 Wenli Huang
Wenli Huang Huiyu Feng2
Huiyu Feng2 Chuan Xiong
Chuan Xiong Qiang Li
Qiang Li- 1Biotechnology and Nuclear Technology Research Institute, Sichuan Academy of Agricultural Sciences, Chengdu, China
- 2Key Laboratory of Coarse Cereal Processing, Ministry of Agriculture and Rural Affairs, School of Food and Biological Engineering, Chengdu University, Chengdu, China
- 3College of Life Sciences, Henan Agricultural University, Zhengzhou, China
The genus of Tricholoma is a group of important ectomycorrhizal fungi. The overlapping of morphological characteristics often leads to the confusion of Tricholoma species classification. In this study, the mitogenomes of five Tricholoma species were sequenced based on the next-generation sequencing technology, including T. matsutake SCYJ1, T. bakamatsutake, T. terreum, T. flavovirens, and T. saponaceum. These five mitogenomes were all composed of circular DNA molecules, with sizes ranging from 49,480 to 103,090 bp. Intergenic sequences were considered to be the main factor contributing to size variations of Tricholoma mitogenomes. Comparative mitogenomic analysis showed that the introns of the Agaricales mitogenome experienced frequent loss/gain events. In addition, potential gene transfer was detected between the mitochondrial and nuclear genomes of the five species of Tricholoma. Evolutionary analysis showed that the rps3 gene of the Tricholoma species was under positive selection or relaxed selection in the evolutionary process. In addition, large-scale gene rearrangements were detected between some Tricholoma species. Phylogenetic analysis using the Bayesian inference and maximum likelihood methods based on a combined mitochondrial gene set yielded identical and well-supported tree topologies. This study promoted the understanding of the genetics, evolution, and phylogeny of the Tricholoma genus and related species.
Introduction
As the “second genome” of eukaryotes, the mitochondrial genome plays an important role in eukaryotic growth and development, stress resistance, energy metabolism, aging, and death (Lang et al., 1999; Latorre-Pellicer et al., 2016). Mutations in the mitochondrial genome have been reported to affect disease development in animals (Gorman et al., 2015). The variation of the genome size, structure, gene content, gene arrangement, repeat sequences, and introns of the mitochondrial genome provides abundant information about the evolution and phylogeny of animals and fungi (Basse, 2010; Cameron, 2014; Li et al., 2020a). Fungi are one of the most diverse eukaryotic kingdoms on Earth (Hibbett et al., 2007). It is estimated that there are over 1.5 million fungal species. However, the mitochondrial genome of fungi is less understood than that of animals, especially Basidiomycetes. As of December 24, 2019, only 654 fungal mitochondrial genomes had been deposited in the National Center for Biotechnology Information (NCBI) database, of which <120 belong to Basidiomycetes. This shows that the mitochondrial genome of fungi is far from being fully understood. According to limited reports, the mitochondrial genome of fungi varies greatly in genome size, structure, gene arrangement, and intron classes, even between closely related species (Barr et al., 2005; Aguileta et al., 2014; Deng et al., 2018; Li et al., 2020b). Highly complex structures, abundant repetitive sequences, and large variations in gene content make it difficult to obtain complete fungal mitochondrial genome sequences, which limits our understanding of the fungal mitochondrial genome (Sandor et al., 2018; Liu et al., 2019; Wang et al., 2020a).
The genus Tricholoma is a group of important ectomycorrhizal fungi, which forms ectomycorrhizal relationships with trees from Fagaceae and Pinaceae (Li et al., 2018a). Through the ectomycorrhiza, plants have access to water and mineral elements and enhance their resistance to pathogens. In return, ectomycorrhizal fungi obtain a carbon source from host plants necessary for growth and fruiting (Vaario et al., 2019). The formation of this symbiotic relationship plays an important role in maintaining the forest ecosystem and promoting the carbon and nitrogen cycles in nature. It is reported that some nuclear genes of fungi play an important role in the adaptation to this symbiotic relationship, such as carbohydrate-degrading enzyme genes and transcription factors (Martin et al., 2008), while the mitochondrial genomes of ectomycorrhizal fungi are less understood.
Some species from the Tricholoma genus are valuable edible fungi, such as the pine mushroom T. matsutake, which are popular in Asia (Li et al., 2016a,b; Heilmann-Clausen et al., 2017). However, some other species from the Tricholoma genus, such as T. bakamatsutake and T. sinoacerbum, are not suitable for eating because of their pungent taste (Hosen et al., 2016). However, the two species have similar morphological characteristics, resulting in possible consumption by consumers (White et al., 2019). It is difficult to classify and identify Tricholoma species precisely because of their limited morphological features and the overlap of some morphological characteristics (Reschke et al., 2018; Endo et al., 2019). The mitochondrial genome is a powerful tool to study the phylogenetic relationships of species, and it has been widely used in the taxonomic study of animals (Boore, 1999). However, no mitochondrial genome has been used to study the phylogenetic relationships of the Tricholoma species.
In this study, the mitochondrial genomes of five species of Tricholoma were sequenced and assembled, including T. matsutake SCYJ1, T. bakamatsutake, T. terreum, T. flavovirens, and T. saponaceum. Comparing them with two published T. matsutake strains from Korea (Yoon et al., 2016) and Japan (LC385608), we revealed the features of Tricholoma mitogenomes and the variations or similarities in genome size, gene content, gene arrangement, and repeat sequences within and among Tricholoma species. The dynamic changes of introns and gene rearrangements in Tricholoma mitogenomes and other Agaricales mitogenomes were also revealed. In addition, the phylogenetic relationships of Basidiomycetes were analyzed based on a combined mitochondrial gene set. The mitogenomes of the five species of Tricholoma improve our understanding of the evolution, taxonomy, and genetics of this important ectomycorrhizal genus.
Materials and Methods
De novo Assembly and Annotation of Mitogenomes
The fruiting bodies of five Tricholoma species were collected from Sichuan, Yunnan, and Jilin provinces. The fruiting bodies were identified according to morphology and rRNA ITS sequences. The genomic DNA of the five Tricholoma species was extracted by a fungal DNA extraction kit (Omega Bio-Tek, Norcross, GA, USA) for sequencing library construction. We constructed sequencing libraries with the genomic DNA according to the instructions of NEBNext® Ultra™ II DNA Library Prep Kit (NEB, Beijing, China). Whole genomic sequencing was performed by the Illumina HiSeq 2500 Platform (Illumina, San Diego, CA, USA). The raw data obtained were first passed through a series of quality control steps, which included removing adapter reads using AdapterRemoval v2 (Schubert et al., 2016) and filtering low-quality sequences using ngsShoRT (Chen et al., 2014) with default parameters. The five mitogenomes were de novo assembled with the obtained clean data using the SPAdes 3.9 (Bankevich et al., 2012) with the k-mer of 17. Gaps among contigs were filled using the software MITObim V1.9 (Hahn et al., 2013). The MFannot tool (Valach et al., 2014) and MITOS (Bernt et al., 2013) were used to annotate the complete mitogenomes of the five Tricholoma species according to our previous described methods (Li et al., 2019a,b; Li et al., 2020c). OGDRAW (Lohse et al., 2007) was used to map the five mitogenomes of Tricholoma.
Sequence Analysis
Base compositions of the five Tricholoma mitogenomes were analyzed using the DNASTAR Lasergene v7.1 (http://www.dnastar.com/). We assessed the strand asymmetry of the five mitogenomes according to the following formulas: AT skew = [A - T] / [A + T] and GC skew = [G - C] / [G + C] (Wang et al., 2017). MEGA v6.06 (Caspermeyer, 2016) was used to calculate genetic distances between each pair of the 15 core protein coding genes (PCGs), including atp6, atp8, atp9, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, and rps3, using the Kimura-2-parameter (K2P) model. DnaSP v6 (Rozas et al., 2017) was used to calculate the nonsynonymous substitution rate (Ka) and the synonymous substitution rate (Ks) for all of the 15 core PCGs in the five Tricholoma mitogenomes. We conducted the codon usage analysis using the Sequence Manipulation Suite (Stothard, 2000), based on the genetic code 4.
Repetitive Elements Analysis
We conducted BLASTn searches of the five mitogenomes against themselves at an E value of <10−10 to determine whether there are intra-genomic duplications of large fragments and interspersed repeats in the five Tricholoma mitogenomes. Tandem Repeats Finder (Benson, 1999) was used to detect tandem repeats (>10 bp in length) in the five mitogenomes. Repeated sequences were also searched by REPuter (Kurtz et al., 2001) to identify forward (direct), reverse, complemented, and palindromic (reverse complemented) repeats in the five mitogenomes. We performed BLASTn searches of the five mitogenomes against their previously published nuclear genomes (Li et al., 2018b) to identify any gene segments that may have transferred between the mitochondrial and nuclear genomes of the five species (acc. Tmat, QMFF00000000.1; Tbak, QLOL00000000.1; Tter, QFEU00000000.1; Tfla, QLOK00000000.1; Tsap, QLOJ00000000.1).
Intron Analysis
Introns of the core PCGs in the five Tricholoma mitogenomes and other Agaricales mitogenomes were classified into different position classes (Pcls) using the Ganoderma calidophilum mitogenome (Li et al., 2019c) as the reference according to the method described by Ferandon et al. (2013). The host genes of introns were aligned with G. calidophilum by Clustal W (Larkin et al., 2007). Each Pcl was constituted by introns inserted at the same position in the coding region of the PCGs. Introns belonging to the same Pcls usually contain high sequence similarities and are considered orthologous (Ferandon et al., 2010). Different Pcls usually show low sequence similarities and contain non-orthologous mobile genetic elements. The Pcls of core PCGs in Agaricales were named by number according to the insert position in the coding region of the host gene. The phylogenetic relationships of 27 Agaricales species were inferred based on the following phylogenetic methods.
