 Hye-Sun Park
Hye-Sun Park Yeon Hee Lee2†
Yeon Hee Lee2† Namki Hong
Namki Hong Dongju Won
Dongju Won Yumie Rhee
Yumie Rhee- 1Department of Internal Medicine, Gangnam Severance Hospital, Yonsei University College of Medicine, Seoul, South Korea
- 2Department of Internal Medicine, Seoul Eco Internal Medicine Clinic, Seoul, South Korea
- 3Department of Internal Medicine, Endocrine Research Institute, Yonsei University College of Medicine, Seoul, South Korea
- 4Department of Laboratory Medicine, Severance Hospital, Yonsei University College of Medicine, Seoul, South Korea
Primary hyperparathyroidism (PHPT) is characterized by overproduction of parathyroid hormone and subsequent hypercalcemia. Approximately 10% of PHPT cases are hereditary, and several genes, such as MEN1, RET, CASR, and CDC73, are responsible for the familial forms of PHPT. However, other genetic mutations involved in the etiology of PHPT are largely unknown. In this study, we identified genetic variants that might be responsible for PHPT, including familial PHPT, benign sporadic PHPT, and sporadic parathyroid cancer, using next-generation sequencing (NGS). A total of 107 patients with PHPT who underwent NGS from 2017 to 2021 at Severance Hospital were enrolled. We reviewed the pathogenic variants, likely pathogenic variants, and variants of uncertain significance (VUS) according to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology criteria. Of the 107 patients (mean age: 47.6 ± 16.1 years, women 73.8%), 12 patients were diagnosed with familial PHPT, 13 with parathyroid cancer, and 82 with benign sporadic PHPT. Using NGS, we identified three pathogenic variants in two genes (CDC73 and MEN1), 10 likely pathogenic variants in six genes (CASR, CDC73, LRP5, MEN1, SDHA, and VHL), and 39 non-synonymous VUS variants that could be related to parathyroid disease. Interestingly, we identified one GCM2 variant (c.1162A>G [p.Lys388Glu]) and five APC variants that were previously reported in familial isolated hyperparathyroidism, benign sporadic PHPT, and parathyroid cancer. We also analyzed the characteristics of subjects with positive genetic test results (pathogenic or likely pathogenic variants), and 76.9% of them had at least one of the following features: 1) age < 40 years, 2) family history of PHPT, 3) multiglandular PHPT, or 4) recurrent PHPT. In this study, we analyzed the NGS data of patients with PHPT and observed variants that could possibly be related to PHPT pathogenesis. NGS screening for selected patients with PHPT might help in the diagnosis and management of the disease.
Introduction
Primary hyperparathyroidism (PHPT) is a common endocrinological disorder with an estimated prevalence of one to seven per 1,000 adults (1). It is characterized by overproduction of the parathyroid hormone (PTH) and hypercalcemia, leading to complications such as osteoporosis and formation of renal stones. (2) The hereditary form of PHPT accounts for approximately 10% of all cases, including multiple endocrine neoplasia (MEN) 1, MEN2A, familial hypocalciuric hypercalcemia (FHH), neonatal severe hyperparathyroidism, hyperparathyroidism jaw tumor syndrome (HPT-JT), and familial isolated hyperparathyroidism (FIHP). (3, 4) Eighty-five percent of PHPT cases are usually sporadic, and < 1% of PHPT cases present as parathyroid cancer, which is commonly associated with severe hypercalcemia and associated clinical manifestations (4).
Several genes have been established as containing disease-causing mutations for the familial PHPT: MEN1 gene for MEN1, RET gene for MEN2A, CASR gene for neonatal severe hyperparathyroidism, and CDC73 gene for HPT-JT. A few other genetic mutations in familial PHPT have been discovered relatively recently. FHH was initially known to be caused by a mutation in CASR. However, GNA11 and AP2S1 mutations were additionally identified as causes of FHH type 2 and type 3, respectively. (5, 6) In addition, GCM2 mutations were recently identified in FIHP, another form of familial PHPT. Several germline mutations in CDC73, MEN1, CASR, and PTH are associated with benign sporadic PHPT or parathyroid cancer. (7) However, data on genetic abnormalities in PHPT are limited. In this study, we identified genetic alterations that may be involved in the pathogenesis of PHPT, using next-generation sequencing (NGS) data.
Methods
Study Participants
We enrolled 107 patients with PHPT who visited the endocrinology clinic at Severance Hospital and underwent NGS from 2017 to 2021. PHPT was diagnosed as inappropriately high intact PTH (normal range: 15–65 pg/mL) with normal or high albumin-corrected serum calcium levels (normal range: 8.5–10.1 mg/dL). We excluded patients who had elevated PTH levels due to secondary causes, such as chronic kidney disease and vitamin D deficiency. We collected baseline information of the study participants, including age, sex, medical history, family history, and PHPT forms. We classified MEN, FHH, and FIHP as familial PHPT. (8) Persistent PHPT was defined as elevated serum calcium levels within 6 months after primary surgery for PHPT, whereas recurrent PHPT was defined as elevated serum calcium levels that presented after 6 months of initial normocalcemia following primary surgery for PHPT. (9) Multiglandular PHPT was defined as the presence of two or more enlarged parathyroid glands. This study was approved by the Institutional Review Board of Severance Hospital, Yonsei University Health System, Seoul, Korea (No.4-2021-1387).
Laboratory Data and Gene Sequencing
Calcium, phosphorus, albumin, alkaline phosphatase, intact PTH, blood urea nitrogen, creatinine, 25-hydroxy vitamin D, ionized calcium, and 24 h urinary calcium levels were measured routine laboratory methods. Albumin-corrected calcium was calculated by the following equation: serum calcium (mg/dL) + 0.8×(4.0-alubmin [g/dL]). (10) The serum intact PTH concentration was measured using a second-generation PTH assay (Elecsys PTH; Roche Diagnostics, Mannheim, Germany) on a Cobas e801 immunoassay analyzer (Roche Diagnostics).
