Paracrine Role of the Endothelium in Metabolic Homeostasis in Health and Nutrient Excess
 Cheukyau Luk
Cheukyau Luk Natalie J. Haywood
Natalie J. Haywood Katherine I. Bridge
Katherine I. Bridge - Leeds Institute of Cardiovascular and Metabolic Medicine, Faculty of Medicine and Health, University of Leeds, Leeds, United Kingdom
The vascular endothelium traditionally viewed as a simple physical barrier between the circulation and tissue is now well-established as a key organ mediating whole organism homeostasis by release of a portfolio of anti-inflammatory and pro-inflammatory vasoactive molecules. Healthy endothelium releases anti-inflammatory signaling molecules such as nitric oxide and prostacyclin; in contrast, diseased endothelium secretes pro-inflammatory signals such as reactive oxygen species, endothelin-1 and tumor necrosis factor-alpha (TNFα). Endothelial dysfunction, which has now been identified as a hallmark of different components of the cardiometabolic syndrome including obesity, type 2 diabetes and hypertension, initiates and drives the progression of tissue damage in these disorders. Recently it has become apparent that, in addition to vasoactive molecules, the vascular endothelium has the potential to secrete a diverse range of small molecules and proteins mediating metabolic processes in adipose tissue (AT), liver, skeletal muscle and the pancreas. AT plays a pivotal role in orchestrating whole-body energy homeostasis and AT dysfunction, characterized by local and systemic inflammation, is central to the metabolic complications of obesity. Thus, understanding and targeting the crosstalk between the endothelium and AT may generate novel therapeutic opportunities for the cardiometabolic syndrome. Here, we provide an overview of the role of the endothelial secretome in controlling the function of AT. The endothelial-derived metabolic regulatory factors are grouped and discussed based on their physical properties and their downstream signaling effects. In addition, we focus on the therapeutic potential of these regulatory factors in treating cardiometabolic syndrome, and discuss areas of future study of potential translatable and clinical significance. The vascular endothelium is emerging as an important paracrine/endocrine organ that secretes regulatory factors in response to nutritional and environmental cues. Endothelial dysfunction may result in imbalanced secretion of these regulatory factors and contribute to the progression of AT and whole body metabolic dysfunction. As the vascular endothelium is the first responder to local nutritional changes and adipocyte-derived signals, future work elucidating the changes in the endothelial secretome is crucial to improve our understanding of the pathophysiology of cardiometabolic disease, and in aiding our development of new therapeutic strategies to treat and prevent cardiometabolic syndrome.
Introduction
Cardiometabolic syndrome (CMS), or metabolic syndrome, is characterized by a combination of metabolic dysfunction, including hyperglycaemia, insulin resistance, hyperlipidaemia, obesity and hypertension. The global prevalence of CMS is continuing to rise and, in 2018 it was estimated to affect 25% of the population (1). To address the differences in clinical definitions of the disease by various health organizations, the International Diabetes Federation proposed the latest accepted worldwide definition of CMS; central obesity and two of following: elevated triglycerides, lower HDL(high-density lipoprotein)-cholesterol, elevated blood pressure and elevated fasting plasma glucose level (2). These metabolic and cardiovascular perturbations individually and in combination, contribute to a substantial increase in cardiovascular disease morbidity and mortality.
Many environmental factors can contribute to the onset and development of central obesity in CMS, notably recent changes in human lifestyle, such as increased sedentary behaviors and consumption of high calorific foods (1). Opposing the conventional view that obesity is purely a metabolic disorder, the concept of “immunometabolism” describes the previously underdetermined relationship between chronic inflammation in metabolic organs and the brain, and energy imbalance in obesity (3). Several pioneering papers revealing the role of inflammation in metabolic organs, and its link to metabolic defects, focussed on the interaction between macrophages and adipocytes in adipose tissue (4–6). The idea of immunometabolism was then explored and expanded further into other organs including liver, skeletal muscle, pancreas, gastrointestinal tract and brain (3, 7). In addition, endothelial dysfunction is believed to play an important role in the pathophysiology of CMS, as it is well-established that obesity and metabolic dysfunction are able to disrupt a number of signaling pathways in the endothelium (8). With specific attention to interactions between the vascular endothelium and adipocytes, we review the role of endothelial-derived factors in mediating metabolic and inflammatory signaling in adipose tissue (AT), both in the context of normal physiology, and in the pathophysiology associated with CMS.
Endothelial Dysfunction in CMS and the Emerging Interest of Endothelial Secretome in Health and Disease
The endothelium, anatomically defined as the inner cellular lining of blood and lymphatic vessels, acts as a physiological barrier, which prevents direct contact between tissues and the circulation (9). It also plays a role in mediating angiogenesis, inflammation, thrombosis, vascular tone and energy homeostasis (8–12). Endothelial dysfunction is often defined as the reduced production or availability of nitric oxide (NO), the elevated release of vasoconstrictors such as endothelin-1 (13) and the upregulation of cell adhesion molecules, associated with enhanced leukocyte adherence, increased cell permeability, and platelet activation (14). Endothelial dysfunction is a well-established response to cardiovascular risk factors and precedes the development of atherosclerosis (15).
The endothelium is a dynamic organ, and is one of the first responders to metabolic and vascular changes (16), such as insulin level (17) and blood flow (18). Due to its intimate relationship with nearby cells, impaired endothelial barrier function, or imbalanced endothelial secretion of metabolites, results in dysregulated signaling pathways of peripheral tissues, which in turn contributes to the development of metabolic dysfunction and chronic tissue inflammation. Time-course experiments on diet-induced obese mice revealed the preceding role of endothelial dysfunction in the development of metabolic dysfunction and chronic inflammation in liver, muscle and AT (19). In obesity, dysfunctional endothelium displays a more pro-inflammatory and pro-fibrotic phenotype, resulting in increased vascular leakage and acceleration of tissue inflammation by recruitment and activation of immune cells (20).
In addition to acting as a physical barrier between local tissues and the circulation, the endothelium plays an important role in signaling. Since the discovery of endothelial-derived relaxing factor (EDRF), now identified as NO, the endothelial secretory profile has gained much attention (21, 22). There is a growing body of evidence of the presence of endothelial-derived small membrane vesicles, which are released to the extracellular space (23). These findings further establish the existence of an endothelial secretome and raise the question of its role in various diseases including obesity in CMS.
Adipose Dysfunction and Its Treatment Options in Obesity
Obesity is a multisystem disorder driven by nutrient excess, leading to perturbation of glucose homeostasis, immune dysregulation, dyslipidaemia and disadvantageous alterations in adipocyte phenotype, which leads to a vicious cycle of cellular dysfunction contributing to CMS (15, 24). During nutrient excess, white AT (WAT) undergoes expansive remodeling, whereby white adipocytes adopt a hypertrophic/hyperplastic phenotype (25), accompanied by neovascularisation to support this remodeling (26, 27). In advanced obesity there is a mismatch between WAT expansion and vascularisation leading to inadequate perfusion (28), adipocyte ischaemia (28), inflammation (28–30), secretion of pro-inflammatory mediators (31) and the requirement to store lipids in tissues ill-equipped to deal with this challenge, all contributing to CMS (32).
AT is crucial in storing excess energy and maintaining thermal homeostasis (33). It is dynamically regulated by a wide range of physiological cues, including norepinephrine, insulin and acetylcholine (33). There are two main types of AT; WAT which is specialized for the storage of energy in the form of triglyceride (34) and thermogenic brown AT (BAT), which unlike WAT expresses uncoupling protein-1 (UCP-1) (35). UCP-1 uncouples cellular respiration from mitochondrial ATP synthesis, affording thermogenic AT the capacity to burn fat and generate heat (36). Recent studies indicate that at least two distinct types of thermogenic adipocyte exist in mammals: a pre-existing form established during development, termed classical BAT, and an inducible form, “beige” adipocytes. Beige adipocyte biogenesis can be stimulated by various environmental cues, such as chronic cold exposure, in a process frequently referred to as “beiging” of white fat (37, 38). The thermogenic capacity of beige adipocytes and the possibility of controlling their expression has stimulated substantial attention due to its potential application in the mitigation of obesity-related disease (39–42). Although extensive efforts have been made pharmacologically to activate AT thermogenesis, these attempts have been unsuccessful due to poor bioavailability and concerns over the side effects of the agents used (43). More importantly, a recent study reported that human brown adipocyte thermogenesis is actually mediated by β2-adrenoceptors, instead of β3-adrenoceptors as in rodents (44), explaining the lack of efficacy of β3-adrenoceptor agonists to activate brown adipose thermogenesis or white adipose beiging. Thus, identifying alternative pathways to promote beige adipose biogenesis could lead to therapeutic interventions which reduce cardiovascular risk.
Alternative therapeutic goals also include reducing AT inflammation and fibrosis (45). Under normal physiology, inflammation is important in maintaining healthy adipogenesis and adipocyte function and helps regulate whole-body glucose tolerance (46). However, chronic adipocyte inflammation can link to metabolic defects such as insulin resistance (46–48). Current therapeutics targeting AT inflammation do not always present positive outcomes in human trials and potential risk of increased susceptibility to infections is yet to be explored (45). On the other hand, we still await clinical studies employing anti-fibrotic agents to determine the efficacy and safety of targeting WAT fibrosis to treat CMS.
Scope of Review
AT is highly vascularized with a capillary network, allowing the reciprocal transport of nutrients, substrates and oxygen. During AT expansion, the capillary network also expands to meet the increasing demand for nutrients and oxygen (49). With particular interest in dissecting the communication between endothelial cells and adipocytes, here we review the role of endothelial-derived molecules in mediating metabolism and inflammation in the pathophysiology of CMS. We focus on the synthesis, release and the paracrine action of the endothelial secretome in AT and discuss how it may be exploited therapeutically in the future. Endothelial-derived molecules are grouped according to their physical properties.
Gaseous Regulators
Nitric Oxide (NO)
Endothelial-derived NO has been traditionally viewed as a key modulator of cardiovascular function, and reduced bioavailability of NO has been associated with inflammation and vascular disease (10, 50). More recently, data suggests that NO also acts as a metabolic modulator, and reduced circulating NO levels have been observed before the development of peripheral insulin resistance in diet-induced obesity (12, 19). Under normal physiological conditions, endothelial nitric oxide synthase (eNOS, also known as NOS3) is active and generates NO transiently. The activity of eNOS is regulated by shear stress within vessels. The synthesis of NO can also be elevated upon stimulation by endogenous signaling molecules such as insulin, insulin-like growth factor 1 (IGF-1), acetylcholine and the pro-inflammatory molecule bradykinin (51–54). Within AT, eNOS-mediated NO generation can be upregulated by adipocyte-secreted adiponectin (55) and leptin (56). Apart from its well-known vasodilatory effect via activating the soluble guanylate cyclase (sGC)-generated cyclic guanosine monophosphate (cGMP) pathway in vascular smooth muscle, NO is thought to be important in regulating oxygen transport and mitochondrial respiration in other tissues (57). In contrast, in diseases such as CMS, eNOS activity is suppressed whereas inducible NOS (iNOS) in the endothelium is activated to generate potentially pathophysiological concentrations of NO, which reacts with superoxide to form peroxynitrite (58). Peroxynitrite is cytotoxic and is a known pro-atherosclerotic factor (58, 59) (reviewed in Peroxynitrite (ONOO–)).
A range of factors, including elevated plasma levels of free fatty acids, pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNFα), oxidized low-density lipoprotein (OxLDL) and asymmetric dimethylarginine, all common features of CMS, are known to inhibit eNOS activity (60–63). Under prolonged high fat diet, mice display profoundly reduced eNOS expression, specifically in AT suggesting diseased adiposity is coupled with a decline in eNOS activity (64). The WAT of diet-induced obese mice demonstrates a reduction in NO coupled with increased AT inflammation, characterized by increased gene expression of the pro-inflammatory TNFα (65). eNOS-deficient mice are insulin-resistant and hyperlipidaemic (66, 67), even on a chow diet, AT from eNOS-deficient mice displays a notably inflammatory profile, characterized by increased gene expression of TNFα, interleukin-6 (IL-6) and monocyte chemoattractant protein-1 (MCP-1) (65). Together these findings suggested endothelial-derived NO is insulin-sensitizing and anti-inflammatory in AT under normal physiological conditions. Promisingly, in eNOS-deficient mice, the addition of dietary nitrate at a dose just enough to restore physiological NO levels, is able to reverse the unfavorable metabolic sequelae, reducing both visceral fat deposition and circulating triglycerides (68). Restoring NO downstream signaling by administration of sildenafil also reduces AT inflammation in high fat diet-challenged mice (65). Complementary studies using mice overexpressing eNOS, demonstrate that eNOS over-expression offers protection against diet-induced obesity, with a more metabolically active AT, characterized by smaller adipocyte size and elevated mitochondrial biogenesis and metabolism (64). Taken together these murine studies show that eNOS activity is important in regulating AT function and inflammation.
Various therapeutic approaches have been employed to determine whether targeting NO can improve AT metabolism in rodents and humans. S-nitrosoacetyl penicillamine (SNAP) an NO donor has been shown to enhance mitochondrial biogenesis in primary murine brown adipocyte precursors and cultured 3T3-L1 white adipocytes, in a process mediated by cGMP (69). Nitrate, which can be utilized to produce NO under hypoxia, has been shown to have beneficial effects in AT in several studies. Nitrate can induce gene expression of beiging markers in cultured primary murine white adipocytes (70, 71), as well as inducing other favorable metabolic changes, including upregulation of mitochondrial respiration, increased uptake and oxidation of fatty acid and glucose (71, 72). Application of nitrite on palmitate-treated primary mouse white adipocytes also induces beiging (73). Although eNOS is not the only source of NO in mammals due to the presence of other isoforms of NOS and dietary nitrate intake, these findings support the hypothesis that eNOS-derived NO promotes adipocyte beiging and mitochondrial biogenesis.
eNOS-derived NO has also been well-characterized as an anti-inflammatory mediator. It inhibits platelet aggregation (74) and acts on the endothelium to suppress expression of adhesion molecules, thus suppressing leukocyte recruitment and accumulation (75). Impaired eNOS-NO signaling may contribute to AT inflammation in obesity, as characterized by increased leukocyte adhesion and platelet aggregation (76).
In rodent models, dietary nitrate resulted in beiging of white AT in rats (71) and more interestingly, in mice challenged with high fat diet, dietary nitrate supplementation reduced weight gain, improved glucose tolerance and attenuated AT inflammation (73, 77). Chronic dietary supplementation of L-arginine (the precursor of NO) reduced weight gain, circulating glucose and triglyceride levels in Zucker diabetic fatty rats (78). Human studies have also confirmed the benefits of targeting NO in CMS. In a randomized, double-blind, placebo-controlled clinical trial, 6 weeks of dietary L-arginine supplementation reduced central adiposity in women with obesity (79). In another clinical trial, 6 months of dietary L-arginine supplementation improved whole body insulin-sensitivity in individuals with obesity (80). However, conflicting results showing lack of efficacy of L-arginine treatment have also been reported in studies of patients with obesity and insulin resistance, raising the doubt of therapeutic potential of L-arginine (81, 82). A recent systematic review and meta-analysis of clinical trials suggested L-arginine supplementation resulted in reduced waist circumference but had no effect in reducing body weight or BMI (83). Future studies determining the appropriate dosage and length of the treatment are needed to improve treatment outcome using L-arginine.
