 Kiranmayi Vemuri
Kiranmayi Vemuri Sarah H. Radi
Sarah H. Radi Frances M. Sladek
Frances M. Sladek Michael P. Verzi1,2
Michael P. Verzi1,2- 1Department of Genetics, Human Genetics Institute of New Jersey, Rutgers, The State University of New Jersey, Piscataway, NJ, United States
- 2Cancer Institute of New Jersey, Rutgers, The State University of New Jersey, New Brunswick, NJ, United States
- 3Department of Molecular, Cell and Systems Biology, University of California, Riverside, Riverside, CA, United States
- 4Department of Biochemistry, University of California, Riverside, Riverside, CA, United States
Hepatocyte nuclear factor 4-alpha (HNF4α) drives a complex array of transcriptional programs across multiple organs. Beyond its previously documented function in the liver, HNF4α has crucial roles in the kidney, intestine, and pancreas. In the intestine, a multitude of functions have been attributed to HNF4 and its accessory transcription factors, including but not limited to, intestinal maturation, differentiation, regeneration, and stem cell renewal. Functional redundancy between HNF4α and its intestine-restricted paralog HNF4γ, and co-regulation with other transcription factors drive these functions. Dysregulated expression of HNF4 results in a wide range of disease manifestations, including the development of a chronic inflammatory state in the intestine. In this review, we focus on the multiple molecular mechanisms of HNF4 in the intestine and explore translational opportunities. We aim to introduce new perspectives in understanding intestinal genetics and the complexity of gastrointestinal disorders through the lens of HNF4 transcription factors.
Introduction
The specification of cell types and patterning of tissues depends on transcription factors which interpret and effectuate regulatory sequences of DNA. In this review, we explore the functions of a key transcription factor family in the intestine, HNF4. The intestinal epithelium is one of the most rapidly self-renewing tissues in mammals. The inner surface of the intestine is lined with a single cell layer of tightly packed, columnar epithelial cells. The epithelium is further organized into finger-like protrusions called villi which increase the surface area of absorption, and invaginations into the submucosa known as the crypts of Lieberkühn (1). The large intestine or colon has crypts but no villi (2). The crypts in both the small intestine and colon are characterized by a population of self-renewing, undifferentiated cells, which give rise to differentiated cell types: goblet cells, enteroendocrine cells, Paneth cells, tuft cells, and enterocytes (1). Goblet cells and enteroendocrine cells secrete mucus and a variety of hormones, respectively, and reside in both villi and crypts. Tuft cells are chemosensory cells which have been postulated to mediate defense against parasitic infection by releasing IL-25 (3, 4). The most abundant cell type in the villi, however, are the enterocytes, which are responsible for nutrient absorption. Each enterocyte has a tightly organized array of 1–2μm microvilli projecting from the apical cell surface that collectively forms the brush border and dramatically increases the surface area of this absorptive epithelium (5).
Meanwhile, in the crypts, Paneth cells occupy the base of the crypts and secrete antimicrobial agents such as α-defensins and lysozyme (6). They also participate in epithelial maintenance by expressing some of the canonical ligands of the Wnt and Notch signaling pathways (7). Additionally, the base of the crypts has a population of undifferentiated, multipotent stem cells marked by Lgr5. These crypt-base-columnar stem cells either renew and remain at the crypt bottom or differentiate into secretory cells or enterocytes which then migrate upward from the crypts towards the tips of the villi (8). Cells at the villus tips ultimately undergo anoikis as they are shed into the intestinal lumen (9, 10). Homeostasis of the epithelium is tightly regulated at the transcriptional level. It has been estimated using RNA-seq that nearly 4,000 genes are differentially expressed between the duodenal crypt and villus compartments (11). Several transcription factor families such as TCF/LEF, KLF, CDX (12–14), GATA (15), and HNF4 (16, 17) have been identified as important regulators involved in the control of these differentially expressed genes. In particular, HNF4 plays pivotal roles in regulating intestinal maturation, development, differentiation and architecture. We aim to provide an overview of HNF4 transcription factors, with specific focus on its role in the intestine.
HNF4-mediated regulation in the gastrointestinal mucosa
Hepatocyte Nuclear Factors (HNFs) were identified based upon their abundance in liver extracts and ability to bind regulatory elements of liver specific genes (18). However, HNF4 is not just liver specific but is broadly expressed throughout the gastrointestinal system (19, 20) (Figure 1). The HNF4α homodimer binds via its DNA binding domain to its canonical DNA recognition site, DR1 (direct repeat 1; AGGTCAxAGGTCA) (21) and recruits co-regulatory proteins which mediate the regulation of its target genes. The first two identified targets of HNF4 activity were Ttr (transerythrin) and Apoc3 (apolipoprotein C3), found using crude nuclear extracts of rat liver (18). Since then, HNF4 has been shown to perform crucial tasks in the liver, intestine, pancreas and kidney during development, differentiation, cell proliferation and for maintaining homeostasis. Tissue-restricted expression is highly conserved among species (22). The existence of two redundant paralogs of HNF4 (HNF4α and HNF4γ), along with their ability to isomerize and form different combinations of homodimers and heterodimers, adds significant complexity to the regulation of HNF4 and its target genes (23–25).
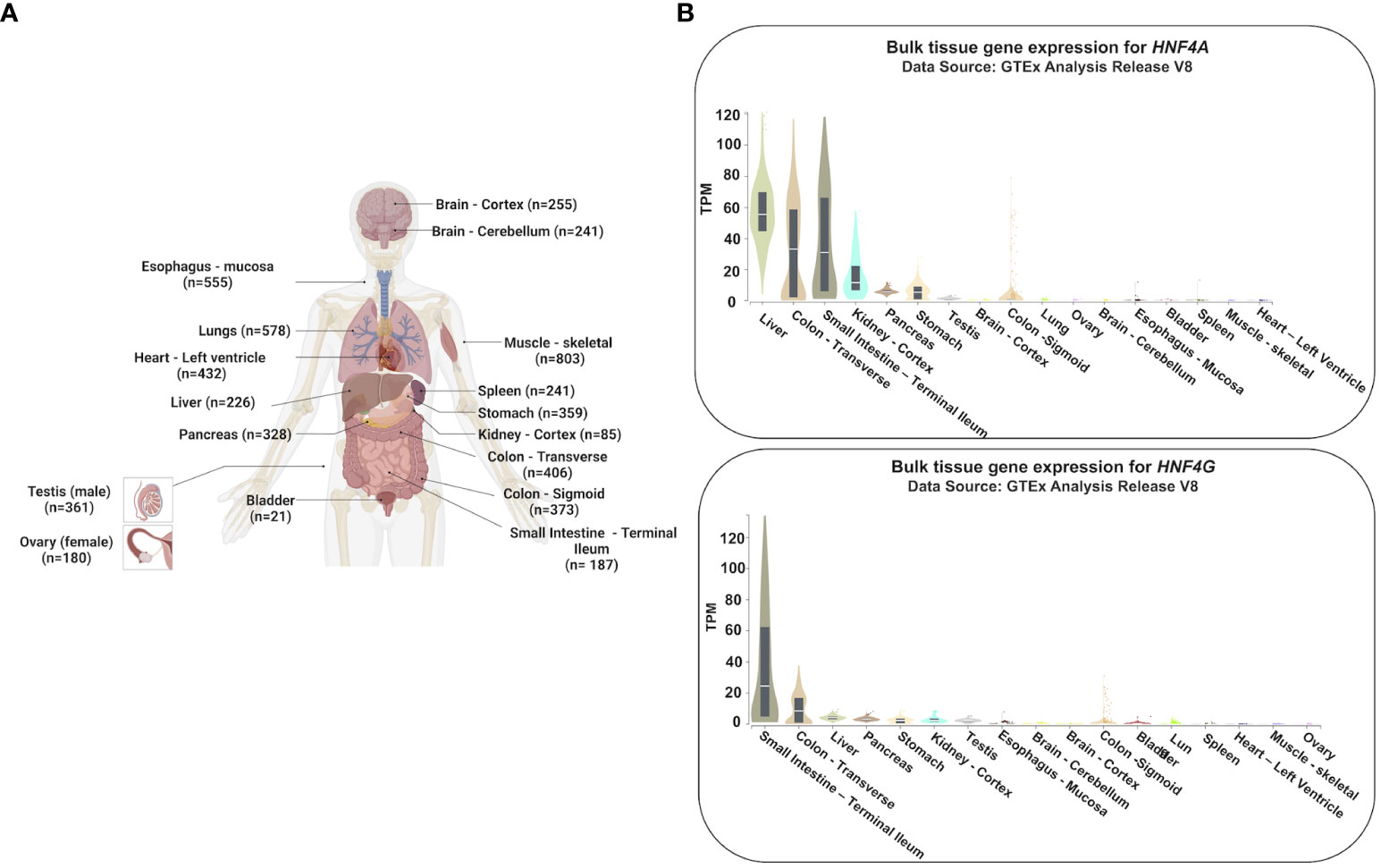
Figure 1 Tissue-specific gene expression profiles of HNF4A and HNF4G. (A) Expression profiles of HNF4A and HNF4G from the Genotype-Tissue Expression Project (GTEx) were evaluated in tissues of healthy individuals (n=number of replicates per tissue examined for gene expression) (20). (B) HNF4A is expressed across multiple tissues of the gastrointestinal tract whereas HNF4G is primarily an intestine-restricted paralog (TPM: Transcripts Per Million). The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from the GTEx Portal on 05/25/23. Adapted from “Human Internal Organs”, by BioRender.com (2023).
HNF4 activates enhancer chromatin and facilitates long-range chromatin interactions at its target genes
Transcription of genes typically requires multiple events: i) the generation of active and accessible chromatin at one or more enhancers, ii) looping of enhancers to transcriptional start sites (promoters), and iii) the recruitment and activation of the RNA polymerase holoenzyme at promoters to generate mRNA transcripts. HNF4 appears to be involved in multiple steps of transcriptional activation of its target genes. An investigation of the HNF4α protein interactome with a BioID quantitative mass spectrometry experiment showed components of the ATP-dependent chromatin remodeling complexes SWI/SNF and NuRD, to be prominent interacting partners (23). This hints at HNF4α potentially mediating a permissive chromatin landscape. In fact, in the adult intestine this was shown to be the case as HNF4 factors maintain accessible, active enhancer chromatin (17). H3K4me3-targeted HiChIP in cells isolated from duodenal crypts and villi show HNF4 regulates its target genes by facilitating enhancer-promoter chromatin looping (11). A significant decrease in chromatin looping was observed upon loss of HNF4 factors, with far more HNF4-dependent chromatin looping events observed in the villus compared to the crypts. A multi-omics analysis incorporating H3K27ac micrococcal nuclease ChIP-seq inferred that HNF4 target genes depend upon HNF4 for chromatin accessibility at distal enhancer elements and for chromatin looping between enhancers and promoters. Functional annotation of these gene sets showed genes with disrupted chromatin loops upon HNF4 depletion were associated with steroid and lipid metabolic processes whereas genes with increased looping events were associated with a stress response. However, while HNF4 is required for local chromatin interactions, this is not a global cellular event, as genes without HNF4 binding did not show an appreciable change in chromatin structure on average (11). Further downstream of enhancer chromatin activation and chromatin looping, a potentially interesting avenue of investigation is the role of HNF4 in the recruitment and regulation of RNA polymerase II. In the intestine, little is known about RNA polymerase dynamics during gene transcription, but regulation of polymerase dynamics has the potential to be a new mechanism through which HNF4-dependent differentiation can be accomplished.
HNF4 paralogs and their splice variants generate diverse expression patterns
The combination of HNF4 paralogs and their splice variants contributes to the complexity and versatility of gene regulation, allowing cells and tissues to fine-tune their gene expression profiles to meet specific requirements during development. The two mammalian paralogs of HNF4 – Hnf4a and Hnf4g are each expressed with distinct patterns across the GI system, and the gene products are further diversified through differential promoter usage and alternative splicing.
Hnf4a encodes multiple isoforms that exhibit variations in their N-termini. These isoforms are generated through alternative splicing events occurring at the 3’ end of the gene, coupled with the utilization of two distinct promoters, P1 (proximal) and P2 (distal), which are located approximately 40 kilobases (kb) apart in both human and mouse genomes (Figure 2B) (26). Notably, the epididymis and the intestine are the only adult tissues which express both P1- and P2- derived HNF4α in humans (28, 29). Studies in mouse and human colonic crypts have demonstrated that P1-driven Hnf4a isoforms are expressed more robustly in the luminal, differentiated colonic epithelium, while P2-driven isoforms are enriched deeper in the proliferative crypt epithelium, co-localizing with the proliferative marker, Ki67 (27, 29) (Figure 2A). Expression analyses of transgenic mouse models expressing either P1 or P2 isoforms (29, 30) demonstrate P1-HNF4α chiefly controls genes involved in differentiation, wound healing, and immune responses, whereas P2-HNF4α controls DNA repair and cell cycle genes (29).
