocytes and the brush border of the renal tubules, while the pathogenic antibody derived from NEP deficiency in the mother. This was the first confirmation that the immune complex antigen of human MN could be an intrinsic component of the podocyte membrane (8, 30). More importantly, in 2009 Beck et al. discovered PLA2R1 and its circulating autoantibodies, the first podocyte-targeted antigen-antibody system found in adult IMN (9) (Table 2). The presence of circulating anti-PLA2R1 antibodies can be detected in 70–80% of IMN (31–33). As with megalin, PLA2R1 has multiple antigenic epitopes (34–36), and the epitopes spread as the disease progresses (36). The second target podocyte antigen, THSD7A, was discovered in 2014 by Tomas et al. (10) (Table 2). These findings not only demonstrate that human MN immune complexes form by the same mechanism as seen in HN, but also gives us reason to believe that there are still undiscovered pathogenic podocyte antigen-autoantibody pairs in IMN (37). Other proteins postulated to serve as autoantigens in adult MN include superoxide dismutase (SOD2), aldose reductase (AR), and alfa-enolase. While these proteins typically localize inside the cell, AR, and SOD2 are also expressed on the plasma membrane of podocytes in patients with MN (24, 38, 39). However, the clinical significance and pathogenic role of these cytoplasmic antigens, and the differences between them and the podocyte membrane antigens (PLA2R1 or THSD7A) in pathogenic autoimmune responses has yet to be clearly demonstrated.
Complement Activation and Podocyte Injury
Another important area of research regarding MN is the mechanism of kidney injury following the formation of immune complexes (Figure 1B). In 1980, Salant et al. found that C3-deficient rats treated with cobra venom factor did not produce proteinuria within 5 days after PHN was induced with sheep anti-rat Fx1A antibody injection (40). Similar results were obtained when HN was established in C6 and C8-deficient rats (41, 42). These findings suggest that complement activation to produce C5b-9 is a key factor in the development of proteinuria in HN. Subsequent studies on experimental MN have further clarified that the pathogenic role of C5b-9 is mainly on podocytes because of its non-dissolving activity. The mechanisms involved include: (1) Inducing podocytes to produce oxygen free radicals; (2) Stimulating podocytes to produce various proteases to cause GBM damage; (3) Influencing a microfilament skeleton structure in podocytes, by separating and redistributing the proteins nephrin and podocin, which are major components of the membrane; (4) Upregulating cyclooxygenase 2 (COX-2) in podocytes, causing damage to the endoplasmic reticulum; (5) Increasing the extracellular matrix by promoting the production of TGF-β by podocytes, leading to GBM thickening and glomerular sclerosis; (6) Promoting podocyte apoptosis and shedding from GBM (43). In addition to the above findings from experimental MN, C5b-9 is also found in the glomeruli and urine of patients with MN, with levels of urine C5b-9 correlating with disease severity and patient prognosis (44, 45), indicating that complement activation is involved in the pathogenesis of human MN. The complement system mainly consists of three early activation pathways (classical pathway, alternative pathway, and lectin pathway), terminal pathway and regulatory system. The formation of C5b-9 is the final step of complement activation (46). Activation of the classical pathway starts with the binding of antibodies to C1q, which can generally be visualized in SMN, and especially lupus-associated MN, by immunofluorescence. However, the subepithelial deposition of immune complexes in IMN mainly consists of IgG4, whose ability to bind C1q is very weak. Indeed, the amount of C1q is very low or undetectable in immune deposits of IMN, indicating that activation of the classical pathway is not the main pathogenic mechanism in IMN (1). Aside from the fact that the presence of IgG1 in early deposits could activate the classical pathway (47), some evidence suggests that the alternative pathway and/or the lectin pathway may be more crucial in the pathogenesis of IMN. Hayashi et al. detected the deposition of mannose-binding lectin (MBL) in glomeruli in IMN, and found that the staining intensity of MBL correlated with the IgG4 staining intensity (48). Bally et al. reported five patients with IMN who had complete MBL deficiency and the complement activation was mainly due to the activity of the alternative pathway (49). Moreover, Luo et al. found that factor B-null mice with established MN did not develop albuminuria or exhibit glomerular deposition of C3c and C5b-9, which suggests that the alternative pathway is necessary in the pathogenicity induced by glomerular subepithelial immune complexes (50).
Revelation From Other Experimental Membranous Nephropathy
Exogenous “Planting” Antigen
There is another possibility for the formation of in situ immune complexes (Figure 1B). In 1980 and 1981, Batsford et al. and Border et al. created an animal model with the typical pathological lesion of MN by intravenous infusion of cationized exogenous serum proteins (51, 52) (Table 2). The glomerular subepithelial deposition of IgG and C3 can only occur after immunization with a cationized antigen. After immunization with an anionic or neutral antigen, deposition occurs simultaneously in the mesangial area. Furthermore, the proteinuria of an animal that is immunized with cationized antigen is more severe (53, 54). This is due to the negative charge of the glomerular capillary wall, which interacts electrostatically with the cationized antigen resulting in the “planting” of the exogenous antigen. This exogenous antigen then binds to circulating antibodies in situ to form the immune complex. In humans, cationic bovine serum albumin (cBSA) is most often the exogenous “planting” antigen found in early childhood MN (55) when the diet of the children is mainly based on milk products. This suggests that some cases of MN may be related to dietary and environmental factors. Moreover, the formation of subepithelial immune complexes in SMN, such as hepatitis B virus-associated MN, is similar (7).
Other Causes of Podocyte Injury
Complement activation is the crucial mechanism of MN podocyte injury, but it is not the only one. After Beck et al. discovered PLA2R1 in 2009, Tomas et al. found THSD7A, the second adult podocyte antigen, in 2014 (9, 10). THSD7A and PLA2R1 are large transmembrane proteins with multiple domains which are both expressed on podocyte membranes. However, in contrast to PLA2R1, THSD7A is also expressed in mouse podocytes (56). Human anti-THSD7A containing sera can be used to immune-precipitate THSD7A from mouse glomeruli in vitro and in vivo injections of human anti-THSD7A can specifically bind to murine THSD7A on podocyte foot processes, inducing proteinuria and initiating a histopathological pattern that is typical of MN (57) (Table 2). However, no C3 deposition has been found in the renal tissue of mice shortly following immunization with rabbit anti-THSD7A antibodies, similar to the absence of complement deposition after the injection of purified human anti-THSD7A antibodies (56, 57). These findings indicate that complement activation is not vital in the initiation of podocyte injury and proteinuria in the THSD7A-dependent mouse model of MN. And further, they demonstrate that human and rabbit anti-THSD7A antibodies are directly pathogenic by altering the architecture of podocytes (57). Furthermore, proteinuria also develops in C6-deficient rats with established Heymann nephritis and in C3-deficient mice with established anti-podocyte glomerulonephritis (58–60). In conclusion, the podocyte injury of MN is the result of a complex multifactorial process, especially in IMN where IgG4 deposition predominates, and the mechanism of podocyte injury beyond complement activation needs to be further explored.