Proteins Mediators
Endothelin-1 (ET-1)
Endothelin was the first potent endothelium-derived vasoconstrictor to be identified. ET-1 was isolated from the culture media of porcine aortic endothelial cells as a 21 amino acid peptide (84). Two other endothelins, ET-2 and ET-3, were later identified. The endothelin family is synthesized and released upon cleavage of its precursors by endothelin-converting enzymes (85). The synthesis of ET-1 can be upregulated by endogenous mediators such as thrombin (84), angiotensin II (86), insulin (87), lipoproteins (88, 89), and suppressed by NO (90) and prostacyclin (91). Secreted endothelin acts on endothelin receptors type A and B (ETAR and ETBR) and mediate glucose and lipid metabolism in the AT (92).
In in vitro experiments using mature 3T3-L1 adipocytes, ET-1 upregulated glycerol release in a dose-dependent manner (93). This effect was prevented by pre-treatment using an ETAR antagonist BQ-610, but not an ETBR antagonist BQ-788, suggesting ET-1-induced lipolysis was mediated by ETAR (93). In time-course experiments the effect of ET-1 on glucose uptake was shown to be biphasic; in mature 3T3-L1 adipocytes, treatment for up to 6 h stimulated ETAR-mediated glucose transporter expression and glucose uptake, whereas longer treatments suppressed glucose uptake (94). In cultured 3T3-L1 adipocytes, prolonged ET-1 treatment has also been shown to cause insulin resistance, in an ETAR-mediated process (95, 96), which is consistent with another study where prolonged ET-1 stimulation enhanced lipolysis in differentiated subcutaneous white adipocytes, again mediated by ETAR (97). Prolonged ET-1 stimulation also results in ETBR-mediated suppression of insulin receptor substrate-1 (IRS-1) expression and insulin-stimulated anti-lipolytic responses in differentiated visceral adipocytes (98). Taken together, these studies demonstrate the detrimental effect that long-term exposure of ET-1 has on AT, which could contribute to the development of CMS in obesity.
From a conventional perspective, ET-1 is vasoconstrictive and pro-inflammatory. There is some evidence suggesting that ET-1 induces immune cell recruitment and activation (99, 100). ET-1 can stimulate monocytes to release IL-8 and MCP-1, which recruit neutrophils and monocytes, respectively (99). ET-1 can also stimulate degranulation of mast cells to release TNFα and VEGF (100). These in vitro findings suggest ET-1 can induce inflammation by activating circulating monocytes or tissue-resident mast cells, which may lead to AT inflammation and fibrosis in obesity (101).
In patients with obesity, ET-1 is upregulated in AT (97, 98). Elevated ET-1 activity has also been reported in patients with type II diabetes (102). Studies support a link between higher basal ET-1 signaling activity and greater adiposity, although whether ET-1 has causal role in excess adiposity is unknown. In support of ET-1 playing a causal role in the pathophysiology of CMS, patients with CMS have an elevated arterial ET-1 level which is reported to be associated with circulating triglyceride level (103). Enhanced ET-1 signaling is also observed in overweight individuals and is preserved in individuals with obesity suggesting heightened ET-1 signaling precedes the development of obesity and its associated complications in patients with CMS (104). In contrast, body mass index has been recently reported to be negatively correlated with ET-1 expression level in omental AT (105). Differences in these findings may be due to varying influence of other co-morbidities such as vascular diseases which were excluded in the former study (104) but not the latter (105), or the complex biphasic actions of ET-1 as described above.
Although whether AT ET-1 signaling plays a causal role in obesity remains inconclusive, preclinical studies suggest inhibiting ET-1 could be another possible therapeutic strategy for CMS. Mice with endothelial-specific overexpression of ET-1 fed with chow diet displayed impaired glucose intolerance and lower systemic energy expenditure (105). However, endothelial-specific ET-1 overexpression did not attenuate Western diet-induced WAT inflammation in mice (105). Pharmacological inhibition of systemic ET-1 signaling, using either the ETAR-specific inhibitor atrasentan or the non-specific ET receptor inhibitor bosentan, lowered fasting blood glucose and enhanced adiponectin secretion in high fat diet-challenged mice (106). Atrasentan administration also lowered circulating triglycerides in high fat diet-challenged mice (106). The improved metabolic phenotype observed in high fat diet-challenged mice treated with of ET receptor blockers may be in part due to attenuated ET receptor-mediated perturbation in AT function (106). Besides, atrasentan administration also alleviated high fat diet-induced AT inflammation, as evidenced in reduced accumulation of immune cells and gene expression of pro-inflammatory cytokines such as TNFα (106). ETAR-selective inhibition may be a promising pharmacological intervention to combat perturbed metabolism and elevated inflammation in AT in CMS.
Platelet-Derived Growth Factor-CC (PDGF-CC)
Platelet-derived Growth Factor (PDGF)-CC is a protein mediator formed by two PDGF-C monomers (107). PDGF-CC is expressed in various cell types, which share a mesenchymal origin, including the endothelium (107, 108). It is produced and secreted in its full length and digested by extracellular proteases, such as plasmin, to release its active growth factor fragment (109). Endothelial release of PDGF-CC can be upregulated by VEGF (108). The growth factor fragment acts on the transmembrane PDGF receptor-alpha (PDGFRα) to mediate various cellular processes including proliferation (109).
A study using genetically modified mice and β3-selective adrenergic stimulation, using a β3 adrenoceptor agonist, CL-316243, demonstrated that the endothelium responds to adipocyte-secreted VEGF to secrete PDGF-CC, which then induces beiging in neighboring white adipocytes (108). Although increasing PDGF-CC signaling may be explored to develop adipocyte beiging therapeutics, it may also pose a risk of inducing WAT fibrosis. Between the two identified subpopulations of PDGFRα-positive adipocyte progenitors, high fat diet activates PDGFRα-positive adipocyte progenitors with high CD9 expression, which are more proliferative and pro-fibrotic in WAT than their low CD9-expressing counterparts (110). Notably, higher levels of PDGFRα-positive high CD9-expressing adipocyte progenitors were identified in patients with obesity and glucose intolerance or diabetes (110). As PDGFRα can also be activated by PDGF-AA in WAT (110), these findings suggest caution on specificity of adipocyte progenitor subset activation is required when designing therapeutics manipulating PDGFRα signaling to treat obesity and diabetes.
Heparanase
Heparanase is a heterodimeric endoglycosidase responsible for degrading cell surface or extracellular heparan sulfate proteoglycans (HSPGs) and is secreted by various mammalian cell types including endothelial cells (111). Heparanase is first synthesized as a 60 kDa protein, before undergoing lysosomal modification to give an active 50 kDa protein. Upon activation by cytokines, such as TNFα and IL-1beta (IL-1β) or OxLDL, endothelial cells secrete heparanase into the extracellular matrix (112). In contrast, vascular endothelial growth factor (VEGF) inhibits basal endothelial heparanase secretion (112).
Upon release into the extracellular matrix, heparanase breaks down matrix or membrane-bound HSPGs. HSPGs are heparan sulfate chains-containing glycoproteins, which function as structural proteins and cell surface receptors to mediate cellular signaling (113). HSPGs bind to a wide variety of molecules such as chemokines, growth factors and extracellular matrix proteins and enable a regulated release of such proteins upon their degradation by heparanase (111). Of interest to this review, endothelial-released heparanase mediates AT metabolism and inflammation. In terms of metabolism, endothelial cells activated by lysophosphatidylcholine, a product of lipoprotein lipase (LPL)-mediated lipolysis, secrete heparanase to modulate adipocyte secretion of LPL (114). LPL is taken up by endothelial cells and expressed at the luminal surface to hydrolyse circulating triglycerides, hence, the heparanase-HSPG signaling serves as a positive feedback loop to luminal LPL expression (114). In addition, as cell surface HSPG and HSPG-bound LPL mediate effective fatty acid uptake and storage in adipocytes (115, 116), depleting adipocyte HSPG and LPL may impair adipocyte lipid uptake and stimulate de novo fatty acid synthesis.
Regarding inflammation, endothelial cell-released heparanase is pro-inflammatory and pro-atherogenic. High levels of heparanase expression are found in atherosclerotic plagues in ApoE-knockout mice, suggesting heparanse may be involved in inflammation (112). As HSPGs govern immune cell recruitment and activation, and extracellular matrix composition (113, 117), elevation in heparanse-HSPG signaling in AT may drive AT inflammation and fibrosis in CMS.
Heparanase levels are significantly higher in both the plasma and the urine of humans with diabetes compared with healthy individuals (118). Higher heparanase gene expression was also found in the visceral AT of hyperglycaemic type 1 diabetic rats (119). Although the regulatory role in lipid metabolism and inflammation of heparanse-HSPG signaling has been extensively studied, the focus was mostly its effect in atherosclerosis and cancer, and not in CMS. Future work examining the AT-specific effect of the signaling pathway is needed to support the suggestion that endothelial-secreted heparanse may be involved in the progression of AT dysfunction in CMS.
Classic Pro-inflammatory Mediators
In obesity, AT switches to a more pro-inflammatory profile, with elevated secretion of pro-inflammatory mediators such as tumor necrosis factor α (TNFα) and interleukin(IL)-6 (120–122). Alongside adipocytes, other cell types such as macrophages and endothelial cells are suggested as the primary source of pro-inflammatory mediators in the AT in obesity (120). Endothelial cells have been shown to secrete a diverse profile of pro-inflammatory mediators in response to adipocyte-derived signals (123). Twenty four-hour treatment using conditioned media from healthy human adipocytes stimulated sustained secretion of pro-inflammatory cytokines including TNFα and IL-6 from human umbilical endothelial cells (HUVECs) (123). Both TNFα and IL-6 have been extensively studied and reviewed as promoters of an inflammatory response, with local recruitment of immune cells, such as macrophages, which contribute to chronic inflammation, and may in turn lead to insulin resistance in AT (122). In addition to their pro-inflammatory actions, these endothelial-derived cytokines can also act directly on adipocytes to mediate various metabolic functions.
Tumor Necrosis Factor-Alpha (TNFα)
TNFα is a member of the TNF family, known to be primarily secreted by monocytes or macrophages (124). TNFα can also be synthesized and secreted by endothelial cells upon stimulation by pro-inflammatory signals such as a combination of interferon-γ, IL-1β and lipopolysaccharide (125), or IL-1α (126). TNFα signals via binding to TNF receptor (TNFR)-1 or TNFR-2 of the TNFR superfamily, thus activating its downstream signaling cascades involving protein partners such as c-Jun N-terminal kinases (JNK) and mitogen-activated protein kinase (MAPK) (124). In adipocytes, TNFα induces insulin resistance and upregulates lipolysis (127, 128). Prolonged exposure to exogenous TNFα for 24 or 72 h inhibited insulin-stimulated glucose uptake and induced glycerol release in cultured primary human adipocytes (127). On a molecular level, 3-day TNFα treatment inhibited insulin-stimulated insulin receptor (IR) and IRS-1 phosphorylation in murine 3T3-F442A adipocytes (128). Consistent with these findings using cultured adipocytes, neutralizing TNFα activity by application of a soluble TNF receptor-IgG protein improved insulin-stimulated IR and IRS-1 phosphorylation in AT and lowered circulating free fatty acid concentration in genetically obese Zucker rats (129). In addition, high fat diet-challenged TNFα-deficient mice had lower circulating free fatty acids and fasting insulin levels compared to control littermates, suggesting suppressing TNFα activity attenuated unfavorable lipolysis and hyperinsulinemia in diet-induced obesity (5). In line with the finding that TNFα production and secretion is upregulated in AT from human patients with obesity (130), these studies highlight the therapeutic potential of targeting TNFα to combat dyslipidemia and insulin resistance in CMS. Unfortunately, current clinical trials have reported conflicting outcomes. Increased adiposity or body weight gain were reported as side effects in a study treating obesity or autoimmune diseases with anti-TNFα antibodies (131). In contrast, the TNFα inhibitor etanercept was shown to lower fasting glucose in patients with obesity over a 6-month period (132). These conflicting findings highlight the need for an alternative therapeutic target of higher specificity in downstream TNFα signaling in adipocytes, or refinement of treatment regimes. Future studies should also determine the effect of targeting TNFα on inflammation or susceptibility to infection in treated patients, as increased risk of infections has been reported in some patients receiving anti-TNF therapy (133).
Interleukin-6 (IL-6)
Similar to TNFα, IL-6 can be synthesized and secreted by endothelial cells upon stimulation by pro-inflammatory cytokines such as IL-1α, and interestingly, TNFα (134). IL-6 signals via binding to a transmembrane receptor complex consisting of a IL-6 receptor subunit and two signal transducing receptor gp130 subunits (135). The activated receptor complex conducts downstream signaling, mediated by signal transducer and activator of transcription (STAT) proteins or mitogen-activated protein kinase (MAPK) (135). In adipocytes, similar to TNFα, prolonged exposure to IL-6 induces insulin resistance and upregulates lipolysis (136, 137). Prolonged IL-6 stimulation enhanced glucose uptake (138), but induced insulin resistance by inhibiting expression of IRS-1, in cultured murine 3T3-L1 adipocytes (136). IL-6 also stimulated glycerol secretion of subcutaneous and omental AT cultures isolated from human donors with obesity in vitro, suggesting IL-6 stimulates basal lipolysis in adipocytes (137). In agreement with these findings, isolated mesenteric adipocytes from high fat diet-challenged obese adipocyte-specific gp130-knockout mice, possess a lower rate of basal lipolysis and leptin secretion compared to isolated adipocytes from control littermates (139). However, it is of note that these mice also demonstrated impaired glucose tolerance, associated with suppressed adipose leptin secretion and intestinal glucagon-like peptide-1 (GLP-1) release, suggesting that adipose IL-6 signaling is important in mediating glucose metabolism in vivo (140). In humans, circulating IL-6 levels are positively correlated with body mass index and percent body fat (141, 142). We have previously reported a negative correlation between serum IL-6 and endothelial-dependent vasodilatation in humans across a range of BMIs (143). In addition, elevated serum IL-6 levels were found in individuals with impaired glucose tolerance and type 2 diabetes (T2DM) (144, 145). Hence, in obesity, endothelial cells activated by pro-inflammatory cytokine-activated may contribute to the already elevated serum IL-6 level by secreting more IL-6, as part of a positive feedback loop. Suppressing IL-6 signaling is shown to improve hepatic insulin sensitivity in obese mice (139). As IL-6 also plays a key role in mediating skeletal muscle glucose disposal and pancreatic insulin secretion (146), future research into targeting downstream IL-6 signaling with greater tissue specificity should be undertaken to develop anti-diabetic treatments of high efficacy.
