 Yan Zhou1†
Yan Zhou1† Xiao Leng
Xiao Leng Yan Li
Yan Li Guixiu Shi
Guixiu Shi Yantang Wang
Yantang Wang- 1Department of Emergency, West China Second University Hospital and Key Laboratory of Obstetric and Gynecologic and Pediatric Diseases and Birth Defects, Ministry of Education, Sichuan University, Chengdu, China
- 2Department of Immunology, School of Basic Medical Sciences, Chengdu Medical College, Chengdu, China
- 3Department of Rheumatology and Clinical Immunology, The First Affiliated Hospital, Xiamen University, Xiamen, China
T helper 17 (Th17) cells are crucial for the pathogenesis of multiple sclerosis (MS) in humans and experimental autoimmune encephalomyelitis (EAE) in animals. High frequency of Th17 cells and low sensitivity to activation-induced cell death (AICD) are detected in MS patients. However, the mechanisms underlying apoptosis resistance of T cells remain unclear. Perp is an apoptosis-associated target of p53 and implicated in the development of cancers. Here, we show that loss of Perp in T cells does not affect Th1, Th17, or Treg cell differentiation, but does significantly increase the resistance of Perp−/− Th17 cells to AICD and anti-Fas in Lck-Cre × Perpfl/fl mice by inhibiting the caspase-dependent apoptotic pathway. Moreover, Lck-Cre × Perpfl/fl mice exhibited earlier onset of EAE and severe spinal cord inflammation and demyelination, accompanied by increased levels of pro-inflammatory cytokines and enlarged population of Th17 cells. Therefore, Perp deletion promoted Th17 responses and exacerbated the development and severity of EAE.
Introduction
T helper 17 (Th17) cells are crucial for the pathogenesis of autoimmune diseases (AID), such as multiple sclerosis (MS) and its experimental model, experimental autoimmune encephalomyelitis (EAE) (1, 2). Initially, IL-17A and IL-22 produced by Th17 cells can disrupt tight junctions at the blood–brain barrier and facilitate the penetration of autoreactive Th17 and Th1 cells into the central nervous system (CNS) (3). Subsequently, autoreactive Th1 and Th17 cells interact with myeloid cells and B cells, together with their secreting pro-inflammatory cytokines, to create a microenvironment, leading to a sustained myelin and axonal damage in the CNS (4). Actually, a high frequency of Th17 cells is detected in peripheral blood and the CNS lesions of patients with active MS (5, 6). In addition, adoptive transfer of myelin-specific Th17 cells induces EAE in naive syngeneic animals (7). Hence, the maintenance of Th17 cell homeostasis is important for the inhibition of autoimmune demyelination. It is well known that activated T cells can undergo apoptosis, or activation-induced cell death (AICD), following repeated T-cell receptor (TCR) stimulation and through the Fas/FasL signaling. Such self-regulation can eliminate excessive and prolonged presence of effector T cells and help in maintaining peripheral tolerance (8). Alternation in AICD of T cells is associated with the development of AID and low levels of AICD in T cells are observed in patients with relapsing-remitting MS (9). However, the precise mechanisms underlying the resistance of Th17 cells to AICD in MS patients have not been clarified.
PERP, an apoptosis-associated target of p53, is a membrane protein of the PMP-22/gas3 family and is implicated in the development of a variety of cancers (10). The Perp−/− mice die postnatally with extensive blistering on the skin and oral mucosa, the lethal phenotype that indicated an important role of PERP in the p63-directed epithelial development (11, 12). Furthermore, PERP also regulates the apoptosis of thymocytes and peripheral T cells through the p53 apoptotic pathway. Activation of murine T cells by antigen stimulation upregulates the expression of PERP, contributing to their apoptosis (13). Modulation of PERP expression by a CD25 blockade decreases the FasL-mediated AICD of human T cells (14). Furthermore, CD4+CD8+ thymocytes from Perp−/− mice are resistant to radiation-induced apoptosis (15). In addition, decreased levels of PERP expression are detected on peripheral blood mononuclear cells (PBMCs) from patients with rheumatic arthritis (RA), and the levels of PERP expression are inversely correlated with IL-17 responses and disease activity (16). Accordingly, we hypothesize that Perp−/− in T cells may inhibit AICD of Th17 cells to exacerbate the development of EAE.
In this study, we generated the Lck-Cre × Perpfl/fl mice with Perp−/− in T cells and examined the impact of Perp−/− in T cells on Th1, Th17, or Treg cell differentiation and apoptosis as well as potential apoptosis pathway in vitro. Furthermore, we investigated the effect of Perp−/− in T cells on the development of EAE in mice. Our data indicated that Perp−/− in T cells did not affect Th1, Th17, or Treg cell differentiation, but did increase the resistance to anti-Fas induced apoptosis in Th17 cells, accompanied by inhibiting the caspase-dependent pathway in vitro. Perp−/− in T cells promoted the early onset and severity of EAE by increased levels of inflammation and demyelination in the CNS, which was associated with enhanced Th17 responses in vivo. Our findings may provide new insights in the regulation of Th17 responses and pathogenesis of EAE.
Materials and Methods
Mice
Perpfl/+ mice were purchased from the Jackson Laboratory (Stock No: 016132, Bar Harbor, ME, USA). Lck-Cre mice were obtained from Model Animal Research Center of Nanjing University (Nanjing, China). The Perpfl/fl and Lck-Cre mice were bred to obtain littermate control, Lck-Cre × Perpfl/+ and Lck-Cre × Perpfl/fl mice. All mice were housed in a specific pathogen-free facility at the Experimental Animal Center of Chengdu Medical College. The experimental protocol was approved by the Animal Care and Use Committee of Chengdu Medical College. C57BL/6 mice with EAE were used as an animal model of human MS.
Breeding and Identification of Lck-Cre × Perpfl/+ and Lck-Cre × Perpfl/fl Mice
To obtain the mice with Perp−/− specific in T cells, male and female Perpfl/+ mice were mated with a ratio of 1:1. The F1 Perpfl/fl mice were identified by PCR and bred with Lck-Cre mice to generate the Lck-Cre × Perpfl/+ mice. Subsequently, the male and female Lck-Cre × Perpfl/+ mice were bred to obtain the Lck-Cre × Perpfl/fl mice. Their genotypes were analyzed by PCR and Western blot. The sequences of PCR primers were forward 5′-AGT CTT CAG GGA TGA CAC AGA-3′ and reverse, 5′-TAC GAA ACT AGA GCA CAG CTA-3′ for Perp; forward 5′-ATT TGC CTG CAT TAC CGG TC-3′ and reverse, 5′-ATT TGC CTG CAT TAC CGG TC-3′ for Lck-Cre.
Murine T Cell Isolation and Differentiation
Naïve splenic CD4+ T cells were isolated from 8-week-old female littermate control, Lck-Cre × Perpfl/+ and Lck-Cre × Perpfl/fl mice as described previously (17) and purified using the Naive CD4+ T Cell Isolation Kit (Miltenyi-Biotec, Bergisch Gladbach, Germany). The purified cells were characterized by FACS. The purified naive CD4+ T cells (1 × 106 cells/ml) were stimulated with immobilized anti-CD3 (2 µg/ml) and soluble anti-CD28 (2 µg/ml, BD PharMingen, San Diego, CA, USA) in RPMI 1640 containing 10% FCS. To evaluate the effect of Perp−/− on the differentiation of Th17 cell, the naïve CD4+ T cells were stimulated with anti-CD3 and anti-CD28 in the presence of recombinant human TGF-β1 (5 ng/ml, PeproTech, Rocky Hill, NJ, USA), IL-6 (20 ng/ml, PeproTech), anti-IL-4 (10 µg/ml), and anti-IFN-γ (20 µg/ml BD PharMingen) for 3 days. For Th1 differentiation, naïve CD4+ T cells were stimulated with anti-CD3 and anti-CD28 in the presence of anti-IL-4 (10 µg/ml), and recombinant mouse IL-12 (10 ng/ml, PeproTech) for 3 days. For Treg differentiation, naïve CD4+ T cells were stimulated with anti-CD3 and anti-CD28 in the presence of anti-IL-4 (10 µg/ml), anti-IFN-γ (20 µg/ml), and recombinant human TGF-β1 (2 ng/ml) for 3 days. The cells were washed with PBS and used for subsequent experiments.