Production and Pathogenesis of Anti-PlA2R1 Antibody
PLA2R1 and Its Epitopes
PLA2R1 is a type I transmembrane receptor that is a member of the mannose receptor family (61, 62). In general, the mannose receptors have endocytic properties and circulate continuously between the plasma membrane and the endosome, with ~70% of the receptors located inside the cell at steady state (61, 63). PLA2R1 is the specific receptor that promotes the internalization of phospholipase A2 (sPLA2), as has been demonstrated in rat vascular smooth muscle cells, rabbit skeletal muscle cells, and human embryonic kidney (HEK 293) cells transfected with rabbit PLA2R1 and human PLA2R1 (64–66). The exact mechanism by which PLA2R1 mediates PLA2 removal has yet to be determined. Therefore, it may act either as a clearance receptor that inhibits, inactivates and removes sPLA2s from the extracellular milieu, or as a signaling receptor that transduces a sPLA2 cellular signal in a manner independent of the catalytic activity of sPLA2 (62). Several studies have shown that, the physiological role of human PLA2R1 in its expression cells is related to sPLA2s (67–69). However, the interaction of the human PLA2R1 to sPLA2 appears very different from that of other mammals (65), and the endocytic properties of human PLA2R1 has yet to be validated by direct experimental evidence. Thus, we can only propose that, hypothetically, the extracellular domain of human PLA2R1 can bind to sPLA2s and transport it into the cell. In this hypothesis, human PLA2R1 is expressed both intra- and extracellularly (Figure 2B). The higher pH value for extracellular conditions relative to the endosome, vesicle or intracellular environment may result in a more extended conformation of human PLA2R1 (70, 71), which may contribute to the exposure of the human PLA2R1 epitope. The observed epitope spreading may relate to the neutralization of antibodies that bind to the initial epitope (72), which may causes human PLA2R1 to lose its hypothetic endocytic activity and become exposed to the extracellular environment long-term. The PH-dependent conformational change of human PLA2R1 may lead to the exposure of internal domains, which would otherwise not be recognized by the immune system, triggering autoimmune responses to different epitopes. Interestingly, similar to megalin, the binding of IMN patient serum anti-PLA2R1 antibodies to PLA2R1 in vitro needs to be carried out under non-reducing conditions, because addition of a reducing agent eliminates the binding of these antibodies (9, 34–36). These facts demonstrate that the epitope(s) to which anti-PLA2R1 antibodies bind is spatial and requires the presence of disulfide bonds in PLA2R1 (Figure 2A). The oxidative extracellular environment may cause PLA2R1 to form or preserve disulfide bonds, resulting in long-term expression of pathogenic epitopes that specifically bind to the anti-PLA2R1 antibodies circulating in the peripheral blood of a patient with IMN (Figure 2C).
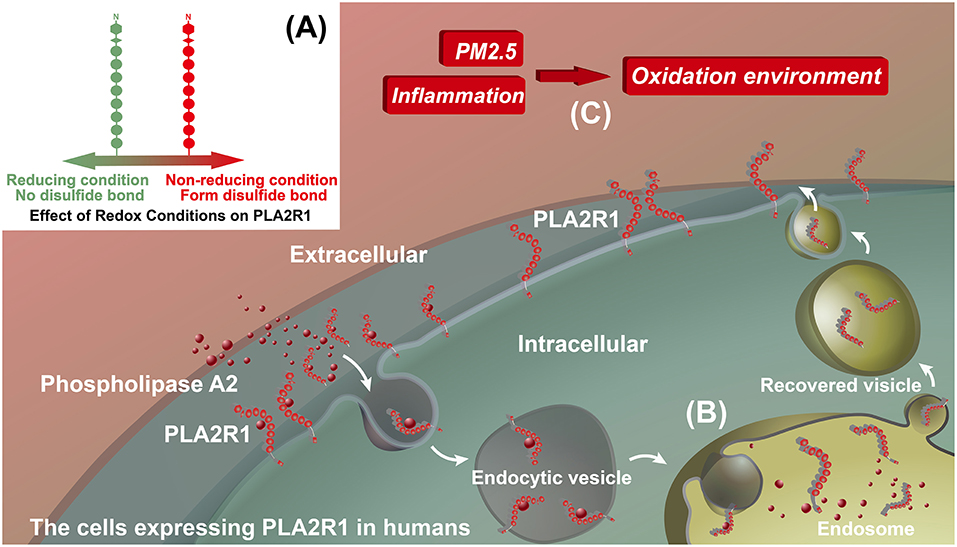
Figure 2. Hypothetical model for the effect of inflammation on PLA2R1. (A) Effect of redox conditions on PLA2R1. PLA2R1 without disulfide bond under reducing condition (Green) and with disulfide bond under non-reducing condition (Red). (B) The potential endocytic property of human PLA2R1. We hypothesize that the extracellular domain of PLA2R1 binds to phospholipase A2 and is transported into the cell by receptor-mediated endocytosis. This model has yet to be supported by any direct evidence. (C) Inflammation, PM2.5, and oxidative microenvironments. We hypothesize that inflammation, including that associated with PM2.5, alters the microenvironment of PLA2R1-expressing cells. PLA2R1 exposed to this oxidative microenvironment may form or retain disulfide bonds resulting the long-term expression of pathogenic epitopes.