Small Molecules
Reactive Oxygen Species (ROS)
Reactive oxygen species (ROS) are highly reactive intermediates of oxygen produced by a range of cellular enzymes, such as NADPH oxidases (NOXs), complexes I and III of the mitochondrial respiratory chain, uncoupled eNOS or xanthine oxidases (XOs) (147). Most of the reactions mediated by these enzymes generate superoxide (), which has a very short half-life and is converted to hydrogen peroxide (H2O2) and peroxynitrite (ONOO−) after reacting with water or NO, respectively. Under normal physiological conditions, intracellular ROS mediate cellular processes such as proliferation and differentiation (148). However, in diseased states, such as insulin resistance where eNOS is uncoupled and generates a large amount of superoxide, which can become stress signals, resulting in increased tissue or vascular inflammation and downregulated adipocyte function (147, 149, 150).
Hydrogen Peroxides (H2O2)
In the endothelium, H2O2 is generated via a number of pathways, including dismutation of spontaneously or catalyzed by superoxide dismutase (SOD) or direct production by XOs and glucose oxidases (151). It has been reported that physiological concentration of H2O2 in human blood is approximately 10 μM (152). H2O2 mediates cellular processes such as cell proliferation and energy metabolism by interacting with different signaling partners including adenosine monophosphate-activated protein kinase (AMPK) (153, 154). Depending on the site of production and the availability of aquaporins to facilitate cross-membrane diffusion, H2O2 can act both intracellularly and intercellularly (154, 155).
Under normal physiological conditions, H2O2 regulates endothelial secretion of vascular endothelial growth factor (VEGF) and eNOS activity (151). Extracellular H2O2 may modulate adipocyte function, H2O2 has been shown to enhance murine 3T3-L1 adipocyte differentiation (156). At high concentrations (400 μM), H2O2 increased conversion of glucose to glycogen by activating glycogen synthase in cultured rat adipocytes (157). H2O2 has also been shown to induce basal lipolysis but inhibit adrenergic or glucagon-stimulated lipolysis in rat adipocytes (158). These studies highlight the potential (albeit complex) role of H2O2 in modulating adipocyte metabolism.
H2O2 is also known to influence vascular tone and inflammation. Depending on the concentration, vascular bed or presence of other vasoactive substances, H2O2 can act as a vasodilator (159, 160) or vasoconstrictor (161, 162). H2O2 also acts as a pro-inflammatory mediator, high concentrations of H2O2 promote leukocyte recruitment by upregulating endothelial expression of leukocyte adhesion molecules (163). H2O2 has also been shown to induce gene expression of pro-inflammatory mediators such as IL-6 and MCP-1 in a dose-dependent manner in cultured 3T3-L1 adipocytes (164). These findings suggests that extracellular H2O2 switches endothelial cells and adipocytes to possess a more pro-inflammatory phenotype.
In patients with obesity, higher levels of H2O2 were determined in visceral AT and were suggested to correlate with insulin resistance (165) as a result of upregulated endothelial release of H2O2. In diet-induced obese mice, endothelial extracellular H2O2 production is upregulated (166), predominantly by NOX4, and is suggested to mediate glucose disposal in BAT (167). We recently reported that endothelial insulin and IGF-1 signaling governs endothelial production of miR-25 which suppresses NOX4 expression (167). Endothelial-specific expression of mutant IGF-1R receptors which form non-functioning hybrid receptors with insulin receptors and IGF-1R improved insulin-stimulated glucose uptake in brown AT and muscle in mice, in a H2O2 –dependent manner (167). Our findings suggest that blunted endothelial NOX4 activity and subsequent blunted endothelial production of H2O2 may contribute to impaired brown adipocyte glucose uptake and development of insulin-resistance in obesity (167).
Despite evidence suggesting some favorable metabolic effects modulated by H2O2, high concentrations of H2O2 are unlikely to be therapeutic. It is important to note that H2O2 can also be generated by NOXs in insulin-stimulated adipocytes or by mitochondrial respiration complexes in stressed adipocytes (168, 169). In vitro evidence has suggested that high intracellular levels of ROS and prolonged hyperinsulinaemia can induce apoptosis in brown adipocytes (170). Accumulation of mitochondrial-generated ROS (including H2O2) is also reported to induce insulin resistance and suppress adiponectin secretion in adipocytes (169). This raises the question of whether H2O2 released from different cellular sources (endothelial cells or adipocytes) or under different circumstances (normal or pathological conditions) may produce different outcomes in mediating adipocyte health and function.
Aiming to counteract H2O2-mediated ill-effects in adipose function, some studies have proposed suppression of H2O2 signaling as a potential therapeutic option in CMS. Treatment with antioxidant NAC (N-acetylcysteine) reversed H2O2-suppressed adiponectin secretion in mature 3T3-L1 adipocytes (164). Treatment with another antioxidant apocynin reduced WAT level of H2O2, enhanced plasma adiponectin levels, reduced glucose and insulin levels and reduced pro-inflammatory TNFα gene expression in obese KKAy mice, suggesting suppression of H2O2 production has favorable metabolic effects and may alleviate WAT inflammation in obesity in vivo (164). Similar therapeutic benefits were observed in apocynin-treated high fat diet-challenged obese C57BL/6 mice (171).
Peroxynitrite (ONOO–)
Obesity, and diabetes-related stress signals such as cytokines and hyperglycaemia, stimulate the production of a large amount of peroxynitrite (ONOO−) as superoxide react with the NO generated by enzymes such as NOXs, uncoupled NO and iNOS (59, 172). ONOO− can react with different molecules such as carbon dioxide to produce reactive nitrogen species or hydroxyl radicals (59). ONOO− and its derivatives either diffuse or are transported via anion channels across the plasma membrane, initiating downstream signaling in neighboring cells (172). ONOO− can react with a wide range of cellular targets, including prostacyclin synthase (173), low-density lipoproteins (174) and DNA (175), making it a very powerful inducer of unfavorable metabolic and pro-inflammatory effects.
In AT, ONOO− induces insulin resistance and cell death. In the submillimolar range, ONOO− increased glucose uptake dose-dependently, whereas at higher concentrations (millimolar range), it reduced viability of cultured 3T3-L1 adipocytes (176). Treatment using higher concentrations of an ONOO− donor (in millimolar range) also inhibited insulin-stimulated glucose uptake by tyrosine nitration and subsequent inhibition of insulin receptor substrate-1 (IRS-1) in cultured 3T3-L1 adipocytes (177). ONOO− can also cause nitration of various proteins, which control energy homeostasis including fatty acid binding protein-4 (FABP-4) in adipocytes (178). As an example, nitrated FABP-4 has reduced binding affinity to lipids (178), which may result in dysregulated lipid release in adipocytes. As FABP-4 plays an important role in modulating lipid release and metabolism in adipocytes via its interaction with hormone-sensitive lipase (179) or PPARγ transcription factors (180), peroxynitrite-mediated nitration of FABP-4 may contribute to the development of dysregulated lipolysis and insulin resistance in adipocytes in obesity.
In addition to inducing insulin resistance, peroxynitrite is known as a pro-inflammatory mediator and is suggested to play a role in driving vascular diseases (59). Of relevance to AT, peroxynitrite may lead to activation of pro-inflammatory signaling cascades in endothelial cells and circulating immune cells, resulting in adipose inflammation. In bovine microvascular endothelial cells, treatment with an ONOO− donor elevated iNOS and NFκB protein expression, but suppressed prostacyclin synthase expression, suggesting an increase in iNOS and NFκB pro-inflammatory signaling but a decrease in anti-inflammatory vasodilator prostacyclin production (181). As NFκB signaling modulates endothelial activation and secretion of pro-inflammatory cytokines (reviewed in 3.4 above), peroxynitrite may contribute to adipose inflammation by enhancing recruitment and activation of immune cells. Moreover, peroxynitrite can activate leukocytes to release IL-8 (182), which attracts neutrophils to sites of inflammation (183). Lastly, peroxynitrite-induced apoptotic adipocytes may also themselves serve as pro-inflammatory signals, and drive tissue inflammation (184).
Despite some evidence from in vitro reports that targeting peroxynitrite may improve AT health and function, studies using systemic intervention seems to suggest otherwise. Global iNOS-knockout mice demonstrated improved glucose tolerance and insulin sensitivity, but the effect was likely limited to skeletal muscle rather than AT (185). More importantly, abrogating ROS using long-term antioxidant supplementation did not seem to lower the risk of developing CMS in humans despite a negative association between serum antioxidant concentration and risk of CMS (186). These findings suggest that perhaps ROS play a smaller role in driving AT dysfunction in CMS and that targeting ROS may not be sufficient to reverse severe metabolic defects and chronic inflammation in humans.
Prostaglandins
Prostaglandins are a family of lipid mediators formed from the conversion of arachidonic acids catalyzed by respective synthases (187). In AT, prostaglandin I2 (PGI2) and E2 (PGE2) are the two most abundant members of the family present (188). Studies have proposed the importance of the presence of both non-fat cells, predominantly endothelial cells, and adipocytes for optimal AT prostaglandin production and secretion (189, 190). Experiments using isolated endothelial cells from human adipose capillaries presented evidences that human AT endothelial cells are capable to secrete both PGI2 and PGE2 (191).
Prostacyclin (PGI2)
Prostacyclin (also called prostaglandin I2 or PGI2) inhibits platelet activation and is also a vasodilator (192). PGI2 is the most dominant prostanoid produced in the endothelium (193). PGI2 is generated by prostacyclin synthase, which uses prostaglandin endoperoxides converted from arachidonic acid by cyclooxygenases (194). The synthesis of PGI2 takes place only upon endothelial cell activation by substances such as thrombin (195), endothelins (196, 197), platelet-derived microparticles (198), angiotensin I (199) or mechanically by shear stress (200). In particular to the AT, endothelium is the main source of PGI2 by utilizing adipocyte-released arachidonic acid (189).
Endothelial-derived PGI2 acts on the prostacyclin receptor (IP) and may influence adipogenesis and adipocyte beiging via both IP-dependent and IP-independent pathways (201). It is reported that IP gene expression is upregulated during adipogenesis, and its activation induces beiging of white adipocytes in culture (201, 202). Treatment using carbaprostacyclin, a PGI2 analog, induced gene transcription of UCP-1 and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) in primary stromal vascular fraction cell cultures isolated from murine white AT, suggesting a role of PGI2 in activating adipocyte precursors to form energy-expending beige adipocytes by upregulating mitochondrial uncoupled respiration and biogenesis (203). Carbaprostacyclin also increased gene expression of beige adipocyte markers in mature human adipocytes (202). Taken together, these studies suggest that endothelial-derived PGI2 contributes to the activation of preadipocytes, giving rise to energy-expending beige adipocytes.
Similar to NO, PGI2 is an anti-inflammatory signaling molecule. Pharmacological activation of IP results in a reduction of cytokine-stimulated expression of cell adhesion molecules and immune cell recruitment in endothelial cells in vitro (204, 205). Treatment with a PGI2 analog iloprost attenuates lymphocyte adhesion and reduces IL-1 stimulated expression of ICAM-1 and E-selectin in HUVECs (204). Treatment using another PGI2 analog beroprost sodium (BPS) reduces VCAM-1 and E-selectin expression in TNFα-stimulated HUVECs (205). It is worth noting that the anti-inflammatory effects of PGI2 were not observed in unstimulated endothelial cells in either study (204, 205), suggesting PGI2 is important in suppressing inflammation only in activated endothelial cells.
In both genetic induced obesity in rats, and high fat diet-induced obese mice, PGI2 synthase activity is impaired (206); subsequent administration of PGI2 was shown to improve metabolic profiles in both of these models (207, 208). Oral treatment with BPS improved glucose tolerance and insulin sensitivity, and attenuated WAT inflammation in high fat diet-challenged obese mice (208). These animal studies suggest increasing PGI2 signaling could lead to beneficial metabolic and anti-inflammatory outcomes.
However, the translational potential of PGI2 application in activating beige AT activity has not been established successfully in human subjects. Twelve-week oral administration of BPS improved insulin sensitivity without altering body weight, BMI, waist circumference or lipid profile in human patients with type 2 diabetes (209), suggesting PGI2 supplementation improves insulin sensitivity without improving AT function. In contrast, another study using 3-year administration of BPS reported no improvement in insulin sensitivity in patients with type 2 diabetes (205). Although IP activation may alleviate inflammation in patients with T2DM (205), it is yet to be verified whether IP activation improves AT function and systemic homeostasis in patients with CMS.
Prostaglandin E2 (PGE2)
Prostaglandin E2 (PGE2) can be produced by prostaglandin E (PDE) synthase-catalyzed derivatization of arachidonic acid in adipocytes (210), preadipocytes (211) and endothelial cells (191). In endothelial cells, PGE2 production can be upregulated by stimuli including histamine (212) and angiotensin I (199). Secreted PGE2 exerts downstream signaling effects via PGE receptors subtype 1–4 (EP1–4) (187). Depending on the subtype of PGE receptors, PGE2 can elicit different metabolic outcomes in recipient cells. Of interest to this review, EP3 and EP4 actions are reviewed below as these two subtypes are better studied.
In terms of metabolism, EP3 suppresses preadipocyte differentiation and lipolysis in adipocytes. EP3 antagonism by L-798106 enhanced adipogenesis in cultured mouse embryonic fibroblasts (213), suggesting EP3 activation suppresses preadipocyte differentiation. In line of agreement, isolated preadipocytes from EP3-deficient mice displayed a higher differentiation potential (213). In addition, EP3-deficient mice possessed higher circulating glycerol level and cultured adipocytes from EP3-deficient mice exhibited higher rate of lipolysis, suggesting EP3 suppresses lipolysis (213).
In contrast, EP4 activation induces lipolysis and insulin resistance. Pharmacological EP4-specific activation by ONO-AE1-329 increased adipose triglyceride lipase-encoding Pnpla2 gene expression and hormone-sensitive lipase phosphorylation and inhibited insulin-stimulated Akt phosphorylation in cultured OP9 adipocytes, suggesting EP4 activation can increase lipolysis and insulin resistance (214). In line of agreement, adipocyte-specific EP4-deficient mice displayed increased body weight, reduced WAT Pnpla2 gene expression and lower circulating free fatty acids under chow diet (214). In addition, it was reported that PGE2 induced leptin secretion in subcutaneous adipose tissue isolated from human donors with obesity (215) and increased beiging potential of murine preadipocytes (216).
Similarly to PGI2, PGE2 exerts anti-inflammatory effects. Pretreatment using PGE2 activated EP4 and suppressed production of pro-inflammatory cytokines such as IL-8, MCP-1, macrophage inflammatory protein-1beta (MIP1β) and interferon gamma-induced protein-10 (IP-10) in lipopolysaccharides (LPS)-activated human macrophages (217). In the same report, MIP1β and MCP-1 production in human macrophages activated by obesity-related stress signal TNFα was also attenuated by PGE2 (212). Similar anti-inflammatory effects were reported in mice studies (218). Pretreatment using PGE2 reduced pro-inflammatory MIP1β and IP-10 production in isolated murine adipocytes as well as control but not EP4-deficient LPS-activated mouse WAT explants (218). Taken together, these reports support the notion that PGE2-EP4 signaling suppresses adipocyte and macrophage inflammatory response upon activation.
