 Rafiye Ciftciler†
Rafiye Ciftciler† Ibrahim Celalettin Haznedaroglu*†
Ibrahim Celalettin Haznedaroglu*†- Department of Hematology, Faculty of Medicine, Hacettepe University, Ankara, Turkey
Circulating renin-angiotensin system (RAS) and local paracrin-autocrin-intracrin tissue-based RAS participate in numerous pathobiological events. Pro-inflammatory, pro-fibrotic, and pro-thrombotic consequences associated with local RAS activation have been detected at cellular and molecular level. Regenerative progenitor cell therapy in response to RAS modulating pharmacotherapy has emerged as an adjunct in the context of endothelial cell injury and regeneration to improve regeneration of the vascular endothelium. Local hematopoietic bone marrow (BM) RAS symbolizes the place of cross-interaction between vascular biology and cellular events from embryogenesis to definitive hematopoiesis underlying vascular atherosclerosis. The BM microenvironment also contains Mas receptors, which control the proliferative role of Ang 1-7 on hematopoietic stem cells. Ang 1-7 is produced from Ang-II or Ang-I with the help of ACE2. Various tissues and organs also have an effect on the RAS system. The leukocytes contain and synthesize immunoreactive angiotensinogen species capable of producing angiotensin in the basal state or after incubation with renin. The significance of RAS employment in atherosclerosis and hypertension was indicated by novel bidirectional Central Nervous System (CNS) RAS–BM RAS communications. Myeloid cells generated within the context of hematopoietic BM RAS are considered as the initiators and decision shapers in atherosclerosis. Macrophages in the atherosclerotic lesions contain angiotensin peptides by which RAS blockers inhibit monocyte activation and adherence. Furthermore, vascular biology in relation to inflammation and neoplasia is also affected by local tissue RAS. The purpose of this article is to outline interactions of circulating and local angiotensin systems, especially local bone marrow RAS, in the vascular pathobiological microenvironment of CNS.
Introduction
As previously known, renin-angiotensin system (RAS) was described as an endocrine system that regulates blood pressure and body electrolyte balance (1). Recently RAS, which is considered as an “ubiquitous” system with various effects on tissue physiology and homeostasis, has been shown to be locally expressed in different tissues (2). With the detection of novel RAS components, the emerging tissue RAS concept has expanded the physiological and clinical location of the RAS (2). Circulating local RAS and local paracrin-autocrin-intracrin tissue-based RAS participate in numerous pathobiological events. Pro-inflammatory, pro-fibrotic, and pro-thrombotic consequences associated with local RAS activation have been detected at cellular and molecular level (2). For these reasons, it is important to know RAS components, tissue-specific expressions of RAS and how they can change under pathological circumstances (2). There is a RAS located in the bone marrow (BM) microenvironment and within the hematopoietic stem cells (HSC). The concept of local BM RAS, which is active in primitive and definitive hematopoiesis, had been proposed by Haznedaroglu and coworkers about three decades ago (3, 4).
RAS molecules, particularly ACE, ACE2, AGT, AGTR1, AGTR2, AKR1C4, AKR1D1, ANPEP, ATP6AP2, CMA1, CPA3, CTSA, CTSD, CTSG, CYP11A1, CYP11B1, CYP11B2, CYP17A1, CYP21A2, DPP3, EGFR, ENPEP, GPER, HSD11B1, HSD11B2, IGF2R, KLK1, LNPEP, MAS1, MME, NR3C1, NR3C2, PREP, REN, RNPEP, THOP1 are available in the BM (5). These molecules affect the entire stem cell-oriented hematopoiesis. Local BM RAS seems to be the major station of cross-talk between hematopoietic stem/progenitor cells and their afferent functions within the heart, kidney and Central Nervous System (CNS) as a (patho)biological trigger for vascular atherosclerosis (6). There is also a local CNS RAS. The importance of RAS and BM-RAS activity in atherosclerosis and hypertension was indicated by novel bidirectional CNS RAS–BM RAS communications (7–9). The purpose of this article is to outline interactions of circulating and local angiotensin systems, especially local bone marrow RAS, in the vascular pathobiological microenvironment of CNS.
Local Bone Marrow Renin Angiotensin System: Definition and a Brief History
Major RAS molecules, including renin, angiotensinogen, angiotensin receptors and ACE, are all found in the BM microenvironment (10). Haznedaroglu et al. first proposed the idea of a local hematopoietic RAS in BM in 1996. The study supposed that there is a locally active RAS in the BM that affects the production, proliferation growth, and differentiation of hematopoietic cells (3). Later, evidence of a local RAS in the BM increased day by day. Locally active BM RAS influences important pathways in physiological and pathological blood cell production by autocrine, paracrine and intracrine routes (11, 12). The development of hematopoietic niche, erythropoiesis, myelopoiesis, thrombopoiesis and other cellular linage is controlled by local BM RAS peptides (13–16). Additionally, many important pathobiological events such as cellular proliferative events, mobilization, angiogenesis, fibrosis, and apoptosis in the cytokine network are affected by RAS molecules (11, 12, 17, 18). Local RAS in the BM stromal niche controls important hematopoietic functions (10, 12–15, 19, 20). The BM stromal microenvironment contains important peptides that are components of the RAS (11, 12).
BM stromal microenvironment includes AT1R and AT2R (angiotensin type 1 and type 2 receptors, respectively) and inhibitory tetrapeptide AcSDKP (N-acetyl-Ser-Asp-Lys-Pro) (11, 12). The major RAS effector agent angiotensin II (Ang II) performs its impacts on the hematopoietic system by activating the AT1Rs and AT2Rs, along with the BM microenvironment (11, 12). As a result of ACE's (CD143) disrupting the inhibitory tetrapetide, AcSDKP, priming of stem cells into S-phase is triggered (16, 21). Additionally, Ang-II stimulates the AT1/AT2 receptors, so it has stimulating or inhibitory effects on erythropoietin, thrombopoietin and other hematopoietic cytokines in normal hematopoiesis and myeloproliferative diseases (16, 22, 23). Multiple clinical studies have been made to evaluate the role of local BM RAS in several diseases (18, 24–30).
Phase I/II clinical trials of a pharmaceutical agent of peptide angiotensin 1-7 (Ang-1-7) have been directed to assess the role of local BM RAS in different diseases (18, 24, 25). Other participating components of Ang II and RAS such as Ang IV [Ang-(3-8)] and Ang-(1-7) play a regulatory role on the cardiovascular system (26). Moreover, ACE2 and Mas receptor are very important components of the option line of RAS and are expressed in Hematopoietic stem/progenitor cells (HSPCs) (27). Activation of the ACE2/Ang-(1-7)/MasR axis stimulates the functions of HSPCs related to vascular repair and repulses dysfunctions caused by chronic pathological situations (27). On the other hand, white blood cells locally produce angiotensin peptides (28–30). Gomez et al. showed that circulating rat leukocytes express the angiotensinogen gene. It showed that leukocytes contain and synthesize immunoreactive angiotensinogen species that can produce angiotensin in the basal state or after incubation with renin. As a system that produces angiotensin, leukocytes can be important in modulating inflammatory responses, tissue damage, and cardiovascular pathology such as hypertension (28).