The patients underwent gene sequencing using either targeted gene sequencing or clinical exome sequencing. A customized NGS panel was used for targeted sequencing, which included 400 genes related to various endocrine disorders. (Supplemenatry Table 1) The other NGS panel was the xGen Inherited Diseases Panel (Integrated DNA Technologies, Coralville, IA, USA) comprising 4,503 genes for clinical exome sequencing. We used this expanded NGS panel since September 2019, and the patients who visited the clinic before September 2019 underwent targeted gene sequencing.
Genomic DNA was extracted from leukocytes of peripheral blood samples using the QIAamp Blood DNA Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. Subsequent sequencing procedures and data analyses were conducted as previously described. (11, 12) The variants were interpreted using the 5-tier classification system recommended by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology guidelines (13).
Data Analysis
We analyzed all reported pathogenic variants, likely pathogenic variants, and variants of uncertain significance (VUSs). Pathogenic and likely pathogenic variants were defined as positive genetic tests. Among VUSs, we prioritized variants that met the following: 1) non-synonymous variants that are missense variants, frameshift variants, or variants at canonical ± 1 or 2 splice sites and 2) variants in genes that were in the candidate gene list of parathyroid disease. We built a list of genes associated with parathyroid disease based on previous studies. We obtained a list of candidate parathyroid genes from Cetani et al. (14) They sorted 118 genes co-occurring with the term ‘parathyroid’ in literature-supported statements from the GeneRIF Biological Term Annotations dataset, and 41 genes from previous studies. (15–17) We then collected candidate genes from previous studies on PHPT. (14, 18) Supplementary Table 2 shows the final 161 candidate gene list of parathyroid disease.
Statistical Analysis
Values are presented as mean with standard deviation for normally distributed continuous variables, or median with interquartile range for non-normally distributed continuous variables. Categorical variables are described as numbers with percentages (%) and compared using the chi-square analysis. The Mann–Whitney test was used for continuous variables with non-normal distribution to compare the differences between groups. Statistical significance was set at p < 0.05. All statistical analyses were conducted using the Statistical Package for Social Sciences for Windows version 26.0 (IBM Corp., Armonk, NY, USA).
Results
The baseline characteristics of the study subjects are shown in Table 1. The mean age of the study subjects was 47.6 ± 16.1 years and 73.8% of them were women. Of the 107 study patients, 12 (11.2%) patients were diagnosed with familial PHPT, comprised of five patients with MEN1, six with FHH, and one with FIHP. Other 95 patients were diagnosed with sporadic PHPT, and 13 of them had pathologically confirmed parathyroid cancer. Mean serum calcium and intact PTH levels were 11.3 ± 1.7 mg/dL and 264.6 ± 385.3 pg/mL, respectively. Seventy-seven subjects underwent gene sequencing with a targeted NGS panel, and 30 subjects underwent clinical exome sequencing. The baseline characteristics did not differ between the groups according to the NGS panel (Supplementary Table 3). Targeted sequencing panel detected 41 variants (one pathogenic variant, seven likely pathogenic variants, and 33 VUSs) and the other panel for clinical exome sequencing identified 11 variants (two pathogenic variants, three likely pathogenic variants and six VUSs).
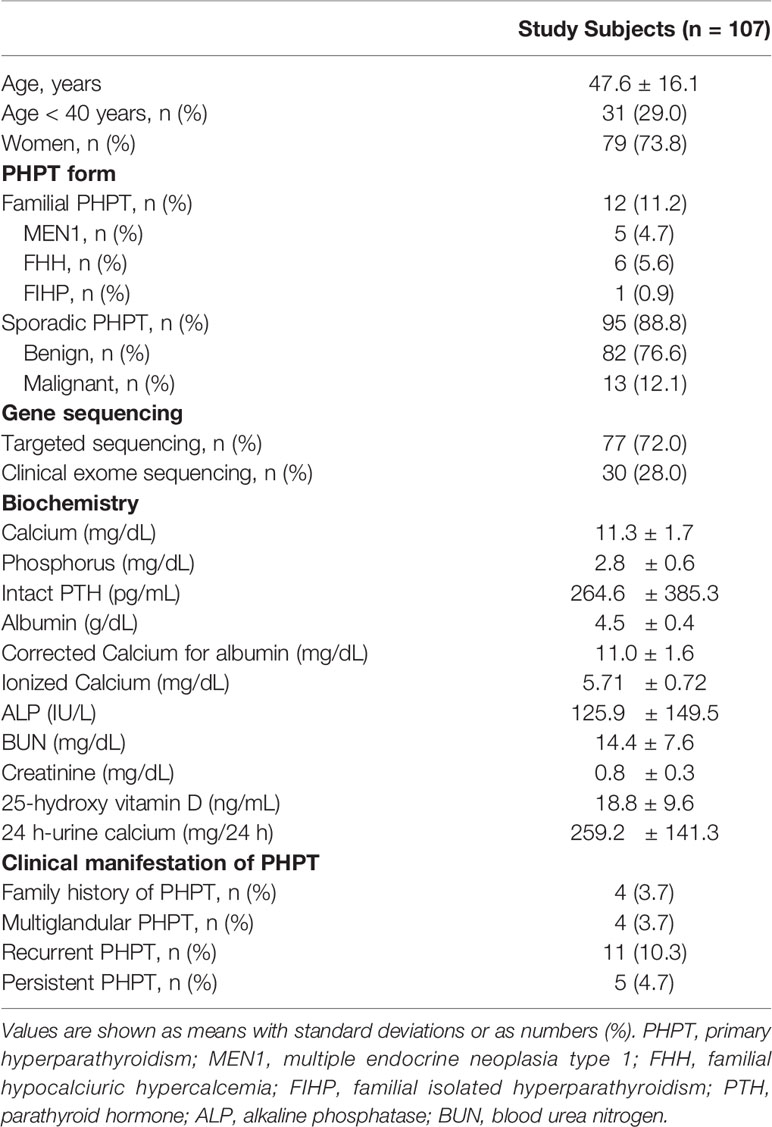
Table 1 Baseline characteristics of study subjects.