In murine models of diet-induced obesity, higher PGE2 and EP4 levels (218) but lower EP3 level (213) were reported in AT. EP4-deficient mice were more susceptible to high fat diet-induced weight gain and adipocyte hypertrophy (214). Higher AT PGE2 level was also reported in humans with obesity (219). Considering the differences in subtype-mediated metabolic outcomes, changes in abundance of receptor subtypes in AT may reflect maladaptive response to overnutrition, as AT may be more sensitive to EP4-mediated lipolysis and insulin resistance and less sensitive to EP3-mediated suppression of lipolysis. Inhibiting EP4-mediated signaling may be a possible therapeutic to treat CMS. However, pharmacological studies activating EP4 seems to suggest the otherwise. Administration of an EP4 agonist, ONO-AE1-329, was reported to elicit beneficial metabolic outcomes and anti-inflammatory effects in db/db mice, as characterized by improved glucose tolerance and insulin sensitivity, and shifted AT macrophages toward anti-inflammatory M2 type (220). Administration of another EP4 agonist, CAY10580, reduced AT IP-10 and MIP-1α levels in high fat diet-challenged mice, suggesting systemic EP4 activation alleviates obesity-associated AT inflammation and such anti-inflammatory effects outweigh potential EP4-mediated metabolic defects (218). Of high therapeutic interest, non-specific activation of PGE2 signaling posed anti-fibrotic and anti-inflammatory effects, induced beiging and suppressed adrenergic-induced lipolysis in isolated human AT explants (219). As the favorable outcomes in this study may be partly contributed by EP1 and EP2 signaling, this finding highlights the need of future studies studying EP1 and EP2 signaling in different cell types in AT to better establish the therapeutic potential of manipulating PGE2 signaling as a treatment goal for CMS.
Peroxisome Proliferator-Activated Receptor Gamma (PPARγ)-Activating Lipids
Upon activation by fatty acids, human AT endothelial cells (hATECs) increase expression of fatty acid transporters including FABP-4 to enhance fatty acid uptake from the circulation (221). hATECs then release fatty acids to adipocytes for storage (221). More importantly, endothelial cells release lipids to activate peroxisome proliferator-activated receptor gamma (PPARγ) signaling in adipocytes (221). Treatment using the lipid fraction of conditioned media from isolated hATECs was able to induce PPARγ activation in primary human adipocytes. PPARγ activation was enhanced further when the hATECs were pre-treated with oleic acid, a fatty acid (221). Human adipocytes are unable to produce PPARγ agonists but are responsive to PPARγ stimuli (221), therefore, these findings demonstrated that the AT endothelial cells are able to respond to circulating fatty acids, and secrete lipids, activating PPARγ signaling in adipocytes. As PPARγ is a key regulator of adipogenesis and adipocyte function, including mitochondrial respiration (222), future studies examining the identity of endothelial-derived PPARγ ligand and elucidating the mechanism by which the activated endothelial cells secrete such ligands may give rise to an alternative drug candidate to combat diseased adiposity in obesity.
Concluding Remarks
Balanced coordination of metabolic processes in the adipose tissue, liver, skeletal muscle and pancreas are crucial to whole-body energy homeostasis. Dysfunction of these highly metabolic organs can lead to the development of the cardiometabolic syndrome. In the past few decades, a considerable amount of interest has been generated in exploiting inter-organ signaling pathways, particularly organ-specific endocrine factors such as adipose-derived adiponectin, and liver-derived fibroblast growth factor 21, to treat metabolic disorders (223–225). However, the localized crosstalk between the heterogeneous endothelium and these metabolic organs is still yet to be fully explored.
AT is crucial in mediating systemic homeostasis. Along with governing energy level by regulating glucose and lipid metabolism, AT secretes adipokines such as adiponectin and leptin which can act on distal organs to modulate insulin sensitivity and food intake. The AT is a sophisticated organ made up of adipocytes, supported by stromal vascular cells composed of endothelial cells, fibroblasts, pericytes, immune cells and adipose progenitors (226). Among different types of stromal vascular cells, endothelial cells serve as the first responder to fluctuations in circulating signaling cues such as hormones or glucose and AT-derived signals such as pro-inflammatory mediators and lipids. Endothelial dysfunction in AT could drive different components of AT dysfunction such as dyslipidaemia, increased fibrosis, chronic inflammation and insulin resistance. It is worth noted that functional heterogeneity exists among different AT depots (227). This review focuses on the role of endothelial-derived mediators in AT depots at classic locations such as subcutaneous and omental AT due to their significance in mediating whole-body energy homeostasis and contribution to the progression of obesity. Recent studies have shed light on the importance of perivascular AT in regulating local energy homeostasis around blood vessels and vital organs such as the heart and the disastrous vascular complications caused by perivascular AT dysfunction (228–230). Future work dissecting the role of endothelial-derived factors in mediating perivascular AT function specifically may provide new insights in our understanding of the paracrine role of endothelium in this depot and its contribution to the vascular components of CMS (231).
Over the past decade, our understanding of the endothelium has evolved, from an inert barrier layer in the vasculature to an active and diverse metabolic organ. Following advances in biological techniques such as multi-omics, it has become possible to separate and identify individual endothelial-derived metabolites, both from different subsets of endothelium, and across species. This deepens our understanding of the role of the endothelium as an active organ, orchestrating the function of neighboring tissue (and of interest to this review, the AT), as well as expanding our knowledge on the consequences of endothelial dysfunction in metabolic and cardiovascular diseases.
Here we have presented an overview of various key endothelial-derived regulatory factors which induce local effects on the metabolism and inflammation of the AT and discussed potential areas of future development on exploiting these pathways to treat cardiometabolic syndrome (Table 1; Figure 1). Among various regulatory factors reviewed here, NO, PGI2, PGE2, PDGF-CC, and the yet-to-be-identified PPARγ-activating lipid, share great therapeutic potential to upregulate energy expenditure by increasing adipocyte beiging, enhancing AT insulin sensitivity, and alleviating undesirable AT inflammation. Our recent report also suggested that endothelial-derived ROS plays a role in enhancing whole-body (including BAT) insulin sensitivity in response to short-term overnutrition (167). However, diseased upregulation of endothelial-derived ET-1, ROS, heparanse and pro-inflammatory mediators can bring detrimental metabolic and inflammatory outcomes by upregulating lipolysis and inducing insulin resistance. It is worth noting that, as some of these regulatory factors (such as NO, PGI2 and ET-1) also play a role in mediating vascular tone and inflammation, the vascular effects of targeting these pathways must also be considered during drug development. For instance, reduced NO bioavailability (232) and ET-1 hyperactivity (233) have been reported in patients with hypertension (233), one of the components of CMS. Exploiting these biologics may bring beneficial therapeutic outcomes to both the metabolic and vascular components of cardiometabolic syndrome.
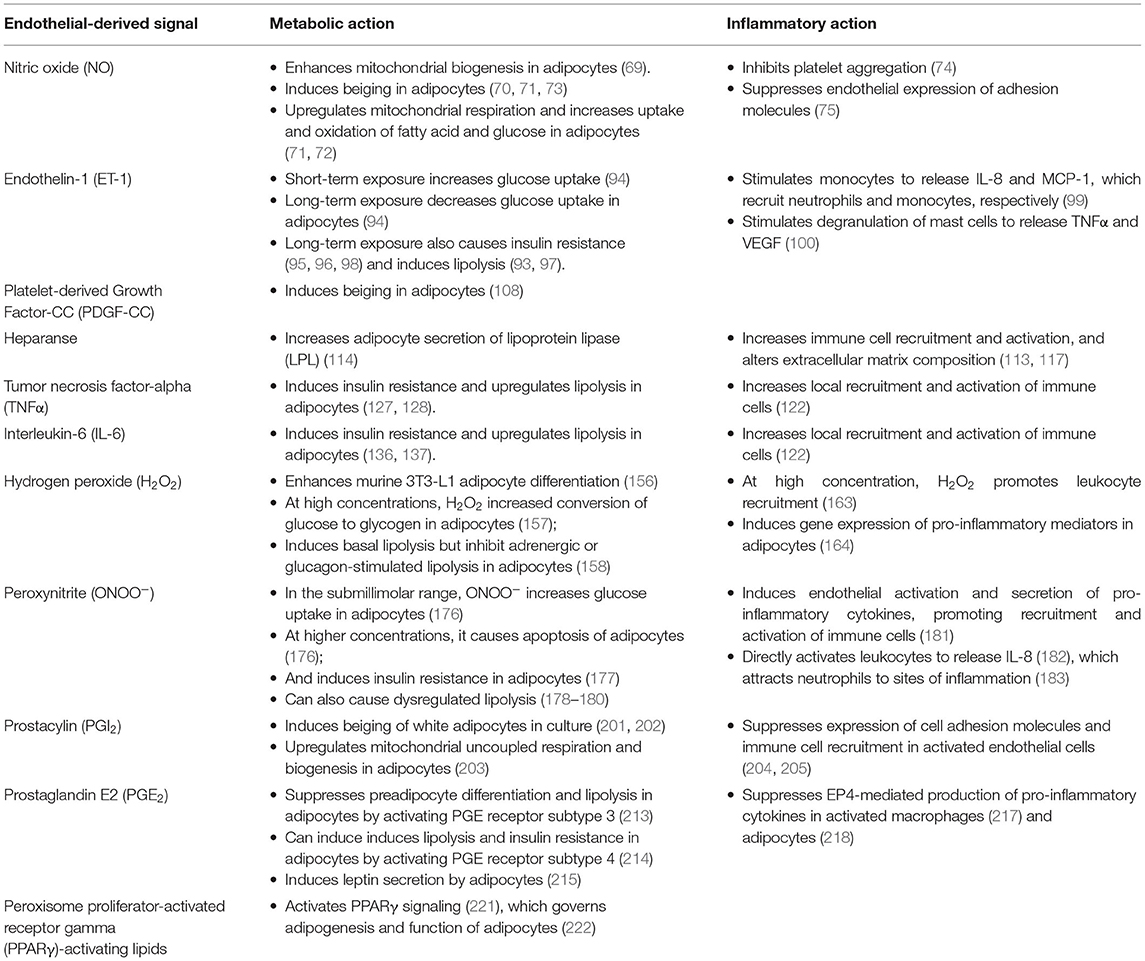
Table 1. Metabolic and inflammatory actions of endothelial-derived signals in AT under normal and pathophysiological conditions in CMS.
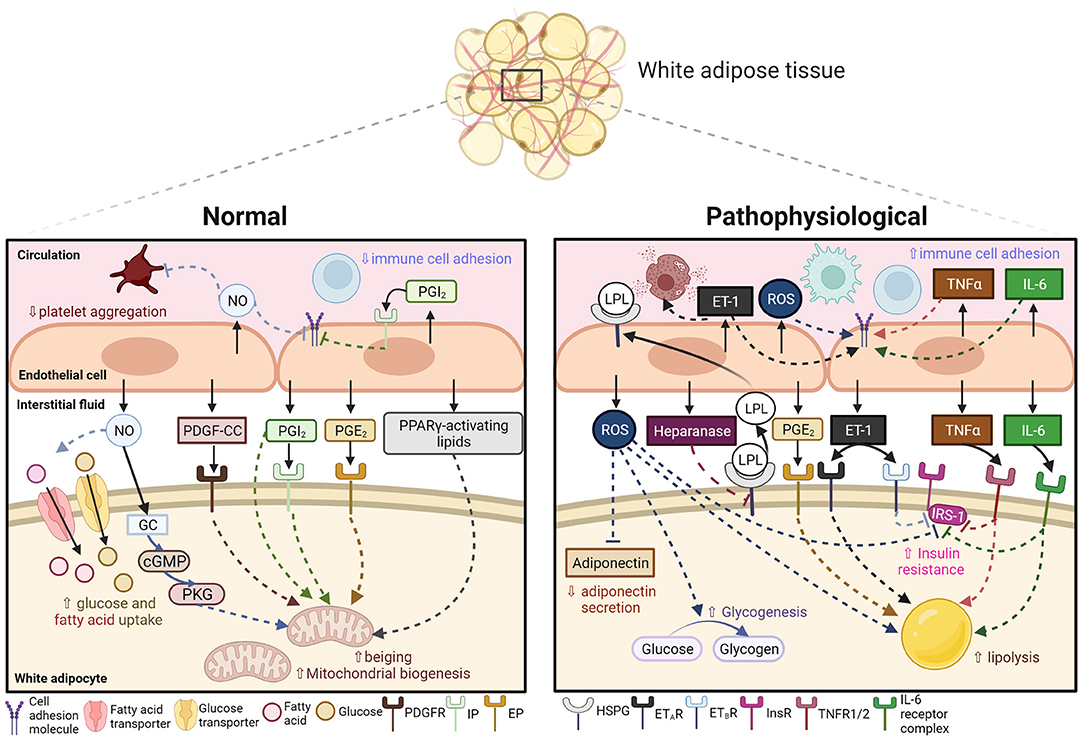
Figure 1. Endothelial-derived signals in mediating AT metabolism and inflammation under normal and pathophysiological conditions in CMS. Under normal conditions, the adipose endothelium releases nitric oxide (NO), platelet-derived growth factor-CC (PDGF-CC), prostacyclin (PGI2), prostaglandin E2 (PGE2) and peroxisome proliferator-activated receptor gamma (PPARγ)-activating lipid in response to changes in microenvironment. These biological signals then act on downstream signaling molecules (soluble guanylate cyclase, GC; cyclic guanine monophosphate, cGMP; protein kinase G, PKG) or receptors (platelet-derived growth factor receptor, PDGFR; prostacyclin receptor, IP; prostaglandin E2 receptor, EP) in neighboring white adipocytes, resulting in increased glucose and fatty acid uptake and upregulated beiging and mitochondrial biogenesis. NO and PGI2 also act on the endothelium and circulating platelets to suppress immune cell adhesion and platelet aggregation. Under pathophysiological conditions, circulating or adipose-derived pro-inflammatory signals stimulate the endothelium to release reactive oxygen species (ROS), heparanase, endothelin-1 (ET-1), tumor necrosis factor-alpha (TNFα) and interleukin-6 (IL-6). These signals act on downstream signaling partners (heparan sulfate proteoglycans, HSPG; endothelin receptors type A and B, ETAR and ETBR; TNF receptor 1/2, TNFR1/2; IL-6 receptor complex) to inhibit adiponectin secretion, increase endothelial expression of lipoprotein lipase (LPL), upregulate glycogenesis and lipolysis, and induce insulin resistance by inhibiting insulin receptor (InsR)-insulin receptor substrate-1 (IRS-1) signaling in white adipocytes. These signals also pose pro-inflammatory effects by increasing recruitment and activation of immune cells. Created with BioRender.com.
In the future, several key areas of research include point (1) exploring further the role of endothelial-derived small molecules and microribonucleic acids (miRNAs) as well as other proteins as local regulators of metabolism in AT and other metabolic organs; (2) investigating the heterogeneity of the endothelium due to differences in environment (tissue), species (rodents or human) and model (cell-lines or primary cells, healthy or diseased models); (3) revisiting current literature on developing therapeutics and evaluating both metabolic and vascular outcomes of such. To conclude, we believe that current understanding on endothelial crosstalk with neighboring cells in metabolic organs will remain a hot topic, and foster the development of therapeutics using biologics to combat metabolic disorders.