PLA2R1 Exposure
In humans, PLA2R1 is expressed not only in podocytes (9), but also in neutrophils (67), pulmonary macrophages (73), airway epithelial cells, and submucosal epithelial cells (69) (Figure 3A). Podocytes are highly differentiated glomerular capillary epithelial cells whose big cell bodies swell into the crude urine space while the foot processes adhere to the GBM, which is interlaced with the foot processes of adjacent podocytes (74). Normally, the filtration barrier, which is composed of endothelial cells, GBMs and podocytes, only allows water and small molecules to pass freely, while retaining larger molecules such as plasma proteins (75). Even if the glomerulus is easily damaged by the immune system, antigen-presenting cells such as macrophages or dendritic cells remain inaccessible to podocytes if endothelial cells and GBMs are not damaged (75). We hypothesize that podocytes or other cells expressing PLA2R1 may become damaged and release extracellular vesicles (EVs), which can be measured in the urinary tract (76, 77), leading to the onset of autoimmune activity and the development of MN. EVs are small vesicles with membrane proteins on their surface derived from the source cells. Though it has not yet been proved experimentally, cells expressing PLA2R1 may release EVs carrying PLA2R1 on the surface after stimulation. In order to meet the precise specificity required for antigen-antibody binding (78), the epitopes exposed on these non-podocyte cells must be consistent with the PLA2R1 epitope expressed in podocytes, theoretically in a non-reducing environment.
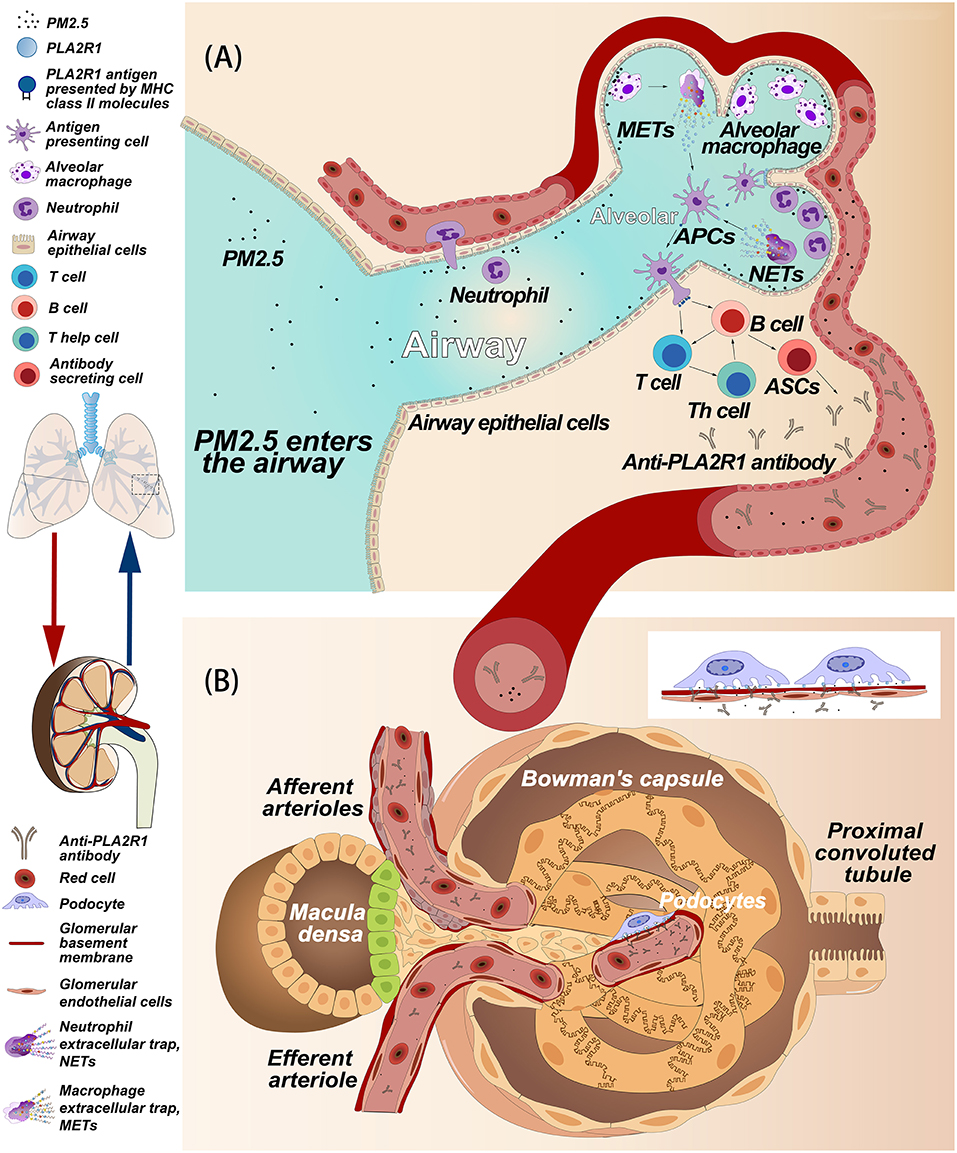
Figure 3. Hypothetical model of the relationship between PM2.5 and membranous nephropathy. (A) Hypothesis of how PM2.5 induces anti-PLA2R1 antibody production. PM2.5 in the airways and alveoli causes an inflammatory response involving neutrophils, alveolar macrophages, and airway epithelial cells. We hypothesize that these cells may express PLA2R1, that due to oxidative stress associated with inflammation, may assume a conformation that includes pathogenic epitopes that contribute to the formation of autoantibodies. Alternatively, PLA2R1 may be released into the inflammatory space during the release of extracellular traps. PLA2R1 may then be bound by antigen presenting cells, triggering the humoral immune response, and producing anti-PLA2R1 antibodies. (B) The hypothetical process of subepithelial immune complex deposition caused by the anti-PLA2R1 antibody exogenous to glomeruli. Both PM2.5 and anti-PLA2R1 antibodies enter blood vessel and circulate into the glomerular capillaries. The anti-PLA2R1 antibodies penetrate the endothelial cells and glomerular basement membrane (GBM), recognize and bind to naturally-expressed PLA2R1 on podocytes to form the immune complex. These complexes then deposit into the space between podocytes and GBM.