The variants identified by NGS are listed on Table 2 according to their clinical diagnosis. Patients with familial PHPT or parathyroid cancer had germline mutation in MEN1, CASR, and CDC73, classified as pathogenic or likely pathogenic variants. The specific criteria used for each pathogenic and likely pathogenic variants are shown in Supplementary Table 4.
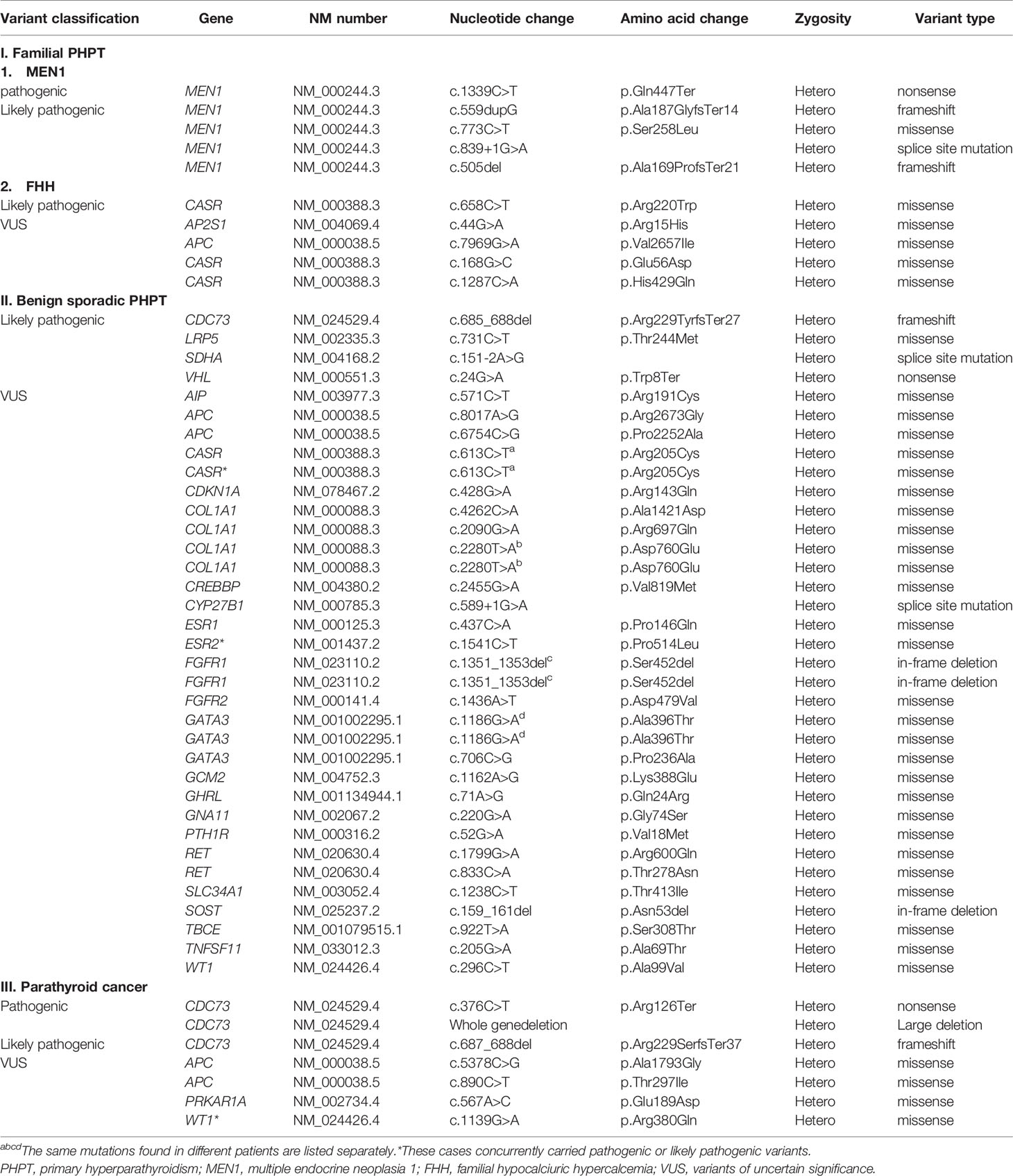
Table 2 Genetic variants identified by next-generation sequencing.
A total of 1,315 VUSs were detected in 484 genes, and we prioritized non-synonymous variants (missense variants, frameshift variants, and variants at canonical ± 1 or 2 splice sites), which were included in our candidate parathyroid gene panel (Supplementary Table 2). Thirty-nine VUSs were selected and are listed in Table 2. Among the 39 variants, three VUSs (WT1, ESR2, and CASR) were accompanied by pathogenic or likely pathogenic variants (CDC73, LRP5, and VHL, respectively). The clinical characteristics of the three cases are shown in Table 3.