Intracellular Staining and Flow Cytometry
The frequency of different subsets of Th cells was characterized by FACS. Briefly, the cells were stained with fluorescein isothiocyanate (FITC)-anti-CD4, fixed, and permeabilized with GolgiPlug™ (BD PharMingen). After being washed, the cells were stained intracellularly with PE-conjugated anti-IFN-γ and Alexa Fluor® 647-conjugated anti-IL-17, followed by FACS analysis. Some splenocytes were stained with FITC-anti-CD4 and APC-anti-CD25 (BD PharMingen), fixed, permeabilized, and stained with PE-anti-Foxp3 (BD PharMingen), followed by FACS analysis of the frequency of Tregs. Some splenocytes were stained with FITC-anti-CD4 and PE-anti-CD44 (BD PharMingen), fixed, permeabilized, and stained with Alexa Fluor® 647-conjugated anti-IL-17 (BD PharMingen), followed by FACS analysis of the frequency of memory Th17 cells.
Apoptosis
The naïve T cells were stimulated with anti-CD3/anti-CD28 in the presence of Th1, Th17, or Treg polarizing cytokine-cocktail and neutralizing antibodies for 5 days. The cells were reactivated with anti-CD3 (2 µg/ml) for 72 h in the presence or absence of 1 µg/ml anti-Fas (BD PharMingen). The percentages of Annexin V+7-AAD− apoptotic Th1, Th17, or Treg cells were analyzed by FACS using an Annexin V apoptosis detection kit (BD PharMingen), according to the manufacturer’s instructions.
Western Blot
The differentiated Th17 cells from littermate control, Lck-Cre × Perpfl/+ and Lck-Cre × Perpfl/fl mice were lysed in RIPA buffer (Millipore, Billerica, MA, USA) and centrifuged. The protein concentrations in the lysates were determined using a BCA Protein Assay Kit (Pierce, Rockford, IL, USA). The mitochondria and cytosol of some differentiated Th17 cells were prepared using the Mitochondria/Cytosol Fractionation kit (Abcam). The cell lysate (50 μg/lane) was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) on 12% gels and transferred onto the polyvinylidene difluoride membrane. The membranes were blocked with 5% non-fat dry milk in PBST and incubated with primary antibodies against PERP (Santa Cruz Biotechnology, Santa Cruz, CA, USA), cleaved caspase-8, Bcl-2, Bcl-xL, cytochrome c (cyto-c), cleaved caspase-3 (Cell Signaling Technology, Beverly, MA, USA), and tubulin (ZSBiO, Beijing, China) at 4°C overnight. After being washed, the bound antibodies were detected with horseradish peroxidase-conjugated goat anti-rabbit IgG (Cell Signaling Technology) and visualized by the ChemiDoc XRS (Bio-Rad, Hercules, CA, USA). The relative levels of target protein to the control tubulin were determined by densitometric analyses using Quantity One software (Bio-Rad).
Induction and Evaluation of EAE
The littermate control, Lck-Cre × Perpfl/+ and Lck-Cre × Perpfl/fl mice at 10 weeks of age were immunized with 200 µg MOG35–55 (Chinese Peptide, Shanghai, China) emulsified in 50% complete Freud’s adjuvant (CFA) containing 4 mg/ml heat-killed Mycobacterium tuberculosis (Chondrex, Redmond, WA, USA) at their dorsal flanks. Individual mice were injected intraperitoneally with pertussis toxin (200 ng/mouse, Millipore) on day 0 and 2. The development and severity of EAE in individual mice were scored daily using the following score system: 0, healthy; 1, tail paralyzed; 2, no coordinated movement; hind limb paresis; 3, both hind limbs paralyzed; 4, forelimbs paralyzed; and 5, moribund state.
Histology
At 23 days post-induction, blood samples were collected from individual mice. The mice were anesthetized by 2% pentobarbital sodium and perfused intracardially with PBS (pH 7.4) followed by 4% paraformaldehyde in PBS. Their spinal cord samples were dissected. Some tissues from each group were fixed in 4% paraformaldehyde overnight and paraffin embedded. The sagittal cervicothoracic spinal cord sections (5 µm) were stained with H&E and Luxol fast blue to examine the degrees of inflammation and demyelination, respectively.
RNA Extraction and Real-Time PCR
Total RNA was extracted from the CNS tissues in individual groups of mice using Trizol (Invitrogen), following the manufacturer’s instructions. Subsequently, 2 µg total RNA of each sample was reversely transcribed to cDNA using the iScript™ Advanced cDNA Synthesis Kit, according to the manufacturer’s protocol (Bio-Rad). The relative levels of TNF-α, IL-6, and IL-17A mRNA transcripts to GAPDH were determined by quantitative real-time PCR (CFX 96, Bio-Rad) using iQ™ SYBR® Green Supermix (Bio-Rad) and specific primers. The sequences of primers were sense 5′-TGGCTAAGGACCAAGACCATCCAA-3′ and antisense 5′-AACGCACTAGGTTTGCCGAGTAGA-3′ for IL-6; sense 5′-GCTCCAGAAGGCCCTCAGA-3′ and antisense 5′-AGCTTTCCCTCCGCATTGA-3′ for IL-17A; sense 5′-CCTCCCTCTCATCAGTTCTATGG-3′ and antisense 5′-CGTGGGCTACAGGCTTGTC-3′ for TNF-α; and sense 5′-GGCAAATTCAACGGCACA-3′ and antisense 5′-GTTAGTGGGGTCTCGCTCTG-3′ for GAPDH. The levels of mRNA transcripts were normalized to GAPDH and analyzed by the 2−ΔΔCt method.
Immunofluorescent Staining
Some spinal cord tissues from each group were fixed in 4% paraformaldehyde overnight, immersed in 30% sucrose, and embedded in OCT on dry ice. The cryostat transversal lumbar spinal cord sections (20 µm) were washed twice with PBS and blocked with 10% normal mouse serum in PBS for 1 h at room temperature. The sections were incubated with goat anti-mouse IL-17A (Santa Cruz Biotechnology) or rat anti-mouse CD4 (BD PharMingen) overnight at 4°C. After being washed, the sections were incubated with PE-anti-goat IgG (Santa Cruz Biotechnology) and Alexa Fluor 488-anti-rat IgG (Abcam). Images were taken under an Olympus Confocal FV1000 microscope.
ELISA
The concentrations of serum IL-1β, IL-6, IL-10, IL-23, TNFα, and IL-17A in individual mice were determined using the cytokine-specific ELISA kits (eBioscience, San Diego, CA, USA), according to the manufacturer’s instructions.
Statistical Analysis
Data are expressed as the mean ± SEM. The difference between two groups was evaluated by two-tailed unpaired Student’s t-test (Gaussian distribution) or two-tailed Mann–Whitney U-test (no Gaussian distribution). The difference among three or more groups was analyzed by repeated ANOVA and post hoc Bonferroni correction. All statistical analyses were performed using PRISM 6.0 software (GraphPad Software, San Diego, CA, USA). A P-value of <0.05 was considered statistically significant.
Results
Propagation and Genotype Identification of the Transgenic Mice
To evaluate the impact of Perp−/− in T cells on the Th17-cell survival and pathogenesis of EAE, the Lck-Cre × Perpfl/+ and Lck-Cre × Perpfl/fl mice were generated and their genotypes on CD4+ T cells were identified by PCR (Figures 1A,C). Western blot analysis indicated that there was almost no detectable PERP expression in CD4+ T cells from Lck-Cre × Perpfl/fl mice (Figure 1B), but the levels of PERP expression in CD4+ T cells from Lck-Cre × Perpfl/+ mice were only slightly lower than that in the littermate control mice (Figure 1B). These two stains of mice were valuable for evaluating the importance of PERP in the survival of Th17 cells and the development of EAE.
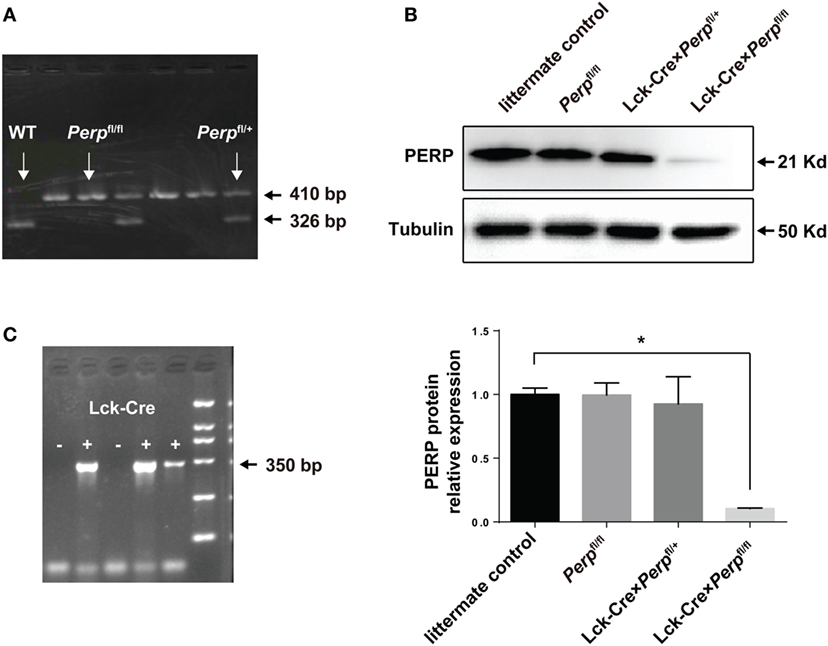
Figure 1. Identification of Lck-Cre × Perpfl/+ and Lck-Cre × Perpfl/fl mice. Following breeding Perpfl/fl mice, the F1 Lck-Cre × Perpfl/+ and Lck-Cre × Perpfl/fl mice were identified by PCR analysis of the genomic DNA from peripheral blood mononuclear cell for the Perp (A) and Lck-Cre (C) genes and Western blot analysis of splenic T cells (B). Data are representative images or expressed as the mean ± SEM of each group from three separate experiments (*P < 0.05).