Inflammation, Oxidative Stress, and PLA2R1
Both neutrophils and pulmonary macrophages are inflammatory cells that can accumulate at the site of inflammation (79, 80). In an inflammatory environment, reactive oxygen species (ROS) are produced in an amount that exceeds the ability of the cell to detoxify itself, leading to an oxidative stress state (81). Under this circumstance, the intracellular environment becomes strongly oxidizing, which results in disulfide bond formation in cytoplasmic proteins (81, 82). Interestingly, there is a significant correlation between the morbidity of MN patients in China and the concentration of PM2.5 (11). Indeed, respirable particulate matter (PM) in polluted air is also associated with various inflammatory lungs diseases (83–85). The occurrence of pulmonary inflammation caused by PM is associated with the production of ROS and oxidative stress (86). Polycyclic aromatic hydrocarbons and transition metals in the PM can directly produce ROS (87, 88). In addition, target cells in the lungs, such as airway epithelial cells, macrophages and neutrophils, can also produce ROS when exposed to PM (87–90). In theory, these cells could aggregate in the airway or alveolar and express PLA2R1 with pathogenic epitopes in response to PM and oxidative stress. PLA2R1 could also then be discharged into the inflammatory space when the neutrophils or macrophages release extracellular traps (91, 92) (Figure 3A). Interestingly, rat PLA2R1 expressed on lymphocytes and granulocytes can be up-regulated by interleukin-1β in vitro (93). However, it is unclear whether this is the case with human PLA2R1. Most of the cells involved with inflammation are also capable of presenting antigens (94). Therefore, in theory, PLA2R1 in an inflammatory environment may be more easily recognized by the immune system, thus triggering an autoimmune response (Figure 3A). In addition, PM2.5 exposure in lung could also be considered as a danger signal that induce activation of autoreactive T cells, which also may a trigger be required for induce autoimmune response. However, further studies are needed to explore these hypotheses.
Risk Genes and Anti-PLA2R1 Antibodies
Two risk alleles (HLA-DQA1 and PLA2R1) were identified in French, Dutch, and British patients with IMN using the genome-wide association study of single nucleotide polymorphism (SNP) Genes (95). Moreover, there is likewise a correlation between HLA-DQA1 and PLA2R1 risk alleles in patients with IMN in the Spanish and Chinese populations (96, 97). Interestingly, anti-PLA2R1 antibodies were detectable in 73% of patients with both PLA2R1 and HLA-DQA1 high-risk genotypes and in none of the patients with both low-risk genotypes (98). HLA-DQA1 produces a receptor protein on antigen presenting cells as part of the major histocompatibility complex, and PLA2R1 produces the specific podocyte target antigen of IMN patients, both of which are related to the triggering of autoimmune responses. Gupta et al. proposed that after PLA2R1 protein has been processed and displayed on the surface of antigen presenting cells as peptides bound to the class II receptor (DQA1) groove, the genetics of DQA1 will shape the amino acid structure of its receptor groove to fit with the peptide sequences available from PLA2R1. Additionally, the genetics of PLA2R1 may control the fragmentation of protein or the level of transcript due to a change in the number of positions of enzyme cut sites, leading to higher levels of peptide (98). The two risk alleles of IMN may be involved in the formation of anti-PLA2R1 antibodies. However, the SNPs identified are common and thus can only partially explain the onset of MN in an individual, even in an individual with multiple risk factors. Therefore, further studies are needed to identifying the triggers or environmental factors that can contribute to MN. It would be especially useful to compare IMN patients with a control population that carries the risk allele (98).
Pathogenesis of Anti-PLA2R1 Antibodies
In the case where non-podocyte cells initiate the autoimmune response, the process by which the circulating autoantibodies locate, recognize, and bind the target antigens on podocytes is similar to the process seen in the PHN animal model. The exogenous antibodies can penetrate endothelial cells and the GBM to bind with the podocyte target antigen (15) (Figure 3B). However, just as megalin glycosylation and spatial conformation determine its pathogenicity in HN (23), antigenicity of PLA2R1 also requires some additional conditions. When the anti-PLA2R1 antibodies are produced by non-podocyte cell sources in and or enter the circulation, the antibodies will only bind to the podocyte PLA2R1 under non-reducing conditions (9, 34–36). As a cellular transmembrane receptor (61, 62), the extracellular domain of PLA2R1 is in contact with the extracellular environment and thus is most likely to be in the naturally expressed spatial conformation (Figure 2B). PM2.5 also causes early kidney damage through oxidative stress or inflammation (99), which may contribute to the development of non-reducing conditions in the renal microenvironment. Autoantibodies binding to podocyte antigens in situ form immune complexes, which result in podocyte injury (1). However, since the immunoglobulins deposited in IMN are mainly IgG4 whose ability to bind C1q is weak, complement may be activated from non-classical pathways (Please refer to the paragraph of “Complement activation and podocyte injury”). Beyond that, antibody binding can also inhibit the normal function of the antigenic protein (72). PLA2R1 may be involved in the adhesion of podocytes to GBM and the serum anti-PLA2R1 antibodies may interfere with adhesion by binding to PLA2R (100), suggesting that serum antibodies binding to podocyte PLA2R1 may cause kidney damage through more than just complement activation.
Conclusions
Although research on MN has greatly improved quality of life in the patients, there are still many unsolved mysteries (Table 3). In this review, we have mainly discussed the pathogenesis of PLA2R1-associated MN, and proposed some hypotheses based on the available research. We believe that further research into these questions will be beneficial to the clinical treatment of IMN patients by further revealing mechanisms behind the development of pathogenic antigens and antibodies and searching for treatments that prevent or inhibit the resultant kidney damage.

Table 3. Known and unknown about the pathogenesis of membranous nephropathy.
Author Contributions
WenL collected most of the material for reviewing and wrote the main part of the review. CG and HD collected the rest of the material for reviewing. YZ, ZD, and YG wrote the rest part of the review. FL, ZZ, and ZL made the figures and tables. WeiL, JS, QL, and BL discusses and modifies the content of the review article.