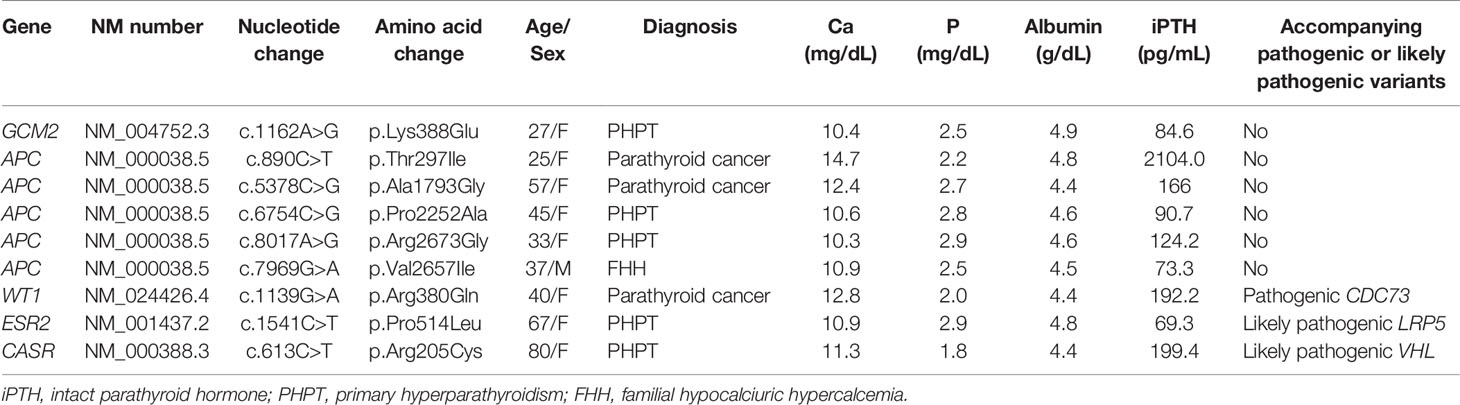
Table 3 Patients with VUSs in GCM2, APC, WT1, ESR2, and CASR genes.
As APC and GCM2 variants were recently reported in PHPT and parathyroid cancer, (15, 17) we analyzed the data of subjects with variants in APC and GCM2 genes (Table 3). One of the patients had a GCM2 variant, and was diagnosed with PHPT at the age of 27. Five subjects had APC variants, and their mean age at the time of diagnosis was 37.3 ± 12.0. Two out of five subjects with APC variants were diagnosed with parathyroid cancer.
We further analyzed the clinical characteristics of the study subjects according to their genetic tests (Table 4). The median age of subjects with positive test results was not significantly different from that of subjects without positive test results. Among the patients with positive test results, 38.5% had the recurrent PHPT, which was significantly higher than those without positive test results (38.5% vs. 11.7%, p < 0.025) We had 11 patients with recurrent PHPT in this study, and 5 of them had positive genetic test results. Four out of these five patients had either MEN1 (n=2) or parathyroid cancer (n=2). In contrast, none of the five patients with persistent PHPT had positive test result. Of the subjects with positive test results, 76.9% met at least one of the following clinical features: 1) age < 40 years, 2) family history of PHPT, 3) multiglandular PHPT, or 4) recurrent PHPT. This proportion was significantly higher in those with positive results than in those without (76.9% vs. 38.3%, p = 0.010). Laboratory findings, such as serum calcium and intact PTH levels, were statistically similar between the groups.
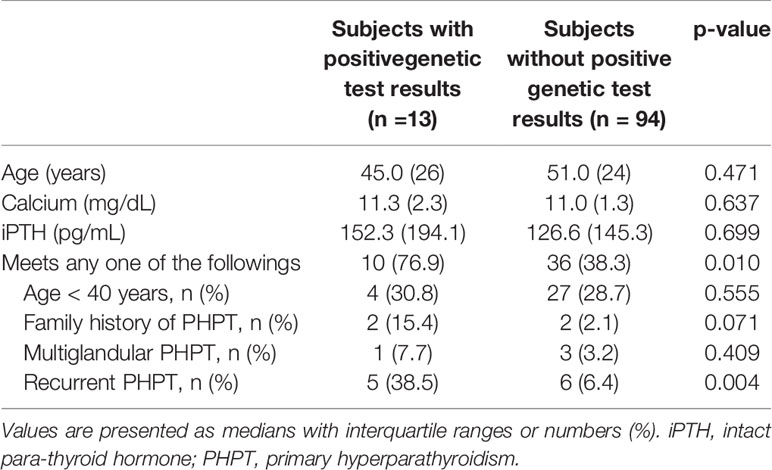
Table 4 Characteristics of subjects according to genetic test results.
Discussion
PHPT is a common endocrinological disorder with relatively well-established diagnosis and management. (2, 10, 19) However, knowledge about the genetic background of PHPT is limited, and genetic testing of PHPT is often overlooked in clinical practice. The development of NGS has helped advance research into the genetics of various types of endocrinological disorders. In addition, NGS is being widely used in clinical settings to detect genetic abnormalities and provide genetic counseling. We analyzed NGS data of 107 patients with PHPT and identified 3 pathogenic and 10 likely pathogenic variants. We further assessed 39 VUSs that could be related to parathyroid disease.
Genetic variants associated with parathyroid cancer and familial PHPT have been reported, and MEN1, CDC73, and RET mutations are known to be associated with the pathogenesis of parathyroid cancer or familial PHPT. (20) Also, in this study, patients with familial PHPT or parathyroid cancer had variants in MEN1 and CDC73 genes reported as pathogenic or likely pathogenic variants. We included 13 patients with parathyroid cancer, and three of them had CDC73 mutations. Additionally, four patients with parathyroid cancer had VUSs: two APC variants (c.5378C>G [p.Ala1793Gly] and c.890C>T [p.Thr297Ile]), one WT1 variant (c.1139G>A [p.Arg380Gln]), and one PRKAR1A variant (c.567A>C [p.Glu189As]). However, since the number of the patients with parathyroid cancer was small, it was hard to conclude the causative role of these variants.