Phylogenetic Analysis
To investigate the phylogenetic status of the five Tricholoma species among the Basidiomycota phylum, we constructed a phylogenetic tree of 67 Basidiomycota species based on the combined mitochondrial gene set, which included 14 core PCGs. We first aligned single mitochondrial genes using MAFFT v7.037 (Katoh et al., 2019) and concatenated these alignments to a gene set using the SequenceMatrix v1.7.8 (Vaidya et al., 2011). Best-fit models of evolution and partitioning schemes for the gene set were determined according to PartitionFinder 2.1.1 (Lanfear et al., 2017). We used MrBayes v3.2.6 (Ronquist et al., 2012) to analyze the phylogenetic relationships of Basidiomycetes using a Bayesian inference (BI) method based on the combined gene set. Two independent runs with four chains (three heated and one cold) each were conducted simultaneously for 2 × 106 generations. Each run was sampled every 100 generations. We assumed that stationarity had been reached when the estimated sample size (ESS) was greater than 100, and the potential scale reduction factor (PSRF) approached 1.0 (the closer the PSRF value is to 1, the better the convergence effect is). The first 25% samples were discarded as burn-in, and the remaining trees were used to calculate Bayesian posterior probabilities (BPP) in a 50% majority-rule consensus tree (Li et al., 2018c). The maximum likelihood (ML) method was also used to assess the phylogenetic relationships of 67 Basidiomycetes using RAxML v8.0.0 (Stamatakis, 2014) with the combined gene set. We assessed bootstrap values (BS) through an ultrafast bootstrap approach, with 10,000 replicates.
Availability of Data
The five Tricholoma mitogenomes, including T. matsutake SCYJ1, T. bakamatsutake, T. terreum, T. flavovirens, and T. saponaceum, were submitted to GenBank under accession numbers MN873034, MN873035, MN873036, MN873037, and MN873038, respectively.
Results
Features of the Five Tricholoma Mitogenomes
The complete mitogenomes of the five Tricholoma species tested were all composed of circular DNA molecules, with sizes ranging from 49,480 to 103,090 bp (Figure 1). T. bakamatsutake contained the largest mitogenome among the five Tricholoma species, followed by T. saponaceum, T. terreum, and T. matsutake. The mitogenome of T. flavovirens was the smallest among the five Tricholoma species. The GC content of mitogenomes in the five Tricholoma species ranged from 20.57 to 23.03% (Supplementary Table 1). The GC content of the T. saponaceum mitogenome was the highest, while that of T. matsutake mitogenome was the lowest. The AT skews in mitogenomes of T. terreum, T. matsutake, and T. bakamatsutake were positive, while those in T. flavovirens and T. saponaceum were negative. GC skews of all five Tricholoma mitogenomes were positive. There were 19–42 non-intronic open-reading frames (ORFs) detected in the five Tricholoma species. Most Tricholoma species contained 14 core PCGs, except T. bakamatsutake, which did not contain the nad1 gene. Non-conserved PCGs in the Tricholoma species mainly encoded DNA polymerase and proteins with unknown functions (Supplementary Table 2). A total of 57 introns were detected in the mitogenomes of five Tricholoma species, 80.70% of which contained intronic ORFs, which encoded LAGLIDADG homing endonuclease, GIY-YIG homing endonuclease, and putative reverse transcriptase. These introns were distributed in cob, cox1, cox2, cox3, nad1, nad4, and nad5 genes. Most of these introns belonged to group I, and only two introns belonged to group II. All five Tricholoma mitogenomes contained two rRNA genes, namely the small subunit ribosomal RNA (rns) and the large subunit ribosomal RNA (rnl). The tRNA genes in the five Tricholoma mitogenomes ranged from 19 to 29.
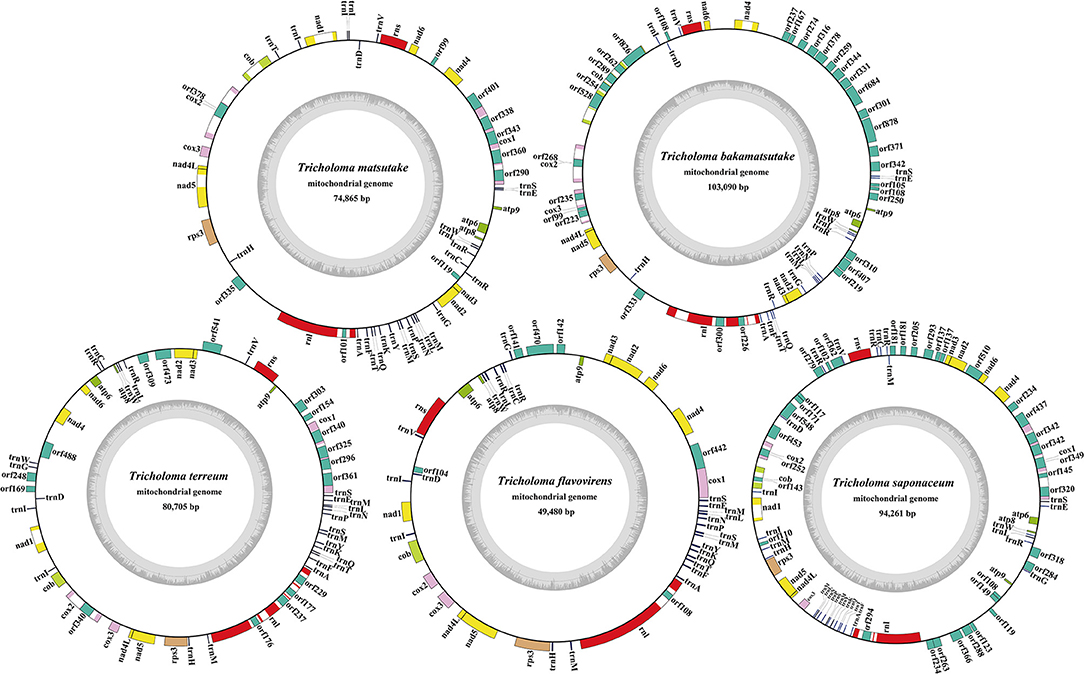
Figure 1. Circular maps of the mitochondrial genomes of five Tricholoma species. Genes are represented by different-colored blocks. Colored blocks outside each ring indicate that the genes are on the direct strand, while colored blocks within the ring indicate that the genes are located on the reverse strand.
Overlapping Genes and Composition of Mitogenomes
We detected two overlapping genes in the mitogenomes of T. matsutake, T. bakamatsutake, T. terreum, and T. flavovirens (Supplementary Table 2). The four mitogenomes all contained a set of overlapping genes located across the neighboring genes nad4L and nad5 (-1 bp). Four sets of overlapping genes were detected in the mitogenome of T. saponaceum, and the largest set of overlapping genes was located between orf234 and orf263 (-43 bp). A total of 34,053 bp, 35,257 bp, 34,505 bp, 20,344 bp, and 37,638 bp of intergenic sequences were detected in the mitogenome of T. matsutake SCYJ1, T. bakamatsutake, T. terreum, T. flavovirens, and T. saponaceum, respectively. The length of intergenic sequences ranged from 19 to 3,660 bp, and the longest intergenic sequence was located between orf335 and rnl in the T. matsutake SCYJ1 mitogenome.
Among the five Tricholoma mitogenomes we tested, the protein-coding region accounts for the largest proportion, accounting for 40.21% of the complete mitogenomes on average, followed by the intergenic region, accounting for 28.68% of the five mitogenomes on average (Figure 2). Introns accounted for an average of 19.52% of the five Tricholoma mitogenomes. The proportion of the RNA region was the smallest, only accounting for 11.59% of the five mitogenomes. In the mitogenome of T. bakamatsutake, containing the largest mitogenome in the five Tricholoma species, introns accounted for the largest proportion (36.97%) of the entire mitogenome. T. flavovirens, which had the smallest mitogenome, contained only 2.35% of the intronic regions. Pearson correlation analysis indicated intergenic region was closely related to the size variation of mitogenome in Tricholoma (P < 0.05).

Figure 2. The protein-coding, intronic, intergenic, and RNA gene region proportions of the entire mitochondrial genomes of the five Tricholoma species (A) and pearson correlation analysis between mitogenome sizes and sequence lengths of 5 Tricholoma species (B).
Codon Usage Analysis
Most of the core PCGs in the five Tricholoma species used ATG as a start codon, while only the cox1 genes of T. matsutake SCYJ1, T. bakamatsutake and T. flavovirens used GTG as a start codon (Supplementary Table 3). The nad1, nad5, and nad6 genes of T. terreum used TAG as a stop codon, and TAG was also used as a stop codon of nad2 in T. matsutake SCYJ1, nad5 in T. flavovirens, and nad6 in T. saponaceum. Other Tricholoma core PCGs used TAA as stop codons.
Codon usage analysis indicated that the most frequently used codons in the five mitogenomes were AAA (for lysine; Lys), TTT (for phenylalanine; Phe), AAT (for asparagine; Asn), TTA (for leucine; Leu), ATT (for isoleucine; Ile), and TAT (for tyrosine; Tyr) (Figure 3 and Supplementary Table 4). The frequent use of A and T in codons contributed to the high AT content in the Tricholoma mitogenomes (average: 77.70%).