The supposition that local autocrine BM RAS may be effective in neoplastic hematopoiesis supports the prominent functions of local RAS in primitive embryonic hematopoiesis (31–33). Likewise the considerable functions of local RAS, which are involved in primitive embryonic hematopoiesis, also reinforce the supposition that local autocrine BM RAS may have an effective role in neoplastic hematopoiesis (19). Critical RAS modulating agents such as renin, ACE, angiotensinogen, and AngII have been previously described in leukemic malignant cells (34–36). Recently, Yamashita et al. showed that angiotensin-(1-12) generation is revealed in the BM of rats. Chymase-mediated Ang II production in BM was importantly higher than ACE-mediated and 280-fold higher than that in the heart (37). CD68 positive myeloid lineage cells, especially myeloid progenitors, have higher chymase expression than CD68 negative lymphoid lineage cells in BM (37).
Local Bone Marrow Renin Angiotensin System and Atherosclerosis
Local BM RAS has significant effects on hematopoietic systems, particularly on myeloid, and erythroid cells (3, 12, 19). Local BM RAS is involved in the regulation of important peptides that control hematopoiesis. With the help of ACE, Ang I transformed into Ang II, while bioactive SP, Ac-SDKP and Ag 1-7 were inactivated by ACE. Also, during this process, substance P (SP) is secreted from nerve endings. BM stromal and hematopoietic cells secrete RAS peptides by the AT1 and NK1 receptors that coordinate the effect of Ang II and SP, respectively as depicted in Figure 1. Additionally, it has been proven that important receptors of Ang 1-7 and MAS are available in the BM stroma (38). The BM microenvironment contains Mas receptors, which control the proliferative role of Ang 1-7 on HSCs. Ang 1-7 is created from Ang-II or Ang-I with the help of ACE2. Hematopoietic recovery after myelosuppression increases with Angiotensin (1-7)(5, 17). Besides all these, RAS plays a role in the pathogenesis of various diseases (42).
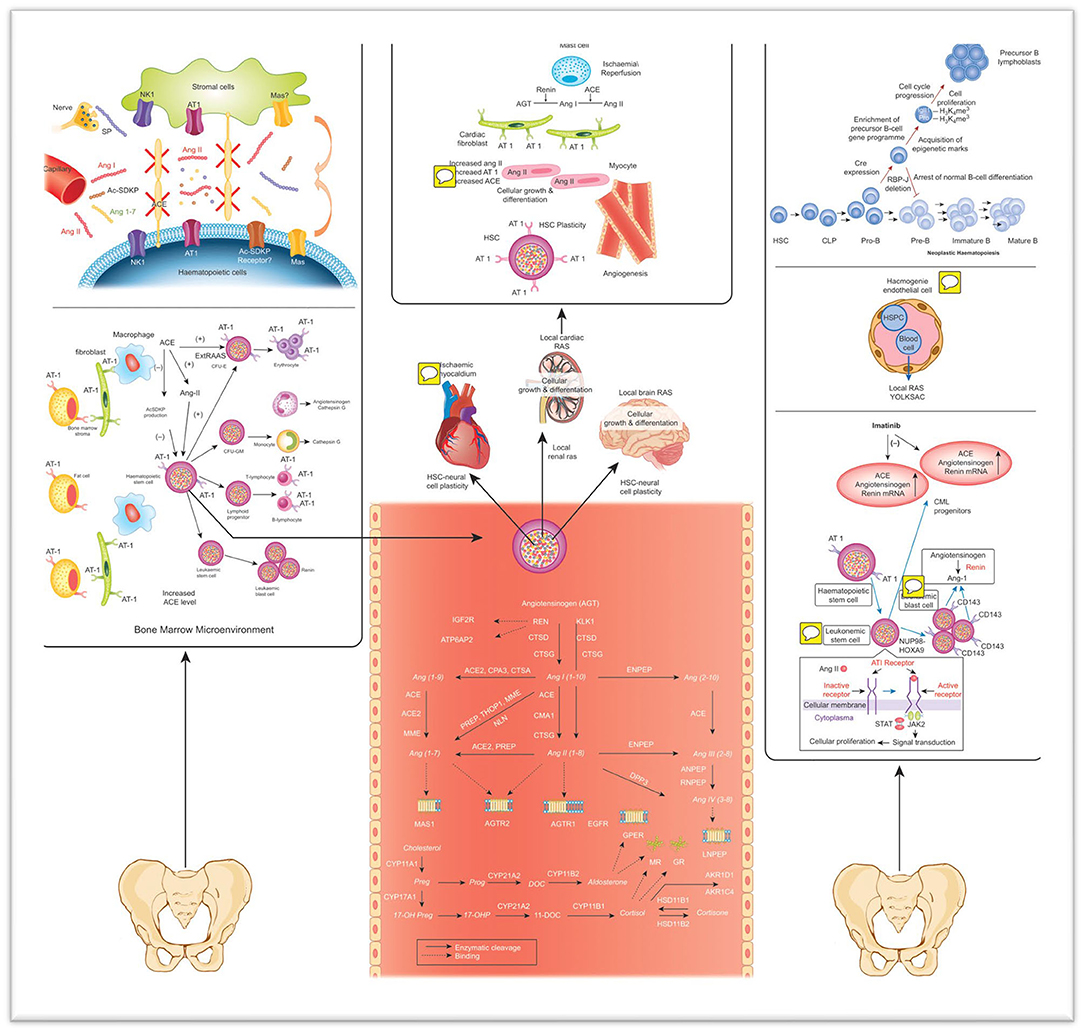
Figure 1. The effect of local RAS on normal and leukemic hematopoiesis, as well as plasticity and systemic circulation, are shown (19). Ang I, Ang II, Ac-SDKP, and Ang 1-7 are secreted in different tissues and transferred to the BM through circulation. SP is secreted from the nerve end protruding into the BM. ACE converts Ang II from Ang I and reduces bioactive SP, Ac-SDKP and Ang 1-7 (38). Mas, with the Ang 1-7 receptor, is detected in the BM. Peptides in RAS can stimulate hematopoietic cells (39). Furthermore, local BM RAS has a function in the development, formation, proliferation, and differentiation of hematopoietic progenitors. Angiotensin peptides and steroid hormones were symbolized in gray. Lymphoid cells cannot differentiate throughout the normal B-cell pathway (40). Therefore, the leader B-cell gene undergoes epigenetic initiation and enrichment of a precursor B-cell gene programme. As a result, cell cycle progression and cell proliferation are observed in mutant cells. This leads to the enlargement of a lymphoblast population and the development of leading B cell leukemia (5, 19, 23, 38, 40, 41). HSPC, Hematopoietic stem/progenitor cells; ACE, Angiotensin converting enzyme; CML, Chronic myeloid leukemia; Ang, Angiotensinogen; CFU-GM/E, Colonyforming units-Granulocyte-Macrophage/erythroid; JAS/STAT, Janus kinase/Signal transduction and transcription; BC, Blood cell; Ang, Angiotensin; Preg, Pregnanolone; Prog, Progesterone; DOC, deoxycortisol; 17-OHP, 17-OH Progesterone; ACE, angiotensin I converting enzyme; ACE2, angiotensin I converting enzyme type 2; AGTR1, angiotensin II type 1 receptor; AGTR2, angiotensin II type 2 receptor; AKRIC4, aldo-ketoreductase family 1 member C4; AKRID1, aldo-ketoreductase family 1 member D1; ANPEP, alanyl-aminopeptidase; ATP6AP2, prorenin/renin receptor; CMA1, chymase 1; CPA3, carboxypeptidase A3; CTSA, cathepsin A; CTSD, cathepsin D; CTSG, cathepsin G; CYP11A1, cytochrome P450 family 11 subfamily A polypeptide 1; CYP11B1, cortisolsynthase; CYP11B2, aldosteronesynthase; CYP17A1, cytochrome P450 family 17 subfamily A polypeptide 1; CYP21A2, cytochrome P450 enzyme family 21 subfamily A polypeptide 2; DPP3, dipeptidyl-peptidase 3; ENPEP, glutamylaminopeptidase (aminopeptidase A); GR, glucocorticoidreceptor; HSD11B1, hydroxysteroid (11-beta) dehydrogenase 1; HSD11B2, hydroxysteroid (11-beta) dehydrogenase 2; IGF2R, insulin-like growth factor 2 receptor; KLK1, tissuekallikrein; LNPEP, leucyl/cystinylaminopeptidase; MAS1, MAS1 proto-oncogene; MME, membranemetallo-endopeptidase; MR, mineralocorticoidreceptor; NLN, neurolysin (metallopeptidase M3 family); PREP, prolylendopeptidase; REN, renin; RNPEP, arginylaminopeptidase (aminopeptidase B); THOP1, thimetoligopeptidase 1. Images of IGF2R36, ATP6AP237, MR38, GR39, G-protein coupled receptors (AGTR1, AGTR2, GPER, and MAS1) 40 and LNPEP41 (19).