Deficiency of Perp Inhibits Th17 Cell Apoptosis, But Does Not Affect Their Differentiation
To investigate the impact of Perp−/− on Th1, Th17, or Treg differentiation and survival, naïve CD4+ T cells from littermate control, Lck-Cre × Perpfl/+ and Lck-Cre × Perpfl/fl mice were purified and stimulated with anti-CD3/anti-CD28 in the presence of cocktails of cytokines and antibodies to induce Th1, Th17, or Treg cell differentiation for 3 days. The percentages of Th1, Th17, or Treg cells were analyzed by FACS. There was no significant difference in the frequency of Th1, Th17, or Treg cells among these groups (Figures 2A–F). Such data indicated that the Perp deficiency did not affect the differentiation of Th1, Th17, or Treg cells in our experimental conditions. Our previous study shows that there is a remarkably inverse correlation between the percentages of IL-17 producing cells and PERP expression in PBMCs from RA patients (16). Meanwhile, the p53 expression was downregulated in PBMC from patients with RA or MS (17). Accordingly, we studied the effect of Perp−/− on the apoptosis of Th1, Th17, or Treg cells by FACS. While there was no significant difference in the frequency of apoptotic Th1 and Treg cells, the percentages of apoptotic Th17 cells from Lck-Cre × Perpfl/fl mice were lower than that from Lck-Cre × Perpfl/+ and littermate control mice, particularly after anti-Fas treatment (Figures 2G–L). Hence, the PERP may positively regulate Th17 cell apoptosis.
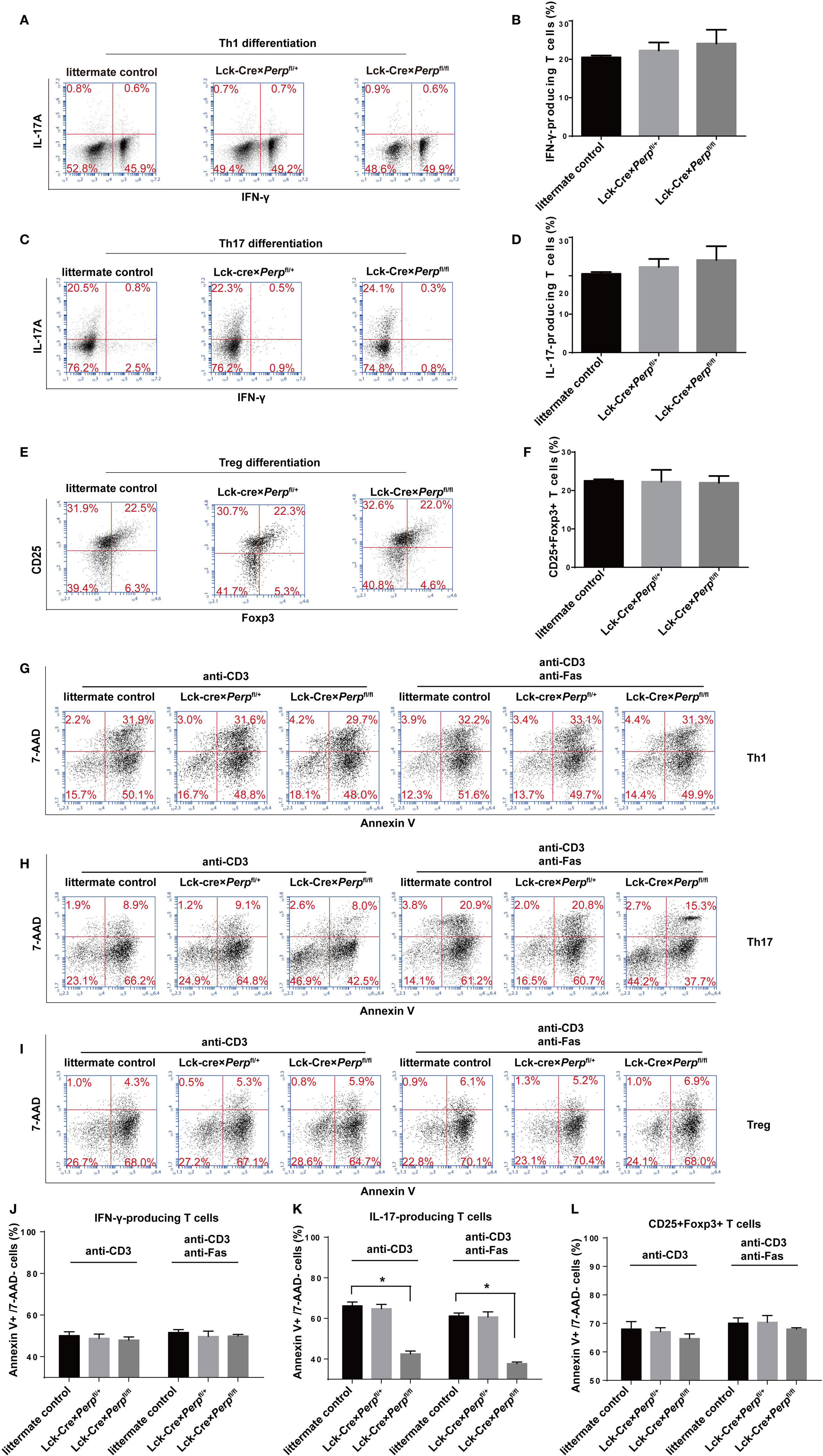
Figure 2. Perp−/− in T cells promotes the resistance to apoptosis in T helper 17 (Th17) cells. CD4+ T cells were isolated from littermate control, Lck-Cre × Perpfl/+ and Lck-Cre × Perpfl/fl mice by negative selection and stimulated with anti-CD3/anti-CD28 in the presence of cocktail of cytokines and antibodies to induce Th1, Th17, or Treg cell differentiation for 3 days. The cells were stained with anti-CD4 and/or anti-CD25, fixed, permeabilized, and intracellularly stained with anti-IFN-γ, anti-IL-17A, or anti-Foxp3. The frequency of Th1, Th17, or Treg cells was determined by FACS. Furthermore, following in vitro differentiation for 3 days, the cells were re-stimulated with anti-CD3/anti-CD28 in the presence or absence of anti-Fas for 72 h. The percentages of apoptotic Th1, Th17, or Treg cells were determined by FACS using Annexin V-PE and 7-AAD staining. Data are representative images or expressed as the mean ± SEM of each group from three separate experiments. (A,B) Th1 cell differentiation. (C,D) Th17 cell differentiation. (E,F) Treg cell differentiation. (G,J) Th1 cell apoptosis. (H,K) Th17 cell apoptosis. (I,L) Th17 cell apoptosis (*P < 0.05).
Perp−/− Reduces the Caspase Activation in Th17 Cells
It is well known that PERP is crucial for caspase activation (18, 19), while Bcl-xL and Bcl-2 are pro-survival factors of Th17 cells (20, 21). To understand the mechanisms underlying the action of Perp−/− in regulating Th17 cell apoptosis, the relative levels of cyto-c, caspase-8, cleaved caspase-3, BcL-xL, and BcL-2 expression in the differentiated Th17 cells from three strains of mice were determined by Western blot. There was no significant difference in the relative levels of these molecule expression between heterozygous and littermate control CD4+ T cells (Figure 3). However, in comparison with that in the Perp−/+ and littermate control CD4+ T cells, significantly decreased levels of cleaved caspase-8, cytosolic cyto-c, cleaved caspase-3, but increased levels of BcL-xL and BcL-2 expression were detected in Perp−/− CD4+ T cells (Figures 3A–E). However, Perp−/− did not change the levels of mitochondrial cyto-c in Th17 cells (Figure 3F). Such data were consistent with previous observation that PERP-induced apoptosis is mediated by the caspase-dependent pathway (18).
