 Guanxi Qiao
Guanxi Qiao Minhui Chen
Minhui Chen Mark J. Bucsek
Mark J. Bucsek Elizabeth A. Repasky
Elizabeth A. Repasky Bonnie L. Hylander
Bonnie L. Hylander- Immunology, Roswell Park Comprehensive Cancer Center, Buffalo, NY, United States
An immune response must be tightly controlled so that it will be commensurate with the level of response needed to protect the organism without damaging normal tissue. The roles of cytokines and chemokines in orchestrating these processes are well known, but although stress has long been thought to also affect immune responses, the underlying mechanisms were not as well understood. Recently, the role of nerves and, specifically, the sympathetic nervous system, in regulating immune responses is being revealed. Generally, an acute stress response is beneficial but chronic stress is detrimental because it suppresses the activities of effector immune cells while increasing the activities of immunosuppressive cells. In this review, we first discuss the underlying biology of adrenergic signaling in cells of both the innate and adaptive immune system. We then focus on the effects of chronic adrenergic stress in promoting tumor growth, giving examples of effects on tumor cells and immune cells, explaining the methods commonly used to induce stress in preclinical mouse models. We highlight how this relates to our observations that mandated housing conditions impose baseline chronic stress on mouse models, which is sufficient to cause chronic immunosuppression. This problem is not commonly recognized, but it has been shown to impact conclusions of several studies of mouse physiology and mouse models of disease. Moreover, the fact that preclinical mouse models are chronically immunosuppressed has critical ramifications for analysis of any experiments with an immune component. Our group has found that reducing adrenergic stress by housing mice at thermoneutrality or treating mice housed at cooler temperatures with β-blockers reverses immunosuppression and significantly improves responses to checkpoint inhibitor immunotherapy. These observations are clinically relevant because there are numerous retrospective epidemiological studies concluding that cancer patients who were taking β-blockers have better outcomes. Clinical trials testing whether β-blockers can be repurposed to improve the efficacy of traditional and immunotherapies in patients are on the horizon.
Introduction
Psychosocial and physical stresses have long been believed to negatively affect health and reduce our resistance to immune-mediated diseases (including cancer), but the mechanisms have been poorly understood (1, 2). A comprehensive review of early work on the effects of stress on tumor growth in animal models highlights the dichotomous results between viral tumors which grow faster with stress and non-viral/chemically induced tumors which grow more slowly during the stress period (3). In the last decade or so, however, we are beginning to understand the myriad mechanisms by which chronic adrenergic stress molds the immune response and promotes tumor growth (2–9). In this brief review, we will first give examples of how adrenergic signaling impacts immune cells and then how it affects tumor growth. We then summarize how preclinical mouse models are used to study the effects of different stress types on tumor growth, survival, and metastasis, highlighting the idea that tumors attract their own innervation in a process akin to angiogenesis. We emphasize the potential ramifications of the baseline cold stress which is imposed on mice by sub-thermoneutral housing and how this impacts our understanding of the capabilities of the endogenous immune response. In addition, in light of recent excitement about the potential of new immunotherapies to treat cancer, we discuss how adrenergic suppression of the antitumor immune response is a targetable checkpoint which may be blocked to increase the efficacy of immunotherapy. Finally, we summarize retrospective clinical epidemiological studies that support the idea of “repurposing” β-blockers for use in oncology.
Neural Regulation of the Immune Response
Stress can impact the immune response through two major neural pathways, the hypothalamic/pituitary/adrenal (HPA) axis and the sympathetic nervous system (SNS); we have recently reviewed these pathways in depth (10). While stimulation of the HPA axis results in the release of glucocorticoids from the adrenal cortex, the SNS innervates the adrenal medulla and stimulates release of the catecholamine neurotransmitters, epinephrine (Epi) and, to a lesser extent, norepinephrine (NE) (11). Perhaps more importantly for the purpose of this review, NE is also released by postganglionic sympathetic neurons which densely innervate both primary and secondary lymphoid organs (12–15) as well as essentially all other organs. Adrenergic receptors (ARs) for NE and Epi are located on the surface of most cells, including immune cells, and thus in addition to the regulatory influences of chemokines and cytokines, NE released from sympathetic nerve endings also plays a critical role in regulating immune cells. In fact, we are now realizing that neural reflexes may reach immune cells in a fraction of the time (milliseconds) that it takes chemokines and cytokines to circulate and have an effect on these cells (16). While activation of the SNS in response to acute stress such as exercise or injury promotes a rapid immune response (17), chronic ongoing stress often is immunosuppressive (18). As discussed below in the Section “Modeling the Tumor-Promoting Effects of Adrenergic Stress/Signaling in Mice”, suppression of the antitumor immune response is one of the consequences of this chronic stress.
Adrenergic receptors are a class of G protein-coupled receptors and are subdivided into two types (α and β) based on their structure, pharmacology, and signaling mechanisms, and these include several subtypes (19). Although there are many studies showing that both α- and β-ARs are expressed by innate immune cells, including neutrophils (20, 21), monocytes (22–24), macrophages (22, 24–26), mature DC (22), NK cells (27–33), and hematopoietic stem cells and progenitors (34), β2-ARs are the most highly expressed subtype on both innate and adaptive immune cells. T and B cells exclusively express β2-AR (35–37); this is also true for hematopoietic stem cells and progenitors (34). Consequently, β2-ARs are regarded as the main mediators of the immune effects of catecholamines (8). Activation of these receptors activates adenylate cyclase to increase intracellular cAMP, which in turn activates PKA which ultimately activates downstream transcription factors (2, 35, 36, 38–40). The details of this signaling pathway have recently been reviewed for both immune cells (41) and cancer cells (9).
Adrenergic Regulation of Innate Immunity
That activity of innate immune cells is regulated by activation of ARs has been clearly demonstrated. Adrenergic signaling affects macrophage polarization and cytokine production. β-AR signaling promotes macrophage differentiations toward an M2 phenotype which produces anti-inflammatory cytokines (42–45). In one study, gut macrophages in the muscularis mucosa, which are in close proximity to the autonomic myenteric plexus, differentially expressed the β2-AR, which mediated alternative activation of these macrophages, resulting in an M2 anti-inflammatory and tissue-protective profile (46). Both in vitro and in vivo studies support the conclusion that at least one way in which β-adrenergic signaling can promote breast cancer progression is by polarizing macrophages toward an M2 phenotype (42, 43). Moreover, in response to LPS stimulation, human monocyte-derived macrophages produce reduced amounts of the inflammatory cytokines TNF-α, IL-1β, CCL2, CCL3, and CCL4 (47–49) and decrease IL-27 secretion in response to acute inflammation (50) while, at the same time, increasing production of the anti-inflammatory cytokines IL-4, IL-10, and IL-13 production (44, 50). In contrast to the effects of β-AR signaling in macrophage, α-AR signaling promotes secretion of pro-inflammatory cytokines (24, 51).
Adrenergic signaling has been shown to impact DCs by impairing their maturation, cytokine production, and antigen presentation. Studies have shown that β2-AR activation prevents differentiation of monocytes into DCs (52) and enhances production of the anti-inflammatory cytokines IL-6, IL-10, and IL-33 while decreasing IL-12 and TNF-α production (53–58) by inhibition of NF-κB and AP-1 (54). In addition to altering cytokine production, Epi has also been shown to activate β1-AR/arrestin2–PI3K–MMP9/CCR7 signaling to inhibit migration of human DCs (59) which could impair migration to lymph nodes. Pretreating bone marrow derived DCs (BMDC) with β2-AR agonists before adoptive transfer reduces migration and responses to chemokines. Both in vitro and in vivo studies show that α2-AR suppresses DC migration by inhibiting type IV collagenase/gelatinase activity (60). The primary function of DCs, antigen presentation, is impaired by treating epidermal Langerhans cells with catecholamines, and this effect is blocked by using a β2-AR antagonist (61). In addition, cross-presentation of proteins by DCs is impaired by activation of β2-AR signaling, which in turn decreases CD8+ T cell proliferation and IL-2 production (62). Our lab has also found that adrenergic stress impacts the phenotype and function of DCs (63) in ways that could suppress their function.
Several studies have demonstrated that chronic adrenergic signaling can suppress NK cell activity. In a restraint stress mouse model, the number of NK cells is reduced in the intraparenchymal region of the lung and circulation (64). Another study showed that activation of β-ARs reduces the activity (65) of murine NK cells and leads to an increase in tumor metastasis. Interestingly, the two arms of the stress response can cooperate to regulate immune cells. De Lorenzo et al. found that during sleep deprivation, glucocorticoids increase expression of β2-AR in NK cells resulting in reduction of NK cell numbers and cytotoxicity (66). In contrast to the effects observed in these models of chronic stress, some studies have concluded that stress can increase NK cell activation and function. In one such study, six episodes of social disruption increased NK cell activity (67). In addition, voluntary exercise reduces the incidence and growth of breast cancer and this effect is mediated by catecholamine induced plasma IL-6, which in this case mobilizes NK cells (17). Enriching the housing environment for mice also increases NK antitumor function (68). Clearly, the effects of stress differ depending on the type and duration.
In addition, there are many reports investigating the regulation of other innate immune cells by adrenergic signaling. For example, a β2-AR agonist was able to prevent eosinophil functions which are induced by exposure to IL-5, LTD4, or IP-10 and which worsen the severity of asthma (69). In another example, the phagocytic efficiency of wound neutrophils was found to be impaired by a pharmacologic dose of NE, in a standard subcutaneous sponge wound model, and this change was found to be mediated by α- and β-ARs and downstream protein kinase A (70).
Adrenergic Regulation of Adaptive Immunity
Norepinephrine also activates ARs on lymphocytes to regulate the differentiation, trafficking/migration, and effector functions of all lymphocyte subpopulations (11). Adrenergic signaling impacts T-cells by directly regulating thymocytes and inhibiting activation, differentiation, and effector function of T-cells directly or indirectly by inhibiting T-cell activating cytokine production by DCs. A study in stressed mice showed that β-AR signaling promotes negative selection in the thymus through the p38 mitogen-activated protein kinase pathway, resulting in decreased numbers of thymocytes (71). In addition, exposure to chronic unpredictable stress reduces the number of double negative thymocytes (72). Adrenergic signaling decreases production of IL-2, IFN-γ, and proliferation of CD4+ T-cell through many mechanisms. β2-AR signaling inhibits cytokines by inhibiting calcineurin in a PKA dependent manner (73). IFN-γ production by CD4+ T-cells can also be affected indirectly by inhibition of cytokine production by DCs. Adrenergic signaling also regulates Th1 and Th2 differentiation (34). Naïve CD4+ T-cells or activated Th1 cells express a detectable level of the β2-AR, while Th2 cells do not, due to differences in histone and DNA modifications within the β2-AR proximal promoter (22, 74). Therefore, Th1 cells can be suppressed by adrenergic signals and β2-AR activation has been found to be involved in directing CD4+ T-cell polarization toward a Th2 phenotype (75). The impact of β2-AR signaling on CD8+ T-cells depends on the stage of differentiation. Memory and effector CD8+ T-cells have significantly higher expression of β2-ARs than naïve T-cells and β2-AR signaling reduces IL-2 and IFN-γ production by memory CD8+ T-cells upon restimulation (76). IL-2 increases β2-AR expression on effector CD8+ T-cells; however, β2-AR signaling suppresses IL-2 production by CD8+ T-cells, which forms a negative-feedback loop in regulating CD8+ T-cell activation and function (77). By contrast, while β2-AR signaling suppresses CD8+ effector T-cell function, β2-AR signaling in regulatory T-cells increases their suppressor activity by increasing expression of the checkpoint molecule CTLA-4 and promotes inducible Tregs by inducing Foxp3 expression in T-cells (55).
Suppression of immune cells by chronic adrenergic stress has long-term negative implications for disease progression and has been well studied in the context of both infectious diseases and autoimmunity. β2-AR signaling impairs CD8+ T-cell-mediated antiviral responses to influenza in vivo and blocking the β2-AR with Nadolol enabled development of a robust antiviral response (78). Similarly, β2-AR signaling reduced the CD8+ T-cell response to the vesicular stomatitis virus (79). Dysregulation of the immune response also underlies many autoimmune diseases. Because autoimmune diseases involve overactive immune responses, adrenergic signaling actually helps to suppress autoimmunity, and ablation of sympathetic nerves worsened disease severity in a mouse model of multiple sclerosis (80). An in depth discussion of these studies is beyond the scope of this review but is the subject of a recent review by our group (81).