RAS is an extremely complex system consisting of a series of enzymes, peptides and receptors known to play a role in the formation of hypertension and atherosclerosis (43). It has been shown that most of the enzymes and peptides described initially as a hormonal system of RAS can be made locally in various organs, including blood vessels (43). RAS has a critical function in the management of blood flow, fluid volume, blood pressure and electrolyte balance (6). The overworking of RAS participates in the pathogenesis of various clinical situations, such as the beginning and progression of atherosclerosis (6). Ang II is the peptide known as the main last effector molecule of the classical RAS line, consisting of ACE, Ang II and angiotensin receptor type I (AT1R) (43). Ang II is produced in the brain, kidney and blood vessels. Ang II triggers hypertension through transcription activation, reactive oxygen species (ROS) production, inflammation and numerous alternative cellular events (44, 45). Ang II takes part in all stages of the pathogenesis, up to early lesion formation, growth, progression and plaque rupture, and as a result causes atherosclerosis (44, 45). Oxidative stress and inflammation make up the majority of the mechanism of action of Ang II on atherosclerosis (46). Inflammatory cells in atherosclerotic lesions are generally thought to originate from BM (15). Angiotensin II improves erythroid differentiation in the BM by interacting with the AT1R (15). Fukuda et al. analyzed a few BM chimeric mice whose BM cells were positive or negative for AT1R (47). They demonstrated that AT1aR in BM cells attend in the pathogenesis of atherosclerosis (47). Another study showed that AT1a receptor was expressed by human BM CD34+, CD38− cells, and lymphocytes (48). Ang II has been reported to stimulate the differentiation of human CD34 + hematopoietic progenitors from cord blood (48).
Besides all these, atherosclerosis due to hypercholesterolemia is affected by the pharmacological antagonism of AT1 receptors or the reduction of the AT1A receptor (49). Hypercholesterolemia induces the production of angiotensin peptides for the effect of AT1A receptor deficiency on atherogenesis (49). There is also a close relation between cardiac RAS and the hematopoietic BM RAS (3, 12). Myocardial tissue healing through HSC plasticity shows the interaction between local cardiac RAS and hematopoietic RAS (23).
Atherosclerosis and Central Nervous System
Atherosclerosis is often assessed as a chronic inflammatory disease, because inflammation has a significant function in all stages of atherosclerosis (50, 51). Studies have shown that atherosclerotic disease is often the cause of the onset of ischemic cerebro-vascular events (52–54). The atherogenic process is attended by flow-mediated inflammatory alterations in endothelial cells (EC) (55). In early-stage atherosclerosis, endothelial damage, abnormal lipid metabolism and hemodynamic damage are the causes of the disease (55). In late-stage atherosclerosis, lots of macrophages and inflammatory cytokines leak into the vascular wall, excrete matrix metalloproteinases (MMPs) and result in plaque rupture, bleeding and thrombosis (56). The harmonious effect of all proinflammatory signals on the plaque increases inflammation and also prevents the regeneration of structural elements that support the mechanical stability of the inflamed tissue (57).
Atherosclerosis is a chronic inflammatory syndrome that affects the unity and activity of major blood vessels which supplies the brain. As a result, chronic inflammation in the vessels impairs cerebral blood flow and neurovascular communication, which is important for cerebrovascular function (58). Chronic atherosclerosis has an effect on the brain throughout the life of people (59–62). Atherosclerosis leads to impaired vascular integrity, causing cognitive decline, stroke and vascular dementia (58). Atherosclerosis is most effective on large and medium-sized arteries, including internal carotid and vertebral arteries. Atherosclerosis and associated cardiovascular diseases can cause a wide variety of vascular diseases and lesions in the brain. These lesions caused by atherosclerosis occur as a result of arterial stiffness and inflammation (63). All these events take place with a complex mechanism.
There is an association between the autonomic nervous system (ANS) and BM cells. This relationship between BM stromal cells, HSCs and nerve terminals has been defined as the ‘neuro-reticular complex’ (64, 65). A distinctive feature of early hypertension is endothelial dysfunction (8). BM-derived endothelial progenitor cells (EPC) participate in the healing of damaged endothelium. Studies have shown that EPC numbers and functions are lower in patients with hypertension and cardiovascular disease (66–69).