Author Contributions
CL performed literature search and wrote this manuscript. NH, KB, and MK reviewed this manuscript. All authors contributed to the article and approved the submitted version.
Funding
CL was funded by a British Heart Foundation studentship (FS/19/59/34896). NH was funded by British Heart Foundation Project grant (PG/18/82/34120). MK holds a British Heart Foundation Chair in Cardiovascular and Diabetes Research (RG/15/7/31521).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Saklayen MG. The global epidemic of the metabolic syndrome. Curr Hypertens Rep. (2018) 20:12. doi: 10.1007/s11906-018-0812-z
2. Alberti KG, Zimmet P, Shaw J. Metabolic syndrome–a new world-wide definition. A consensus statement from the International Diabetes Federation. Diabet Med. (2006) 23:469–80. doi: 10.1111/j.1464-5491.2006.01858.x
3. Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature. (2017) 542:177–85. doi: 10.1038/nature21363
4. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. (1993) 259:87–91. doi: 10.1126/science.7678183
5. Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking tnf-alpha function. Nature. (1997) 389:610–4. doi: 10.1038/39335
6. Pekala P, Kawakami M, Vine W, Lane MD, Cerami A. Studies of insulin resistance in adipocytes induced by macrophage mediator. J Exp Med. (1983) 157:1360–5. doi: 10.1084/jem.157.4.1360
7. Lee YS, Olefsky J. Chronic tissue inflammation and metabolic disease. Genes Dev. (2021) 35:307–28. doi: 10.1101/gad.346312.120
8. Tziomalos K, Athyros VG, Karagiannis A, Mikhailidis DP. Endothelial dysfunction in metabolic syndrome: prevalence, pathogenesis and management. Nutr Metab Cardiovasc Dis. (2010) 20:140–6. doi: 10.1016/j.numecd.2009.08.006
9. Aird WC. Phenotypic heterogeneity of the endothelium: I. structure, function, and mechanisms. Circ Res. (2007) 100:158–73. doi: 10.1161/01.RES.0000255691.76142.4a
10. Rubanyi GM. The role of endothelium in cardiovascular homeostasis and diseases. J Cardiovasc Pharmacol. (1993) 22(Suppl. 4):S1–14. doi: 10.1097/00005344-199322004-00002
11. Hasan SS, Fischer A. The endothelium: an active regulator of lipid and glucose homeostasis. Trends Cell Biol. (2021) 31:37–49. doi: 10.1016/j.tcb.2020.10.003
12. Pi X, Xie L, Patterson C. Emerging roles of vascular endothelium in metabolic homeostasis. Circ Res. (2018) 123:477–94. doi: 10.1161/CIRCRESAHA.118.313237
13. Schwartz BG, Economides C, Mayeda GS, Burstein S, Kloner RA. The endothelial cell in health and disease: its function, dysfunction, measurement and therapy. Int J Impot Res. (2010) 22:77–90. doi: 10.1038/ijir.2009.59
14. Anderson TJ. Assessment and treatment of endothelial dysfunction in humans. J Am Coll Cardiol. (1999) 34:631–8. doi: 10.1016/s0735-109700259-4
15. Imrie H, Abbas A, Kearney M. Insulin resistance, lipotoxicity and endothelial dysfunction. Biochim Biophys Acta. (2010) 1801:320–6. doi: 10.1016/j.bbalip.2009.09.025
16. Galley HF, Webster NR. Physiology of the endothelium. Br J Anaesth. (2004) 93:105–13. doi: 10.1093/bja/aeh163
17. Barrett EJ, Liu Z. The endothelial cell: an “Early Responder” in the development of insulin resistance. Rev Endocr Metab Disord. (2013) 14:21–7. doi: 10.1007/s11154-012-9232-6
18. Zhang J, Friedman MH. Adaptive response of vascular endothelial cells to an acute increase in shear stress magnitude. Am J Physiol Heart Circ Physiol. (2012) 302:H983–91. doi: 10.1152/ajpheart.00168.2011
19. Kim F, Pham M, Maloney E, Rizzo NO, Morton GJ, Wisse BE, et al. Vascular inflammation, insulin resistance, and reduced nitric oxide production precede the onset of peripheral insulin resistance. Arterioscler Thromb Vasc Biol. (2008) 28:1982–8. doi: 10.1161/ATVBAHA.108.169722
20. Kwaifa IK, Bahari H, Yong YK, Noor SM. Endothelial dysfunction in obesity-induced inflammation: molecular mechanisms and clinical implications. Biomolecules. (2020) 10. doi: 10.3390/biom10020291
21. Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. (1980) 288:373–6. doi: 10.1038/288373a0
22. Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. (1987) 327:524–6. doi: 10.1038/327524a0
23. Dignat-George F, Boulanger CM. The many faces of endothelial microparticles. Arterioscler Thromb Vasc Biol. (2011) 31:27–33. doi: 10.1161/ATVBAHA.110.218123
24. Cohen P, Spiegelman BM. Cell biology of fat storage. Mol Biol Cell. (2016) 27:2523–7. doi: 10.1091/mbc.E15-10-0749
25. Pellegrinelli V, Carobbio S, Vidal-Puig A. Adipose tissue plasticity: how fat depots respond differently to pathophysiological cues. Diabetologia. (2016) 59:1075–88. doi: 10.1007/s00125-016-3933-4
26. Seki T, Hosaka K, Fischer C, Lim S, Andersson P, Abe M, et al. Ablation of endothelial vegfr1 improves metabolic dysfunction by inducing adipose tissue browning. J Exp Med. (2018) 215:611–26. doi: 10.1084/jem.20171012
27. Shimizu I, Aprahamian T, Kikuchi R, Shimizu A, Papanicolaou KN, MacLauchlan S, et al. Vascular rarefaction mediates whitening of brown fat in obesity. J Clin Invest. (2014) 124:2099–112. doi: 10.1172/JCI71643
28. Goossens GH, Bizzarri A, Venteclef N, Essers Y, Cleutjens JP, Konings E, et al. Increased adipose tissue oxygen tension in obese compared with lean men is accompanied by insulin resistance, impaired adipose tissue capillarization, and inflammation. Circulation. (2011) 124:67–76. doi: 10.1161/CIRCULATIONAHA.111.027813
29. Pasarica M, Sereda OR, Redman LM, Albarado DC, Hymel DT, Roan LE, et al. Reduced adipose tissue oxygenation in human obesity: evidence for rarefaction, macrophage chemotaxis, and inflammation without an angiogenic response. Diabetes. (2009) 58:718–25. doi: 10.2337/db08-1098
30. Rausch ME, Weisberg S, Vardhana P, Tortoriello DV. Obesity in C57bl/6j mice is characterized by adipose tissue hypoxia and cytotoxic T-cell infiltration. Int J Obes. (2008) 32:451–63. doi: 10.1038/sj.ijo.0803744
31. Ye J, Gao Z, Yin J, He Q. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. Am J Physiol Endocrinol Metab. (2007) 293:E1118–28. doi: 10.1152/ajpendo.00435.2007
32. Fuster JJ, Ouchi N, Gokce N, Walsh K. Obesity-induced changes in adipose tissue microenvironment and their impact on cardiovascular disease. Circ Res. (2016) 118:1786–807. doi: 10.1161/CIRCRESAHA.115.306885
33. Chouchani ET, Kajimura S. Metabolic adaptation and maladaptation in adipose tissue. Nat Metab. (2019) 1:189–200. doi: 10.1038/s42255-018-0021-8
34. Peirce V, Carobbio S, Vidal-Puig A. the different shades of fat. Nature. (2014) 510:76–83. doi: 10.1038/nature13477
35. Lee YH, Mottillo EP, Granneman JG. Adipose tissue plasticity from wat to bat and in between. Biochim Biophys Acta. (2014) 1842:358–69. doi: 10.1016/j.bbadis.2013.05.011
36. Kajimura S, Spiegelman BM, Seale P. Brown and beige fat: physiological roles beyond heat generation. Cell Metab. (2015) 22:546–59. doi: 10.1016/j.cmet.2015.09.007
37. Kajimura S, Saito M. A new era in brown adipose tissue biology: molecular control of brown fat development and energy homeostasis. Annu Rev Physiol. (2014) 76:225–49. doi: 10.1146/annurev-physiol-021113-170252
38. Wu J, Cohen P, Spiegelman BM. Adaptive thermogenesis in adipocytes: is beige the new brown? Genes Dev. (2013) 27:234–50. doi: 10.1101/gad.211649.112
39. Yoneshiro T, Aita S, Matsushita M, Kayahara T, Kameya T, Kawai Y, et al. Recruited brown adipose tissue as an antiobesity agent in humans. J Clin Invest. (2013) 123:3404–8. doi: 10.1172/JCI67803
40. Hanssen MJ, Hoeks J, Brans B, van der Lans AA, Schaart G, van den Driessche JJ, et al. Short-term cold acclimation improves insulin sensitivity in patients with type 2 diabetes mellitus. Nat Med. (2015) 21:863–5. doi: 10.1038/nm.3891
41. Bartelt A, Bruns OT, Reimer R, Hohenberg H, Ittrich H, Peldschus K, et al. Brown adipose tissue activity controls triglyceride clearance. Nat Med. (2011) 17:200–5. doi: 10.1038/nm.2297
42. Stanford KI, Middelbeek RJ, Townsend KL, An D, Nygaard EB, Hitchcox KM, et al. Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J Clin Invest. (2013) 123:215–23. doi: 10.1172/JCI62308
43. Arch JR. Challenges in beta -adrenoceptor agonist drug development. Ther Adv Endocrinol Metab. (2011) 2:59–64. doi: 10.1177/2042018811398517
44. Blondin DP, Nielsen S, Kuipers EN, Severinsen MC, Jensen VH, Miard S, et al. Human brown adipocyte thermogenesis is driven by beta2-ar stimulation. Cell Metab. (2020) 32:287–300 e7. doi: 10.1016/j.cmet.2020.07.005
45. Kusminski CM, Bickel PE, Scherer PE. Targeting adipose tissue in the treatment of obesity-associated diabetes. Nat Rev Drug Discov. (2016) 15:639–60. doi: 10.1038/nrd.2016.75
46. Wernstedt Asterholm I, Tao C, Morley TS, Wang QA, Delgado-Lopez F, Wang ZV, et al. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. (2014) 20:103–18. doi: 10.1016/j.cmet.2014.05.005
47. Zeyda M, Farmer D, Todoric J, Aszmann O, Speiser M, Gyori G, et al. Human adipose tissue macrophages are of an anti-inflammatory phenotype but capable of excessive pro-inflammatory mediator production. Int J Obes. (2007) 31:1420–8. doi: 10.1038/sj.ijo.0803632
48. Permana PA, Menge C, Reaven PD. Macrophage-secreted factors induce adipocyte inflammation and insulin resistance. Biochem Biophys Res Commun. (2006) 341:507–14. doi: 10.1016/j.bbrc.2006.01.012
49. Hausman GJ, Richardson RL. Adipose tissue angiogenesis. J Anim Sci. (2004) 82:925–34. doi: 10.2527/2004.823925x
50. Grandl G, Wolfrum C. Hemostasis, endothelial stress, inflammation, and the metabolic syndrome. Semin Immunopathol. (2018) 40:215–24. doi: 10.1007/s00281-017-0666-5
51. Zeng G, Quon MJ. Insulin-stimulated production of nitric oxide is inhibited by wortmannin. Direct measurement in vascular endothelial cells. J Clin Invest. (1996) 98:894–8. doi: 10.1172/JCI118871
52. Repetto S, Salani B, Maggi D, Cordera R. Insulin and Igf-I phosphorylate Enos in huvecs by a caveolin-1 dependent mechanism. Biochem Biophys Res Commun. (2005) 337:849–52. doi: 10.1016/j.bbrc.2005.09.125
53. Chataigneau T, Feletou M, Huang PL, Fishman MC, Duhault J, Vanhoutte PM. Acetylcholine-induced relaxation in blood vessels from endothelial nitric oxide synthase knockout mice. Br J Pharmacol. (1999) 126:219–26. doi: 10.1038/sj.bjp.0702300
54. Michel T, Li GK, Busconi L. Phosphorylation and subcellular translocation of endothelial nitric oxide synthase. Proc Natl Acad Sci USA. (1993) 90:6252–6. doi: 10.1073/pnas.90.13.6252
55. Chen H, Montagnani M, Funahashi T, Shimomura I, Quon MJ. Adiponectin stimulates production of nitric oxide in vascular endothelial cells. J Biol Chem. (2003) 278:45021–6. doi: 10.1074/jbc.M307878200
56. Vecchione C, Maffei A, Colella S, Aretini A, Poulet R, Frati G, et al. Leptin effect on endothelial nitric oxide is mediated through Akt-endothelial nitric oxide synthase phosphorylation pathway. Diabetes. (2002) 51:168–73. doi: 10.2337/diabetes.51.1.168
57. Sansbury BE, Hill BG. Regulation of obesity and insulin resistance by nitric oxide. Free Radic Biol Med. (2014) 73:383–99. doi: 10.1016/j.freeradbiomed.2014.05.016
58. Kolluru GK, Bir SC, Kevil CG. Endothelial dysfunction and diabetes: effects on angiogenesis, vascular remodeling, and wound healing. Int J Vasc Med. (2012) 2012:918267. doi: 10.1155/2012/918267
59. Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. (2007) 87:315–424. doi: 10.1152/physrev.00029.2006
60. Wang XL, Zhang L, Youker K, Zhang MX, Wang J, LeMaire SA, et al. Free fatty acids inhibit insulin signaling-stimulated endothelial nitric oxide synthase activation through upregulating pten or inhibiting Akt kinase. Diabetes. (2006) 55:2301–10. doi: 10.2337/db05-1574
61. Yoshizumi M, Perrella MA, Burnett JC Jr., Lee ME. Tumor necrosis factor downregulates an endothelial nitric oxide synthase mrna by shortening its half-life. Circ Res. (1993) 73:205–9. doi: 10.1161/01.res.73.1.205
62. Blair A, Shaul PW, Yuhanna IS, Conrad PA, Smart EJ. Oxidized low density lipoprotein displaces endothelial nitric-oxide synthase (Enos) from plasmalemmal caveolae and impairs enos activation. J Biol Chem. (1999) 274:32512–9. doi: 10.1074/jbc.274.45.32512
63. Boger RH, Sydow K, Borlak J, Thum T, Lenzen H, Schubert B, et al. Ldl cholesterol upregulates synthesis of asymmetrical dimethylarginine in human endothelial cells: involvement of S-adenosylmethionine-dependent methyltransferases. Circ Res. (2000) 87:99–105. doi: 10.1161/01.res.87.2.99
64. Sansbury BE, Cummins TD, Tang Y, Hellmann J, Holden CR, Harbeson MA, et al. Overexpression of endothelial nitric oxide synthase prevents diet-induced obesity and regulates adipocyte phenotype. Circ Res. (2012) 111:1176–89. doi: 10.1161/CIRCRESAHA.112.266395
65. Handa P, Tateya S, Rizzo NO, Cheng AM, Morgan-Stevenson V, Han CY, et al. Reduced vascular nitric oxide-cgmp signaling contributes to adipose tissue inflammation during high-fat feeding. Arterioscler Thromb Vasc Biol. (2011) 31:2827–35. doi: 10.1161/ATVBAHA.111.236554
66. Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, et al. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc Natl Acad Sci USA. (1996) 93:13176–81. doi: 10.1073/pnas.93.23.13176
67. Duplain H, Burcelin R, Sartori C, Cook S, Egli M, Lepori M, et al. Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation. (2001) 104:342–5. doi: 10.1161/01.cir.104.3.342
68. Carlstrom M, Larsen FJ, Nystrom T, Hezel M, Borniquel S, Weitzberg E, et al. Dietary inorganic nitrate reverses features of metabolic syndrome in endothelial nitric oxide synthase-deficient mice. Proc Natl Acad Sci USA. (2010) 107:17716–20. doi: 10.1073/pnas.1008872107
69. Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. (2003) 299:896–9. doi: 10.1126/science.1079368
70. Lundberg JO, Gladwin MT, Ahluwalia A, Benjamin N, Bryan NS, Butler A, et al. Nitrate and nitrite in biology, nutrition and therapeutics. Nat Chem Biol. (2009) 5:865–9. doi: 10.1038/nchembio.260
71. Roberts LD, Ashmore T, Kotwica AO, Murfitt SA, Fernandez BO, Feelisch M, et al. Inorganic nitrate promotes the browning of white adipose tissue through the nitrate-nitrite-nitric oxide pathway. Diabetes. (2015) 64:471–84. doi: 10.2337/db14-0496
72. McNally BD, Moran A, Watt NT, Ashmore T, Whitehead A, Murfitt SA, et al. Inorganic nitrate promotes glucose uptake and oxidative catabolism in white adipose tissue through the Xor-catalyzed nitric oxide pathway. Diabetes. (2020) 69:893–901. doi: 10.2337/db19-0892
73. Peleli M, Ferreira DMS, Tarnawski L, McCann Haworth S, Xuechen L, Zhuge Z, et al. Dietary nitrate attenuates high-fat diet-induced obesity via mechanisms involving higher adipocyte respiration and alterations in inflammatory status. Redox Biol. (2020) 28:101387. doi: 10.1016/j.redox.2019.101387
74. Radomski MW, Palmer RM, Moncada S. The anti-aggregating properties of vascular endothelium: interactions between prostacyclin and nitric oxide. Br J Pharmacol. (1987) 92:639–46. doi: 10.1111/j.1476-5381.1987.tb11367.x
75. Lefer DJ, Jones SP, Girod WG, Baines A, Grisham MB, Cockrell AS, et al. Leukocyte-endothelial cell interactions in nitric oxide synthase-deficient mice. Am J Physiol. (1999) 276:H1943–50. doi: 10.1152/ajpheart.1999.276.6.H1943
76. Nishimura S, Manabe I, Nagasaki M, Seo K, Yamashita H, Hosoya Y, et al. In vivo imaging in mice reveals local cell dynamics and inflammation in obese adipose tissue. J Clin Invest. (2008) 118:710–21. doi: 10.1172/JCI33328
77. Brunetta HS, Politis-Barber V, Petrick HL, Dennis K, Kirsh AJ, Barbeau PA, et al. Nitrate attenuates high fat diet-induced glucose intolerance in association with reduced epididymal adipose tissue inflammation and mitochondrial reactive oxygen species emission. J Physiol. (2020) 598:3357–71. doi: 10.1113/JP279455
78. Fu WJ, Haynes TE, Kohli R, Hu J, Shi W, Spencer TE, et al. Dietary L-arginine supplementation reduces fat mass in zucker diabetic fatty rats. J Nutr. (2005) 135:714–21. doi: 10.1093/jn/135.4.714
79. Alizadeh M, Safaeiyan A, Ostadrahimi A, Estakhri R, Daneghian S, Ghaffari A, et al. Effect of L-arginine and selenium added to a hypocaloric diet enriched with legumes on cardiovascular disease risk factors in women with central obesity: a randomized, double-blind, placebo-controlled trial. Ann Nutr Metab. (2012) 60:157–68. doi: 10.1159/000335470
80. Suliburska J, Bogdanski P, Szulinska M, Pupek-Musialik D, Jablecka A. Changes in mineral status are associated with improvements in insulin sensitivity in obese patients following L-arginine supplementation. Eur J Nutr. (2014) 53:387–93. doi: 10.1007/s00394-013-0533-7
81. Fazelian S, Hoseini M, Namazi N, Heshmati J, Sepidar Kish M, Mirfatahi M, et al. Effects of L-arginine supplementation on antioxidant status and body composition in obese patients with pre-diabetes: a randomized controlled clinical trial. Adv Pharm Bull. (2014) 4(Suppl. 1):449–54. doi: 10.5681/apb.2014.066
82. Boon MR, Hanssen MJW, Brans B, Hulsman CJM, Hoeks J, Nahon KJ, et al. Effect of L-arginine on energy metabolism, skeletal muscle and brown adipose tissue in south asian and europid prediabetic men: a randomised double-blinded crossover study. Diabetologia. (2019) 62:112–22. doi: 10.1007/s00125-018-4752-6
83. Mousavi SM, Milajerdi A, Fatahi S, Rahmani J, Zarezadeh M, Ghaedi E, et al. The effect of L-arginine supplementation on obesity-related indices: a systematic review and meta-analysis of randomized clinical trials. Int J Vitam Nutr Res. (2021) 91:164–74. doi: 10.1024/0300-9831/a000523
84. Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. (1988) 332:411–5. doi: 10.1038/332411a0
85. Russell FD, Davenport AP. Secretory pathways in endothelin synthesis. Br J Pharmacol. (1999) 126:391–8. doi: 10.1038/sj.bjp.0702315
86. Dohi Y, Hahn AW, Boulanger CM, Buhler FR, Luscher TF. Endothelin stimulated by angiotensin ii augments contractility of spontaneously hypertensive rat resistance arteries. Hypertension. (1992) 19:131–7. doi: 10.1161/01.hyp.19.2.131
87. Hu RM, Levin ER, Pedram A, Frank HJ. Insulin stimulates production and secretion of endothelin from bovine endothelial cells. Diabetes. (1993) 42:351–8. doi: 10.2337/diab.42.2.351
88. Hu RM, Chuang MY, Prins B, Kashyap ML, Frank HJ, Pedram A, et al. High density lipoproteins stimulate the production and secretion of endothelin-1 from cultured bovine aortic endothelial cells. J Clin Invest. (1994) 93:1056–62. doi: 10.1172/JCI117055
89. Boulanger CM, Tanner FC, Bea ML, Hahn AW, Werner A, Luscher TF. Oxidized low density lipoproteins induce mrna expression and release of endothelin from human and porcine endothelium. Circ Res. (1992) 70:1191–7. doi: 10.1161/01.res.70.6.1191
90. Boulanger C, Luscher TF. Release of endothelin from the porcine aorta. Inhibition by endothelium-derived nitric oxide. J Clin Invest. (1990) 85:587–90. doi: 10.1172/JCI114477
91. Prins BA, Hu RM, Nazario B, Pedram A, Frank HJ, Weber MA, et al. Prostaglandin E2 and prostacyclin inhibit the production and secretion of endothelin from cultured endothelial cells. J Biol Chem. (1994) 269:11938–44.
92. Davenport AP, Hyndman KA, Dhaun N, Southan C, Kohan DE, Pollock JS, et al. Endothelin. Pharmacol Rev. (2016) 68:357–418. doi: 10.1124/pr.115.011833
93. Juan CC, Chang CL, Lai YH, Ho LT. Endothelin-1 induces lipolysis in 3t3-L1 adipocytes. Am J Physiol Endocrinol Metab. (2005) 288:E1146–52. doi: 10.1152/ajpendo.00481.2004
94. Ishibashi K, Imamura T, Sharma PM, Ugi S, Olefsky JM. The acute and chronic stimulatory effects of endothelin-1 on glucose transport are mediated by distinct pathways in 3t3-L1 adipocytes. Endocrinology. (2000) 141:4623–8. doi: 10.1210/endo.141.12.7820
95. Ishibashi KI, Imamura T, Sharma PM, Huang J, Ugi S, Olefsky JM. Chronic endothelin-1 treatment leads to heterologous desensitization of insulin signaling in 3t3-L1 adipocytes. J Clin Invest. (2001) 107:1193–202. doi: 10.1172/JCI11753
96. Chien Y, Lai YH, Kwok CF, Ho LT. Endothelin-1 suppresses long-chain fatty acid uptake and glucose uptake via distinct mechanisms in 3t3-L1 adipocytes. Obesity. (2011) 19:6–12. doi: 10.1038/oby.2010.124
97. Eriksson AK, van Harmelen V, Stenson BM, Astrom G, Wahlen K, Laurencikiene J, et al. Endothelin-1 stimulates human adipocyte lipolysis through the Et a receptor. Int J Obes. (2009) 33:67–74. doi: 10.1038/ijo.2008.212
98. van Harmelen V, Eriksson A, Astrom G, Wahlen K, Naslund E, Karpe F, et al. Vascular peptide endothelin-1 links fat accumulation with alterations of visceral adipocyte lipolysis. Diabetes. (2008) 57:378–86. doi: 10.2337/db07-0893
99. Helset E, Sildnes T, Konopski ZS. Endothelin-1 stimulates monocytes in vitro to release chemotactic activity identified as interleukin-8 and monocyte chemotactic protein-1. Mediators Inflamm. (1994) 3:155–60. doi: 10.1155/S0962935194000207
100. Matsushima H, Yamada N, Matsue H, Shimada S. The effects of endothelin-1 on degranulation, cytokine, and growth factor production by skin-derived mast cells. Eur J Immunol. (2004) 34:1910–9. doi: 10.1002/eji.200424912
101. Hirai S, Ohyane C, Kim YI, Lin S, Goto T, Takahashi N, et al. Involvement of mast cells in adipose tissue fibrosis. Am J Physiol Endocrinol Metab. (2014) 306:E247–55. doi: 10.1152/ajpendo.00056.2013
102. Cardillo C, Campia U, Bryant MB, Panza JA. Increased activity of endogenous endothelin in patients with type II diabetes mellitus. Circulation. (2002) 106:1783–7. doi: 10.1161/01.cir.0000032260.01569.64
103. Piatti PM, Monti LD, Conti M, Baruffaldi L, Galli L, Phan CV, et al. Hypertriglyceridemia and hyperinsulinemia are potent inducers of endothelin-1 release in humans. Diabetes. (1996) 45:316–21. doi: 10.2337/diab.45.3.316
104. Weil BR, Westby CM, Van Guilder GP, Greiner JJ, Stauffer BL, DeSouza CA. Enhanced endothelin-1 system activity with overweight and obesity. Am J Physiol Heart Circ Physiol. (2011) 301:H689–95. doi: 10.1152/ajpheart.00206.2011
105. Jurrissen TJ, Grunewald ZI, Woodford ML, Winn NC, Ball JR, Smith TN, et al. Overproduction of endothelin-1 impairs glucose tolerance but does not promote visceral adipose tissue inflammation or limit metabolic adaptations to exercise. Am J Physiol Endocrinol Metab. (2019) 317:E548–58. doi: 10.1152/ajpendo.00178.2019
106. Rivera-Gonzalez O, Wilson NA, Coats LE, Taylor EB, Speed JS. Endothelin receptor antagonism improves glucose handling, dyslipidemia, and adipose tissue inflammation in obese mice. Clin Sci. (2021) 135:1773–89. doi: 10.1042/CS20210549
107. Fredriksson L, Li H, Eriksson U. The Pdgf family: four gene products form five dimeric isoforms. Cytokine Growth Factor Rev. (2004) 15:197–204. doi: 10.1016/j.cytogfr.2004.03.007
108. Seki T, Hosaka K, Lim S, Fischer C, Honek J, Yang Y, et al. Endothelial Pdgf-Cc regulates angiogenesis-dependent thermogenesis in Beige fat. Nat Commun. (2016) 7:12152. doi: 10.1038/ncomms12152
109. Li X, Ponten A, Aase K, Karlsson L, Abramsson A, Uutela M, et al. Pdgf-C is a new protease-activated ligand for the Pdgf alpha-receptor. Nat Cell Biol. (2000) 2:302–9. doi: 10.1038/35010579
110. Marcelin G, Ferreira A, Liu Y, Atlan M, Aron-Wisnewsky J, Pelloux V, et al. A Pdgfralpha-mediated switch toward Cd9(High) adipocyte progenitors controls obesity-induced adipose tissue fibrosis. Cell Metab. (2017) 25:673–85. doi: 10.1016/j.cmet.2017.01.010
111. Vreys V, David G. Mammalian heparanase: what is the message? J Cell Mol Med. (2007) 11:427–52. doi: 10.1111/j.1582-4934.2007.00039.x
112. Chen G, Wang D, Vikramadithyan R, Yagyu H, Saxena U, Pillarisetti S, et al. Inflammatory cytokines and fatty acids regulate endothelial cell heparanase expression. Biochemistry. (2004) 43:4971–7. doi: 10.1021/bi0356552
113. Sarrazin S, Lamanna WC, Esko JD. Heparan sulfate proteoglycans. Cold Spring Harb Perspect Biol. (2011) 3:a004952. doi: 10.1101/cshperspect.a004952
114. Pillarisetti S, Paka L, Sasaki A, Vanni-Reyes T, Yin B, Parthasarathy N, et al. Endothelial cell heparanase modulation of lipoprotein lipase activity. Evidence that heparan sulfate oligosaccharide is an extracellular chaperone. J Biol Chem. (1997) 272:15753–9. doi: 10.1074/jbc.272.25.15753
115. Wilsie LC, Chanchani S, Navaratna D, Orlando RA. Cell surface heparan sulfate proteoglycans contribute to intracellular lipid accumulation in adipocytes. Lipids Health Dis. (2005) 4:2. doi: 10.1186/1476-511X-4-2
116. Gonzales AM, Orlando RA. Role of adipocyte-derived lipoprotein lipase in adipocyte hypertrophy. Nutr Metab. (2007) 4:22. doi: 10.1186/1743-7075-4-22
117. Pessentheiner AR, Ducasa GM, Gordts P. Proteoglycans in obesity-associated metabolic dysfunction and meta-inflammation. Front Immunol. (2020) 11:769. doi: 10.3389/fimmu.2020.00769
118. Shafat I, Ilan N, Zoabi S, Vlodavsky I, Nakhoul F. Heparanase levels are elevated in the urine and plasma of type 2 diabetes patients and associate with blood glucose levels. PLoS ONE. (2011) 6:e17312. doi: 10.1371/journal.pone.0017312
119. Arfian N, Setyaningsih WAW, Romi MM, Sari DCR. Heparanase upregulation from adipocyte associates with inflammation and endothelial injury in diabetic condition. BMC Proc. (2019) 13(Suppl. 11):17. doi: 10.1186/s12919-019-0181-x
120. Fain JN. Release of inflammatory mediators by human adipose tissue is enhanced in obesity and primarily by the nonfat cells: a review. Mediat Inflamm. (2010) 2010:513948. doi: 10.1155/2010/513948
121. Monteiro R, Azevedo I. Chronic inflammation in obesity and the metabolic syndrome. Mediators Inflamm. (2010) 2010:289645. doi: 10.1155/2010/289645
122. Reilly SM, Saltiel AR. Adapting to obesity with adipose tissue inflammation. Nat Rev Endocrinol. (2017) 13:633–43. doi: 10.1038/nrendo.2017.90
123. Sommer G, Kralisch S, Stangl V, Vietzke A, Kohler U, Stepan H, et al. Secretory products from human adipocytes stimulate proinflammatory cytokine secretion from human endothelial cells. J Cell Biochem. (2009) 106:729–37. doi: 10.1002/jcb.22068