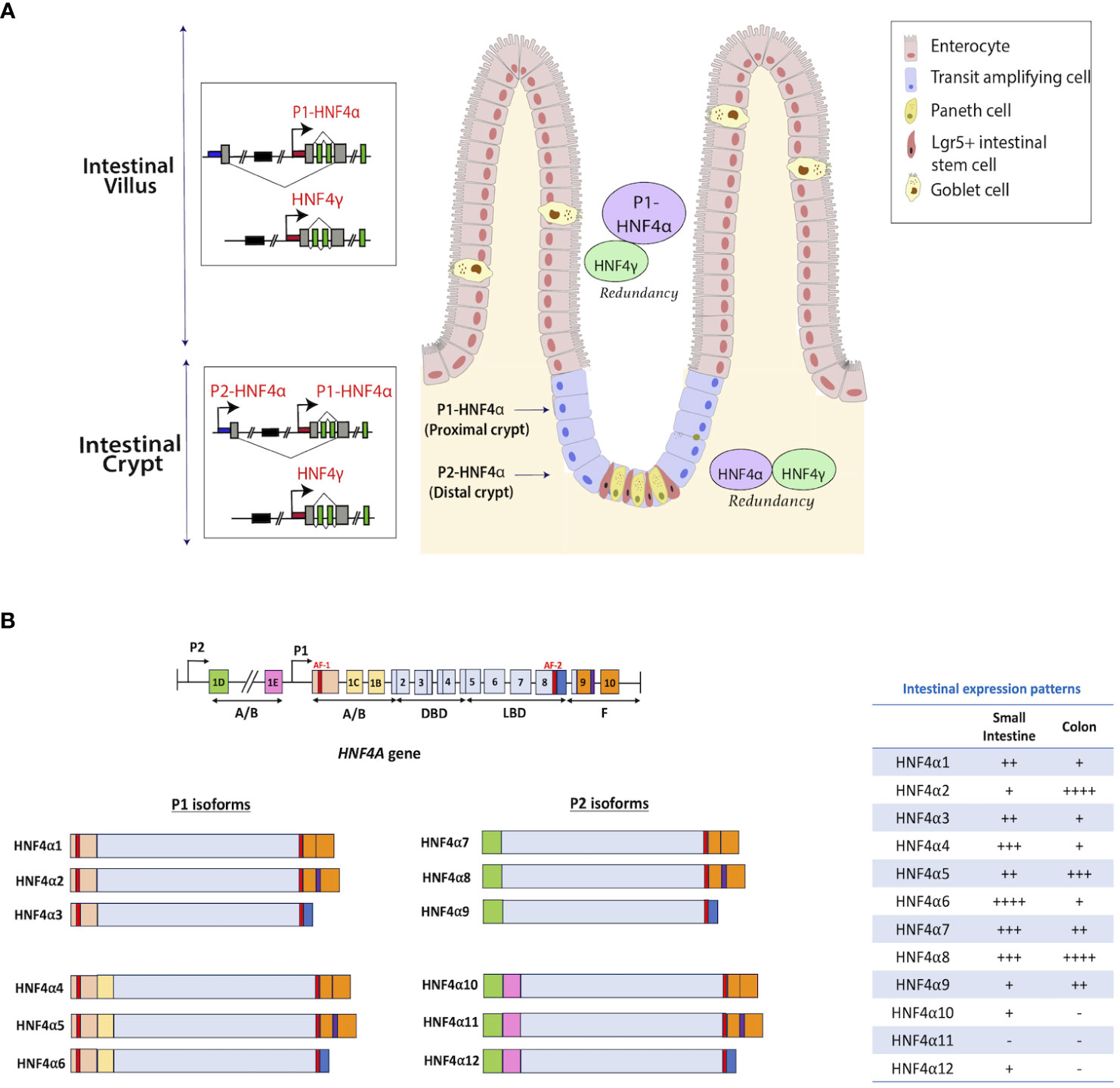
Figure 2 Distribution of HNF4 isoforms along the intestinal epithelium. (A) Isoforms of Hnf4a are generated through alternative splicing and usage of alternative promoters (proximal P1 and distal P2) that are forty kilobases (kb) apart in human and mouse (26). Enterocytes in the villi express high levels of P1 isoforms, whereas crypt stem cells express lower amounts (27). Studies in mouse and human colonic crypts show P1-driven HNF4α isoforms are expressed more robustly in the luminal, differentiated colonic epithelium, while P2-driven isoforms are enriched deeper in the proliferative crypt epithelium, co-localizing with the proliferative marker, Ki67. HNF4γ is expressed in both villus and crypt. (B) There are 12 isoforms of the HNF4A gene that are produced through the utilization of alternative promoters – P1 and P2. HNF4α1-6 are generated from the P1 promoter, while HNF4α7-12 are generated from the P2 promoter. The table shows the mRNA expression patterns of each isoform in the intestine from adult human tissue (25). Figure panel modeled off of the work of (25).
Isoforms from promoter 1 (P1) include HNF4α1- HNF4α6 and are predominantly seen in the liver and small intestine with some expression also seen in the testis and kidney. Promoter 2 (P2) driven isoforms include HNF4α7- HNF4α12 and are seen in the pancreas, bile duct, stomach and GI tract (25). In both the small and large intestine, each isoform has distinct patterns of expression (Figure 2B). In the small intestine, HNF4α4, 6, 7, and 8 are expressed at higher levels, while HNF4α11 is not detected (25). Conversely, in the colon, HNF4α2, 5, and 8 show elevated expression, whereas HNF4α10-12 isoforms are absent (Figure 2B) (25). Differences were also identified in the transcriptional activation capabilities of the 12 isoforms. In HCT116 cells, upon individual expression of each HNF4α isoform using a tetracycline-inducible system, HNF4α1 and HNF4α2 demonstrated the highest effectiveness in regulating gene expression. Conversely, the transcriptional potential of HNF4α4, HNF4α5, and HNF4α6 isoforms was found to be diminished (23). Electrophoretic mobility shift assays showed the likely reason for the reduced transcriptional activity of HNF4α4, α5, and α6 is their inability to bind to the HNF4 consensus DNA binding sequence, DR1. The same study also showed that P2- isoforms exhibit a lower transactivation capacity than HNF4α1 and α2 (23). Interactions among isoforms can generate a repertoire of dimer combinations which influence numerous cellular processes.
In mammals, HNF4γ also exhibits two splice variants, HNF4γ1 and HNF4γ2. It is unclear if these are the products of differential promoter usage or alternative splicing. Expression of Hnf4g1 is enriched in the kidney, intestine, and pancreas, whereas Hnf4g2 is mostly intestine-restricted with the highest expression seen in the small intestine (31).
Functional differences between HNF4α and HNF4γ proteins can be attributed to structural differences
HNFα and HNF4γ bind to many of the same sites in ChIP-seq studies of mouse intestine (17), and the two paralogs also have the ability to control specific target genes (17, 31). In humans, HNF4α ranges from a 392 to 474 amino acid protein depending on the isoform, whereas HNF4γ1 is made up of 408 amino acids (32) (Figure 3). In mice, the two paralogs, HNF4α and HNF4γ, share common features such as the ~200 amino acid ligand binding domain (LBD) and the 76 amino acid DNA binding domain (DBD), which are typical of nuclear receptors. However, HNF4γ1 lacks an N-terminal transactivation domain found in HNF4α and other HNF4γ isoforms. The C-terminal transactivation domain involved in coactivator binding, the AF-2 domain, is 100% conserved between the paralogs. The remainder of HNF4γ1 is highly similar to HNF4α, with a 94% similarity between the DBDs and an 80% similarity between the LBDs (34). In contrast, murine HNF4γ2 is a 448 amino acid protein with a functional AF-1 domain and no repressor domain (F domain). In many instances, the activity of the AF-2 region is suppressed by the repressor F domain (35). Hence, the lack of the F domain with a functioning AF-1 domain suggests that HNF4γ2 is a stronger transactivator than HNF4γ1 (31).
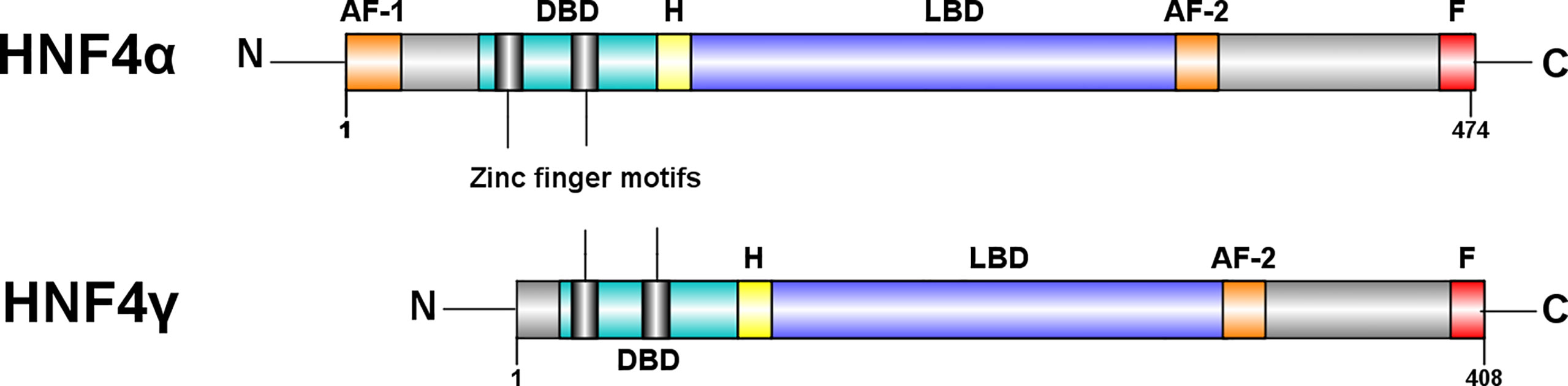
Figure 3 Structural differences between HNF4α and HNF4γ. HNF4α is typically a 474 amino acid protein amino acid protein, whereas HNF4γ1 is a 408 amino acid protein. Both paralogs have a DNA binding domain (DBD), a ligand binding domain (LBD), an AF-2 transactivation domain and a proline rich repressor domain (F) at the C-terminal. There is a 94% similarity between the DBDs and an 80% similarity between the LBDs (31). However, HNF4γ is missing an N-terminal, AF-1 transactivator domain. Figure generated using Illustrator for Biological Sequences, Version 1.0 (33).
The lack of a specific ligand makes HNF4 an unorthodox nuclear receptor. Crystallographic structures of bacterially expressed HNF4α and HNF4γ showed their LBDs constitutively bind endogenous fatty acids (36, 37). In the bacterial-expressed HNF4 system, the bound fatty acids do not readily exchange other fatty acids or ligands and cannot be displaced without denaturing the protein (37). This suggests the fatty acid is trapped in the binding pocket during protein folding and cannot be separated from it. After translation, HNF4 spontaneously adopts a transcriptionally active conformation upon binding of an endogenous fatty acid in its ligand binding cleft. The fatty acid is presumed to act as a structural co-factor for HNF4 by facilitating the formation of a hydrophobic cleft in the LBD and stabilizes the α helical conformation of the protein, which allows it to bind to DNA and regulate its target genes (37, 38). However, in mammalian cells, HNF4α’s LBD was found to be reversibly occupied by linoleic acid (38, 39). There have also been reports of HNF4 binding to fatty acyl-coA thioesters, but their effect on HNF4’s regulatory mechanisms are uncertain (40). Expression profiling in HCT116 colon cancer cells revealed ligand occupancy does not impact the transactivation potential of HNF4α; in fact the presence of a ligand modestly represses HNF4α activity (39). Hence it is unlikely that HNF4 is dynamically regulated by ligands, but rather by post-translational modifications and interactions with co-regulators.
Post-transcriptional modifications of HNF4 alter its regulatory landscape
Post-translational modifications increase the diversity and modify the regulatory capabilities of HNF4 proteins (Figure 4). Several prominent post-translational modifications HNF4 undergoes are:
(i) Phosphorylation: Phosphorylation of serine and threonine residues by several enzymes including protein kinase C, AMP-activated kinase, ERK1/2 kinase, protein kinase A, Src, and p38 kinase can modify HNF4 function (41–46). Much like other nuclear receptors, HNF4 is phosphorylated in the AF-1 domain, the LBD or the DBD. Based on the domain where this modification occurs, the functional impact on HNF4 activity can vary between changes in dimerization, transactivation, DNA binding and cofactor recruitment. As the nuclear localization signals and export signals are in these domains, HNF4’s transport into the nucleus could also be affected by phosphorylation. For instance, phosphorylation of P1-HNF4α by Src kinase causes mislocalization of the protein to the cytoplasm and complete loss of protein stability (46).
(ii) Acetylation: Acetylation at DBD residues K97, K99, K117 and K118 is mediated by the CREB-binding protein (CBP), in association with co-activators, p300 and DDX3, and increases the DNA binding capacity of HNF4α. The region of acetylation overlaps with the NLS, suggesting that acetylation is required for the nuclear retention of the protein (47).
(iii) Methylation: Methylation of R91 in the DBD by protein arginine methyltransferase 1 (PRMT1) also increases the transcriptional activity of HNF4α. PRMT1 also functions as a HNF4α co-activator and functions synergistically with other co-activators at HNF4α target promoters (50).
(iv) SUMOylation: In addition, HNF4α also undergoes ubiquitination and SUMOylation. Successive SUMOylations at the C-terminal residues K365 and D367 destabilize HNF4α in a ubiquitin-dependent manner (48).
(v) Ubiquitylation: Ubiquitylation occurs at residues K234 and K307, and targets HNF4 for proteasomal degradation (49).