Central Nervous System and Hypertension
All components of RAS are found in the brain (70, 71). The first finding that Ang (1-7) could be produced in areas of the CNS was obtained from studies of the hydrolysis of [125I]-Ang I in brain homogenates (72). It has been shown as immunostaining for Ang (1-7) in the paraventricular, supraoptic and suprachiasmatic nuclei of the hypothalamus, in the stria terminalis bed nucleus, substantia innominata, median exclusivity and neurohypophysis (73), (74). Other studies have shown the effect of the immune system and neuroimmune pathways in hypertensive patients (9, 75).
Hypertension is a very significant risk factor for cardiovascular diseases. It remains a global public health problem (75). Various effects such as salt sensitivity and high systemic RAS activity are included in the pathophysiology of hypertension (76, 77). Moreover, studies have shown that disruptions in activity within the cardiovascular CNS fields as a result of increased sympathetic and reduced parasympathetic impulse to the peripheral organs cause end organ injury, vascular/endothelial dysfunction, and hormonal instability (78, 79). Recently, significant advances have been made in the treatment of hypertension using ACE inhibitors or AT1 receptor blockers, diuretics, α-adrenoreceptor antagonists, RAS inhibitors and calcium channel blockers (75). Molecular and neuronal changes at the brainstem and hypothalamus level have been shown to contribute to neurogenic hypertension. Studies have shown that sympathetic activation not only starts hypertension but also protects it (75). The first evidence of the significance of the autonomic nervous system for BM cell homeostasis was obtained from studies demonstrating the regulation of BM cell activity (80). It was showed that the release of BM HSPCs is rhythmically regulated in a circadian manner, for which sympathetic drive is essential (80–82).
Local Bone Marrow Renin Angiotensin System and Central Nervous System Within the Context of Essential Hypertension
BM has an essential function in hematopoiesis regulation (19). BM HSPCs are in communication with cells of secondary lymphoid organs, such as the spleen, which control HSPC differentiation and maturation (83, 84). The relationship of BM and HSPCs with hypertension has recently attracted interest. It is thought to be a bidirectional brain-BM communication hypothesis, and this hypothesis is based on various evidences (7, 9).
Ang II related HT lead to a recommendation that activation of microglia in the pre-sympathetic cardio-regulatory brain fields may precede both the activation of the sympathetic drive and the increase in blood pressure (9, 75). Prohypertensive signals such as increased Ang II cause neurovascular-glial inflammation in the cardio-regulator areas of the brain (7). Dysfunctional ANS output is characterized by increment in the sympathetic and a reduction in the parasympathetic impulse to the periphery, including BM (7). This process induces an increment in peripheral Ang II (7). Thus, it results in a permanent increment in the inflammatory cells and a decrease in the endothelial progenitor cells (7). Increased inflammatory cells contribute to vascular and tissue damage, while reduced endothelial progenitor cells contribute to repair of this harm, leading to cardiorenal pathology (7). Extravasation of inflammatory cells into pre-sympathetic brain areas and increased somatic afferent input from BM to the brain contribute to neurovascular-glial inflammation (7). So, it directs the dysfunctional ANS output to the environment. These pathways are circumstances that cause cardiovascular and kidney pathophysiology and resistant hypertension (7). On the other hand, in Ang II-induced hypertension, BM-induced AT1R receptors limit mononuclear cell aggregation in the kidney. Thus, it reduces the chronic hypertensive response, possibly through the arrangement of vasoactive cytokines (85). Dysfunctional ANS output is characterized by a sympathetic increase and a decrease in parasympathetic impulses and is involved in the pathophysiology of hypertension (7, 9).
One study demonstrated that a dysfunctional BM ANS is correlated with imbalanced EPCs and inflammatory cells in hypertension (80). They demonstrated that in their study presympathetic neuronal activation in a spontaneously hypertensive rat was related with an accelerated retrograde transfer of the gren fluorescent protein–labeled pseudorabies virus from the BM in mangasese-enhanced MRI (80). Pro-hypertensive markers, such as increased brain and systemic RAS, cause neurovascular-glial inflammation in the brain cardio-regulator sites (27). All this evidence supports the bidirectional brain-BM interaction hypothesis for cardiovascular system homeostasis and hypertension. The effect of somatic afferent input from the BM to the CNS continues to be investigated (27).
BM has an important role in neurogenic hypertension. Memory T cells are present in the BM. T-cell activation has a significant function in hypertension (9). The BM is the primary source of EPC, which play a significant function in endothelial repair in arterial or renal injury (86, 87). Chronic elevation in BM norepinephrine may disrupt the role of EPCs and this may be important in the terms of hypertension. These interactions strengthen the connection between the CNS and BM (9). Cross-communication between ANS and BM vasculature may be an important mechanism of the pathophysiology of hypertension. NE has a vasoconstrictor role in the BM and plays a significant function in controlling blood flow (9, 88). Prohypertensive signals such as Ang II cause neuro-vascular-glial inflammation in the cardio-regulator sites of the brain (7–9). Dysfunctional ANS output is represented by an increase in sympathetic and a reduction in parasympathetic impulses to the environment, including BM. As a result of these events, there is a permanent increase in inflammatory cells and a reduce in EPCs. As a result, these increased inflammatory cells cause vascular and tissue damage, while the reduction of endothelial progenitor cells causes a reduction in repair of this damage, leading to cardio-renal pathologies (7, 9). The combination of increased somatic afferent input from BM to the brain through the extravasation of inflammatory cells into pre-sympathetic brain areas and activation of TRPV1 channels contributes to neuro-vascular-glial inflammation (89–91). All these processes sustain the emerging cardiovascular and kidney pathophysiology and resistant hypertension (7).
New Pharmacological Approaches for RAS
There are lots of described important therapeutic usages for Angiotensin (1-7) and analogs on treating cardiovascular diseases and atherosclerosis. Some studies showed new pharmacological approaches. One of them investigated a BM-specific adrenergic beta 1 and beta 2 knock out mouse chimera (AdrB1.B2 KO) to research how sympathetic impulse to the bone influences transcripts and miRNAs in the hypothalamic paraventricular nucleus (92). The results showed that there are molecular axes involved in neural-immune interactions that can serve as targets of therapeutic treatment for a dysfunctional ANS (92). Ahmari et al. produced a mouse chimera in which the BM was irradiated and exactly reconstituted with BM from beta 1 and 2 adrenergic receptor KO mice (93). The study showed that genetic ablation of beta 1 and 2 adrenergic receptors in the BM directs an impressive modification in the BM immune system mediators. This results in decreased circulation levels of a subtype of T cells, neutrophils and macrophages (93). To research Ang-(1-7)-dependent Mas receptor function, Yang et al. used apoE-KO and apoE/Mas-KO mice with Ang-(1-7) or saline for 6 weeks (94). To check whether Ang-(1-7) regulates atherosclerosis through a NO-dependent pathway, apoE-KO mice were used with the NO synthase inhibitor in the presence or lack of Ang-(1-7) (94). Ang-(1-7) has been shown to have protective vascular effects through Mas receptor activation (94).