124. MacEwan DJ. Tnf Ligands and receptors–a matter of life and death. Br J Pharmacol. (2002) 135:855–75. doi: 10.1038/sj.bjp.0704549
125. Ranta V, Orpana A, Carpen O, Turpeinen U, Ylikorkala O, Viinikka L. Human vascular endothelial cells produce tumor necrosis factor-alpha in response to proinflammatory cytokine stimulation. Crit Care Med. (1999) 27:2184–7. doi: 10.1097/00003246-199910000-00019
126. Imaizumi T, Itaya H, Fujita K, Kudoh D, Kudoh S, Mori K, et al. Expression of tumor necrosis factor-alpha in cultured human endothelial cells stimulated with lipopolysaccharide or interleukin-1alpha. Arterioscler Thromb Vasc Biol. (2000) 20:410–5. doi: 10.1161/01.atv.20.2.410
127. Hauner H, Petruschke T, Russ M, Rohrig K, Eckel J. Effects of tumour necrosis factor alpha (Tnf alpha) on glucose transport and lipid metabolism of newly-differentiated human fat cells in cell culture. Diabetologia. (1995) 38:764–71. doi: 10.1007/s001250050350
128. Hotamisligil GS, Murray DL, Choy LN, Spiegelman BM. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc Natl Acad Sci USA. (1994) 91:4854–8. doi: 10.1073/pnas.91.11.4854
129. Hotamisligil GS, Budavari A, Murray D, Spiegelman BM. Reduced tyrosine kinase activity of the insulin receptor in obesity-diabetes. Central role of tumor necrosis factor-alpha. J Clin Invest. (1994) 94:1543–9. doi: 10.1172/JCI117495
130. Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest. (1995) 95:2409–15. doi: 10.1172/JCI117936
131. Peluso I, Palmery M. The relationship between body weight and inflammation: lesson from anti-Tnf-alpha antibody therapy. Hum Immunol. (2016) 77:47–53. doi: 10.1016/j.humimm.2015.10.008
132. Stanley TL, Zanni MV, Johnsen S, Rasheed S, Makimura H, Lee H, et al. Tnf-alpha antagonism with etanercept decreases glucose and increases the proportion of high molecular weight adiponectin in obese subjects with features of the metabolic syndrome. J Clin Endocrinol Metab. (2011) 96:E146–50. doi: 10.1210/jc.2010-1170
133. Ali T, Kaitha S, Mahmood S, Ftesi A, Stone J, Bronze MS. Clinical use of anti-Tnf therapy and increased risk of infections. Drug Healthc Patient Saf. (2013) 5:79–99. doi: 10.2147/DHPS.S28801
134. May LT, Torcia G, Cozzolino F, Ray A, Tatter SB, Santhanam U, et al. Interleukin-6 gene expression in human endothelial cells: Rna start sites, multiple Il-6 proteins and inhibition of proliferation. Biochem Biophys Res Commun. (1989) 159:991–8. doi: 10.1016/0006-291x92206-7
135. Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (Il)-6-type cytokine signalling and its regulation. Biochem J. (2003) 374(Pt. 1):1–20. doi: 10.1042/BJ20030407
136. Rotter V, Nagaev I, Smith U. Interleukin-6 (Il-6) induces insulin resistance in 3t3-L1 adipocytes and is, like Il-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects. J Biol Chem. (2003) 278:45777–84. doi: 10.1074/jbc.M301977200
137. Trujillo ME, Sullivan S, Harten I, Schneider SH, Greenberg AS, Fried SK. Interleukin-6 regulates human adipose tissue lipid metabolism and leptin production in vitro. J Clin Endocrinol Metab. (2004) 89:5577–82. doi: 10.1210/jc.2004-0603
138. Stouthard JM, Oude Elferink RP, Sauerwein HP. Interleukin-6 enhances glucose transport in 3t3-L1 adipocytes. Biochem Biophys Res Commun. (1996) 220:241–5. doi: 10.1006/bbrc.1996.0389
139. Wueest S, Item F, Lucchini FC, Challa TD, Muller W, Bluher M, et al. Mesenteric fat lipolysis mediates obesity-associated hepatic steatosis and insulin resistance. Diabetes. (2016) 65:140–8. doi: 10.2337/db15-0941
140. Wueest S, Laesser CI, Boni-Schnetzler M, Item F, Lucchini FC, Borsigova M, et al. Il-6-type cytokine signaling in adipocytes induces intestinal Glp-1 secretion. Diabetes. (2018) 67:36–45. doi: 10.2337/db17-0637
141. Mohamed-Ali V, Goodrick S, Rawesh A, Katz DR, Miles JM, Yudkin JS, et al. Subcutaneous adipose tissue releases interleukin-6, but not tumor necrosis factor-alpha, in vivo. J Clin Endocrinol Metab. (1997) 82:4196–200. doi: 10.1210/jcem.82.12.4450
142. Straub RH, Hense HW, Andus T, Scholmerich J, Riegger GA, Schunkert H. Hormone replacement therapy and interrelation between serum interleukin-6 and body mass index in postmenopausal women: a population-based study. J Clin Endocrinol Metab. (2000) 85:1340–4. doi: 10.1210/jcem.85.3.6355
143. Williams IL, Chowienczyk PJ, Wheatcroft SB, Patel A, Sherwood R, Momin A, et al. Effect of fat distribution on endothelial-dependent and endothelial-independent vasodilatation in healthy humans. Diabetes Obes Metab. (2006) 8:296–301. doi: 10.1111/j.1463-1326.2005.00505.x
144. Muller S, Martin S, Koenig W, Hanifi-Moghaddam P, Rathmann W, Haastert B, et al. Impaired glucose tolerance is associated with increased serum concentrations of interleukin 6 and co-regulated acute-phase proteins but not tnf-alpha or its receptors. Diabetologia. (2002) 45:805–12. doi: 10.1007/s00125-002-0829-2
145. Kado S, Nagase T, Nagata N. Circulating levels of interleukin-6, its soluble receptor and interleukin-6/interleukin-6 receptor complexes in patients with type 2 diabetes mellitus. Acta Diabetol. (1999) 36:67–72. doi: 10.1007/s005920050147
146. Qu D, Liu J, Lau CW, Huang Y. Il-6 in diabetes and cardiovascular complications. Br J Pharmacol. (2014) 171:3595–603. doi: 10.1111/bph.12713
147. Incalza MA, D'Oria R, Natalicchio A, Perrini S, Laviola L, Giorgino F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul Pharmacol. (2018) 100:1–19. doi: 10.1016/j.vph.2017.05.005
148. Cho SH, Lee CH, Ahn Y, Kim H, Kim H, Ahn CY, et al. Redox regulation of Pten and protein tyrosine phosphatases in H O mediated cell signaling. FEBS Lett. (2004) 560:7–13. doi: 10.1016/s0014-579300112-7
149. Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res. (2018) 122:877–902. doi: 10.1161/CIRCRESAHA.117.311401
150. Castro JP, Grune T, Speckmann B. The two faces of reactive Oxygen Species (Ros) in adipocyte function and dysfunction. Biol Chem. (2016) 397:709–24. doi: 10.1515/hsz-2015-0305
151. Cai H. Hydrogen peroxide regulation of endothelial function: origins, mechanisms, and consequences. Cardiovasc Res. (2005) 68:26–36. doi: 10.1016/j.cardiores.2005.06.021
152. Rao PS, Rujikarn N, Luber JM Jr. High-performance liquid chromatographic method for the direct quantitation of oxy radicals in myocardium and blood by means of 1,3-dimethylthiourea and dimethyl sulfoxide. J Chromatogr. (1988) 459:269–73. doi: 10.1016/s0021-967382036-7
153. Zmijewski JW, Banerjee S, Bae H, Friggeri A, Lazarowski ER, Abraham E. Exposure to hydrogen peroxide induces oxidation and activation of Amp-activated protein kinase. J Biol Chem. (2010) 285:33154–64. doi: 10.1074/jbc.M110.143685
154. Breton-Romero R, Lamas S. Hydrogen peroxide signaling in vascular endothelial cells. Redox Biol. (2014) 2:529–34. doi: 10.1016/j.redox.2014.02.005
155. Bienert GP, Chaumont F. Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide. Biochim Biophys Acta. (2014) 1840:1596–604. doi: 10.1016/j.bbagen.2013.09.017
156. Lee H, Lee YJ, Choi H, Ko EH, Kim JW. Reactive oxygen species facilitate adipocyte differentiation by accelerating mitotic clonal expansion. J Biol Chem. (2009) 284:10601–9. doi: 10.1074/jbc.M808742200
157. Lawrence JC Jr., Larner J. Activation of glycogen synthase in rat adipocytes by insulin and glucose involves increased glucose transport and phosphorylation. J Biol Chem. (1978) 253:2104–13.
158. Little SA, de Haen C. Effects of hydrogen peroxide on basal and hormone-stimulated lipolysis in perifused rat fat cells in relation to the mechanism of action of insulin. J Biol Chem. (1980) 255:10888–95.
159. Thomas G, Ramwell P. Induction of vascular relaxation by hydroperoxides. Biochem Biophys Res Commun. (1986) 139:102–8. doi: 10.1016/s0006-291x80085-7
160. Matoba T, Shimokawa H, Nakashima M, Hirakawa Y, Mukai Y, Hirano K, et al. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J Clin Invest. (2000) 106:1521–30. doi: 10.1172/JCI10506
161. Heinle H. Vasoconstriction of carotid artery induced by hydroperoxides. Arch Int Physiol Biochim. (1984) 92:267–71. doi: 10.3109/13813458409071166
162. Suvorava T, Lauer N, Kumpf S, Jacob R, Meyer W, Kojda G. Endogenous vascular hydrogen peroxide regulates arteriolar tension in vivo. Circulation. (2005) 112:2487–95. doi: 10.1161/CIRCULATIONAHA.105.543157
163. Bradley JR, Johnson DR, Pober JS. Endothelial activation by hydrogen peroxide. Selective increases of intercellular adhesion molecule-1 and major histocompatibility complex class I. Am J Pathol. (1993) 142:1598–609.
164. Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. (2004) 114:1752–61. doi: 10.1172/JCI21625
165. Akl MG, Fawzy E, Deif M, Farouk A, Elshorbagy AK. Perturbed adipose tissue hydrogen peroxide metabolism in centrally obese men: association with insulin resistance. PLoS ONE. (2017) 12:e0177268. doi: 10.1371/journal.pone.0177268
166. Noronha BT, Li JM, Wheatcroft SB, Shah AM, Kearney MT. Inducible nitric oxide synthase has divergent effects on vascular and metabolic function in obesity. Diabetes. (2005) 54:1082–9. doi: 10.2337/diabetes.54.4.1082
167. Viswambharan H, Yuldasheva NY, Imrie H, Bridge K, Haywood NJ, Skromna A, et al. Novel paracrine action of endothelium enhances glucose uptake in muscle and fat. Circ Res. (2021) 129:720–34. doi: 10.1161/CIRCRESAHA.121.319517
168. Krieger-Brauer HI, Medda PK, Kather H. Insulin-induced activation of Nadph-dependent H2o2 generation in human adipocyte plasma membranes is mediated by Galphai2. J Biol Chem. (1997) 272:10135–43. doi: 10.1074/jbc.272.15.10135
169. Wang CH, Wang CC, Huang HC, Wei YH. Mitochondrial dysfunction leads to impairment of insulin sensitivity and adiponectin secretion in adipocytes. FEBS J. (2013) 280:1039–50. doi: 10.1111/febs.12096
170. Porras A, Zuluaga S, Valladares A, Alvarez AM, Herrera B, Fabregat I, et al. Long-term treatment with insulin induces apoptosis in brown adipocytes: role of oxidative stress. Endocrinology. (2003) 144:5390–401. doi: 10.1210/en.2003-0622
171. Meng R, Zhu DL, Bi Y, Yang DH, Wang YP. Apocynin improves insulin resistance through suppressing inflammation in high-fat diet-induced obese mice. Mediat Inflamm. (2010) 2010:858735. doi: 10.1155/2010/858735
172. Stadler K. Peroxynitrite-driven mechanisms in diabetes and insulin resistance - the latest advances. Curr Med Chem. (2011) 18:280–90. doi: 10.2174/092986711794088317
173. Bachschmid M, Thurau S, Zou MH, Ullrich V. Endothelial cell activation by endotoxin involves superoxide/no-mediated nitration of prostacyclin synthase and thromboxane receptor stimulation. FASEB J. (2003) 17:914–6. doi: 10.1096/fj.02-0530fje
174. Graham A, Hogg N, Kalyanaraman B, O'Leary V, Darley-Usmar V, Moncada S. Peroxynitrite modification of low-density lipoprotein leads to recognition by the macrophage scavenger receptor. FEBS Lett. (1993) 330:181–5. doi: 10.1016/0014-579380269-z
175. Yu H, Venkatarangan L, Wishnok JS, Tannenbaum SR. Quantitation of four guanine oxidation products from reaction of dna with varying doses of peroxynitrite. Chem Res Toxicol. (2005) 18:1849–57. doi: 10.1021/tx050146h
176. Guzman-Grenfell AM, Garcia-Macedo R, Gonzalez-Martinez MT, Hicks JJ, Medina-Navarro R. Peroxynitrite activates glucose uptake in 3t3-L1 adipocytes through a Pi3-K-dependent mechanism. Front Biosci. (2005) 10:47–53. doi: 10.2741/1505
177. Nomiyama T, Igarashi Y, Taka H, Mineki R, Uchida T, Ogihara T, et al. Reduction of insulin-stimulated glucose uptake by peroxynitrite is concurrent with tyrosine nitration of insulin receptor substrate-1. Biochem Biophys Res Commun. (2004) 320:639–47. doi: 10.1016/j.bbrc.2004.06.019
178. Koeck T, Willard B, Crabb JW, Kinter M, Stuehr DJ, Aulak KS. Glucose-mediated tyrosine nitration in adipocytes: targets and consequences. Free Radic Biol Med. (2009) 46:884–92. doi: 10.1016/j.freeradbiomed.2008.12.010
179. Smith AJ, Thompson BR, Sanders MA, Bernlohr DA. Interaction of the adipocyte fatty acid-binding protein with the hormone-sensitive lipase: regulation by fatty acids and phosphorylation. J Biol Chem. (2007) 282:32424–32. doi: 10.1074/jbc.M703730200
180. Adida A, Spener F. Adipocyte-type fatty acid-binding protein as inter-compartmental shuttle for peroxisome proliferator activated receptor gamma agonists in cultured cell. Biochim Biophys Acta. (2006) 1761:172–81. doi: 10.1016/j.bbalip.2006.02.006
181. Cooke CL, Davidge ST. Peroxynitrite increases inos through Nf-Kappab and decreases prostacyclin synthase in endothelial cells. Am J Physiol Cell Physiol. (2002) 282:C395–402. doi: 10.1152/ajpcell.00295.2001
182. Zouki C, Jozsef L, Ouellet S, Paquette Y, Filep JG. Peroxynitrite mediates cytokine-induced Il-8 gene expression and production by human leukocytes. J Leukoc Biol. (2001) 69:815–24. doi: 10.1189/jlb.69.5.815