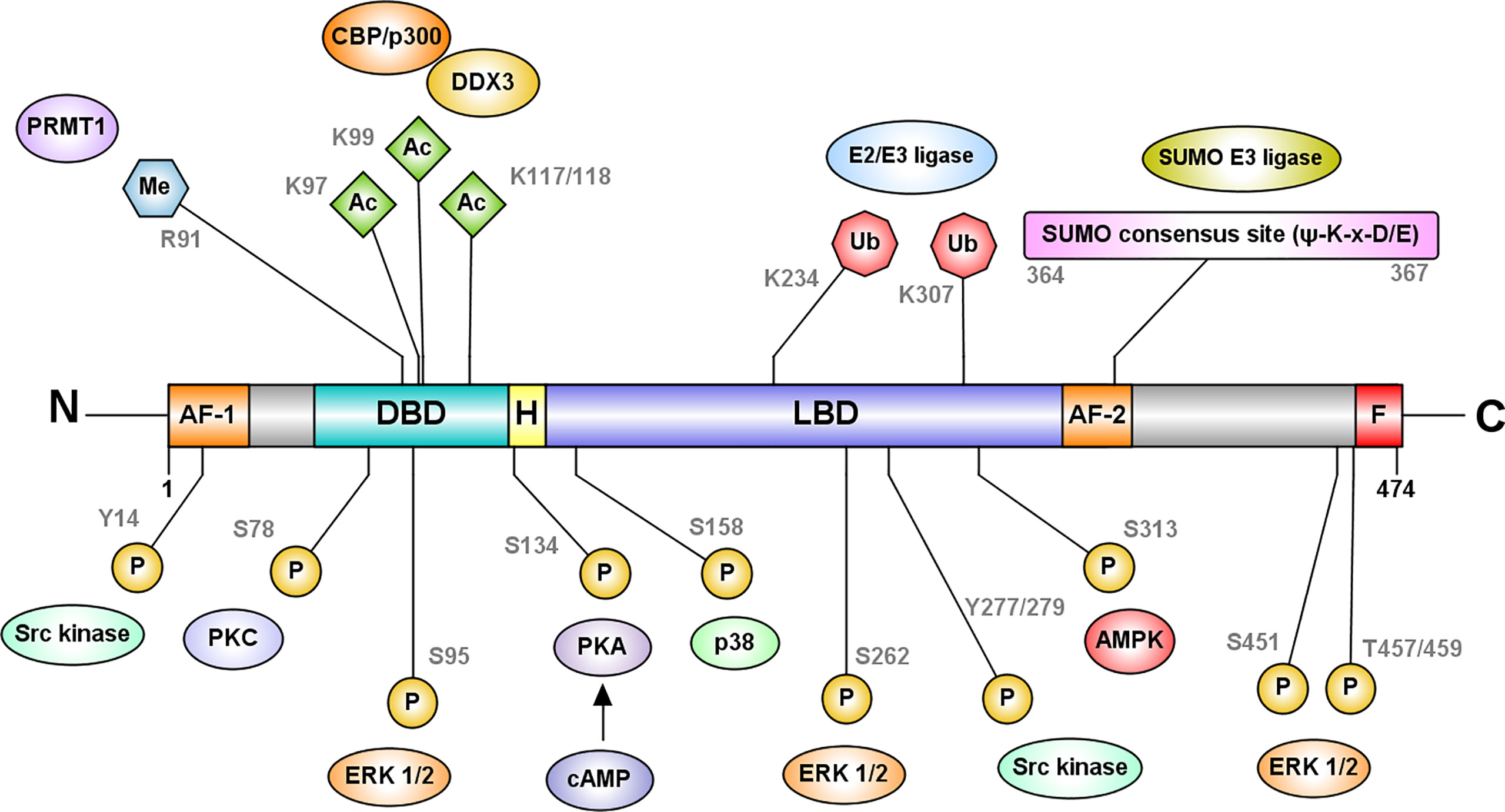
Figure 4 HNF4α is regulated at the protein level by post-translational modifications. The most frequently recurring protein modification is phosphorylation of serine and threonine residues by several enzymes including protein kinase C, AMP-activated kinase, ERK1/2 kinase, protein kinase A, and p38 kinase (41–45). Phosphorylation at the DBD residue S78, by protein kinase C impairs the DNA binding capacity of HNF4α and its nuclear localization (44). cAMP-induced protein kinase A phosphorylates S134 in the DBD and inhibits HNF4α’s recruitment to target genes (41). AMP activated kinase phosphorylates HNFα at S313 in the LBD and destabilizes the protein (42). ERK1/2 kinase can phosphorylate multiple residues such as S95, S262/S265, S451, and T457/T459, which reduce the transactivation capacity of HNF4α (45). Src kinase phosphorylates Y14 followed by Y277/279 of P1-HNF4α causing mislocalization of the protein to the cytoplasm (46). In contrast, p38 kinase mediated phosphorylation at S158 increases the transactivation potential of HNF4α (43). Further modifications include acetylation at DBD residues K97, K99, K117 and K118 mediated by the CREB-binding protein (CBP), in association with co-activators, p300 and DDX3 and methylation of R91 in the DBD by protein arginine methyltransferase 1 (PRMT1). Acetylation is likely required for the nuclear retention of the protein (47). HNF4α also undergoes SUMOylation at the C-terminal consensus site (Ψ-K-x-D/E) which destabilizes HNF4α in a ubiquitin-dependent manner (48). Ubiquitylation of HNF4α occurs at residues K234 and K307, and targets HNF4 for proteasomal degradation (49). Figure generated using Illustrator for Biological Sequences, Version 1.0 (33).
Considering that post-translational modifications in other proteins are generally isoform-specific, we can assume that the same paradigm might apply to HNF4. Understanding this can be a focal point for future investigations.
An intriguing form of regulation: intestinal microbiota suppress HNF4α
The intestinal microbiota exerts a fascinating form of regulation by suppressing the expression and activity of Hnf4α (hepatocyte nuclear factor 4 alpha) in the intestines of various organisms, including zebrafish and mice.
In zebrafish, studies have shown that following microbial colonization, the transcriptional expression of hundreds of genes is suppressed, with nearly half of them being dependent on Hnf4α (51, 52). Interestingly, reduced Hnf4α binding, as measured by ChIP, is a key feature of microbial-suppressed genes, suggesting that Hnf4α activity itself is suppressed by the microbiome. Researchers have found that Hnf4α can mediate the reactivation of microbiota-suppressed genes by specifically binding to and activating transcriptional enhancers, such as the in3.4 enhancer located at the angptl4 gene in zebrafish, as demonstrated through GFP reporter assays. Similar effects have been observed in genomic analyses of gnotobiotic mice, where microbial colonization leads to broad changes in enhancer activation and a reduction in the occupancy of HNF4α and HNF4γ on their respective target genes. The interaction between HNF44α and other putative transcription factors, including GATA, PDX1, and HOXC9, may play a role in mediating the microbial control of intestinal gene expression (51). The downstream effects of the alliance between HNF4α and the microbiota have been explored in mouse jejunal epithelial cells. Through multi-omics analyses, researchers have illustrated that suppression of Hnf4a by a combination of the microbiota and a high-fat meal promotes a proliferative phenotype, leading to a loss of homeostatic balance (53). Moreover, interspecies meta-analysis connected these findings to humans with the discovery that HNF4α-activated microbiota-suppressed gene sets were associated with obesity related traits and inflammatory bowel diseases (51). Clearly there exists a conserved regulatory relationship between HNF4α, its transcription factor network and microbiota in maintaining intestinal homeostasis. The microbiota-HNF4α relationship may have important implications for understanding and addressing human intestinal diseases.
Unraveling the role of HNF4 in the intestine: key perspectives and findings
HNF4 is of paramount importance in the regulation of multiple facets of intestinal development, function, and homeostasis. In the following section, we explore and elucidate some of the major roles it plays in these critical processes.
HNF4 plays an essential role during intestinal development
E8.5 is the inception point for mouse intestinal development. At this stage, the visceral endoderm invaginates to form a common gut tube that includes the foregut and the hindgut. Expression of CDX2 is subsequently required to specify the intestine from the more antral GI tissues (54–56). Until E14.5, the developing intestine is comprised of rapidly proliferating, pseudostratified epithelium. Between E14.5 and E16.5, villus morphogenesis and maturation occur. Analysis of chromatin landscapes using ATAC-seq at different time points suggest a transition in genome occupancy across this developmental transition and indicates that HNF4 binding is abundant at accessible genomic regions upon intestinal maturation. Facilitated by the Shh-Cre driver, inactivation of both HNF4 paralogs in the embryonic endoderm demonstrates that HNF4 factors are largely dispensable in the developing intestine until villus elongation and maturation at E16.5 (57). It should be noted that the intestine remains properly specified in the Shh-Cre, Hnf4 double mutants, indicating that HNF4 factors function only in the maturation of the tissue, rather than its developmental specification, a role unmistakably attributed to the transcription factor, CDX2 (54, 57). CDX2 functions upstream of HNF4, with decreased Hnf4 expression in the Cdx2 knockout intestine. The HNF4α-CDX2 pair occupy shared genomic regulatory sites to promote chromatin accessibility and gene expression in the maturing intestine. Together they control genes responsible for formation of the apical brush border (Figure 5) and absorption of dietary lipids in the intestine (62). Further studies in human cell lines in vitro reveal the HNF4 regulatory cascade is balanced by either COUP-TFII or HNF3α, both of which can repress gene expression of HNF4A (64, 65). Retinoic acid receptors RXR-RAR compete for the same binding site as COUP-TFII and can alleviate the repressive effects (65).
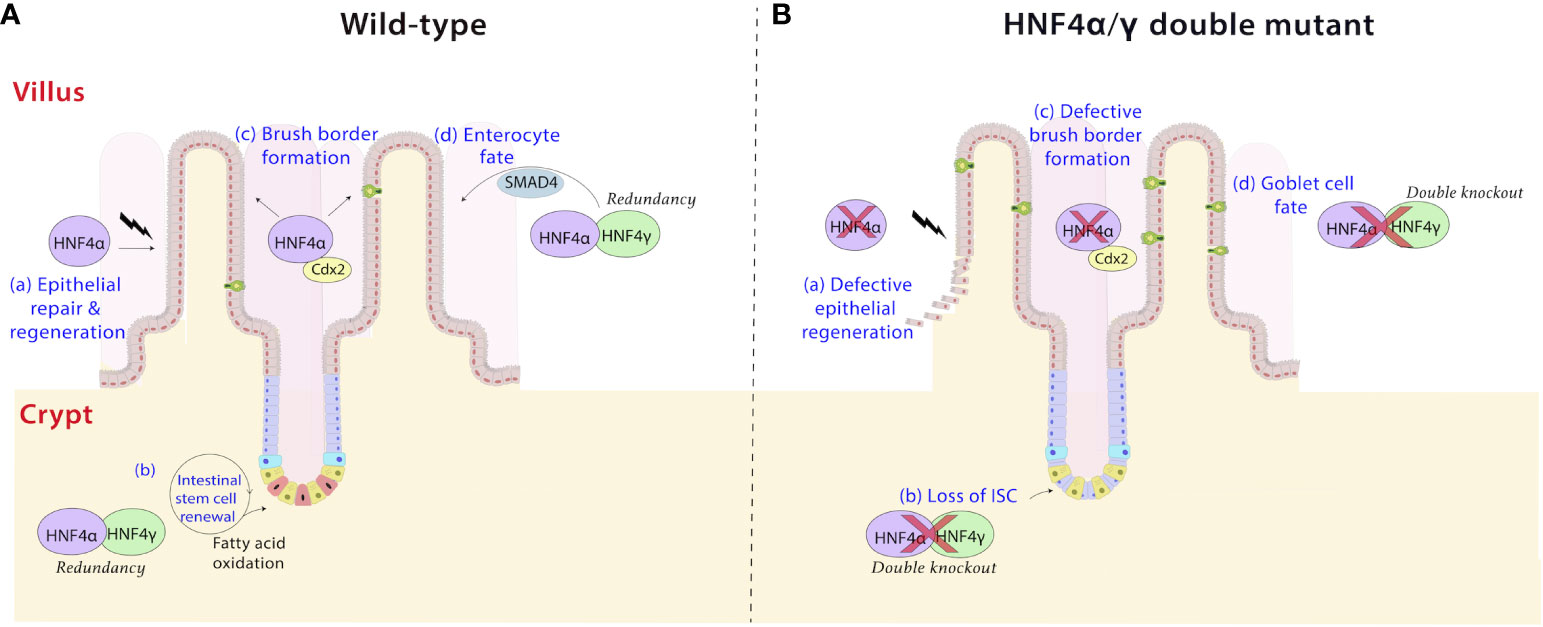
Figure 5 HNF4 plays diverse roles in the intestine. (a) HNF4α is a driver of repair and regeneration mechanisms in the intestine (58). It controls crypt survival and proliferating cell survival, which would allow epithelial repopulation (A), a phenomenon which is lost in HNF4α mutants (B). (b) HNF4α and HNF4γ are highly expressed in Lgr5+ intestinal stem cells (ISCs) and are required for ISC maintenance and renewal (A) (59). Dysregulation of fatty acid oxidation in stem cells causes exhaustion, loss of ISC and premature lineage commitment (Panel B) (60, 61). This effect is pronounced in HNF4α/γ double mutants, implying redundancy masks the phenotype in single mutants. (c) The HNF4α-CDX2 pair controls genes responsible for formation of the apical brush border in the intestine (62). HNF4 functions as a conserved and universal regulator of brush border genes by likely operating as a mechanosignaling sensor detecting changes in actin fibers and brush border transcripts upon mechanical stress (63). Electron microscopy cross sections through microvilli reveal a larger brush border diameter and shorter height in HNF4α/γ double mutant mice compared to wild type (A, B). (d) HNF4 paralogs, in association with the BMP/SMAD pathway mediate the formation of enterocytes in the villi. HNF4 and SMAD4 reciprocally activate each other’s expression in villi in vivo and in organoid models ex vivo, and disruption of this feed-forward loop results in loss of enterocyte fate in favor of a goblet cell fate (B) (17).
During later stages of development, several signaling cascades which control patterning of the gut have been shown to be influenced by HNF4 (66, 67). For instance, in crypts, which develop postnatally in mice, HNF4α sequesters TCF4, an effector of Wnt/β-catenin signaling, whose inactivation is required to control excessive proliferation (16, 68). Similarly, the activation of the Notch pathway induces the transcription factors HES1 and MATH1, to commit epithelial cells to the absorptive or secretory lineages, respectively. Ablation of HNF4 skews the phenotype towards secretory cells by increasing MATH1 levels (16). Further, during colon development, use of the Foxa3-Cre driver, which is active before the onset of Shh-Cre, found that HNFα is required for the formation of colonic crypts. This phenotype was not observed in the Shh-Cre model (57), which could either reflect differences in the onset of Cre activity between the two models, or secondary consequences of loss of HNF4 in the liver of the Foxa3-Cre model (69, 70). Differences in HNF4α sequence determinants, binding motifs and absolute dosage among different species may, in some measure, account for inter-species developmental variations among tissues of the gastrointestinal system (71, 72).
HNF4 is a key regulator of intestinal metabolism
The first and foremost role of the intestine is digestion and absorption of nutrients, primarily dietary lipids. This process primarily occurs in enterocytes of the villi (73). HNF4α controls fatty acid uptake by enterocytes, cellular lipid transport, and apolipoprotein synthesis (74, 75). In Drosophila, dHNF4 functions as a sensor for free fatty acids, and is essential for fatty acid oxidation (FAO). dHNF4 null mutant larvae retain increased levels of long-chain fatty acids in their midgut and fat body, suggesting an inability to mobilize stored fat for energy (76). In higher mammals, HNF4 binds intermediate metabolites of lipid metabolic pathways. Genes associated with acyl coenzyme A metabolism were also identified as putative HNF4α targets (21, 77). In fact, the loss of HNF4 factors in mice leads to decreased transcript levels of genes involved in almost every step of FAO (59). This activity of HNF4 factors in regulating FAO is important for supporting intestinal stem cell renewal (further explored below).
HNF4α also affects incretin metabolism. Incretins are two hormones produced by the enteroendocrine cells of the villi, which augment insulin release by pancreatic β-cells. Incretins have broad roles in glucose metabolism, cardiovascular function, bone metabolism, and triglyceride storage in adipose tissue (78). The precise impact of HNF4α on one of the incretins, glucagon-like peptide-1 (GLP-1), is not yet well understood. However, studies in intestinal Hnf4a-deficient mice have revealed that HNF4α regulates the production of the other incretin, glucose-dependent insulinotropic polypeptide (GIP) in association with GATA-4 (79). Interestingly, the deletion of intestinal Hnf4a does not directly affect enterocyte lipid metabolism but has a broader whole-body effect, resulting in a resistance to diet-induced obesity (DIO) (80). Intestinal Hnf4a-mutant mice showed a preference for utilizing fat as an energy substrate and experienced significant changes in energy metabolism within white adipose tissue (WAT). Notably, the WAT underwent beiging, a process where it acquired characteristics similar to brown adipose tissue. When a stabilized analog of GIP was introduced, it was able to rescue the DIO resistance phenotype. This suggests that the impairment in fat-induced GIP release is a key mechanism contributing to the DIO resistance phenotype associated with intestinal HNF4α deletion. The reintroduction of the GIP analog restores the proper signaling and metabolic response, resulting in the normalization of the mice’s response to a high-fat diet (80).