Future Prospects and Hypotheses
In light of all these data, it is thought that the autonomous control of the BM has an important function in hypertension and there is bidirectional communication between the brain and the BM in this procedure (7). It is thought that there will be important developments that will provide new therapeutic targets in the therapy of hypertension with new studies in this area. It is obvious that there will be innovations at genetic level in the treatment of hypertension with the clarification of this process. Angiotensin peptides on HSCs can be edited with clustered regularly interspaced short palindromic repeats (CRISPR). Cellular therapy and gene edition can contribute to the treatment of essential hypertension. It can change the biology of HSCs that will go to the CNS in an anti-atherogenic and anti-hypertensive direction by genomic edition with CRISPR, by down-regulating AT1 receptors on the HSCs and upregulating MAS receptors. Another therapeutic target will be active microglia, which can affect the activity of neurons in the cardio-regulatory regions of the brain, which can control BM and effect the role of BM HSPCs (7). In addition to the known effects of RAS in many areas, future research and clinical studies are required to explain the different roles of local tissue RAS, including local BM RAS, and to use them as therapeutic targets.
Author Contributions
RC was responsible for the writing of the article. IH served as scientific adviser, drafted the article, and revised it critically for important intellectual content. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bader M. Tissue renin-angiotensin-aldosterone systems: targets for pharmacological therapy. Ann Rev Pharmacol Toxicol. (2010) 50:439–65. doi: 10.1146/annurev.pharmtox.010909.105610
2. Nehme A, Zouein FA, Deris Zayeri Z, Zibara K. An update on the tissue renin angiotensin system and its role in physiology and pathology. J Cardiovasc Develop Dis. (2019) 6:14. doi: 10.3390/jcdd6020014
3. Haznedaroglu I, Tuncer S, Gürsoy MJ. A local renin-angiotensin system in the bone marrow. Med Hypotheses. (1996) 46:507–10. doi: 10.1016/S0306-9877(96)90122-X
4. Haznedaroglu IJEH. A local renin-angiotensin system in the bone marrow still awaits its Christopher Columbus. Exp Hematol. (1998) 26:279.
5. Haznedaroglu IC, Malkan UY. Local bone marrow renin-angiotensin system in the genesis of leukemia and other malignancies. Eur Rev Med Pharmacol. (2016) 20:4089–111.
6. Beyazit Y, Purnak T, Guven GS, Haznedaroglu IC. Local bone marrow renin-angiotensin system and atherosclerosis. Caridiol Res Pract. (2011). 2011:714515. doi: 10.4061/2011/714515
7. Zubcevic J, Santisteban MM, Pitts T, Baekey DM, Perez PD, Bolser DC, et al. Functional neural–bone marrow pathways: implications in hypertension and cardiovascular disease. Hypertension. (2014) 63:e129–39. doi: 10.1161/HYPERTENSIONAHA.114.02440
8. Jun JY, Zubcevic J, Qi Y, Afzal A, Carvajal JM, Thinschmidt JS, et al. Brain-mediated dysregulation of the bone marrow activity in angiotensin II–induced hypertension. Hypertension. (2012) 60:1316–23. doi: 10.1161/HYPERTENSIONAHA.112.199547
9. Santisteban MM, Zubcevic J, Baekey DM, Raizada MK. Dysfunctional brain-bone marrow communication: a paradigm shift in the pathophysiology of hypertension. Curr Hypertension Rep. (2013) 15:377–89. doi: 10.1007/s11906-013-0361-4
10. Strawn WB, Richmond RS, Ann Tallant E, Gallagher PE, Ferrario CM. Renin–angiotensin system expression in rat bone marrow haematopoietic and stromal cells. Br J Haematol. (2004) 126:120–6. doi: 10.1111/j.1365-2141.2004.04998.x
11. Haznedaroglu IC, Beyazit YJJotR-A-AS. Pathobiological aspects of the local bone marrow renin-angiotensin system: a review. J Renin Angiotensin Syst. (2010) 11:205–13. doi: 10.1177/1470320310379876
12. Haznedaroglu IC, Oztürk MA. Towards the understanding of the local hematopoietic bone marrow renin-angiotensin system. Int J Biochem Cell Biol. (2003) 35:867–80. doi: 10.1016/S1357-2725(02)00278-9
13. Hubert C, Savary K, Gasc J-M, Corvol P. The hematopoietic system: a new niche for the renin–angiotensin system. Nat Clin Cardiovasc Med. (2006) 3:80–5. doi: 10.1038/ncpcardio0449
14. Lin C, Datta V, Okwan-Duodu D, Chen X, Fuchs S, Alsabeh R, et al. Angiotensin-converting enzyme is required for normal myelopoiesis. FASEB J. (2011) 25:1145–55. doi: 10.1096/fj.10-169433
15. Kato H, Ishida J, Imagawa S, Saito T, Suzuki N, Matsuoka T, et al. Enhanced erythropoiesis mediated by activation of the renin-angiotensin system via angiotensin II type 1a receptor. FASEB J. (2005) 19:2023–5. doi: 10.1096/fj.05-3820fje
16. Kwiatkowski BA, Richard RE. Angiotensin II receptor-associated protein (AGTRAP) synergizes with Mpl signaling to promote survival and to increase proliferation rate of hematopoietic cells. Blood. (2009) 114:3606. doi: 10.1182/blood.V114.22.3606.3606
17. Durik M, Sevá Pessôa B, Roks AJJCS. The renin–angiotensin system, bone marrow and progenitor cells. Clin Sci. (2012) 123:205–23. doi: 10.1042/CS20110660
18. Heringer-Walther S, Eckert K, Schumacher S-M, Uharek L, Wulf-Goldenberg A, Gembardt F, et al. Angiotensin-(1–7) stimulates hematopoietic progenitor cells in vitro and in vivo. Haematologica. (2009) 94:857–60. doi: 10.3324/haematol.2008.000034
19. Haznedaroglu IC, Beyazit Y. Local bone marrow renin–angiotensin system in primitive, definitive and neoplastic haematopoiesis. Clin Sci. (2013) 124:307–23. doi: 10.1042/CS20120300