Funding
This work was supported by grants from the National Natural Science Foundation of China (no. 81673907, 81470301, 81670128), Natural Science Foundation of Beijing Municipality (no. 7182070), Beijing Municipal Science and Technology Project (no. Z161100000516024), and Beijing Municipal Administration of Hospitals Clinical Medicine Development of special funding support, code: XLMX201833.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ronco P, Debiec H. Pathophysiological advances in membranous nephropathy: time for a shift in patient's care. Lancet. (2015) 385:1983–92. doi: 10.1016/S0140-6736(15)60731-0
2. McGrogan A, Franssen CF, de Vries CS. The incidence of primary glomerulonephritis worldwide: a systematic review of the literature. Nephrol Dial Transplant. (2011) 26:414–30. doi: 10.1093/ndt/gfq665
3. von Haxthausen F, Reinhard L, Pinnschmidt HO, Rink M, Soave A, Hoxha E, et al. Antigen-specific IgG subclasses in primary and malignancy-associated membranous nephropathy. Front Immunol. (2018) 9:3035. doi: 10.3389/fimmu.2018.03035
4. Hoxha E, Beck LH Jr, Wiech T, Tomas NM, Probst C, Mindorf S, et al. An indirect immunofluorescence method facilitates detection of thrombospondin type 1 domain-containing 7A-specific antibodies in membranous nephropathy. J Am Soc Nephrol. (2017) 28:520–31. doi: 10.1681/ASN.2016010050
5. Hoxha E, Wiech T, Stahl PR, Zahner G, Tomas NM, Meyer-Schwesinger C, et al. A mechanism for cancer-associated membranous nephropathy. N Engl J Med. (2016) 374:1995–6. doi: 10.1056/NEJMc1511702
6. Fogo AB, Lusco MA, Najafian B, Alpers CE. AJKD atlas of renal pathology: membranous nephropathy. Am J Kidney Dis. (2015) 66:e15–7. doi: 10.1053/j.ajkd.2015.07.006
7. Bhimma R, Coovadia HM. Hepatitis B virus-associated nephropathy. Am J Nephrol. (2004) 24:198–211. doi: 10.1159/000077065
8. Debiec H, Guigonis V, Mougenot B, Haymann JP, Bensman A, Deschenes G, et al. Antenatal membranous glomerulonephritis with vascular injury induced by anti-neutral endopeptidase antibodies: toward new concepts in the pathogenesis of glomerular diseases. J Am Soc Nephrol. (2003) 14 (Suppl. 1):S27–32. doi: 10.1097/01.ASN.0000067649.64849.75
9. Beck LH Jr, Bonegio RG, Lambeau G, Beck DM, Powell DW, Cummins TD, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. (2009) 361:11–21. doi: 10.1056/NEJMoa0810457
10. Tomas NM, Beck LH Jr, Meyer-Schwesinger C, Seitz-Polski B, Ma H, Zahner G, et al. Thrombospondin type-1 domain-containing 7A in idiopathic membranous nephropathy. N Engl J Med. (2014) 371:2277–87. doi: 10.1056/NEJMoa1409354
11. Xu X, Wang G, Chen N, Lu T, Nie S, Xu G, et al. Long-term exposure to air pollution and increased risk of membranous nephropathy in China. J Am Soc Nephrol. (2016) 27:3739–46. doi: 10.1681/ASN.2016010093
12. Glassock RJ. The pathogenesis of idiopathic membranous nephropathy: a 50-year odyssey. Am J Kidney Dis. (2010) 56:157–67. doi: 10.1053/j.ajkd.2010.01.008
13. Sinico RA, Mezzina N, Trezzi B, Ghiggeri GM, Radice A. Immunology of membranous nephropathy: from animal models to humans. Clin Exp Immunol. (2016) 183:157–65. doi: 10.1111/cei.12729
14. Heymann W, Hackel DB, Harwood S, Wilson SG, Hunter JL. Production of nephrotic syndrome in rats by Freund's adjuvants and rat kidney suspensions. 1951. J Am Soc Nephrol. (2000) 11:183–8. doi: 10.3181/00379727-100-24736
15. Barabas AZ, Lannigan R Jr. Induction of an autologous immune complex glomerulonephritis in the rat by intravenous injection of heterologous anti-rat kidney tubular antibody. II. Early glomerular lesions. Br J Exp Pathol. (1974) 55:282–90.
16. Edgington TS, Glassock RJ, Dixon FJ. Autologous immune-complex pathogenesis of experimental allergic glomerulonephritis. Science. (1967) 155:1432–4. doi: 10.1126/science.155.3768.1432
17. Naruse T, Fukasawa T, Hirokawa N, Oike S, Miyakawa Y. The pathogenesis of experimental membranous glomerulonephritis induced with homologous nephritogenic tubular antigen. J Exp Med. (1976) 144:1347–62. doi: 10.1084/jem.144.5.1347
18. Abrass CK, Border WA, Glassock RJ. Circulating immune complexes in rats with autologous immune complex nephritis. Lab Invest. (1980) 43:18–27.
19. Couser WG, Steinmuller DR, Stilmant MM, Salant DJ, Lowenstein LM. Experimental glomerulonephritis in the isolated perfused rat kidney. J Clin Invest. (1978) 62:1275–87. doi: 10.1172/JCI109248
20. Van Damme BJ, Fleuren GJ, Bakker WW, Vernier RL, Hoedemaeker PJ. Experimental glomerulonephritis in the rat induced by antibodies directed against tubular antigens. V. Fixed glomerular antigens in the pathogenesis of heterologous immune complex glomerulonephritis. Lab Invest. (1978) 38:502–10.
21. Makker SP. Evidence that the antigen of autologous immune complex glomerulonephritis of rats is a mannose- or glucose-containing glycoprotein. Proc Soc Exp Biol Med. (1980) 163:95–9. doi: 10.3181/00379727-163-40729
22. Kerjaschki D, Farquhar MG. The pathogenic antigen of Heymann nephritis is a membrane glycoprotein of the renal proximal tubule brush border. Proc Natl Acad Sci USA. (1982) 79:5557–61. doi: 10.1073/pnas.79.18.5557
23. Tramontano A, Makker SP. Conformation and glycosylation of a megalin fragment correlate with nephritogenicity in Heymann nephritis. J Immunol. (2004) 172:2367–73. doi: 10.4049/jimmunol.172.4.2367
24. Makker SP, Tramontano A. Idiopathic membranous nephropathy: an autoimmune disease. Semin Nephrol. (2011) 31:333–40. doi: 10.1016/j.semnephrol.2011.06.004
25. Farquhar MG, Saito A, Kerjaschki D, Orlando RA. The Heymann nephritis antigenic complex: megalin (gp330) and RAP. J Am Soc Nephrol. (1995) 6:35–47.