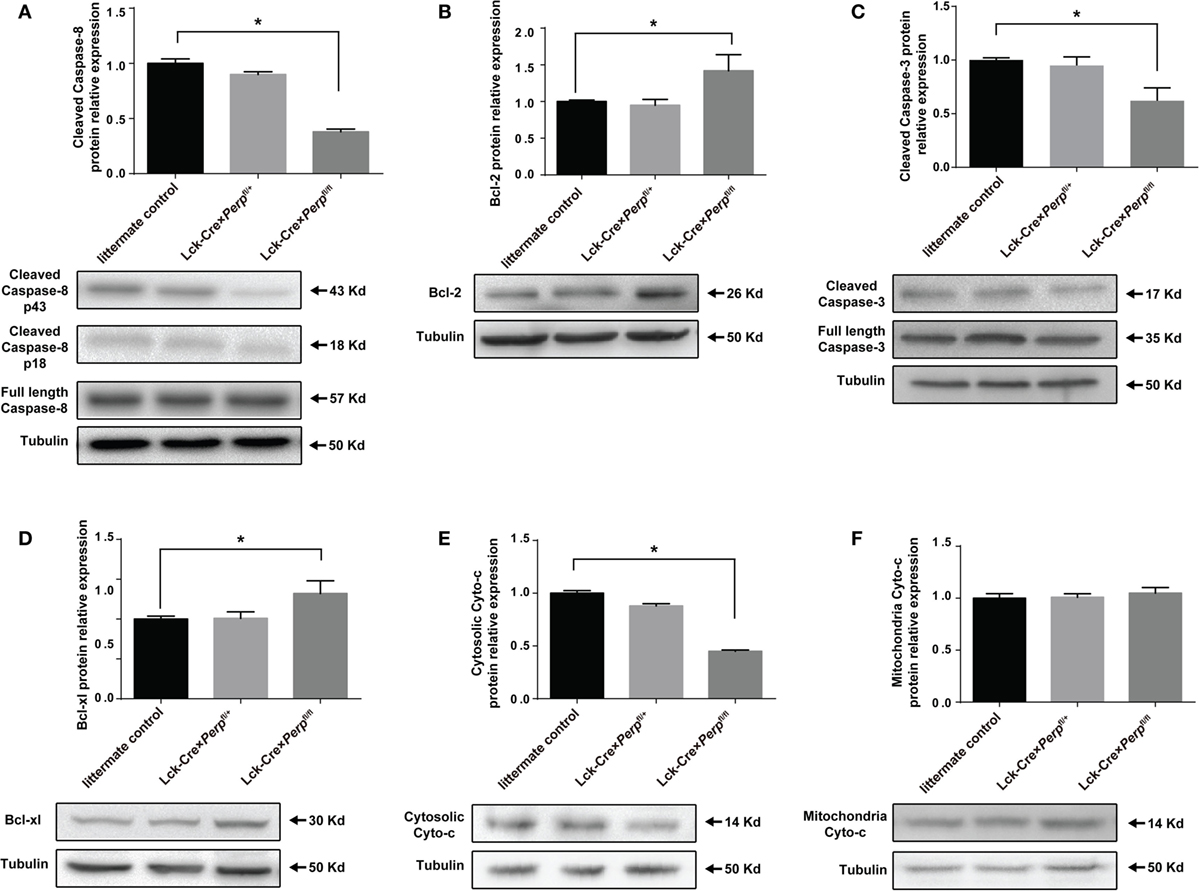
Figure 3. Perp−/− in T cells promotes the resistance of T helper 17 (Th17) cells to apoptosis in a caspase-dependent manner. Following induction of apoptosis, the relative levels of cleaved caspase-8 (A), Bcl-2 (B), cleaved caspase-3 (C), Bcl-xL (D), cytosolic cytochrome (cyto-c) (E), and mitochondrial cyto-c (F) to the control tubulin expression in the different groups of Th17 cells were determined by Western blot. Data are representative images or expressed as the mean ± SEM of each group from three separate experiments (*P < 0.05).
Perp−/− Enhances the Development of EAE
T helper 17 cells play a key role in the development of EAE (22). Given that Perp−/− decreased Th17 cell apoptosis we investigated that whether Perp−/− would exacerbate the development of EAE. Littermate control, Lck-Cre × Perpfl/+ and Lck-Cre × Perpfl/fl mice were immunized with MOG35–55 in 50% CFA to induce EAE and the severity of EAE in individual groups of mice was monitored daily (Figure 4A). At 23 days post-immunization, the scores of EAE in the Lck-Cre × Perpfl/fl mice were significantly higher than that in the littermate control and Lck-Cre × Perpfl/+ mice. The mean time for disease onset in the Lck-Cre × Perpfl/fl mice was significantly earlier than that in the littermate control and Lck-Cre × Perpfl/+ mice (Figure 4B), and the mean maximum scores the Lck-Cre × Perpfl/fl mice were significantly higher than that in the littermate control and Lck-Cre × Perpfl/+ mice (Figure 4C). Such data indicated that Perp−/− in T cells exacerbated the development and severity of EAE in mice. Histopathological examinations revealed more obvious demyelination and inflammatory infiltrates in the spinal cord tissues from the Lck-Cre × Perpfl/fl mice than that in the littermate control and Lck-Cre × Perpfl/+ mice (Figures 4D–G).
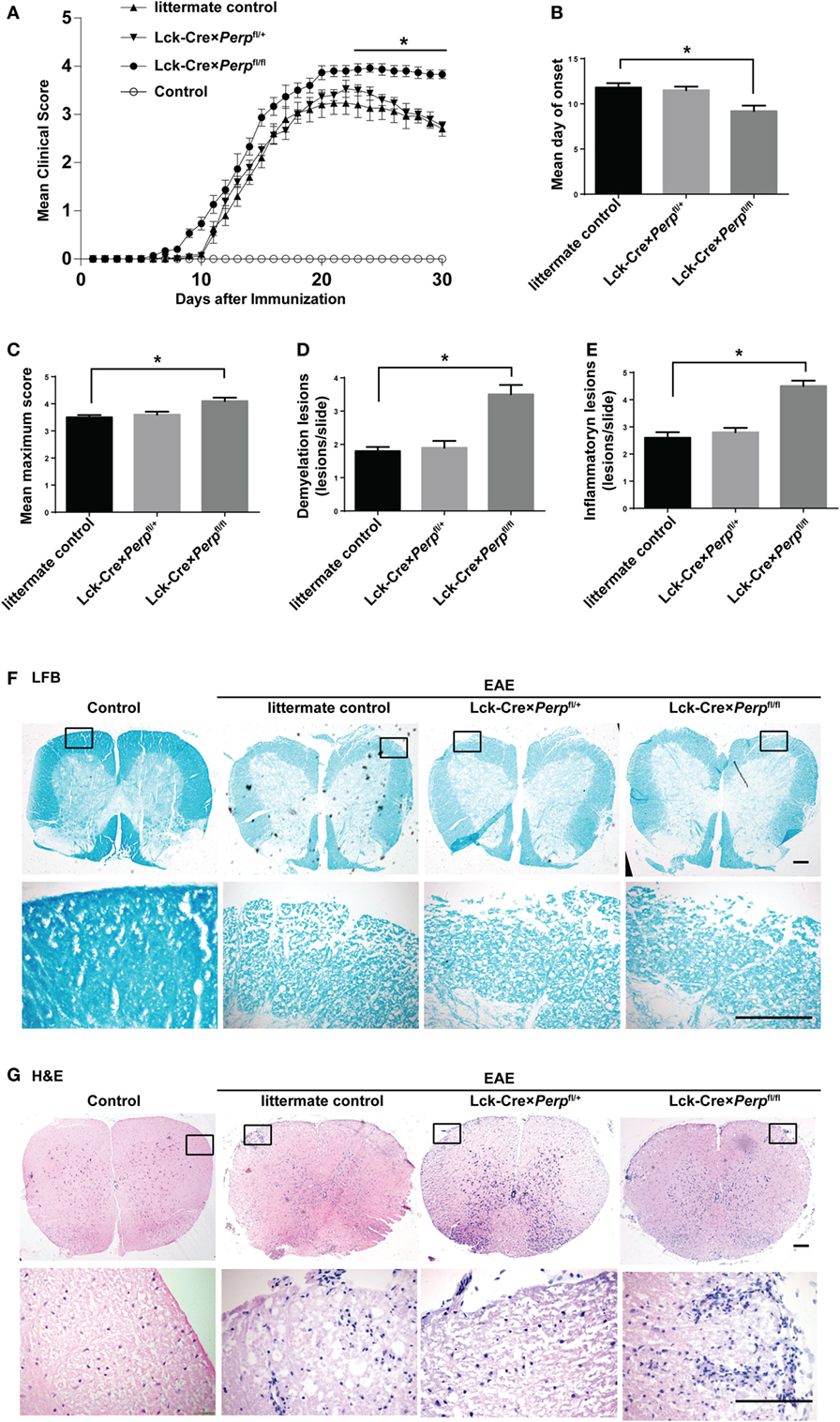
Figure 4. Perp−/− in T cells exacerbates the development of experimental autoimmune encephalomyelitis (EAE) in mice. Littermate control, Lck-Cre × Perpfl/+ and Lck-Cre × Perpfl/fl mice (n = 10 per group) were injected with MOG35–55 in 50% complete Freud’s adjuvant and the development and severity of EAE in individual groups of mice were monitored daily (A). The mean time for EAE onset (B) and the maximum EAE scores (C) in individual groups of mice were calculated. At end of the experiment, the mice were perfused and their spinal cord tissues were dissected, fixed, and stained with LFB (F) and H&E (G). The number of lesions with demyelination (D) and inflammatory infiltrates (E) were quantified in spinal cord section of each mouse. Data are representative images or expressed as the mean ± SEM of each group. Scale bar, 200 µm.