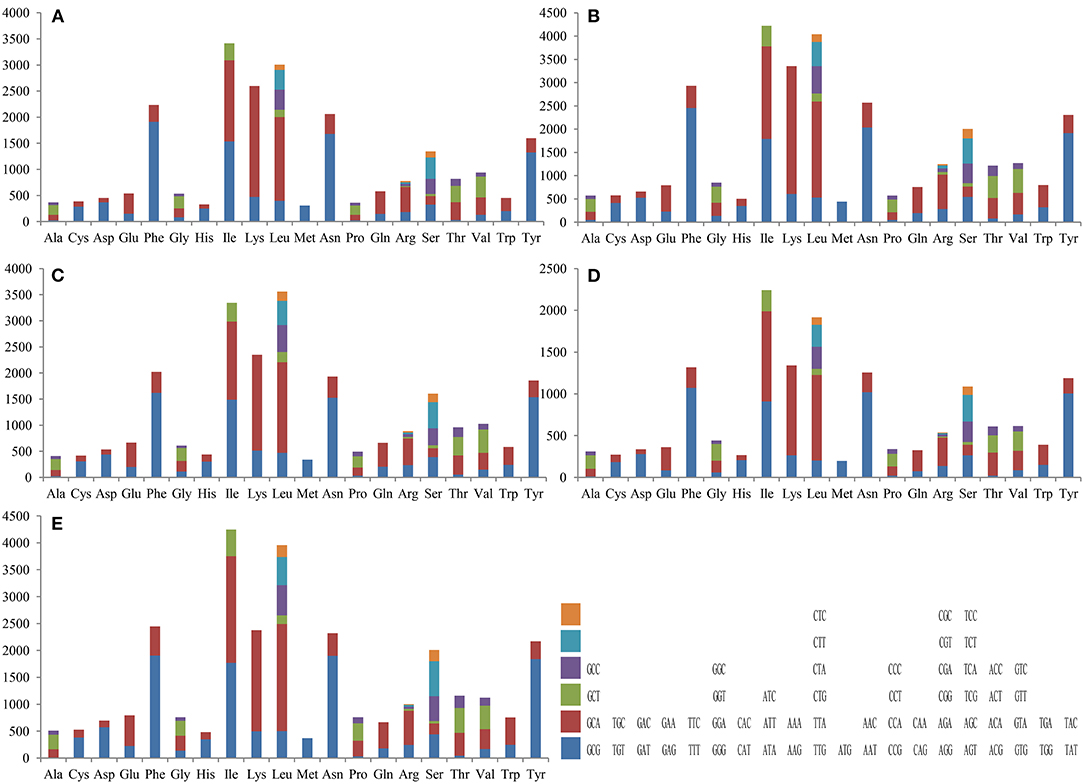
Figure 3. Codon usage in the mitochondrial genomes of five Tricholoma species. Frequency of codon usage is plotted on the y-axis: (A) T. matsutake SCYJ1; (B) T. bakamatsutake; (C) T. terreum; (D) T. flavovirens; and (E) T. saponaceum.
Repetitive Sequences Analysis
Comparing the whole mitogenomes of the five Tricholoma species with themselves via BLASTn searches, we identified nine repeat sequences in the mitogenome of T. matsutake SCYJ1, seven repeat sequences in T. bakamatsutake, 15 in T. terreum, 8 in T. flavovirens, and 31 in T. saponaceum (Supplementary Table 5). The length of repeat sequences in the five Tricholoma mitogenomes ranged from 28 to 1,139 bp, with pair-wise nucleotide similarities ranging from 76.21 to 100%. The largest repeats were detected in the protein-coding region of orf362 and intergenic region between trnV and orf362, as well as in the protein-coding region of orf366 and intergenic region between orf263 and orf366 in the T. saponaceum mitogenome. Repetitive sequences accounted for 1.10–6.86% of the whole mitogenomes of the five Tricholoma species. The mitogenome of T. saponaceum had the highest proportion of repeat sequences, followed by that of T. terreum, while T. bakamatsutake had the lowest proportion of repeat sequences.
A total of 154, 188, 67, 34, and 131 tandem repeats were detected in the mitogenomes of T. matsutake SCYJ1, T. bakamatsutake, T. terreum, T. flavovirens, and T. saponaceum, respectively (Supplementary Table 6). The longest tandem repeat sequence was observed in the mitogenome of T. matsutake SCYJ1, comprising of 247 bp. Most of the tandem repeats in the five Tricholoma mitogenomes were duplicated once or twice, with the highest replication number (115) in the T. bakamatsutake mitogenome. Tandem repeat sequences accounted for 3.20–12.78% of the five Tricholoma mitogenomes. Using REPuter, we identified 6 complemented, 17 forward, 5 palindromic, and 22 reverse repeats in the mitogenome of T. matsutake SCYJ1, accounting for 2.88% of the entire mitogenome (Supplementary Table 7). Repeats identified by REPuter accounted for 3.50, 3.44, 3.19, and 4.48% of the T. bakamatsutake, T. terreum, T. flavovirens, and T. saponaceum mitogenomes, respectively.
To detect if there were any gene segments that may had been transferred between the mitochondrial and nuclear genomes, we blasted the five mitogenomes against their nuclear genomes. A total of 75, 357, 104, 110, and 55 aligned fragments were detected in the mitogenome of T. matsutake SCYJ1, T. bakamatsutake, T. terreum, T. flavovirens, and T. saponaceum, respectively (Supplementary Table 8). The length of these aligned fragments ranged from 32 to 32,436 bp, with sequence similarities between 74.92 and 100%. The largest aligned fragment was located between the orf548 and rnl genes, which encompassed the protein-coding regions of several conserved and non-conserved genes, as well as coding regions of several tRNA genes in the T. saponaceum mitogenome. The similarity of this large alignment is 99.87% between the nuclear sequence (acc. QLOJ01012658.1) and the sequence, with 17 mismatches and one gap (Supplementary Table 8). The large aligned sequences between respective mitochondrial and nuclear genomes of the five Tricholoma species indicated that genetic transfer between the mitochondrial and nuclear genomes may have occurred during the evolution of Tricholoma species.
Variation, Genetic Distance, and Evolutionary Rates of Core Genes
Among the 15 core PCGs we detected, the length of eight core PCGs varied between different Tricholoma species, including atp6, cob, cox2, nad2, nad3, nad5, nad6, and rps3 genes (Figure 4). Among the genes with length variation, the rps3 gene had the largest length variation, and no two Tricholoma species had an rps3 gene of the same length. The GC content of atp9 was the highest, and atp8 was the lowest among the 15 core protein-coding genes. The GC content of all the core PCGs was different between different Tricholoma species, which indicated that there were frequent base variations in the core PCGs of Tricholoma species. Most core PCGs exhibited negative AT skews, except for atp9 and rps3, which exhibited positive AT skews in one or all the five mitogenomes, respectively. The GC skews of core PCGs in the five mitogenomes were variable. The atp8, nad2, nad3, nad4, and nad6 genes contained negative GC skews in the five Tricholoma species. However, GC skews in atp9, cob, cox1, cox2, cox3, nad4L, nad5, and rps3 genes of the five mitogenomes were positive.
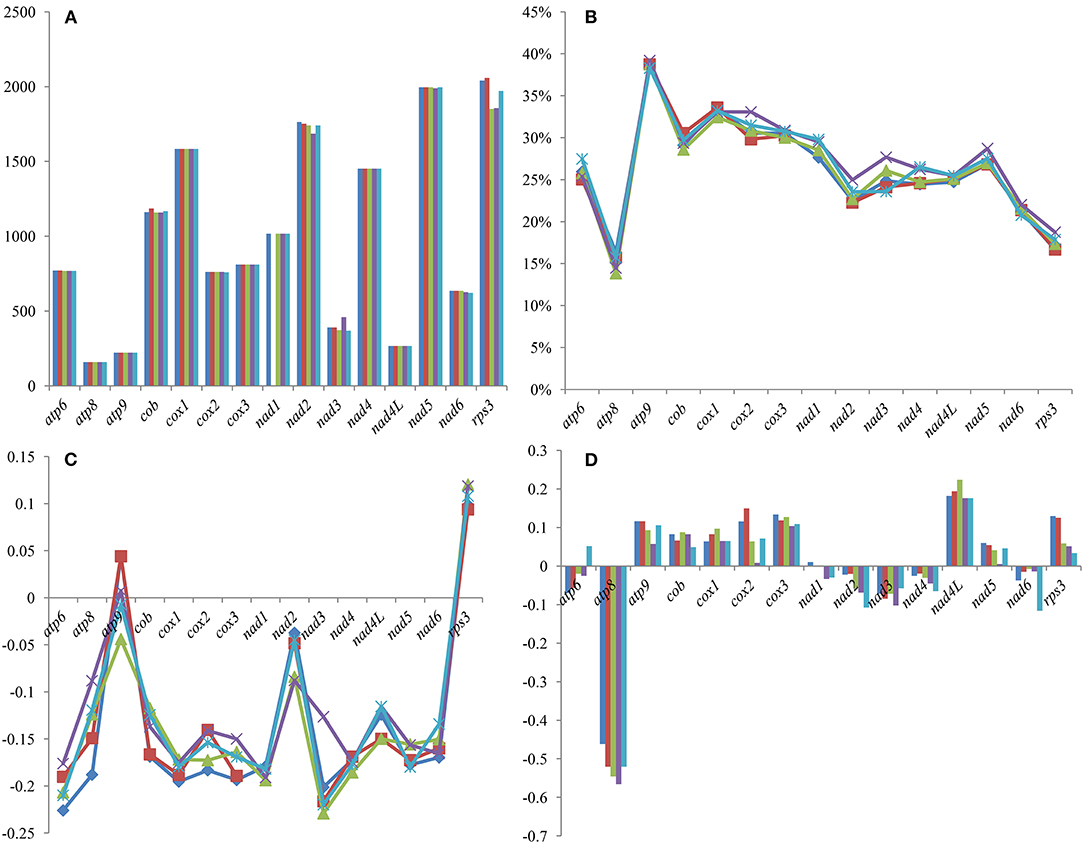
Figure 4. Variation in the length and base composition of 15 protein-coding genes (PCGs) between the five Tricholoma mitogenomes. (A) PCG length variation; (B) GC content of the PCGs; (C) AT skew; and (D) GC skew.