26. Ronco P, Debiec H. Molecular dissection of target antigens and nephritogenic antibodies in membranous nephropathy: towards epitope-driven therapies. J Am Soc Nephrol. (2006) 17:1772–4. doi: 10.1681/ASN.2006050497
27. Tramontano A, Knight T, Vizzuso D, Makker SP. Nested N-terminal megalin fragments induce high-titer autoantibody and attenuated Heymann nephritis. J Am Soc Nephrol. (2006) 17:1979–85. doi: 10.1681/ASN.2005101144
28. Shah P, Tramontano A, Makker SP. Intramolecular epitope spreading in Heymann nephritis. J Am Soc Nephrol. (2007) 18:3060–6. doi: 10.1681/ASN.2007030342
29. Shankland SJ. New insights into the pathogenesis of membranous nephropathy. Kidney Int. (2000) 57:1204–5. doi: 10.1046/j.1523-1755.2000.00950.x
30. Ronco P, Debiec H. Molecular pathomechanisms of membranous nephropathy: from Heymann nephritis to alloimmunization. J Am Soc Nephrol. (2005) 16:1205–13. doi: 10.1681/ASN.2004121080
31. Bobart SA, De Vriese AS, Pawar AS, Zand L, Sethi S, Giesen C, et al. Noninvasive diagnosis of primary membranous nephropathy using phospholipase A2 receptor antibodies. Kidney Int. (2019) 95:429–38. doi: 10.1016/j.kint.2018.10.021
32. De Vriese AS, Glassock RJ, Nath KA, Sethi S, Fervenza FC. A proposal for a serology-based approach to membranous nephropathy. J Am Soc Nephrol. (2017) 28:421–30. doi: 10.1681/ASN.2016070776
33. Couser WG. Primary membranous nephropathy. Clin J Am Soc Nephrol. (2017) 12:983–97. doi: 10.2215/CJN.11761116
34. Fresquet M, Jowitt TA, Gummadova J, Collins R, O'Cualain R, McKenzie EA, et al. Identification of a major epitope recognized by PLA2R autoantibodies in primary membranous nephropathy. J Am Soc Nephrol. (2015) 26:302–13. doi: 10.1681/ASN.2014050502
35. Kao L, Lam V, Waldman M, Glassock RJ, Zhu Q. Identification of the immunodominant epitope region in phospholipase A2 receptor-mediating autoantibody binding in idiopathic membranous nephropathy. J Am Soc Nephrol. (2015) 26:291–301. doi: 10.1681/ASN.2013121315
36. Seitz-Polski B, Dolla G, Payre C, Girard CA, Polidori J, Zorzi K, et al. Epitope spreading of autoantibody response to PLA2R associates with poor prognosis in membranous nephropathy. J Am Soc Nephrol. (2016) 27:1517–33. doi: 10.1681/ASN.2014111061
37. Beck LH Jr. PLA2R and THSD7A: disparate paths to the same disease? J Am Soc Nephrol. (2017) 28:2579–89. doi: 10.1681/ASN.2017020178
38. Bruschi M, Carnevali ML, Murtas C, Candiano G, Petretto A, Prunotto M, et al. Direct characterization of target podocyte antigens and auto-antibodies in human membranous glomerulonephritis: alfa-enolase and borderline antigens. J Proteomics. (2011) 74:2008–17. doi: 10.1016/j.jprot.2011.05.021
39. Prunotto M, Carnevali ML, Candiano G, Murtas C, Bruschi M, Corradini E, et al. Autoimmunity in membranous nephropathy targets aldose reductase and SOD2. J Am Soc Nephrol. (2010) 21:507–19. doi: 10.1681/ASN.2008121259
40. Salant DJ, Belok S, Madaio MP, Couser WG. A new role for complement in experimental membranous nephropathy in rats. J Clin Invest. (1980) 66:1339–50. doi: 10.1172/JCI109987
41. Cybulsky AV, Rennke HG, Feintzeig ID, Salant DJ. Complement-induced glomerular epithelial cell injury. Role of the membrane attack complex in rat membranous nephropathy. J Clin Invest. (1986) 77:1096–107. doi: 10.1172/JCI112408
42. Baker PJ, Ochi RF, Schulze M, Johnson RJ, Campbell C, Couser WG. Depletion of C6 prevents development of proteinuria in experimental membranous nephropathy in rats. Am J Pathol. (1989) 135:185–94.
43. Nangaku M, Shankland SJ, Couser WG. Cellular response to injury in membranous nephropathy. J Am Soc Nephrol. (2005) 16:1195–204. doi: 10.1681/ASN.2004121098
44. Schulze M, Donadio JV Jr, Pruchno CJ, Baker PJ, Johnson RJ, Stahl RA, et al. Elevated urinary excretion of the C5b-9 complex in membranous nephropathy. Kidney Int. (1991) 40:533–8. doi: 10.1038/ki.1991.242
45. Kon SP, Coupes B, Short CD, Solomon LR, Raftery MJ, Mallick NP, et al. Urinary C5b-9 excretion and clinical course in idiopathic human membranous nephropathy. Kidney Int. (1995) 48:1953–8. doi: 10.1038/ki.1995.496
46. Hadders MA, Beringer DX, Gros P. Structure of C8alpha-MACPF reveals mechanism of membrane attack in complement immune defense. Science. (2007) 317:1552–4. doi: 10.1126/science.1147103
47. Ma H, Sandor DG, Beck LH Jr. The role of complement in membranous nephropathy. Semin Nephrol. (2013) 33:531–42. doi: 10.1016/j.semnephrol.2013.08.004
48. Hayashi N, Okada K, Matsui Y, Fujimoto K, Adachi H, Yamaya H, et al. Glomerular mannose-binding lectin deposition in intrinsic antigen-related membranous nephropathy. Nephrol Dial Transplant. (2018) 33:832–40. doi: 10.1093/ndt/gfx235
49. Bally S, Debiec H, Ponard D, Dijoud F, Rendu J, Faure J, et al. Phospholipase A2 receptor-related membranous nephropathy and mannan-binding lectin deficiency. J Am Soc Nephrol. (2016) 27:3539–44. doi: 10.1681/ASN.2015101155
50. Luo W, Olaru F, Miner JH, Beck LH Jr, van der Vlag J, Thurman JM, et al. Alternative pathway is essential for glomerular complement activation and proteinuria in a mouse model of membranous nephropathy. Front Immunol. (2018) 9:1433. doi: 10.3389/fimmu.2018.01433
51. Batsford S, Oite T, Takamiya H, Vogt A. Anionic binding sites in the glomerular basement membrane: possible role in the pathogenesis of immune complex glomerulonephritis. Ren Physiol. (1980) 3:336–40. doi: 10.1159/000172780
52. Border WA, Kamil ES, Ward HJ, Cohen AH. Antigenic changes as a determinant of immune complex localization in the rat glomerulus. Lab Invest. (1981) 45:442–9.