HNF4 and SMAD4 stabilize enterocyte identity
In the small intestine, enterocytes in the villi predominantly express P1-HNF4α while both P1 and P2 related isoforms are seen in the crypts (Figure 2A). Differential expression can be a contributing factor to architectural and functional variations between the two compartments. In villi, HNF4 paralogs, in association with the BMP/SMAD pathway mediate the differentiation of enterocytes from crypt progenitor cells (Figure 5). Transcriptome analyses between Villin-CreERT2; Hnf4af/f; Hnf4g-/- compound mutant mice and Villin-CreERT2; SMAD4f/f mutant mice reveals 541 downregulated genes in common upon both SMAD4 and HNF4 inactivation. These genes are strongly associated with enterocyte functions. HNF4 and SMAD4 reciprocally activate each other’s expression in villi in vivo and in organoid models ex vivo and disruption of this feed-forward loop results in loss of enterocyte fate in favor of a goblet cell fate (17). Additional evidence that HNF4α precludes a secretory state is the decrease in functional enteroendocrine cells in intestinal Hnf4a-deficient mice. In this mouse model, the transcription factor NGN3 drives production of enteroendocrine cells but the lack of HNF4α prevents their full terminal differentiation (16, 81). Further, HNF4α influences the distribution of enteroendocrine cells along the crypt-villus axis driving them more towards villi than crypts (16).
HNF4 governs key aspects of intestinal architecture while ensuring intestinal barrier integrity
The cellular organization and architecture of the intestinal epithelium is also dependent on HNF4α. HNF4α depletion causes a wider spread of intercellular tight junctions, as a result of a decline in tight junction proteins, ZO-1 and claudins 4 and 7 (16). This likely explains why mucosal erosion, loss of paracellular permeability, and susceptibility to IBD are so high in HNF4α dysfunction disorders. Likewise, HNF4α is required for the translocation of E-cadherin, an epithelial adhesion molecule, to membrane surfaces (16). Early loss of E-cadherin in enterocytes triggers anoikis in the epithelia (9, 10). Interestingly, ectopic induction of Hnf4a in F9 murine embryonic cells has been shown to drive the expression of tight junction proteins, occludin and claudin-7. This consequently promotes the de novo formation of functional tight junctions, thus maintaining epithelial integrity (82). This highlights the role of HNF4α in the regulation of tight junction proteins and its ability to preserve the barrier function of epithelial cells. It’s likely that this association with barrier integrity is a major reason why mutations in the human HNF4A locus are associated with heritable risk for ulcerative colitis (83–85).
Additionally, HNF4 controls an extensive battery of genes associated with the brush border (Figure 5). Recent studies show expression of brush border markers, such as alkaline phosphatase and Espn, are downregulated upon loss of both intestinal Hnf4 factors in mice. Electron microscopy cross sections through microvilli reveal a larger diameter and shorter height in Hnf4a/g double mutant mice compared to the organized, uniform microvillar arrays in the wild type. Mouse genetic experiments also show that HNF4α controls expression of brush border genes in other tissues in vivo, including the proximal tubules of the kidney and in the villous structures protruding from the proximal murine yolk sac (63). Clearly, HNF4 functions as a conserved and universal regulator of brush border genes. Brush border anomalies are reported in many human intestinal disorders, including Crohn’s disease, celiac disease, and congenital sodium diarrhea (86–88).
Epithelium renewal and repair in the intestine is dependent on HNF4
The renewal of the intestinal epithelium is accomplished by the Lgr5+ stem cell population located in the crypts. These cells divide every 24 hours, generating rapidly proliferative progenitors, which differentiate into various cell types of the intestine, a process which requires high energy expenditure. Hnf4a and Hnf4g are highly expressed in intestinal stem cells (ISC) and have been determined to be necessary for ISC maintenance and renewal (59). HNF4α and HNF4γ bind to endogenous fatty acids (36, 37), such as linoleic acid in the case of HNF4α (39), and activate genes corresponding to the fatty acid oxidation (FAO) pathway; a process essential for ISC renewal (89, 90). ChIP-seq experiments conducted in wild-type mice have provided evidence that HNF4 directly binds to genes involved in FAO, including Abcd1, Acox1, and Ehhadh (17, 59). Further, RNA-seq experiments in Hnf4a/g double mutant mice revealed a significant decrease in the expression of FAO genes, indicating that HNF4 is indeed involved in activating their expression (17, 59). Loss of HNF4 paralogs impairs ISC renewal, instead pushing ISCs towards lineage commitment leading to an increased number of transit-amplifying cells and an escalation of cell proliferation. In intestinal organoid assays, metabolic intervention with acetate or dichloroacetate has been shown to have a beneficial effect on ISC loss in Hnf4a/g double mutants. By providing these exogenous 2-carbon fatty acids, the metabolic intervention helps to compensate for the deficiency in FAO observed in the Hnf4a/g double mutants. The homeostasis of ISCs and other concomitant intestinal cell types, is therefore heavily dependent on HNF4 and its regulation of metabolic pathways (59).
The intestinal epithelium exhibits remarkable plasticity, with the ability to regenerate within 72 hours following damage (58). HNF4α is a likely driver of these repair mechanisms in intestinal organoids and mutant mouse models (58) (Figure 5). Studies ex vivo in organoids show HNF4α helps establish organoids from clusters of broken up epithelial cells during passaging. Further, in vivo, HNF4 was shown to control epithelial repopulation after irradiation in mice. Intestinal damage inflicted by sublethal radiation leads to a reduction in the rate of amino acid and lipid metabolism, both of which are processes dependent on HNF4 in the intestine. There could be connections in the intestine between activation of p53 and repression of HNF4 activity to support this change in damaged intestine, as this connection has been documented in liver cells (91, 92). As expected, intestinal Hnf4a mutant mice had fewer proliferating cells per surviving crypt. This and previous studies of HNF4 on stem cell renewal illustrate the critical role HNF4α orchestrates in crypt and proliferating cell survival (59, 76).
Paneth cell homeostasis is dependent on HNF4
HNF4α plays a pivotal role in maintaining Paneth cell homeostasis and epithelial renewal in the intestine. Recent studies have provided further insights into the dependence of these processes on HNF4α and its relationship with Wnt signaling.
Studies using murine jejunal enteroids lacking Hnf4a have shown that the addition of exogenous Wnt3a or co-culture with mesenchymal cells can rescue the phenotype (93). Notably, immunofluorescence assays for lysozyme showed a reduction of Paneth cells in the Hnf4a-deficient enteroids co-cultured with mesenchyme but a restoration of the Paneth cell signature in those rescued with Wnt3a. The likely reason for this is the dependence of Paneth cells on Wnt signaling (4). Transcriptomic analyses of Hnf4a-deficient enteroids have revealed that both Wnt3a supplementation and mesenchymal cell co-culture can rescue a significant proportion of the transcriptomic changes observed in the absence of HNF4α - Wnt3a rescue accounted for approximately 89% of the changes, while mesenchymal cell co-culture rescued around 91%. Further, in studies using engineered intestinal epithelial cell lines expressing HNF4α2, researchers have demonstrated the direct binding of HNF4α2 to the Wnt3 gene. Overall, these findings highlight the intricate relationship between HNF4α, Wnt signaling, and Paneth cell development. HNF4α acts as an upstream transcriptional regulator of the Wnt3 gene, affecting autocrine epithelial Wnt3 signaling and contributing to Paneth cell homeostasis in the intestine (7, 93).
HNF4α and HNF4γ exhibit genetic redundancy in the intestine
While HNF4α is a key factor in maintaining intestinal homeostasis, its study is incomplete without decoding the role of its intestine-restricted paralog HNF4γ (Figure 1). HNF4γ functionally overlaps with HNF4α and compensates for its loss, pointing to genetic redundancy between the two factors. In mice, HNF4α and HNF4γ show similar DNA binding profiles, along with overlapping in situ expression patterns in the intestine. Ablation of either Hnf4a or Hnf4g alone yield fertile, developmentally normal offspring, indicating each individual paralog is largely dispensable. However, a tamoxifen-inducible double knockout of Hnf4a and Hnf4g results in striking changes in intestinal structure and function (17). Apart from demonstrating an emaciated phenotype, the Hnf4a/g double mutant mice develop fluid-filled intestines and die within 4-5 days of phenotype onset, suggesting redundant functions between the HNF4 paralogs (17).
Loss of either HNF4α (16, 94) or HNF4γ (95, 96) paralogs in mice show relatively modest phenotypic changes due to compensation by the other, thus buffering against fluctuations in overall Hnf4 gene expression. Redundancy would allow one paralog to accumulate mutations if the other is functionally intact. Loss of both paralogs, however, would cause detrimental changes in intestinal structure and function with loss in homeostatic balance (17). In this section, we delve into the functionalities and impacts of HNF4 redundancy within the intestinal system.
Redundancy during zebrafish gut development
In zebrafish larvae, hnf4a and hnf4g show partial genetic redundancy. Germline loss of hnf4a alone has a stronger impact on intestinal processes than loss of hnf4g and these changes are further exacerbated in a hnf4a/hnf4g double mutant model. The double mutant ultimately results in larval lethality between 6 to 14 days post fertilization (97). Moreover, zebrafish and other lower vertebrates like Xenopus laevis express a third paralog, hnf4b (98, 99). hnf4a and hnf4b are reported to redundantly regulate genes involved in yolk lipid mobilization to the embryonic body during early zebrafish development; a function which might be conserved in other egg-laying vertebrates but lost in higher vertebrates due to the lack of hnf4b. Double mutants of hnf4a and hnf4b or triple mutants of hnf4a, hnf4g and hnf4b in zebrafish are not viable, further pointing to multiple layers of redundancy between paralogs (97).
Redundancy during murine intestinal development
HNF4 paralogs are also redundant in the developing gut. HNF4 DNA-binding motifs are more prevalent at accessible chromatin regions at E14.5-E18.5, which corresponds to the period of villus morphogenesis, compared to earlier stages of intestinal development. Ablating Hnf4a in a Hnf4g-/- background using an Shh-Cre driver leads to shorter villi, indicating HNF4 is redundantly required for villus elongation and extension into the gut lumen. As mentioned previously, brush border formation is also affected in the double mutant (57). Loss of HNF4α alone in the developing intestinal epithelium does not result in the same pronounced morphological effects (69, 94).
Redundancy is required to maintain intestinal homeostasis
The redundant functions of HNF4 paralogs have also been elucidated in the context of intestinal homeostasis. As previously discussed, HNF4α and HNF4γ are necessary for ISC maintenance and renewal (59) (Figure 5). Single knockouts of either Hnf4a or Hnf4g show only minor phenotypic changes with no effect on ISC maintenance. Conversely, Hnf4a/g double mutants show compromised β-oxidation and increased apoptosis of ISCs in the crypt base, causing disruption in their renewal (59). It is noteworthy that the redundancy of HNF4 paralogs clearly extends beyond the fetal stage and coordinates metabolic regulation in the mature intestine.
Redundancy during intestinal cellular differentiation programs
Furthermore, HNF4 paralogs redundantly regulate cellular identity in the intestine. HNF4α acts redundantly with HNF4γ, to activate and maintain distal enhancer chromatin and upregulate programs of cellular differentiation. As the most abundant intestinal cell type, enterocytes are a key beneficiary of such mechanisms. Indeed, as mentioned previously, a positive regulatory circuit between HNF4 and the BMP/SMAD signaling pathway promotes enterocyte identity. Disruption of this module requires ablation of Smad4 or both Hnf4 paralogs for the increased goblet cell phenotype to be established (17) (Figure 5). Cellular proliferation and differentiation are thus tightly regulated in the intestinal epithelium via mechanisms of feedback and functional redundancy.
Redundancy during intestinal immune signaling
Recent studies in mice have highlighted the crucial role of HNF4α in mediating communication between intraepithelial lymphocytes (IELs) and epithelial cells (100). This communication relies on the HNF4α-dependent regulation of immune signaling molecules, including Btnl1, Btnl6, H2-T3, and Clec2e. In the colon and distal small intestine, the expression of Btnl1 and Btnl6 is controlled by HNF4α. However, in the duodenum and jejunum, HNF4γ takes over the regulatory role. ChIP-qPCR analyses have revealed that HNF4γ strongly binds to the promoters of Btnl1 and Btnl6, even in the absence of HNF4α. Furthermore, in duodenal epithelial cells lacking both Hnf4a and Hnf4g, there is a significant reduction in the levels of Btnl1 and Btnl6 compared to single mutant models of Hnf4a or Hnf4g (17, 100). This suggests that HNF4γ acts redundantly with HNF4α, serving as a substitute in the regulation of genes involved in the communication between IELs and intestinal epithelial cells.
HNF4 redundancy may have a regulatory role in modulating circadian behaviors
Of note, HNF4α and HNF4γ contribute to the maintenance of tissue-specific circadian oscillations in both intestinal and liver cells, by trans-repressing the activity of the master circadian transcription factor CLOCK:BMAL1 (101). This suggests that HNF4 transcription factors may be redundantly involved in shaping the circadian clock’s molecular mechanisms. However, whether this non-canonical activity is redundant or not is debatable. Nevertheless, the redundancy between HNF4α and HNF4γ may provide a selective advantage for HNF4 in the control of intestinal gene expression.