Among the 15 core PCGs detected, rps3 gene had the largest K2P genetic distance between the five Tricholoma species on average, followed by the nad3 gene (Figure 5). The nad4L gene had the smallest mean K2P genetic distance between the five Tricholoma species, indicating that this gene was highly conserved between Tricholoma species. The mean nonsynonymous substitution rate (Ka) of the atp9 gene was the smallest, while that of the rps3 gene was the largest in the Tricholoma species. The atp9 gene had the smallest synonymous substitution rate (Ks) and the rps3 gene had the largest Ks value among the 15 core PCGs. The Ka/Ks values of the 14 core PCGs used for energy metabolism were <1, indicating that these genes were subjected to purifying selection. However, the rps3 gene contained a Ka/Ks value greater than one, indicating that this gene was subjected to positive selection or relaxed selection in some Tricholoma species.
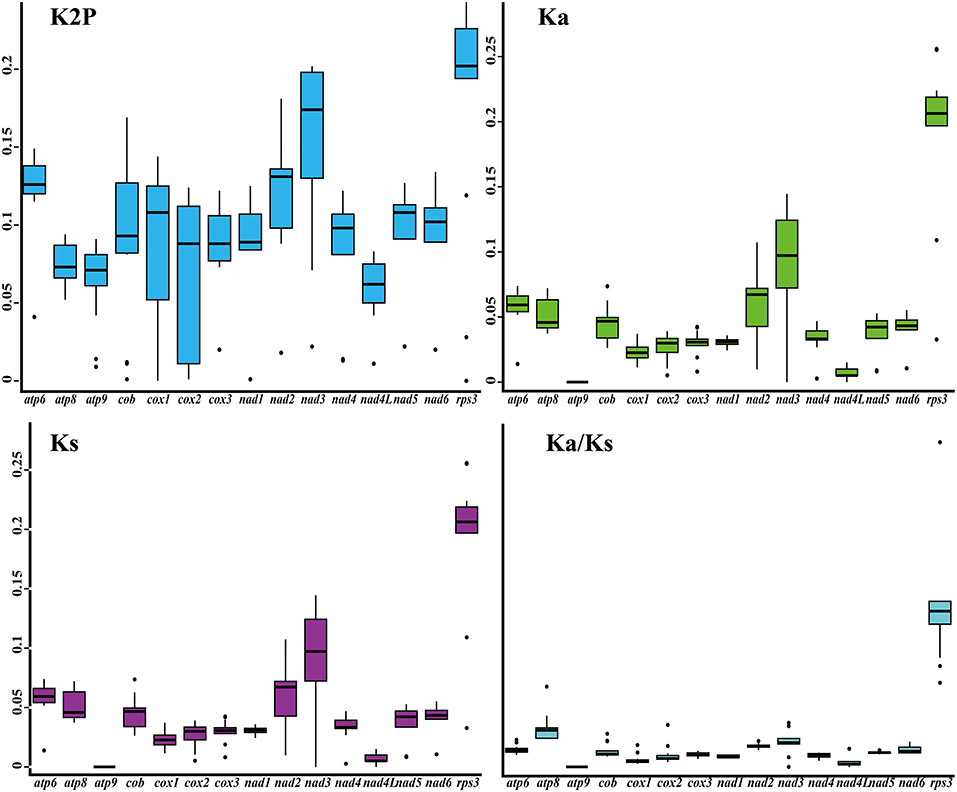
Figure 5. Genetic analysis of 15 protein-coding genes conserved in the five Tricholoma mitogenomes. K2P, the Kimura-2-parameter distance; Ka, the mean number of non-synonymous substitutions per non-synonymous site; Ks, the mean number of synonymous substitutions per synonymous site.
Intron Dynamics of PCGs in Agaricales
A total of 289 introns were detected in the core PCGs of 27 Agaricales species we tested. The number of introns in the core PCGs of each Agaricales species ranged from 0 to 41, indicating that the loss or gain of introns occurred frequently in the Agaricales species (Figure 6). Introns were found to be distributed in cob, cox1, cox2, cox3, nad1, nad2, nad4, nad4L, and nad5 genes of Agaricales, and the dynamic change of introns promoted the large variation of mitogenome size in Agaricales. The cox1 gene was the largest host gene of these introns, harboring 140 introns (48.44%), followed by the cob gene, which harbored 64 introns (22.15%). Only one intron was detected in the nad2 and nad4L genes. According to the insertion site of introns in the protein-coding region of the host gene, we divide the introns into different position classes (Pcls). The introns from the same PCG belonging to the same Pcl were considered to be orthologous introns, which had high sequence similarity and usually contain orthologous intronic ORFs (Ferandon et al., 2010). Among the 27 Agaricales species tested, 84 Pcls were detected in core PCGs, with 32 in the cox1 gene and 20 in the cob gene. Only one Pcl was detected in the nad2 and nad4L genes. Pcls P10, P22, P16, P32, and P13 were widely distributed introns in the cox1 gene, which was distributed in more than 10 out of the 27 Agaricales species. However, P2, P3, P5, P7, and P14 were only detected in one of the 27 Agaricales species, which were considered to be rare Pcls in Agaricales. These rare Pcls were also detected in distant species, such as Austropuccinia psidii (MN018834), Heterobasidion irregular (KF957635), and Paxillus rubicundulus (Li et al., 2020c), indicating that potential intron transfers might occur in the mitogenomes of Agaricales or the intron insertions were convergent in distant species. Pcl P19 was the most widely distributed Pcl in the cob gene, which was distributed in 12 of the 27 species, while Pcls P1, P4, and P5 were rare Pcls in the cob gene and only distributed in one of the 27 Agaricales species.

Figure 6. Position class (Pcl) information of core protein coding genes (PCGs) of the 27 Agaricales species. The phylogenetic positions of 27 Agaricales species were established using the Bayesian inference (BI) method and maximum likelihood (ML) method based on 15 concatenated mitochondrial core proteins. Species information is provided in Supplementary Table 9.
Among the 7 Tricholoma species tested, we found that the number and position class of introns varied significantly within or between species (Figure 6). T. bakamatsutake had the largest number of introns in the core PCGs among the seven Tricholoma species, while T. flavovirens did not contain any intron in core PCGs. Several Pcls, including P22 in the cox1 gene, P3 in the cox2 gene, and P3 in the nad1 gene, were considered widely distributed Pcls in Tricholoma species. Interestingly, within the T. matsutake species, T. matsutake SCYJ1, which was collected from the Sichuan province, China, lost the Pcl P9 of the cox1 gene compared with the T. matsutake species collected from Korea and Japan.
Gene Arrangement and Phylogenetic Analyses
The arrangement of mitochondrial genes could provide reference information for understanding the phylogenetic relationships between species (Sankoff et al., 1992). In this study, we found that the gene arrangement of the 27 Agaricales species varied significantly at the family level, indicating that large-scale gene rearrangements occurred in the evolution of Agaricales species (Figure 7). Identical gene arrangements were only observed between some species from the same genus, such as the Moniliophthora (Formighieri et al., 2008; Costa et al., 2012), Pleurotus (Li et al., 2018d), and Armillaria (Kolesnikova et al., 2019). In the genus Tricholoma, we found that the gene arrangement of the three T. matsutake species collected from different regions was identical, and large-scale gene rearrangements were detected between different species of Tricholoma, including gene transfer, insertion, deletion, and inversion events. In addition, we found that the T. bakamatsutake had a similar gene order with T. matsutake species, indicating that they were closely related, which was confirmed by phylogenetic analysis based on the combined mitochondrial gene set.