IL-23, IL-6, and IL-1β are crucial for Th17 cell differentiation and IL-17A and TNF-α are necessary for the development of EAE (23–25). However, IL-10 can inhibit the pathogenicity of Th17 cell-related EAE in mice (26). To further understand the impact of Perp−/− in T cells on the development of EAE, the levels of serum IL-1β, TNF-α, IL-23, IL-10, IL-6, and IL-17A in individual groups of mice were measured by ELISA. The results showed that the levels of serum TNF-α, IL-6, and IL-17A, but not IL-1β, IL-23, and IL-10, in the Lck-Cre × Perpfl/fl mice were significantly higher than that in the littermate control and Lck-Cre × Perpfl/+ mice (Figures 5A–F). Further quantitative real-time PCR indicated that the relative levels of TNF-α, IL-6, and IL-17A mRNA transcripts to GAPDH in the brain of Lck-Cre × Perpfl/fl mice were significantly higher than that in the littermate control and Lck-Cre × Perpfl/+ mice (Figures 5G–I). In addition, some 10-month-old Lck-Cre × Perpfl/fl mice, but not age-matched littermates, developed low degrees of EAE-like symptoms (Figure 6A) and mild demyelination and inflammatory infiltrates in their spinal cord tissues (Figures 6B–E). Together, the early onset, higher disease scores, severer pathogenic lesions, and elevated levels of pro-inflammatory TNF-α, IL-6, and IL-17A demonstrated that Perp−/− in T cells exacerbated the development and severity of EAE in mice.
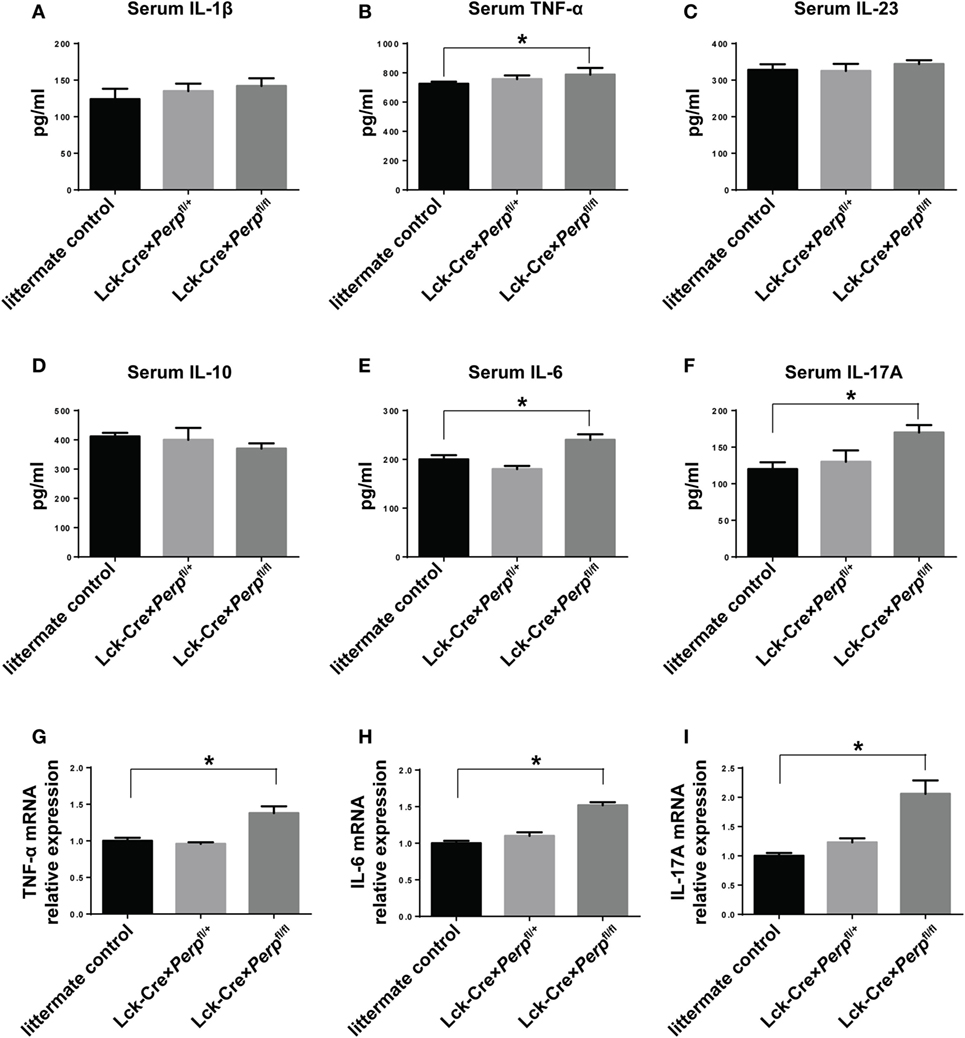
Figure 5. Perp−/− in T cells increases the levels of pro-inflammatory cytokines in mice following induction of experimental autoimmune encephalomyelitis. At 23 days post-induction, peripheral blood samples and central nervous system tissues were obtained from individual mice. The relative levels of serum IL-1β (A), TNF-α (B), IL-23 (C), IL-10 (D), IL-6 (E), and IL-17A (F) were determined by ELISA. The relative levels of TNF-α (G), IL-6 (H), and IL-17A (I) mRNA transcripts to GAPDH were determined by quantitative real-time PCR. Data are expressed as the mean ± SEM of each group (n = 8 per group) from three separate experiments (*P < 0.05).