Neural Regulation of Tumor Growth
The early observations of Levi Montalcini and collaborators first clearly demonstrated that tumor cells secrete nerve growth factors and can induce neural outgrowth from both sensory and sympathetic ganglia (82). As evidence of the tumor promoting effects of adrenergic signaling accumulated, Entschladen and colleagues hypothesized that in a process analogous to angiogenesis and lymphangiogenesis, tumors might secrete neurotrophic factors to attract their own innervation (83). But it was the elegant study of Magnon et al. (84) that clearly demonstrated both the ability of a developing tumor to attract autonomic innervation and the critical role such innervation plays in development and progression of a tumor. Using an orthotopic xenograft model (PC-3 prostate cells), they showed that the developing tumor attracted new autonomic innervation and that if they severed the hypogastric nerves or chemically sympathectomized the mice, tumor growth was significantly inhibited. Furthermore, they showed that parasympathetic nerves promoted the later stages of invasion and metastasis. Others have shown that NE levels in tumors are elevated by stress that activates the SNS and that NE is produced locally within tumors (as evidenced by tyrosine hydroxylase positive cells) (81, 85–88).
In the last decade or so there has been a growing effort to understand how adrenergic signaling promotes tumor growth and we now know that β-AR signaling acts through multiple mechanisms (Figure 1). ARs are expressed by multiple cell types found in the tumor microenvironment and, therefore, the effects of adrenergic signaling that support tumor survival, growth, and metastasis are complex.
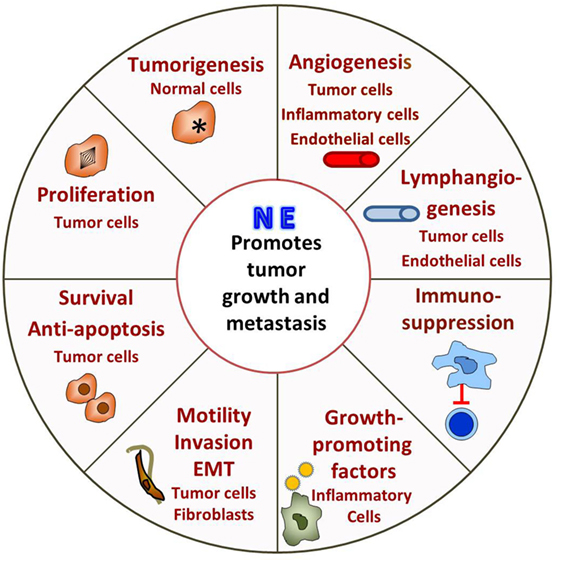
Figure 1. Adrenergic signaling promotes tumor survival, growth, and metastasis. The tumor is innervated by postganglionic nerves of the sympathetic nervous system and, in response to stress, these nerves secrete norepinephrine (NE). Many cells in the tumor microenvironment express adrenergic receptors, and their responses support tumor growth. See text for discussion.
Direct effects include promoting tumorigenesis, tumor cell proliferation, antiapoptotic mechanisms, and promoting metastasis by inducing epithelial to mesenchymal transformation (EMT), motility and invasion. β-Adrenergic signaling activates p53 degradation and DNA damage through the β-AR/ARRB1/PKA pathway (89–91) and activation of oncogenes, such as Src and Her2 (92) and may promote tumorigenesis. In addition, increased psychological stress is associated with decreased telomere length (93), which is associated with increased cancer risk (94). Adrenergic signaling promotes tumor cell proliferation both in vitro and in vivo (95–101), while blocking adrenergic signaling leads to G1/S phase cell cycle arrest and apoptosis (102, 103). Antiapoptotic mechanisms are upregulated in tumor cells both in vitro and in mouse xenografts (84, 85, 104–106). Our lab found that adrenergic signaling increases expression of the antiapoptotic proteins BAD, BCL-2, and MCP-1 in tumors and blocking adrenergic signaling significantly reduces antiapoptotic protein expression and tumor growth (85). In addition, in vitro, NE activates the TGF-β pathway in treated cancer cells enhancing migration and invasion (107, 108). Mesenchymal markers such as α-SMA, vimentin, and snail are also increased by β-adrenergic signaling (109, 110). More recently, analysis of exosomes isolated from ovarian patients with high levels of adrenergic pathway activation (associated with low social support) shows upregulation of mesenchymal-characteristic gene transcripts and downregulation of epithelial-characteristic gene transcripts, and this is mediated by the ADRB/cAMP/PKA pathway (111). Adrenergic signaling plays a vital role in promoting of metastasis (112–122) and could directly induce invasive genes such as MMPs by tumor cells (123) and indirectly by causing cancer-associated fibroblasts to produce high levels of collagen and extracellular matrix components, which facilitate tumor invasion and dissemination (124).
Adrenergic signaling also affects other cells in the tumor microenvironment, which in turn support tumor growth, survival and metastasis. Studies show that adrenergic signaling promotes angiogenesis through increasing VEGF, IL-8, IL-6, PEG2 expression in tumor cells (87, 119, 125–130), and decreasing expression of TSP1, a potent angiogenesis inhibitor (129). A recent study from Zahalka et al. demonstrates that adrenergic signaling also directly alters the metabolism of endothelial cells in the TME, leading to increased angiogenesis (131). In addition, studies demonstrate that adrenergic signaling promotes lymphangiogenesis by inducing VEGFC production by tumor cells and/or macrophages (112, 132). Formation of a pre-metastatic niche in the lung is induced by adrenergic signaling in monocyte/macrophage cells in a breast cancer model (133). In another study, stimulation of bone marrow stromal cells by adrenergic signaling increases breast cancer bone colonization (115). Finally, it is becoming clear that the immunosuppressive effects of adrenergic signaling that have been seen in infection models (78, 79) are also playing a major role in suppressing the antitumor immune response. This inhibits tumor infiltration and function of cytotoxic T-cells (134–136) and promotes infiltration of immunosuppressive cells such as MDSC and Tregs (134, 136–139).
Modeling the Tumor-Promoting Effects of Adrenergic Stress/Signaling in Mice
Although the concept of stress is generally understood, it is challenging to define. The original concept of stress, as proposed by Selye, was the response of an organism to a stimulus that challenged its well-being and he called this the General Adaptation Syndrome (140). Briefly, he proposed that an organism had the same response to many different noxious stimuli (swelling of the adrenal cortex, atrophy of the thymus, gastric ulcers) and that people suffering from various diseases had the same symptoms or syndrome. Eventually, he coined the term “stressor” to refer to the stimulus and the “stress response” to refer to the reaction. Stressors can be psychological (such as anxiety, fear, depression), physical (such as injury, surgery), or physiological (environmental factors including temperature) and exposure to stressors can be either acute (a single short-term exposure—minutes to hours) or chronic (long term, ongoing, or repeated). In general, an acute stress such as an injury leads to a beneficial response including activation of immune cells, whereas chronic exposure is most often deleterious and immunosuppressive (141). Dhabhar (18) has discussed the idea of categorizing stress as “protective, pathological, or regulatory” depending on their effects on overall health.
There is a general view that stress is detrimental to health and in ovarian carcinoma patients with little social support, it has been shown that the stress of social isolation elevates tumor and ascites NE while Epi was undetectable; this elevated NE correlates with more severe disease stages (86). To study how stress impacts tumor growth in preclinical models, protocols for inducing stress in mice have been developed (Table 1). It is important to point out that the same method can be used to induce both acute and chronic stress and these are distinguished by the duration of exposure. A good example of this is a study that used restraint stress to compare changes in the hippocampal transcriptome following acute vs. chronic stress by looking at samples taken after 1, 8, or 13 days of stress exposure (142).
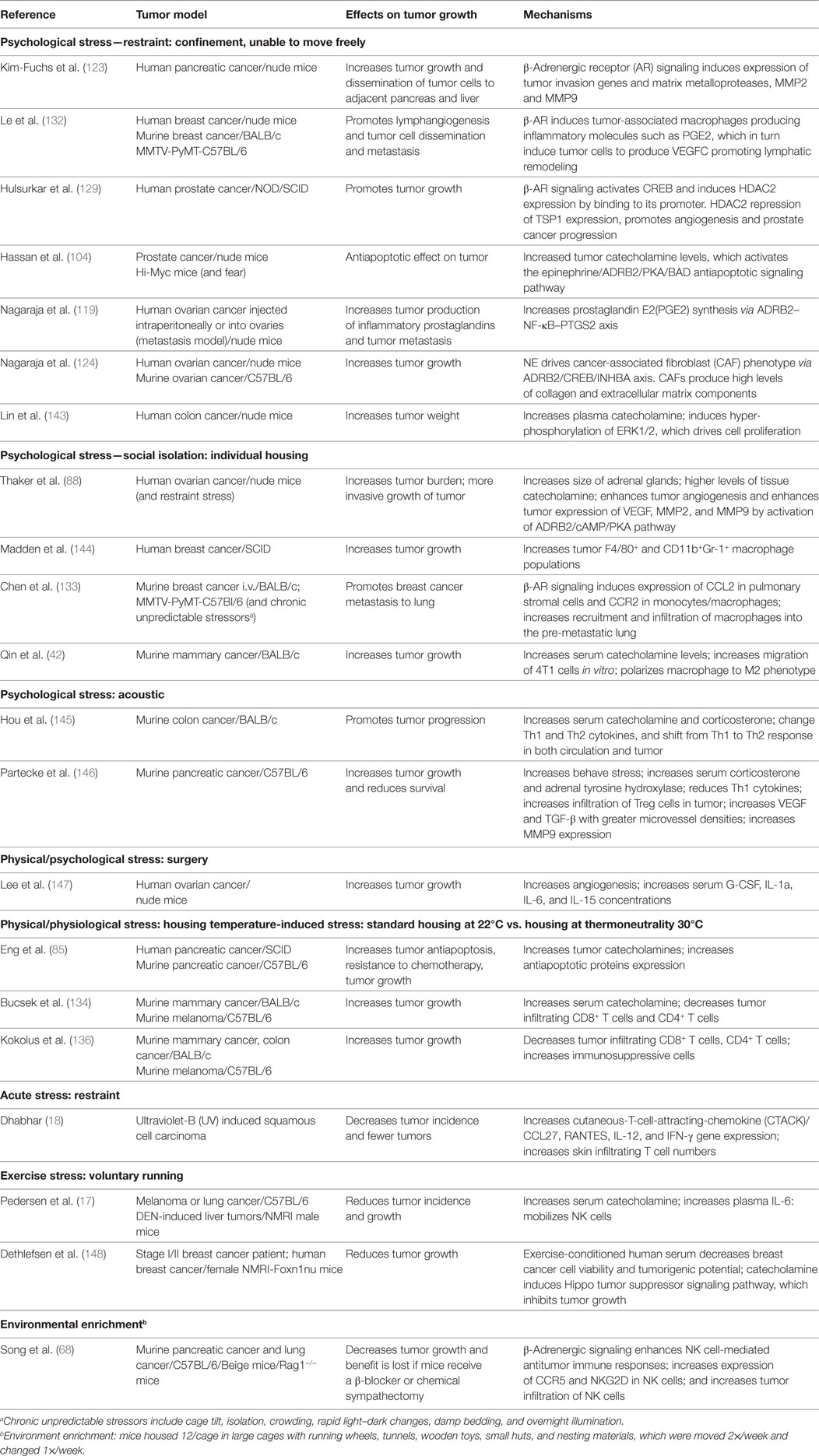
Table 1. Effects of different models of stress on tumor growth in mice.