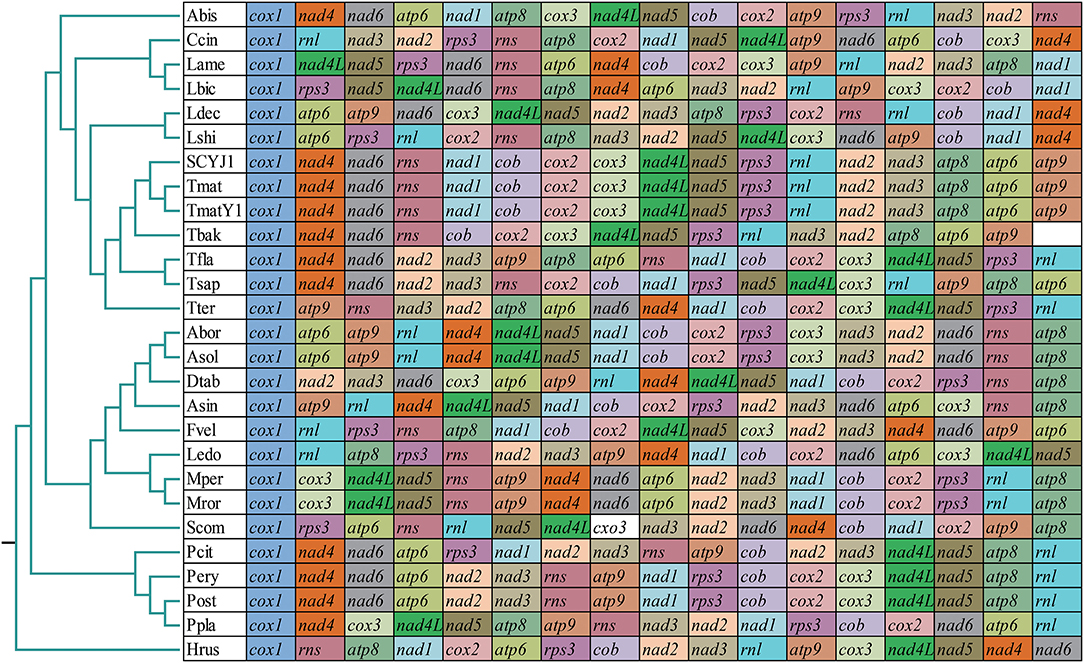
Figure 7. Gene order comparison between 27 Agaricales mitogenomes. Genes are represented with different-colored blocks. All genes are shown in order of occurrence in the mitochondrial genome, starting from cox1. Fourteen core protein-coding genes, one rps3 gene, and two rRNA genes were included in the gene arrangement analysis. Species and NCBI accession number used for gene arrangement analysis in this study are listed in Supplementary Table 9.
Phylogenetic analysis using ML and Bayesian inference (BI) methods based on the combined mitochondrial gene set (14 core PCGs) yielded identical and well-supported tree topologies (Figure 8). All major clades within the trees were well-supported (BPP ≥ 0.99; BS ≥ 98). According to the phylogenetic tree, the 67 Basidiomycota species could be divided into 12 major clades, corresponding to the orders Tremellales, Trichosporonales, Microstromatales, Ustilaginales, Tilletiales, Microbotryales, Sporidiobolales, Pucciniales, Agaricales, Russulales, Polyporales, and Cantharellales. The 27 Agaricales species could be divided into four groups, wherein the first comprised only one species forming the Hygrophorus genus (Li et al., 2019d), and the second group comprised four species within the Pleurotus genus (Li et al., 2018d). The phylogenetic analyses indicated that T. matsutake was a sister species to T. bakamatsutake, and T. flavovirens was a sister species to T. saponaceum. The analyses also indicated that the Tricholoma genus showed close relationships with the Lyophyllum genus (Li et al., 2019e).
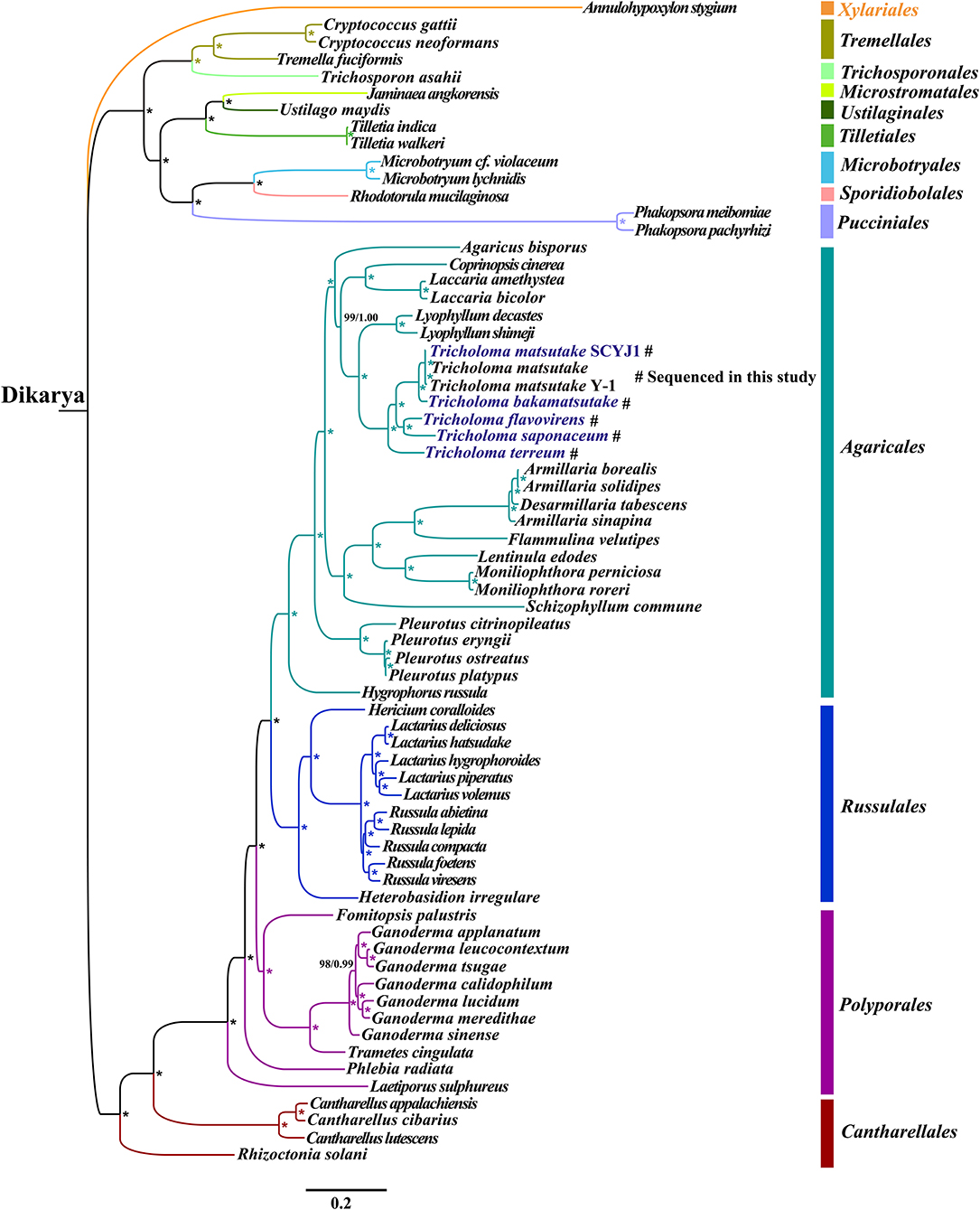
Figure 8. Molecular phylogeny of 67 Basidiomycota species based on Bayesian inference (BI) and maximum likelihood (ML) analysis of 15 protein-coding genes. Support values are Bayesian posterior probabilities (BPP; before slash) and bootstrap (BS) values. The asterisk indicates that the BS and BPP values on the evolutionary branch are 100 and 1.00, respectively. Species and NCBI accession numbers for genomes used in the phylogenetic analysis are provided in Supplementary Table 9.
Discussion
Size Variations of Mitogenomes in Tricholoma Species
The mitogenome of fungi vary greatly in size, possibly caused by the accumulation of repeat sequences, transferred genes, and intron variations (Li et al., 2018e,f). In the present study, we found that the mitogenome size of Tricholoma species also varied greatly, and the largest Tricholoma mitogenome was 2.08 greater than the smallest mitogenome. We found that T. bakamatsutake with the largest mitogenome among Tricholoma species contained 22 introns, while T. flavovirens, containing the smallest mitogenome of Tricholoma, had only one intron. The results indicated that the dynamic changes of introns were one of the main factors contributing to the size variations of the mitogenomes in Tricholoma. Intergenic regions also played an important role in the size variation of Tricholoma mitogenomes. In addition, within the T. matsutake species, we found that the length of T. matsutake collected from Sichuan was 1,224 and 1,172 bp smaller than that from Japan (LC385608) and Korea (Yoon et al., 2016), respectively. Comparative mitogenomic analysis indicated that the T. matsutake collected from Sichuan has lost intron P9 (1,290 bp) in the cox1 gene compared with that from Korea and Japan, indicating that the loss of the intron contributed to the contraction of the mitogenome in T. matsutake from Sichuan compared with that from Korea and Japan.
Mitochondrial Content Evolution of Tricholoma
The natural gene transfer between nuclear and mitochondrial genomes plays an important role in the evolution of species (Adams and Palmer, 2003; Baris et al., 2017). Some of the mitochondrial genes were transferred to the nuclear genome, while some were retained (Adams and Palmer, 2003; Allen, 2015). In the present study, we observed several aligned fragments between nuclear and mitochondrial genomes of the five Tricholoma species, respectively, indicating that gene transfer may have occurred in Tricholoma during evolution. In addition, we found that the core PCGs in Tricholoma species varied in length and base composition, and the 14 core PCGs for energy metabolism were subjected to purifying selection. Interestingly, the rps3 gene, a conserved gene in Tricholoma involved in the translation of the mRNA, demonstrated positive selection or relaxed selection between some Tricholoma species (Bullerwell et al., 2000). The rps3 gene is an ancient gene in fungi, which evolved within the endosymbiotic model and presents varied evolutionary routes (Korovesi et al., 2018). The rps3 gene was frequently observed to experience positive selection or relaxed selection in fungi (Wang et al., 2020b,c), and the selection pressure on rps3 gene needs to be further verified. Most mitochondrial genes have been transferred to the nuclear genome during the evolution of eukaryotic lineages, which is considered to have many advantages (Bjorkholm et al., 2015). However, in the present study, we found some non-conserved PCGs in the Tricholoma species, most of which had unknown functions (Allen, 2015; Bjorkholm et al., 2015). Some non-conserved PCGs in the Tricholoma species encoded DNA polymerases, which are likely to be derived from mitochondrial plasmids (Wu et al., 2021). The results indicated that there are still some unknown functional proteins in Tricholoma species to be revealed, which would promote the understanding of evolution and function of fungal mitogenomes.