20. Park TS, Zambidis ETJH. A role for the renin-angiotensin system in hematopoiesis. Haematologica. (2009) 94:745. doi: 10.3324/haematol.2009.006965
21. Comte L, Lorgeot V, Bignon J, Volkov L, Dupuis F, Wdzieczak-Bakala J, et al. In vivo modifications of AcSDKP metabolism and haematopoiesis in mice treated with 5-fluorouracil and Goralatide. Eur J Clin Invest. (1998) 28:856–63. doi: 10.1046/j.1365-2362.1998.00356.x
22. Arici M, Büyükaşik YJM. A unifying hypothesis for the renin–angiotensin system and hematopoiesis: sticking the pieces together with the JAK-STAT pathway. Med Hypotheses. (2000) 54:80–3. doi: 10.1054/mehy.1998.0830
23. Öztürk MA, Güven GS, Haznedaroglu IC. How hematopoietic stem cells know and act in cardiac microenvironment for stem cell plasticity? Impact of local renin–angiotensin systems. Med Hypotheses. (2004) 63:866–74. doi: 10.1016/j.mehy.2004.04.011
24. Rodgers K, Xiong S. Effect of angiotensin II and angiotensin (1–7) on hematopoietic recovery after intravenous chemotherapy. Cancer Chemother Pharmacol. (2003) 51:97–106. doi: 10.1007/s00280-002-0509-4
25. Rodgers KE, Oliver J. Phase I/II dose escalation study of angiotensin 1-7 [A (1-7)] administered before and after chemotherapy in patients with newly diagnosed breast cancer. Cancer Chemother Pharmacol. (2006) 57:559–68. doi: 10.1007/s00280-005-0078-4
26. Ferrario CM. Angiotensin I, angiotensin II and their biologically active peptides. J Hypertension. (2002) 20:805–7. doi: 10.1097/00004872-200205000-00004
27. Jarajapu YP. Targeting ACE2/angiotensin-(1-7)/mas receptor axis in the vascular progenitor cells for cardiovascular diseases. Mol Pharmacol. (2020) 98:mol.119.117580. doi: 10.1124/mol.119.117580
28. Gomez RA, Norling LL, Wilfong N, Isakson P, Lynch KR, Hock R, et al. Leukocytes synthesize angiotensinogen. Hypertension. (1993) 21:470–5. doi: 10.1161/01.HYP.21.4.470
29. Dezsö B, Jacobsen J, Poulsen K. Evidence for the presence of angiotensins in normal, unstimulated alveolar macrophages and monocytes. J Hypertension. (1989) 7:5–11. doi: 10.1097/00004872-198901000-00002
30. Ehlers MR, Riordan JF. Angiotensin-converting enzyme: new concepts concerning its biological role. Biochemistry. (1989) 28:5311–8. doi: 10.1021/bi00439a001
31. Jokubaitis VJ, Sinka L, Driessen R, Whitty G, Haylock DN, Bertoncello I, et al. Angiotensin-converting enzyme (CD143) marks hematopoietic stem cells in human embryonic, fetal, and adult hematopoietic tissues. Blood. (2008) 111:4055–63. doi: 10.1182/blood-2007-05-091710
32. Tavian M, Biasch K, Sinka L, Vallet J, Péault B. Embryonic origin of human hematopoiesis. Int J Dev Boil. (2010) 54:1061–5. doi: 10.1387/ijdb.103097mt
33. Sinka L, Biasch K, Khazaal I, Péault B, Tavian MJB. Angiotensin-converting enzyme (CD143) specifies emerging lympho-hematopoietic progenitors in the human embryo. Blood. (2012) 119:3712–23. doi: 10.1182/blood-2010-11-314781
34. Aksu S, Beyazit Y, Haznedaroglu IC, Canpinar H, Kekilli M, Uner A, et al. Over-expression of angiotensin-converting enzyme (CD 143) on leukemic blasts as a clue for the activated local bone marrow RAS in AML. Leuk Lymphoma. (2006) 47:891–6. doi: 10.1080/10428190500399250
35. Beyazit Y, Aksu S, Haznedaroglu IC, Kekilli M, Misirlioglu M, Tuncer S, et al. Overexpression of the local bone marrow renin-angiotensin system in acute myeloid leukemia. J Natl Med Assoc. (2007) 99:57.
36. Wulf GG, Jahns-Streubel G, Nobiling R, Strutz F, Hemmerlein B, Hiddemann W, et al. Renin in acute myeloid leukaemia blasts. Br J Haematol. (1998) 100:335–7. doi: 10.1046/j.1365-2141.1998.00565.x
37. Yamashita T, Ahmad S, Wright KN, Roberts DJ, VonCannon JL, Wang H, et al. Noncanonical mechanisms for direct bone marrow generating Ang II (angiotensin II) predominate in CD68 positive myeloid lineage cells. Hypertension. (2020) 75:500–9. doi: 10.1161/HYPERTENSIONAHA.119.13754
38. Shen XZ, Bernstein KE. The peptide network regulated by angiotensin converting enzyme (ACE) in hematopoiesis. Cell Cycle. (2011) 10:1363–9. doi: 10.4161/cc.10.9.15444
39. Gallagher PE, Tallant EA, Ferrario CM. Angiotensin converting enzyme 2: a critical regulator of the renin-angiotensin system. Circ Res. (2006) 126:129–42. doi: 10.1007/0-387-27826-5_11
40. Belyea BC, Xu F, Pentz ES, Medrano S, Li M, Hu Y, et al. Identification of renin progenitors in the mouse bone marrow that give rise to B-cell leukaemia. Nat Commun. (2014) 5:1–14. doi: 10.1038/ncomms4273
41. Nehme A, Cerutti C, Dhaouadi N, Gustin MP, Courand P-Y, Zibara K, et al. Atlas of tissue renin-angiotensin-aldosterone system in human: a transcriptomic meta-analysis. Sci Rep. (2015) 5:10035. doi: 10.1038/srep10035
42. Tanaka K, Sata M. Role of vascular progenitor cells in cardiovascular disease. Curr Pharm Design. (2009) 15:2760–8. doi: 10.2174/138161209788923868
43. de Souza-Neto FP, Santuchi MC, e Silva MdM, Campagnole-Santos MJ, da Silva RF. Angiotensin-(1–7) and alamandine on experimental models of hypertension and atherosclerosis. Curr Hypertens Rep. (2018) 20:17. doi: 10.1007/s11906-018-0798-6
44. Mazzolai L, Hayoz DJ. The renin-angiotensin system and atherosclerosis. Curr Hyperten Rep. (2006) 8:47–53. doi: 10.1007/s11906-006-0040-9
45. Durante A, Peretto G, Laricchia A, Ancona F, Spartera M, Mangieri A, et al. Role of the renin-angiotensin-aldosterone system in the pathogenesis of atherosclerosis. Curr Pharm Des. (2012) 18:981–1004. doi: 10.2174/138161212799436467