To model psychological stress, mice have most often been subjected to social isolation or restraint stress. Over 10 years ago, using a mouse xenograft model of ovarian cancer, Thaker’s group showed that restraint stress resulted in significant elevation of NE (255–358%) and corticosterone (488–789%) in organs adjacent to the peritoneal cavity in tumor-bearing mice. Tumor growth and metastasis were also significantly promoted in these mice. Mice are social animals and are normally housed at four to five per cage; in fact, special permission is needed if mice are going to be housed singly and in that case, enrichment materials are required. In addition to restraint stress, Thaker’s group also subjected mice to social isolation and found enlargement of the adrenal glands as well as confirming the tumor promoting effects of stress. In addition, they showed that these effects could be duplicated by treating mice with the β2-AR agonist terbutaline and that the tumor promoting effects of both restraint and social isolation stress could be blocked by the pan β-AR antagonist propranolol. Altogether, these results clearly defined a role for adrenergic signaling in tumor growth and this was shown to be mediated by tumor VEGF production and increased angiogenesis (88). Since these seminal studies, the role of adrenergic signaling in tumor progression has been characterized in other models. Other groups have noted increased levels of catecholamines in response to psychosocial stressors. Qin et al. found that Epi was elevated and breast cancer progression was promoted in association with an M2 skewed macrophage population in a social isolation model; however, NE levels were not reported (42). Partecke et al. found that in response to a combination of acoustic and chronic stress, animals’ measures of behavioral stress as well as levels of stress hormones (steroids and adrenal tyrosine hydroxylase, the rate limiting enzyme in NE synthesis) were significantly elevated and furthermore, that orthotopic, syngeneic pancreatic tumors grew faster in these mice and there were indications that their immune response was suppressed, with a trend toward fewer CD4 and increased intratumoral Tregs present (146). Treatment of these mice with propranolol reduced tumor growth and improved overall survival (OS). These studies and several others (see Table 1) demonstrate that psychosocial stress promotes tumor growth through various mechanisms and that adrenergic signaling can be blocked by administration of β-AR or specific β2-AR antagonists.
In laboratory studies, the effects of physical/physiological environmental stressors on catecholamine levels and tumor growth have not been as well studied as the effects of psychological stresses, but environmental factors have great capacity to induce a stress response and alter internal metabolism and physiology (149). Our lab has recently defined a role for chronic mild cold stress in promoting tumor growth. This environmental stressor is universally imposed on mice as a result of mandated, cool ambient housing temperatures (150), and the impact of this cold stress on the modeling of several non-tumor mouse models of disease has been recently reviewed (151–153). The increasing awareness of this problem is reflected in several additional publications over the last 2–3 years reporting on how ambient housing temperatures affect disease outcomes in atherosclerosis (154–157), Alzheimer’s (158), monocyte mobilization into the blood (159), and obesity (160–162). Newer reviews have also drawn attention to this situation (163, 164). We first reported the unexpected discovery that when mice are housed at thermoneutrality (30°C), tumor growth is significantly inhibited in comparison to mice housed at standard temperatures (22°C) even though mice under both conditions maintain a normal core body temperature of 37°C (136). We also demonstrated that this difference in tumor growth was dependent on the adaptive immune response which was significantly suppressed when mice were housed at 22°C. Furthermore, we found that although the effects of cold stress are commonly studied by exposing mice to 4°C, just housing mice at the sub-thermoneutral 22°C was sufficient to cause chronic cold stress and significant elevation of both plasma and tumor NE levels (85, 134). Thus we found that even what was considered mild cold stress at 22°C, is biologically sufficient to induce SNS activation to produce NE and drive non-shivering thermogenesis to maintain a normal body temperature. Furthermore, we have shown that chronic adrenergic signaling in mice housed at 22°C promotes tumor growth in two different ways, both by induction of antiapoptotic signaling molecules and resistance to cytotoxic therapies in tumor cells (85) and by profound immunosuppression of the antitumor immune response which is associated with increases in immunosuppressive cell populations and inhibition of CD8+ T-cell effector phenotypes (134). Therefore, not only are the tumor cells more resistant to being killed, but the ability of the immune cells to kill tumor cells is much less robust when NE levels are increased by baseline housing stress; both of these situations are reversed by administration of the β-blocker propranolol. Interestingly, the recent studies of Wrobel et al. (135) agree with our findings; this group administered propranolol to MT/Ret mice and found inhibition of tumor development, increased CD8+ T-cells in the tumors, and decreased MDSC. However, these authors did not associate this baseline stress with housing temperature and the temperature at which their mice were housed was not reported.
We are particularly concerned about this immunosuppression by baseline adrenergic stress in experimental mice because both the long-term outcome of traditional therapies (165–170) and immunotherapies (171) depend on the development of a robust antitumor immune response. We tested whether the efficacy of immunotherapy, specifically checkpoint inhibitor therapies, was limited by adrenergic stress. In mouse models of melanoma and breast cancer, we used three separate approaches: housing mice at 30°C, treating mice housed at 22°C with β-blockers, or testing anti-PD-1 in ADRB2−/− mice at 22°C and asked how this impacted in efficacy of anti-PD-1. In each case, it is clear that adrenergic signaling reduces responses to checkpoint inhibitor therapy and the efficacy is significantly improved by reducing this stress.
Taken together, it is clear that the majority of studies on the effects of stress are performed at 22°C when the “control” mice are already “stressed” in comparison to the relatively reduced levels they would be experiencing at 30°C (Figure 2). Therefore, restraint stress or other forms of psychosocial stress are imposed on already stressed mice, so that measures of tumor growth, response to therapy or the capabilities of the antitumor immune response in the controls are somewhat compromised. In the future, it will be important to determine if, and/or how, baseline cold stress may be affecting the results of these stress experiments. Although due to the thermal comfort requirements of personnel, it seems unlikely that the ambient temperatures in animal facilities will be changed to 30°C, it may be possible to reduce cold stress in experimental animals by providing nesting materials or constructing special cages in which mice can behaviorally regulate their thermal comfort (172–174).
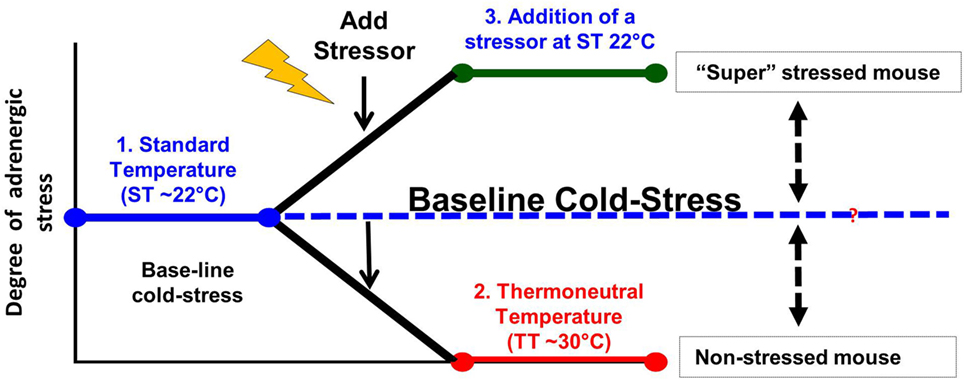
Figure 2. Modeling adrenergic stress in mouse models. 1. Mandated housing of mice at ~22°C imposes chronic cold stress and results in elevated norepinephrine (NE) levels which drive thermogenesis to maintain a normal body temperature of 37°. This “baseline cold stress” is sufficient to inhibit the development of an immune response in several disease models including cancer. 2. Reduction of NE levels and reversal of immunosuppression can be achieved by housing mice at thermoneutrality (~30°C) or administration of β-blockers. Reduction of baseline adrenergic stress significantly improves the antitumor immune response in preclinical models compared with the response in mice housed at 22°C. The improved immune response correlates with a significant improvement in the efficacy of immunotherapy. 3. The majority of studies compare the impaired immune response seen at 22°C with that observed after an additional source of stress is imposed on animals at 22°C. This approach may compromise a full understanding of the capabilities of the endogenous immune response and could also lead to a misunderstanding of the efficacy of therapies depending on an immune response in these models.
Clinical Relevance of Preclinical Findings
How do these findings relate to patients? Studies have demonstrated that several types of patient tumors express ARs including pediatric (175, 176), pancreatic (103), lung (177), melanoma (178), and prostate (179) cancers. More provocatively, as it became apparent that blockade of adrenergic signaling could inhibit tumor growth in preclinical models, researchers looked for evidence of benefit in patients. Several retrospective epidemiological studies have supported the conclusion that patients who are taking β-AR antagonists (“β-blockers”) and receiving conventional therapies have better outcomes in terms of both progression-free survival and OS than those who are not (Table 2). It should be noted, however, that there are other analyses that failed to identify any benefit of β-blockers (180–186). There are several potential reasons for why some studies show that cancer patients benefit from taking β-blockers while other studies conclude there is no benefit. One important reason is that the type of β-blocker being taken by the patient likely influences the patient’s response as well as the overall outcome of the study. There are studies showing that patients taking non-selective β-blockers (blockade of β1 and β2) had improved OS compared with patients taking β1-specific blockers which are the most commonly prescribed β-blockers (180, 183, 184, 187). The heterogeneity of ARs expressed by the tumors themselves may also account for the different responses in cancer patients (180, 184). Besides the type of β-blocker, it is possible that the dosage needed to elicit a favorable antitumor effect is higher than the normal prescription dose for treating cardiovascular diseases (182, 185). In addition, the patient parameters are variable among the studies. Although the most important potential confounders such as sex, stage, treatment were usually adjusted for, there are others, such as socioeconomic status which is proven to be associated with cancer progression, which were not taken into consideration (182, 185). The timing of β-blocker exposure is different between studies. Some studies recruited patients already taking β-blockers before cancer diagnosis, while some studies investigated patients who began taking β-blockers after cancer diagnosis (180, 181, 183, 184). In addition, there are limited records showing whether patients complied with the prescriptions (182, 185). The study design may also influence the result. Some studies used all-cause of mortality as the endpoint, which makes it hard to study the relationship of β-blocker usage and cancer-specific mortality (183). In other studies, patients taking β-blockers who died within 3 months after cancer diagnosis were still included, which is thought to be too short an interval to assess benefit (181–184).
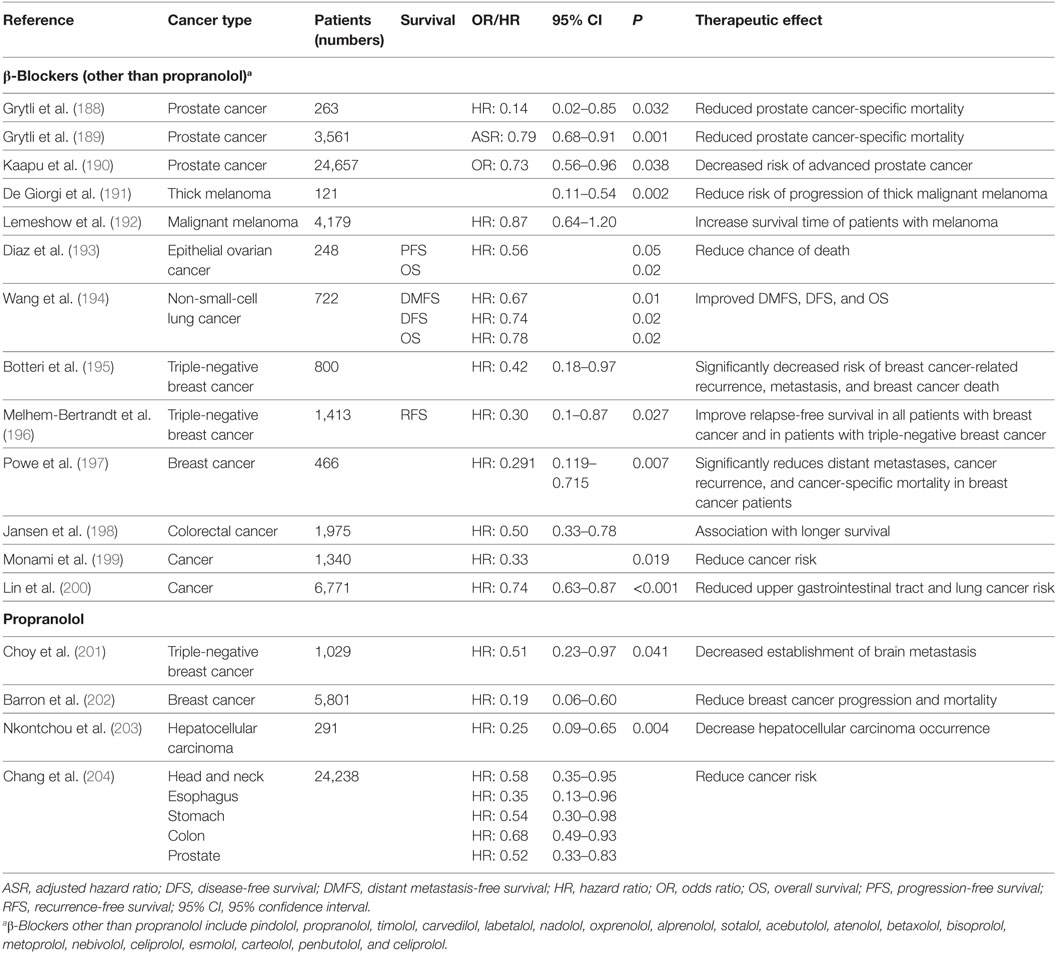
Table 2. A summary of retrospective studies assessing the beneficial effects of β-blockers, including Propranolol, in patients with different cancers who were also taking β-blockers.