Dynamics of Introns in Agaricales
The variation of introns was one of the main factors contributing to the size variation of the mitogenome in Agaricales (Hamari et al., 2002). In the present study, the characterization and dynamic changes of introns in the mitogenome of Agaricales were analyzed. Comparative intron analysis showed that the number of introns in the mitochondrial core PCGs of Agaricales varied greatly, ranging from 0 to 46, while most of these introns were located in cox1 and cob genes, which may be due to the diverse insertion sites of the two genes. Introns could be divided into different Pcls according to their precise insertion site in the protein-coding region (Ferandon et al., 2013). Introns belonging to the same Pcl were considered to be orthologous (Ferandon et al., 2010). In the present study, we found that the Pcl of Agaricales introns varied greatly. Some introns were widely distributed in Agaricales, such as the P10 and P22 in the cox1 gene and P19 in the cob gene. These introns may be inherited from the ancestors of Agaricales. However, some rare introns were only found in one of the 27 Agaricales species. Introns with the same insert sites were detected in distant species from other taxa (Himmelstrand et al., 2014), indicating that the potential transfer of introns or differential retention and loss occurred in the evolution of Agaricales. This phenomenon may also be due to the convergence of intron insertion sites in distant species. In addition, we also found the loss/gain of introns in the evolution of Tricholoma species. Compared with T. matsutake from Korea and Japan, T. matsutake from China lost the P9 intron of the cox1 gene, indicating that even within the species of T. matsutake, the intron also varied in type or quantity. The impact of this phenomenon on the ecological adaptation and species differentiation of T. matsutake needs to be further analyzed.
Gene Rearrangements and Phylogenetic Analysis
The arrangement of mitochondrial genes can provide important reference information for revealing the phylogeny and evolutionary status of eukaryotes (Sankoff et al., 1992). Reports indicated that the arrangement of mitogenome in fungi varied greatly, and the variation frequency was significantly higher than that of animals (Aguileta et al., 2014). Mitochondrial gene rearrangements in animals have been widely studied, and several models have been proposed to reveal the mechanism of mitochondrial rearrangements, including the tandem duplication-random loss (TDLR) (Xia et al., 2016) and duplication and nonrandom loss model (Lavrov et al., 2002). However, mitochondrial gene rearrangements in fungi have not been fully studied. In this study, we found that the mitochondrial gene arrangement varied greatly in Agaricales, even between closely related species. Large-scale gene rearrangements between Tricholoma species were detected, including mitochondrial gene transfer, insertion, deletion, and inversion events. The gene arrangement of the three T. matsutake species was consistent. T. bakamatsutake was found containing similar gene arrangement with T. matsutake, indicating a close phylogenetic relationship between the two species.
Mitochondrial genes have been widely used to study the phylogenetic relationships of eukaryotes, because of their single parent inheritance and rapid evolution rate (Beaudet et al., 2013; Andersen and Balding, 2018). However, due to the insufficiency of complete mitogenomes of fungi, the study on phylogenetic relationships of Basidiomycetes based on mitochondrial genomes was limited. So far, only one complete mitogenome from the Tricholoma genus has been published (Yoon et al., 2016), which limits the understanding of the evolution and phylogeny of Tricholoma species. The rapid development of the next generation sequencing technology promoted the acquisition of the mitogenome. As a supplement of the rDNA ITS, RPB2, and EF1α (Heilmann-Clausen et al., 2017; Reschke et al., 2018), mitochondrial gene molecular markers provide more abundant genetic information for reconstructing the phylogenetic tree of fungi or analyzing the evolution of fungi (Li et al., 2021). In addition, many Basidiomycete species have limited morphological features for recognition, and some morphological features are easy to overlap, leading to the confusion of Basidiomycetes classification. Phylogenetic analysis based on mitochondrial genes effectively promotes the classification and species identification of Basidiomycetes. In this study, we obtained identical and well-supported tree topologies of Basidiomycota based on the combined mitochondrial gene set using both ML and BI methods. Tricholoma species have a relatively close phylogenetic relationship with Lyophyllum species (Li et al., 2019e), which was consistent with previous phylogenetic results based on nuclear gene markers (Moncalvo et al., 2000). The results showed that the mitogenome was an effective molecular marker to analyze the phylogenetic relationships of Basidiomycetes. More Basidiomycetes mitogenomes are needed to reveal the phylogenetic relationships of all Basidiomycetes.
Conclusion
In this study, five mitogenomes from the Tricholoma genus were sequenced, assembled, and compared. Comparative mitogenomic analyses indicated that intron was one of the main factors contributing to size variations of Tricholoma mitogenomes, even within species. The introns of Agaricales mitogenomes experienced frequent loss/gain events. T. matsutake collected from Sichuan had lost one intron P9 (1,290 bp) in the cox1 gene compared with that from Korea and Japan, contributing to the contraction of the mitogenome in T. matsutake from Sichuan. In addition, large aligned fragments were detected between respective Tricholoma mitogenomes and their nuclear genomes, indicating that potential gene transfers may have occurred in the evolution of Tricholoma species. Evolutionary analysis showed that the core PCGs for energy metabolism were subject to a purifying selection, while the rps3 gene was subject to positive selection or relaxed selection. In addition, large-scale gene rearrangements were detected between some Tricholoma species and other related Agaricales species. Phylogenetic analysis using the BI and ML methods based on a combined mitochondrial gene set yielded identical and well-supported tree topologies, and the Tricholoma genus showed close relationships with the Lyophyllum genus.This study promoted the understanding of the genetics, evolution, and phylogeny of the Tricholoma genus and related species.
Data Availability Statement
Publicly available datasets were analyzed in this study. This data can be found at: The five Tricholoma mitogenomes, including T. matsutake SCYJ1, T. bakamatsutake, T. terreum, T. flavovirens, and T. saponaceum, were submitted to GenBank under accession numbers MN873034, MN873035, MN873036, MN873037, and MN873038, respectively.
Author Contributions
QL and WH: conceived and designed experiments. PL, CX, HF, WT, and XJ: performed the experiments and analyze the data. QL and XW: wrote and revise the paper. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by the outstanding youth fund of Sichuan Province (2019JDJQ0034), the Talent fund of Sichuan Academy of Agricultural Sciences (2019QYXK003), the Foundation for Young Scholars of Sichuan Academy of Agricultural Sciences (2018QNJJ-025), and the High-tech field expansion project of Sichuan Academy of Agricultural Sciences (2018GXTZ-001).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.534871/full#supplementary-material
References
Adams, K. L., and Palmer, J. D. (2003). Evolution of mitochondrial gene content: gene loss and transfer to the nucleus. Mol. Phylogenet. Evol. 29, 380–395. doi: 10.1016/S1055-7903(03)00194-5
Aguileta, G., de Vienne, D. M., Ross, O. N., Hood, M. E., Giraud, T., Petit, E., et al. (2014). High variability of mitochondrial gene order among fungi. Genome Biol. Evol. 6, 451–465. doi: 10.1093/gbe/evu028
Allen, J. F. (2015). Why chloroplasts and mitochondria retain their own genomes and genetic systems: colocation for redox regulation of gene expression. Proc. Natl. Acad. Sci. U. S. A. 112, 10231–10238. doi: 10.1073/pnas.1500012112
Andersen, M. M., and Balding, D. J. (2018). How many individuals share a mitochondrial genome? PLoS Genetics 14:e1007774. doi: 10.1371/journal.pgen.1007774