53. Border WA, Ward HJ, Kamil ES, Cohen AH. Induction of membranous nephropathy in rabbits by administration of an exogenous cationic antigen. J Clin Invest. (1982) 69:451–61. doi: 10.1172/JCI110469
54. Adler SG, Wang H, Ward HJ, Cohen AH, Border WA. Electrical charge. Its role in the pathogenesis and prevention of experimental membranous nephropathy in the rabbit. J Clin Invest. (1983) 71:487–99. doi: 10.1172/JCI110793
55. Debiec H, Lefeu F, Kemper MJ, Niaudet P, Deschenes G, Remuzzi G, et al. Early-childhood membranous nephropathy due to cationic bovine serum albumin. N Engl J Med. (2011) 364:2101–10. doi: 10.1056/NEJMoa1013792
56. Tomas NM, Hoxha E, Reinicke AT, Fester L, Helmchen U, Gerth J, et al. Autoantibodies against thrombospondin type 1 domain-containing 7A induce membranous nephropathy. J Clin Invest. (2016) 126:2519–32. doi: 10.1172/JCI85265
57. Tomas NM, Meyer-Schwesinger C, von Spiegel H, Kotb AM, Zahner G, Hoxha E, et al. A heterologous model of thrombospondin type 1 domain-containing 7A-associated membranous nephropathy. J Am Soc Nephrol. (2017) 28:3262–77. doi: 10.1681/ASN.2017010030
58. Spicer ST, Tran GT, Killingsworth MC, Carter N, Power DA, Paizis K, et al. Induction of passive Heymann nephritis in complement component 6-deficient PVG rats. J Immunol. (2007) 179:172–8. doi: 10.4049/jimmunol.179.1.172
59. Leenaerts PL, Hall BM, Van Damme BJ, Daha MR, Vanrenterghem YF. Active Heymann nephritis in complement component C6 deficient rats. Kidney Int. (1995) 47:1604–14. doi: 10.1038/ki.1995.224
60. Meyer-Schwesinger C, Dehde S, Klug P, Becker JU, Mathey S, Arefi K, et al. Nephrotic syndrome and subepithelial deposits in a mouse model of immune-mediated anti-podocyte glomerulonephritis. J Immunol. (2011) 187:3218–29. doi: 10.4049/jimmunol.1003451
61. East L, Isacke CM. The mannose receptor family. Biochim Biophys Acta. (2002) 1572:364–86. doi: 10.1016/S0304-4165(02)00319-7
62. Lambeau G, Lazdunski M. Receptors for a growing family of secreted phospholipases A2. Trends Pharmacol Sci. (1999) 20:162–70. doi: 10.1016/S0165-6147(99)01300-0
63. Lew DB, Songu-Mize E, Pontow SE, Stahl PD, Rattazzi MC. A mannose receptor mediates mannosyl-rich glycoprotein-induced mitogenesis in bovine airway smooth muscle cells. J Clin Invest. (1994) 94:1855–63. doi: 10.1172/JCI117535
64. Hanasaki K, Arita H. Characterization of a high affinity binding site for pancreatic-type phospholipase A2 in the rat. Its cellular and tissue distribution. J Biol Chem. (1992) 267:6414–20.
65. Ancian P, Lambeau G, Mattei MG, Lazdunski M. The human 180-kDa receptor for secretory phospholipases A2. Molecular cloning, identification of a secreted soluble form, expression, and chromosomal localization. J Biol Chem. (1995) 270:8963–70. doi: 10.1074/jbc.270.15.8963
66. Zvaritch E, Lambeau G, Lazdunski M. Endocytic properties of the M-type 180-kDa receptor for secretory phospholipases A2. J Biol Chem. (1996) 271:250–7. doi: 10.1074/jbc.271.1.250
67. Silliman CC, Moore EE, Zallen G, Gonzalez R, Johnson JL, Elzi DJ, et al. Presence of the M-type sPLA(2) receptor on neutrophils and its role in elastase release and adhesion. Am J Physiol Cell Physiol. (2002) 283:C1102–13. doi: 10.1152/ajpcell.00608.2001
68. Pan Y, Wan J, Liu Y, Yang Q, Liang W, Singhal PC, et al. sPLA2 IB induces human podocyte apoptosis via the M-type phospholipase A2 receptor. Sci Rep. (2014) 4:6660. doi: 10.1038/srep06660
69. Nolin JD, Ogden HL, Lai Y, Altemeier WA, Frevert CW, Bollinger JG, et al. Identification of epithelial phospholipase A2 receptor 1 as a potential target in asthma. Am J Respir Cell Mol Biol. (2016) 55:825–36. doi: 10.1165/rcmb.2015-0150OC
70. Mellman I. Endocytosis and molecular sorting. Annu Rev Cell Dev Biol. (1996) 12:575–625. doi: 10.1146/annurev.cellbio.12.1.575
71. Dong Y, Cao L, Tang H, Shi X, He Y. Structure of human M-type phospholipase A2 receptor revealed by cryo-electron microscopy. J Mol Biol. (2017) 429:3825–35. doi: 10.1016/j.jmb.2017.10.019
72. Casadevall A, Pirofski LA. A new synthesis for antibody-mediated immunity. Nat Immunol. (2011) 13:21–8. doi: 10.1038/ni.2184
73. Granata F, Petraroli A, Boilard E, Bezzine S, Bollinger J, Del Vecchio L, et al. Activation of cytokine production by secreted phospholipase A2 in human lung macrophages expressing the M-type receptor. J Immunol. (2005) 174:464–74. doi: 10.4049/jimmunol.174.1.464
74. Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. (2003) 83:253–307. doi: 10.1152/physrev.00020.2002
75. Mathieson PW. What has the immune system got against the glomerular podocyte? Clin Exp Immunol. (2003) 134:1–5. doi: 10.1046/j.1365-2249.2003.02236.x
76. Burger D, Thibodeau JF, Holterman CE, Burns KD, Touyz RM, Kennedy CR. Urinary podocyte microparticles identify prealbuminuric diabetic glomerular injury. J Am Soc Nephrol. (2014) 25:1401–7. doi: 10.1681/ASN.2013070763
77. EL Andaloussi S, Mager I, Breakefield XO, Wood MJ. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov. (2013) 12:347–57. doi: 10.1038/nrd3978