Several somatic mutations have been identified in benign sporadic PHPTs. Somatic MEN1 gene mutations occur in 12% to 35% of sporadic PHPT (21–24), and somatic mutations in the CCND1 gene are also observed in 20%–40% of sporadic PHPT. (25–27) However, as the studies on germline mutations in sporadic PHPT are limited, variants found in subjects with sporadic PHPT are usually classified as VUSs. In this study, 32 non-synonymous variants in genes that could be related to parathyroid disease were classified as VUSs in patients with benign sporadic PHPT.
One of the patients had a GCM2 variant (c.1162A>G [p.Lys388Glu]), classified as VUS. GCM2 is mainly expressed in the parathyroid gland and regulates its development. (15) Germline mutations in GCM2 have recently been described as causative genetic alterations in FIHP. The specific genetic cause of FIHP, one of the hereditary forms of PHPT, was unclear until 2016, when Guan et al. demonstrated that GCM2 mutation can cause FIHP. (15) They found rare variants located in the GCM2 C-terminal conserved inhibitory domain (CCID) in 7 of the 40 kindreds with FIHP. GCM2 variants can be found in various functional domains of the human GCM2 protein, but those with transcription-activating functions are usually located within the CCID region. (15, 18) GCM2 variants have been reported not only in FIHP but also in sporadic PHPT. (28, 29) The prevalence of GCM2 variants in sporadic PHPT ranges from approximately 1.5% to 26.9% depending on ethnicity, and is particularly high in the Ashkenazi Jewish population. (18, 28, 30) There are limited studies on the Asian population, and one study found that the prevalence of GCM2 mutation with trans-activating function in Chinese PHPT patients was 1.3%. (18) In our study, one patient out of 107 had the GCM2 variant (c.1162A>G [p.Lys388Glu]), which is located in the CCID region. This same variant was previously reported in a study of Chinese patients with sporadic PHPT. (18) They screened 232 patients diagnosed with PHPT and found two cases with the variant c.1162A>G (p.Lys388Glu) of GCM2. Cases with variant c.1162A>G (p.Lys388Glu) had carcinoma pathology. Considering its prevalence, location, and transcription activity, we speculated that this variant in our patient could be associated with the development of PHPT.
There were concerns that PHPT patients with GCM2 variant could have an aggressive clinical phenotype, a high rate of multiglandular disease, and a low rate of biochemical cure. (31) Our patient with GCM2 variant was diagnosed with PHPT during health check-up and did not have any clinical manifestations, including renal stones and low bone mass. The patient underwent right inferior parathyroidectomy, and the pathology revealed parathyroid adenoma. After the surgery, the patient achieved biochemical cure and was under routine follow-up without recurrence. However, the patient’s age at diagnosis was 27 years, indicating possible involvement of genetic components in disease development. On NGS testing, no pathogenic or likely pathogenic variant was found, and variant c.1162A>G (p.Lys388Glu) in GCM2 was reported as VUS.
APC gene mutations are a well-known pathogenic mutations of familial adenomatous polyposis (FAP). Notably, germline mutations in the APC gene were identified in a patient with sporadic MEN1, metastatic papillary thyroid cancer, and FAP. (32) It has been suggested that APC gene variant might be involved in the pathogenesis of tumors in the parathyroid and thyroid glands. APC gene variants have also been found in parathyroid cancers. (17) The aberrant WNT/β-catenin signaling in parathyroid cancer could be due to a loss of expression or alteration of the APC gene (33).
In this study, two missense variants c.6754C>G (p.Pro2252Ala) and c.8017A>G (p.Arg2673Gly) of the APC gene were identified and classified as VUSs in patients with benign sporadic PHPT. The patients were diagnosed with PHPT at relatively young ages of 33 and 45 years. Of interest, APC mutations were also observed in patients with parathyroid cancer and in those with FHH. Among the 13 parathyroid cancer patients in this study, two patients carried APC variants (c.890C>T [p.Thr297Ile] and c.5378C>G (p.Ala1793Gly]) and did not harbor any other pathogenic or likely pathogenic variants. The other patient with APC variant (c.7969G>A[p.Val2657Ile]) was diagnosed with FHH and concurrently had the AP2S1 variant, classified as VUS. These five patients with APC variants were relatively young at the time of diagnosis, and two were diagnosed with parathyroid cancer. It is possible that the APC variants in these patients are involved in the development of PHPT.
Three patients with VUSs concurrently carried pathogenic or likely pathogenic variants. One patient with a WT1 variant (c.1139G>A [p.Arg380Gln]), reported as VUS, had a pathogenic CDC73 mutation (c.376C>T [p.Arg126Ter]) and was diagnosed with parathyroid cancer. Pathogenic CDC73 mutation is suspected to cause parathyroid cancer, and the WT1 variant could be an incidental finding from NGS.
The second patient carried the likely pathogenic LRP5 mutation (c.731C>T [p.Thr244Met]) and ESR2 variant (c.1541C>T [p.Pro514Leu]), classified as VUS. LRP5 mutation was not only reported in osteoporosis (34), but has also been associated with parathyroid tumors. (21, 35) In this patient, the LRP5 mutation could have played a role in the development of PHPT and osteoporosis. In addition, based on a study reporting estrogen receptor involvement in parathyroid adenoma, (36) there is a possibility that ESR2 variant also played a role in the development of PHPT in this case.
Lastly, a patient with CASR variant (c.613C>T [p.Arg205Cys]), reported as VUS, also had likely pathogenic VHL mutation (c.24G>A [p.Trp8Ter]). Although VHL mutation was reported as a likely pathogenic variant, the phenotype of VHL mutation is unclear in this patient, which might be due to variable expression of VHL mutation. (37) In contrast, CASR variant is associated with PHPT in previous studies. (38–40) Therefore, we speculated that although it was reported as VUS, CASR variant was involved in the development of PHPT and VHL mutation did not present its phenotype in this patient.