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Baris, T. Z., Wagner, D. N., Dayan, D. I., Du, X., Blier, P. U., Pichaud, N., et al. (2017). Evolved genetic and phenotypic differences due to mitochondrial-nuclear interactions. PLoS Genetics 13:e1006517. doi: 10.1371/journal.pgen.1006517
Barr, C. M., Neiman, M., and Taylor, D. R. (2005). Inheritance and recombination of mitochondrial genomes in plants, fungi and animals. New Phytol. 168, 39–50. doi: 10.1111/j.1469-8137.2005.01492.x
Basse, C. W. (2010). Mitochondrial inheritance in fungi. Curr. Opin. Microbiol. 13, 712–719. doi: 10.1016/j.mib.2010.09.003
Beaudet, D., Nadimi, M., Iffis, B., and Hijri, M. (2013). Rapid mitochondrial genome evolution through invasion of mobile elements in two closely related species of arbuscular mycorrhizal fungi. PLoS ONE 8:e60768. doi: 10.1371/journal.pone.0060768
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucl. Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bernt, M., Donath, A., Juhling, F., Externbrink, F., Florentz, C., Fritzsch, G., et al. (2013). MITOS: improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319. doi: 10.1016/j.ympev.2012.08.023
Bjorkholm, P., Harish, A., Hagstrom, E., Ernst, A. M., and Andersson, S. G. (2015). Mitochondrial genomes are retained by selective constraints on protein targeting. Proc. Natl. Acad. Sci. U. S. A. 112, 10154–10161. doi: 10.1073/pnas.1421372112
Boore, J. L. (1999). Animal mitochondrial genomes. Nucl. Acids Res. 27, 1767–1780. doi: 10.1093/nar/27.8.1767
Bullerwell, C. E., Burger, G., and Lang, B. F. (2000). A novel motif for identifying rps3 homologs in fungal mitochondrial genomes. Trends Biochem. Sci. 25, 363–365. doi: 10.1016/S0968-0004(00)01612-1
Cameron, S. L. (2014). Insect mitochondrial genomics: implications for evolution and phylogeny. Annu. Rev. Entomol. 59, 95–117. doi: 10.1146/annurev-ento-011613-162007
Caspermeyer, J. (2016). MEGA evolutionary software re-engineered to handle today's big data demands. Mol. Biol. Evol. 33:1887. doi: 10.1093/molbev/msw074
Chen, C., Khaleel, S. S., Huang, H., and Wu, C. H. (2014). Software for pre-processing Illumina next-generation sequencing short read sequences. Source Code Biol. Med. 9:8. doi: 10.1186/1751-0473-9-8
Costa, G. G., Cabrera, O. G., Tiburcio, R. A., Medrano, F. J., Carazzolle, M. F., Thomazella, D. P., et al. (2012). The mitochondrial genome of Moniliophthora roreri, the frosty pod rot pathogen of cacao. Fungal Biol. 116, 551–562. doi: 10.1016/j.funbio.2012.01.008
Deng, Y., Hsiang, T., Li, S., Lin, L., Wang, Q., Chen, Q., et al. (2018). Comparison of the mitochondrial genome sequences of six annulohypoxylon stygium isolates suggests short fragment insertions as a potential factor leading to larger genomic size. Front. Microbiol. 9:2079. doi: 10.3389/fmicb.2018.02079
Endo, N., Ushijima, S., Nagasawa, E., Sugawara, R., Okuda, Y., Sotome, K., et al. (2019). Taxonomic reconsideration of Tricholoma foliicola (Agaricales, Basidiomycota) based on basidiomata morphology, living culture characteristics, and phylogenetic analyses. Mycoscience 60, 323–330. doi: 10.1016/j.myc.2019.07.002
Ferandon, C., Moukha, S., Callac, P., Benedetto, J. P., Castroviejo, M., and Barroso, G. (2010). The Agaricus bisporus cox1 gene: the longest mitochondrial gene and the largest reservoir of mitochondrial group I introns. PLoS ONE 5:e14048. doi: 10.1371/journal.pone.0014048
Ferandon, C., Xu, J., and Barroso, G. (2013). The 135 kbp mitochondrial genome of Agaricus bisporus is the largest known eukaryotic reservoir of group I introns and plasmid-related sequences. Fungal Genet. Biol. 55, 85–91. doi: 10.1016/j.fgb.2013.01.009
Formighieri, E. F., Tiburcio, R. A., Armas, E. D., Medrano, F. J., Shimo, H., Carels, N., et al. (2008). The mitochondrial genome of the phytopathogenic basidiomycete Moniliophthora perniciosa is 109 kb in size and contains a stable integrated plasmid. Mycol. Res. 112, 1136–1152. doi: 10.1016/j.mycres.2008.04.014
Gorman, G. S., Schaefer, A. M., Ng, Y., Gomez, N., Blakely, E. L., Alston, C. L., et al. (2015). Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 77, 753–759. doi: 10.1002/ana.24362
Hahn, C., Bachmann, L., and Chevreux, B. (2013). Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads–a baiting and iterative mapping approach. Nucl. Acids Res. 41:e129. doi: 10.1093/nar/gkt371
Hamari, Z., Juhasz, A., and Kevei, F. (2002). Role of mobile introns in mitochondrial genome diversity of fungi (a mini review). Acta Microbiol. Immunol. Hung. 49, 331–335. doi: 10.1556/AMicr.49.2002.2-3.22
Heilmann-Clausen, J., Christensen, M., Froslev, T. G., and Kjoller, R. (2017). Taxonomy of Tricholoma in northern Europe based on ITS sequence data and morphological characters. Persoonia 38, 38–57. doi: 10.3767/003158517X693174
Hibbett, D. S., Binder, M., Bischoff, J. F., Blackwell, M., Cannon, P. F., Eriksson, O. E., et al. (2007). A higher-level phylogenetic classification of the Fungi. Mycol. Res. 111, 509–547. doi: 10.1016/j.mycres.2007.03.004
Himmelstrand, K., Olson, A., Brandstrom Durling, M., Karlsson, M., and Stenlid, J. (2014). Intronic and plasmid-derived regions contribute to the large mitochondrial genome sizes of Agaricomycetes. Curr. Genet. 60, 303–313. doi: 10.1007/s00294-014-0436-z
Hosen, M. I., Li, T. H., Li, T., Zhong, X. J., and Chen, Y. (2016). Tricholoma sinoacerbum, a bitter species from Guangdong Province of China. Mycoscience 57, 233–238. doi: 10.1016/j.myc.2016.02.002
Katoh, K., Rozewicki, J., and Yamada, K. D. (2019). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Kolesnikova, A. I., Putintseva, Y. A., Simonov, E. P., Biriukov, V. V., Oreshkova, N. V., Pavlov, I. N., et al. (2019). Mobile genetic elements explain size variation in the mitochondrial genomes of four closely-related Armillaria species. BMC Genom. 20:351. doi: 10.1186/s12864-019-5732-z
Korovesi, A. G., Ntertilis, M., and Kouvelis, V. N. (2018). Mt-rps3 is an ancient gene which provides insight into the evolution of fungal mitochondrial genomes. Mol. Phylogenet. Evol. 127, 74–86. doi: 10.1016/j.ympev.2018.04.037
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucl. Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T., and Calcott, B. (2017). PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34, 772–773. doi: 10.1093/molbev/msw260
Lang, B. F., Gray, M. W., and Burger, G. (1999). Mitochondrial genome evolution and the origin of eukaryotes. Annu. Rev. Genet. 33, 351–397. doi: 10.1146/annurev.genet.33.1.351
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Latorre-Pellicer, A., Moreno-Loshuertos, R., Lechuga-Vieco, A. V., Sanchez-Cabo, F., Torroja, C., Acin-Perez, R., et al. (2016). Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 535, 561–565. doi: 10.1038/nature18618
Lavrov, D. V., Boore, J. L., and Brown, W. M. (2002). Complete mtDNA sequences of two millipedes suggest a new model for mitochondrial gene rearrangements: duplication and nonrandom loss. Mol. Biol. Evol. 19, 163–169. doi: 10.1093/oxfordjournals.molbev.a004068
Li, H., Wu, S., Ma, X., Chen, W., Zhang, J., Duan, S., et al. (2018b). The genome sequences of 90 mushrooms. Sci. Rep. 8:9982. doi: 10.1038/s41598-018-28303-2
Li, Q., Chen, C., Xiong, C., Jin, X., Chen, Z., and Huang, W. (2018d). Comparative mitogenomics reveals large-scale gene rearrangements in the mitochondrial genome of two Pleurotus species. Appl. Microbiol. Biotechnol. 102, 6143–6153. doi: 10.1007/s00253-018-9082-6
Li, Q., He, X., Ren, Y., Xiong, C., Jin, X., Peng, L., et al. (2020b). Comparative mitogenome analysis reveals mitochondrial genome differentiation in ectomycorrhizal and asymbiotic Amanita species. Front. Microbiol. 11:1382. doi: 10.3389/fmicb.2020.01382
Li, Q., Li, S. H., Huang, W. L., Liu, C. Y., Xiong, C., Li, X. L., et al. (2016a). Mineral constituents of a prized edible mushroom (Tricholoma matsutake) and soils beneath the fruiting bodies from the production areas across China. J. Mountain Sci. 13, 2046–2052. doi: 10.1007/s11629-015-3568-9
Li, Q., Liao, M., Yang, M., Xiong, C., Jin, X., Chen, Z., et al. (2018f). Characterization of the mitochondrial genomes of three species in the ectomycorrhizal genus Cantharellus and phylogeny of Agaricomycetes. Int. J. Biol. Macromol. 118, 756–769. doi: 10.1016/j.ijbiomac.2018.06.129
Li, Q., Ren, Y., Shi, X., Peng, L., Zhao, J., Song, Y., et al. (2019b). Comparative mitochondrial genome analysis of two ectomycorrhizal fungi (rhizopogon) reveals dynamic changes of intron and phylogenetic relationships of the subphylum agaricomycotina. Int. J. Mol. Sci. 20:5167. doi: 10.3390/ijms20205167