78. Michnick SW, Sidhu SS. Submitting antibodies to binding arbitration. Nat Chem Biol. (2008) 4:326–9. doi: 10.1038/nchembio0608-326
79. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. (2013) 13:159–75. doi: 10.1038/nri3399
80. Lambrecht BN. Alveolar macrophage in the driver's seat. Immunity. (2006) 24:366–8. doi: 10.1016/j.immuni.2006.03.008
81. Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. (2008) 454:455–62. doi: 10.1038/nature07203
82. Chakravarthi S, Jessop CE, Bulleid NJ. The role of glutathione in disulphide bond formation and endoplasmic-reticulum-generated oxidative stress. EMBO Rep. (2006) 7:271–5. doi: 10.1038/sj.embor.7400645
83. Kim KH, Kabir E, Kabir S. A review on the human health impact of airborne particulate matter. Environ Int. (2015) 74:136–43. doi: 10.1016/j.envint.2014.10.005
84. Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. (2004) 350:2645–53. doi: 10.1056/NEJMoa032158
85. Ghio AJ, Devlin RB. Inflammatory lung injury after bronchial instillation of air pollution particles. Am J Respir Crit Care Med. (2001) 164:704–8. doi: 10.1164/ajrccm.164.4.2011089
86. Nel A. Atmosphere. Air pollution-related illness: effects of particles. Science. (2005) 308:804–6. doi: 10.1126/science.1108752
87. Li N, Sioutas C, Cho A, Schmitz D, Misra C, Sempf J, et al. Ultrafine particulate pollutants induce oxidative stress and mitochondrial damage. Environ Health Perspect. (2003) 111:455–60. doi: 10.1289/ehp.6000
88. Kadiiska MB, Mason RP, Dreher KL, Costa DL, Ghio AJ. In vivo evidence of free radical formation in the rat lung after exposure to an emission source air pollution particle. Chem Res Toxicol. (1997) 10:1104–8. doi: 10.1021/tx970049r
89. Nel AE, Diaz-Sanchez D, Ng D, Hiura T, Saxon A. Enhancement of allergic inflammation by the interaction between diesel exhaust particles and the immune system. J Allergy Clin Immunol. (1998) 102:539–54. doi: 10.1016/S0091-6749(98)70269-6
90. Lim HB, Ichinose T, Miyabara Y, Takano H, Kumagai Y, Shimojyo N, et al. Involvement of superoxide and nitric oxide on airway inflammation and hyperresponsiveness induced by diesel exhaust particles in mice. Free Radic Biol Med. (1998) 25:635–44. doi: 10.1016/S0891-5849(98)00073-2
91. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. (2018) 18:134–47. doi: 10.1038/nri.2017.105
92. von Kockritz-Blickwede M, Goldmann O, Thulin P, Heinemann K, Norrby-Teglund A, Rohde M, et al. Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood. (2008) 111:3070–80. doi: 10.1182/blood-2007-07-104018
93. Beck S, Beck G, Ostendorf T, Floege J, Lambeau G, Nevalainen T, et al. Upregulation of group IB secreted phospholipase A2 and its M-type receptor in rat ANTI-THY-1 glomerulonephritis. Kidney Int. (2006) 70:1251–60. doi: 10.1038/sj.ki.5001664
94. Batista FD, Harwood NE. The who, how and where of antigen presentation to B cells. Nat Rev Immunol. (2009) 9:15–27. doi: 10.1038/nri2454
95. Stanescu HC, Arcos-Burgos M, Medlar A, Bockenhauer D, Kottgen A, Dragomirescu L, et al. Risk HLA-DQA1 and PLA(2)R1 alleles in idiopathic membranous nephropathy. N Engl J Med. (2011) 364:616–26. doi: 10.1056/NEJMoa1009742
96. Bullich G, Ballarin J, Oliver A, Ayasreh N, Silva I, Santin S, et al. HLA-DQA1 and PLA2R1 polymorphisms and risk of idiopathic membranous nephropathy. Clin J Am Soc Nephrol. (2014) 9:335–43. doi: 10.2215/CJN.05310513
97. Lv J, Hou W, Zhou X, Liu G, Zhou F, Zhao N, et al. Interaction between PLA2R1 and HLA-DQA1 variants associates with anti-PLA2R antibodies and membranous nephropathy. J Am Soc Nephrol. (2013) 24:1323–9. doi: 10.1681/ASN.2012080771
98. Gupta S, Kottgen A, Hoxha E, Brenchley P, Bockenhauer D, Stanescu HC, et al. Genetics of membranous nephropathy. Nephrol Dial Transplant. (2018) 33:1493–502. doi: 10.1093/ndt/gfx296
99. Aztatzi-Aguilar OG, Uribe-Ramirez M, Narvaez-Morales J, De Vizcaya-Ruiz A, Barbier O. Early kidney damage induced by subchronic exposure to PM2.5 in rats. Part Fibre Toxicol. (2016) 13:68. doi: 10.1186/s12989-016-0179-8
Keywords: membranous nephropathy, immunological pathogenesis, PLA2R1, PM2.5, lung, kidney
Citation: Liu W, Gao C, Dai H, Zheng Y, Dong Z, Gao Y, Liu F, Zhang Z, Liu Z, Liu W, Liu B, Liu Q and Shi J (2019) Immunological Pathogenesis of Membranous Nephropathy: Focus on PLA2R1 and Its Role. Front. Immunol. 10:1809. doi: 10.3389/fimmu.2019.01809
Received: 06 February 2019; Accepted: 17 July 2019;
Published: 06 August 2019.
Edited by:
Marie-Agnes Dragon-Durey, Université Paris Descartes, FranceReviewed by:
Renato Alberto Sinico, University of Milano-Bicocca, ItalyBarbara Seitz-Polski, Centre Hospitalier Universitaire de Nice, France
Copyright © 2019 Liu, Gao, Dai, Zheng, Dong, Gao, Liu, Zhang, Liu, Liu, Liu, Liu and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Baoli Liu, liubaoli@bjzhongyi.com; Qingquan Liu, liuqingquan2003@126.com; Jialan Shi, jialan_shi@hms.harvard.edu
†These authors have contributed equally to this work