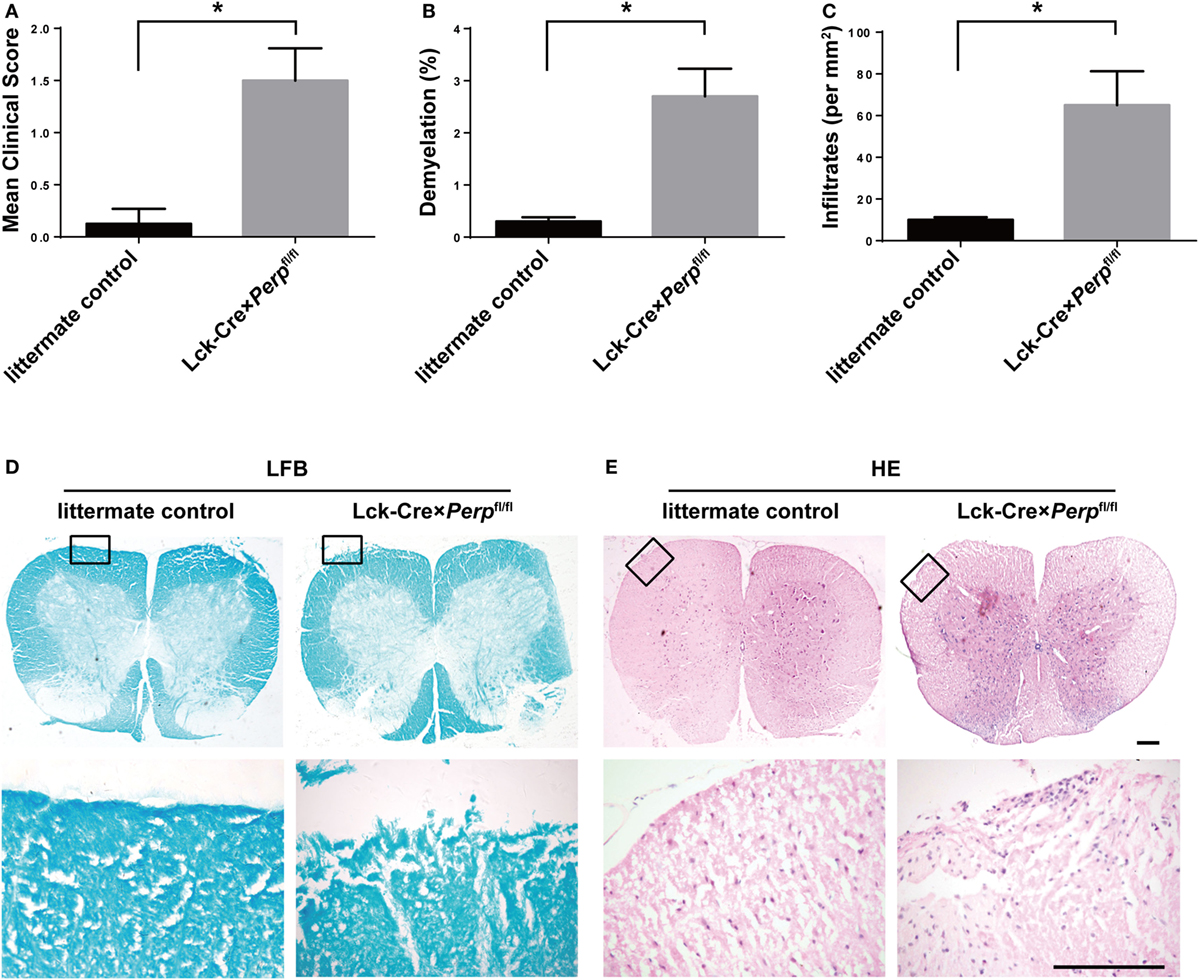
Figure 6. Aged Lck-Cre × Perpfl/fl mice exhibit mild experimental autoimmune encephalomyelitis (EAE)-like symptoms. (A) The EAE scores of aged Littermate control and Lck-Cre × Perpfl/fl mice (n = 8 per group, 10 months of age) were calculated. The mice were perfused after symptom onset and their spinal cord tissues were dissected, fixed, and stained with LFB (D) and H&E (E). The number of lesions with demyelination (B) and infiltrates (C) were quantified in three spinal cord tissue sections of each mouse. Data are representative images or expressed as the mean ± SEM of each group. Scale bar, 200 µm.
Perp−/− in T Cells Increases the Frequency of Th17 Cells During the Process of EAE
To understand the mechanisms by which Perp−/− in T cells exacerbated the development of EAE, the percentages of splenic Th1, Th17 cells, and Tregs in the different groups of mice were characterized by FACS. The results showed that the percentages of Th17 cells, but not Th1 cells and Tregs, in the Lck-Cre × Perpfl/fl mice were almost twofold higher than that in the littermate control and Lck-Cre × Perpfl/+ mice (Figures 7A–E). Further immunofluorescent analysis revealed more frequent IL17A+ cells in the spinal cord tissues of the Lck-Cre × Perpfl/fl mice than that of the littermate control and Lck-Cre × Perpfl/+ mice (Figure 7F). Furthermore, the percentages of splenic memory Th17 cells (CD4+CD44hiIL-17+) in the Lck-Cre × Perpfl/fl mice were also significantly higher than that in the littermate control and Lck-Cre × Perpfl/+ mice (Figures 7G,H). Therefore, Perp−/− in T cells promoted Th17 responses, exacerbating the development of EAE in mice.
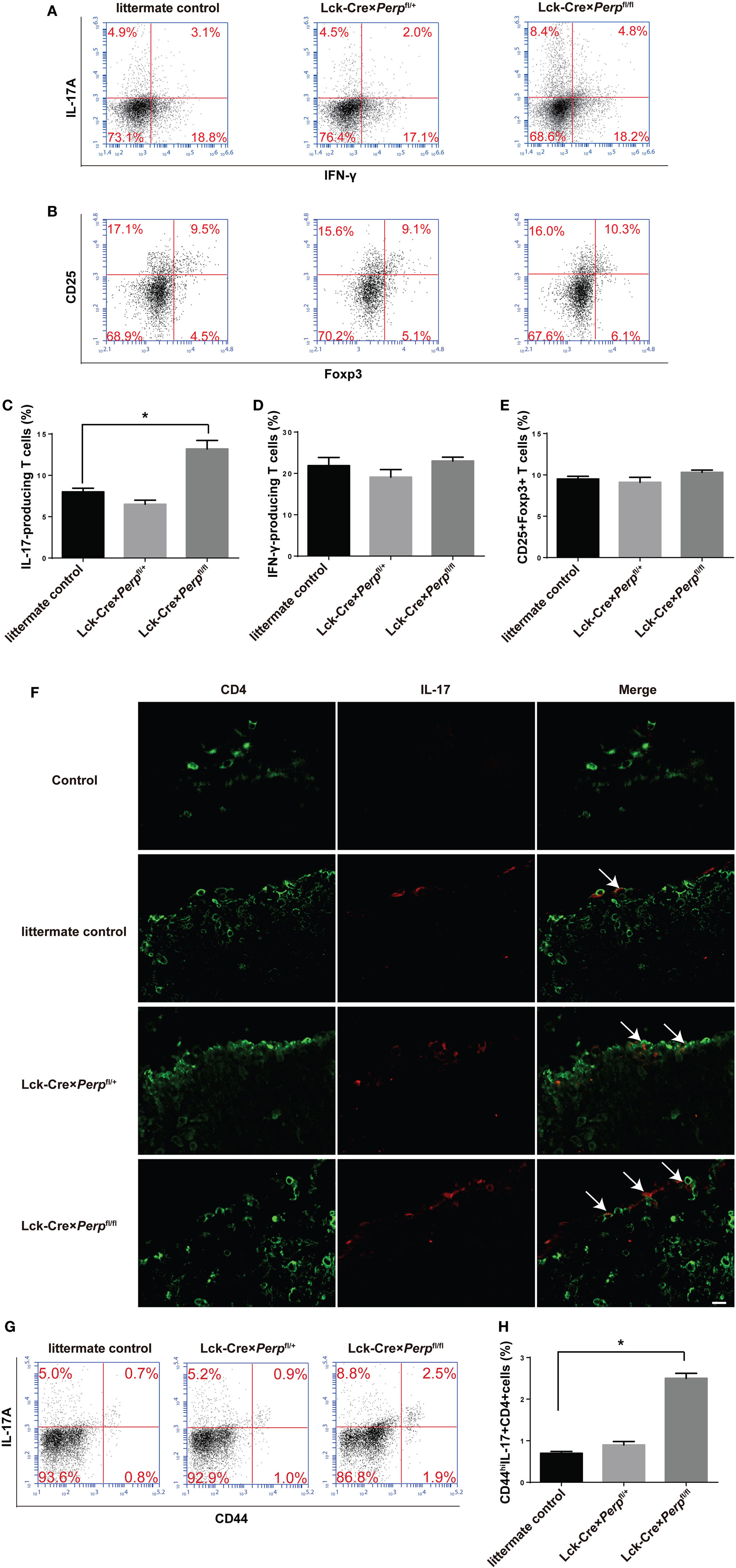
Figure 7. Increased frequency of T helper (Th17) cells in the peripheral spleen and spinal cord tissues in Lck-Cre × Perpfl/fl mice. At 23 days post-immunization, splenic mononuclear cells were isolated from littermate control, Lck-Cre × Perpfl/+ and Lck-Cre × Perpfl/fl mice. The cells were stained with anti-CD4, fixed, permeabilized, and intracellularly stained with anti-IFN-γ and anti-IL-17A. Some cells were stained with anti-CD4, anti-CD25, fixed, permeabilized, and intracellularly stained with anti-Foxp3. The percentages of Th1, Th17 cells, and Tregs were determined by FACS. In addition, the presence of Th17 cells in the spinal cord tissues was characterized by immunofluorescent assay. The cryostat spinal cord tissue sections were stained with anti-CD4 (Alexa Fluor 488) and anti-IL-17A (PE), followed by imaging under a confocal microscope. Some splenic cells were stained with anti-CD4, anti-CD44, fixed, permeabilized, and intracellularly stained with anti-IL-17A. The percentages of splenic memory Th17 cells (CD4+CD44hiIL-17A+) were determined by FACS. Data are representative images or expressed as the mean ± SEM of each group (n = 8 per group) from three separate experiments. (A,C,D) The frequency of Th1 and Th17 cells. (B,E) The frequency of Tregs (*P < 0.05). (F) Immunofluorescence of CD4 (green) and IL-17 (red) in the spinal cord tissues. Calibration bar, 20 µm. (G,H) The frequency of splenic memory Th17 cells.