To date, there has been a single report of a prospective study treating melanoma patients with propranolol. In this study, De Giorgi and colleagues offered patients the choice of “off-label” treatment with propranolol and found an 80% risk reduction for recurrence for the 19 of 53 patients who chose to receive it (205). Ultimately, the ability of β-blockers to support the development of an antitumor immune response and to improve the efficacy of traditional and immunotherapies will have be determined in blinded, prospective clinical trials, but the results of this study in melanoma lend support for the “repurposing” of β-blockers in oncology.
β-Blockers have been used extensively in the clinic to treat patients with hypertension, angina and anxiety. In general, β-blockers have a good toxicity profile and are well tolerated (206); common side effects include nausea, vomiting, diarrhea, insomnia, weakness and fatigue. However, the safety of using of β-blockers in cancer patients needs to be considered. Although most cancer patients do not have hypertension and/or angina, they do often have increased levels of adrenergic stress and so in that way may be considered to have abnormal adrenergic signaling. In several studies in which cancer patients were taking β-blockers as an off-label treatment, either no drug related adverse effects were recorded (205, 207) or small number of patients had minor side effects which did not require discontinuation (203, 208, 209). However, an understanding of the comparative safety of β-blockers in combination with other cancer therapies remains to be evaluated in the context of clinical trials.
Conclusion
Recently developed immunotherapies are showing great promise in cancer treatment, yet many tumor types are not responding and even in sensitive tumors such as melanoma and lung cancer, the majority of patients are not benefiting. Therefore, developing new strategies for improving the therapeutic efficacy of cancer immunotherapies is critical. Immunotherapies and traditional therapies that have an immune-mediated component depend on the development of a robust antitumor immune response, but immunosuppressive factors often limit this. Therefore, a promising strategy to improve response to immunotherapy is to develop additional approaches to reverse immunosuppression that can be used in combination therapies.
A newly identified mechanism of immunosuppression is by stress-induced sympathetic adrenergic signaling, which suppresses many aspects of the immune system including development, differentiation, activation, and function of many types of immune cells. Adrenergic signaling has been shown to inhibit immune responses in both autoimmune diseases and infection models. Sympathetic neurons innervate both primary and secondary lymphoid organs supporting the idea that adrenergic signaling can impact immune cells locally. Recently, it has become clear that adrenergic signaling also affects tumor progression. By using mouse models of both psychosocial and physical stress stress-induced adrenergic signaling was found to be tumor-promoting in many types of cancers. The accumulated evidence demonstrates that adrenergic stress regulates tumor growth directly through multiple mechanisms, including development, proliferation, and protection of tumor cells from treatments. Although less well studied, recent findings show that adrenergic signaling also impacts tumor growth indirectly by regulating antitumor immunity. Our lab recently demonstrated that chronic mild cold stress induces SNS activation which promotes tumor growth; blocking this stress by either housing mice at thermoneutral temperature or adding β-blockers slows tumor growth. These effects were found to be meditated by CD8+ T-cells. In addition, combining a pan-β-blocker with an immune checkpoint inhibitor (anti-PD-1) significantly increased the therapeutic efficacy of anti-PD-1. Because cancer patients who were already taking β-blockers were found to have better outcomes in several retrospective studies, the preclinical studies provide a rationale for testing this combination in prospective clinical trials. Finally, accumulating evidence shows that external environmental stressors, i.e., housing temperature, creates a level of baseline adrenergic stress that affects experimental outcomes in mouse models. In the future, when designing and analyzing experiments, it will be important to factor in this potentially confounding experimental variable to better understand the biology underlying the results and to improve rigor and reproducibility.
Author Contributions
GQ, MC, MB, ER, and BH developed the concepts for this review and were involved in identifying the references and writing and editing the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This research was supported by The Breast Cancer Coalition of Rochester, NY; The Peter T. Rowley Breast Cancer Research Grant C028252; The Harry J. Lloyd Charitable Trust; The Roswell Park Alliance Foundation; The National Institute of Health Grant 5-T32CA085183 and used Shared Resources supported by the Roswell Park Cancer Institute’s Comprehensive Cancer Center Support Grant CA016056.
References
1. Calcagni E, Elenkov I. Stress system activity, innate and T helper cytokines, and susceptibility to immune-related diseases. Ann N Y Acad Sci (2006) 1069:62–76. doi:10.1196/annals.1351.006
2. Antoni MH, Lutgendorf SK, Cole SW, Dhabhar FS, Sephton SE, McDonald PG, et al. The influence of bio-behavioural factors on tumour biology: pathways and mechanisms. Nat Rev Cancer (2006) 6:240–8. doi:10.1038/nrc1820
3. Justice A. Review of the effects of stress on cancer in laboratory animals: importance of time of stress application and type of tumor. Psychol Bull (1985) 98:108–38. doi:10.1037/0033-2909.98.1.108
4. Padgett DA, Glaser R. How stress influences the immune response. Trends Immunol (2003) 24:444–8. doi:10.1016/S1471-4906(03)00173-X
5. Glaser R, Kiecolt-Glaser JK. Stress-induced immune dysfunction: implications for health. Nat Rev Immunol (2005) 5:243–51. doi:10.1038/nri1571
6. Elenkov IJ, Chrousos GP. Stress system – organization, physiology and immunoregulation. Neuroimmunomodulation (2006) 13:257–67. doi:10.1159/000104853
7. Nance DM, Sanders VM. Autonomic innervation and regulation of the immune system (1987–2007). Brain Behav Immun (2007) 21:736–45. doi:10.1016/j.bbi.2007.03.008
8. Marino F, Cosentino M. Adrenergic modulation of immune cells: an update. Amino Acids (2013) 45:55–71. doi:10.1007/s00726-011-1186-6
9. Cole SW, Sood AK. Molecular pathways: beta-adrenergic signaling in cancer. Clin Cancer Res (2012) 18:1201–6. doi:10.1158/1078-0432.CCR-11-0641
10. Eng JW, Kokolus KM, Reed CB, Hylander BL, Ma WW, Repasky EA. A nervous tumor microenvironment: the impact of adrenergic stress on cancer cells, immunosuppression, and immunotherapeutic response. Cancer Immunol Immunother (2014) 63:1115–28. doi:10.1007/s00262-014-1617-9
11. Bellinger DL, Lorton D. Autonomic regulation of cellular immune function. Auton Neurosci (2014) 182:15–41. doi:10.1016/j.autneu.2014.01.006
12. Bellinger DL, Lorton D, Felten SY, Felten DL. Innervation of lymphoid organs and implications in development, aging, and autoimmunity. Int J Immunopharmacol (1992) 14:329–44. doi:10.1016/0192-0561(92)90162-E
13. Felten DL, Felten SY, Bellinger DL, Carlson SL, Ackerman KD, Madden KS, et al. Noradrenergic sympathetic neural interactions with the immune system: structure and function. Immunol Rev (1987) 100:225–60. doi:10.1111/j.1600-065X.1987.tb00534.x
14. Felten DL, Livnat S, Felten SY, Carlson SL, Bellinger DL, Yeh P. Sympathetic innervation of lymph nodes in mice. Brain Res Bull (1984) 13:693–9. doi:10.1016/0361-9230(84)90230-2
15. Felten SY, Felten DL, Bellinger DL, Carlson SL, Ackerman KD, Madden KS, et al. Noradrenergic sympathetic innervation of lymphoid organs. Prog Allergy (1988) 43:14–36.
16. Chavan SS, Tracey KJ. Essential neuroscience in immunology. J Immunol (2017) 198:3389–97. doi:10.4049/jimmunol.1601613
17. Pedersen L, Idorn M, Olofsson GH, Lauenborg B, Nookaew I, Hansen RH, et al. Voluntary running suppresses tumor growth through epinephrine- and IL-6-dependent NK cell mobilization and redistribution. Cell Metab (2016) 23:554–62. doi:10.1016/j.cmet.2016.01.011
18. Dhabhar FS. Effects of stress on immune function: the good, the bad, and the beautiful. Immunol Res (2014) 58:193–210. doi:10.1007/s12026-014-8517-0
19. Hieble JP, Bondinell WE, Ruffolo RR Jr. Alpha- and beta-adrenoceptors: from the gene to the clinic. 1. Molecular biology and adrenoceptor subclassification. J Med Chem (1995) 38:3415–44. doi:10.1021/jm00018a001
20. Scanzano A, Schembri L, Rasini E, Luini A, Dallatorre J, Legnaro M, et al. Adrenergic modulation of migration, CD11b and CD18 expression, ROS and interleukin-8 production by human polymorphonuclear leukocytes. Inflamm Res (2015) 64:127–35. doi:10.1007/s00011-014-0791-8
21. Brunskole Hummel I, Reinartz MT, Kalble S, Burhenne H, Schwede F, Buschauer A, et al. Dissociations in the effects of beta2-adrenergic receptor agonists on cAMP formation and superoxide production in human neutrophils: support for the concept of functional selectivity. PLoS One (2013) 8:e64556. doi:10.1371/journal.pone.0064556
22. Padro CJ, Sanders VM. Neuroendocrine regulation of inflammation. Semin Immunol (2014) 26:357–68. doi:10.1016/j.smim.2014.01.003
23. Fragala MS, Kraemer WJ, Mastro AM, Denegar CR, Volek JS, Hakkinen K, et al. Leukocyte beta2-adrenergic receptor expression in response to resistance exercise. Med Sci Sports Exerc (2011) 43:1422–32. doi:10.1249/MSS.0b013e31820b88bc
24. Grisanti LA, Woster AP, Dahlman J, Sauter ER, Combs CK, Porter JE. alpha1-adrenergic receptors positively regulate toll-like receptor cytokine production from human monocytes and macrophages. J Pharmacol Exp Ther (2011) 338:648–57. doi:10.1124/jpet.110.178012
25. Spengler RN, Allen RM, Remick DG, Strieter RM, Kunkel SL. Stimulation of alpha-adrenergic receptor augments the production of macrophage-derived tumor necrosis factor. J Immunol (1990) 145:1430–4.
26. Hjemdahl P, Larsson K, Johansson MC, Zetterlund A, Eklund A. Beta-adrenoceptors in human alveolar macrophages isolated by elutriation. Br J Clin Pharmacol (1990) 30:673–82. doi:10.1111/j.1365-2125.1990.tb03835.x
27. Maisel AS, Harris T, Rearden CA, Michel MC. Beta-adrenergic receptors in lymphocyte subsets after exercise. Alterations in normal individuals and patients with congestive heart failure. Circulation (1990) 82:2003–10. doi:10.1161/01.CIR.82.6.2003
28. Whalen MM, Bankhurst AD. Effects of beta-adrenergic receptor activation, cholera toxin and forskolin on human natural killer cell function. Biochem J (1990) 272:327–31. doi:10.1042/bj2720327
29. Xiao J, Huang HW, Peng YP, Bao JY, Huang Y, Qiu YH. Modulation of natural killer cell function by alpha-adrenoreceptor-coupled signalling. Neuro Endocrinol Lett (2010) 31:635–44.
30. Takamoto T, Hori Y, Koga Y, Toshima H, Hara A, Yokoyama MM. Norepinephrine inhibits human natural killer cell activity in vitro. Int J Neurosci (1991) 58:127–31. doi:10.3109/00207459108987189
31. Jetschmann JU, Benschop RJ, Jacobs R, Kemper A, Oberbeck R, Schmidt RE, et al. Expression and in-vivo modulation of alpha- and beta-adrenoceptors on human natural killer (CD16+) cells. J Neuroimmunol (1997) 74:159–64. doi:10.1016/S0165-5728(96)00221-4
32. Benschop RJ, Oostveen FG, Heijnen CJ, Ballieux RE. Beta 2-adrenergic stimulation causes detachment of natural killer cells from cultured endothelium. Eur J Immunol (1993) 23:3242–7. doi:10.1002/eji.1830231230
33. Schedlowski M, Hosch W, Oberbeck R, Benschop RJ, Jacobs R, Raab HR, et al. Catecholamines modulate human NK cell circulation and function via spleen-independent beta 2-adrenergic mechanisms. J Immunol (1996) 156:93–9.