46. Husain K, Hernandez W, Ansari RA, Ferder L. Inflammation, oxidative stress and renin angiotensin system in atherosclerosis. World J Biol Chem. (2015) 6:209. doi: 10.4331/wjbc.v6.i3.209
47. Fukuda D, Sata M. Role of bone marrow renin–angiotensin system in the pathogenesis of atherosclerosis. Pharmacol Ther. (2008) 118:268–76. doi: 10.1016/j.pharmthera.2008.02.007
48. Rodgers KE, Xiong S, Steer R, DiZerega GS. Effect of angiotensin II on hematopoietic progenitor cell proliferation. Stem Cells. (2000) 18:287–94. doi: 10.1634/stemcells.18-4-287
49. Daugherty A, Rateri DL, Lu H, Inagami T, Cassis LA. Hypercholesterolemia stimulates angiotensin peptide synthesis and contributes to atherosclerosis through the AT1A receptor. Circulation. (2004) 110:3849–57. doi: 10.1161/01.CIR.0000150540.54220.C4
50. Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med. (1999) 340:115–26. doi: 10.1056/NEJM199901143400207
51. Libby P, Ridker PM, Hansson GKJN. Progress and challenges in translating the biology of atherosclerosis. Nature. (2011). 473:317–25. doi: 10.1038/nature10146
52. Flaherty ML, Kissela B, Khoury JC, Alwell K, Moomaw CJ, Woo D, et al. Carotid artery stenosis as a cause of stroke. Neuroepidemiology. (2013) 40:36–41. doi: 10.1159/000341410
53. Banerjee C, Chimowitz MI. Stroke caused by atherosclerosis of the major intracranial arteries. Circ Res. (2017) 120:502–13. doi: 10.1161/CIRCRESAHA.116.308441
54. Arenillas JF. Intracranial atherosclerosis: current concepts. Stroke. (2011) 42(Suppl. 1):S20–3. doi: 10.1161/STROKEAHA.110.597278
55. Tabas I, García-Cardeña G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol. (2015) 209:13–22. doi: 10.1083/jcb.201412052
56. Liu Y, Yu H, Zhang Y, Zhao Y. TLRs are important inflammatory factors in atherosclerosis and may be a therapeutic target. Med Hypotheses. (2008) 70:314–6. doi: 10.1016/j.mehy.2007.05.030
57. Hansson GK, Libby P, Tabas I. Inflammation and plaque vulnerability. J Intern Med. (2015) 278:483–93. doi: 10.1111/joim.12406
58. Shabir O, Berwick J, Francis SE. Neurovascular dysfunction in vascular dementia, Alzheimer's and atherosclerosis. BMC Neurosci. (2018) 19:62. doi: 10.1186/s12868-018-0465-5
59. Group FSoAPiS. Atherosclerotic disease of the aortic arch as a risk factor for recurrent ischemic stroke. N Engl J Med. (1996) 334:1216–21. doi: 10.1056/NEJM199605093341902
60. Ohira T, Shahar E, Chambless LE, Rosamond WD, Mosley TH, Folsom AR. Risk factors for ischemic stroke subtypes: the atherosclerosis risk in communities study. Stroke. (2006) 37:2493–8. doi: 10.1161/01.STR.0000239694.19359.88
61. Chambless LE, Folsom AR, Clegg LX, Sharrett AR, Shahar E, Nieto FJ, et al. Carotid wall thickness is predictive of incident clinical stroke: the Atherosclerosis Risk in Communities (ARIC) study. Am J Epidemiol. (2000) 151:478–87. doi: 10.1093/oxfordjournals.aje.a010233
62. Iadecola C. The overlap between neurodegenerative and vascular factors in the pathogenesis of dementia. Acta Neuropathol. (2010) 120:287–96. doi: 10.1007/s00401-010-0718-6
63. Saji N, Toba K, Sakurai TJP. Cerebral small vessel disease and arterial stiffness: tsunami effect in the brain. Pulse. (2015) 3:182–9. doi: 10.1159/000443614
64. Yamazaki K, Allen TD. Ultrastructural morphometric study of efferent nerve terminals on murine bone marrow stromal cells, and the recognition of a novel anatomical unit: The “neuro-reticular complex”. Am J Anatomy. (1990) 187:261–76. doi: 10.1002/aja.1001870306
65. Miyan J, Broome C, Whetton A. Neural regulation of bone marrow. Blood. (1998) 92:2971–3. doi: 10.1182/blood.V92.8.2971b.splL2_2971_2972
66. MacEneaney OJ, DeSouza CA, Weil BR, Kushner EJ, Van Guilder GP, Mestek M, et al. Prehypertension and endothelial progenitor cell function. J Hum Hypertension. (2011) 25:57–62. doi: 10.1038/jhh.2010.31
67. Giannotti G, Doerries C, Mocharla PS, Mueller MF, Bahlmann FH, Horvàth T, et al. Impaired endothelial repair capacity of early endothelial progenitor cells in prehypertension: relation to endothelial dysfunction. Hypertension. (2010) 55:1389–97. doi: 10.1161/HYPERTENSIONAHA.109.141614
68. Yao EH, Yu Y, Fukuda N. Oxidative stress on progenitor and stem cells in cardiovascular diseases. Curr Pharmac Biotechnol. (2006) 7:101–8. doi: 10.2174/138920106776597685
69. Imanishi T, Hano T, Nishio I. Estrogen reduces endothelial progenitor cell senescence through augmentation of telomerase activity. J Hypertension. (2005) 23:1699–706. doi: 10.1097/01.hjh.0000176788.12376.20
70. Ganten D, Speck G. The brain renin-angiotensin system: a model for the synthesis of peptides in the brain. Biochem Pharmacol. (1978) 27:2379–89. doi: 10.1016/0006-2952(78)90348-9
71. Karamyan VT, Speth RC. Enzymatic pathways of the brain renin-angiotensin system: unsolved problems and continuing challenges. Regul Pept. (2007) 143:15–27. doi: 10.1016/j.regpep.2007.03.006
72. Santos R, Brosnihan KB, Chappell MC, Pesquero J, Chernicky CL, Greene LJ, et al. Converting enzyme activity and angiotensin metabolism in the dog brainstem. Hypertension. (1988) 11(2 Pt 2):I153. doi: 10.1161/01.HYP.11.2_Pt_2.I153
73. Block C, Santos R, Brosnihan K, Ferrario CJP. Immunocytochemical localization of angiotensin-(1–7) in the rat forebrain. Peptides. (1988) 9:1395–401. doi: 10.1016/0196-9781(88)90208-2
74. Brosnihan KB, Schiavone M, Sprunger A, Chappell MC, Rizzo M, Ferrario CMJH. In vivo release of angiotensin II from the rat hypothalamus. Hypertension. (1988) 11(2 Pt 2):I158. doi: 10.1161/01.HYP.11.2_Pt_2.I158