Discussion
Physiologically, PERP functions to regulate the formation of desmosome complex and apoptosis (27, 28). Currently, there is little information on that PERP regulates immune responses because of the postnatal lethality of Perp−/− mice (27). Our previous study has shown that decreased levels of PERP expression are inversely correlated with disease activity and IL-17A expression in PBMC from patients with RA (16). In this study, we generated the Lck-Cre × Perpfl/fl mice with Perp−/− in T cells and demonstrated that Perp−/− Th17 cells were relatively resistant to AICD and anti-Fas mediated apoptosis in vitro and exacerbated the development of EAE in vivo by enhanced Th17 responses. Besides, some aged Lck-Cre × Perpfl/fl mice developed mild EAE symptoms. Such data suggest that PERP may promote Th17 cell apoptosis and attenuate the pathogenesis of EAE. Therefore, our findings may provide new insights into the pathogenesis of EAE.
T helper 17 responses are crucial for the pathogenesis of AID, such as MS, systemic lupus erythematosus (SLE), and RA (29, 30). Activated Th17 cells not only exist in peripheral blood but also infiltrate into the inflamed organs during the pathogenic process of SLE (31), rheumatoid synovium (32), and MS (5). It is assumed that the enhanced Th17 responses may be caused by prolonged survival or enhanced differentiation of Th17 cells. In this study, we found that Perp−/− in T cells did not affect the differentiation of Th1, Th17, or Treg cells. However, Perp−/− Th17 cells were resistant to apoptosis induced by chronic TCR stimulation or anti-Fas in a caspase-dependent manner. Evidentially, significantly lower levels of cleaved caspase-8 and cleaved caspase-3 were detected in Perp−/− Th17 cells. Such data support the notion that PERP can activate caspase-8 and caspase-3 to induce apoptosis (18). In addition, we detected significantly higher levels of Bcl-2 and Bcl-xL in Perp−/− Th17 cells, which should contribute to the survival of Perp−/− Th17 cells and the exacerbation of EAE (20, 21). It is possible that Perp−/− may promote the activity of Aiolos and/or c-Myb, which together with the TCR activation-mediated NF-κB activation, enhances Bcl-2 and Bcl-xl expression to support the survival of Perp−/− Th17 cells (33, 34).
Currently, the survival period of activated Th17 cells remains controversial (35, 36). In this study, we found that Perp−/− Th17 cells were resistant to apoptosis induced by chronic TCR stimulation and anti-Fas, and increased frequency of peripheral blood Th17 cells was detected in EAE Lck-Cre × Perpfl/fl mice. Hence, Perp−/− supports the survival of Th17 cells although surrounding cells express FasL. These findings may provide a new explanation why decreased levels of PERP expression in PBMC are inversely correlated with Th17 responses in patients with RA (16). More importantly, Lck-Cre × Perpfl/fl mice developed severer inflammation and remarkable demyelination in the spinal cord tissues, accompanied by increased levels of pro-inflammatory cytokines and higher frequency of Th17 cells, as well as memory Th17 cells. Given that Perp−/− in T cells did not affect the differentiation and survival of Th1 cells, it is possible that Perp−/− in T cells may preferably support the survival of activated Th17 cells, contributing to the development and progression of EAE. It is notable that autoreactive Th17 cells participate in the pathogenesis of several types of AID. Hence, we speculate that Perp−/− in T cells may also enhance Th17 responses, contributing to the pathogenic process of other AID.
Although Perp is a direct transcriptional target of p53, previous studies have reported that p53−/− CD4+ T cells differentiate into Th17 more efficiently and p53 is disposable for inducing the development of Th17 cell-associated autoimmune arthritis (37, 38). Our data showed that Perp−/− Th17 cells were resistant to AICD and apoptosis induced by anti-Fas. It is notable that p53−/− mice are prone to development of AID and have strong Th17 responses (39). Actually, a previous study has shown that p53 negatively regulates AID by inhibiting the Stat3 pathway and suppressing Th17 cell differentiation (40). However, Perp−/− in T cells did not affect the differentiation of Th17 cells. Apparently, the p53/Perp pathway regulates the different stages of the development of Th17 responses in a collaborative manner to control autoreactive Th17 responses and inflammation. Conceptually, enhancement of PERP expression and activity may be valuable for control of autoreactive Th17 responses and AID.
In conclusion, our data indicated that Perp−/− in T cells did not affect the differentiation of Th1, Th17, or Treg cells, but did promote the resistance of Th17 cells, but not Th1 and Treg cells, to apoptosis induced by chronic TCR stimulation and anti-Fas in a caspase-dependent manner in vitro. Perp−/− in T cells increased the levels of pro-inflammatory cytokines and exacerbated the development of EAE by increased frequency of Th17 cells and severer inflammation and demyelination in the spinal cord tissues of mice. Our findings may highlight a novel function of PERP in the maintenance of Th17 cell homeostasis and new insights in the pathogenesis of EAE. Potentially, enhancement of PERP expression and activity may be a valuable strategy to control AID.
Ethics Statement
The animal experiment was carried out in accordance with the recommendations of “The Ethics Review Committee for Animal Experimentation at Chengdu Medical College.” The protocol was approved by the “The Ethics Review Committee for Animal Experimentation at Chengdu Medical College.”
Author Contributions
YZ carried out the histological analysis, the animal experiment and drafted the manuscript. XL and YH carried out the cell culture and FACS. YL (Yan Li) participated in the animal experiment. YL (Yuan Liu) performed Western blotting and ELISA. YL (Yang Liu) and QZ participated in the design of the study and performed the statistical analysis. YW and GS conceived of the study, participated in its design and coordination, and helped to draft the manuscript. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Medjaden Bioscience Limited for its assistance in preparing this manuscript.
Funding
This study was supported by the grants from the National Natural Science Foundation of China (NSFC 81202363 to YW) and the Research Fund of Development and Regeneration Key Laboratory of Sichuan Province (SYS16-002 to YW).