34. Muthu K, Iyer S, He LK, Szilagyi A, Gamelli RL, Shankar R, et al. Murine hematopoietic stem cells and progenitors express adrenergic receptors. J Neuroimmunol (2007) 186:27–36. doi:10.1016/j.jneuroim.2007.02.007
35. Cremaschi GA, Fisher P, Boege F. Beta-adrenoceptor distribution in murine lymphoid cell lines. Immunopharmacology (1991) 22:195–206. doi:10.1016/0162-3109(91)90044-Y
36. Kohm AP, Sanders VM. Suppression of antigen-specific Th2 cell-dependent IgM and IgG1 production following norepinephrine depletion in vivo. J Immunol (1999) 162:5299–308.
37. Van Tits LJ, Michel MC, Grosse-Wilde H, Happel M, Eigler FW, Soliman A, et al. Catecholamines increase lymphocyte beta 2-adrenergic receptors via a beta 2-adrenergic, spleen-dependent process. Am J Physiol (1990) 258:E191–202.
38. Bartik MM, Bauman GP, Brooks WH, Roszman TL. Costimulatory signals modulate the antiproliferative effects of agents that elevate cAMP in T cells. Cell Immunol (1994) 158:116–30. doi:10.1006/cimm.1994.1261
39. Bauman GP, Bartik MM, Brooks WH, Roszman TL. Induction of cAMP-dependent protein kinase (PKA) activity in T cells after stimulation of the prostaglandin E2 or the beta-adrenergic receptors: relationship between PKA activity and inhibition of anti-CD3 monoclonal antibody-induced T cell proliferation. Cell Immunol (1994) 158:182–94. doi:10.1006/cimm.1994.1266
40. Kasprowicz DJ, Kohm AP, Berton MT, Chruscinski AJ, Sharpe A, Sanders VM. Stimulation of the B cell receptor, CD86 (B7-2), and the beta 2-adrenergic receptor intrinsically modulates the level of IgG1 and IgE produced per B cell. J Immunol (2000) 165:680–90. doi:10.4049/jimmunol.165.2.680
41. Lorton D, Bellinger DL. Molecular mechanisms underlying beta-adrenergic receptor-mediated cross-talk between sympathetic neurons and immune cells. Int J Mol Sci (2015) 16:5635–65. doi:10.3390/ijms16035635
42. Qin JF, Jin FJ, Li N, Guan HT, Lan L, Ni H, et al. Adrenergic receptor beta2 activation by stress promotes breast cancer progression through macrophages M2 polarization in tumor microenvironment. BMB Rep (2015) 48:295–300. doi:10.5483/BMBRep.2015.48.5.008
43. Sloan EK, Priceman SJ, Cox BF, Yu S, Pimentel MA, Tangkanangnukul V, et al. The sympathetic nervous system induces a metastatic switch in primary breast cancer. Cancer Res (2010) 70:7042–52. doi:10.1158/0008-5472.CAN-10-0522
44. Lamkin DM, Ho HY, Ong TH, Kawanishi CK, Stoffers VL, Ahlawat N, et al. beta-adrenergic-stimulated macrophages: comprehensive localization in the M1-M2 spectrum. Brain Behav Immun (2016) 57:338–46. doi:10.1016/j.bbi.2016.07.162
45. Grailer JJ, Haggadone MD, Sarma JV, Zetoune FS, Ward PA. Induction of M2 regulatory macrophages through the beta2-adrenergic receptor with protection during endotoxemia and acute lung injury. J Innate Immun (2014) 6:607–18. doi:10.1159/000358524
46. Gabanyi I, Muller PA, Feighery L, Oliveira TY, Costa-Pinto FA, Mucida D. Neuro-immune interactions drive tissue programming in intestinal macrophages. Cell (2016) 164:378–91. doi:10.1016/j.cell.2015.12.023
47. Victoni T, Salvator H, Abrial C, Brollo M, Porto LCS, Lagente V, et al. Human lung and monocyte-derived macrophages differ with regard to the effects of beta2-adrenoceptor agonists on cytokine release. Respir Res (2017) 18:126. doi:10.1186/s12931-017-0613-y
48. Sharma M, Patterson L, Chapman E, Flood PM. Salmeterol, a long-acting beta2-adrenergic receptor agonist, inhibits macrophage activation by lipopolysaccharide from Porphyromonas gingivalis. J Periodontol (2017) 88:681–92. doi:10.1902/jop.2017.160464
49. Gill SK, Marriott HM, Suvarna SK, Peachell PT. Evaluation of the anti-inflammatory effects of beta-adrenoceptor agonists on human lung macrophages. Eur J Pharmacol (2016) 793:49–55. doi:10.1016/j.ejphar.2016.11.005
50. Roewe J, Higer M, Riehl DR, Gericke A, Radsak MP, Bosmann M. Neuroendocrine modulation of IL-27 in macrophages. J Immunol (2017) 199:2503–14. doi:10.4049/jimmunol.1700687
51. Huang JL, Zhang YL, Wang CC, Zhou JR, Ma Q, Wang X, et al. Enhanced phosphorylation of MAPKs by NE promotes TNF-alpha production by macrophage through alpha adrenergic receptor. Inflammation (2012) 35:527–34. doi:10.1007/s10753-011-9342-4
52. Wu H, Chen J, Song S, Yuan P, Liu L, Zhang Y, et al. beta2-adrenoceptor signaling reduction in dendritic cells is involved in the inflammatory response in adjuvant-induced arthritic rats. Sci Rep (2016) 6:24548. doi:10.1038/srep24548
53. Chen G, Le Y, Zhou L, Gong L, Li X, Li Y, et al. Dexmedetomidine inhibits maturation and function of human cord blood-derived dendritic cells by interfering with synthesis and secretion of IL-12 and IL-23. PLoS One (2016) 11:e0153288. doi:10.1371/journal.pone.0153288
54. Takenaka MC, Araujo LP, Maricato JT, Nascimento VM, Guereschi MG, Rezende RM, et al. Norepinephrine controls effector T cell differentiation through beta2-adrenergic receptor-mediated inhibition of NF-kappaB and AP-1 in dendritic cells. J Immunol (2016) 196:637–44. doi:10.4049/jimmunol.1501206
55. Giordani L, Cuzziol N, Del Pinto T, Sanchez M, Maccari S, Massimi A, et al. beta2-Agonist clenbuterol hinders human monocyte differentiation into dendritic cells. Exp Cell Res (2015) 339:163–73. doi:10.1016/j.yexcr.2015.10.032
56. Yanagawa Y, Matsumoto M, Togashi H. Adrenoceptor-mediated enhancement of interleukin-33 production by dendritic cells. Brain Behav Immun (2011) 25:1427–33. doi:10.1016/j.bbi.2011.04.012
57. Goyarts E, Matsui M, Mammone T, Bender AM, Wagner JA, Maes D, et al. Norepinephrine modulates human dendritic cell activation by altering cytokine release. Exp Dermatol (2008) 17:188–96. doi:10.1111/j.1600-0625.2007.00677.x
58. Maestroni GJ, Mazzola P. Langerhans cells beta 2-adrenoceptors: role in migration, cytokine production, Th priming and contact hypersensitivity. J Neuroimmunol (2003) 144:91–9. doi:10.1016/j.jneuroim.2003.08.039
59. Yang H, Du RZ, Qiu JP, Tang YQ, Chen SC. Bisoprolol reverses epinephrine-mediated inhibition of cell emigration through increases in the expression of beta-arrestin 2 and CCR7 and PI3K phosphorylation, in dendritic cells loaded with cholesterol. Thromb Res (2013) 131:230–7. doi:10.1016/j.thromres.2012.12.009
60. Ueshima H, Inada T, Shingu K. Suppression of phagosome proteolysis and Matrigel migration with the alpha2-adrenergic receptor agonist dexmedetomidine in murine dendritic cells. Immunopharmacol Immunotoxicol (2013) 35:558–66. doi:10.3109/08923973.2013.822509
61. Seiffert K, Hosoi J, Torii H, Ozawa H, Ding W, Campton K, et al. Catecholamines inhibit the antigen-presenting capability of epidermal Langerhans cells. J Immunol (2002) 168:6128–35. doi:10.4049/jimmunol.168.12.6128
62. Herve J, Dubreil L, Tardif V, Terme M, Pogu S, Anegon I, et al. beta2-adrenoreceptor agonist inhibits antigen cross-presentation by dendritic cells. J Immunol (2013) 190:3163–71. doi:10.4049/jimmunol.1201391
63. Kokolus KM, Spangler HM, Povinelli BJ, Farren MR, Lee KP, Repasky EA. Stressful presentations: mild cold stress in laboratory mice influences phenotype of dendritic cells in naive and tumor-bearing mice. Front Immunol (2014) 5:23. doi:10.3389/fimmu.2014.00023
64. Kanemi O, Zhang X, Sakamoto Y, Ebina M, Nagatomi R. Acute stress reduces intraparenchymal lung natural killer cells via beta-adrenergic stimulation. Clin Exp Immunol (2005) 139:25–34. doi:10.1111/j.1365-2249.2005.02672.x
65. Shakhar G, Ben-Eliyahu S. In vivo beta-adrenergic stimulation suppresses natural killer activity and compromises resistance to tumor metastasis in rats. J Immunol (1998) 160:3251–8.
66. De Lorenzo BH, de Oliveira Marchioro L, Greco CR, Suchecki D. Sleep-deprivation reduces NK cell number and function mediated by beta-adrenergic signalling. Psychoneuroendocrinology (2015) 57:134–43. doi:10.1016/j.psyneuen.2015.04.006
67. Tarr AJ, Powell ND, Reader BF, Bhave NS, Roloson AL, Carson WE III, et al. beta-Adrenergic receptor mediated increases in activation and function of natural killer cells following repeated social disruption. Brain Behav Immun (2012) 26:1226–38. doi:10.1016/j.bbi.2012.07.002
68. Song Y, Gan Y, Wang Q, Meng Z, Li G, Shen Y, et al. Enriching the housing environment for mice enhances their NK cell antitumor immunity via sympathetic nerve-dependent regulation of NKG2D and CCR5. Cancer Res (2017) 77:1611–22. doi:10.1158/0008-5472.CAN-16-2143
69. Noguchi T, Nakagome K, Kobayashi T, Ueda Y, Soma T, Nakamoto H, et al. Effect of beta2-adrenergic agonists on eosinophil adhesion, superoxide anion generation, and degranulation. Allergol Int (2015) 64(Suppl):S46–53. doi:10.1016/j.alit.2015.05.009
70. Gosain A, Gamelli RL, DiPietro LA. Norepinephrine-mediated suppression of phagocytosis by wound neutrophils. J Surg Res (2009) 152:311–8. doi:10.1016/j.jss.2008.05.001
71. Lajevic MD, Suleiman S, Cohen RL, Chambers DA. Activation of p38 mitogen-activated protein kinase by norepinephrine in T-lineage cells. Immunology (2011) 132:197–208. doi:10.1111/j.1365-2567.2010.03354.x
72. Monteiro S, Roque S, de Sa-Calcada D, Sousa N, Correia-Neves M, Cerqueira JJ. An efficient chronic unpredictable stress protocol to induce stress-related responses in C57BL/6 mice. Front Psychiatry (2015) 6:6. doi:10.3389/fpsyt.2015.00006
73. Riether C, Kavelaars A, Wirth T, Pacheco-Lopez G, Doenlen R, Willemen H, et al. Stimulation of beta(2)-adrenergic receptors inhibits calcineurin activity in CD4(+) T cells via PKA-AKAP interaction. Brain Behav Immun (2011) 25:59–66. doi:10.1016/j.bbi.2010.07.248
74. McAlees JW, Smith LT, Erbe RS, Jarjoura D, Ponzio NM, Sanders VM. Epigenetic regulation of beta2-adrenergic receptor expression in T(H)1 and T(H)2 cells. Brain Behav Immun (2011) 25:408–15. doi:10.1016/j.bbi.2010.10.019
75. Heijink IH, Vellenga E, Borger P, Postma DS, Monchy JG, Kauffman HF. Polarized Th1 and Th2 cells are less responsive to negative feedback by receptors coupled to the AC/cAMP system compared to freshly isolated T cells. Br J Pharmacol (2003) 138:1441–50. doi:10.1038/sj.bjp.0705193
76. Slota C, Shi A, Chen G, Bevans M, Weng NP. Norepinephrine preferentially modulates memory CD8 T cell function inducing inflammatory cytokine production and reducing proliferation in response to activation. Brain Behav Immun (2015) 46:168–79. doi:10.1016/j.bbi.2015.01.015
77. Wahle M, Stachetzki U, Krause A, Pierer M, Hantzschel H, Baerwald CG. Regulation of beta2-adrenergic receptors on CD4 and CD8 positive lymphocytes by cytokines in vitro. Cytokine (2001) 16:205–9. doi:10.1006/cyto.2001.0965
78. Grebe KM, Hickman HD, Irvine KR, Takeda K, Bennink JR, Yewdell JW. Sympathetic nervous system control of anti-influenza CD8+ T cell responses. Proc Natl Acad Sci U S A (2009) 106:5300–5. doi:10.1073/pnas.0808851106
79. Estrada LD, Agac D, Farrar JD. Sympathetic neural signaling via the beta2-adrenergic receptor suppresses T-cell receptor-mediated human and mouse CD8(+) T-cell effector function. Eur J Immunol (2016) 46:1948–58. doi:10.1002/eji.201646395
80. Simonini MV, Polak PE, Sharp A, McGuire S, Galea E, Feinstein DL. Increasing CNS noradrenaline reduces EAE severity. J Neuroimmune Pharmacol (2010) 5:252–9. doi:10.1007/s11481-009-9182-2
81. Bucsek MJ, Giridharan T, MacDonald CR, Hylander BL, Repasky EA. An overview of the role of sympathetic regulation of immune responses in infectious disease and autoimmunity. Int J Hyperthermia (2018).