Genetic testing is usually indicated in some patients with PHPT who are at high risk of carrying a mutation: those with familial PHPT or parathyroid cancer (41) However, other clinical indications for genetic testing are still unclear, and the role of genetic testing in PHPT is often overlooked. In this study, 13 out of 107 study subjects (12.1%) had pathogenic or likely pathogenic variants, and 76.9% of the subjects with positive genetic test results had at least one of these clinical characteristics: 1) age < 40 years, 2) family history of PHPT, 3) multiglandular PHPT, or 4) recurrent PHPT. These clinical characteristic are generally consistent with previous studies, (3) and this implies that patients with these clinical characteristics need to undergo genetic testing.
Interestingly, 38.5% of patients with positive genetic testing had recurrent PHPT, but none of them had persistent PHPT. This different prevalence of recurrent or persistent PHPT in patients with positive test genetic testing might be due to the different clinical or genetic characteristics between the two. In our study, the patients who showed persistent PHPT were mainly due to the residual tissues of parathyroid adenoma or hyperplasia after the surgery or ectopic tissue which was not identified at the first operation. In contrast, the patients with recurrent PHPT were more likely to have genetic alterations, such as MEN1 or parathyroid cancer, so that they had the recurred disease even after the complete resection of the initial parathyroid tumor. In clinical practice, recurrent or persistent PHPT has been considered from the same point of view. However, based on the results in this study, patients with recurrent PHPT are at higher risk of having genetic alterations, thereby, should be urged to undergo genetic study. However, since this study included only small number of the patients, further study with large number of patients with recurrent or persistent PHPT is required.
The age of the subjects at the time of diagnosis did not differ between the groups according to the genetic test results. This might be due to a selection bias. Patients with classical sporadic form of PHPT and aged above 50 years would be less likely to undergo genetic testing and thus were not included in this study. In contrast, young patients were easily suspected to have genetic abnormalities, underwent genetic testing, and were enrolled in this study retrospectively. Therefore, there might be selection bias and the difficulty in determining the age difference between the groups with and without positive test results. In addition, the number of subjects with positive genetic test results was relatively small, leading to statistically insignificant results.
There are several other limitations to this study. This study included heterogeneous groups of PHPT, familial PHPT, benign sporadic PHPT, and parathyroid cancer. However, the number of subjects with familial PHPT and parathyroid cancer may not be sufficient to detect novel gene mutations. Second, the NGS panel used in this study did not include all candidate genes related to parathyroid disease. Lastly, because two different sequencing panels were used among the study patients, it might have affected the results.
In this study, we analyzed the NGS results of patients with PHPT. We speculated that although some variants were reported as VUSs, they could be associated with the development of the disease. In particular, as well as patients with familial PHPT or parathyroid cancer, which are classical indications for NGS, patients with young age or recurrent disease should be urged to undergo genetic testing. Advances in genetics and the declining cost of genetic testing may lead to its wider utilization in the future, thus helping in the diagnosis and management of PHPT patients and their relatives. We believe that this study provides insights into the genetic background of PHPT and provides a better approach for genetic counseling. Further studies are warranted to investigate the genetic abnormalities in PHPT pathogenesis and the role of NGS in PHPT in clinical practice.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Institutional Review Board of Severance Hospital, Yonsei University Health System, Seoul, Korea. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Author Contributions
Conceptualization: H-SP, NH, and YR. Methodology: H-SP, NH, DW, and YR. Formal analysis: H-SP and YR. Investigation: H-SP, YL, NH, and YR. Writing—original draft preparation: H-SP, and YL. Writing—review and editing: H-SP, YL, NH, DW, and YR. Supervision: YR. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by Hanim Precision Medicine Center of Yonsei University Health System under Grant number (6-2021-0208).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Editage (www.editage.co.kr) for English language editing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2022.853171/full#supplementary-material
Supplementary Table 1 | Gene panel list: targeted gene sequencing. Genes which are included in our candidate gene list (Supplementary Table 2) are shown in yellow box.
Supplementary Table 2 | List of candidate genes related to parathyroid diseases.
Supplementary Table 4 | The criteria used for each pathogenic and likely pathogenic variants.