HNF4 variants are associated with disease phenotypes in humans
Mutations in HNF4 are a pathophysiological feature of many human diseases (Figure 5). While the mutational spectrum tends to be variable, most reported cases of disease-associated mutations are often deleterious and involve loss of function. Understanding HNF4 disease phenotypes is further complicated by the functional redundancy between HNF4α and HNFγ, as phenotypes might only manifest under a specified set of circumstances. Nevertheless, a comprehensive review of clinical phenotypes attributed to HNF4 mutations reveals common themes of loss of function and haploinsufficiency. Some of the most well-known manifestations of HNF4α mutations are seen as perturbations of glucose metabolism, such as in Type 1 maturity onset diabetes of the young (MODY1) and non-insulin dependent diabetes mellitus (NIDDM) (Figure 6). MODY is an adolescent-onset, monogenic and hereditary form of Type 1 diabetes (104), whereas NIDDM is adult-onset, multifactorial and occurs due to an imbalance between insulin sensitivity and secretion (105). In MODY1, a low frequency missense mutation in HNF4α, Q268X, results in the deletion of 187 C-terminal amino acids, leading to mislocalization of the protein, loss of transactivation activity and failure to dimerize and bind DNA (109, 110). In NIDDM, a V393I substitution in HNF4α results in a decrease in transcriptional activity and insulin secretion (107). Additionally, Fanconi renotubular syndrome is a unique phenotype comprising both MODY1 and atypical Fanconi syndrome, which occurs due to a heterozygous missense mutation, R76W in HNF4α. Such patients present with fetal macrosomia and neonatal hypoglycemia associated with hyperinsulinemia (108). The differential gene expression patterns of HNF4 across various organs also helps account for the wide range of disease manifestations.
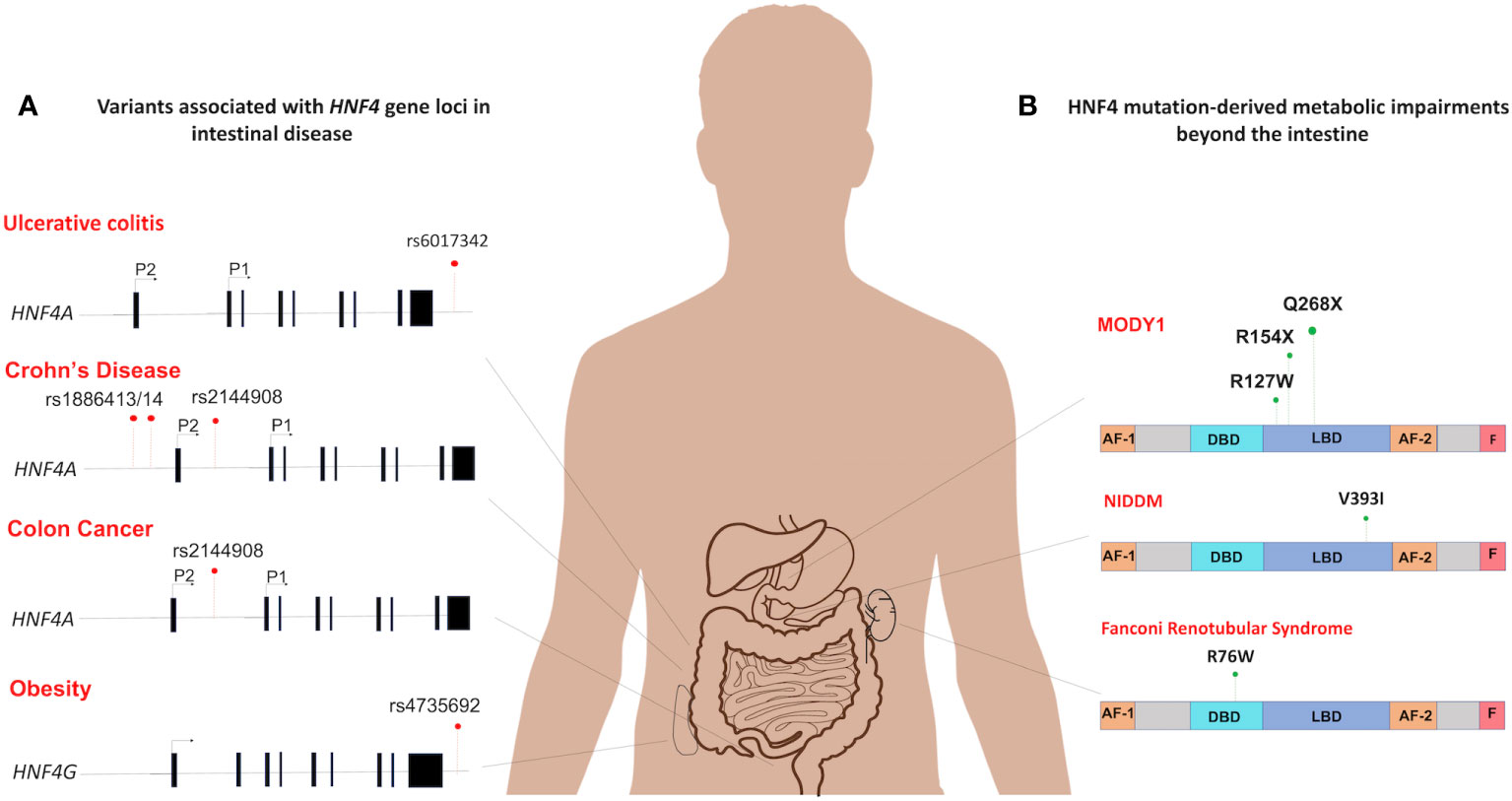
Figure 6 Spectrum of HNF4 dysfunctions in the intestine and beyond. (A) The mutational distribution of HNF4 generates 4 main disease phenotypes. An SNP in in the 3’-UTR of HNF4A has been associated with ulcerative colitis (rs6017342) (83–85). Three SNPs each are significantly associated with increased susceptibility to childhood-onset Crohn’s disease (102) (rs2144908, rs1884613 and rs1884614) and colon cancer (46) (rs6031602, rs1063239, and rs6093980). A single SNP in the human locus of HNF4G (rs4735692) has been associated with obesity (103). (B) At the protein level, HNF4α dysfunctions are seen as metabolic impairments such as MODY1 (104), and Non-insulin dependent diabetes mellitus (NIDDM) (105). In MODY1, a low frequency missense mutation in HNF4α, Q268X, results in the deletion of 187 C-terminal amino acids (106), whereas a V393I substitution causes increase in susceptibility to NIDDM (107). Also, Fanconi renotubular syndrome is a unique phenotype comprising both MODY1 and atypical Fanconi syndrome, which occurs due to a heterozygous missense mutation, R76W in HNF4α (108).
HNF4-related diseases specifically affecting the intestine are primarily associated with mutations or variations in the HNF4 gene, leading to impaired regulation of various aspects of intestinal development, function, and homeostasis. The specific manifestations of HNF4-related intestinal diseases can vary and may include disruptions in metabolism, architecture, epithelial cell differentiation, and other essential processes within the intestine. In this section, we explore different diseases and their connection to HNF4 in the context of intestinal health.
HNF4 is required to avert a chronic inflammatory state in the intestine
In the intestine, dysregulation of HNF4α underlies a multitude of disease phenotypes, notably, inflammatory bowel disease (IBD). Indeed, Hnf4a is considered an IBD susceptibility gene (83, 111, 112). Crohn’s disease and ulcerative colitis are the most common forms of IBD and HNF4α has been implicated in both. Previous studies using a DSS-induced colitis mouse model have provided evidence that conditional deletion of Hnf4a leads to a decrease in body mass and an increase in intestinal permeability (110). However, another study has shown that HNF4α deficiency did not result in clinical disease or dysbiosis in young mice. Instead, it caused an early disruption of intestinal homeostasis, which subsequently increased the susceptibility to colitis in older animals (100). HNF4α, is therefore necessary to maintain the integrity of the mucosal epithelial barrier. Separate studies in exon swap mice genetically engineered to express only P1-Hnf4a or P2-Hnf4a show that intestinal barrier functions mediated by HNF4α are isoform-specific. Mice producing P1-HNF4α proteins were less susceptible to colitis whereas mice ectopically expressing only P2-HNF4α were more inclined to develop colitis. The P2-Hnf4α mice had higher levels of RELMβ, a cytokine which activates innate immune responses upon disruption of intestinal barrier function (29). Such mice also exhibit imbalances in electrogenic sodium and chloride secretion and impaired water absorption in the colonic mucosa, two of the main triggers of the diarrheal symptoms of colitis (29, 113). Results implicating HNF4α in IBD are bolstered by evidence from genome-wide association studies in geographically diverse cohorts of ulcerative colitis patients, which identify HNF4A as a major susceptibility locus. A single nucleotide polymorphism (SNP), rs6017342, in the 3’-UTR of HNF4A causes increased susceptibility to ulcerative colitis (83–85) (Figure 6). Furthermore, three additional SNPs (rs2144908, rs1884613 and rs1884614) are significantly associated with increased susceptibility to childhood-onset Crohn’s disease (102) (Figure 6). In addition, human intestinal biopsies from patients with IBD show decreased expression of HNF4α (111). Recent work demonstrating a potential treatment strategy against IBD uses HNF4α to induce NHE3 (Na+/H+ exchanger isoform 3), which can restore Na+ absorption in the intestine (114). HNF4α integrity is thus crucial and plays a protective role against predisposition to IBD following chronic intestinal inflammation.
HNF4 alterations can increase the risk of colon carcinomas
A diagnosis of IBD is a significant risk factor for developing colorectal cancer (CRC) and colitis-associated cancer (CAC), predominantly due to the pro-neoplastic effects of chronic inflammatory insults. Thus, by default, the protective effect of HNFα in IBD can be extended to intestinal cancers as well. In a mouse model of CAC, expression of only P1-HNF4α is associated with lower tumor burden, while the opposite effect is seen during ectopic expression of only P2-HNF4α. The tumor-inducing effect of P2-HNF4α is attributed to the absence of P1-HNF4α in these mice, thus, suggesting a protective role for P1-HNF4α against cancer (29). A more direct relationship between HNF4α and CRC in humans can be seen by three functional variants of HNF4A (rs6031602, rs1063239, and rs6093980), the expression of which could increase an individual’s susceptibility to Src-kinase mediated CRC (46) (Figure 6). Investigation into the molecular connections between HNF4 and Src-kinase mediated CRC reveals that HNF4α again mediates susceptibility in an isoform dependent. Phosphorylation of three residues in P1-HNF4α by Src kinase leads to protein instability and transcriptional dysregulation. This action by Src is specific to P1-HNF4α as P2-HNF4α lacks a Y14 residue in the AF-1 which is the first residue phosphorylated followed by 2 residues in the LBD (46). Three SNPs in the human HNF4α protein, two of which are in the HNF4α F domain which interacts with the Src SH3 domain, increase phosphorylation by Src and decrease HNF4α protein stability and function, suggesting that individuals with those variants may be more susceptible to Src-mediated effects. Indeed, there is an 80% loss of nuclear P1-HNF4α in >450 analyzed human Stage III colon tumors which correlates with active Src (46). Further, a loss of P1- but not P2-HNF4α has been observed in many other human cancers, including renal carcinoma and hepatocellular carcinoma (28, 112, 115, 116). Rather than an active role promoting cell proliferation, P2-HNF4α is merely thought to play a permissive role, consistent with its expression in the distal proliferative compartment of the colonic crypt (29).
Vuong et al. also explored the differential effects of the HNF4α isoforms on tumor growth in human CRC (117). They used Tet-On-Inducible HCT116 colon cancer cell lines expressing either HNF4α2 or HNF4α8 isoforms that were injected in immunocompromised mice. After 8 days of development, mice were fed a diet supplemented with doxycycline to induce HNF4α expression. Tumors with HNF4α2 were significantly smaller compared to the parental controls suggesting that HNF4α2 is an active tumor suppressor in CRC. Conversely, the HNF4α8 induced tumors were larger and showed higher invasive index, making it permissive of the cancer state. Ontology analyses on genes specifically upregulated by HNF4α2 showed terms such as cell death and growth inhibition, whereas HNF4α8 upregulated genes were involved in cell proliferation. Mechanistically, HNF4α8 is believed to preferentially modulate the Wnt/β-catenin/TCF4 and AP-1 pathways. Taken together, these results help explain the contradictory notion that HNF4α is both a tumor suppressor and at least somewhat oncogenic; the isoforms perform distinct roles in the HCT116 cells by interacting with different co-regulators.
HNF4 regulates intestinal entry of SARS-CoV2
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative organism of coronavirus disease 2019 (COVID-19), has been implicated in triggering gastrointestinal symptoms by infecting the epithelial cells lining the gastrointestinal tract (118). ACE2 functions as the viral receptor in absorptive enterocytes and TMPRSS2 is involved in viral spike protein priming (119). Using epigenomic approaches Chen et al. identified HNF4 factors, in association with CDX2, SMAD4, and GATA factors, impact Ace2 and Tmprss2 gene expression (120). All four factors coordinately alter chromatin structure and activate intestinal Ace2 expression, while conversely suppressing Tmprss2. Further, H3K4me3-targeted HiChIP assays demonstrate multiple contacts between COVID-19-related gene promoters and these regulatory elements (120). Differential usage of HNF4 and its accessory elements, along with variability in their binding sites could be one possible explanation for the wide-ranging disparity in the susceptibility to COVID-19. Better understanding of intestinal regulatory mechanisms may help in the development of therapies to reduce the systemic effects of COVID-19 and lower the severity of its associated symptoms.
Disease phenotypes attributed to HNF4γ are ill-defined
Interestingly, very few reports exist of disease phenotypes attributed to HNF4γ alterations. A single nucleotide variant in the human locus of the HNF4G gene (rs4735692) has been associated with obesity (103) (Figure 6). Systematic association mapping of individuals with IBD reveals a mild association with a SNP of the HNF4G intron (121). Moreover, in a clinical study of ulcerative colitis patients, HNF4γ2 is significantly downregulated, hinting at a protective role for HNF4γ2 against ulcerative colitis (31, 122). A homozygous deletion of HNF4G in mice resulted in animals with a higher body weight despite having reduced intake of food and water. Additionally, they exhibited less nocturnal activity, and were less inclined to build nests, compared to their wild-type littermates (95). Also, studies have shown HNF4G expression increases in some bladder cancer phenotypes and in intestinal metaplasia, which is highly associated with gastric cancer (123, 124). One study has named HNF4γ as an oncoprotein in bladder and lung carcinomas (125). It remains poorly understood how the principle of redundancy between HNF4α and HNF4γ can be applied to such disease phenotypes.
HNF4 has diagnostic and therapeutic potential in the intestine and beyond
HNF4 has diagnostic potential in the intestine as a marker for intestinal differentiation and function. Its expression and activity can serve as indicators of the intestinal cellular state and proper functionality. Moreover, the regulatory roles of HNF4 in gene expression and metabolic pathways make it an enticing target for therapeutic interventions in a wide range of disease states.