Li, Q., Ren, Y., Xiang, D., Shi, X., Zhao, J., Peng, L., et al. (2020c). Comparative mitogenome analysis of two ectomycorrhizal fungi (Paxillus) reveals gene rearrangement, intron dynamics, and phylogeny of basidiomycetes. IMA Fungus 11:12. doi: 10.1186/s43008-020-00038-8
Li, Q., Wang, Q., Chen, C., Jin, X., Chen, Z., Xiong, C., et al. (2018e). Characterization and comparative mitogenomic analysis of six newly sequenced mitochondrial genomes from ectomycorrhizal fungi (Russula) and phylogenetic analysis of the Agaricomycetes. Int. J. Biol. Macromol. 119, 792–802. doi: 10.1016/j.ijbiomac.2018.07.197
Li, Q., Wang, Q., Jin, X., Chen, Z., Xiong, C., Li, P., et al. (2019a). Characterization and comparative analysis of six complete mitochondrial genomes from ectomycorrhizal fungi of the Lactarius genus and phylogenetic analysis of the Agaricomycetes. Int. J. Biol. Macromol. 121, 249–260. doi: 10.1016/j.ijbiomac.2018.10.029
Li, Q., Wang, Q., Jin, X., Chen, Z., Xiong, C., Li, P., et al. (2019e). Characterization and comparison of the mitochondrial genomes from two Lyophyllum fungal species and insights into phylogeny of Agaricomycetes. Int. J. Biol. Macromol. 121, 364–372. doi: 10.1016/j.ijbiomac.2018.10.037
Li, Q., Wang, Q. F., Jin, X., Chen, Z. Q., Xiong, C., Li, P., et al. (2019d). The first complete mitochondrial genome from the family Hygrophoraceae (Hygrophorus russula) by next-generation sequencing and phylogenetic implications. Int. J. Biol. Macromol. 122, 1313–1320. doi: 10.1016/j.ijbiomac.2018.09.091
Li, Q., Wu, P., Li, L., Feng, H., Tu, W., Bao, Z., et al. (2021). The first eleven mitochondrial genomes from the ectomycorrhizal fungal genus (Boletus) reveal intron loss and gene rearrangement. Int. J. Biol. Macromol. 172, 560–572. doi: 10.1016/j.ijbiomac.2021.01.087
Li, Q., Xiang, D., Wan, Y., Wu, Q., Wu, X., Ma, C., et al. (2019c). The complete mitochondrial genomes of five important medicinal Ganoderma species: features, evolution, and phylogeny. Int. J. Biol. Macromol. 139, 397–408. doi: 10.1016/j.ijbiomac.2019.08.003
Li, Q., Xiong, C., Li, X., Jin, X., and Huang, W. (2018a). Ectomycorrhization of Tricholoma matsutake with Quercus aquifolioides affects the endophytic microbial community of host plant. J. Basic Microbiol. 58, 238–246. doi: 10.1002/jobm.201700506
Li, Q., Yang, L., Xiang, D., Wan, Y., Wu, Q., Huang, W., et al. (2020a). The complete mitochondrial genomes of two model ectomycorrhizal fungi (Laccaria): features, intron dynamics and phylogenetic implications. Int. J. Biol. Macromol. 145, 974–984. doi: 10.1016/j.ijbiomac.2019.09.188
Li, Q., Yang, M., Chen, C., Xiong, C., Jin, X., Pu, Z., et al. (2018c). Characterization and phylogenetic analysis of the complete mitochondrial genome of the medicinal fungus Laetiporus sulphureus. Sci. Rep. 8:9104. doi: 10.1038/s41598-018-27489-9
Li, Q., Zhang, L., Li, W. H., Li, X. L., Huang, W. L., Yang, H., et al. (2016b). Chemical compositions and volatile compounds of Tricholoma matsutake from different geographical areas at different stages of maturity. Food Sci. Biotechnol. 25, 71–77. doi: 10.1007/s10068-016-0010-1
Liu, W., Cai, Y., Zhang, Q., Chen, L., Shu, F., Ma, X., et al. (2019). The mitochondrial genome of Morchella importuna (272.2 kb) is the largest among fungi and contains numerous introns, mitochondrial non-conserved open reading frames and repetitive sequences. Int. J. Biol. Macromol. 143, 373–381. doi: 10.1016/j.ijbiomac.2019.12.056
Lohse, M., Drechsel, O., and Bock, R. (2007). OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 52, 267–274. doi: 10.1007/s00294-007-0161-y
Martin, F., Aerts, A., Ahren, D., Brun, A., Danchin, E. G., Duchaussoy, F., et al. (2008). The genome of Laccaria bicolor provides insights into mycorrhizal symbiosis. Nature 452, 88–92. doi: 10.1038/nature06556
Moncalvo, J. M., Lutzoni, F. M., Rehner, S. A., Johnson, J., and Vilgalys, R. (2000). Phylogenetic relationships of agaric fungi based on nuclear large subunit ribosomal DNA sequences. Syst. Biol. 49, 278–305. doi: 10.1093/sysbio/49.2.278
Reschke, K., Popa, F., Yang, Z. L., and Kost, G. (2018). Diversity and taxonomy of Tricholoma species from Yunnan, China, and notes on species from Europe and North America. Mycologia 110, 1081–1109. doi: 10.1080/00275514.2018.1512295
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Hohna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Rozas, J., Ferrer-Mata, A., Sanchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Sandor, S., Zhang, Y., and Xu, J. (2018). Fungal mitochondrial genomes and genetic polymorphisms. Appl. Microbiol. Biotechnol. 102, 9433–9448. doi: 10.1007/s00253-018-9350-5
Sankoff, D., Leduc, G., Antoine, N., Paquin, B., Lang, B. F., and Cedergren, R. (1992). Gene order comparisons for phylogenetic inference: evolution of the mitochondrial genome. Proc. Natl. Acad. Sci. U. S. A. 89, 6575–6579. doi: 10.1073/pnas.89.14.6575
Schubert, M., Lindgreen, S., and Orlando, L. (2016). AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res. Notes 9:88. doi: 10.1186/s13104-016-1900-2
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stothard, P. (2000). The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 28:1104. doi: 10.2144/00286ir01
Vaario, L. M., Sah, S. P., Norisada, M., Narimatsu, M., and Matsushita, N. (2019). Tricholoma matsutake may take more nitrogen in the organic form than other ectomycorrhizal fungi for its sporocarp development: the isotopic evidence. Mycorrhiza 29, 51–59. doi: 10.1007/s00572-018-0870-8
Vaidya, G., Lohman, D. L., and Meier, R. (2011). SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 27, 171–180. doi: 10.1111/j.1096-0031.2010.00329.x
Valach, M., Burger, G., Gray, M. W., and Lang, B. F. (2014). Widespread occurrence of organelle genome-encoded 5S rRNAs including permuted molecules. Nucl. Acids Res. 42, 13764–13777. doi: 10.1093/nar/gku1266
Wang, J., Zhang, L., Zhang, Q. L., Zhou, M. Q., Wang, X. T., Yang, X. Z., et al. (2017). Comparative mitogenomic analysis of mirid bugs (Hemiptera: Miridae) and evaluation of potential DNA barcoding markers. PeerJ 5:e3661. doi: 10.7717/peerj.3661
Wang, X., Jia, L., Wang, M., Yang, H., Chen, M., Li, X., et al. (2020c). The complete mitochondrial genome of medicinal fungus Taiwanofungus camphoratus reveals gene rearrangements and intron dynamics of Polyporales. Sci. Rep. 10:16500. doi: 10.1038/s41598-020-73461-x
Wang, X., Song, A., Wang, F., Chen, M., Li, X., Li, Q., et al. (2020a). The 206 kbp mitochondrial genome of Phanerochaete carnosa reveals dynamics of introns, accumulation of repeat sequences and plasmid-derived genes. Int. J. Biol. Macromol. 162, 209–219. doi: 10.1016/j.ijbiomac.2020.06.142
Wang, X., Wang, Y. J., Yao, W., Shen, J. W., Chen, M. Y., Gao, M., et al. (2020b). The 256 kb mitochondrial genome of Clavaria fumosa is the largest among phylum Basidiomycota and is rich in introns and intronic ORFs. Ima Fungus 11:26. doi: 10.1186/s43008-020-00047-7
White, J., Weinstein, S. A., De Haro, L., Bedry, R., Schaper, A., Rumack, B. H., et al. (2019). Mushroom poisoning: a proposed new clinical classification. Toxicon 157, 53–65. doi: 10.1016/j.toxicon.2018.11.007
Wu, P., Bao, Z., Tu, W., Li, L., Xiong, C., Jin, X., et al. (2021). The mitogenomes of two saprophytic Boletales species (Coniophora) reveals intron dynamics and accumulation of plasmid-derived and non-conserved genes. Comput. Struct. Biotechnol. J. 19, 401–414. doi: 10.1016/j.csbj.2020.12.041
Xia, Y., Zheng, Y., Murphy, R. W., and Zeng, X. (2016). Intraspecific rearrangement of mitochondrial genome suggests the prevalence of the tandem duplication-random loss (TDLR) mechanism in Quasipaa boulengeri. BMC Genom. 17:965. doi: 10.1186/s12864-016-3309-7
Keywords: Tricholoma, mitochondrial genome, intron, gene rearrangement, evolution, phylogenetic analysis
Citation: Huang W, Feng H, Tu W, Xiong C, Jin X, Li P, Wang X and Li Q (2021) Comparative Mitogenomic Analysis Reveals Dynamics of Intron Within and Between Tricholoma Species and Phylogeny of Basidiomycota. Front. Genet. 12:534871. doi: 10.3389/fgene.2021.534871
Received: 03 June 2020; Accepted: 18 January 2021;
Published: 15 February 2021.
Edited by:
Denis Baurain, University of Liège, BelgiumReviewed by:
Baojun Wu, Clark University, United StatesGerard Barroso, Université de Bordeaux, France
Copyright © 2021 Huang, Feng, Tu, Xiong, Jin, Li, Wang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiang Li, leeq110@126.com; Xu Wang, xuwang@henau.edu.cn
†Present address: Xu Wang and Qiang Li, School of Food and Biological Engineering, Chengdu University, Chengdu, China