82. Cohen S, Levi-Montalcini R, Hamburger V. A nerve growth-stimulating factor isolated from Sarcom AS 37 and 180. Proc Natl Acad Sci U S A (1954) 40:1014–8. doi:10.1073/pnas.40.10.1014
83. Entschladen F, Palm D, Lang K, Drell TLT, Zaenker KS. Neoneurogenesis: tumors may initiate their own innervation by the release of neurotrophic factors in analogy to lymphangiogenesis and neoangiogenesis. Med Hypotheses (2006) 67:33–5. doi:10.1016/j.mehy.2006.01.015
84. Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ, et al. Autonomic nerve development contributes to prostate cancer progression. Science (2013) 341:1236361. doi:10.1126/science.1236361
85. Eng JW, Reed CB, Kokolus KM, Pitoniak R, Utley A, Bucsek MJ, et al. Housing temperature-induced stress drives therapeutic resistance in murine tumour models through beta-adrenergic receptor activation. Nat Commun (2015) 6:6426. doi:10.1038/ncomms7426
86. Lutgendorf SK, DeGeest K, Dahmoush L, Farley D, Penedo F, Bender D, et al. Social isolation is associated with elevated tumor norepinephrine in ovarian carcinoma patients. Brain Behav Immun (2011) 25:250–5. doi:10.1016/j.bbi.2010.10.012
87. Szpunar MJ, Belcher EK, Dawes RP, Madden KS. Sympathetic innervation, norepinephrine content, and norepinephrine turnover in orthotopic and spontaneous models of breast cancer. Brain Behav Immun (2016) 53:223–33. doi:10.1016/j.bbi.2015.12.014
88. Thaker PH, Han LY, Kamat AA, Arevalo JM, Takahashi R, Lu C, et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med (2006) 12:939–44. doi:10.1038/nm1447
89. Hara MR, Kovacs JJ, Whalen EJ, Rajagopal S, Strachan RT, Grant W, et al. A stress response pathway regulates DNA damage through beta2-adrenoreceptors and beta-arrestin-1. Nature (2011) 477:349–53. doi:10.1038/nature10368
90. Hara MR, Sachs BD, Caron MG, Lefkowitz RJ. Pharmacological blockade of a beta(2)AR-beta-arrestin-1 signaling cascade prevents the accumulation of DNA damage in a behavioral stress model. Cell Cycle (2013) 12:219–24. doi:10.4161/cc.23368
91. Flint MS, Baum A, Episcopo B, Knickelbein KZ, Liegey Dougall AJ, Chambers WH, et al. Chronic exposure to stress hormones promotes transformation and tumorigenicity of 3T3 mouse fibroblasts. Stress (2013) 16:114–21. doi:10.3109/10253890.2012.686075
92. Cole SW, Nagaraja AS, Lutgendorf SK, Green PA, Sood AK. Sympathetic nervous system regulation of the tumour microenvironment. Nat Rev Cancer (2015) 15:563–72. doi:10.1038/nrc3978
93. Mathur MB, Epel E, Kind S, Desai M, Parks CG, Sandler DP, et al. Perceived stress and telomere length: a systematic review, meta-analysis, and methodologic considerations for advancing the field. Brain Behav Immun (2016) 54:158–69. doi:10.1016/j.bbi.2016.02.002
94. Wentzensen IM, Mirabello L, Pfeiffer RM, Savage SA. The association of telomere length and cancer: a meta-analysis. Cancer Epidemiol Biomarkers Prev (2011) 20:1238–50. doi:10.1158/1055-9965.EPI-11-0005
95. Huang XY, Wang HC, Yuan Z, Huang J, Zheng Q. Norepinephrine stimulates pancreatic cancer cell proliferation, migration and invasion via beta-adrenergic receptor-dependent activation of P38/MAPK pathway. Hepatogastroenterology (2012) 59:889–93.
96. Wu FQ, Fang T, Yu LX, Lv GS, Lv HW, Liang D, et al. ADRB2 signaling promotes HCC progression and sorafenib resistance by inhibiting autophagic degradation of HIF1alpha. J Hepatol (2016) 65:314–24. doi:10.1016/j.jhep.2016.04.019
97. Coelho M, Soares-Silva C, Brandao D, Marino F, Cosentino M, Ribeiro L. beta-Adrenergic modulation of cancer cell proliferation: available evidence and clinical perspectives. J Cancer Res Clin Oncol (2017) 143:275–91. doi:10.1007/s00432-016-2278-1
98. Dal Monte M, Fornaciari I, Nicchia GP, Svelto M, Casini G, Bagnoli P. beta3-adrenergic receptor activity modulates melanoma cell proliferation and survival through nitric oxide signaling. Naunyn Schmiedebergs Arch Pharmacol (2014) 387:533–43. doi:10.1007/s00210-014-0969-1
99. Lin X, Luo K, Lv Z, Huang J. Beta-adrenoceptor action on pancreatic cancer cell proliferation and tumor growth in mice. Hepatogastroenterology (2012) 59:584–8. doi:10.5754/hge11271
100. Shan T, Ma Q, Zhang D, Guo K, Liu H, Wang F, et al. beta2-adrenoceptor blocker synergizes with gemcitabine to inhibit the proliferation of pancreatic cancer cells via apoptosis induction. Eur J Pharmacol (2011) 665:1–7. doi:10.1016/j.ejphar.2011.04.055
101. Zhang D, Ma Q, Shen S, Hu H. Inhibition of pancreatic cancer cell proliferation by propranolol occurs through apoptosis induction: the study of beta-adrenoceptor antagonist’s anticancer effect in pancreatic cancer cell. Pancreas (2009) 38:94–100. doi:10.1097/MPA.0b013e318184f50c
102. Zhou C, Chen X, Zeng W, Peng C, Huang G, Li X, et al. Propranolol induced G0/G1/S phase arrest and apoptosis in melanoma cells via AKT/MAPK pathway. Oncotarget (2016) 7:68314–27. doi:10.18632/oncotarget.11599
103. Zhang D, Ma Q, Wang Z, Zhang M, Guo K, Wang F, et al. beta2-adrenoceptor blockage induces G1/S phase arrest and apoptosis in pancreatic cancer cells via Ras/Akt/NFkappaB pathway. Mol Cancer (2011) 10:146. doi:10.1186/1476-4598-10-146
104. Hassan S, Karpova Y, Baiz D, Yancey D, Pullikuth A, Flores A, et al. Behavioral stress accelerates prostate cancer development in mice. J Clin Invest (2013) 123:874–86. doi:10.1172/JCI63324
105. Pasquier E, Ciccolini J, Carre M, Giacometti S, Fanciullino R, Pouchy C, et al. Propranolol potentiates the anti-angiogenic effects and anti-tumor efficacy of chemotherapy agents: implication in breast cancer treatment. Oncotarget (2011) 2:797–809. doi:10.18632/oncotarget.343
106. Sood AK, Armaiz-Pena GN, Halder J, Nick AM, Stone RL, Hu W, et al. Adrenergic modulation of focal adhesion kinase protects human ovarian cancer cells from anoikis. J Clin Invest (2010) 120:1515–23. doi:10.1172/JCI40802
107. Zhang J, Deng YT, Liu J, Wang YQ, Yi TW, Huang BY, et al. Norepinephrine induced epithelial-mesenchymal transition in HT-29 and A549 cells in vitro. J Cancer Res Clin Oncol (2016) 142:423–35. doi:10.1007/s00432-015-2044-9
108. Pu J, Zhang X, Luo H, Xu L, Lu X, Lu J. Adrenaline promotes epithelial-to-mesenchymal transition via HuR-TGFbeta regulatory axis in pancreatic cancer cells and the implication in cancer prognosis. Biochem Biophys Res Commun (2017) 493:1273–9. doi:10.1016/j.bbrc.2017.09.146
109. Lu YJ, Geng ZJ, Sun XY, Li YH, Fu XB, Zhao XY, et al. Isoprenaline induces epithelial-mesenchymal transition in gastric cancer cells. Mol Cell Biochem (2015) 408:1–13. doi:10.1007/s11010-015-2477-0
110. Shan T, Cui X, Li W, Lin W, Li Y, Chen X, et al. Novel regulatory program for norepinephrine-induced epithelial-mesenchymal transition in gastric adenocarcinoma cell lines. Cancer Sci (2014) 105:847–56. doi:10.1111/cas.12438
111. Lutgendorf SK, Thaker PH, Arevalo JM, Goodheart MJ, Slavich GM, Sood AK, et al. Biobehavioral modulation of the exosome transcriptome in ovarian carcinoma. Cancer (2018) 124(3):580–86. doi:10.1002/cncr.31078
112. Le CP, Sloan EK. Stress-driven lymphatic dissemination: an unanticipated consequence of communication between the sympathetic nervous system and lymphatic vasculature. Mol Cell Oncol (2016) 3:e1177674. doi:10.1080/23723556.2016.1177674
113. Armaiz-Pena GN, Allen JK, Cruz A, Stone RL, Nick AM, Lin YG, et al. Src activation by beta-adrenoreceptors is a key switch for tumour metastasis. Nat Commun (2013) 4:1403. doi:10.1038/ncomms2903
114. Benish M, Bartal I, Goldfarb Y, Levi B, Avraham R, Raz A, et al. Perioperative use of beta-blockers and COX-2 inhibitors may improve immune competence and reduce the risk of tumor metastasis. Ann Surg Oncol (2008) 15:2042–52. doi:10.1245/s10434-008-9890-5
115. Campbell JP, Karolak MR, Ma Y, Perrien DS, Masood-Campbell SK, Penner NL, et al. Stimulation of host bone marrow stromal cells by sympathetic nerves promotes breast cancer bone metastasis in mice. PLoS Biol (2012) 10:e1001363. doi:10.1371/journal.pbio.1001363
116. Elefteriou F. Chronic stress, sympathetic activation and skeletal metastasis of breast cancer cells. Bonekey Rep (2015) 4:693. doi:10.1038/bonekey.2015.61
117. Glasner A, Avraham R, Rosenne E, Benish M, Zmora O, Shemer S, et al. Improving survival rates in two models of spontaneous postoperative metastasis in mice by combined administration of a beta-adrenergic antagonist and a cyclooxygenase-2 inhibitor. J Immunol (2010) 184:2449–57. doi:10.4049/jimmunol.0903301
118. Magnon C. Role of the autonomic nervous system in tumorigenesis and metastasis. Mol Cell Oncol (2015) 2:e975643. doi:10.4161/23723556.2014.975643
119. Nagaraja AS, Dorniak PL, Sadaoui NC, Kang Y, Lin T, Armaiz-Pena G, et al. Sustained adrenergic signaling leads to increased metastasis in ovarian cancer via increased PGE2 synthesis. Oncogene (2016) 35:2390–7. doi:10.1038/onc.2015.302
120. Palm D, Lang K, Niggemann B, Drell TLT, Masur K, Zaenker KS, et al. The norepinephrine-driven metastasis development of PC-3 human prostate cancer cells in BALB/c nude mice is inhibited by beta-blockers. Int J Cancer (2006) 118:2744–9. doi:10.1002/ijc.21723
121. Zhao L, Xu J, Liang F, Li A, Zhang Y, Sun J. Effect of chronic psychological stress on liver metastasis of colon cancer in mice. PLoS One (2015) 10:e0139978. doi:10.1371/journal.pone.0139978
122. Creed SJ, Le CP, Hassan M, Pon CK, Albold S, Chan KT, et al. beta2-adrenoceptor signaling regulates invadopodia formation to enhance tumor cell invasion. Breast Cancer Res (2015) 17:145. doi:10.1186/s13058-015-0655-3
123. Kim-Fuchs C, Le CP, Pimentel MA, Shackleford D, Ferrari D, Angst E, et al. Chronic stress accelerates pancreatic cancer growth and invasion: a critical role for beta-adrenergic signaling in the pancreatic microenvironment. Brain Behav Immun (2014) 40:40–7. doi:10.1016/j.bbi.2014.02.019
124. Nagaraja AS, Dood RL, Armaiz-Pena G, Kang Y, Wu SY, Allen JK, et al. Adrenergic-mediated increases in INHBA drive CAF phenotype and collagens. JCI Insight (2017) 2. doi:10.1172/jci.insight.93076
125. Park SY, Kang JH, Jeong KJ, Lee J, Han JW, Choi WS, et al. Norepinephrine induces VEGF expression and angiogenesis by a hypoxia-inducible factor-1alpha protein-dependent mechanism. Int J Cancer (2011) 128:2306–16. doi:10.1002/ijc.25589