HNF4 is a biomarker for gastrointestinal disease
Owing to its pivotal role in multiple regulatory pathways and disease states, HNF4 is an attractive target for the clinic, either as a diagnostic tool or a therapeutic strategy. A fair amount of recent work has supported the idea of HNF4 as a biomarker in gastrointestinal diseases. Studies show HNF4α expression as a potential diagnostic tool to distinguish between primary cancers and metastases (126, 127). Another study put forth P1-HNF4α as a prognostic indicator in Duke’s Stage C and D colon cancer patients (112). Additionally, genetic profiling of HNF4α can provide an understanding of whether polymorphisms in the human HNF4 gene segregate with disease phenotypes. Certain individuals with SNP variants of HNF4A may be more susceptible to Src kinase-mediated colon cancer (46). Similarly, other SNP variants of HNF4A and HNFG can cause increased susceptibility to ulcerative colitis, Crohn’s disease and obesity (83–85, 102, 121). While HNF4 clearly has diagnostic potential, additional investigation is required to understand the complex role of the different HNF4 paralogs and isoforms in human disease.
HNF4 shows promising potential for therapeutic applications
HNF4 is a nuclear receptor with a large, hydrophobic ligand-binding pocket potentially making it conducive for accepting antagonistic small molecules. However, its pleiotropic functions across diverse tissues could cause unintended side effects, so caution should be exercised when designing drugs against HNF4. In mammalian systems, initial evidence of the druggability of HNF4 was uncovered when a fatty acid ligand of HNF4α, linoleic acid, was found to be exchangeable and capable of reducing HNF4α’s transactivation capacity (39). Further studies in the T6PNE human pancreatic cell which stably expresses a HNF4α-dependent insulin promoter, shows this effect is limited to medium and long chain fatty acids (128, 129). Using the same in vitro system, a high-throughput screen of compounds identified two putative HNF4α antagonists, BIM5078 and BI6015, which bind in the same orientation as fatty acids. Both compounds bind to HNF4α with high affinity and altered expression of nearly 36% of HNF4 target genes. Further, both compounds are selectively cytotoxic to a panel of neoplastic cell lines but not their untransformed counterparts. At a dosage of 30mg/kg body weight, BI6015, in particular, is effective in mice, yet with sub-optimal pharmacokinetic properties (129). Extensive studies conducted on BI6015 have revealed that it mediates its inhibitory effects by modulating oncogenic Wnt signaling (130). Further studies have revealed HNF4A gene expression is downregulated by AMPK signaling and the AMPK agonist metformin, which also brings about changes in the Wnt signaling pathway. Thus, combining BI6015 with antagonists against the AMPK-HNF4α-Wnt signaling cascade represents another, possibly more efficacious, targetable pathway for drug development (131). This opens the possibility for a future where diseases mediated by dysregulations in HNF4α, could be remedied by intervention with HNF4α antagonists. Of course, there are potential applications for HNF4 agonists that could also be explored.
HNF4 also plays a more direct therapeutic role by being a component of cell-based therapies. Injection of encapsulated immortalized human hepatocytes overexpressing HNF4A promotes hepatic differentiation and improves liver function and survival in rat models with acute liver failure (132). Likewise, injection of mesenchymal stem cells expressing HNF4A in orthotopic human hepatomas in athymic, nude mice results in smaller tumors and decreased metastases through downregulation of Wnt signaling (133). However, none of these therapies, thus far, have been applied to the intestine.
Concluding perspectives
HNF4 is clearly a hub of cellular function. Its significance lies in the fact that it is a highly conserved transcription factor with multifaceted roles across different organs of the gastrointestinal tract. The heterogeneity of its target genes can be accounted for by variations in chromatin looping patterns, post-translational modifications, interaction with coregulators and temporal changes in protein levels in different cell types and conditions. In the intestine, HNF4 plays pivotal roles in its maturation, regeneration, nutrient metabolism, and overall homeostasis. Clearly, the substantial contributions of HNF4 extend beyond its namesake organ. In the future, it will be important to study the transcriptional dynamics of HNF4 in organs such as the pancreas or kidney, to identify drugs that target HNF4, and to interpret the variability of clinical phenotypes arising from HNF4 mutations and SNPs.
While the body of research on HNF4 is large, it is also sometimes contradictory, revealing many unanswered questions. Future areas of investigation could include a deeper study into how promoter switching and alternative splicing are mediated. Isoform-specific effects of HNF4-dependent gene regulation, along with understanding the mechanisms of HNF4γ and how redundancy between HNF4 paralogs potentially works in organs other than the intestine are also important areas of future investigation. Use of conditional knockout rodent models has been fruitful in revealing HNF4 functions in the past, and combinations of mouse genetic tools with dietary interventions and other disease models associated with HNF4 activity should be similarly productive. These studies will be complemented by organoid technologies and improving methodologies in single-cell –omics approaches. CRISPR-based modifications of HNF4-target gene risk alleles and epithelial transplantations could be another area of therapeutic focus. A major challenge in the upcoming era of HNF4 research will be to distill wide-ranging diagnostic and therapeutic studies and translate the knowledge gleaned from them into clinical applications.
Author contributions
KV wrote the draft and revised the review. KV, SR, FS, and MV revised and wrote the final version. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by grants from the National Institutes of Health (NIH) (R01DK121915 and R01DK126446 to MV). KV is supported by an American Heart Association predoctoral fellowship grant (906006).
Acknowledgments
The authors would like to thank the Verzi Lab for helpful comments and feedback.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Barker N, van de Wetering M, Clevers H. The intestinal stem cell. Genes Dev (2008) 22(14):1856–64. doi: 10.1101/gad.1674008
2. van der Wath RC, Gardiner BS, Burgess AW, Smith DW. Cell organisation in the colonic crypt: A theoretical comparison of the pedigree and niche concepts. PloS One (2013) 8(9):e73204. doi: 10.1371/journal.pone.0073204
3. Howitt MR, Lavoie S, Michaud M, Blum AM, Tran SV, Weinstock JV, et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science (2016) 351(6279):1329–33. doi: 10.1126/science.aaf1648
4. Von Moltke J, Ji M, Liang H-E, Locksley RM. Tuft-cell-derived il-25 regulates an intestinal Ilc2–epithelial response circuit. Nature (2016) 529(7585):221–5. doi: 10.1038/nature16161
5. Crawley SW, Mooseker MS, Tyska MJ. Shaping the intestinal brush border. J Cell Biol (2014) 207(4):441–51. doi: 10.1083/jcb.201407015
6. Porter EM, Bevins CL, Ghosh D, Ganz T. The multifaceted paneth cell. Cell Mol Life Sci (2002) 59(1):156–70. doi: 10.1007/s00018-002-8412-z
7. Cray P, Sheahan BJ, Dekaney CM. Secretory sorcery: paneth cell control of intestinal repair and homeostasis. Cell Mol Gastroenterol Hepatol (2021) 12(4):1239–50. doi: 10.1016/j.jcmgh.2021.06.006
8. Wright NA, Alison M. The Biology of Epithelial Cell Populations. USA: Oxford University Press (1984).
9. Fouquet S, Lugo-Martinez VH, Faussat AM, Renaud F, Cardot P, Chambaz J, et al. Early loss of E-cadherin from cell-cell contacts is involved in the onset of anoikis in enterocytes. J Biol Chem (2004) 279(41):43061–9. doi: 10.1074/jbc.M405095200
10. Lugo-Martinez VH, Petit CS, Fouquet S, Le Beyec J, Chambaz J, Pincon-Raymond M, et al. Epidermal growth factor receptor is involved in enterocyte anoikis through the dismantling of E-cadherin-mediated junctions. Am J Physiol Gastrointest Liver Physiol (2009) 296(2):G235–44. doi: 10.1152/ajpgi.90313.2008
11. Chen L, Cao W, Aita R, Aldea D, Flores J, Gao N, et al. Three-dimensional interactions between enhancers and promoters during intestinal differentiation depend upon Hnf4. Cell Rep (2021) 34(4):108679. doi: 10.1016/j.celrep.2020.108679
12. Suh E, Traber PG. An intestine-specific homeobox gene regulates proliferation and differentiation. Mol Cell Biol (1996) 16(2):619–25. doi: 10.1128/MCB.16.2.619
13. Beck F, Chawengsaksophak K, Luckett J, Giblett S, Tucci J, Brown J, et al. A study of regional gut endoderm potency by analysis of Cdx2 null mutant chimaeric mice. Dev Biol (2003) 255(2):399–406. doi: 10.1016/S0012-1606(02)00096-9
14. Verzi MP, Shin H, San ROman AK, Liu XS, Shivdasani RA. Intestinal master transcription factor Cdx2 controls chromatin access for partner transcription factor binding. Mol Cell Biol (2013) 33(2):281–92. doi: 10.1128/mcb.01185-12
15. Gao X, Sedgwick T, Shi Y-B, Evans T. Distinct functions are implicated for the Gata-4,-5, and-6 transcription factors in the regulation of intestine epithelial cell differentiation. Mol Cell Biol (1998) 18(5):2901–11. doi: 10.1128/MCB.18.5.2901
16. Cattin A-L, Le Beyec J, Barreau F, Saint-Just S, Houllier A, Gonzalez FJ, et al. Hepatocyte nuclear factor 4α, a key factor for homeostasis, cell architecture, and barrier function of the adult intestinal epithelium. Mol Cell Biol (2009) 29(23):6294–308. doi: 10.1128/mcb.00939-09
17. Chen L, Toke NH, Luo S, Vasoya RP, Fullem RL, Parthasarathy A, et al. A reinforcing Hnf4-Smad4 feed-forward module stabilizes enterocyte identity. Nat Genet (2019) 51(5):777–85. doi: 10.1038/s41588-019-0384-0
18. Sladek FM, Zhong WM, Lai E, Darnell JE Jr. Liver-enriched transcription factor Hnf-4 is a novel member of the steroid hormone receptor superfamily. Genes Dev (1990) 4(12b):2353–65. doi: 10.1101/gad.4.12b.2353
19. Guo S, Lu H. Novel mechanisms of regulation of the expression and transcriptional activity of hepatocyte nuclear factor 4alpha. J Cell Biochem (2019) 120(1):519–32. doi: 10.1002/jcb.27407
20. The GTE consortium atlas of genetic regulatory effects across human tissues. Science (2020) 369(6509):1318–30. doi: 10.1126/science.aaz1776
21. Fang B, Mane-Padros D, Bolotin E, Jiang T, Sladek FM. Identification of a binding motif specific to Hnf4 by comparative analysis of multiple nuclear receptors. Nucleic Acids Res (2012) 40(12):5343–56. doi: 10.1093/nar/gks190
22. Duncan SA, Manova K, Chen WS, Hoodless P, Weinstein DC, Bachvarova RF, et al. Expression of transcription factor Hnf-4 in the extraembryonic endoderm, gut, and nephrogenic tissue of the developing mouse embryo: Hnf-4 is a marker for primary endoderm in the implanting blastocyst. Proc Natl Acad Sci (1994) 91(16):7598–602. doi: 10.1073/pnas.91.16.7598
23. Lambert E, Babeu JP, Simoneau J, Raisch J, Lavergne L, Levesque D, et al. Human hepatocyte nuclear factor 4-alpha encodes isoforms with distinct transcriptional functions. Mol Cell Proteomics (2020) 19(5):808–27. doi: 10.1074/mcp.RA119.001909
24. Drewes T, Senkel S, Holewa B, Ryffel GU. Human hepatocyte nuclear factor 4 isoforms are encoded by distinct and differentially expressed genes. Mol Cell Biol (1996) 16(3):925–31. doi: 10.1128/mcb.16.3.925
25. Ko HL, Zhuo Z, Ren EC. Hnf4alpha combinatorial isoform heterodimers activate distinct gene targets that differ from their corresponding homodimers. Cell Rep (2019) 26(10):2549–57.e3. doi: 10.1016/j.celrep.2019.02.033
26. Taraviras S, Monaghan AP, Schütz G, Kelsey G. Characterization of the mouse hnf-4 gene and its expression during mouse embryogenesis. Mech Dev (1994) 48(2):67–79. doi: 10.1016/0925-4773(94)90017-5
27. Babeu J-P, Jones C, Geha S, Carrier JC, Boudreau F. P1 promoter-driven Hnf4α Isoforms are specifically repressed by B-catenin signaling in colorectal cancer cells. J Cell Sci (2018) 131(13):jcs214734. doi: 10.1242/jcs.214734
28. Tanaka T, Jiang S, Hotta H, Takano K, Iwanari H, Sumi K, et al. Dysregulated expression of P1 and P2 promoter-driven hepatocyte nuclear factor-4alpha in the pathogenesis of human cancer. J Pathol (2006) 208(5):662–72. doi: 10.1002/path.1928
29. Chellappa K, Deol P, Evans JR, Vuong LM, Chen G, Briançon N, et al. Opposing roles of nuclear receptor Hnf4α Isoforms in colitis and colitis-associated colon cancer. Elife (2016) 5. doi: 10.7554/eLife.10903