References
1. Yeh MW, Ituarte PH, Zhou HC, Nishimoto S, Liu IL, Harari A, et al. Incidence and Prevalence of Primary Hyperparathyroidism in a Racially Mixed Population. J Clin Endocrinol Metab (2013) 98(3):1122–9. doi: 10.1210/jc.2012-4022
2. Khan AA, Hanley DA, Rizzoli R, Bollerslev J, Young JE, Rejnmark L, et al. Primary Hyperparathyroidism: Review and Recommendations on Evaluation, Diagnosis, and Management. A Canadian and International Consensus. Osteoporos Int (2017) 28(1):1–19. doi: 10.1007/s00198-016-3716-2
3. El Lakis M, Nockel P, Gaitanidis A, Guan B, Agarwal S, Welch J, et al. Probability of Positive Genetic Testing Results in Patients With Family History of Primary Hyperparathyroidism. J Am Coll Surg (2018) 226(5):933–8. doi: 10.1016/j.jamcollsurg.2018.01.007
4. Wei Z, Sun B, Wang ZP, He JW, Fu WZ, Fan YB, et al. Whole-Exome Sequencing Identifies Novel Recurrent Somatic Mutations in Sporadic Parathyroid Adenomas. Endocrinology (2018) 159(8):3061–8. doi: 10.1210/en.2018-00246
5. Nesbit MA, Hannan FM, Howles SA, Babinsky VN, Head RA, Cranston T, et al. Mutations Affecting G-Protein Subunit Alpha11 in Hypercalcemia and Hypocalcemia. N Engl J Med (2013) 368(26):2476–86. doi: 10.1056/NEJMoa1300253
6. Nesbit MA, Hannan FM, Howles SA, Reed AA, Cranston T, Thakker CE, et al. Mutations in AP2S1 Cause Familial Hypocalciuric Hypercalcemia Type 3. Nat Genet (2013) 45(1):93–7. doi: 10.1038/ng.2492
7. Brewer K, Costa-Guda J, Arnold A. Molecular Genetic Insights Into Sporadic Primary Hyperparathyroidism. Endocr-relat Cancer (2019) 26(2):R53–72. doi: 10.1530/ERC-18-0304
8. Blau JE, Simonds WF. Familial Hyperparathyroidism. Front Endocrinol (Lausanne) (2021) 12:623667. doi: 10.3389/fendo.2021.623667
9. Guerin C, Paladino NC, Lowery A, Castinetti F, Taieb D, Sebag F. Persistent and Recurrent Hyperparathyroidism. Updates Surg (2017) 69(2):161–9. doi: 10.1007/s13304-017-0447-7
10. Bilezikian JP, Brandi ML, Eastell R, Silverberg SJ, Udelsman R, Marcocci C, et al. Guidelines for the Management of Asymptomatic Primary Hyperparathyroidism: Summary Statement From the Fourth International Workshop. J Clin Endocrinol Metab (2014) 99(10):3561–9. doi: 10.1210/jc.2014-1413
11. Kim SH, Kim B, Lee JS, Kim HD, Choi JR, Lee S-T, et al. Proband-Only Clinical Exome Sequencing for Neurodevelopmental Disabilities. Pediatr Neurol (2019) 99:47–54. doi: 10.1016/j.pediatrneurol.2019.02.017
12. Rim JH, Kim SH, Hwang IS, Kwon SS, Kim J, Kim HW, et al. Efficient Strategy for the Molecular Diagnosis of Intractable Early-Onset Epilepsy Using Targeted Gene Sequencing. BMC Med Genomics (2018) 11(1):1–10. doi: 10.1186/s12920-018-0320-7
13. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
14. Cetani F, Pardi E, Aretini P, Saponaro F, Borsari S, Mazoni L, et al. Whole Exome Sequencing in Familial Isolated Primary Hyperparathyroidism. J Endocrinol Invest (2020) 43(2):231–45. doi: 10.1007/s40618-019-01107-5
15. Guan B, Welch JM, Sapp JC, Ling H, Li Y, Johnston JJ, et al. GCM2-Activating Mutations in Familial Isolated Hyperparathyroidism. Am J Hum Genet (2016) 99(5):1034–44. doi: 10.1016/j.ajhg.2016.08.018
16. Rouillard AD, Gundersen GW, Fernandez NF, Wang Z, Monteiro CD, McDermott MG, et al. The Harmonizome: A Collection of Processed Datasets Gathered to Serve and Mine Knowledge About Genes and Proteins. Database (Oxf) (2016) 2016:1–16. doi: 10.1093/database/baw100
17. Pandya C, Uzilov AV, Bellizzi J, Lau CY, Moe AS, Strahl M, et al. Genomic Profiling Reveals Mutational Landscape in Parathyroid Carcinomas. JCI Insight (2017) 2(6):e92061. doi: 10.1172/jci.insight.92061
18. Song A, Yang Y, Wang Y, Liu S, Nie M, Jiang Y, et al. Germline GCM2 Mutation Screening in Chinese Primary Hyperparathyroidism Patients. Endocr Pract (2020) 26(10):1093–104. doi: 10.4158/EP-2020-0132
19. Bilezikian JP. Primary Hyperparathyroidism. J Clin Endocrinol Metab (2018) 103(11):3993–4004. doi: 10.1210/jc.2018-01225
20. Bilezikian JP, Cusano NE, Khan AA, Liu JM, Marcocci C, Bandeira F. Primary Hyperparathyroidism. Nat Rev Dis Primers (2016) 2:16033. doi: 10.1038/nrdp.2016.33
21. Cromer MK, Starker LF, Choi M, Udelsman R, Nelson-Williams C, Lifton RP, et al. Identification of Somatic Mutations in Parathyroid Tumors Using Whole-Exome Sequencing. J Clin Endocrinol Metab (2012) 97(9):E1774–81. doi: 10.1210/jc.2012-1743
22. Heppner C, Kester MB, Agarwal SK, Debelenko LV, Emmert-Buck MR, Guru SC, et al. Somatic Mutation of the MEN1 Gene in Parathyroid Tumours. Nat Genet (1997) 16(4):375–8. doi: 10.1038/ng0897-375
23. Newey PJ, Nesbit MA, Rimmer AJ, Attar M, Head RT, Christie PT, et al. Whole-Exome Sequencing Studies of Nonhereditary (Sporadic) Parathyroid Adenomas. J Clin Endocrinol Metab (2012) 97(10):E1995–2005. doi: 10.1210/jc.2012-2303
24. Segiet OA, Deska M, Michalski M, Gawrychowski J, Wojnicz R. Molecular Profiling in Primary Hyperparathyroidism. Head Neck (2015) 37(2):299–307. doi: 10.1002/hed.23656
25. Hsi ED, Zukerberg LR, Yang WI, Arnold A. Cyclin D1/PRAD1 Expression in Parathyroid Adenomas: An Immunohistochemical Study. J Clin Endocrinol Metab (1996) 81(5):1736–9. doi: 10.1210/jcem.81.5.8626826
26. Tominaga Y, Tsuzuki T, Uchida K, Haba T, Otsuka S, Ichimori T, et al. Expression of PRAD1/cyclin D1, Retinoblastoma Gene Products, and Ki67 in Parathyroid Hyperplasia Caused by Chronic Renal Failure Versus Primary Adenoma. Kidney Int (1999) 55(4):1375–83. doi: 10.1046/j.1523-1755.1999.00396.x
27. Vasef MA, Brynes RK, Sturm M, Bromley C, Robinson RA. Expression of Cyclin D1 in Parathyroid Carcinomas, Adenomas, and Hyperplasias: A Paraffin Immunohistochemical Study. Mod Pathol (1999) 12(4):412–6.