126. Yang EV, Kim SJ, Donovan EL, Chen M, Gross AC, Webster Marketon JI, et al. Norepinephrine upregulates VEGF, IL-8, and IL-6 expression in human melanoma tumor cell lines: implications for stress-related enhancement of tumor progression. Brain Behav Immun (2009) 23:267–75. doi:10.1016/j.bbi.2008.10.005
127. Yang EV, Sood AK, Chen M, Li Y, Eubank TD, Marsh CB, et al. Norepinephrine up-regulates the expression of vascular endothelial growth factor, matrix metalloproteinase (MMP)-2, and MMP-9 in nasopharyngeal carcinoma tumor cells. Cancer Res (2006) 66:10357–64. doi:10.1158/0008-5472.CAN-06-2496
128. Chen H, Liu D, Yang Z, Sun L, Deng Q, Yang S, et al. Adrenergic signaling promotes angiogenesis through endothelial cell-tumor cell crosstalk. Endocr Relat Cancer (2014) 21:783–95. doi:10.1530/ERC-14-0236
129. Hulsurkar M, Li Z, Zhang Y, Li X, Zheng D, Li W. Beta-adrenergic signaling promotes tumor angiogenesis and prostate cancer progression through HDAC2-mediated suppression of thrombospondin-1. Oncogene (2017) 36:1525–36. doi:10.1038/onc.2016.319
130. Wu W, Murata J, Murakami K, Yamaura T, Hayashi K, Saiki I. Social isolation stress augments angiogenesis induced by colon 26-L5 carcinoma cells in mice. Clin Exp Metastasis (2000) 18:1–10. doi:10.1023/A:1026548715669
131. Zahalka AH, Arnal-Estape A, Maryanovich M, Nakahara F, Cruz CD, Finley LWS, et al. Adrenergic nerves activate an angio-metabolic switch in prostate cancer. Science (2017) 358:321–6. doi:10.1126/science.aah5072
132. Le CP, Nowell CJ, Kim-Fuchs C, Botteri E, Hiller JG, Ismail H, et al. Chronic stress in mice remodels lymph vasculature to promote tumour cell dissemination. Nat Commun (2016) 7:10634. doi:10.1038/ncomms10634
133. Chen H, Liu D, Guo L, Cheng X, Guo N, Shi M. Chronic psychological stress promotes lung metastatic colonization of circulating breast cancer cells by decorating a pre-metastatic niche through activating beta-adrenergic signaling. J Pathol (2018) 244:49–60. doi:10.1002/path.4988
134. Bucsek MJ, Qiao G, MacDonald CR, Giridharan T, Evans L, Niedzwecki B, et al. β-adrenergic signaling in mice housed at standard temperatures suppresses an effector phenotype in CD8+ T cells and undermines checkpoint inhibitor therapy. Cancer Res (2017) 77(20):5639–51. doi:10.1158/0008-5472.CAN-17-0546
135. Wrobel JL, Bod L, Lengagne R, Kato M, Prevost-Blondel A, Le Gal FA. Propranolol induces a favourable shift of anti-tumor immunity in a murine spontaneous model of melanoma. Oncotarget (2016) 7:77825–37. doi:10.18632/oncotarget.12833
136. Kokolus KM, Capitano ML, Lee CT, Eng JW, Waight JD, Hylander BL, et al. Baseline tumor growth and immune control in laboratory mice are significantly influenced by subthermoneutral housing temperature. Proc Natl Acad Sci U S A (2013) 110:20176–81. doi:10.1073/pnas.1304291110
137. Schmidt D, Peterlik D, Reber SO, Lechner A, Mannel DN. Induction of suppressor cells and increased tumor growth following chronic psychosocial stress in male mice. PLoS One (2016) 11:e0159059. doi:10.1371/journal.pone.0159059
138. Zhou L, Li Y, Li X, Chen G, Liang H, Wu Y, et al. Propranolol attenuates surgical stress-induced elevation of the regulatory T cell response in patients undergoing radical mastectomy. J Immunol (2016) 196:3460–9. doi:10.4049/jimmunol.1501677
139. Jin J, Wang X, Wang Q, Guo X, Cao J, Zhang X, et al. Chronic psychological stress induces the accumulation of myeloid-derived suppressor cells in mice. PLoS One (2013) 8:e74497. doi:10.1371/journal.pone.0074497
140. Szabo S, Tache Y, Somogyi A. The legacy of Hans Selye and the origins of stress research: a retrospective 75 years after his landmark brief “letter” to the editor# of nature. Stress (2012) 15(5):472–78.
142. Stankiewicz AM, Goscik J, Majewska A, Swiergiel AH, Juszczak GR. The effect of acute and chronic social stress on the hippocampal transcriptome in mice. PLoS One (2015) 10:e0142195. doi:10.1371/journal.pone.0142195
143. Lin Q, Wang F, Yang R, Zheng X, Gao H, Zhang P. Effect of chronic restraint stress on human colorectal carcinoma growth in mice. PLoS One (2013) 8:e61435. doi:10.1371/journal.pone.0061435
144. Madden KS, Szpunar MJ, Brown EB. Early impact of social isolation and breast tumor progression in mice. Brain Behav Immun (2013) 30(Suppl):S135–41. doi:10.1016/j.bbi.2012.05.003
145. Hou N, Zhang X, Zhao L, Zhao X, Li Z, Song T, et al. A novel chronic stress-induced shift in the Th1 to Th2 response promotes colon cancer growth. Biochem Biophys Res Commun (2013) 439:471–6. doi:10.1016/j.bbrc.2013.08.101
146. Partecke LI, Speerforck S, Kading A, Seubert F, Kuhn S, Lorenz E, et al. Chronic stress increases experimental pancreatic cancer growth, reduces survival and can be antagonised by beta-adrenergic receptor blockade. Pancreatology (2016) 16:423–33. doi:10.1016/j.pan.2016.03.005
147. Lee JW, Shahzad MM, Lin YG, Armaiz-Pena G, Mangala LS, Han HD, et al. Surgical stress promotes tumor growth in ovarian carcinoma. Clin Cancer Res (2009) 15:2695–702. doi:10.1158/1078-0432.CCR-08-2966
148. Dethlefsen C, Hansen LS, Lillelund C, Andersen C, Gehl J, Christensen JF, et al. Exercise-induced catecholamines activate the hippo tumor suppressor pathway to reduce risks of breast cancer development. Cancer Res (2017) 77:4894–904. doi:10.1158/0008-5472.CAN-16-3125
149. Demas GE, Carlton ED. Ecoimmunology for psychoneuroimmunologists: considering context in neuroendocrine-immune-behavior interactions. Brain Behav Immun (2015) 44:9–16. doi:10.1016/j.bbi.2014.09.002
150. National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals. 8th ed. Washington, DC: National Academies (2011).
151. Hylander BL, Repasky EA. Thermoneutrality, mice and cancer: a heated opinion. Trends Cancer (2016) 2:166–75. doi:10.1016/j.trecan.2016.03.005
152. Karp CL. Unstressing intemperate models: how cold stress undermines mouse modeling. J Exp Med (2012) 209:1069–74. doi:10.1084/jem.20120988
153. Messmer MN, Kokolus KM, Eng JW, Abrams SI, Repasky EA. Mild cold-stress depresses immune responses: implications for cancer models involving laboratory mice. Bioessays (2014) 36:884–91. doi:10.1002/bies.201400066
154. Giles DA, Moreno-Fernandez ME, Stankiewicz TE, Graspeuntner S, Cappelletti M, Wu D, et al. Thermoneutral housing exacerbates nonalcoholic fatty liver disease in mice and allows for sex-independent disease modeling. Nat Med (2017) 23:829–38. doi:10.1038/nm.4346
155. Giles DA, Ramkhelawon B, Donelan EM, Stankiewicz TE, Hutchison SB, Mukherjee R, et al. Modulation of ambient temperature promotes inflammation and initiates atherosclerosis in wild type C57BL/6 mice. Mol Metab (2016) 5:1121–30. doi:10.1016/j.molmet.2016.09.008
156. Tian XY, Ganeshan K, Hong C, Nguyen KD, Qiu Y, Kim J, et al. Thermoneutral housing accelerates metabolic inflammation to potentiate atherosclerosis but not insulin resistance. Cell Metab (2015) 23(1):165–78. doi:10.1016/j.cmet.2015.10.003
157. Woollard KJ, Murphy AJ. The endless summer: thermoneutrality prevents monocytosis and reduces atherosclerosis. Circ Res (2017) 121:596–8. doi:10.1161/CIRCRESAHA.117.311721
158. Vandal M, White PJ, Tournissac M, Tremblay C, St-Amour I, Drouin-Ouellet J, et al. Impaired thermoregulation and beneficial effects of thermoneutrality in the 3xTg-AD model of Alzheimer’s disease. Neurobiol Aging (2016) 43:47–57. doi:10.1016/j.neurobiolaging.2016.03.024
159. Williams JW, Elvington A, Ivanov S, Kessler S, Luehmann H, Baba O, et al. Thermoneutrality but not UCP1 deficiency suppresses monocyte mobilization into blood. Circ Res (2017) 121:662–76. doi:10.1161/CIRCRESAHA.117.311519
160. Cui X, Nguyen NL, Zarebidaki E, Cao Q, Li F, Zha L, et al. Thermoneutrality decreases thermogenic program and promotes adiposity in high-fat diet-fed mice. Physiol Rep (2016) 4. doi:10.14814/phy2.12799
161. Razzoli M, Frontini A, Gurney A, Mondini E, Cubuk C, Katz LS, et al. Stress-induced activation of brown adipose tissue prevents obesity in conditions of low adaptive thermogenesis. Mol Metab (2016) 5:19–33. doi:10.1016/j.molmet.2015.10.005
162. Xiao C, Goldgof M, Gavrilova O, Reitman ML. Anti-obesity and metabolic efficacy of the beta3-adrenergic agonist, CL316243, in mice at thermoneutrality compared to 22 degrees C. Obesity (Silver Spring) (2015) 23:1450–9. doi:10.1002/oby.21124
163. Ganeshan K, Chawla A. Warming the mouse to model human diseases. Nat Rev Endocrinol (2017) 13:458–65. doi:10.1038/nrendo.2017.48
164. Gordon CJ. The mouse thermoregulatory system: its impact on translating biomedical data to humans. Physiol Behav (2017) 179:55–66. doi:10.1016/j.physbeh.2017.05.026
165. Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell (2015) 28:690–714. doi:10.1016/j.ccell.2015.10.012
166. Ngiow SF, Young A, Jacquelot N, Yamazaki T, Enot D, Zitvogel L, et al. A threshold level of intratumor CD8+ T-cell PD1 expression dictates therapeutic response to anti-PD1. Cancer Res (2015) 75:3800–11. doi:10.1158/0008-5472.CAN-15-1082
167. Zitvogel L, Apetoh L, Ghiringhelli F, Andre F, Tesniere A, Kroemer G. The anticancer immune response: indispensable for therapeutic success? J Clin Invest (2008) 118:1991–2001. doi:10.1172/JCI35180
168. Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity (2013) 39:74–88. doi:10.1016/j.immuni.2013.06.014
169. Demaria S, Formenti SC. Role of T lymphocytes in tumor response to radiotherapy. Front Oncol (2012) 2:95. doi:10.3389/fonc.2012.00095
170. Pilones KA, Vanpouille-Box C, Demaria S. Combination of radiotherapy and immune checkpoint inhibitors. Semin Radiat Oncol (2015) 25:28–33. doi:10.1016/j.semradonc.2014.07.004
171. Galon J, Fox BA, Bifulco CB, Masucci G, Rau T, Botti G, et al. Immunoscore and immunoprofiling in cancer: an update from the melanoma and immunotherapy bridge 2015. J Transl Med (2016) 14:273. doi:10.1186/s12967-016-1029-z
172. Gordon CJ, Puckett ET, Repasky ES, Johnstone AF. A device that allows rodents to behaviorally thermoregulate when housed in vivariums. J Am Assoc Lab Anim Sci (2017) 56:173–6.