30. Briançon N, Weiss MC. In vivo role of the Hnf4alpha Af-1 activation domain revealed by exon swapping. EMBO J (2006) 25(6):1253–62. doi: 10.1038/sj.emboj.7601021
31. Sasaki S, Urabe M, Maeda T, Suzuki J, Irie R, Suzuki M, et al. Induction of hepatic metabolic functions by a novel variant of hepatocyte nuclear factor 4γ. Mol Cell Biol (2018) 38(24):e00213–18. doi: 10.1128/mcb.00213-18
32. Plengvidhya N, Antonellis A, Wogan LT, Poleev A, Borgschulze M, Warram JH, et al. Hepatocyte nuclear factor-4gamma: cdna sequence, gene organization, and mutation screening in early-onset autosomal-dominant type 2 diabetes. Diabetes (1999) 48(10):2099–102. doi: 10.2337/diabetes.48.10.2099
33. Liu W, Xie Y, Ma J, Luo X, Nie P, Zuo Z, et al. Ibs: an illustrator for the presentation and visualization of biological sequences. Bioinformatics (2015) 31(20):3359–61. doi: 10.1093/bioinformatics/btv362
34. Taraviras S, Mantamadiotis T, Dong-Si T, Mincheva A, Lichter P, Drewes T, et al. Primary structure, chromosomal mapping, expression and transcriptional activity of murine hepatocyte nuclear factor 4γ. Biochim Biophys Acta (BBA)-Gene Structure Expression (2000) 1490(1-2):21–32. doi: 10.1016/S0167-4781(99)00232-8
35. Iyemere VP, Davies NH, Brownlee GG. The activation function 2 domain of hepatic nuclear factor 4 is regulated by a short C-terminal proline-rich repressor domain. Nucleic Acids Res (1998) 26(9):2098–104. doi: 10.1093/nar/26.9.2098
36. Dhe-Paganon S, Duda K, Iwamoto M, Chi YI, Shoelson SE. Crystal structure of the Hnf4 alpha ligand binding domain in complex with endogenous fatty acid ligand. J Biol Chem (2002) 277(41):37973–6. doi: 10.1074/jbc.C200420200
37. Wisely GB, Miller AB, Davis RG, Thornquest AD Jr., Johnson R, Spitzer T, et al. Hepatocyte nuclear factor 4 is a transcription factor that constitutively binds fatty acids. Structure (2002) 10(9):1225–34. doi: 10.1016/s0969-2126(02)00829-8
38. Beinsteiner B, Billas IML, Moras D. Structural insights into the Hnf4 biology. Front Endocrinol (Lausanne) (2023) 14:1197063. doi: 10.3389/fendo.2023.1197063
39. Yuan X, Ta TC, Lin M, Evans JR, Dong Y, Bolotin E, et al. Identification of an endogenous ligand bound to a native orphan nuclear receptor. PloS One (2009) 4(5):e5609. doi: 10.1371/journal.pone.0005609
40. Hertz R, Magenheim J, Berman I, Bar-Tana J. Fatty Acyl-Coa thioesters are ligands of hepatic nuclear factor-4alpha. Nature (1998) 392(6675):512–6. doi: 10.1038/33185
41. Viollet B, Kahn A, Raymondjean M. Protein kinase a-dependent phosphorylation modulates DNA-binding activity of hepatocyte nuclear factor 4. Mol Cell Biol (1997) 17(8):4208–19. doi: 10.1128/mcb.17.8.4208
42. Hong YH, Varanasi US, Yang W, Leff T. Amp-activated protein kinase regulates Hnf4α Transcriptional activity by inhibiting dimer formation and decreasing protein stability. J Biol Chem (2003) 278(30):27495–501. doi: 10.1074/jbc.M304112200
43. Guo H, Gao C, Mi Z, Wai PY, Kuo PC. Phosphorylation of Ser158 regulates inflammatory redox-dependent hepatocyte nuclear factor-4alpha transcriptional activity. Biochem J (2006) 394(Pt 2):379–87. doi: 10.1042/bj20051730
44. Sun K, Montana V, Chellappa K, Brelivet Y, Moras D, Maeda Y, et al. Phosphorylation of a conserved serine in the deoxyribonucleic acid binding domain of nuclear receptors alters intracellular localization. Mol Endocrinol (2007) 21(6):1297–311. doi: 10.1210/me.2006-0300
45. Vető B, Bojcsuk D, Bacquet C, Kiss J, Sipeki S, Martin L, et al. The transcriptional activity of hepatocyte nuclear factor 4 alpha is inhibited via phosphorylation by Erk1/2. PloS One (2017) 12(2):e0172020. doi: 10.1371/journal.pone.0172020
46. Chellappa K, Jankova L, Schnabl JM, Pan S, Brelivet Y, Fung CL, et al. Src tyrosine kinase phosphorylation of nuclear receptor Hnf4alpha correlates with isoform-specific loss of Hnf4alpha in human colon cancer. Proc Natl Acad Sci U.S.A. (2012) 109(7):2302–7. doi: 10.1073/pnas.1106799109
47. Soutoglou E, Katrakili N, Talianidis I. Acetylation regulates transcription factor activity at multiple levels. Mol Cell (2000) 5(4):745–51. doi: 10.1016/S1097-2765(00)80253-1
48. Zhou W, Hannoun Z, Jaffray E, Medine CN, Black JR, Greenhough S, et al. Sumoylation of Hnf4α Regulates protein stability and hepatocyte function. J Cell Sci (2012) 125(15):3630–5. doi: 10.1242/jcs.102889
49. Yokoyama A, Katsura S, Ito R, Hashiba W, Sekine H, Fujiki R, et al. Multiple post-translational modifications in hepatocyte nuclear factor 4alpha. Biochem Biophys Res Commun (2011) 410(4):749–53. doi: 10.1016/j.bbrc.2011.06.033
50. Barrero MJ, Malik S. Two functional modes of a nuclear receptor-recruited arginine methyltransferase in transcriptional activation. Mol Cell (2006) 24(2):233–43. doi: 10.1016/j.molcel.2006.09.020
51. Davison JM, Lickwar CR, Song L, Breton G, Crawford GE, Rawls JF. Microbiota regulate intestinal epithelial gene expression by suppressing the transcription factor hepatocyte nuclear factor 4 alpha. Genome Res (2017) 27(7):1195–206. doi: 10.1101/gr.220111.116
52. Camp JG, Frank CL, Lickwar CR, Guturu H, Rube T, Wenger AM, et al. Microbiota modulate transcription in the intestinal epithelium without remodeling the accessible chromatin landscape. Genome Res (2014) 24(9):1504–16. doi: 10.1101/gr.165845.113
53. Lickwar CR, Davison JM, Kelly C, Mercado GP, Wen J, Davis BR, et al. Transcriptional integration of distinct microbial and nutritional signals by the small intestinal epithelium. Cell Mol Gastroenterol Hepatol (2022) 14(2):465–93. doi: 10.1016/j.jcmgh.2022.04.013
54. Gao N, White P, Kaestner KH. Establishment of intestinal identity and epithelial-mesenchymal signaling by Cdx2. Dev Cell (2009) 16(4):588–99. doi: 10.1016/j.devcel.2009.02.010
55. Kumar N, Tsai YH, Chen L, Zhou A, Banerjee KK, Saxena M, et al. The lineage-specific transcription factor Cdx2 navigates dynamic chromatin to control distinct stages of intestine development. Development (2019) 146(5). doi: 10.1242/dev.172189
56. Banerjee K, Saxena M, Kumar N, Chen L, Cavazza A, Toke N, et al. Enhancer, transcriptional, and cell fate plasticity precedes intestinal determination during endoderm development. Genes Dev (2018) 32:21–2. doi: 10.1101/gad.318832.118
57. Chen L, Toke NH, Luo S, Vasoya RP, Aita R, Parthasarathy A, et al. Hnf4 factors control chromatin accessibility and are redundantly required for maturation of the fetal intestine. Development (2019) 146(19):dev179432. doi: 10.1242/dev.179432
58. Montenegro-MIranda PS, van der Meer JHM, Jones C, Meisner S, Vermeulen JLM, Koster J, et al. A novel organoid model of damage and repair identifies Hnf4α as a critical regulator of intestinal epithelial regeneration. Cell Mol Gastroenterol Hepatol (2020) 10(2):209–23. doi: 10.1016/j.jcmgh.2020.02.007
59. Chen L, Vasoya RP, Toke NH, Parthasarathy A, Luo S, Chiles E, et al. Hnf4 regulates fatty acid oxidation and is required for renewal of intestinal stem cells in mice. Gastroenterology (2020) 158(4):985–99.e9. doi: 10.1053/j.gastro.2019.11.031
60. Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE, et al. A pml-Ppar-delta pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med (2012) 18(9):1350–8. doi: 10.1038/nm.2882
61. Beyaz S, Mana MD, Roper J, Kedrin D, Saadatpour A, Hong SJ, et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature (2016) 531(7592):53–8. doi: 10.1038/nature17173
62. San ROman AK, Aronson BE, Krasinski SD, Shivdasani RA, Verzi MP. Transcription factors Gata4 and Hnf4a control distinct aspects of intestinal homeostasis in conjunction with transcription factor Cdx2. J Biol Chem (2015) 290(3):1850–60. doi: 10.1074/jbc.M114.620211
63. Chen L, Luo S, Dupre A, Vasoya RP, Parthasarathy A, Aita R, et al. The nuclear receptor Hnf4 drives a brush border gene program conserved across murine intestine, kidney, and embryonic yolk sac. Nat Commun (2021) 12(1):2886. doi: 10.1038/s41467-021-22761-5
64. Duncan SA, Navas MA, Dufort D, Rossant J, Stoffel M. Regulation of a transcription factor network required for differentiation and metabolism. Science (1998) 281(5377):692–5. doi: 10.1126/science.281.5377.692
65. Hatzis P, Talianidis I. Regulatory mechanisms controlling human hepatocyte nuclear factor 4alpha gene expression. Mol Cell Biol (2001) 21(21):7320–30. doi: 10.1128/mcb.21.21.7320-7330.2001
66. Sancho E, Batlle E, Clevers H. Signaling pathways in intestinal development and cancer. Annu Rev Cell Dev Biol (2004) 20:695–723. doi: 10.1146/annurev.cellbio.20.010403.092805
67. Stainier DY. No organ left behind: tales of gut development and evolution. Science (2005) 307(5717):1902–4. doi: 10.1126/science.1108709
68. van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, et al. The Beta-catenin/Tcf-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell (2002) 111(2):241–50. doi: 10.1016/s0092-8674(02)01014-0
69. Garrison WD, Battle MA, Yang C, Kaestner KH, Sladek FM, Duncan SA. Hepatocyte nuclear factor 4alpha is essential for embryonic development of the mouse colon. Gastroenterology (2006) 130(4):1207–20. doi: 10.1053/j.gastro.2006.01.003
70. Parviz F, Matullo C, Garrison WD, Savatski L, Adamson JW, Ning G, et al. Hepatocyte nuclear factor 4alpha controls the development of a hepatic epithelium and liver morphogenesis. Nat Genet (2003) 34(3):292–6. doi: 10.1038/ng1175
71. Schmidt D, Wilson MD, Ballester B, Schwalie PC, Brown GD, Marshall A, et al. Five-vertebrate chip-seq reveals the evolutionary dynamics of transcription factor binding. Science (2010) 328(5981):1036–40. doi: 10.1126/science.1186176
72. Harries LW, Brown JE, Gloyn AL. Species-specific differences in the expression of the Hnf1a, Hnf1b and Hnf4a genes. PloS One (2009) 4(11):e7855. doi: 10.1371/journal.pone.0007855
73. Stegmann A, Hansen M, Wang Y, Larsen JB, Lund LR, Ritié L, et al. Metabolome, transcriptome, and bioinformatic cis-element analyses point to hnf-4 as a central regulator of gene expression during enterocyte differentiation. Physiol Genomics (2006) 27(2):141–55. doi: 10.1152/physiolgenomics.00314.2005
74. Marcil V, Seidman E, Sinnett D, Boudreau F, Gendron F-P, Beaulieu J-F, et al. Modification in oxidative stress, inflammation, and lipoprotein assembly in response to hepatocyte nuclear factor 4α Knockdown in intestinal epithelial cells. J Biol Chem (2010) 285(52):40448–60. doi: 10.1074/jbc.M110.155358
75. Frochot V, Alqub M, Cattin A-L, Carrière V, Houllier A, Baraille F, et al. The transcription factor Hnf-4α: A key factor of the intestinal uptake of fatty acids in mouse. Am J Physiology-Gastrointestinal Liver Physiol (2012) 302(11):G1253–G63. doi: 10.1152/ajpgi.00329.2011
76. Palanker L, Tennessen JM, Lam G, Thummel CS. Drosophila Hnf4 regulates lipid mobilization and Beta-oxidation. Cell Metab (2009) 9(3):228–39. doi: 10.1016/j.cmet.2009.01.009
77. Petrescu AD, Payne HR, Boedecker A, Chao H, Hertz R, Bar-Tana J, et al. Physical and functional interaction of Acyl-Coa-binding protein with hepatocyte nuclear factor-4α. J Biol Chem (2003) 278(51):51813–24. doi: 10.1074/jbc.M303858200
78. Nauck MA, Meier JJ. Incretin hormones: their role in health and disease. Diabetes Obes Metab (2018) 20(S1):5–21. doi: 10.1111/dom.13129
79. Girard R, Darsigny M, Jones C, Maloum-Rami F, Gélinas Y, Carpentier AC, et al. Hnf4α Is a novel regulator of intestinal glucose-dependent insulinotropic polypeptide. Sci Rep (2019) 9(1):4200. doi: 10.1038/s41598-019-41061-z
80. Girard R, Tremblay S, Noll C, St-Jean S, Jones C, Gélinas Y, et al. The transcription factor hepatocyte nuclear factor 4a acts in the intestine to promote white adipose tissue energy storage. Nat Commun (2022) 13(1):224. doi: 10.1038/s41467-021-27934-w
81. López-Díaz L, Jain RN, Keeley TM, VanDussen KL, Brunkan CS, Gumucio DL, et al. Intestinal neurogenin 3 directs differentiation of a bipotential secretory progenitor to endocrine cell rather than goblet cell fate. Dev Biol (2007) 309(2):298–305. doi: 10.1016/j.ydbio.2007.07.015
82. Chiba H, Gotoh T, Kojima T, Satohisa S, Kikuchi K, Osanai M, et al. Hepatocyte nuclear factor (Hnf)-4alpha triggers formation of functional tight junctions and establishment of polarized epithelial morphology in F9 embryonal carcinoma cells. Exp Cell Res (2003) 286(2):288–97. doi: 10.1016/s0014-4827(03)00116-2
83. Barrett JC, Lee JC, Lees CW, Prescott NJ, Anderson CA, Phillips A, et al. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the Hnf4a region. Nat Genet (2009) 41(12):1330–4. doi: 10.1038/ng.483
84. van Sommeren S, Visschedijk MC, Festen EA, de Jong DJ, Ponsioen CY, Wijmenga C, et al. Hnf4alpha and Cdh1 are associated with ulcerative colitis in a dutch cohort. Inflammation Bowel Dis (2011) 17(8):1714–8. doi: 10.1002/ibd.21541