28. Guan B, Welch JM, Vemulapalli M, Li Y, Ling H, Kebebew E, et al. Ethnicity of Patients With Germline GCM2-Activating Variants and Primary Hyperparathyroidism. J Endocr Soc (2017) 1(5):488–99. doi: 10.1210/js.2017-00043
29. D'Agruma L, Coco M, Guarnieri V, Battista C, Canaff L, Salcuni AS, et al. Increased Prevalence of the GCM2 Polymorphism, Y282D, in Primary Hyperparathyroidism: Analysis of Three Italian Cohorts. J Clin Endocrinol Metab (2014) 99(12):E2794–8. doi: 10.1210/jc.2014-2857
30. Riccardi A, Aspir T, Shen L, Kuo CL, Brown TC, Korah R, et al. Analysis of Activating GCM2 Sequence Variants in Sporadic Parathyroid Adenomas. J Clin Endocrinol Metab (2019) 104(6):1948–52. doi: 10.1210/jc.2018-02517
31. El Lakis M, Nockel P, Guan B, Agarwal S, Welch J, Simonds WF, et al. Familial Isolated Primary Hyperparathyroidism Associated With Germline GCM2 Mutations Is More Aggressive and has a Lesser Rate of Biochemical Cure. Surgery (2018) 163(1):31–4. doi: 10.1016/j.surg.2017.04.027
32. Sakai Y, Koizumi K, Sugitani I, Nakagawa K, Arai M, Utsunomiya J, et al. Familial Adenomatous Polyposis Associated With Multiple Endocrine Neoplasia Type 1-Related Tumors and Thyroid Carcinoma: A Case Report With Clinicopathologic and Molecular Analyses. Am J Surg Pathol (2002) 26(1):103–10. doi: 10.1097/00000478-200201000-00014
33. Svedlund J, Auren M, Sundstrom M, Dralle H, Akerstrom G, Bjorklund P, et al. Aberrant WNT/beta-Catenin Signaling in Parathyroid Carcinoma. Mol Cancer (2010) 9:294. doi: 10.1186/1476-4598-9-294
34. Cui Y, Niziolek PJ, MacDonald BT, Zylstra CR, Alenina N, Robinson DR, et al. Lrp5 Functions in Bone to Regulate Bone Mass. Nat Med (2011) 17(6):684–91. doi: 10.1038/nm.2388
35. Bjorklund P, Akerstrom G, Westin G. An LRP5 Receptor With Internal Deletion in Hyperparathyroid Tumors With Implications for Deregulated WNT/beta-Catenin Signaling. PloS Med (2007) 4(11):e328. doi: 10.1371/journal.pmed.0040328
36. Haglund F, Ma R, Huss M, Sulaiman L, Lu M, Nilsson IL, et al. Evidence of a Functional Estrogen Receptor in Parathyroid Adenomas. J Clin Endocrinol Metab (2012) 97(12):4631–9. doi: 10.1210/jc.2012-2484
37. Maher ER, Webster AR, Richards FM, Green JS, Crossey PA, Payne SJ, et al. Phenotypic Expression in Von Hippel-Lindau Disease: Correlations With Germline VHL Gene Mutations. J Med Genet (1996) 33(4):328–32. doi: 10.1136/jmg.33.4.328
38. Thakker RV. Genetics of Parathyroid Tumours. J Intern Med (2016) 280(6):574–83. doi: 10.1111/joim.12523
39. Starker LF, Akerstrom T, Long WD, Delgado-Verdugo A, Donovan P, Udelsman R, et al. Frequent Germ-Line Mutations of the MEN1, CASR, and HRPT2/CDC73 Genes in Young Patients With Clinically Non-Familial Primary Hyperparathyroidism. Horm Cancer (2012) 3(1-2):44–51. doi: 10.1007/s12672-011-0100-8
40. Hannan FM, Nesbit MA, Christie PT, Lissens W, Van Der Schueren B, Bex M, et al. A Homozygous Inactivating Calcium-Sensing Receptor Mutation, Pro339Thr, Is Associated With Isolated Primary Hyperparathyroidism: Correlation Between Location of Mutations and Severity of Hypercalcaemia. Clin Endocrinol (Oxf) (2010) 73(6):715–22. doi: 10.1111/j.1365-2265.2010.03870.x
Keywords: sporadic primary hyperparathyroidism, familial primary hyperparathyroidism, parathyroid cancer, variants of unknown significance (VUS), next-generation sequencing, germline mutation
Citation: Park H-S, Lee YH, Hong N, Won D and Rhee Y (2022) Germline Mutations Related to Primary Hyperparathyroidism Identified by Next-Generation Sequencing. Front. Endocrinol. 13:853171. doi: 10.3389/fendo.2022.853171
Received: 12 January 2022; Accepted: 29 March 2022;
Published: 28 April 2022.
Edited by:
Daniela Merlotti, University of Siena, ItalyReviewed by:
Federica Saponaro, University of Pisa, ItalyJessica Costa-Guda, University of Connecticut, United States
Copyright © 2022 Park, Lee, Hong, Won and Rhee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yumie Rhee, YUMIE@yuhs.ac
†These authors have contributed equally to this work and share first authorship