75. Zubcevic J, Waki H, Raizada MK, Paton JFJH. Autonomic-immune-vascular interaction: an emerging concept for neurogenic hypertension. Hypertension. (2011) 57:1026–33. doi: 10.1161/HYPERTENSIONAHA.111.169748
76. Bardgett ME, Holbein WW, Herrera-Rosales M, Toney GM. Ang II–salt hypertension depends on neuronal activity in the hypothalamic paraventricular nucleus but not on local actions of tumor necrosis factor-α. Hypertenion. (2014) 63:527–34. doi: 10.1161/HYPERTENSIONAHA.113.02429
77. Toney GM, Pedrino GR, Fink GD, Osborn JW. Does enhanced respiratory–sympathetic coupling contribute to peripheral neural mechanisms of angiotensin II–salt hypertension? Exp Physiol. (2010) 95:587–94. doi: 10.1113/expphysiol.2009.047399
78. Shan Z, Zubcevic J, Shi P, Jun JY, Dong Y, Murça TM, et al. Chronic knockdown of the nucleus of the solitary tract AT1 receptors increases blood inflammatory-endothelial progenitor cell ratio and exacerbates hypertension in the spontaneously hypertensive rat. Hypertension. (2013) 61:1328–33. doi: 10.1161/HYPERTENSIONAHA.111.00156
79. Capone C, Faraco G, Peterson JR, Coleman C, Anrather J, Milner TA, et al. Central cardiovascular circuits contribute to the neurovascular dysfunction in angiotensin II hypertension. J Neurosci. (2012) 32:4878–86. doi: 10.1523/JNEUROSCI.6262-11.2012
80. Zubcevic J, Jun JY, Kim S, Perez PD, Afzal A, Shan Z, et al. Altered inflammatory response is associated with an impaired autonomic input to the bone marrow in the spontaneously hypertensive rat. Hypertension. (2014) 63:542–50. doi: 10.1161/HYPERTENSIONAHA.113.02722
81. Casanova-Acebes M, Pitaval C, Weiss LA, Nombela-Arrieta C, Chèvre R, Noelia A, et al. Rhythmic modulation of the hematopoietic niche through neutrophil clearance. Cell. (2013) 153:1025–35. doi: 10.1016/j.cell.2013.04.040
82. Méndez-Ferrer S, Lucas D, Battista M, Frenette PS. Haematopoietic stem cell release is regulated by circadian oscillations. Nature. (2008) 452:442–7. doi: 10.1038/nature06685
83. Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. (2009) 325:612–6. doi: 10.1126/science.1175202
84. Malhotra D, Fletcher AL, Turley SJ. Stromal and hematopoietic cells in secondary lymphoid organs: partners in immunity. Immunol Rev. (2013) 251:160–76. doi: 10.1111/imr.12023
85. Crowley SD, Song Y-S, Sprung G, Griffiths R, Sparks M, Yan M, et al. A role for angiotensin II type 1 receptors on bone marrow-derived cells in the pathogenesis of angiotensin II–dependent hypertension. Hypertension. (2010) 55:99–108. doi: 10.1161/HYPERTENSIONAHA.109.144964
86. Hristov M, Zernecke A, Bidzhekov K, Liehn EA, Shagdarsuren E, Ludwig A, et al. Importance of CXC chemokine receptor 2 in the homing of human peripheral blood endothelial progenitor cells to sites of arterial injury. Circ Res. (2007) 100:590–7. doi: 10.1161/01.RES.0000259043.42571.68
87. Zhu XY, Urbieta-Caceres V, Krier JD, Textor SC, Lerman A, Lerman LO. Mesenchymal stem cells and endothelial progenitor cells decrease renal injury in experimental swine renal artery stenosis through different mechanisms. Stem Cells. (2013) 31:117–25. doi: 10.1002/stem.1263
88. Feitelson J, Kulenovic E, Beck D, Harris P, Passmore J, Malkani A, et al. Endogenous norepinephrine regulates blood flow to the intact rat tibia. J Orthopaedic Res. (2002) 20:391–6. doi: 10.1016/S0736-0266(01)00121-8
89. Hollis M, Wang DH. Transient receptor potential vanilloid in blood pressure regulation. Curr Opin Nephrol Hypertension. (2013) 22:170–6. doi: 10.1097/MNH.0b013e32835c8d4c
90. Wang Y, Wang DH. Neural control of blood pressure: focusing on capsaicin-sensitive sensory nerves. Cardiovasc Haematol Disorders Drug Targets. (2007) 7:37–46. doi: 10.2174/187152907780059100
91. Yu S-q, Wang DH. Enhanced salt sensitivity following shRNA silencing of neuronal TRPV1 in rat spinal cord. Acta Pharmacol Sinica. (2011) 32:845–52. doi: 10.1038/aps.2011.43
92. Martyniuk CJ, Martinez R, Kostyniuk DJ, Mennigen JA, Zubcevic JJPG. Genetic ablation of bone marrow beta-adrenergic receptors in mice modulates miRNA-transcriptome networks of neuroinflammation in the paraventricular nucleus. Physiol Genomics. (2020) 52:169–77. doi: 10.1152/physiolgenomics.00001.2020
93. Ahmari N, Schmidt JT, Krane GA, Malphurs W, Cunningham BE, Owen JL, et al. Loss of bone marrow adrenergic beta 1 and 2 receptors modifies transcriptional networks, reduces circulating inflammatory factors, and regulates blood pressure. Physiol Genomics. (2016) 48:526–36. doi: 10.1152/physiolgenomics.00039.2016
Keywords: renin-angiotensin system, bone marrow, atherosclerosis, hypertension, central nervous system
Citation: Ciftciler R and Haznedaroglu IC (2020) Pathobiological Interactions of Local Bone Marrow Renin-Angiotensin System and Central Nervous System in Systemic Arterial Hypertension. Front. Endocrinol. 11:425. doi: 10.3389/fendo.2020.00425
Received: 24 March 2020; Accepted: 28 May 2020;
Published: 07 August 2020.
Edited by:
Ana Cristina Simões E. Silva, Federal University of Minas Gerais, BrazilReviewed by:
Eric Lazartigues, Louisiana State University, United StatesSérgio Henrique Sousa Santos, Federal University of Minas Gerais, Brazil
Copyright © 2020 Ciftciler and Haznedaroglu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ibrahim Celalettin Haznedaroglu, ichaznedaroglu@gmail.com
†ORCID: Rafiye Ciftciler orcid.org/0000-0001-5687-8531
Ibrahim Celalettin Haznedaroglu orcid.org/0000-0003-2020-2745