85. Yang SK, Jung Y, Kim H, Hong M, Ye BD, Song K. Association of Fcgr2a, Jak2 or Hnf4a variants with ulcerative colitis in koreans. Dig Liver Dis (2011) 43(11):856–61. doi: 10.1016/j.dld.2011.07.006
86. VanDussen KL, Stojmirović A, Li K, Liu TC, Kimes PK, Muegge BD, et al. Abnormal small intestinal epithelial microvilli in patients with crohn’s disease. Gastroenterology (2018) 155(3):815–28. doi: 10.1053/j.gastro.2018.05.028
87. Shiner M, Birbeck MS. The microvilli of the small intestinal surface epithelium in coeliac disease and in idiopathic steatorrhoea. Gut (1961) 2(3):277–84. doi: 10.1136/gut.2.3.277
88. Fell JM, Miller MP, Finkel Y, Booth IW. Congenital sodium diarrhea with a partial defect in jejunal brush border membrane sodium transport, normal rectal transport, and resolving diarrhea. J Pediatr Gastroenterol Nutr (1992) 15(2):112–6. doi: 10.1097/00005176-199208000-00002
89. Rodriguez-Colman MJ, Schewe M, Meerlo M, Stigter E, Gerrits J, Pras-Raves M, et al. Interplay between metabolic identities in the intestinal crypt supports stem cell function. Nature (2017) 543(7645):424–7. doi: 10.1038/nature21673
90. Schell JC, Wisidagama DR, Bensard C, Zhao H, Wei P, Tanner J, et al. Control of intestinal stem cell function and proliferation by mitochondrial pyruvate metabolism. Nat Cell Biol (2017) 19(9):1027–36. doi: 10.1038/ncb3593
91. Maeda Y, Seidel SD, Wei G, Liu X, Sladek FM. Repression of hepatocyte nuclear factor 4alpha tumor suppressor P53: involvement of the ligand-binding domain and histone deacetylase activity. Mol Endocrinol (2002) 16(2):402–10. doi: 10.1210/mend.16.2.0769
92. Maeda Y, Hwang-Verslues WW, Wei G, Fukazawa T, Durbin ML, Owen LB, et al. Tumour suppressor P53 down-regulates the expression of the human hepatocyte nuclear factor 4alpha (Hnf4alpha) gene. Biochem J (2006) 400(2):303–13. doi: 10.1042/bj20060614
93. Jones C, Avino M, Giroux V, Boudreau F. Hnf4α Acts as upstream functional regulator of intestinal Wnt3 and paneth cell fate. Cell Mol Gastroenterol Hepatol (2023) 15(3):593–612. doi: 10.1016/j.jcmgh.2022.11.010
94. Babeu JP, Darsigny M, Lussier CR, Boudreau F. Hepatocyte nuclear factor 4alpha contributes to an intestinal epithelial phenotype in vitro and plays a partial role in mouse intestinal epithelium differentiation. Am J Physiol Gastrointest Liver Physiol (2009) 297(1):G124–34. doi: 10.1152/ajpgi.90690.2008
95. Gerdin AK, Surve VV, Jonsson M, Bjursell M, Bjorkman M, Edenro A, et al. Phenotypic screening of hepatocyte nuclear factor (Hnf) 4-gamma receptor knockout mice. Biochem Biophys Res Commun (2006) 349(2):825–32. doi: 10.1016/j.bbrc.2006.08.103
96. Baraille F, Ayari S, Carriere V, Osinski C, Garbin K, Blondeau B, et al. Glucose tolerance is improved in mice invalidated for the nuclear receptor Hnf-4gamma: A critical role for enteroendocrine cell lineage. Diabetes (2015) 64(8):2744–56. doi: 10.2337/db14-0993
97. Heppert JK, Lickwar CR, Tillman MC, Davis BR, Davison JM, Lu H-Y, et al. Conserved roles for Hnf4 family transcription factors in zebrafish development and intestinal function. Genetics (2022) 222(4). doi: 10.1093/genetics/iyac133
98. Holewa B, Zapp D, Drewes T, Senkel S, Ryffel GU. Hnf4beta, a new gene of the Hnf4 family with distinct activation and expression profiles in oogenesis and embryogenesis of Xenopus laevis. Mol Cell Biol (1997) 17(2):687–94. doi: 10.1128/mcb.17.2.687
99. Bertrand S, Brunet FG, Escriva H, Parmentier G, Laudet V, Robinson-Rechavi M. Evolutionary genomics of nuclear receptors: from twenty-five ancestral genes to derived endocrine systems. Mol Biol Evol (2004) 21(10):1923–37. doi: 10.1093/molbev/msh200
100. Lei X, Ketelut-Carneiro N, Shmuel-Galia L, Xu W, Wilson R, Vierbuchen T, et al. Epithelial hnf4a shapes the intraepithelial lymphocyte compartment via direct regulation of immune signaling molecules. J Exp Med (2022) 219(8). doi: 10.1084/jem.20212563
101. Qu M, Duffy T, Hirota T, Kay SA. Nuclear receptor Hnf4a transrepresses clock : Bmal1 and modulates tissue-specific circadian networks. Proc Natl Acad Sci (2018) 115(52):E12305–E12. doi: 10.1073/pnas.1816411115
102. Marcil V, Sinnett D, Seidman E, Boudreau F, Gendron FP, Beaulieu JF, et al. Association between genetic variants in the Hnf4a gene and childhood-onset crohn’s disease. Genes Immun (2012) 13(7):556–65. doi: 10.1038/gene.2012.37
103. Berndt SI, Gustafsson S, Magi R, Ganna A, Wheeler E, Feitosa MF, et al. Genome-wide meta-analysis identifies 11 new loci for anthropometric traits and provides insights into genetic architecture. Nat Genet (2013) 45(5):501–12. doi: 10.1038/ng.2606
104. Gardner DS, Tai ES. Clinical features and treatment of maturity onset diabetes of the young (Mody). Diabetes Metab Syndr Obes (2012) 5:101–8. doi: 10.2147/dmso.S23353
105. DeFronzo RA, Bonadonna RC, Ferrannini E. Pathogenesis of niddm: A balanced overview. Diabetes Care (1992) 15(3):318–68. doi: 10.2337/diacare.15.3.318
106. Yamagata K, Furuta H, Oda N, Kaisaki PJ, Menzel S, Cox NJ, et al. Mutations in the hepatocyte nuclear factor-4alpha gene in maturity-onset diabetes of the young (Mody1). Nature (1996) 384(6608):458–60. doi: 10.1038/384458a0
107. Hani EH, Suaud L, Boutin P, Chevre JC, Durand E, Philippi A, et al. A missense mutation in hepatocyte nuclear factor-4 alpha, resulting in a reduced transactivation activity, in human late-onset non-insulin-dependent diabetes mellitus. J Clin Invest (1998) 101(3):521–6. doi: 10.1172/jci1403
108. Hamilton AJ, Bingham C, McDonald TJ, Cook PR, Caswell RC, Weedon MN, et al. The hnf4a R76w mutation causes atypical dominant fanconi syndrome in addition to a beta cell phenotype. J Med Genet (2014) 51(3):165–9. doi: 10.1136/jmedgenet-2013-102066
109. Stoffel M, Duncan SA. The maturity-onset diabetes of the young (Mody1) transcription factor Hnf4alpha regulates expression of genes required for glucose transport and metabolism. Proc Natl Acad Sci U.S.A. (1997) 94(24):13209–14. doi: 10.1073/pnas.94.24.13209
110. Sladek FM, Dallas-Yang Q, Nepomuceno L. Mody1 mutation Q268x in hepatocyte nuclear factor 4alpha allows for dimerization in solution but causes abnormal subcellular localization. Diabetes (1998) 47(6):985–90. doi: 10.2337/diabetes.47.6.985
111. Ahn SH, Shah YM, Inoue J, Morimura K, Kim I, Yim S, et al. Hepatocyte nuclear factor 4alpha in the intestinal epithelial cells protects against inflammatory bowel disease. Inflammation Bowel Dis (2008) 14(7):908–20. doi: 10.1002/ibd.20413
112. Oshima T, Kawasaki T, Ohashi R, Hasegawa G, Jiang S, Umezu H, et al. Downregulated P1 promoter-driven hepatocyte nuclear factor-4alpha expression in human colorectal carcinoma is a new prognostic factor against liver metastasis. Pathol Int (2007) 57(2):82–90. doi: 10.1111/j.1440-1827.2006.02061.x
113. Barkas F, Liberopoulos E, Kei A, Elisaf M. Electrolyte and acid-base disorders in inflammatory bowel disease. Ann Gastroenterol (2013) 26(1):23–8.
114. Muthusamy S, Jeong JJ, Cheng M, Bonzo JA, Kumar A, Gonzalez FJ, et al. Hepatocyte nuclear factor 4α Regulates the expression of intestinal epithelial na+/H+ Exchanger isoform 3. Am J Physiology-Gastrointestinal Liver Physiol (2018) 314(1):G14–21. doi: 10.1152/ajpgi.00225.2017
115. Ning B-F, Ding J, Yin C, Zhong W, Wu K, Zeng X, et al. Hepatocyte nuclear factor 4α Suppresses the development of hepatocellular carcinoma. Cancer Res (2010) 70(19):7640–51. doi: 10.1158/0008-5472.Can-10-0824
116. Hatziapostolou M, Polytarchou C, Aggelidou E, Drakaki A, Poultsides GA, Jaeger SA, et al. An Hnf4α-mirna inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell (2011) 147(6):1233–47. doi: 10.1016/j.cell.2011.10.043
117. Vuong LM, Chellappa K, Dhahbi JM, Deans JR, Fang B, Bolotin E, et al. Differential effects of hepatocyte nuclear factor 4α Isoforms on tumor growth and T-cell factor 4/ap-1 interactions in human colorectal cancer cells. Mol Cell Biol (2015) 35(20):3471–90. doi: 10.1128/MCB.00030-15
118. Lamers MM, Beumer J, van der Vaart J, Knoops K, Puschhof J, Breugem TI, et al. Sars-cov-2 productively infects human gut enterocytes. Science (2020) 369(6499):50–4. doi: 10.1101/2020.04.25.060350
119. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. Sars-cov-2 cell entry depends on ace2 and Tmprss2 and is blocked by a clinically proven protease inhibitor. Cell (2020) 181(2):271–80.e8. doi: 10.1016/j.cell.2020.02.052
120. Chen L, Marishta A, Ellison CE, Verzi MP. Identification of transcription factors regulating Sars-Cov-2 entry genes in the intestine. Cell Mol Gastroenterol Hepatol (2020) 11(1):181–4. doi: 10.1016/j.jcmgh.2020.08.005
121. Franke A, Hampe J, Rosenstiel P, Becker C, Wagner F, Hasler R, et al. Systematic association mapping identifies nell1 as a novel Ibd disease gene. PloS One (2007) 2(8):e691. doi: 10.1371/journal.pone.0000691
122. Bueno-Hernández N, Sánchez-Muñoz F, Barreto-Zuñiga R, Dominguez-López A, Yamamoto-Furusho JK. Expression of Hnf4γ Is downregulated in patients with active ulcerative colitis (Uc) compared to uc patients in remission and healthy controls. Inflamm Bowel Dis (2011) 17(8):E91–E. doi: 10.1002/ibd.21753
123. Okegawa T, Ushio K, Imai M, Morimoto M, Hara T. Orphan nuclear receptor Hnf4g promotes bladder cancer growth and invasion through the regulation of the hyaluronan synthase 2 gene. Oncogenesis (2013) 2(7):e58. doi: 10.1038/oncsis.2013.25
124. Sousa JF, Nam KT, Petersen CP, Lee HJ, Yang HK, Kim WH, et al. Mir-30-hnf4γ and Mir-194-Nr2f2 regulatory networks contribute to the upregulation of metaplasia markers in the stomach. Gut (2016) 65(6):914–24. doi: 10.1136/gutjnl-2014-308759
125. Wang J, Zhang J, Xu L, Zheng Y, Ling D, Yang Z. Expression of Hnf4g and its potential functions in lung cancer. Oncotarget (2018) 9(26):18018–28. doi: 10.18632/oncotarget.22933
126. Jucá P, Corrêa S, Vignal GM, Accioly M, Lustosa SAS, Abdelhay E, et al. Hnf4a expression as a potential diagnostic tool to discriminate primary gastric cancer from breast cancer metastasis in a Brazilian cohort. Diagn Pathol (2017) 12(1):43. doi: 10.1186/s13000-017-0635-2
127. van der Post RS, Bult P, Vogelaar IP, Ligtenberg MJ, Hoogerbrugge N, van Krieken JH. Hnf4a immunohistochemistry facilitates distinction between primary and metastatic breast and gastric carcinoma. Virchows Arch (2014) 464(6):673–9. doi: 10.1007/s00428-014-1574-x
128. Kiselyuk A, Farber-Katz S, Cohen T, Lee S-H, Geron I, Azimi B, et al. Phenothiazine neuroleptics signal to the human insulin promoter as revealed by a novel high-throughput screen. J Biomol Screening (2010) 15(6):663–70. doi: 10.1177/1087057110372257
129. Kiselyuk A, Lee S-H, Farber-Katz S, Zhang M, Athavankar S, Cohen T, et al. Hnf4α Antagonists discovered by a high-throughput screen for modulators of the human insulin promoter. Chem Biol (2012) 19(7):806–18. doi: 10.1016/j.chembiol.2012.05.014
130. Kim J-H, Eom HJ, Lim G, Park S, Lee J, Nam S, et al. Differential effects, on oncogenic pathway signalling, by derivatives of the Hnf4 A Inhibitor Bi6015. Br J Cancer (2019) 120(5):488–98. doi: 10.1038/s41416-018-0374-5
131. Chang HR, Nam S, Kook M-C, Kim K-T, Liu X, Yao H, et al. Hnf4α Is a therapeutic target that links Ampk to Wnt signalling in early-stage gastric cancer. Gut (2016) 65(1):19–32. doi: 10.1136/gutjnl-2014-307918
132. Hang HL, Liu XY, Wang HT, Xu N, Bian JM, Zhang JJ, et al. Hepatocyte nuclear factor 4a improves hepatic differentiation of immortalized adult human hepatocytes and improves liver function and survival. Exp Cell Res (2017) 360(2):81–93. doi: 10.1016/j.yexcr.2017.08.020
133. Wu N, Zhang YL, Wang HT, Li DW, Dai HJ, Zhang QQ, et al. Overexpression of hepatocyte nuclear factor 4α in human mesenchymal stem cells suppresses hepatocellular carcinoma development through Wnt/B-catenin signaling pathway downregulation. Cancer Biol Ther (2016) 17(5):558–65. doi: 10.1080/15384047.2016.1177675
Keywords: HNF4, transcription factor, intestine, redundancy, intestinal differentiation, intestinal regeneration, colon cancer, inflammatory bowel disease
Citation: Vemuri K, Radi SH, Sladek FM and Verzi MP (2023) Multiple roles and regulatory mechanisms of the transcription factor HNF4 in the intestine. Front. Endocrinol. 14:1232569. doi: 10.3389/fendo.2023.1232569
Received: 31 May 2023; Accepted: 24 July 2023;
Published: 10 August 2023.
Edited by:
Wendong Huang, City of Hope, United StatesReviewed by:
Nisha Sharma, City of Hope National Medical Center, United StatesMingjie Fan, Shandong First Medical University, China
Copyright © 2023 Vemuri, Radi, Sladek and Verzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kiranmayi Vemuri, kv257@scarletmail.rutgers.edu