173. Gaskill BN, Garner JP. Stressed out: providing laboratory animals with behavioral control to reduce the physiological effects of stress. Lab Anim (NY) (2017) 46:142–5. doi:10.1038/laban.1218
174. Gaskill BN, Gordon CJ, Pajor EA, Lucas JR, Davis JK, Garner JP. Impact of nesting material on mouse body temperature and physiology. Physiol Behav (2013) 11(0–111):87–95. doi:10.1016/j.physbeh.2012.12.018
175. Sardi I, Giunti L, Bresci C, Buccoliero AM, Degl’innocenti D, Cardellicchio S, et al. Expression of beta-adrenergic receptors in pediatric malignant brain tumors. Oncol Lett (2013) 5:221–5. doi:10.3892/ol.2012.989
176. Whitsett JA, Burdsall J, Workman L, Hollinger B, Neely J. beta-Adrenergic receptors in pediatric tumors: uncoupled beta 1-adrenergic receptor in Ewing’s sarcoma. J Natl Cancer Inst (1983) 71:779–86.
177. Kondratenko TY, Zacharova IV, Kuzina NV, Katukov V, Severin ES, Kornilova Z, et al. Alterations in human lung adrenergic receptors in cancer. Biochem Mol Biol Int (1993) 29:123–30.
178. Moretti S, Massi D, Farini V, Baroni G, Parri M, Innocenti S, et al. beta-adrenoceptors are upregulated in human melanoma and their activation releases pro-tumorigenic cytokines and metalloproteases in melanoma cell lines. Lab Invest (2013) 93:279–90. doi:10.1038/labinvest.2012.175
179. Nagmani R, Pasco DS, Salas RD, Feller DR. Evaluation of beta-adrenergic receptor subtypes in the human prostate cancer cell line-LNCaP. Biochem Pharmacol (2003) 65:1489–94. doi:10.1016/S0006-2952(03)00105-9
180. Cardwell CR, Coleman HG, Murray LJ, Entschladen F, Powe DG. Beta-blocker usage and breast cancer survival: a nested case-control study within a UK Clinical Practice Research Datalink cohort. Int J Epidemiol (2013) 42:1852–61. doi:10.1093/ije/dyt196
181. Cardwell CR, Coleman HG, Murray LJ, O’Sullivan JM, Powe DG. Beta-blocker usage and prostate cancer survival: a nested case-control study in the UK Clinical Practice Research Datalink cohort. Cancer Epidemiol (2014) 38:279–85. doi:10.1016/j.canep.2014.03.011
182. Hicks BM, Murray LJ, Powe DG, Hughes CM, Cardwell CR. beta-Blocker usage and colorectal cancer mortality: a nested case-control study in the UK Clinical Practice Research Datalink cohort. Ann Oncol (2013) 24:3100–6. doi:10.1093/annonc/mdt381
183. Livingstone E, Hollestein LM, van Herk-Sukel MP, van de Poll-Franse L, Nijsten T, Schadendorf D, et al. beta-Blocker use and all-cause mortality of melanoma patients: results from a population-based Dutch cohort study. Eur J Cancer (2013) 49:3863–71. doi:10.1016/j.ejca.2013.07.141
184. McCourt C, Coleman HG, Murray LJ, Cantwell MM, Dolan O, Powe DG, et al. Beta-blocker usage after malignant melanoma diagnosis and survival: a population-based nested case-control study. Br J Dermatol (2014) 170:930–8. doi:10.1111/bjd.12894
185. Parker WP, Lohse CM, Zaid HB, Cheville JC, Boorjian SA, Leibovich BC, et al. Evaluation of beta-blockers and survival among hypertensive patients with renal cell carcinoma. Urol Oncol (2017) 35:36.e31–6. doi:10.1016/j.urolonc.2016.08.013
186. Yang P, Deng W, Han Y, Shi Y, Xu T, Shi J, et al. Analysis of the correlation among hypertension, the intake of beta-blockers, and overall survival outcome in patients undergoing chemoradiotherapy with inoperable stage III non-small cell lung cancer. Am J Cancer Res (2017) 7:946–54.
187. Kokolus KM, Zhang Y, Sivik JM, Schmeck C, Zhu J, Repasky EA, et al. Beta blocker use correlates with better overall survival in metastatic melanoma patients and improves the efficacy of immunotherapies in mice. Oncoimmunology (2017) 7(3). doi:10.1080/2162402X.2017.1405205
188. Grytli HH, Fagerland MW, Fossa SD, Tasken KA, Haheim LL. Use of beta-blockers is associated with prostate cancer-specific survival in prostate cancer patients on androgen deprivation therapy. Prostate (2013) 73:250–60. doi:10.1002/pros.22564
189. Grytli HH, Fagerland MW, Fossa SD, Tasken KA. Association between use of beta-blockers and prostate cancer-specific survival: a cohort study of 3561 prostate cancer patients with high-risk or metastatic disease. Eur Urol (2014) 65:635–41. doi:10.1016/j.eururo.2013.01.007
190. Kaapu KJ, Ahti J, Tammela TL, Auvinen A, Murtola TJ. Sotalol, but not digoxin is associated with decreased prostate cancer risk: a population-based case-control study. Int J Cancer (2015) 137:1187–95. doi:10.1002/ijc.29470
191. De Giorgi V, Grazzini M, Gandini S, Benemei S, Lotti T, Marchionni N, et al. Treatment with beta-blockers and reduced disease progression in patients with thick melanoma. Arch Intern Med (2011) 171:779–81. doi:10.1001/archinternmed.2011.131
192. Lemeshow S, Sorensen HT, Phillips G, Yang EV, Antonsen S, Riis AH, et al. beta-Blockers and survival among Danish patients with malignant melanoma: a population-based cohort study. Cancer Epidemiol Biomarkers Prev (2011) 20:2273–9. doi:10.1158/1055-9965.EPI-11-0249
193. Diaz ES, Karlan BY, Li AJ. Impact of beta blockers on epithelial ovarian cancer survival. Gynecol Oncol (2012) 127:375–8. doi:10.1016/j.ygyno.2012.07.102
194. Wang HM, Liao ZX, Komaki R, Welsh JW, O’Reilly MS, Chang JY, et al. Improved survival outcomes with the incidental use of beta-blockers among patients with non-small-cell lung cancer treated with definitive radiation therapy. Ann Oncol (2013) 24:1312–9. doi:10.1093/annonc/mds616
195. Botteri E, Munzone E, Rotmensz N, Cipolla C, De Giorgi V, Santillo B, et al. Therapeutic effect of beta-blockers in triple-negative breast cancer postmenopausal women. Breast Cancer Res Treat (2013) 140:567–75. doi:10.1007/s10549-013-2654-3
196. Melhem-Bertrandt A, Chavez-Macgregor M, Lei X, Brown EN, Lee RT, Meric-Bernstam F, et al. Beta-blocker use is associated with improved relapse-free survival in patients with triple-negative breast cancer. J Clin Oncol (2011) 29:2645–52. doi:10.1200/JCO.2010.33.4441
197. Powe D, Voss M, Zänker K, Habashy H, Green A, Ellis I, et al. Beta-blocker drug therapy reduces secondary cancer formation in breast cancer and improves cancer specific survival. Oncotarget (2010) 1:628–38. doi:10.18632/oncotarget.101009
198. Jansen L, Hoffmeister M, Arndt V, Chang-Claude J, Brenner H. Stage-specific associations between beta blocker use and prognosis after colorectal cancer. Cancer (2014) 120:1178–86. doi:10.1002/cncr.28546
199. Monami M, Filippi L, Ungar A, Sgrilli F, Antenore A, Dicembrini I, et al. Further data on beta-blockers and cancer risk: observational study and meta-analysis of randomized clinical trials. Curr Med Res Opin (2013) 29:369–78. doi:10.1185/03007995.2013.772505
200. Lin CS, Lin WS, Lin CL, Kao CH. Carvedilol use is associated with reduced cancer risk: a nationwide population-based cohort study. Int J Cardiol (2015) 184:9–13. doi:10.1016/j.ijcard.2015.02.015
201. Choy C, Raytis JL, Smith DD, Duenas M, Neman J, Jandial R, et al. Inhibition of beta2-adrenergic receptor reduces triple-negative breast cancer brain metastases: the potential benefit of perioperative beta-blockade. Oncol Rep (2016) 35:3135–42. doi:10.3892/or.2016.4710
202. Barron TI, Connolly RM, Sharp L, Bennett K, Visvanathan K. Beta blockers and breast cancer mortality: a population-based study. J Clin Oncol (2011) 29:2635–44. doi:10.1200/JCO.2010.33.5422
203. Nkontchou G, Aout M, Mahmoudi A, Roulot D, Bourcier V, Grando-Lemaire V, et al. Effect of long-term propranolol treatment on hepatocellular carcinoma incidence in patients with HCV-associated cirrhosis. Cancer Prev Res (Phila) (2012) 5:1007–14. doi:10.1158/1940-6207.CAPR-11-0450
204. Chang PY, Huang WY, Lin CL, Huang TC, Wu YY, Chen JH, et al. Propranolol reduces cancer risk: a population-based cohort study. Medicine (Baltimore) (2015) 94:e1097. doi:10.1097/MD.0000000000001097
205. De Giorgi V, Grazzini M, Benemei S, Marchionni N, Botteri E, Pennacchioli E, et al. Propranolol for off-label treatment of patients with melanoma: results from a cohort study. JAMA Oncol (2017). doi:10.1001/jamaoncol.2017.2908
206. Pantziarka P, Bouche G, Sukhatme V, Meheus L, Rooman I, Sukhatme VP. Repurposing drugs in oncology (ReDO)-propranolol as an anti-cancer agent. Ecancermedicalscience (2016) 10:680. doi:10.3332/ecancer.2016.680
207. Albinana V, Escribano RMJ, Soler I, Padial LR, Recio-Poveda L, Villar Gomez de Las Heras K, et al. Repurposing propranolol as a drug for the treatment of retinal haemangioblastomas in von Hippel-Lindau disease. Orphanet J Rare Dis (2017) 12:122. doi:10.1186/s13023-017-0664-7
208. Leaute-Labreze C, Boccara O, Degrugillier-Chopinet C, Mazereeuw-Hautier J, Prey S, Lebbe G, et al. Safety of oral propranolol for the treatment of infantile hemangioma: a systematic review. Pediatrics (2016) 138. doi:10.1542/peds.2016-0353
Keywords: adrenergic, norepinephrine, antitumor immune response, temperature, stress, β-blocker
Citation: Qiao G, Chen M, Bucsek MJ, Repasky EA and Hylander BL (2018) Adrenergic Signaling: A Targetable Checkpoint Limiting Development of the Antitumor Immune Response. Front. Immunol. 9:164. doi: 10.3389/fimmu.2018.00164
Received: 12 October 2017; Accepted: 18 January 2018;
Published: 06 February 2018
Edited by:
Willem Van Eden, Utrecht University, NetherlandsReviewed by:
Grzegorz Sowa, University of Missouri, United StatesJo A. Van Ginderachter, Vrije Universiteit Brussel, Belgium
Copyright: © 2018 Qiao, Chen, Bucsek, Repasky and Hylander. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bonnie L. Hylander, bonnie.hylander@roswellpark.org