183. Bickel M. The role of interleukin-8 in inflammation and mechanisms of regulation. J Periodontol. (1993) 64:456–60.
184. Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res. (2005) 46:2347–55. doi: 10.1194/jlr.M500294-JLR200
185. Perreault M, Marette A. Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nat Med. (2001) 7:1138–43. doi: 10.1038/nm1001-1138
186. Czernichow S, Vergnaud AC, Galan P, Arnaud J, Favier A, Faure H, et al. Effects of long-term antioxidant supplementation and association of serum antioxidant concentrations with risk of metabolic syndrome in adults. Am J Clin Nutr. (2009) 90:329–35. doi: 10.3945/ajcn.2009.27635
187. Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. (2011) 31:986–1000. doi: 10.1161/ATVBAHA.110.207449
188. Richelsen B. Release and effects of prostaglandins in adipose tissue. Prostaglandins Leukot Essent Fatty Acids. (1992) 47:171–82. doi: 10.1016/0952-327890235-b
189. Parker J, Lane J, Axelrod L. Cooperation of adipocytes and endothelial cells required for catecholamine stimulation of Pgi2 production by rat adipose tissue. Diabetes. (1989) 38:1123–32. doi: 10.2337/diab.38.9.1123
190. Chatzipanteli K, Rudolph S, Axelrod L. Coordinate control of lipolysis by prostaglandin E2 and prostacyclin in rat adipose tissue. Diabetes. (1992) 41:927–35. doi: 10.2337/diab.41.8.927
191. Taylor L, Foxall T, Auger K, Heinsohn C, Polgar P. Comparison of prostaglandin synthesis by endothelial cells from blood vessels originating in the rat, baboon, calf and human. Atherosclerosis. (1987) 65:227–36. doi: 10.1016/0021-915090038-4
192. Vane J, Corin RE. Prostacyclin: a vascular mediator. Eur J Vasc Endovasc Surg. (2003) 26:571–8. doi: 10.1016/s1078-588400385-x
193. Weksler BB, Marcus AJ, Jaffe EA. Synthesis of prostaglandin I2 (Prostacyclin) by cultured human and bovine endothelial cells. Proc Natl Acad Sci USA. (1977) 74:3922–6. doi: 10.1073/pnas.74.9.3922
194. Moncada S, Gryglewski R, Bunting S, Vane JR. An enzyme isolated from arteries transforms prostaglandin endoperoxides to an unstable substance that inhibits platelet aggregation. Nature. (1976) 263:663–5. doi: 10.1038/263663a0
195. Weksler BB, Ley CW, Jaffe EA. Stimulation of endothelial cell prostacyclin production by thrombin, trypsin, and the ionophore a 23187. J Clin Invest. (1978) 62:923–30. doi: 10.1172/JCI109220
196. de Nucci G, Thomas R, D'Orleans-Juste P, Antunes E, Walder C, Warner TD, et al. Pressor effects of circulating endothelin are limited by its removal in the pulmonary circulation and by the release of prostacyclin and endothelium-derived relaxing factor. Proc Natl Acad Sci USA. (1988) 85:9797–800. doi: 10.1073/pnas.85.24.9797
197. Emori T, Hirata Y, Marumo F. Endothelin-3 stimulates prostacyclin production in cultured bovine endothelial cells. J Cardiovasc Pharmacol. (1991) 17(Suppl. 7):S140–2. doi: 10.1097/00005344-199100177-00038
198. Barry OP, Pratico D, Lawson JA, FitzGerald GA. Transcellular activation of platelets and endothelial cells by bioactive lipids in platelet microparticles. J Clin Invest. (1997) 99:2118–27. doi: 10.1172/JCI119385
199. Jaiswal N, Diz DI, Chappell MC, Khosla MC, Ferrario CM. Stimulation of endothelial cell prostaglandin production by angiotensin peptides. Characterization of receptors. Hypertension. (1992) 19:II49–55. doi: 10.1161/01.hyp.19.2_suppl.ii49
200. Frangos JA, Eskin SG, McIntire LV, Ives CL. Flow effects on prostacyclin production by cultured human endothelial cells. Science. (1985) 227:1477–9. doi: 10.1126/science.3883488
201. Rahman MS. Prostacyclin: a major prostaglandin in the regulation of adipose tissue development. J Cell Physiol. (2019) 234:3254–62. doi: 10.1002/jcp.26932
202. Ghandour RA, Giroud M, Vegiopoulos A, Herzig S, Ailhaud G, Amri EZ, et al. Ip-receptor and Ppars trigger the conversion of human white to brite adipocyte induced by carbaprostacyclin. Biochim Biophys Acta. (2016) 1861:285–93. doi: 10.1016/j.bbalip.2016.01.007
203. Vegiopoulos A, Muller-Decker K, Strzoda D, Schmitt I, Chichelnitskiy E, Ostertag A, et al. Cyclooxygenase-2 controls energy homeostasis in mice by de novo recruitment of brown adipocytes. Science. (2010) 328:1158–61. doi: 10.1126/science.1186034
204. Della Bella S, Molteni M, Mocellin C, Fumagalli S, Bonara P, Scorza R. Novel mode of action of iloprost: in vitro down-regulation of endothelial cell adhesion molecules. Prostaglandins Other Lipid Mediat. (2001) 65:73–83. doi: 10.1016/s0090-698000131-9
205. Goya K, Otsuki M, Xu X, Kasayama S. Effects of the prostaglandin I2 analogue, beraprost sodium, on vascular cell adhesion molecule-1 expression in human vascular endothelial cells and circulating vascular cell adhesion molecule-1 level in patients with type 2 diabetes mellitus. Metabolism. (2003) 52:192–8. doi: 10.1053/meta.2003.50025
206. Du X, Edelstein D, Obici S, Higham N, Zou MH, Brownlee M. Insulin resistance reduces arterial prostacyclin synthase and enos activities by increasing endothelial fatty acid oxidation. J Clin Invest. (2006) 116:1071–80. doi: 10.1172/JCI23354
207. Sato N, Kaneko M, Tamura M, Kurumatani H. The prostacyclin analog beraprost sodium ameliorates characteristics of metabolic syndrome in obese zucker (fatty) rats. Diabetes. (2010) 59:1092–100. doi: 10.2337/db09-1432
208. Inoue E, Ichiki T, Takeda K, Matsuura H, Hashimoto T, Ikeda J, et al. Beraprost sodium, a stable prostacyclin analogue, improves insulin resistance in high-fat diet-induced obese mice. J Endocrinol. (2012) 213:285–91. doi: 10.1530/JOE-12-0014
209. Katsuyama H, Kubota N, Kubota T, Haraguchi M, Obata A, Takamoto I, et al. Effects of beraprost sodium, an oral prostacyclin analog, on insulin resistance in patients with type 2 diabetes. Diabetol Int. (2015) 6:39–45. doi: 10.1007/s13340-014-0169-8
210. Richelsen B, Borglum JD, Sorensen SS. Biosynthetic capacity and regulatory aspects of prostaglandin e2 formation in adipocytes. Mol Cell Endocrinol. (1992) 85:73–81. doi: 10.1016/0303-720790126-q
211. Michaud A, Lacroix-Pepin N, Pelletier M, Daris M, Biertho L, Fortier MA, et al. Expression of genes related to prostaglandin synthesis or signaling in human subcutaneous and omental adipose tissue: depot differences and modulation by adipogenesis. Mediat Inflamm. (2014) 2014:451620. doi: 10.1155/2014/451620
212. Alhenc-Gelas F, Tsai SJ, Callahan KS, Campbell WB, Johnson AR. Stimulation of prostaglandin formation by vasoactive mediators in cultured human endothelial cells. Prostaglandins. (1982) 24:723–42. doi: 10.1016/0090-698090040-5
213. Xu H, Fu JL, Miao YF, Wang CJ, Han QF, Li S, et al. Prostaglandin E2 receptor Ep3 regulates both adipogenesis and lipolysis in mouse white adipose tissue. J Mol Cell Biol. (2016) 8:518–29. doi: 10.1093/jmcb/mjw035
214. Inazumi T, Yamada K, Shirata N, Sato H, Taketomi Y, Morita K, et al. Prostaglandin E2-Ep4 axis promotes lipolysis and fibrosis in adipose tissue leading to ectopic fat deposition and insulin resistance. Cell Rep. (2020) 33:108265. doi: 10.1016/j.celrep.2020.108265
215. Fain JN, Leffler CW, Cowan GS Jr., Buffington C, Pouncey L, et al. Stimulation of leptin release by arachidonic acid and prostaglandin E in adipose tissue from obese humans. Metabolism. (2001) 50:921–8. doi: 10.1053/meta.2001.24927
216. Garcia-Alonso V, Lopez-Vicario C, Titos E, Moran-Salvador E, Gonzalez-Periz A, Rius B, et al. Coordinate functional regulation between microsomal prostaglandin E synthase-1 (Mpges-1) and peroxisome proliferator-activated receptor gamma (Ppargamma) in the conversion of white-to-brown adipocytes. J Biol Chem. (2013) 288:28230–42. doi: 10.1074/jbc.M113.468603
217. Takayama K, Garcia-Cardena G, Sukhova GK, Comander J, Gimbrone MA Jr., et al. Prostaglandin E2 suppresses chemokine production in human macrophages through the Ep4 receptor. J Biol Chem. (2002) 277:44147–54. doi: 10.1074/jbc.M204810200
218. Tang EH, Cai Y, Wong CK, Rocha VZ, Sukhova GK, Shimizu K, et al. Activation of prostaglandin E2-Ep4 signaling reduces chemokine production in adipose tissue. J Lipid Res. (2015) 56:358–68. doi: 10.1194/jlr.M054817
219. Garcia-Alonso V, Titos E, Alcaraz-Quiles J, Rius B, Lopategi A, Lopez-Vicario C, et al. Prostaglandin E2 exerts multiple regulatory actions on human obese adipose tissue remodeling, inflammation, adaptive thermogenesis and lipolysis. PLoS ONE. (2016) 11:e0153751. doi: 10.1371/journal.pone.0153751
220. Yasui M, Tamura Y, Minami M, Higuchi S, Fujikawa R, Ikedo T, et al. The prostaglandin E2 receptor Ep4 regulates obesity-related inflammation and insulin sensitivity. PLoS ONE. (2015) 10:e0136304. doi: 10.1371/journal.pone.0136304
221. Gogg S, Nerstedt A, Boren J, Smith U. Human adipose tissue microvascular endothelial cells secrete ppargamma ligands and regulate adipose tissue lipid uptake. JCI Insight. (2019) 4:e125914. doi: 10.1172/jci.insight.125914
222. Christodoulides C, Vidal-Puig A. Ppars and adipocyte function. Mol Cell Endocrinol. (2010) 318:61–8. doi: 10.1016/j.mce.2009.09.014
223. Yamada T, Oka Y, Katagiri H. Inter-organ metabolic communication involved in energy homeostasis: potential therapeutic targets for obesity and metabolic syndrome. Pharmacol Ther. (2008) 117:188–98. doi: 10.1016/j.pharmthera.2007.09.006
224. Castillo-Armengol J, Fajas L, Lopez-Mejia IC. Inter-organ communication: a gatekeeper for metabolic health. EMBO Rep. (2019) 20:e47903. doi: 10.15252/embr.201947903
225. Priest C, Tontonoz P. Inter-organ cross-talk in metabolic syndrome. Nat Metab. (2019) 1:1177–88. doi: 10.1038/s42255-019-0145-5
226. Ramakrishnan VM, Boyd NL. The adipose stromal vascular fraction as a complex cellular source for tissue engineering applications. Tissue Eng Part B Rev. (2018) 24:289–99. doi: 10.1089/ten.TEB.2017.0061
227. Lee MJ, Wu Y, Fried SK. Adipose tissue heterogeneity: implication of depot differences in adipose tissue for obesity complications. Mol Aspects Med. (2013) 34:1–11. doi: 10.1016/j.mam.2012.10.001
228. Iacobellis G, Ribaudo MC, Assael F, Vecci E, Tiberti C, Zappaterreno A, et al. Echocardiographic epicardial adipose tissue is related to anthropometric and clinical parameters of metabolic syndrome: a new indicator of cardiovascular risk. J Clin Endocrinol Metab. (2003) 88:5163–8. doi: 10.1210/jc.2003-030698
229. Britton KA, Fox CS. Perivascular adipose tissue and vascular disease. Clin Lipidol. (2011) 6:79–91. doi: 10.2217/clp.10.89
230. Akoumianakis I, Tarun A, Antoniades C. Perivascular adipose tissue as a regulator of vascular disease pathogenesis: identifying novel therapeutic targets. Br J Pharmacol. (2017) 174:3411–24. doi: 10.1111/bph.13666
231. Nava E, Llorens S. The paracrine control of vascular motion. A historical perspective. Pharmacol Res. (2016) 113(Pt. A):125–45. doi: 10.1016/j.phrs.2016.08.003
232. Hermann M, Flammer A, Luscher TF. Nitric oxide in hypertension. J Clin Hypertens. (2006) 8(Suppl. 4):17–29. doi: 10.1111/j.1524-6175.2006.06032.x
Keywords: endothelium, adipose tissue, paracrine signaling, cardiometabolic syndrome, obesity, metabolism, endothelial dysfunction, adipose tissue dysfunction
Citation: Luk C, Haywood NJ, Bridge KI and Kearney MT (2022) Paracrine Role of the Endothelium in Metabolic Homeostasis in Health and Nutrient Excess. Front. Cardiovasc. Med. 9:882923. doi: 10.3389/fcvm.2022.882923
Received: 24 February 2022; Accepted: 04 April 2022;
Published: 26 April 2022.
Edited by:
Fawaz Alzaid, Sorbonne Universités, FranceReviewed by:
Eduardo Nava, University of Castilla-La Mancha, SpainManfredi Tesauro, University of Rome Tor Vergata, Italy
Copyright © 2022 Luk, Haywood, Bridge and Kearney. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark T. Kearney, m.t.kearney@leeds.ac.uk