References
1. Zhang X, Tao Y, Chopra M, Dujmovic-Basuroski I, Jin J, Tang Y, et al. IL-11 induces Th17 cell responses in patients with early relapsing-remitting multiple sclerosis. J Immunol (2015) 194(11):5139–49. doi:10.4049/jimmunol.1401680
2. Jadidi-Niaragh F, Mirshafiey A. Th17 cell, the new player of neuroinflammatory process in multiple sclerosis. Scand J Immunol (2011) 74(1):1–13. doi:10.1111/j.1365-3083.2011.02536.x
3. Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, et al. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med (2007) 13(10):1173–5. doi:10.1038/nm1651
4. Mishra MK, Yong VW. Myeloid cells – targets of medication in multiple sclerosis. Nat Rev Neurol (2016) 12(9):539–51. doi:10.1038/nrneurol.2016.110
5. Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, et al. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol (2008) 172(1):146–55. doi:10.2353/ajpath.2008.070690
6. Durelli L, Conti L, Clerico M, Boselli D, Contessa G, Ripellino P, et al. T-helper 17 cells expand in multiple sclerosis and are inhibited by interferon-beta. Ann Neurol (2009) 65(5):499–509. doi:10.1002/ana.21652
7. Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med (2008) 205(7):1535–41. doi:10.1084/jem.20080159
8. Green DR, Droin N, Pinkoski M. Activation-induced cell death in T cells. Immunol Rev (2003) 193:70–81. doi:10.1034/j.1600-065X.2003.00051.x
9. Moreno M, Negrotto L, Rio J, Moubarak R, Martin I, Bustamante MF, et al. Activation-induced cell death in T lymphocytes from multiple sclerosis patients. J Neuroimmunol (2014) 272(1–2):51–5. doi:10.1016/j.jneuroim.2014.04.007
10. Attardi LD, Reczek EE, Cosmas C, Demicco EG, McCurrach ME, Lowe SW, et al. PERP, an apoptosis-associated target of p53, is a novel member of the PMP-22/gas3 family. Genes Dev (2000) 14(6):704–18. doi:10.1101/gad.14.6.704
11. Dusek RL, Bascom JL, Vogel H, Baron S, Borowsky AD, Bissell MJ, et al. Deficiency of the p53/p63 target Perp alters mammary gland homeostasis and promotes cancer. Breast Cancer Res (2012) 14(2):R65. doi:10.1186/bcr3171
12. Awais R, Spiller DG, White MR, Paraoan L. p63 is required beside p53 for PERP-mediated apoptosis in uveal melanoma. Br J Cancer (2016) 115(8):983–92. doi:10.1038/bjc.2016.269
13. Wan YY, DeGregori J. The survival of antigen-stimulated T cells requires NFkappaB-mediated inhibition of p73 expression. Immunity (2003) 18(3):331–42. doi:10.1016/S1074-7613(03)00053-0
14. Richter GH, Mollweide A, Hanewinkel K, Zobywalski C, Burdach S. CD25 blockade protects T cells from activation-induced cell death (AICD) via maintenance of TOSO expression. Scand J Immunol (2009) 70(3):206–15. doi:10.1111/j.1365-3083.2009.02281.x
15. Ihrie RA, Reczek E, Horner JS, Khachatrian L, Sage J, Jacks T, et al. Perp is a mediator of p53-dependent apoptosis in diverse cell types. Curr Biol (2003) 13(22):1985–90. doi:10.1016/j.cub.2003.10.055
16. Du Y, Deng L, Li Y, Gan L, Wang Y, Shi G. Decreased PERP expression on peripheral blood mononuclear cells from patient with rheumatoid arthritis negatively correlates with disease activity. Clin Dev Immunol (2013) 2013:256462. doi:10.1155/2013/256462
17. Takatori H, Kawashima H, Suzuki K, Nakajima H. Role of p53 in systemic autoimmune diseases. Crit Rev Immunol (2014) 34(6):509–16. doi:10.1615/CritRevImmunol.2014012193
18. Davies L, Gray D, Spiller D, White MR, Damato B, Grierson I, et al. P53 apoptosis mediator PERP: localization, function and caspase activation in uveal melanoma. J Cell Mol Med (2009) 13(8B):1995–2007. doi:10.1111/j.1582-4934.2008.00590.x
19. Hallstrom KN, Srikanth CV, Agbor TA, Dumont CM, Peters KN, Paraoan L, et al. PERP, a host tetraspanning membrane protein, is required for Salmonella-induced inflammation. Cell Microbiol (2015) 17(6):843–59. doi:10.1111/cmi.12406
20. Banuelos J, Shin S, Cao Y, Bochner BS, Morales-Nebreda L, Budinger GR, et al. BCL-2 protects human and mouse Th17 cells from glucocorticoid-induced apoptosis. Allergy (2016) 71(5):640–50. doi:10.1111/all.12840
21. Liu X, Leung S, Wang C, Tan Z, Wang J, Guo TB, et al. Crucial role of interleukin-7 in T helper type 17 survival and expansion in autoimmune disease. Nat Med (2010) 16(2):191–7. doi:10.1038/nm.2077
22. Murugaiyan G, da Cunha AP, Ajay AK, Joller N, Garo LP, Kumaradevan S, et al. MicroRNA-21 promotes Th17 differentiation and mediates experimental autoimmune encephalomyelitis. J Clin Invest (2015) 125(3):1069–80. doi:10.1172/JCI74347
23. Reppert S, Zinser E, Holzinger C, Sandrock L, Koch S, Finotto S. NFATc1 deficiency in T cells protects mice from experimental autoimmune encephalomyelitis. Eur J Immunol (2015) 45(5):1426–40. doi:10.1002/eji.201445150
24. Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature (2010) 467(7318):967–71. doi:10.1038/nature09447
25. Luchtman DW, Ellwardt E, Larochelle C, Zipp F. IL-17 and related cytokines involved in the pathology and immunotherapy of multiple sclerosis: current and future developments. Cytokine Growth Factor Rev (2014) 25(4):403–13. doi:10.1016/j.cytogfr.2014.07.013
26. Guo B. IL-10 modulates Th17 pathogenicity during autoimmune diseases. J Clin Cell Immunol (2016) 7(2):400. doi:10.4172/2155-9899.1000400
27. Ihrie RA, Marques MR, Nguyen BT, Horner JS, Papazoglu C, Bronson RT, et al. Perp is a p63-regulated gene essential for epithelial integrity. Cell (2005) 120(6):843–56. doi:10.1016/j.cell.2005.01.008
28. Chen K, Luo Z, Li Z, Liu Y, Zhao Q. PERP gene therapy attenuates lung cancer xenograft via inducing apoptosis and suppressing VEGF. Cancer Biol Ther (2011) 12(12):1114–9. doi:10.4161/cbt.12.12.18435
29. Yazid S, Gardner PJ, Carvalho L, Chu CJ, Flower RJ, Solito E, et al. Annexin-A1 restricts Th17 cells and attenuates the severity of autoimmune disease. J Autoimmun (2015) 58:1–11. doi:10.1016/j.jaut.2014.12.004
30. Tabarkiewicz J, Pogoda K, Karczmarczyk A, Pozarowski P, Giannopoulos K. The role of IL-17 and Th17 lymphocytes in autoimmune diseases. Arch Immunol Ther Exp (2015) 63(6):435–49. doi:10.1007/s00005-015-0344-z
31. Yang J, Chu Y, Yang X, Gao D, Zhu L, Yang X, et al. Th17 and natural Treg cell population dynamics in systemic lupus erythematosus. Arthritis Rheum (2009) 60(5):1472–83. doi:10.1002/art.24499
32. Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N, et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med (2007) 204(12):2803–12. doi:10.1084/jem.20071397
33. Salomoni P, Perrotti D, Martinez R, Franceschi C, Calabretta B. Resistance to apoptosis in CTLL-2 cells constitutively expressing c-Myb is associated with induction of BCL-2 expression and Myb-dependent regulation of bcl-2 promoter activity. Proc Natl Acad Sci U S A (1997) 94(7):3296–301. doi:10.1073/pnas.94.7.3296
34. Kurland JF, Kodym R, Story MD, Spurgers KB, McDonnell TJ, Meyn RE. NF-kappaB1 (p50) homodimers contribute to transcription of the bcl-2 oncogene. J Biol Chem (2001) 276(48):45380–6. doi:10.1074/jbc.M108294200
35. Shi G, Ramaswamy M, Vistica BP, Cox CA, Tan C, Wawrousek EF, et al. Unlike Th1, Th17 cells mediate sustained autoimmune inflammation and are highly resistant to restimulation-induced cell death. J Immunol (2009) 183(11):7547–56. doi:10.4049/jimmunol.0900519
36. Cencioni MT, Santini S, Ruocco G, Borsellino G, De Bardi M, Grasso MG, et al. FAS-ligand regulates differential activation-induced cell death of human T-helper 1 and 17 cells in healthy donors and multiple sclerosis patients. Cell Death Dis (2015) 6:e1785. doi:10.1038/cddis.2015.100
37. Simelyte E, Rosengren S, Boyle DL, Corr M, Green DR, Firestein GS. Regulation of arthritis by p53: critical role of adaptive immunity. Arthritis Rheum (2005) 52(6):1876–84. doi:10.1002/art.21099
38. Leech M, Xue JR, Dacumos A, Hall P, Santos L, Yang Y, et al. The tumour suppressor gene p53 modulates the severity of antigen-induced arthritis and the systemic immune response. Clin Exp Immunol (2008) 152(2):345–53. doi:10.1111/j.1365-2249.2008.03629.x
39. Fray MA, Bunnell SC. p53 keeps bystanders at the gates. Immunity (2014) 40(5):633–5. doi:10.1016/j.immuni.2014.05.001
Keywords: Perp, T helper 17 cell, activation-induced cell death, experimental autoimmune encephalomyelitis/multiple sclerosis, caspase activation
Citation: Zhou Y, Leng X, He Y, Li Y, Liu Y, Liu Y, Zou Q, Shi G and Wang Y (2018) Loss of Perp in T Cells Promotes Resistance to Apoptosis of T Helper 17 Cells and Exacerbates the Development of Experimental Autoimmune Encephalomyelitis in Mice. Front. Immunol. 9:842. doi: 10.3389/fimmu.2018.00842
Received: 25 November 2017; Accepted: 05 April 2018;
Published: 23 April 2018
Edited by:
Song Guo Zheng, Penn State Milton S. Hershey Medical Center, United StatesReviewed by:
Li Huabin, Fudan University, ChinaJixin Zhong, Case Western Reserve University, United States
Copyright: © 2018 Zhou, Leng, He, Li, Liu, Liu, Zou, Shi and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guixiu Shi, guixiu.shi@gmail.com;
Yantang Wang, yt-wang@hotmail.com
†Cofirst authors, contributed equally, and shared the first authorship of this work.